 Ji-Yue Wen1†
Ji-Yue Wen1† Mei Wang
Mei Wang Zhi-Wu Chen
Zhi-Wu Chen- 1Department of Pharmacology, Anhui Medical University, Hefei, China
- 2Department of Pharmacy, Children's Hospital of Soochow University, Suzhou, China
This study was undertaken to demonstrate the vascular protection of exogenous and endogenous hydrogen sulfide (H2S) on cerebral ischemia/reperfusion (I/R) injury. The effect of H2S on cerebrovascular dysfunction in middle cerebral artery (MCA) and neuronal damage were measured after cerebral I/R induced by transient middle cerebral artery occlusion (MCAO) in cystathionine c-lyase (CSE) knockdown and wild-type rats. The effect of sodium hydrosulfide (NaHS, donor of exogenous H2S), L-cysteine (L-Cys, substrate of endogenous H2S), and endothelium cells on the responses of isolated MCA derived from non-ischemic rats was also evaluated to assess the underlying mechanism of H2S-mediate cerebral vasodilation. The results revealed that the contraction and dilation of MCA profoundly decreased after cerebral I/R. The vascular dysfunction became more grievous in CSE knockdown rats than in wild-type rats. Interestingly, this vascular dysfunction was significantly alleviated by NaHS supplementation. Moreover, both NaHS and L-cysteine could induce remarkable relaxation in the isolated MCA, which was eliminated by co-application of potassium channel blockers ChTx and Apamin, or endothelial removal. By contrast, adding endothelium cells cultured in vitro together with ACh into the luminal perfusate could mimic non-NO and non-PGI2 relaxation in endothelium-denuded MCA, once CSE was knocked down from endothelium cells, and its effect on vasorelaxation was abolished. Furthermore, the indexes of neuronal injury were measured after cerebral I/R to confirm the neuroprotection of H2S, and we found that the neurological scores, cerebral infarction volume, brain water content, malondialdehyde content, and serum lactate dehydrogenase activity (a marker of cellular membrane integrity) were significantly higher in CSE knockdown rats than in normal control rats. It is not surprising that NaHS could alleviate the cerebral injury. These findings revealed that H2S has a protective effect on cerebral I/R injury via its upregulation of the endothelium-dependent contraction and dilation function of cerebral vessels, which may be related to activating potassium channel.
Introduction
Ischemic stroke is one of the most common cerebrovascular diseases with high mortality and disability rate. Previous studies have shown that ischemia could reduce the cerebral vascular response and change the tension of the vessels, which is the leading cause of disruption of the cerebral blood flow around the ischemic area, and the following hypotension and hypercapnia induced by ischemia could induce vascular dysfunction and finally neuronal injury (1, 2). Autoregulation of cerebral blood vessels is of great importance to protect the neuron against ischemia injury during the hypercapnia and hypotension condition (3, 4). Therefore, the effective treatment for ischemic stroke depends on a functional and patent vasculature, and hence vascular protection is regarded as an important therapeutic approach to reduce stroke damage (5).
Hydrogen sulfide (H2S) is regarded as the third endogenous gasotransmitter (6), following carbon monoxide (CO) and nitric oxide (NO). Accumulated evidence indicates that H2S plays a much more active and important role against ischemia/reperfusion (I/R) injury, such as kidney I/R injury (7), myocardial I/R injury (8), and cerebral I/R injury (9). Endogenous H2S is mainly produced from L-cysteine (L-Cys) in intracytoplasm by cystathionine r-lyase (CSE), cystathionine β-synthase (CBS), and β-mercaptopyruvic acid in mitochondria by 3-mercaptopyruvate sulfurtransferase (3-MST) (10). In the vasculature, the endogenous H2S is mainly produced from L-cysteine by CSE in endothelium (11).
H2S plays a number of roles in the central nervous system (CNS) under pathological and physiological states such as anti-inflammation, cytoprotection, antiapoptosis, and antioxidation (12–14). In our previous studies, we found that H2S mediated the hyperpolarization and dilation of rat cerebral arteries including the MCA and the basilar artery (BA) (15, 16). However, the effect of H2S on the cerebrovascular dysfunction after cerebral I/R is still unclear. In addition, we previously also found that intravenous injection with CSE-siRNA and atelocollagen in rats could remarkably knock down the CSE mRNA and protein expression in vivo in cerebral vessels and reduce the production of H2S. Moreover, we have revealed that NaHS could augment the KCa current in CBA vascular smooth muscle cells (17). Therefore, we tested the hypothesis in this study, whether H2S could attenuate the cerebrovascular dysfunction and the neuronal damage that follows cerebral I/R. Likewise, we followed the same CSE-siRNA- transfection approach to knockdown the CSE expression and reduce the H2S production for investigating the effect of endogenous H2S on cerebrovascular dysfunction and neuronal damage. In addition, we also sought to explore the role of exogenous H2S on cerebral I/R injury and further investigate the underlying mechanism of vascular protection of H2S.
Materials and Methods
Reagents
CSE-siRNA and negative siRNA were purchased from GenePharma (Shanghai, China), and atelocollagen was purchased from KOKEN (Tokyo, Japan); CSE antibody was purchased from Santa Cruz (Delaware Ave, USA); NaHS, Acetylcholine(ACh), bradykinin, 9, 11-dideoxy-11α, 9α-epoxy-methanoprostaglandin F2α (U46619), and Vinpocetine were purchased from Sigma Chemicals (St. Louis, USA); lactate dehydrogenase (LDH) and malondialdehyde (MDA) assay kits were purchased from Nanjing Jiancheng Biological Co (Nanjing, China). ChTx, Apamin, L-Cys, L-NG-nitroarginine methyl ester (L-NAME), and indomethacin (Indo) were purchased from sigma Chemicals (St. Louis, USA); Krebs solution (comprising the following (mM): NaCl 118, KCl 3.4, CaCl2 2.5, KH2PO4 1.2, MgSO4 1.2, NaHCO3 25, and glucose 11.1) was aerated with a mixture of 95% O2 and 5% CO2 and oxygenated during the incubation period.
Experimental Animals
Adult male Sprague-Dawley (SD) rats, weighing between 250 and 300 g, were obtained from the Experimental Animal Center of Anhui Medical University. The animals were allowed free access to water and rodent chow. All experimental procedures were approved by the Ethics Review Committee of Anhui Medical University, which comply with the Guide for the Care and Use of laboratory Animals published by the US National Institutes of Health (NIH publication no. 85-23, revised 2011).
Cell Cultures
Human umbilical vein endothelial cells, EAhy926, were purchased from the Cell Bank, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, and were cultured with high glucose Dulbecco's Modified Eagle Medium containing 10% heat-inactivated fetal bovine serum (Gibco), and were transfected by siRNA to knock down the expression of CSE according to the previous research (18).
CSE-siRNA Transfection and Cerebral I/R Injury Model in Rats
As described in our previous study (17), the CSE was knocked down with siRNA-transfection technique. The decrease of CSE and its mRNA expression in MCA was used as the indicator of CSE knockdown, measured by western blot and real-time PCR analysis. At 48 h after siRNA-transfection, the cerebral I/R injury of rats was induced by MCAO under chloral hydrate anesthesia (350 mg/kg, ip) (19). Briefly, a 4-0 nylon monofilament suture (total length: 30 mm; diameter: 0.23 mm) was dipped in melted hard wax at the head end, slightly inserted into the right common carotid artery, and pushed ~18–22 mm from the carotid bifurcation to the internal carotid; blood flow of MCA was then blocked at the origin. After 2 h of ischemia, the suture was carefully withdrawn for reperfusion for 24 h. Rats of the non-CSE-siRNA transfected experiment were grouped as: (1) Sham (n = 10); (2) MCAO (n = 10); (3) MCAO+1 × 10−5 mol/kg NaHS (n = 10); (4) MCAO+1 × 10−6 mol/kg NaHS (n = 10); (5) MCAO+1 × 10−7 mol/kg NaHS (n = 10). Sham group animals were also subjected to the above procedures, except for suture insertion. Rats of CSE-siRNA transfected experiment were grouped as: (1) Sham (n = 10); (2) Control (n = 10); (3) CSE-siRNA (n = 10); (4) CSE-siRNA+NaHS (n = 10). Sham group animals were also subjected to the above procedures, except for suture insertion. NaHS was injected into the tail vein of rats after ischemia, while the sham and control rats were injected with saline.
Cerebral Vessel Experiment
As described previously (17), the brains of MCAO or non-ischemic rats were rapidly removed after sacrifice under anesthesia and placed in precooled Krebs solution. MCA was carefully isolated immediately and cut into serial segments of 3 mm in length. Subsequently, both ends of the vessel segment were cannulated with glass micropipettes, secured with a nylon monofilament suture and then placed in a perfusion chamber. Thereafter, the segments were equilibrated with 37°C Krebs solutions and continuously aerated with a gas mixture of 95% O2 and 5% CO2 and then pressurized to 85 mmHg. The luminal flow was then adjusted to 150 μl/min. After 60 min of equilibrium, 1 × 10−7 mol/l U46619 or 30 mmol/L KCl was added to the luminal perfusate until a stable contraction was obtained. The diameter of the artery of non-ischemic rats was continually measured utilizing E-rule software, and MCA tension of MCAO rats was measured by myograph (17). The percentage of maximum diameter (% Dmax) was calculated and used to evaluate the vascular dilation of non-ischemic rats using the following formula: Dilation (%) = (Dx – Dmin)/(Dmax – Dmin) × 100%, where Dx is the diameter after administration of NaHS, L-Cys, or endothelial cells, Dmin is the stable diameter of artery precontracted with U46619 or KCl, and Dmax is the initial diameter. The maximum rate of vascular dilation of MCAO rats was calculated using the following formula: Dilation (%) = (Tmin-Tx)/(Tmin – Tmax) × 100%, where Tmin is the stably tension of artery precontracted with U46619, Tx is the vascular tension after administration of ACh or vinpocetine, and Tmax is the initially vascular tension.
Evaluation of Neurological Score
Neurological score (20) of rats was evaluated at 24 h after reperfusion. It was scored on a five-point scale: (1) score 0: no neurologic deficit; (2) score 1: a mild focal neurologic deficit (failure to extend left forepaw fully); (3) score 2: a moderate focal neurologic deficit (circling to the left); (4) score 3: a severe focal deficit (falling to the left); (5) score 4: rat could not walk spontaneously and had a depressed level of consciousness.
Determination of Infarction Volume and Brain Water Content
At the end of the neurological score test, the rats were sacrificed with anesthesia. The brains were rapidly removed and sliced coronally at a 2 mm interval. The brain slices were then incubated in the dark in 2% TTC in phosphate-buffered solution (PBS) at 37°C for 30 min for staining. Subsequently, the stained slices were placed in 4% paraformaldehyde for 10 min. All the stained brain slices were photographed subsequently to delineate the area of infarct size using Image J, version 1.6 (National Institutes of Health, Bethesda, MD, USA). As described previously (9), the percentage of infarction volume was determined by normalizing the whole brain.
The dry-wet approach was used to measure the brain water content (21). In short, the fresh slices of each brain were weighed to attain the wet weight. The fresh tissues were then dried in an oven at 105°C for 48 h and weighed again to obtain the dry weight. Brain water content was calculated using the following formula:
Brain water content (%) = (wet weight – dry weight)/wet weight × 100%.
Measurement of Serum LDH Activity and MDA Level
Briefly, serum and supernatant of brain tissue homogenate of rats were collected and transferred to 96 well plates for LDH activity and MDA level analysis, using the biochemistry assay kit (Jiancheng Bioengineering Ltd, Nanjing, China) and abiding by the manufacturer's manual.
Statistical Analysis
Statistical analysis was performed by one-way analysis of variance (ANOVA) followed by the Duncan test to determine the difference between groups. Blood vessel data is presented as mean ± SD, and the other data are expressed as mean ± SEM. The p < 0.05 are considered significant.
Results
Effect of H2S on Cerebrovascular Function of Rats
Exogenous H2S Attenuated Cerebrovascular Dysfunction Induced by Cerebral I/R
Changes of vascular tension in MCA from MCAO rats were examined after cerebral I/R. As shown in Figure 1, in the sham group, 1 × 10−7 mmol/L U46619 evoked significant constriction in MCA with maximum response (Emax) of 1.88 ± 0.19 mN. The contraction in MCA from MCAO rats to U46619 was profoundly decreased and the Emax was reduced to 0.39 ± 0.12 mN, but the reduction was significantly ameliorated by 1 × 10−5 ~ 1 × 10−7 mol/kg NaHS supplement. Furthermore, the ACh-mediated relaxations in MCA were also remarkably inhibited by MCAO, with the Emax being reduced from 68.27 ± 3.71% of the sham group rats to 8.91 ± 3.66% of the model group rats, and the dilation dysfunction in MCA was also attenuated by 1 × 10−5 ~ 1 × 10−7 mol/kg NaHS supplement. In addition, vinpocetine-mediated non-endothelium-dependent relaxation in MCA was also significantly attenuated in the model group (Emax: 68.44 ± 12.21% in the model group and 151.48 ± 25.32% in the sham group). Interestingly, the decrease of vascular relaxation to vinpocetine injured by cerebral I/R was similarly ameliorated by 1 × 10−6 ~ 1 × 10−7 mol/kg NaHS supplement. These results indicated that exogenous H2S has a protective effect on the cerebrovascular dysfunction injured by cerebral I/R.
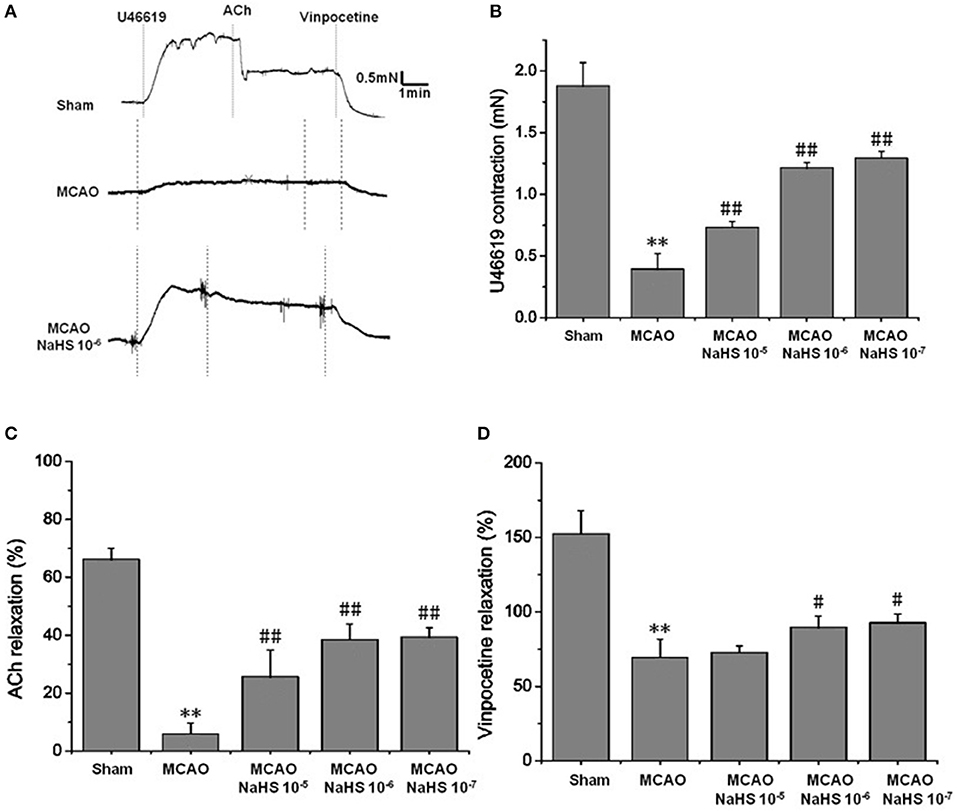
Figure 1. Exogenous H2S attenuated cerebrovascular dysfunction induced by cerebral I/R in rats (mean ± SD, n = 8). (A) Original tracings of U46619-, ACh-, vinpocetine- and NaHS-induced responses of MCA derived from MCAO rats. (B) The effect of NaHS on U46619 meditated MCA contraction. (C) The effect of NaHS on ACh-meditated relaxation in MCA preconstricted with U46619. (D) The effect of NaHS on vinpocetine-meditated relaxation in MCA preconstricted with U46619. **P < 0.01 vs. sham, ##P < 0.01 vs. MCAO, #P < 0.05 vs. MCAO.
Effect of Endogenous H2S on Cerebrovascular Dysfunction Injured by Cerebral I/R
In order to clarify the effect of endogenous H2S on cerebrovascular dysfunction injured by cerebral I/R, we examined the changes of vascular tension in MCA from CSE knocked down rats after cerebral I/R. As shown in Figure 2, MCA almost had no contractile response to U-46619 (Emax: 0.11 ± 0.01 mN) in CSE knock down rats after cerebral I/R, which could be significantly elevated by 1 × 10−6 mol/kg NaHS supplement (Emax: 0.59 ± 0.03 mN). Similarly, CSE knockdown attenuated both ACh- and vinpocetine-mediated relaxation of MCA from MCAO rats. Emax of ACh-mediated relaxation was reduced from 8.91 ± 3.66% in the MCAO group of wild-type rats to 2.48 ± 2.65% in the CSE-siRNA group; Emax of vinpocetine-mediated relaxation was reduced from 68.44 ± 12.21% in the MCAO group of wild-type rats to 8.42 ± 3.47% in the CSE-siRNA group. Interestingly, supplementing with NaHS could also further elevate the ACh- and vinpocetine-mediated vascular relaxation of MCAO rats. These results indicate that the CSE knockdown could induce significant vascular dysfunction, which can be ameliorated by exogenous H2S.
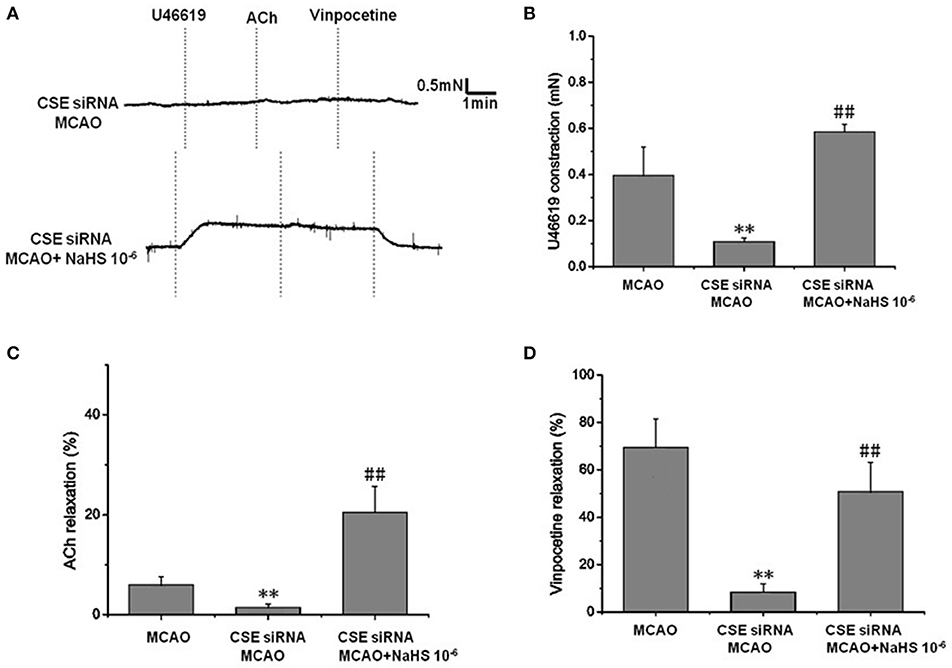
Figure 2. Effect of endogenous H2S on cerebrovascular dysfunction injured by cerebral I/R (mean ± SD, n = 8). (A) Original tracings of U46619-, ACh-, vinpocetine-, and NaHS-induced responses of MCA derived from CSE knocked down rats after MCAO. (B) The effect of CSE knockdown and NaHS on U46619-meditated contraction. (C) The effect of CSE knockdown on and NaHS- or ACh-meditated relaxation. (D) The effect of CSE knockdown and NaHS on vinpocetine-meditated relaxation. **P < 0.01 vs. sham, ##P < 0.01 vs. MCAO.
Effect of Ca2+-Activated K+ (KCa) Channel Blockers on H2S-Mediated Relaxation of MCA
We next sought to demonstrate further the effect of H2S on MCA and explore the underlying mechanism using KCa channel blockers CTX and Apa. The results of changes of vascular diameter as shown in Figure 3A and Table 1, the NaHS could induce concentration-dependent dilation in MCA precontracted with U46619 from non-ischemic rats, which was obviously abolished by co-application of CTX and Apa, Emax of vascular relaxation being reduced from 76.23 ± 7.4 to 8.75 ± 1.7% after co-application of CTX and Apa. Moreover, L-Cys, the substrate of endogenous H2S-producing enzyme similarly induced a concentration-dependent dilation in MCA precontracted with U46619 (Figure 3B, Emax: 79.28 ± 5.4%, p < 0.01 vs. the vehicle group). However, the relaxation in MCA to L-Cys was also obviously abolished by co-application of CTX and Apa, Emax being reduced from 79.3 ± 5.4 to 9.7 ± 2.0% (Figure 3D). These data suggested that KCa channel might be involved in the H2S-induced cerebrovascular relaxation.
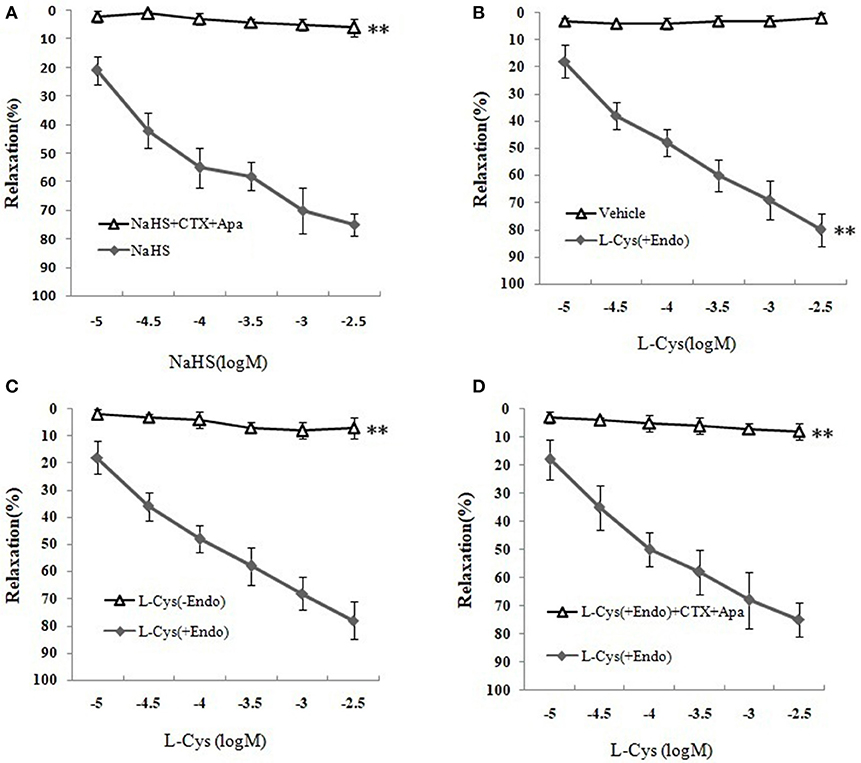
Figure 3. Effects of KCa channel blockers CTX and Apa on H2S-mediated relaxation in MCA of normal rats (mean ± SD, n = 8). (A) Effects of CTX and Apa on NaHS-induced relaxation. (B) Effect of L-Cys on relaxation in U46619-preconstricted rat MCA. (C) Effects of the endothelium removal on the L-Cys- induced relaxation. **P < 0.01 vs. L-Cys (+Endo). (D) Effect of CTX and Apa on L-Cys-induced relaxation in MCA. **P < 0.01 vs. NaHS or vehicle or L-Cys (+Endo).

Table 1. Effects of CTX plus Apa on relaxation of MCA to NaHS or L-Cys, and role of vascular endothelium in L-Cys-induced relaxation in the MCA (Mean ± SD, n = 8).
Effect of Vascular Endothelium on H2S-Mediated Relaxation of MCA
As shown in Figures 3B,C and Table 1, the removal of vascular endothelium significantly reduced the relaxation of MCA to L-Cys, with Emax being reduced to 8.8 ± 3.8%. Co-adding of ACh and endothelium cells (EAhy926 cells) cultured in vitro into luminal perfusate could induce a non-NO and non-PGI2 relaxation in endothelium-denuded rat MCA precontracted with KCl (Figure 4, Emax: 66.1 ± 1.6%). However, co-application of ACh and EAhy926 cells of CSE knockdown cannot induce the relaxation in the endothelium-denuded MCA. These results further suggest that vascular endothelium participated in the relaxation in rat MCA, and that endothelial H2S might mediate vasodilation in the blood vessel.
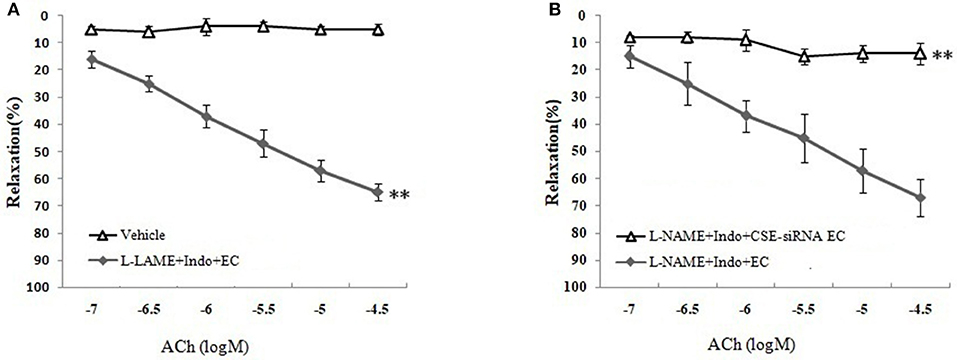
Figure 4. Role of endothelial CSE in ACh-induced non-NO- non-PGI2 relaxation in endothelium-denuded rat MCA (mean ± SD, n = 8). (A) Effect of EAhy926 cells (endothelial cell, EC) on ACh-induced non-NO- non-PGI2 relaxation in KCl-preconstricted endothelium-denuded rat MCA. (B) Effect of EAhy926 cells with CSE knockdown (CSE-siRNA EC) on ACh-induced non-NO- non-PGI2 relaxation in KCl-preconstricted endothelium-denuded rat MCA. **P < 0.01 vs. vehicle or L-NAME+Indo+EC.
Effects of H2S on Neuronal Injury Induced by MCAO in Rats
The rats were transfected with siRNA to knock down the expression of CSE and used to investigate the role of endogenous H2S on neuronal injury induced by MCAO.
Effect of H2S on Neurological Score
The neurologic deficit scores of rats are presented in Figure 5A. No neurologic deficits were observed in the sham group. Moderate neurologic deficits (average score: 3) were observed at 24 h after reperfusion in control group rats, while in the CSE-siRNA group, the rats had significant neurologic deficits (average score: 3.5), and interestingly, the neurologic deficits were remarkably inhibited by 1 × 10−6 mol/kg NaHS supplementation within 30 min after ischemia.
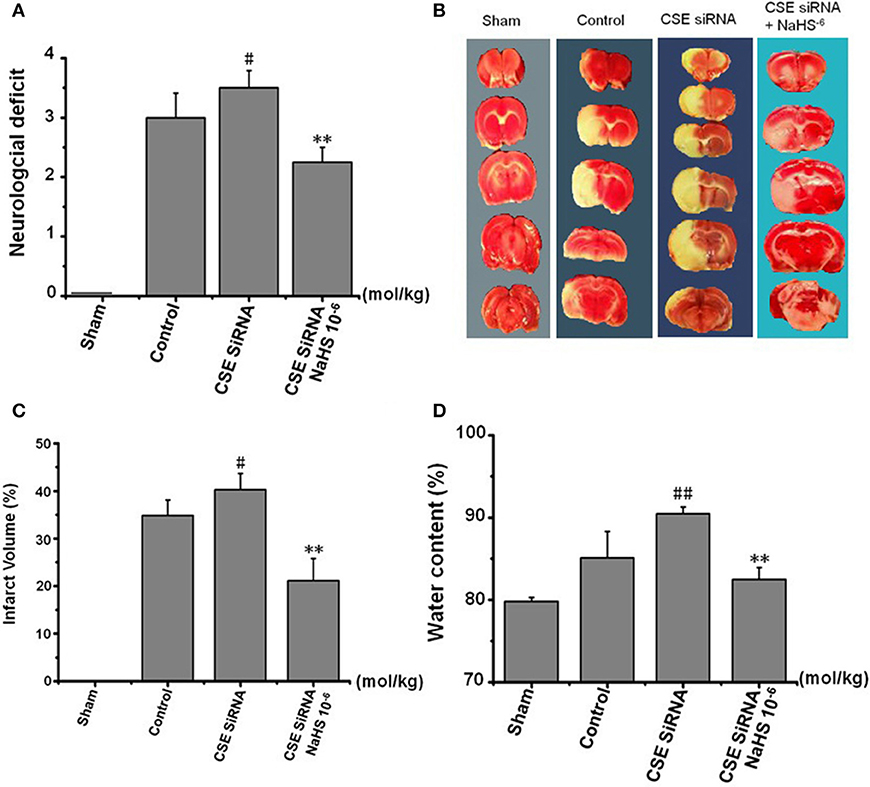
Figure 5. Effect of CSE knockdown (CSE siRNA) on neuronal injury induced by MCAO in rats (Mean ± SEM, n = 8). (A) Neurological deficits. (B) Infarct volume. (C) Quantitative analysis of brain infarct volume. (D) Brain water content. #P < 0.05 vs. control, ##P < 0.01 vs. control, **P < 0.01 vs. CSE siRNA.
Effect of H2S on the Infarction Volume
As shown in Figures 5B,C, I/R remarkably induced cerebral infarction in rats. However, the increase of the infarction volume in the CSE knockdown rats was more significant than that in the control group rats. NaHS (1 × 10−6 mol/kg) supplementation significantly reduced the infarction volume in CSE knockdown rats alike.
Effect of H2S on Brain Water Content in Rats
Brain water content among the other factors is regarded as being responsible for the neuronal dysfunction after brain ischemia (22) and can be used as an indicator of brain edema (21). The results (Figure 5D) showed that MCAO markedly increased the brain water content in CSE knockdown rats when compared to untreated rats of the control group, which could be significantly inhibited by NaHS (1 × 10−6 mol/kg) supplementation.
Serum LDH Activity and MDA Level in Brain Tissue
LDH leakage from cells to serum and MDA, a product of lipid peroxidation, are major indexes of ischemia injury. In the control group (Figure 6) there was a significant increase of LDH activity in serum and MDA content in cerebral tissue induced by cerebral I/R, and the results indicated that I/R could induce significant cerebral injury. However, the injury was more remarkable in CSE knockdown rats than in the control group (p < 0.01) and was significantly inhibited by NaHS (1 × 10−6 mol/kg) supplementation.
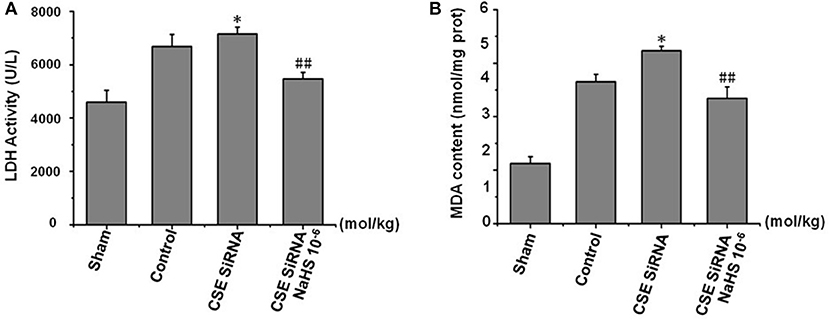
Figure 6. Effect of CSE knockdown (CSE siRNA) on MCAO rat serum LDH activity and MDA content in brain tissue (mean ± SEM, n = 8). (A) Serum LDH activity. (B) MDA content in brain tissue. *P < 0.05 vs. control; ##P < 0.01 vs. CSE isRNA.
These results confirmed that H2S has a remarkable protective effect on MCAO-induced neuronal injury.
Discussion
Cerebral I/R injury is a serious and common clinical disease. The tissue plasminogen activator (t-PA) is the only FDA-approved treatment for acute brain ischemia. However, only a small proportion of brain ischemia patients are eligible to receive tPA treatment because it carries a high risk of secondary impairments, such as bleeding/hemorrhagic transformation and severe neurodegeneration (23, 24). Thus, the main priority is to explore the neuroprotective strategies and find new drugs for possible clinical use.
Previous studies have reported that cerebral I/R decreases endothelial vasoreactivity, impairs blood flow restoration, and also causes further brain injury (5). H2S is a novel vasoactivator, and research has pointed out that H2S is helpful for cerebral ischemic injury. Although the role of H2S on cerebral I/R injury has attracted the interest of many researchers, the mechanism involved in the effect of H2S on cerebral I/R injury is still not completely clear (9). The aim of this study was designed to clarify the vascular protection of H2S on neurovascular dysfunction after cerebral I/R. The results showed that the contraction and dilation of MCA profoundly decrease after cerebral I/R. The reduction in the contraction and dilation was significantly ameliorated by 1 × 10−5 ~ 1 × 10−7 mol/kg NaHS supplement. Not surprisingly, in CSE knockdown rats, MCA almost loses its dilation to ACh or vinpocetion, and constriction to U46619 after cerebral I/R, which could also be significantly ameliorated by 1 × 10−6 mol/kg NaHS supplement. These data indicated that H2S had a significant protection on cerebrovascular dysfunction induced by cerebral I/R.
We next sought to investigate the mechanism of MCA relaxation on to H2S using KCa channel blockers and endothelial removal. As has previously been established, H2S has been classified as a novel gasotransmitter signaling molecule in CNS, which is involved in various signal transmissions such as the regulation of ion channels (14). Furthermore, the cerebral endothelium has a key role in the regulation of vascular tone because the endothelium could release H2S and other relaxing factors such as NO and PGI2 to relax vascular smooth muscle cells (VSMC) (17). In the present study, we found that the relaxation of isolated MCA to H2S donor NaHS and L-Cys (substrate of endogenous H2S-producing enzyme) was abolished by the co-application of the intermediate-conductance KCa channel blocker CTX and small-conductance KCa channel blocker Apa. In parallel, similar dilation of MCA elicited by L-Cys was blocked by endothelial removal. However, adding endothelium cells (EAhy926) cultured in vitro to luminal perfusate could mediate non-NO and non-PGI2 vasorelaxation to ACh in endothelium-denuded MCA, but the relaxation was abolished by CSE knockdown in EAhy926 cells. These results provided solid evidence that the vasodilation of cerebral vessels to H2S is endothelium-dependent and might relate to activate the KCa channel.
To further confirm the protective effects of H2S on neuronal injury after cerebral I/R, the MCAO was still used as a model of focal cerebral I/R and associated with an increase of infarction volume, brain water content, and neurological scoring (25). Our data revealed that cerebral I/R injury led to a significant increase of cerebral infarction, brain edema, and neurological deficits, thereby suggesting an eminently neuronal injury. In addition, it is widely accepted that oxygen-free radicals in neurocytes induced by cerebral I/R injury and subsequent lipid peroxidation play a key role in the pathophysiology of I/R injury. Hence, like MDA, a product of lipid peroxidation, LDH leakage has also been applied to assess cerebral I/R injury (9, 26). In agreement with the previous result, we found that cerebral I/R injury led to a significant increase of serum LDH activity and MDA content in MCAO rats. However, all the above injury indicators occurred more grievously in CSE knockdown rats than in the normal control group, and could be remarkably inhibited by 1 × 10−6 mol/kg NaHS supplementation. Together with treatment, the effect of H2S donor NaHS on the injury suggests that H2S could inhibit cerebral I/R-induced increases of cerebral infarction, brain edema and neurological deficits, LDH leakage, and lipid peroxidation. These findings provide more details and demonstrate that H2S has a protective effect on neuronal injury induced by MCAO.
In conclusion, our study is the first to show the multifaceted vasoprotection of H2S on cerebral I/R injury. We found that (1) both endogenous and exogenous H2S had eminent protection on vasomotor dysfunction induced by MCAO in rats; (2) KCa channel might be involved in the cerebrovascular relaxation to H2S; (3) the cerebrovascular relaxation to H2S is endothelium-dependent; (4) both endogenous and exogenous H2S had a protective effect on neuronal injury after cerebral I/R in rats.
Author Contributions
Z-WC, J-YW, and MW participated in research design and experiments. H-HJ and X-JS contributed new reagents and analytical tools. H-HJ and X-JS performed data analysis. Z-WC, J-YW, and Y-NL contributed to writing of the manuscript.
Funding
This work was supported by grant from the National Natural Science Foundation of China (Nos. 81173596 and 81374002).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Suh JY, Shim WH, Cho G, Fan X, Kwon SJ, Kim JK, et al. Reduced microvascular volume and hemispherically deficient vasoreactivity to hypercapnia in acute ischemia: MRI study using permanent middle cerebral artery occlusion rat model. J Cereb Blood Flow Metab. (2015) 35:1033–43. doi: 10.1038/jcbfm.2015.22
2. Schroder HC, Steffen R, Wenger R, Ugarkovic D, Muller WE. Age-dependent increase of DNA topoisomerase II activity in quail oviduct; modulation of the nuclear matrix-associated enzyme activity by protein phosphorylation and poly(ADP-ribosyl)ation. Mutat Res. (1989) 219:283–94. doi: 10.1016/0921-8734(89)90030-1
3. Levine AB, Punihaole D, Levine TB. Characterization of the role of nitric oxide and its clinical applications. Cardiology (2012) 122:55–68. doi: 10.1159/000338150
4. Jujo K, Ii M, Sekiguchi H, Klyachko E, Misener S, Tanaka T, Tongers J, et al. CXC-chemokine receptor 4 antagonist AMD3100 promotes cardiac functional recovery after ischemia/reperfusion injury via endothelial nitric oxide synthase-dependent mechanism. Circulation (2013) 127:63–73. doi: 10.1161/CIRCULATIONAHA.112.099242
5. Palomares SM, Cipolla MJ. Vascular protection following cerebral ischemia and reperfusion. J Neurol Neurophysiol. (2011). p S1–004. doi: 10.4172/2155-9562.S1-004
6. Chen J, Gao J, Sun W, Li L, Wang Y, Bai S, et al. Involvement of exogenous H2S in recovery of cardioprotection from ischemic post-conditioning via increase of autophagy in the aged hearts. Int J Cardiol. (2016) 220:681–92. doi: 10.1016/j.ijcard.2016.06.200
7. Lobb I, Jiang J, Lian D, Liu W, Haig A, Saha MN, et al. Hydrogen sulfide protects renal grafts against prolonged cold ischemia-reperfusion injury via specific mitochondrial actions. Am J Transplant. (2017) 17:341–352. doi: 10.1111/ajt.14080
8. Donnarumma E, Ali MJ, Rushing AM, Scarborough AL, Bradley JM, Organ CL, et al. Zofenopril protects against myocardial ischemia-reperfusion injury by increasing nitric oxide and hydrogen sulfide bioavailability. J Am Heart Assoc. (2016) 5:e003531. doi: 10.1161/JAHA.116.003531
9. Jiang WW, Huang BS, Han Y, Deng LH, Wu LX. Sodium hydrosulfide attenuates cerebral ischemia/reperfusion injury by suppressing overactivated autophagy in rats. FEBS Open Bio. (2017) 7:1686–1695. doi: 10.1002/2211-5463.12301
10. Guo W, Cheng ZY, Zhu YZ. Hydrogen sulfide and translational medicine. Acta Pharmacol Sin. (2013) 34:1284–91. doi: 10.1038/aps.2013.127
11. Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science (2008) 322:587–90. doi: 10.1126/science.1162667
12. Hu LF, Wong PT, Moore PK, Bian JS. Hydrogen sulfide attenuates lipopolysaccharide-induced inflammation by inhibition of p38 mitogen-activated protein kinase in microglia. J Neurochem. (2007) 100:1121–8. doi: 10.1111/j.1471-4159.2006.04283.x
13. Kimura H. Physiological role of hydrogen sulfide and polysulfide in the central nervous system. Neurochem Int. (2013) 63:492–7. doi: 10.1016/j.neuint.2013.09.003
14. Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. (2012) 92:791–896. doi: 10.1152/physrev.00017.2011
15. Cai SN FY, Chen ZW. The hyperpolorization produced by H2S in VSMC from middle cerebral artery of rat. Chin J Clin Pharmacol Ther. (2011) 16:5.
16. Fan YF, Chen ZW, Guo Y, Wang QH, Song B. Cellular mechanisms underlying Hyperin-induced relaxation of rat basilar artery. Fitoterapia (2011) 82:626–31. doi: 10.1016/j.fitote.2011.01.023
17. Wang M, Hu Y, Fan Y, Guo Y, Chen F, Chen S, et al. Involvement of hydrogen sulfide in endothelium-derived relaxing factor-mediated responses in rat cerebral arteries. J Vasc Res (2016) 53:172–185. doi: 10.1159/000448712
18. Xuanjun S, Youyang H, Zhiwu C. The effects of CSEsiRNA on the expression of CSE and H2 S in EAhy926 cells. Acta Universitatis Medicinalis Anhui (2012) 47:904–907.
19. Wen JY, Chen ZW. Protective effect of pharmacological preconditioning of total flavones of abelmoschl manihot on cerebral ischemic reperfusion injury in rats. Am J Chin Med. (2007) 35:653–61. doi: 10.1142/S0192415X07005144
20. Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke (1989) 20:84–91. doi: 10.1161/01.STR.20.1.84
21. Yu T, Chen QE, Chen ZW, Xiong Z, Ye M. Protective effects of total flavones of rhododendra against global cerebral ischemia reperfusion injury. Am J Chin Med. (2009) 37:877–87. doi: 10.1142/S0192415X09007284
22. Brouns R, De Deyn PP. The complexity of neurobiological processes in acute ischemic stroke. Clin Neurol Neurosurg. (2009) 111:483–95. doi: 10.1016/j.clineuro.2009.04.001
23. Kaur J, Zhao Z, Klein GM, Lo EH, Buchan AM. The neurotoxicity of tissue plasminogen activator? J Cereb Blood Flow Metab. (2004) 24:945–63. doi: 10.1097/01.WCB.0000137868.50767.E8
24. Wang T, Duan S, Wang H, Sun S, Han B, Fu F. Neurological function following cerebral ischemia/reperfusion is improved by the Ruyi Zhenbao pill in a rats. Biomed Rep. (2016) 4:161–166. doi: 10.3892/br.2016.568
25. Huang JL, Liu WW, Sun XJ. Hydrogen inhalation improves mouse neurological outcomes after cerebral ischemia/reperfusion independent of anti-necroptosis. Med Gas Res. (2018) 8:1–5. doi: 10.4103/2045-9912.229596
Keywords: hydrogen sulfide, ischemia/reperfusion, vascular function, neuronal injury, Kca channel
Citation: Wen J-Y, Wang M, Li Y-N, Jiang H-H, Sun X-J and Chen Z-W (2018) Vascular Protection of Hydrogen Sulfide on Cerebral Ischemia/Reperfusion Injury in Rats. Front. Neurol. 9:779. doi: 10.3389/fneur.2018.00779
Received: 28 June 2018; Accepted: 29 August 2018;
Published: 19 October 2018.
Edited by:
Pei Jiang, Jining Medical University, ChinaReviewed by:
Kevin Donald Broad, University College London, United KingdomSulev Kõks, University of Tartu, Estonia
Copyright © 2018 Wen, Wang, Li, Jiang, Sun and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhi-Wu Chen, Y2hwaGFybXp3QDE2My5jb20=
†These authors have contributed equally to this work