 Clara D. M. van Karnebeek1,2,3*
Clara D. M. van Karnebeek1,2,3* Bryan Sayson4Jessica J. Y. Lee2
Bryan Sayson4Jessica J. Y. Lee2 Laura A. Tseng3Nenad Blau5,6
Laura A. Tseng3Nenad Blau5,6 Gabriella A. Horvath4
Gabriella A. Horvath4 Carlos R. Ferreira7,8*
Carlos R. Ferreira7,8*- 1Department of Pediatrics, University of British Columbia, Vancouver, BC, Canada
- 2Centre for Molecular Medicine and Therapeutics, BC Children's Hospital Research Institute, University of British Columbia, Vancouver, BC, Canada
- 3Departments of Pediatrics and Clinical Genetics, Emma Children's Hospital, Amsterdam University Medical Centres, Amsterdam, Netherlands
- 4Division of Biochemical Diseases, Department of Pediatrics, BC Children's Hospital Research Institute, University of British Columbia, Vancouver, BC, Canada
- 5Dietmar-Hopp Metabolic Center, University Children's Hospital, Heidelberg, Germany
- 6Division of Metabolism, University Children's Hospital, Zurich, Switzerland
- 7Division of Genetics and Metabolism, Children's National Health System, Washington, DC, United States
- 8National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, United States
Although inborn errors of metabolism do not represent the most common cause of seizures, their early identification is of utmost importance, since many will require therapeutic measures beyond that of common anti-epileptic drugs, either in order to control seizures, or to decrease the risk of neurodegeneration. We translate the currently-known literature on metabolic etiologies of epilepsy (268 inborn errors of metabolism belonging to 21 categories, with 74 treatable errors), into a 2-tiered diagnostic algorithm, with the first-tier comprising accessible, affordable, and less invasive screening tests in urine and blood, with the potential to identify the majority of treatable conditions, while the second-tier tests are ordered based on individual clinical signs and symptoms. This resource aims to support the pediatrician, neurologist, biochemical, and clinical geneticists in early identification of treatable inborn errors of metabolism in a child with seizures, allowing for timely initiation of targeted therapy with the potential to improve outcomes.
Introduction
The occurrence of epilepsy in the general population is relatively common. Just in the United States, it affects 1.2% of the population, with 3 million adults and 470,000 children suffering from it (1). In cases of neonatal seizures, inborn errors of metabolism (IEMs) account for 1.1% (2) −7.4% (3) of all cases. Although IEMs are responsible for only a fraction of these cases, it is imperative to identify them, as there is often treatment available which can mitigate or even prevent major neurological sequelae (4). Elucidating the etiology of a seizure disorder can also prompt the investigation of extra-neurologic systems commonly affected by IEMs (i.e., arrhythmias in mitochondrial disease, liver disease, hearing or retinal involvement in peroxisomal disorders, etc.). It is important to consider the possibility of an IEM in a patient presenting with seizures of an unknown cause not only due to the amenability to causal treatment of the seizures and the possible co-morbidities, but also in order to adequately perform genetic counseling and to more accurately predict the disease trajectory and prognosis. Finally, a timely diagnosis will not only avoid unnecessary delay and burden to the patient, it might also prevent failed attempts to control seizure with standard antiepileptic drugs, often not without side effects, and it has the potential to decrease the number and cost of unnecessary tests.
Even when seizures are the major presenting feature of an IEM, such as in pyridoxine-dependent epilepsy (PDE), diagnosis is often difficult due to the broad differential diagnoses varying from environmental to non-metabolic genetic etiologies (e.g., hypoxic-ischemic encephalopathy or genetic channelopathy) (5). In addition, there is a great number of metabolic epilepsies, posing a considerable challenge in identification based upon primary symptomology; hence, standardized screening for a subset for IEMs is advisable. Finally, the clinical picture even for a specific metabolic condition may be often complex and mask an otherwise evident IEM. An example is PDE, which can present in the neonatal period not only with an epileptic encephalopathy but also with metabolic acidosis, electrolyte disturbances, abdominal distension mimicking sepsis, or structural brain anomalies that might be considered a sufficient explanation for seizures (5).
Patients with early-onset epilepsy can be evaluated in several different departments within a given hospital, which necessitates a standardized testing protocol in order to facilitate effective coordination of the various departments, as well as to avoid unnecessary diagnostic delay. Standardized testing will prevent the administration of repeat tests, increasing the efficacy of laboratory resources as well as diminishing cost. Finally, the use of a standardized approach will avoid missed diagnoses, especially for clinicians with less experience.
An indirect benefit from establishing an early diagnosis stems from the fact that increased awareness of these IEMs can further expand the knowledge base and clinical phenotype. For example, pyridox(am)ine 5′-phosphate oxidase (PNPO) deficiency has, in the past, been reported to have high mortality in the few cases in which it was reported. However, awareness and subsequent prompt diagnosis has increased survivability as well as expanded the phenotype (6).
Methods
Given the multitude of aforementioned benefits from establishing an early diagnosis of IEMs, we propose a diagnostic approach in patients with epilepsy, with emphasis on early identification of those amenable to treatment. The algorithm is structured according to the TIDE protocol for diagnosis of treatable intellectual disabilities (7, 8). All metabolic conditions included in the IEMbase tool [www.IEMbase.org; (9)] and in the proposed Nosology (10) were individually searched along with the terms “seizures,” “epilepsy,” or “convulsions” in Pubmed (1966-December 2017) to identify all IEMs for which epilepsy is a feature, and for which therapy targeting the underlying pathophysiology is available and deemed at least partially effective or preventive, with sufficient evidence (level IV or higher according to the Center for Evidence-Based Medicine (CEBM).
Results
Our systematic review identified 268 IEMs with epilepsy as a primary or secondary feature, listed in Supplementary Table 1 following the nomenclature of IEMs of the recently-proposed Nosology (10). Of these, 74 conditions are treatable. These IEMs include disorders of amino acids (n = 43, 12 treatable); sterols and bile acids (n = 7, 1 of which is treatable); creatine (n = 3, all treatable); fatty aldehydes (n = 1, not treatable); fatty acids, carnitine and ketones (n = 9, 6 treatable); complex lipids (n = 8, none treatable); carbohydrates and polyols (n = 8, half of them treatable); lysosomes (n = 23, none treatable); lipofuscin storage (n = 9, 1 treatable); metals (n = 5, 3 treatable); mitochondria (n = 26, 7 treatable); autophagy (n = 2, neither of which is treatable); neurotransmitters (n = 4, all treatable); organic acids (n = 10, 6 treatable); peroxisomes (n = 7, 1 treatable); purines and pyrimidines (n = 5, 2 treatable); urea cycle (n = 9, all treatable); vitamins/co-factors (n = 23, 12 treatable); glutathione (n = 1, not treatable), heme (n = 2, 1 treatable); and glycans and glycolipids (n = 63, 1 treatable). The therapeutic modalities available for these IEMs include: “sick–day” management, medical diets, cofactor/vitamin supplements; substrate inhibition, stem cell transplant, and gene therapy. Therapeutic effects vary from complete control, improvement or prevention of epilepsy both clinically and on EEG. Secondary outcomes include improvement and/or stabilization of psychomotor/cognitive development; behavior/psychiatric disturbances; seizures; and neurologic and systemic manifestations (11). Supplementary Table 1 provides more information on these IEMs, among others whether or not included in newborn screening panels.
When to Suspect Metabolic Epilepsies
The presence of any of the following features should raise suspicion for a metabolic etiology of epilepsy (12):
• Dysmorphic features (e.g., peroxisomal disorders, CDGs, crotonase deficiency)
• Organomegaly
• Positive family history for similar condition or death of unknown etiology
• Parental consanguinity
• Developmental regression after a period of apparent normalcy
• Fluctuating course of illness
• Ophthalmological problems (cataracts, retinitis pigmentosa, cherry red spot, optic nerve atrophy)
• High-anion gap metabolic acidosis
• Neonatal ketonuria
• Unusual body fluid odor
• Seizures worsening with fasting (GLUT1) or with high protein meals (urea cycle defects)
• Seizure worsening with anti-epileptic drugs
• Progressive myoclonic epilepsy phenotype in adolescence or young adulthood.
The age of presentation can be a diagnostic indication of the form IEM affecting the patient. Therefore, it is practical to organize epilepsies caused by IEM according to age, which results in a distribution of epilepsies occurring in the neonatal or infantile period, in early childhood, or those that present in late childhood and adolescence. Table 1 outlines this organization. This information may be useful in ordering Tier 2 testing.

Table 1. Metabolic epilepsies classified according to age of onset.
The particular type of seizure can occasionally raise suspicion for a specific disorder. For example, epilepsia partialis continua can be the first clue toward a diagnosis of a POLG-related disorder (13), patients with certain forms of congenital disorders of glycosylation can present with migrating partial seizures of infancy (14), while patients with guanidinoacetate methyltransferase deficiency can have drop attacks and Lennox-Gastaut syndrome (15). For other conditions, the evolution of seizure over time have been well defined, such as for Menkes disease, in which pattern of seizures can be divided in three stages: an early stage at around 3 months showing focal clonic status epilepticus, an intermediate stage at about 10 months showing intractable infantile spasms, and a late stage at about 25 months showing multifocal seizures, tonic spasms and myoclonus (16). In the majority of metabolic epilepsies, however, the particular etiology of seizures cannot be predicted from its semiology. A perfect example of this is that of GLUT1 deficiency, in which the type of seizures is quite variable, being mixed in 68%, generalized tonic-clonic in 53%, absence in 49%, complex partial in 37%, myoclonic in 27%, drop attacks in 26%, tonic in 12%, simple partial in 3%, and spasms in 3% (17).
Although many IEMs will manifest with epilepsy refractory to multiple standard anti-epileptic drugs (AEDs), a favorable response to these drugs does not rule out metabolic epilepsies. For example, patients with GLUT1 deficiency can show response to standard AEDs (17, 18), and patients with PDE can also show some response (5), or can even show lack of seizures after a period of time without pyridoxine supplementation (19). On the other hand, the lack of response to standard treatment for these conditions can also not be used as a criteria to rule out the diagnosis: patients with GLUT1 deficiency can still show recurrence of seizures despite adequate ketosis (20), and in fact up to one third of patients respond poorly to a ketogenic diet (21), while patients with PDE might not show an instant and obvious response to pyridoxine administration (5).
Finally, the presence of specific electroencephalographic (EEG) or brain neuroimaging findings can also provide important clues toward the right diagnosis. Some IEMs can have characteristic EEG changes, such as the comb-like rhythm seen in patients with maple syrup urine disease (22). Certain IEMs can be accompanied by structural brain anomalies identifiable by brain MRI, such as a dysgenetic corpus callosum seen commonly in patients with pyruvate dehydrogenase complex deficiency or glycine encephalopathy (23). Brain magnetic resonance spectroscopy can sometimes point toward the right diagnosis, as has been reported in patients with GABA transaminase deficiency (24), fatty acid oxidation disorders (25), mitochondrial disease or glycine encephalopathy (26). Table 2 provides a summary of the salient clinical, laboratory, electrographic, and neuroimaging finding of the most common metabolic epilepsies.
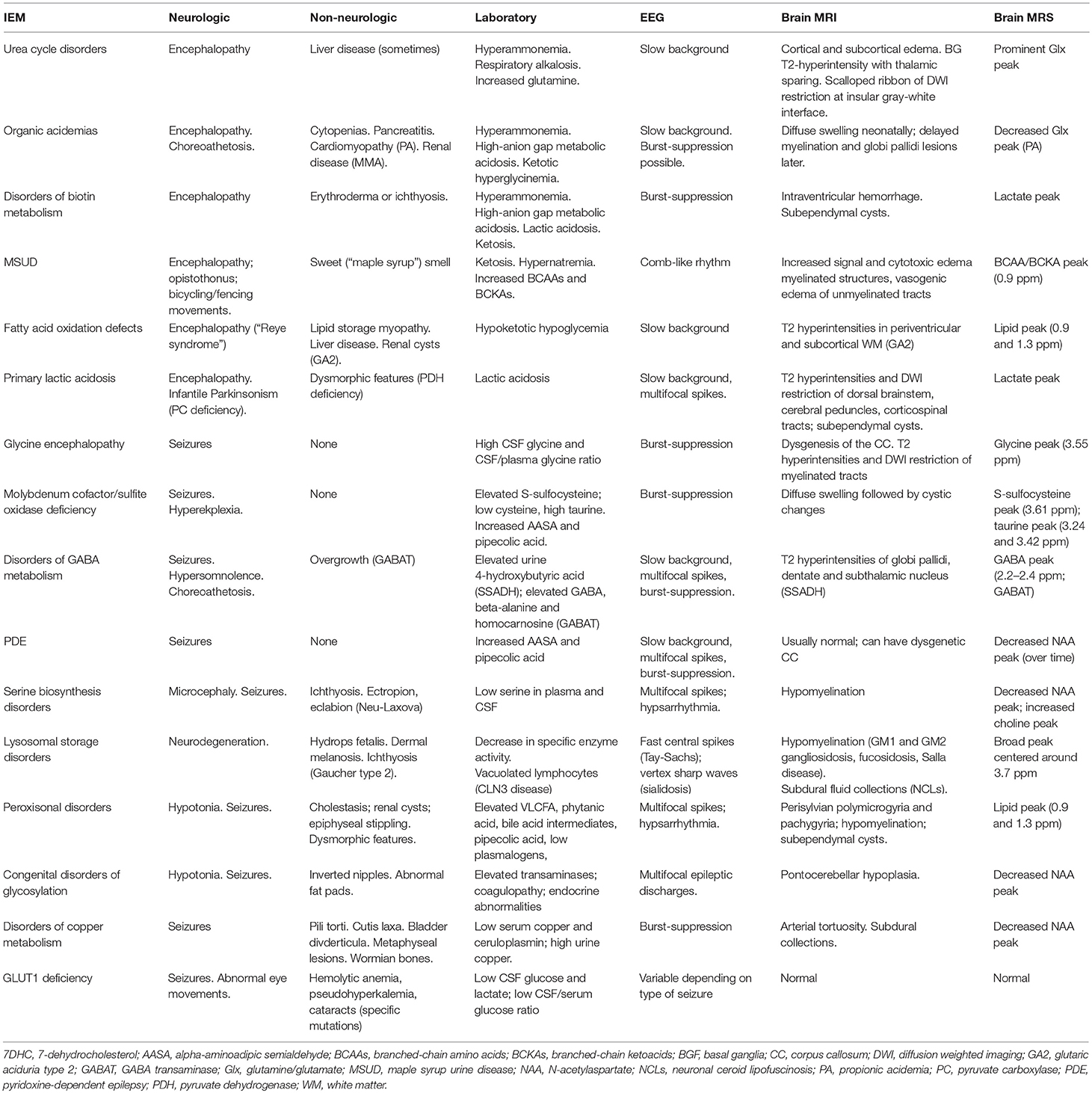
Table 2. Summary of clinical, laboratory, EEG, and neuroimaging finding of metabolic epilepsy.
Metabolic Investigational Algorithm for Patients With Idiopathic or Early Onset Epilepsy
Tier 1: Initial Screening in All Patients
We propose that the following initial screening investigations be carried out for patients in whom epilepsy remains a prominent feature of unknown cause. The tests included in the 1st tier: (a) have the potential to identify 2 or more treatable metabolic epilepsies; (b) are accessible via standard clinical chemistry and biochemical genetics laboratories; (c) are relatively affordable and covered by insurance; and (d) are non-invasive, requiring only blood or dried blood spots and urine collection. Refer to Table 3 for test included in the first tier, and the abnormalities that can be found in those tests in different IEMs.

Table 3. IEMs identified by each of the Tier 1 diagnostic tests.
Diagnostic therapies
Additionally, diagnostic therapy should be simultaneously applied in infants, which will supplement testing in the determination of etiology, and avoid therapeutic delay. For PDE, 100 mg of pyridoxine should be given intravenously (on day 1–3); samples can be collected after initiation of treatment. It is important to emphasize that this should be administered in a controlled setting (with resuscitation equipment available), since up to 20% of patients will develop cerebral depression, with the consequent risk of apnea. If responsive, then therapy should be continued with pyridoxine 15–30 mg/kg/day orally or enteral until diagnostic confirmation (elevated AASA in urine, blood and/or CSF and absence of urine sulfocysteine and at least 1 disease-causing mutation in the ALDH7A1 gene). If seizures are unresponsive to pyridoxine during 3 or more days, then consider switching to pyridoxal 5′-phosphate (30 mg/kg/day orally divided in 4 doses) to treat PNPO deficiency. Patients who can swallow pills should do so, because aqueous PLP is subject to photodegradation, and these degradation products can activate hepatic stellate cells and might lead to hepatic complications (27). For younger children, the crushed tablet or content of the capsule should be dissolved immediately before administration, and the solution should be protected from light, to decrease the chance of photodegradation. Not all brands of PLP have the advertised amount of drug, with some brands have less or more content than advertised; in the UK PLP tablets from SolgarⓇ were found to be the most reliable (28).
Alternatively, start with pyridoxine 15–30 mg/kg/day orally or enterally until results become available (if intravenous pyridoxine administration is not possible); clinical improvement of seizures is seen within minutes of intravenous pyridoxine administration, while EEG improvement can be delayed (seen within hours), and clinical improvement with oral pyridoxine can take 3–7 days to manifest. For biotinidase and holocarboxylase synthetase deficiencies, start infants on biotin 10 mg daily (until diagnosis is ruled out).
Folinic acid-responsive seizures have been shown to be of similar genetic origin to PDE (29); based upon this finding, we have decided not to include folinic acid in the initial diagnostic therapy step. Additionally, administration of folinic acid prior to extraction of a CSF sample may confound diagnostic accuracy by masking deficiency of methyltetrahydrofolate.
Tier 2: Additional Testing Under the Right Clinical Circumstances
Additional tests may be ordered by the clinician according to symptomatology and index of suspicion. This can be done in parallel to the 1st tier mentioned above, or sequentially. The list of these tests can be found in Table 4, along with the main clinical findings that might elicit suspicion of one of the treatable or non-treatable IEMs diagnosed with these tests.

Table 4. IEMs identified by Tier 2 diagnostic tests.
Given the large number of treatable IEMs presenting with epilepsy in infancy, and given that there are no clear clinical indicators that would set them apart from other epilepsy syndromes, we recommend that all infants presenting with epilepsy should undergo a lumbar puncture and CSF biochemical testing. In fact, the younger the infant, the more likely a lumbar puncture will be performed in order to rule out meningitis or viral encephalitis. It should be noted that the blood sample for glucose should always be obtained before the CSF sample is obtained, as otherwise the catecholaminergic response to the lumbar puncture would increase the serum glucose and alter the ratio. Similarly, the lumbar puncture should be collected while the patient is off intravenous dextrose, and fasting. Specific cutoffs for establishing the diagnosis of GLUT1 deficiency based on CSF glucose, CSF lactate, and CSF-to-blood glucose ratio have been published (30). Since the blood sample would be obtained before the lumbar puncture, a sample for plasma amino acids should be collected at the same time. For sampling of CSF neurotransmitters, it is important to note that a rostrocaudal gradient exists for metabolites derived from substances produced in the brain; this means that the more CSF that is drawn, the higher the values of HVA and 5HIAA will go–in fact, values can double with every 5 ml of CSF drawn (31). The reference ranges are established by each laboratory using a specific CSF fraction. Thus, clinicians should adhere to a sampling protocol with numbered tubes, following the specific instructions provided by the laboratory performing the test. Gradients also exist for certain amino acids, with GABA and taurine exhibiting a rostrocaudal gradient, while alanine and asparagine show a reverse gradient (32). Additionally, tetrahydrobiopterin (BH4) is extremely sensitive to oxygen and light, so the tube used to collect BH4 should contain antioxidants and stored in dark to protect the sample integrity. Finally, the CSF samples should be immediately frozen at the bedside. However, if the lumbar puncture was traumatic, blood cells should be removed by centrifugation (to avoid metabolite oxidation by products of hemolyzed RBCs), and only clear CSF should be frozen in new tubes (31). Although neurotransmitter analysis may still be possible, the serum component of the blood is not removed by centrifugation, and thus the CSF amino acid analysis would still have major artifacts in the setting of a traumatic lumbar puncture. Hence, a repeat lumbar puncture might be needed in this setting.
Genomics, Metabolomics, and Multi-Omics Approaches
During the past few years, the field of genomics has seen the advent of faster and cheaper technologies that have revolutionized clinical care. In the field of epilepsy in particular, a recent paper described a diagnostic yield of 14% (49 diagnosed patients out of 349 patients with drug-resistant pediatric epilepsy) when 30 genes were sequenced, while expanding the sequencing panel to 95 genes increased the diagnostic yield to 20.3% (71/349 patients) (33). It is important that if any such epilepsy sequencing panel is offered, that all genes known to be associated with our list of treatable metabolic epilepsies be included, given the important management implications.
A recent study described a small cohort of 32 patients with epileptic encephalopathy who had undergone prior testing including neurologic and genetics consultations, metabolic screening, neuroimaging, neurophysiologic studies, chromosomal microarray, and targeted genetic testing. In this cohort, the diagnostic yield of whole exome sequencing was 50% (16/32 patients), and it was the most cost-effective way to reach a diagnosis, with a cost approximately 10 times less than a standard diagnostic approach (34). There are, however, limitations to the use of either a sequencing panel or whole exome sequencing. As an example, a recent study integrated alternative transcripts for known neonatal epilepsy genes with RNA-Seq, and found that in 30% of cases (89 brain-expressed alternative coding regions in 292 neonatal epilepsy genes) the alternative transcripts corresponded to a noncoding segment in the canonical transcript analyzed by standard clinical tests (35). This is important because in some cases, pathogenic variants were found in these non-canonical brain transcripts; furthermore, half to two-thirds of these alternative coding regions are captured by common exon capture kits, even though these normally fall out of the scope of analysis. Another limitation of the next-generation sequencing approach is that in general the turnaround time for results is longer than for the standard biochemical tests mentioned so far; thus, the biochemical workup for treatable etiologies of metabolic epilepsies should not be postponed, and should occur previously to or concurrently with the genomic workup. Also, the results can be complimentary, with biochemical findings confirming or ruling out the functional effect of variants of unknown significance, while in other cases with negative exome results, biochemical abnormalities may point to the perturbed pathway enabling a targeted or manual inspection of candidate genes for any missed or cryptic variants.
Another diagnostic possibility available in this day and age is that of metabolomics, which is widely available on a research basis, but is only offered on a clinical basis by a single laboratory in the US. In this laboratory, the use of clinical metabolomics led to the diagnosis of three patients with GABA transaminase deficiency, siblings with L-2-hydroxyglutaric aciduria, one patient with aromatic amino acid decarboxylase deficiency, and one patient with adenylosuccinase deficiency in a single year (36). In addition to diagnostics, the use of metabolomics and the combined approach of genomics and metabolomics approaches can lead to the identification of novel biomarkers, and can be used to confirm pathogenicity of variants of unknown significance (37).
Referral to Metabolic Diseases Specialist
For any patient who shows abnormalities on any of the tier 1 or 2 metabolic tests (even before confirmation of an IEM), the biochemical physician on-call should be contacted as soon as possible for further diagnostic testing and management. Other features that should prompt referral to a metabolic specialist include
• Progressive symptoms suggestive of neurodegeneration
• Developmental regression or plateauing
• Significant behavioral deterioration from baseline pattern
• Refractory seizures
• Unexplained movement disorder
• MRI/S of the brain showing unexplained abnormalities
• Recurrent/unexplained emesis
• Coarse facial features
• Hepato- and/or splenomegaly
• X-ray evidence of skeletal dysplasia
• Documented episodes of:
- Hypoglycemia
- Significant metabolic acidosis
- Ketonuria (unusual for clinical circumstances).
The full diagnostic algorithm can be found in Figure 1.
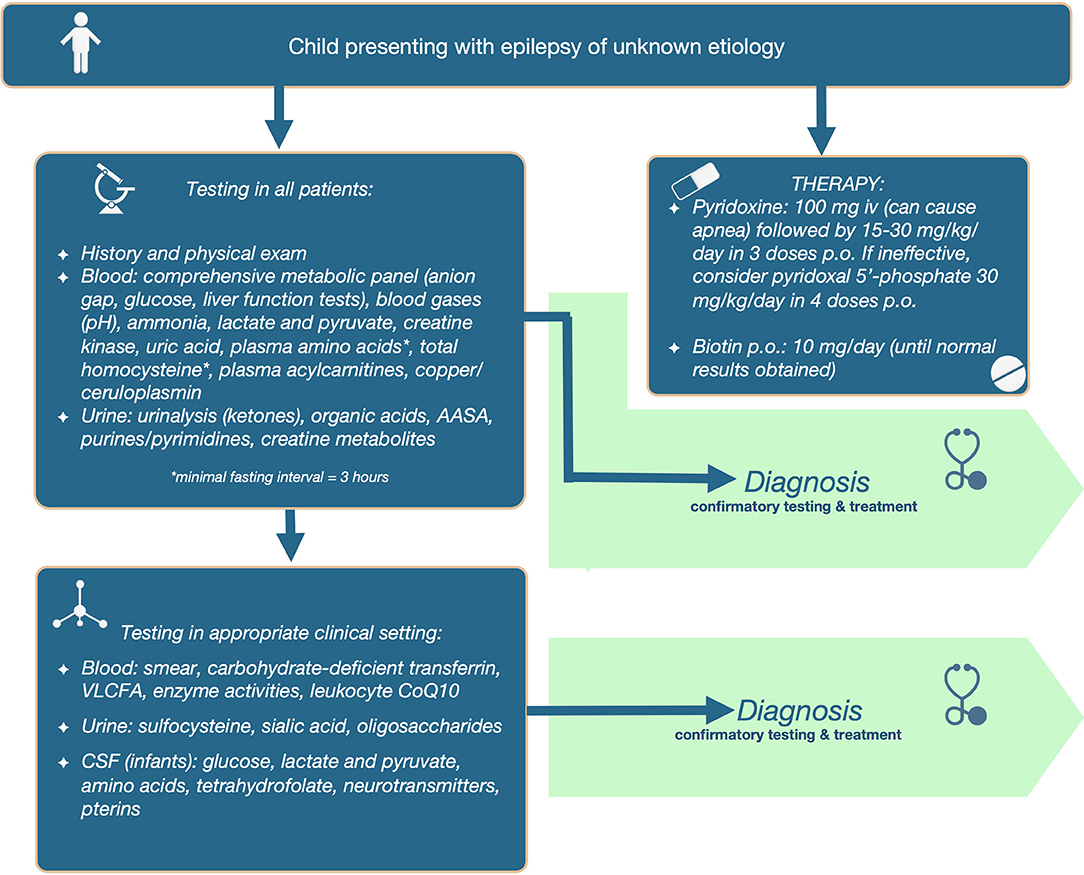
Figure 1. Diagnostic algorithm for metabolic epilepsies.
Conclusion
Based on a literature review, we provide an updated algorithm for the diagnosis of metabolic epilepsies, taking into account the description of novel IEMs in recent years up to 2018. The clinician's expertise and insights into the patient's clinical presentation remain paramount in the diagnostic work-up, as reflected in our algorithm. In fact, for several IEMs, treatment should be started even before confirmation of diagnosis. Our protocol includes tests chosen according to amenability to treatment of the IEMs potentially identified by the test, diagnostic yield, availability, and affordability. This algorithm is meant to provide a structure to the diagnostic workup, but does not claim to be exhaustive. In fact, the current protocol should be regularly updated in order to accommodate newly discovered IEMs and treatments in the future.
To conclude, this protocol has been developed as a standardized testing procedure to increase diagnostic efficacy of those IEMs amenable to treatment as well as reduce costs and diagnostic delay. While this protocol has been organized based upon literature and clinical observations, it is ultimately reliant upon the attending clinician's best judgment to implement an optimal investigative plan according to established parameters. In fact, the current protocol should be regularly updated in order to accommodate newly discovery IEMs, such as uridine-responsive encephalopathy due to biallelic CAD mutations and vitamin B6-responsive epileptic encephalopathy due to biallelic PLPBP mutations (38, 39). Aside from exome sequencing, entry into the clinical arena of integrated multi-omics analyses such as metabolomics and transcriptomics will certainly catalyze such discoveries further (40). Finally, approval of novel treatments such as gene therapy for metachromatic leukodystrophy (41, 42) will require regular updates of the current protocol, and this is favorable news for our patients and families.
Author Contributions
CvK and GH designed the study. LT, JL, and BS collected data. CvK, GH, and CF wrote the manuscript and designed the algorithm. All authors contributed the lit review, edited and approved manuscript for publication.
Funding
CvK is supported by a Stichting Metakids salary award.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge our colleague Dr. Sylvia Stockler (Vancouver, CA) for her contribution to the TIDE diagnostic algorithm, which served as a basis for the algorithm described here.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2018.01016/full#supplementary-material
Supplementary Table 1. Comprehensive list of “metabolic epilepsies,” defined as inborn errors of metabolism presenting with epilepsy.
References
1. Zack MM, Kobau R. National and state estimates of the numbers of adults and children with active epilepsy–United States, 2015. MMWR Morb Mortal Wkly Rep. (2017) 66:821–5. doi: 10.15585/mmwr.mm6631a1
2. Tekgul H, Gauvreau K, Soul J, Murphy L, Robertson R, Stewart J, et al. The current etiologic profile and neurodevelopmental outcome of seizures in term newborn infants. Pediatrics (2006) 117:1270–80. doi: 10.1542/peds.2005-1178
3. Mastrangelo M, Van Lierde A, Bray M, Pastorino G, Marini A, Mosca F. Epileptic seizures, epilepsy and epileptic syndromes in newborns: a nosological approach to 94 new cases by the 2001 proposed diagnostic scheme for people with epileptic seizures and with epilepsy. Seizure (2005) 14:304–11. doi: 10.1016/j.seizure.2005.04.001
4. Valayannopoulos V, Poll-The BT. Diagnostic work-up in acute conditions of inborn errors of metabolism and storage diseases. Handb Clin Neurol. (2013) 113:1553–62. doi: 10.1016/B978-0-444-59565-2.00025-3
5. Mills PB, Footitt EJ, Mills KA, Tuschl K, Aylett S, Varadkar S, et al. Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency). Brain J Neurol. (2010) 133:2148–59. doi: 10.1093/brain/awq143
6. Guerriero RM, Patel AA, Walsh B, Baumer FM, Shah AS, Peters JM, et al. Systemic manifestations in Pyridox(am)ine 5'-phosphate oxidase deficiency. Pediatr Neurol. (2017) 76:47–53. doi: 10.1016/j.pediatrneurol.2017.05.024
7. van Karnebeek CDM, Stockler S. Treatable inborn errors of metabolism causing intellectual disability: a systematic literature review. Mol Genet Metab. (2012) 105:368–81. doi: 10.1016/j.ymgme.2011.11.191
8. van Karnebeek CD, Stockler-Ipsiroglu S. Early identification of treatable inborn errors of metabolism in children with intellectual disability: the treatable intellectual disability endeavor protocol in British Columbia. Paediatr Child Health (2014) 19:469–71. doi: 10.1093/pch/19.9.469
9. Lee JJY, Wasserman WW, Hoffmann GF, van Karnebeek CDM, Blau N. Knowledge base and mini-expert platform for the diagnosis of inborn errors of metabolism. Genet Med. (2018) 20:151–8. doi: 10.1038/gim.2017.108
10. Ferreira CR, van Karnebeek CDM, Vockley J, Blau N. A proposed nosology of inborn errors of metabolism. Genet Med. (2018). doi: 10.1038/s41436-018-0022-8. [Epub ahead of print].
11. van Karnebeek CDM, Shevell M, Zschocke J, Moeschler JB, Stockler S. The metabolic evaluation of the child with an intellectual developmental disorder: diagnostic algorithm for identification of treatable causes and new digital resource. Mol Genet Metab. (2014) 111:428–38. doi: 10.1016/j.ymgme.2014.01.011
12. Sharma S, Prasad AN. Inborn errors of metabolism and epilepsy: current understanding, diagnosis, and treatment approaches. Int J Mol Sci. (2017) 18:1384. doi: 10.3390/ijms18071384
13. Lagan NC, Gorman KM, Shahwan A, King MD. Teaching video neuroimages: epilepsia partialis continua in an adolescent with preexisting focal epilepsy. Neurology (2017) 89:e274–5. doi: 10.1212/WNL.0000000000004713
14. Barba C, Darra F, Cusmai R, Procopio E, Dionisi Vici C, Keldermans L, et al. Congenital disorders of glycosylation presenting as epileptic encephalopathy with migrating partial seizures in infancy. Dev Med Child Neurol. (2016) 58:1085–91. doi: 10.1111/dmcn.13141
15. Mikati AG, Abu Gheida I, Shamseddine A, Mikati MA, Karam PE. Epileptic and electroencephalographic manifestations of guanidinoacetate-methyltransferase deficiency. Epileptic Disord. (2013) 15:407–16. doi: 10.1684/epd.2013.0609
16. Bahi-Buisson N, Kaminska A, Nabbout R, Barnerias C, Desguerre I, De Lonlay P, et al. Epilepsy in menkes disease: analysis of clinical stages. Epilepsia (2006) 47:380–6. doi: 10.1111/j.1528-1167.2006.00432.x
17. Pong AW, Geary BR, Engelstad KM, Natarajan A, Yang H, De Vivo DC. Glucose transporter type I deficiency syndrome: epilepsy phenotypes and outcomes. Epilepsia (2012) 53:1503–10. doi: 10.1111/j.1528-1167.2012.03592.x
18. Arsov T, Mullen SA, Rogers S, Phillips AM, Lawrence KM, Damiano JA, et al. Glucose transporter 1 deficiency in the idiopathic generalized epilepsies. Ann Neurol. (2012) 72:807–15. doi: 10.1002/ana.23702
19. Baumgart A, Spiczak Sv, Verhoeven-Duif NM, Møller RS, Boor R, Muhle H, et al. Atypical vitamin B6 deficiency: a rare cause of unexplained neonatal and infantile epilepsies. J Child Neurol. (2014) 29:704–7. doi: 10.1177/0883073813505354
20. Klepper J, Scheffer H, Leiendecker B, Gertsen E, Binder S, Leferink M, et al. Seizure control and acceptance of the ketogenic diet in GLUT1 deficiency syndrome: a 2- to 5-year follow-up of 15 children enrolled prospectively. Neuropediatrics (2005) 36:302–8. doi: 10.1055/s-2005-872843
21. Pascual JM, Liu P, Mao D, Kelly DI, Hernandez A, Sheng M, et al. Triheptanoin for glucose transporter type I deficiency (G1D): modulation of human ictogenesis, cerebral metabolic rate, and cognitive indices by a food supplement. JAMA Neurol. (2014) 71:1255–65. doi: 10.1001/jamaneurol.2014.1584
22. Tharp BR. Unique EEG pattern (comb-like rhythm) in neonatal maple syrup urine disease. Pediatr Neurol. (1992) 8:65–8. doi: 10.1016/0887-8994(92)90056-5
23. Nissenkorn A, Michelson M, Ben-Zeev B, Lerman-Sagie T. Inborn errors of metabolism: a cause of abnormal brain development. Neurology (2001) 56:1265–72. doi: 10.1212/WNL.56.10.1265
24. Tsuji M, Aida N, Obata T, Tomiyasu M, Furuya N, Kurosawa K, et al. A new case of GABA transaminase deficiency facilitated by proton MR spectroscopy. J Inherit Metab Dis. (2010) 33:85–90. doi: 10.1007/s10545-009-9022-9
25. Ferreira CR, Silber MH, Chang T, Murnick JG, Kirmse B. Cerebral lipid accumulation detected by MRS in a child with carnitine palmitoyltransferase 2 deficiency: a case report and review of the literature on genetic etiologies of lipid peaks on MRS. JIMD Rep. (2016) 28:69–74. doi: 10.1007/8904_2015_506
26. McAdams RM, Richards TL. Detection of nonketotic hyperglycinemia in a neonate using proton magnetic resonance spectroscopy. Radiol Case Rep. (2009) 4:310. doi: 10.2484/rcr.v4i4.310
27. Sudarsanam A, Singh H, Wilcken B, Stormon M, Arbuckle S, Schmitt B, et al. Cirrhosis associated with pyridoxal 5'-phosphate treatment of pyridoxamine 5'-phosphate oxidase deficiency. JIMD Rep. (2014) 17:67–70. doi: 10.1007/8904_2014_338
28. Mohamed-Ahmed AHA, Wilson MP, Albuera M, Chen T, Mills PB, Footitt EJ, et al. Quality and stability of extemporaneous pyridoxal phosphate preparations used in the treatment of paediatric epilepsy. J Pharm Pharmacol. (2017) 69:480–8. doi: 10.1111/jphp.12701
29. Gallagher RC, Van Hove JLK, Scharer G, Hyland K, Plecko B, Waters PJ, et al. Folinic acid-responsive seizures are identical to pyridoxine-dependent epilepsy. Ann Neurol. (2009) 65:550–6. doi: 10.1002/ana.21568
30. Leen WG, Wevers RA, Kamsteeg EJ, Scheffer H, Verbeek MM, Willemsen MA. Cerebrospinal fluid analysis in the workup of GLUT1 deficiency syndrome: a systematic review. JAMA Neurol. (2013) 70:1440–4. doi: 10.1001/jamaneurol.2013.3090
31. Hyland K. Cerebrospinal fluid analysis in the diagnosis of treatable inherited disorders of neurotransmitter metabolism. Future Neurol. (2006) 1:593–603. doi: 10.2217/14796708.1.5.593
32. Crawford PM, Lloyd KG, Chadwick DW. CSF gradients for amino acid neurotransmitters. J Neurol Neurosurg Psychiatr. (1988) 51:1193–200. doi: 10.1136/jnnp.51.9.1193
33. Parrini E, Marini C, Mei D, Galuppi A, Cellini E, Pucatti D, et al. Diagnostic targeted resequencing in 349 patients with drug-resistant pediatric epilepsies identifies causative mutations in 30 different genes. Hum Mutat. (2017) 38:216–25. doi: 10.1002/humu.23149
34. Palmer EE, Schofield D, Shrestha R, Kandula T, Macintosh R, Lawson JA, et al. Integrating exome sequencing into a diagnostic pathway for epileptic encephalopathy: evidence of clinical utility and cost effectiveness. Mol Genet Genomic Med. (2018) 6:186–99. doi: 10.1002/mgg3.355
35. Bodian DL, Kothiyal P, Hauser NS. Pitfalls of clinical exome and gene panel testing: alternative transcripts. Genet Med. (2018). doi: 10.1038/s41436-018-0319-7. [Epub ahead of print].
36. Wagle M, Donti T, Sun Q, Elsea S, Emrick L. Clinical metabolomic profiling for the diagnosis of neurometabolic disorders for global developmental delay, Seizures (I15.002). Neurology (2016) 86:I15.002. Available online at: http://n.neurology.org/content/86/16_Supplement/I15.002/tab-article-info
37. Crowther LM, Poms M, Plecko B. Multiomics tools for the diagnosis and treatment of rare neurological disease. J Inherit Metab Dis. (2018) 41:425–34. doi: 10.1007/s10545-018-0154-7
38. Koch J, Mayr JA, Alhaddad B, Rauscher C, Bierau J, Kovacs-Nagy R, et al. CAD mutations and uridine-responsive epileptic encephalopathy. Brain J Neurol. (2017) 140:279–86. doi: 10.1093/brain/aww300
39. Darin N, Reid E, Prunetti L, Samuelsson L, Husain RA, Wilson M, et al. Mutations in PROSC disrupt cellular pyridoxal phosphate homeostasis and cause vitamin-B6-dependent epilepsy. Am J Hum Genet. (2016) 99:1325–37. doi: 10.1016/j.ajhg.2016.10.011
40. van Karnebeek CDM, Wortmann SB, Tarailo-Graovac M, Langeveld M, Ferreira CR, van de Kamp JM, et al. The role of the clinician in the multi-omics era: are you ready? J Inherit Metab Dis. (2018) 41:571–82. doi: 10.1007/s10545-017-0128-1
41. Sessa M, Lorioli L, Fumagalli F, Acquati S, Redaelli D, Baldoli C, et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet Lond Engl. (2016) 388:476–87. doi: 10.1016/S0140-6736(16)30374-9
Keywords: inborn errors of metabolism, metabolic epilepsy, seizures, diagnostic algorithm, treatment
Citation: van Karnebeek CDM, Sayson B, Lee JJY, Tseng LA, Blau N, Horvath GA and Ferreira CR (2018) Metabolic Evaluation of Epilepsy: A Diagnostic Algorithm With Focus on Treatable Conditions. Front. Neurol. 9:1016. doi: 10.3389/fneur.2018.01016
Received: 07 August 2018; Accepted: 12 November 2018;
Published: 03 December 2018.
Edited by:
Giovanni Stevanin, INSERM U1127 Institut du Cerveau et de la Moelle épinière, FranceReviewed by:
Filippo M. Santorelli, Fondazione Stella Maris (IRCCS), ItalyEric LeGuern, INSERM, France
Copyright © 2018 van Karnebeek, Sayson, Lee, Tseng, Blau, Horvath and Ferreira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clara D. M. van Karnebeek, Yy5kLnZhbmthcm5lYmVla0BhbWMudXZhLm5s
Carlos R. Ferreira, ZmVycmVpcmFjckBtYWlsLm5paC5nb3Y=