 Maria-Letizia Campanari
Maria-Letizia Campanari Annis-Rayan Bourefis
Annis-Rayan Bourefis Edor Kabashi
Edor Kabashi- 1Sorbonne Université, Université Pierre et Marie Curie, Université de Paris 06, Unité Mixte 75, Institut National de la Santé et de la Recherche Médicale (INSERM) Unité 1127, Centre National de la Recherche Scientifique, Unité Mixte de Recherche 7225 Institut du Cerveau et de la Moelle Épinière, Paris, France
- 2Imagine Institute, INSERM Unité 1163, Paris Descartes Université, Paris, France
Amyotrophic lateral sclerosis (ALS) represents the major adult-onset motor neuron disease. Both human and animal studies reveal the critical implication of muscle and neuromuscular junctions (NMJs) in the initial phase of this disease. Despite the common efforts, ALS diagnosis remains particularly challenging since many other disorders can overlap yielding similar clinical phenotypic features. A combination of further research on the NMJ parameters that are specific for this disease and laboratory tests are crucial for the early determination of specific changes in the muscle, as well as in motor neuron and the prediction of ALS progression. Also, it could provide a powerful tool in the discrimination of particular ALS and ALS-mimic cases and increase the efficacy of therapeutic treatments.
Amyotrophic Lateral Sclerosis
Amyotrophic Lateral Sclerosis (ALS) is a disease characterized by a progressive degeneration of upper motor neurons (MNs) in the motor cortex and lower motor neurons in the brainstem and the spinal cord. The death of these neurons leads to spasticity, weakness, and atrophy of the muscles, progressing to paralysis. The incidence of ALS in Europe is 2–16 per 100,000 each year (1), with respiratory failure being the predominant mode of death in patients within 3 years of diagnosis (2). The onset of the disease occurs prevalently during adulthood (peak age of 58–63 years) (3), though with a small proportion of early-onset disease in certain patients (before 35 years of age). ALS also shares neuropathological and genetic features with another neurodegenerative disorder, frontotemporal dementia (FTD) (4, 5), with many ALS patients showing some cognitive or behavioral changes. This has led to consider ALS and FTD as the ends of the same spectrum of disease (6).
Although the majority of ALS cases occur sporadically (sALS), there is a Mendelian inheritance in about 10% of the cases (familial ALS, fALS), mainly in an autosomal, dominant fashion (7). The two are clinically indistinguishable and a variety of genetic defects in more than 20 genetic loci have been linked with the ALS phenotype (8), with new genes constantly being identified in subsets of ALS patients (9–11). Four major genes which mutations are known to cause ALS are the f ollowing: chromosome 9 open reading frame 72 (C9orf72), superoxide dismutase 1 (SOD1), transactive response DNA-binding protein (TARDBP) and fused in sarcoma (FUS) (12–15). C9orf72 has an important role in membrane trafficking and autophagy (16), and SOD1 primary function is thought to be as a cytosolic and mitochondrial antioxidant enzyme, converting superoxide to molecular oxygen and hydrogen peroxide (17). TARDBP and FUS encode nucleic acid-binding proteins that reside in the nucleus, and are involved in multiple aspects of RNA processing, such as transcription and splicing [reviewed in (18)].
NMJ Involvement in ALS
Despite the progress in our understanding of the molecular pathogenesis linked to these genes, it is still unclear where the motor neuron dysfunction begins and the extrinsic factors that accelerate motor neuron degeneration. This led to the consideration of ALS as either a dying forward process that proposes an anterograde degeneration of motor neurons by glutamate excitotoxicity from the cortex, or a dying back phenomenon in which motor neuron degeneration starts distally at the nerve terminal or at the neuromuscular junction (NMJ) and progresses toward the cell body (3, 19). The NMJ is a tripartite synapse composed by the presynaptic motor neuron, the postsynaptic muscle and the synapse-associated glial cells (terminal Schwann cells, TSC) and allows the transmission of action potentials from motor neurons to muscles [reviewed in (20)]. In this complex structure, besides motor neuron degeneration, glial cells, and muscle fibers play also a major role in ALS onset and progression.
The muscle contribution in ALS development, through NMJs disassembly, is still a matter of debate. Nonetheless, increasing evidence points to the critical role of NMJ defects in the early stage of the disease in ALS patients [reviewed in (21)] and a variety of animal models have permitted important advances into exploring this hypothesis.
The human SOD1G93A transgenic mouse, the first and most studied ALS model, is the one that has yielded the majority of information about the muscular deficits in ALS (22). Spatiotemporal analysis of NMJs in SOD1G93A mouse revealed end-plates denervation before the appearance of clinical symptoms and neuron cell body loss (23), with the fast-fatigable synapses being more vulnerable to denervation (24). Because of its high expression in ALS muscle biopsies, the neurite outgrowth inhibitor Nogo-A was proposed as a factor responsible for motor nerve terminals repulsion and destabilization at the NMJ at very early asymptomatic stages (25, 26). This hypothesis was then confirmed in SOD1G93A mouse model, where genetic ablation of Nogo-A in muscle led to marked reduction of muscle denervation and prolonged survival (27). Morphological observation of NMJs in SOD1G93A also contributed to reinforce the dying back hypothesis, showing more detailed NMJ alterations prior to functional symptom onset (28). A detailed overview of the findings concerning neuromuscular defects in the SOD1G93A mouse model has been reviewed by Dupuis and colleagues (22).
Despite the predominant use of rodent models for studying pathomechanisms and potential therapeutic targets in ALS, the use of smaller animal models, like Drosophila melanogaster and zebrafish (Danio rerio), is continually increasing. Their advantages lie in their fast development allowing quick generation of lines, their availability and the ease in manipulating gene expression and in drug screening. In drosophila, studies showed locomotor defects, reduced life span, and anatomical defects at the NMJ, causing impairments in synaptic transmission, in loss and gain of function models of TARDBP (29, 30). Similar results were found for FUS. Gene deficiency and overexpression of FUS in Drosophila models caused decreased synaptic transmission, reduced number of presynaptic active zones, altered postsynaptic glutamate receptor subunit composition at the NMJ, motor neuron degeneration and impaired motor behavior (31, 32). Zebrafish studies have highlighted gain and loss of function mechanisms for TARDBP and FUS, demonstrating shorter axonal projections from motor neurons, premature and excessive branching, impaired synaptic transmission at the NMJ leading to swimming defects (33–35). C9orf72 gene has also been modeled in zebrafish in a loss-of-function model that displayed behavioral and cellular deficits related to locomotion (36). For more details about the different models, all the ALS gene mutations that have been modeled are summarized in a recent review by Van Damme et al. (37).
Altogether, fundamental research supports the crucial role that NMJ could play in ALS pathogenesis and its possible employment as efficient early marker of the disease.
ALS Diagnostic Challenges
The difficulty to diagnose ALS resides mainly in the existence of several mimic syndromes, unrelated to ALS but which present similar clinical features (38, 39).
Motor neuron diseases (MNDs) are classified in four main groups in which ALS represents the most common form (Table 1). Although these diseases affect people in different ways, they share several symptoms due to motor neuron loss of function. All of them present progressive weakening of skeletal muscles, which eventually affects the ability to speak, swallow and breathe. ALS diagnosis is even more difficult if we add to the list other neurological conditions unrelated to MNDs which can mimic its early symptoms. Moreover, increasing evidences point to a possible direct implication of muscle in the early stage of the disease, adding myopathies to the list of ALS-mimic pathologies (Table 1).
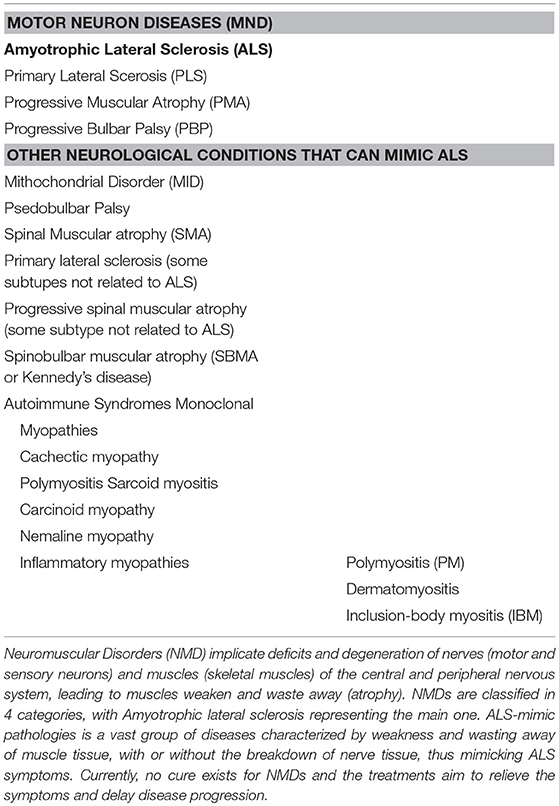
Table 1. General overview of neuromuscular diseases and ALS-mimic pathologies.
Standard diagnostic criteria for ALS have been established in 1991 [El Escorial criteria [EEC] (40)] and were revised in 1997 [AirlieHouse criteria [AHC] or El Escorial Revisited (41)]. Even though the essential requirements for ALS diagnosis were defined by these criteria, many neurologists and neuromuscular clinicians were missing the diagnosis, proving the low clinical accuracy of these diagnostic roles (42).
In 2008, electrodiagnostic studies, known as the Awaji criteria (43), were included in the clinical procedure to allow earlier and more accurate assessment of ALS diagnosis. However, the application of those sets of defined features are still insufficient to rule out other similar and related diseases (44, 45).
Methods for Diagnosis
Although the main ALS evaluation remains the clinical one, laboratory testing, based on advanced techniques of electrodiagnosis, neuroimaging, immunobiochemistry, and neurogenetics, is required for accurate ALS diagnosis.
Tests to rule out other neuromuscular conditions may include:
Electromyogram (EMG)
The needle EMG is the most important study in determining diagnostic certainty of ALS (46). During this test, a needle electrode is inserted through the skin into various muscles, starting with the most severely involved limb (Figure 1). The examination then progresses through four anatomical region: bulbar, cervical, thoracic, and lumbar. At least three anatomical regions have to be positive to this test to define ALS. The fasciculation potential (FP) has been included in Awaji criteria as a hallmark of ALS muscular denervation. In general, a decreased number of motor unit recruitment, with long duration of the motor unit potential, and abnormal spontaneous activity, are measured at the EMG in ALS patients [reviewed in (47)].
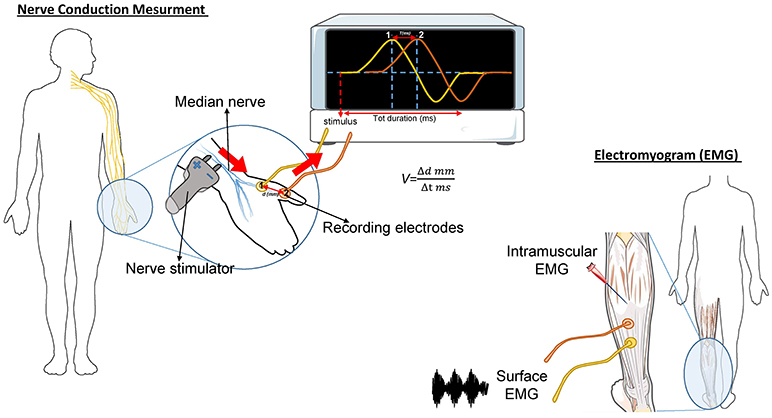
Figure 1. Schematic representation of the nerve conduction and muscle contraction studies. Nerve Conduction Velocity (NCV - left) measures the velocity and the quality of conduction of the electrical signal in a nerve. During the test, your nerve is stimulated, with an electrode attached to your skin. One or two more electrodes patches are placed on the skin over your nerve. The electrical impulse of the stimulated nerve pass from the stimulator to the other receiving electrode. The time (in milliseconds) spent by the impulse to move from a point to another, on the order of millimeters, represent the Velocity. In ALS, the impulse conduction is slower respect with control cases and is worsened by the increase of axonal degeneration. The electromyogram (EMG-right) measures the electrical activity of the muscles at rest and during contraction. There are two kinds of EMG: surface EMG and intramuscular EMG. In the first one the muscle activity is recorded by one or more electrodes patched on the skin and it asses the contractile response of superficial muscles. This approach presents several limitation since the result signal is influenced by the depth of the subcutaneous tissue at the site of the recording and by the discharges of adjacent muscles. With the intramuscular EMG, specific deep muscle activity is recorded by using one needle electrode inserted into the muscle. EMG and NCV tests are often done together to give more complete information.
Nerve Conduction Study (NCS)
This test measures how fast an electrical impulse moves through the nerve (Figure 1). During the test, one electrode placed on the skin stimulates the nerve of interest with a very mild electrical impulse. Variations in time spent to reach a second electrode can help in identifying a nerve damage. Whereas, EMG measures the electrical activity in the muscles, the nerve conduction study is specific for nerves and helps to localize the disorder among nerve, neuromuscular junction, and muscle. NCS is a powerful tool to discriminate ALS from axonal demyelination or conduction block impairments (48). NCS parameters are generally normal in ALS, albeit the presence of prolonged distal motor latency and slowed conduction velocity could be consistent with the diagnosis of ALS (49, 50). These changes suggest loss of large myelinated fibers, but also motor axons regeneration phenomena (50).
Magnetic Resonance Imaging (MRI)
This technique is able to produce detailed images of the brain and spinal cord, the latter with the advantage of simultaneously investigating the upper and lower motor neurons. During several years, its application was related to the exclusion of other disorders, as tumors or hernias that can display certain of the ALS-mimic symptoms (51). The evolution and improvement of this multimodal tool has recently become essential for the diagnosis of ALS. MRI scans can show cerebral degeneration and gray/white matter atrophy [reviewed in (52)], and also detect abnormalities in ALS muscle, likely due to denervation atrophy process (53).
Blood and Urine Tests
Testing hematological factors is helpful to exclude diseases that are capable of mimicking ALS symptoms. Recently, a population-based study, proposed serum albumin, creatinine levels, and lymphocyte count as markers for ALS, indicating muscle waste and inflammation respectively (54). Other markers potentially related to a better ALS outcome have been proposed: LDL/HDL levels, which are elevated in ALS plasma and represent a general unexplained hypermetabolism (55, 56); serum uric acid levels, which are decreased among ALS patients, further demonstrating the possible role of oxidative stress in the induction and propagation of the disease (57); serum ferritin levels which are elevated in ALS patients and could reflect perturbation in iron metabolism (58); concentrations of certain amino acids, which are decreased in ALS (59); levels of serum proinflammatory cytokines, such as IL-6, which are increased in ALS (60). Finally, high level of circulating AChE and metalloproteinases (MMP) have been reported in ALS plasma (61, 62) and although the exact source of these two classes of enzymes remains uncertain, it could in part reflect a disruption of extracellularly bound AChE at the NMJ and early change in the nerve-muscle integrity.
Spinal Tap (Lumbar Puncture)
Using this particular test, a small amount of cerebrospinal fluid (CSF) is taken from the lower back of the patient for laboratory tests. Thanks to its proximity to the central nervous system, the CSF is considered one preferred tissue to search for ALS biomarkers [reviewed in (63)]. Several markers for ALS have been identified in CSF such as Tau, TDP43, Nefl, and MMP levels [reviewed in (64)]. In particular, MMPs with their ability to digest collagen, proteoglycan, and laminin (65), may reflect ongoing destruction of the matrix which wraps synapses (66) and pathological changes at the brain-blood barrier (62).
Muscle Biopsy
With this technique, a small portion of muscle is removed by needle biopsy and sent to a laboratory for histopathological analysis. Rarely performed because of its painful and invasive nature, this tool is useful when ALS diagnosis is in doubt. Generally, ALS muscles present signs of active denervation/reinnervation and an increased number of atrophic fibers (67).
Genetic Testing
People with familial ALS (fALS) background can get an efficient diagnosis through genetic testing (68, 69). This technique may help ALS patients to understand the basis of their condition, and improve the genotype-specific treatments (70). Unluckily, there is a lack of consensus among clinicians above the definition of fALS, since newly genes related to ALS are continuously found (71). Nowadays, genetic testing is not wildly used because of its high cost and the belief that ALS genetics is not well-enough understood to provide a better treatment plan, as reported in 2017 in a study which involved 167 clinicians from 21 different countries around the world (71).
Examples of NMJ Pathologies ALS-mimic
Here we report some examples of ALS-mimic pathologies. The Spinal muscular atrophy (SMA) is an inherited MND that prevalently affects children. Its incidence is 1 per 11,000 live births (72). All forms of SMA are caused by the loss of SMN1, a gene implicated in axonal mRNA transport and snRNP biogenesis (73). Studies involving mice and fly mutants demonstrated that the probable origin of this pathology resides in the early loss of sensory information from proprioceptive neurons (74), which in turn causes degeneration of α motor neurons. In consequence, progressive muscle weakness, and, in severe cases, respiratory failure appear (75). Despite being considered a child's illness, the SMA type 4, that has an adult onset, overlaps with ALS diagnosis (76). Furthermore, like in ALS, several studies reveal the early implication of NMJs in SMA, with synaptic pathology prior to the appearance of clinical symptoms (77–81). However, the first evidence of neuromuscular pathology occurred at different time points of the disease progression, with presynaptic pathology preceding morphological changes at the endplate in ALS, and simultaneous pre and post-synaptic pathologies in SMA, suggesting the possibility to study this particular zone in diagnosis (81). Histochemical skin biopsy comparison was suggested as a powerful diagnostic tool in differentiating ALS and SMA, since the small collagen fibrils and the increased amount of amorphous material, which are characteristic of ALS, are not in SMA (82).
The Spinobulbar muscular atrophy [(SBMA) Kennedy's disease] is a X-linked hereditary lower motor neuron disease, where the expanded trinucleotide repeat (CAG > 37) in the androgen receptor gene (AR) causes its nuclear inclusions and impairment of its function (83, 84). The disease affects 1 per 200,000 males in Europe and Asia each year (85). In this pathology, degeneration of anterior horn cells of the spinal cord, where androgen receptors are widely expressed, is observed (86, 87). Although SBMA patients exhibit facial weakness as first sign of the disease, they progressively develop myopathic features, such as muscle atrophy and necrotic myofibers (88). Like ALS, SBMA disease reveals mixed pathological findings, with both myopathy and neurogenic atrophy features, which is the cause of misdiagnosis at the early stage of the disease.
Among the autoimmune syndromes, myasthenia gravis (MG) overlaps ALS syndrome. The annual incidence ranges from 3 to 30 per 1,000,000 people (89). In fact, the binding of autoantibodies to components of the NMJ in MG causes a characterized muscle weakness and fatigability (90). Even if acetylcholine receptor antibodies are considered to be highly specific for the diagnosis of MG, ALS patients can also present these autoantibodies at the blood test (91, 92). In these cases, it is very difficult to define the false positive cases and an experimental treatment with AChE inhibitors is necessary to differentiate MG from ALS (93).
The skeletal muscle disorders are represented with the term Myopathies. Myopathies hold a list of pathologies (Table 1) where muscle weakness can begin in the hands and feet (distal muscles) as well as in the muscles near the center of the body (proximal muscles) sometimes mimicking ALS features, confusing the diagnostic and the treatment decision. Among them, Inclusion Body Myositis (IBM) (94) is the most common ALS-mimic disease. It is the most common adult myopathy in 50 year-old persons and older, and its incidence is 3.5 per 100,000 (95). It is characterized by inflammatory cells surrounding and invading non-necrotic muscle fibers, rimmed vacuoles, congophilic inclusions, and protein aggregates in muscle (96, 97). In this case, the unique way to exclude ALS is the muscle biopsy combined with quantitative electromyographic analysis, especially in those patients where disease progression is slow and atypical (98).
Conclusion
Amyotrophic Lateral Sclerosis (ALS) and MNDs are not yet curable. However, accurate diagnosis is crucial to provide adequate counseling and information about the prognosis and disease course, and to avoid inappropriate therapy. Moreover, a good diagnosis could furnish a more equal stratification of cases and be important in the choice of additional medical support, as for example nutritional intervention strategies or physical therapy.
Currently, there is not a common consensus in the use of laboratory analysis for ALS diagnosis. Basically, clinicians decide for the application of certain techniques based on their experience, expertise and hospital practice. Progress in molecular genetics and identification of specific biomarkers is ongoing, which will translate to a refined diagnostic certitude. Therefore, there is the emerging need to establish a widely accepted protocol for laboratory tests to discriminate the majority of cases that present clinical features resembling ALS.
Increasing human and animal evidence proposed NMJ impairments as possible biomarkers for detection and discrimination of ALS and mimic diseases in an early, preclinical stage. However, further studies are needed to understand how these impairments could be monitored and specifically treated.
Author Contributions
M-LC and A-RB contributed to conception and drafting. EK provided useful comments and reviewed the manuscript.
Funding
This study was funded by the ERC Consolidator Grant, ANR ToFU, E-rare ERA-NET program, ANR AFM, ARSLA, and the program Investissements d'avenir ANR-10-IAIHU-06 (EK).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Logroscino G, Traynor BJ, Hardiman O, Chiò A, Mitchell D, Swingler RJ, et al. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry (2010) 81:385–90. doi: 10.1136/jnnp.2009.183525
2. Chiò A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, et al. Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler. (2009) 10:310–23. doi: 10.3109/17482960802566824
3. Kiernan MC1, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, et al. Amyotrophic lateral sclerosis. Lancet (2011) 377:942–55. doi: 10.1016/S0140-6736(10)61156-7
4. Morita M, Al-Chalabi A, Andersen PM, Hosler B, Sapp P, Englund E, et al. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology (2006) 66:839–44. doi: 10.1212/01.wnl.0000200048.53766.b4
5. Vance C, Al-Chalabi A, Ruddy D, Smith BN, Hu X, Sreedharan J, et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain (2006) 129:868–76. doi: 10.1093/brain/awl030
6. Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. (2013) 14:248–64. doi: 10.1038/nrn3430
7. Strong MJ, Hudson AJ, Alvord WG. Familial amyotrophic lateral sclerosis, 1850-1989 : a statistical analysis of the world literature. Can J Neurol Sci. (1991) 18:45–58.
8. Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. (2017) 377:162–72. doi: 10.1056/NEJMra1603471
9. Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. (2015) 18:631–6. doi: 10.1038/nn.4000
10. Brenner D, Müller K, Wieland T, Weydt P, Böhm S, Lulé D et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain (2016) 139:e28. doi: 10.1093/brain/aww033
11. Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, et al. TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron (2017) 95:808–16.e9. doi: 10.1016/j.neuron.2017.07.025
12. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature (1993) 362:59–62. doi: 10.1038/362059a0
13. Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron (2011) 72:257–68. doi: 10.1016/j.neuron.2011.09.010
14. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science (2008) 319:1668–72. doi: 10.1126/science.1154584
15. Kwiatkowski TJ, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science (2009) 547:1205–9. doi: 10.1126/science.1166066
16. Nassif M, Woehlbier U, Manque PA. The enigmatic role of C9ORF72 in autophagy. Front Neurosci. (2017) 11:442. doi: 10.3389/fnins.2017.00442
17. Bunton-Stasyshyn RKA, Saccon RA, Fratta P, Fisher EMC. SOD1 Function and its implications for amyotrophic lateral sclerosis pathology: new and renascent themes. Neuroscientist (2015) 21:519–29. doi: 10.1177/1073858414561795
18. Guerrero EN, Wang H, Mitra J, Hegde PM, Stowell SE, Liachko NF, et al. TDP-43/FUS in motor neuron disease: complexity and challenges. Prog Neurobiol. (2016) 145–6:78–97. doi: 10.1016/j.pneurobio.2016.09.004
19. Dadon-Nachum M, Melamed E, Offen D. The ‘dying-back' phenomenon of motor neurons in ALS. J Mol Neurosci. (2011) 43:470–477. doi: 10.1007/s12031-010-9467-1
20. Campanari M-L, García-Ayllón M-S, Ciura S, Sáez-Valero J, Kabashi E. Neuromuscular junction impairment in amyotrophic lateral sclerosis: reassessing the role of acetylcholinesterase. Front Mol Neurosci. (2016) 9:160. doi: 10.3389/fnmol.2016.00160
21. Cappello V, Francolini M. Neuromuscular junction dismantling in amyotrophic lateral sclerosis. Int J Mol Sci. (2017) 18:E2092. doi: 10.3390/ijms18102092
22. Dupuis L, Loeffler J-P. Neuromuscular junction destruction during amyotrophic lateral sclerosis: insights from transgenic models. Curr Opin Pharmacol. (2009) 9:341–6. doi: 10.1016/j.coph.2009.03.007
23. Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. (2004) 185:232–40. doi: 10.1016/j.expneurol.2003.10.004
24. Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. (2000) 20:2534–42. doi: 10.1523/JNEUROSCI.20-07-02534.2000
25. Jokic N, Gonzalez de Aguilar JL, Pradat PF, Dupuis L, Echaniz-Laguna A, Muller A, et al. Nogo expression in muscle correlates with amyotrophic lateral sclerosis severity. Ann Neurol. (2005) 57:553–6. doi: 10.1002/ana.20420
26. Bruneteau G, Bauché S, Gonzalez de Aguilar JL, Brochier G, Mandjee N, Tanguy ML, et al. Endplate denervation correlates with Nogo-A muscle expression in amyotrophic lateral sclerosis patients. Ann Clin Transl Neurol. (2015) 2:362–72. doi: 10.1002/acn3.179
27. Jokic N, Gonzalez de Aguilar JL, Dimou L, Lin S, Fergani A, Ruegg MA, et al. The neurite outgrowth inhibitor Nogo-A promotes denervation in an amyotrophic lateral sclerosis model. EMBO Rep. (2006) 7:1162–7. doi: 10.1038/sj.embor.7400826
28. Clark JA, Southam KA, Blizzard CA, King AE, Dickson TC. Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. J Chem Neuroanat. (2016) 76(Pt A):35–47. doi: 10.1016/j.jchemneu.2016.03.003
29. Feiguin F, Godena VK, Romano G, D'Ambrogio A, Klima R, Baralle FE, et al. Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. (2009) 583:1586–92. doi: 10.1016/j.febslet.2009.04.019
30. Diaper DC, Adachi Y, Sutcliffe B, Humphrey DM, Elliott CJ, Stepto A, et al. Loss and gain of Drosophila TDP-43 impair synaptic efficacy and motor control leading to age-related neurodegeneration by loss-of-function phenotypes. Hum Mol Genet. (2013) 22:1539–57. doi: 10.1093/hmg/ddt005
31. Sasayama H, Shimamura M, Tokuda T, Azuma Y, Yoshida T, Mizuno T, et al. Knockdown of the Drosophila fused in sarcoma (FUS) homologue causes deficient locomotive behavior and shortening of motoneuron terminal branches. PLoS ONE (2012) 7:e39483. doi: 10.1371/journal.pone.0039483
32. Machamer JB, Collins SE, Lloyd TE. The ALS gene FUS regulates synaptic transmission at the Drosophila neuromuscular junction. Hum Mol Genet. (2014) 23:3810–22. doi: 10.1093/hmg/ddu094
33. Kabashi E, Lin L, Tradewell ML, Dion PA, Bercier V, Bourgouin P, et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet. (2009) 19:671–83. doi: 10.1093/hmg/ddp534
34. Kabashi E, Bercier V, Lissouba A, Liao M, Brustein E, Rouleau GA, et al. Fus and tardbp but not sod1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet. (2011) 7:e1002214. doi: 10.1371/journal.pgen.1002214
35. Armstrong GAB, Drapeau P. Loss and gain of FUS function impair neuromuscular synaptic transmission in a genetic model of ALS. Hum Mol Genet. (2013) 22:4282–92. doi: 10.1093/hmg/ddt278
36. Ciura S, Lattante S, Le Ber I, Latouche M, Tostivint H, Brice A, et al. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol. (2013) 74:180–7 doi: 10.1002/ana.23946
37. Van Damme P, Robberecht W, Van Den Bosch L. Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis Model Mech. (2017) 10:537–49. doi: 10.1242/dmm.029058
39. Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman O. Amyotrophic lateral sclerosis mimic syndromes. Arch Neurol. (2000) 57:109–13. doi: 10.1001/archneur.57.1.109
40. Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2000) 1:293–9. doi: 10.1080/146608200300079536
41. Miller RG, Munsat TL, Swash M, Brooks BR. Consensus guidelines for the design and implementation of clinical trials in ALS. World Federation of Neurology committee on Research. J Neurol Sci. (1999) 169:2–12. doi: 10.1016/S0022-510X(99)00209-9
42. Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman OM. Clinical features of amyotrophic lateral sclerosis according to the El Escorial and airlie house diagnostic criteria. Arch. Neurol. (2000) 57:1171–6. doi: 10.1001/archneur.57.8.1171
43. De Carvalho M, Swash M. Awaji diagnostic algorithm increases sensitivity of El Escorial criteria for ALS diagnosis. Amyotroph Lateral Scler. (2009) 10:53–57. doi: 10.1080/17482960802521126
44. Haverkamp LJ, Appel V, Appel SH. Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain (1995) 118(Pt 3):707–19.
45. Davenport RJ, Swingler RJ, Chancellor AM, Warlow CP. Avoiding false positive diagnoses of motor neuron disease: lessons from the Scottish Motor Neuron Disease Register. Neurol Neurosurg Psychiatry (1996) 60:147–51.
46. Daube JR. Electrophysiologic studies in the diagnosis and prognosis of motor neuron diseases. Neurol Clin. (1985) 3:473–93. doi: 10.1016/S0733-8619(18)31017-X
47. Joyce NC, Carter GT. Electrodiagnosis in persons with amyotrophic lateral sclerosis. PM R (2013) 5:S89–95. doi: 10.1016/j.pmrj.2013.03.020
48. Duleep A, Shefner J. Electrodiagnosis of motor neuron disease. Phys Med Rehabil Clin N Am. (2013) 24:139–5. doi: 10.1016/j.pmr.2012.08.022
49. Argyriou AA, Polychronopoulos P, Talelli P, Chroni E. F wave study in amyotrophic lateral sclerosis: assessment of balance between upper and lower motor neuron involvement. Clin Neurophysiol. (2006) 117:1260–5. doi: 10.1016/j.clinph.2006.03.002
50. de Carvalho M. Nerve conduction studies in amyotrophic lateral sclerosis. Muscle Nerve (2000) 23:344–52. doi: 10.1002/(SICI)1097-4598(200003)23:3<344::AID-MUS5>3.0.CO;2-N
51. Filippi M, Agosta F, Abrahams S, Fazekas F, Grosskreutz J, Kalra S, et al. EFNS guidelines on the use of neuroimaging in the management of motor neuron diseases. Eur J Neurol. (2010) 17:526–e20. doi: 10.1111/j.1468-1331.2010.02951.x
52. Turner MR, Modo M. Advances in the application of MRI to amyotrophic lateral sclerosis. Expert Opin Med Diagn. (2010) 4:483–96. doi: 10.1517/17530059.2010.536836
53. Staff NP, Amrami KK, Howe BM. Magnetic resonance imaging abnormalities of peripheral nerve and muscle are common in amyotrophic lateral sclerosis and share features with multifocal motor neuropathy. Muscle Nerve (2015) 52:137–9. doi: 10.1002/mus.24630
54. Chiò A, Calvo A, Bovio G, Canosa A, Bertuzzo D, Galmozzi F, et al. Amyotrophic lateral sclerosis outcome measures and the role of albumin and creatinine. JAMA Neurol. (2014) 71:1134–42. doi: 10.1001/jamaneurol.2014.1129
55. Dupuis L, Corcia P, Fergani A, Gonzalez De Aguilar JL, Bonnefont-Rousselot D, Bittar R, et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology (2008) 70:1004–9. doi: 10.1212/01.wnl.0000285080.70324.27
56. Dorst J, Kühnlein P, Hendrich C, Kassubek J, Sperfeld AD, Ludolph AC. Patients with elevated triglyceride and cholesterol serum levels have a prolonged survival in amyotrophic lateral sclerosis. J Neurol. (2011) 258:613–7. doi: 10.1007/s00415-010-5805-z
57. Keizman D, Ish-Shalom M, Berliner S, Maimon N, Vered Y, Artamonov I, et al. Low uric acid levels in serum of patients with ALS: Further evidence for oxidative stress? J Neurol Sci. (2009) 285:95–9. doi: 10.1016/j.jns.2009.06.002
58. Qureshi M, Brown RH, Rogers JT, Cudkowicz ME. Serum ferritin and metal levels as risk factors for amyotrophic lateral sclerosis. Open Neurol J. (2008) 2:51–4. doi: 10.2174/1874205X00802010051
59. Ilzecka J, Stelmasiak Z, Solski J, Wawrzycki S, Szpetnar M. Plasma amino acids concentration in amyotrophic lateral sclerosis patients. Amino Acids (2003) 25:69–73. doi: 10.1007/s00726-002-0352-2
60. Ono S, Hu J, Shimizu N, Imai T, Nakagawa H. Increased interleukin-6 of skin and serum in amyotrophic lateral sclerosis. J Neurol Sci. (2001) 187:27–34. doi: 10.1016/S0022-510X(01)00514-7
61. Festoff BW, Fernandez HL. Plasma and red blood cell acetylcholinesterase in amyotrophic lateral sclerosis. Muscle Nerve (1981) 4:41–7. doi: 10.1002/mus.880040108
62. Niebroj-Dobosz I, Janik P, Sokołowska B, Kwiecinski H. Matrix metalloproteinases and their tissue inhibitors in serum and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Eur J Neurol. (2010) 17:226–31. doi: 10.1111/j.1468-1331.2009.02775.x
63. Barschke P, Oeckl P, Steinacker P, Ludolph A, Otto M. Proteomic studies in the discovery of cerebrospinal fluid biomarkers for amyotrophic lateral sclerosis. Expert Rev Proteomics (2017) 14:769–77. doi: 10.1080/14789450.2017.1365602
64. Shaw PJ, Williams R. Serum and cerebrospinal fluid biochemical markers of ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. (2000) 1(Suppl. 2):S61–7. doi: 10.1080/14660820050515773-1
65. Vincenti MP, Brinckerhoff CE. Signal transduction and cell-type specific regulation of matrix metalloproteinase gene expression: can MMPs be good for you? J Cell Physiol. (2007) 213:355–64. doi: 10.1002/jcp.21208
66. Lim GP, Backstrom JR, Cullen MJ, Miller CA, Atkinson RD, Tökés ZA. Matrix metalloproteinases in the neocortex and spinal cord of amyotrophic lateral sclerosis patients. J Neurochem. (1996) 67:251–9. doi: 10.1046/j.1471-4159.1996.67010251.x
67. Jensen L, Jørgensen LH, Bech RD, Frandsen U, Schrøder HD. Skeletal muscle remodelling as a function of disease progression in amyotrophic lateral sclerosis. Biomed Res Int. (2016) 2016:5930621. doi: 10.1155/2016/5930621
68. Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat. (2013) 34:812–26. doi: 10.1002/humu.22319
69. Ciura S, Sellier C, Campanari M-L, Charlet-Berguerand N, Kabashi E. The most prevalent genetic cause of ALS-FTD, C9orf72 synergizes the toxicity of ATXN2 intermediate polyglutamine repeats through the autophagy pathway. Autophagy (2016) 12:1406–8. doi: 10.1080/15548627.2016.1189070
70. Roggenbuck J, Quick A, Kolb SJ. Genetic testing and genetic counseling for amyotrophic lateral sclerosis: an update for clinicians. Genet Med. (2017) 19:267–74. doi: 10.1038/gim.2016.107
71. Vajda A, McLaughlin RL, Heverin M, Thorpe O, Abrahams S, Al-Chalabi A, et al. Genetic testing in ALS: a survey of current practices. Neurology (2017) 88:991–9. doi: 10.1212/WNL.0000000000003686
72. Kolb SJ, Kissel JT. Spinal muscular atrophy. Neurol Clin. (2015) 33:831–46. doi: 10.1016/j.ncl.2015.07.004
73. Burghes AHM, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. (2009) 10:597–609. doi: 10.1038/nrn2670
74. Fletcher EV, Mentis GZ. Motor circuit dysfunction in spinal muscular atrophy. Spinal Muscular Atrophy. (2017) 153–65. doi: 10.1016/B978-0-12-803685-3.00009-4
75. Crawford TO, Pardo CA. The Neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. (1996) 3:97–110. doi: 10.1006/nbdi.1996.0010
76. Farrar MA, Kiernan MC. The genetics of spinal muscular atrophy: progress and challenges. Neurotherapeutics (2015) 12:290–302. doi: 10.1007/s13311-014-0314-x
77. Cifuentes-Diaz C, Nicole S, Velasco ME, Borra-Cebrian C, Panozzo C, Frugier T, et al. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum Mol Genet. (2002) 11:1439–47. doi: 10.1093/hmg/11.12.1439
78. Kariya S, Park GH, Maeno-Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, et al. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet. (2008) 17:2552–69. doi: 10.1093/hmg/ddn156
79. Murray LM, Comley LH, Thomson D, Parkinson N, Talbot K, Gillingwater TH. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum Mol Genet. (2008) 17:949–62. doi: 10.1093/hmg/ddm367
80. Ling KKY, Gibbs RM, Feng Z, Ko C-P. Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum Mol Genet. (2012) 21:185–95. doi: 10.1093/hmg/ddr453
81. Comley LH, Nijssen J, Frost-Nylen J, Hedlund E. Cross-disease comparison of amyotrophic lateral sclerosis and spinal muscular atrophy reveals conservation of selective vulnerability but differential neuromuscular junction pathology. J Comp Neurol. (2016) 524:1424–42. doi: 10.1002/cne.23917
82. Ono S, Mannen T, Toyokura Y. Differential diagnosis between amyotrophic lateral sclerosis and spinal muscular atrophy by skin involvement. J Neurol Sci. (1989) 91:301–10. doi: 10.1016/0022-510X(89)90059-2
83. Li M, Miwa S, Kobayashi Y, Merry DE, Yamamoto M, Tanaka F, et al. Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol. (1998) 44:249–54. doi: 10.1002/ana.410440216
84. Takeyama K, Ito S, Yamamoto A, Tanimoto H, Furutani T, Kanuka H, et al. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron (2002) 35:855–64. doi: 10.1016/S0896-6273(02)00875-9
85. Fischbeck KH. Kennedy disease. J Inherit Metab Dis. (1997) 20:152–8. doi: 10.1023/A:1005344403603
86. Diaz-Abad M, Porter NC. Rapidly worsening bulbar symptoms in a patient with spinobulbar muscular atrophy. Neurol Int. (2013) 5:21. doi: 10.4081/ni.2013.e21
87. MacLean HE, Warne GL, Zajac JD. Spinal and bulbar muscular atrophy: androgen receptor dysfunction caused by a trinucleotide repeat expansion. J Neurol Sci. (1996) 135:149–57. doi: 10.1016/0022-510X(95)00284-9
88. Cortes CJ, Ling SC, Guo LT, Hung G, Tsunemi T, Ly L, et al. Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron (2014) 82:295–307. doi: 10.1016/j.neuron.2014.03.001
89. Casetta I, Groppo E, De Gennaro R, Cesnik E, Piccolo L, Volpato S, et al. Myasthenia gravis: a changing pattern of incidence. J Neurol. (2010) 257:2015–9. doi: 10.1007/s00415-010-5651-z
90. Tai H, Cui L, Guan Y, Liu M, Li X, Huang Y, et al. Amyotrophic lateral sclerosis and myasthenia gravis overlap syndrome: a review of two cases and the associated literature. Front Neurol. (2017) 8:218. doi: 10.3389/fneur.2017.00218
91. Abbott RJ, Holden D, Currie S. False positive anti-acetylcholine receptor antibodies in motorneurone disease. Lancet (1986) 1:906–7. doi: 10.1016/S0140-6736(86)91005-6
92. Ashizawa T. False positive anti-acetylcholine receptor antibodies in motorneurone disease. Lancet (1986) 1:1272. doi: 10.1016/S0140-6736(86)91408-X
93. Gold R, Hohlfeld R, Toyka KV. Progress in the treatment of myasthenia gravis. Ther Adv Neurol Disord. (2008) 1:36–51. doi: 10.1177/1756285608093888
94. Naddaf E, Barohn RJ, Dimachkie MM. Inclusion body myositis: update on pathogenesis and treatment. Neurotherapeutics (2018) 15:995–1005. doi: 10.1007/s13311-018-0658-8
95. Dimachkie MM, Barohn RJ. Inclusion body myositis. Neurol Clin. (2014) 32:629–46. doi: 10.1016/j.ncl.2014.04.001
96. Chahin N, Engel AG. Correlation of muscle biopsy, clinical course, and outcome in PM and sporadic IBM. Neurology (2008) 70:418–24. doi: 10.1212/01.wnl.0000277527.69388.fe
97. Chilingaryan A, Rison RA, Beydoun SR. Misdiagnosis of inclusion body myositis: two case reports and a retrospective chart review. J Med Case Rep. (2015) 9:169. doi: 10.1186/s13256-015-0647-z
Keywords: amyotrophic lateral sclerosis (ALS), axonopathy, neuromuscular junction (NMJ), dying back hypothesis, ALS-mimic diseases
Citation: Campanari M-L, Bourefis A-R and Kabashi E (2019) Diagnostic Challenge and Neuromuscular Junction Contribution to ALS Pathogenesis. Front. Neurol. 10:68. doi: 10.3389/fneur.2019.00068
Received: 29 October 2018; Accepted: 17 January 2019;
Published: 06 February 2019.
Edited by:
Pierre-francois Pradat, Hôpitaux Universitaires Pitié Salpêtrière, FranceReviewed by:
Robert M. Brownstone, University College London, United KingdomStephanie Rose Marie Duguez, Ulster University, United Kingdom
Copyright © 2019 Campanari, Bourefis and Kabashi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria-Letizia Campanari, bWFyaWEtbGV0aXppYS5jYW1wYW5hcmlAaW5zdGl0dXRpbWFnaW5lLm9yZw==
Edor Kabashi, ZWRvci5rYWJhc2hpQGluc3RpdHV0aW1hZ2luZS5vcmc=
†These authors have contributed equally to this work