 Shanshan Zhang
Shanshan Zhang Dongli Yuan
Dongli Yuan Ge Tan
Ge Tan- 1Department of Neurology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China
- 2The Institute of Medical Information, Chongqing Medical University, Chongqing, China
Primary systemic vasculitis can affect every structure in both the central and peripheral nervous system, causing varied neurological manifestations of neurological dysfunction. Early recognition of the underlying causes of the neurological symptoms can facilitate timely treatment and improve the prognosis. This review highlights the clinical manifestations of primary systemic vasculitis in the nervous system.
Introduction
Primary systemic vasculitis (PSV) can be defined as a heterogeneous group of uncommon diseases characterized by blood vessel inflammation and necrosis. Unlike secondary systemic vasculitis, which usually results from infections, connective tissue diseases, neoplasms, and drugs, PSV is considered to occur with no identified etiology (1). Nevertheless, recent researches have demonstrated that PSV might be associated with farming (2), silica exposure (2, 3), solvent exposure and hydrocarbons (2, 4, 5), allergy and family history of atopy (2, 6). Despite of unknown causes, two mechanisms, deposition of immune complexes and cell-mediated immunity, were found to possibly participate in the pathophysiology of PSV by causing immunological inflammation and necrosis of the vessel wall (7). And according to the classification criteria revised in 1990 by The American College of Rheumatology (8) and in 1994 and 2012 by the Chapel Hill Consensus Conferences (9, 10), the classification of PSV is based primarily on the major size of the affected vessels, although these disease-affected arteries may have overlaps in diameter.
Many neurologists have limited knowledge of PSV because most of these patients are treated by rheumatologists. However, both the central nervous system (CNS) and peripheral nervous system (PNS) are major targets in PSV and may become involved in the earliest stages; thus, patients may be referred to a neurologist first. Since the clinical manifestations of PSV are often non-specific, the differential diagnosis may be challenging.
CNS damage was reported to occur in 24% of cases with PSV, commonly due to antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis and polyarteritis nodosa. The range of clinical expressions of PSV's CNS damage is relatively wide, including cerebrovascular manifestations such as hemorrhagic and ischemic stroke caused by intra/extra-cerebral vascular stenosis, aneurysm, and sinus venous thrombosis, meningeal and brain parenchymal involvement resulted from granulomatosis and perivasculitis, and encephalopathy due to cytokine damage.
Compared with the frequency of central vasculitic involvement, vasculitic peripheral neuropathies are more common and have been reported to occur in 37% of patients with PSV (11), and 25% of patients experience symptoms of peripheral neuropathy at initial presentation (12). Generally, vasculitic peripheral neuropathies result from the inflammation of precapillary arteries in the nerves, such as eosinophilic granulomatosis with polyangiitis (60–80% of patients), polyarteritis nodosa (50–100% of patients), granulomatosis with polyangiitis (Wegener's granulomatosis, 13–26.6% of cases), cryoglobulinemia (70% of cases), and microscopic polyangiitis (6–18% of cases), usually presenting in a uniform pattern such as multiplex mononeuropathy (accounting for 59–85% of cases) and symmetric polyneuropathy (41% of cases) (11–13). Nerve conduction studies have revealed a much higher frequency of peripheral neurological involvement than that in clinical case series and studies. Asymptomatic vasculitic neuropathy was detected in 21 of 270 cases (7.8%) of biopsy-proven vasculitis (14).
In addition, the introduction of nerve sonography, single-photon emission computed tomography (SPECT), magnetic resonance imaging (MRI), magnetic resonance angiography (MRA), magnetic resonance spectroscopy (MRS), and apparent diffusion coefficient (ADC) have greatly improved the diagnostic rate and accuracy of vasculitic neuropathy.
Search Strategy
We performed an advanced literature search in PubMed and EMBASE for the period January 1, 1990, through September 1, 2017, using as a search query “systemic vasculitis” “nervous system.” Eligible studies were included according to the following criteria. Only human studies that were written in English were considered. All patients included in the studies were diagnosed according to the classification criteria defined by the American College of Rheumatology or the Chapel Hill Consensus Conference. Retrospective and prospective studies were included to determine the frequency of neurological manifestations. Case reports and case series were added in the reference list only when unusual features were described. Pediatric studies were excluded because the clinical features of children and newborns are different.
Large-Vessel Vasculitis
Large vessels were defined as the aorta, vena cava, and their major branches. Large-vessel vasculitis is composed of Takayasu arteritis and giant cell arteritis.
Takayasu Arteritis
Takayasu arteritis (TA) is a rare type of primary chronic inflammatory disease, causing stenosis, occlusion and aneurysm of arteries. TA mainly affects large-caliber arteries such as the aorta and its major branches in patients younger than 40 years old (more than 90% of patients are younger than 40), with a female predominance (female to male ratio is 10:1), and is more common in Asians (1). Studies have shown that more than half of patients with TA show neurological manifestations (15, 16) that range from dizziness/headaches to ischemic or hemorrhagic stroke.
Dizziness and headache are the most common complaints in patients with TA with neurological complications, accounting for ~78.1% (214/274) and 25.5% (70/274), respectively (16). However, headache and dizziness do not necessarily indicate CNS involvement. Visual disturbances affect 4.6–59.3% of patients, and 4–21.9% of TA patients with neurological manifestations were found to have syncope as the main manifestation.
Cerebrovascular complications have always been the focus of researchers. Transient ischemic attack accounts for 3–22.2% of the nervous system manifestations caused by TA. Stroke, as the most severe complication of TA, was found to occur in 10–20% of cases (16–18). Due to lack of knowledgement, the median and average delay between symptom onset and the diagnosis of TA are 2 years (19) and 52.4 months (16), respectively, which delays the treatment of vasculitis. The majority of the strokes caused by TA are ischemic strokes, mostly due to multiple and severe stenosis or occlusive lesions in the aortic arch and its major branches. Couture and colleagues (19) evaluated the impact of stroke on the prognosis of TA patients, and after a median of seven years of follow-up, 59% of the patients had neurological deficits, 35% suffered from stroke recurrence and 24% had epilepsy. However, cerebral aneurysms and subarachnoid hemorrhage are rarely observed in TA patients. The incidence of aneurysm in TA patients is no higher than that in the general population. However, when such incidents occur, aneurysms in TA patients have a high rate of multiplicity and commonly occur in the vertebrobasilar arterial system (20). Posterior reversible encephalopathy syndrome (PRES) is an uncommon neurological complication in TA patients, mainly affecting women under the age of 30, presenting as headache, epilepsy, and neurological deficits in most patients. The pathophysiological mechanism has been suggested to be both endothelial injury and hypertension caused by TA, and the use of immunosuppressants such as cyclosporine and tacrolimus may also be a factor in the development of PRES since they can also induce endothelial dysfunction (21).
In addition, rare complications such as Horner's syndrome and intracranial granulomatosis have been reported as initial manifestations of TA. Other unusual neurological manifestations of TA that have been reported include brachial plexus palsy due to axillary artery aneurysm, unilateral sensorineural hearing loss, subclavian stealing syndrome, Moya-Moya syndrome, multiple cranial nerve palsies, cavernous sinus syndrome, and hypertrophic pachymeningitis.
Giant Cell Arteritis
Giant cell arteritis (GCA) is the most common large vasculitis in Western countries and mainly affects extracranial branches of the carotid artery and/or the aorta and its large arterial branches, usually affecting people over the age of 50 (22).
Neuro-ophthalmological manifestation is the most frequent and serious event among neurological complications of GCA, accounting for 20–28.8% of affected patients (23, 24), with 15% of them suffering from permanent visual loss (25). Visual symptoms may be caused by ischemic optic neuropathy and retinal ischemia, among which anterior ischemic optic neuropathy accounts for the largest proportion.
Cerebrovascular disease is another severe neurological manifestation, occurring in ~5% of all GCA-related neurological complications. According to recent studies, the risk ratio of cerebrovascular accident in patients with GCA vs. non-GCA comparators was 1.40 (26), and the risk of cerebrovascular complications was higher during the first year (or month) after GCA diagnosis (27, 28), which means that cerebrovascular events often occur during the active period of the disease. Epidemiological study shows that stroke is observed in 1–3% of patients (25), among which cerebral infarction is the most common (58%), followed by subarachnoid hemorrhage (24%), and cerebral hemorrhage (18%) (29). The main reasons are considered to be, on the one hand, ischemia or occlusion caused by direct involvement of carotid and vertebrobasilar arteries and, on the other hand, atherosclerotic changes of the vessels caused by chronic inflammation. The incidence of vertebrobasilar artery vascular accident in patients with GCA (35%) has been shown to be higher than that of the general population (which is usually <15%) (27, 28), and one study showed that audiovestibular dysfunction is not uncommon in GCA patients (30). It is interesting to note that in a retrospective study (25), researchers found that most GCA patients who experienced a vertebrobasilar stroke had neurological symptom onset after the start of corticosteroid treatment. However, it is still being debated whether this situation is due to a direct effect of vasculitis or vascular occlusion promoted by the effects of corticosteroids.
Peripheral nervous system involvement was found in 1–14% of GCA patients, presenting as cranial neuropathies, multiple mononeuropathy, or polyneuropathies (24); existing case reports include multiple cranial nerve palsy, trigeminal autonomic cephalalgia, occipital neuralgia, Horner syndrome, peroneal nerve palsy, cervical radiculopathy, brachial plexopathy, hypertrophic pachymeningitis, and cavernous sinus syndrome.
Medium-Vessel Vasculitis
Medium vessels refer to the main visceral arteries and veins and their initial branches. Medium-vessel vasculitis consists of polyarteritis nodosa and Kawasaki disease.
Polyarteritis Nodosa
Polyarteritis nodosa (PAN) is a rare systemic necrotizing vasculitis mainly involving medium and small vessels, causing microaneurysm, stenosis, and thrombosis, therefore leading to ischemia or hemorrhage of the supplied tissues. PAN affects women more often than men, usually with an onset between 40 and 50 years of age (31). The condition may affect any organ; however, the most frequently affected organs are the peripheral nerves, followed by muscles, joints, kidneys, skin, gastrointestinal tract, and heart (32).
Involvement of the CNS has been reported to exist in 20–40% of PAN cases (33) and mainly occurs at late stages (2–3 years) (34). CNS involvement has been recognized as a sign of a poor prognosis, and the expected mortality rate at 5 years is 26% (35, 36). The most common manifestation of CNS involvement includes diffuse encephalopathy and a deficit of focal neurological function. Diffuse encephalopathy may present as new onset seizure and headache, reduced level of consciousness or altered vision, among which some of the cases also suffer from reversible encephalopathy syndrome (37–39). The main manifestations of focal CNS involvement include cerebral infarction (which occurs in 13–17% of PAN patients), hemorrhage, multifocal encephalopathy and episodes of neurological dysfunction that mimic multiple sclerosis (33, 40). Intracranial hemorrhage is a rare complication of PAN, and subarachnoid hemorrhage and intracerebral hemorrhage caused by aneurysms have been reported in PAN cases. The features of these aneurysms were found to be multiple, small in size, and equally located in the infratentorial and supratentorial arteries (41).
Reichart et al. (33) investigated early lacunar strokes complicating PAN and revealed that 33.3% (5/15) of strokes occurred within 1 month after the onset of PAN and 15% (2/13) shortly after the initiation of corticosteroids; ischemic strokes occurred during corticosteroid treatment in 77% (10/13) of the patients, 80% (8/10) of which occurred within 6 months after corticosteroid initiation and 50% (5/10) within 3 weeks. Combined with the results of prior studies showing that all strokes occurred while the patients were under adequate corticosteroid treatment, it was suggested that the promoting effect of corticosteroids in stroke might work by two mechanisms (33). For one thing, corticosteroids promote platelet aggregation in cerebral medium-sized arteries by increasing thromboxane A2, quickly causing a lacunar stroke. Lacunar strokes appearing 1 or more months after initiation of corticosteroids can be explained by the promoting effect of corticosteroids on thrombotic microangiopathies.
PNS involvement is the most common complication of PAN and occurs in 60–70% of cases; it has an onset early in the course of the disease, mostly within a few months of diagnosis (42). The common patterns of PNS involvement include mononeuropathy, polyneuropathy and mononeuritis multiplex, the pathophysiology of which has been recognized as vasculitis of the vasa nervorum. Sudden onset asymmetrical sensorimotor mononeuritis multiplex, which mainly affects the lower extremities, and insidious symmetrical peripheral neuropathy are typical and common in PAN (31). In addition, patients with PAN may also develop pachymeningitis (43) and present with headache or cranial nerve palsy. Sudden bilateral hearing loss (44) and visual alterations caused by optic neuropathy (45) have also been reported.
Kawasaki Disease
Kawasaki disease (KD) is a systemic vasculitis that mainly affects medium-sized vessels, occurring predominately in children under 5 years of age. Neurological complications of KD have been described in children, such as encephalopathy, seizures, cerebral infarction, intracranial hemorrhage, ataxia, and cranial nerve palsy (46). However, although rare cases of first onset have also been reported in adult patients, most reported cases are in young adults and the manifestations are due to late-onset sequelae instead of new-onset active disease. We found one case (47) describing a 20-year-old young adult with a history of KD suffering from subarachnoid hemorrhage due to an intracranial aneurysm with a stalk-like narrow neck, located at the trunk of the middle cerebral artery. As the researchers mentioned, coronary aneurysms in KD also arise in places where no branch exists. Therefore, non-bifurcation intracranial aneurysm might be related to KD and might cause intracranial hemorrhage in young adults as a late sequela.
Small-Vessel Vasculitis
Small-vessel vasculitis refers to necrotizing inflammation in the wall of small intraparenchymal arteries, arterioles, capillaries, and venules, and to a secondary degree in medium-size arteries. Vascular and perivascular inflammation leads to fibrinoid necrosis and consequently vascular necrosis, occlusion and thrombosis (10). The disease consists of two major groups, ANCA-associated vasculitis and immune complex small-vessel vasculitis.
ANCA-Associated Vasculitis
ANCA-associated vasculitis is defined as necrotizing small-vessel vasculitis with few or no immune deposits, mostly associated with myeloperoxidase (MPO) ANCA or proteinase 3 (PR3) ANCA (10). Among the cases of ANCA-associated vasculitis, CNS involvement is more common in granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) than it is in eosinophilic granulomatosis with polyangiitis (EGPA) (48). Nevertheless, peripheral nerve involvement is more prevalent in EGPA than are MPA and GPA (49).
Granulomatosis With Polyangiitis (Wegener's)
Granulomatosis with polyangiitis (GPA) is characterized by necrotizing granulomatous inflammation usually associated with antibodies against PR3 and most commonly involves the upper and lower respiratory tract, and sinonasal inflammation and necrotizing glomerulonephritis are frequently present (10). Neurological complications have been shown to be present in 29–50% of all cases (50–52).
CNS involvement occurs in 7–11% of GPA patients (50, 53, 54), and when isolated cranial nerve palsies are included, the rate is 8–28% (51, 54). Since GPA can commonly involve sinonasal structures and cause granulomatous inflammation, which is capable of invading neighboring structures, the orbit, mastoid, optic nerve, chiasma, cranial nerves, meninges, and pituitary gland might be compromised, presenting as oculomotor dysfunction, mastoiditis, visual disturbances, cranial nerve palsy, headache, and endocrine dysfunction. In addition, small and medium arteries in the cranium can be involved, causing ischemia, or hemorrhage in different vascular territories (55). Cranial nerve palsy was reported to occur in 4.7–6% of GPA patients, among which the most commonly affected cranial nerves are II, VI, and VII (50, 51). The pituitary has been reported to be affected in 1.1–1.3% of GPA patients (56, 57). Meningeal pachymeningitis usually occurs i4n the early stages of GPA, with lower incidences of accompanying systemic symptoms (58), which is perhaps due to granulomatous erosion of the skull base in the early phase and may lead to a delay of diagnosis (52). The most common symptom is headache, followed by cranial nerve palsy and symptoms due to venous sinus occlusion. Vasculitis involvement can cause ischemic and hemorrhagic complications of the brain and spinal cord (58). Dysfunction of intracranial arteries may cause PRES, presenting as various symptoms (59–62). Hypophysitis has been widely reported as one of the CNS complications of GPA, and its clinical manifestations vary according to the specific sites involved. Panhypopituitarism might occur as a consequence of granulomatous inflammation of both the anterior and posterior pituitary (56). Anterior pituitary involvement may influence the release of antidiuretic hormone and present as diabetes insipidus, which was shown to occur in 47 of 58 GPA patients (81%) with sellar involvement, according to an analysis of cases performed by Peters et al. (63). Posterior pituitary inflammation may cause diminished secretion of many hormones, leading to secondary hypogonadism (occurred in 32 out of 58 sellar involved GPA patients, 55%), secondary hypothyroidism (20 out of 58 sellar involved GPA patients, 34.5%), secondary adrenal insufficiency, secondary growth hormone deficiency (19 of 58 sellar involved GPA patients, 32.8%). Compression of the pituitary stalk can result in hyperprolactinemia (5 of 58 sellar involved GPA patients, 8.6%) and galactorrhea, and visual alteration may occur if the optic chiasm is compressed (56, 57).
PNS complications have been reported to occur in 11–44% of GPA patients (54), which occurs milder and later in the course of the disease than in other ANCA-associated vasculitis (64), presenting mainly as recurrent mononeuropathies, mononeuritis multiplex, or symmetric polyneuropathy due to ischemia caused by vasculitic inflammation of the vasa nervorum. As the most frequent manifestation, distal symmetrical sensory neuropathy was reported in 7–10% of cases, followed by motor mononeuritis multiplex in 2–12% (51, 53). Commonly, peroneal (90–95%), tibial (38–55%), ulnar (35–45%), and median (26–36%) nerves are involved (29, 51).
Eosinophilic Granulomatosis With Polyangiitis (Churg-Strauss)
The characteristics of eosinophilic granulomatosis with polyangiitis (EGPA) is an eosinophil-rich and granulomatous inflammation that often involves the respiratory tract and is associated with asthma and eosinophilia (10).
Neurological involvement is common in EGPA, accounting for up to 60% of cases and usually manifesting as peripheral neuropathy that tends to present before visceral involvement (29). Therefore, early detection of peripheral nerve involvement is fundamental for early diagnosis and treatment of EGPA. The most common peripheral neurological manifestation is multiple mononeuropathy (accounting for 68% of cases with peripheral neuropathy), followed by distal symmetric polyneuropathy (28%) and asymmetric polyneuropathy (4%) (65). Since mononeuritis multiplex is typical in acute systemic vasculitis, the initial symptoms commonly present as dysesthesia, paresthesia, and edema in distal limbs, especially the lower limbs. The condition will gradually progress into asymmetrical polyneuropathy and may affect motor nerves, leading to muscle atrophy. Electrophysiological studies have shown decreases in the amplitude of sensory nerve action potentials and compound muscle action potentials, indicating the presence of axonal injury (66).
CNS involvement in EGPA is rather rare, accounting for only 6–10% of all cases (67). Cerebral infarctions and intracerebral hemorrhage resulting from intracranial vasculitis are the most common CNS presentations of EGPA. In addition, rarer manifestations have been noted, such as subarachnoid hemorrhage, cranial nerve palsy, encephalopathy, epilepsy, hydrocephalus, headache, sinus venous thrombosis, spinal hemorrhage, meningeal involvement, and optic neuropathy.
Microscopic Polyangiitis
Microscopic polyangiitis (MPA) presents with non-granulomatous inflammation with few or no immune deposits in the walls of the affected vessels, mainly associated with antibodies against MPO. Necrotizing glomerulonephritis and alveolar hemorrhage are common complications (10). Peripheral neurological involvement occurs in 55 to 79% of cases with MPA, with no special manifestations compared with those of other ANCA-associated types of vasculitis (mainly presenting as polyneuropathy and mononeuropathy); (64). CNS involvement in MPA is rare and was reported to be able to manifest as intracerebral infarction or hemorrhage, subarachnoid hemorrhage, hypertrophic pachymeningitis, PRES or spinal cord involvement (68).
Immune Complex Small-Vessel Vasculitis
Immune complex small-vessel vasculitis is characterized by moderate to marked deposits of immunoglobulin and/or complement components in the small vessel walls.
Antiglomerular Basement Membrane Disease
Antiglomerular basement membrane (anti-GBM) disease, also called Goodpasture syndrome, is a vasculitis affecting glomerular capillaries and/or pulmonary capillaries with deposition of anti-GBM autoantibodies on GBM, accounting for 5% of adult patients with glomerulonephritis and leading to acute renal failure in approximately half of the patients. Lung involvement results in pulmonary hemorrhage, and renal involvement leads to glomerulonephritis with necrosis and crescents, which are the classic presentations of anti-GBM disease (10, 69). Neurological involvement is uncommon. According to the rare cases reported (69–73), PRES is the most frequent neurological complication. All patients reported are younger than 40 and they all manifested as hypertension, renal failure, and seizures. Destruction of the blood-brain barrier resulting from inflammatory endothelial injury and increased capillary filtration due to hypertension are important factors in the pathophysiology of PRES in GBM patients.
Cryoglobulinemic Vasculitis
Cryoglobulinemic vasculitis (CV) is defined as vasculitis with cryoglobulin immune deposits affecting small vessels (predominantly capillaries, venules, or arterioles) caused by chronic inflammation, autoimmune disorders, and lymphoproliferative disorders, frequently involving skin, glomeruli, and peripheral nerves (10). Most cases of cryoglobulinemic vasculitis result from infection, B-cell lymphoproliferative disorders and autoimmune diseases, among which hepatitis C virus infection is the cause in ~80% of cases (74). In a study of 242 cases with non-infectious mixed cryoglobulinemia vasculitis (75), 117 patients (48%) were found to have no identified causal factor, also defined as idiopathic. However, no study has evaluated the neurological involvement of idiopathic cryoglobulinemia vasculitis alone. For patients with mixed CV, PNS involvement is not uncommon and typically starts with polyneuropathy that affects the sensory nerves initially and later the motor nerves, predominantly involving the lower limbs. CNS involvement is rare, and studies have shown that mixed CV may cause cerebrovascular events, PRES, hydrocephalus and intracranial hypertension (29, 76).
IgA Vasculitis (Henoch-Schönlein Purpura)
IgA vasculitis (IgAV) is characterized by IgA1-dominant immune deposits affecting small vessels, often involving the skin and gastrointestinal tract and frequently causing arthritis (10). The majority (75%) of cases occur in young people no more than 20 years of age (77), and CNS involvement has been reported to manifest as headache, decreased consciousness, seizures, focal neurological deficits, and visual and verbal abnormalities. However, PNS involvement has been documented as neuropathies of the brachial plexus and facial, peroneal, femoral and ulnar nerve, and in addition, Guillain–Barre syndrome and mononeuritis multiplex (78). Rare cases of IgAV-induced neurological manifestations have been reported in adult patients as ischemic stroke, axonal sensorimotor polyneuropathy (79, 80), mononeuritis multiplex (81, 82), acoustic neuritis, facial nerve palsy (83), intracerebral hemorrhage (84), anterior ischemic optic neuropathy (85), focal seizures (86), and encephalopathy (87).
Hypocomplementemic Urticarial Vasculitis (anti-C1q Vasculitis)
Hypocomplementemic urticarial vasculitis (HUV) is a kind of vasculitis accompanied by urticaria and hypocomplementemia affecting cutaneous small vessels and is associated with anti-C1q antibodies, commonly causing urticaria and multiorgan involvement such as glomerulonephritis, arthritis, obstructive pulmonary disease, and ocular inflammation (10). HUV may be induced by connective tissue diseases, infections (such as hepatitis B, hepatitis C and infectious mononucleosis), neoplasms and drugs; however, most cases of HUV are idiopathic (88). Neurological involvement has been occasionally reported as pseudotumor cerebri (89), lower cranial nerve (VIII, IX, and X) palsies (90) and peripheral nerve involvement such as asymmetrical multifocal axonal sensory neuropathy.
Variable-Vessel Vasculitis
Variable-vessel vasculitis refers to vasculitis with no predominant type of vessel involved that can affect vessels of any size (small, medium, and large) and type (arteries, veins, and capillaries) (10).
Behçet's Syndrome
Behçet's syndrome is a vasculitis occurring in patients with Behçet's disease (BD) and can affect arteries or veins and lead to a relapsing inflammatory disorder in almost any tissue, with oral and genital ulcerations and uveitis as its most typical symptoms (41). Neurological manifestations of BD, which are called neuro-BD (NBD), occur in 1.3–59% of all cases and usually affect patients aged between 20 and 40 years, with a male predominance (the male to female ratio is 2.8:1) (91–95). NBD commonly appears 3–6 years after other systemic involvement (92, 96, 97) and is considered to be caused by perivasculitis.
The manifestations of neurological involvement can be caused by either CNS parenchymal inflammation or vascular complications in the nervous system, with a reported prevalence of 67–76 vs. 12–20% in all NBD cases (93, 98). The former is considered to be caused by small vasculitis, leading to axonal damage and gliosis, frequently affecting the brain stem (occurs in 25–50% of all parenchymal NBD cases) (92, 93, 99, 100), thalamus and basal ganglia, and, only rarely, the white matter and spinal cord. The main clinical manifestations are subacute cranial neuropathy, ophthalmoparesis, meningoencephalitis, and alteration of cerebellar, pyramidal and extrapyramidal function. Uncommon symptoms such as subcortical dementia, stroke and transverse myelitis have also been reported (91, 101). Vascular complications mainly result from cerebral venous thrombosis (CVT) due to large vessels endothelial cell activation, and rarely, aneurysms due to perivasculitis to the vasa nervorum (91). CVT in BD mainly causes focal neurological deficits and seizures in young men (102). However, not all epileptic seizures in NBD patients result from CVT, and brainstem lesions may also lead to complex partial seizures (103).
In addition, perivasculitis to the vasa nervorum may also cause PNS involvement. However, this condition is extremely rare and was found in merely 8 out of 1,031 cases (0.8%); thus, its relationship with BD is still doubtful (91).
Cogan Syndrome
Cogan syndrome (CS) is a sequence of clinical manifestations due to a chronic immunological inflammatory multisystem disease of unknown origin, characterized by recurrent episodes of keratitis, vestibuloauditory dysfunction, and systemic vasculitis, frequently leading to visual loss, vertigo, sensorineural hearing loss, tinnitus and systemic manifestations (10). The syndrome mainly affects children and young adults with an average age at onset of 38 years (SD, 15.1 years; range, 9–70 years) (104).
It is worth noting that complications of the eyes and ears do not always occur at the same time (sometimes up to 9 years apart) (105) and in 20% of cases, vestibuloauditory symptoms can be the only presentation (104). Therefore, although the vestibuloauditory involvement in Cogan syndrome is mainly due to inflammation of inner ear structures rather than neurological involvement (105), its manifestations can be easily misdiagnosed as central acute vestibular syndrome of vascular origin, Ménière's disease or vestibular migraine, which are common in neurological out-patients. According to a study that enrolled 60 cases of CS at Mayo Clinic (104), all cases suffered from hearing loss, 90% of the cases experienced vertigo, and tinnitus (80%), ataxia (53%), and oscillopsia (25%) were also common during the course of the disease. Hearing loss is typically sudden, mainly involves bilateral high frequencies, and the decline in hearing fluctuates and progresses gradually. Vestibular function is commonly damaged bilaterally (accounting for 74% of cases that underwent a caloric test) and vestibular symptoms may last for days to weeks (sometimes indefinitely) with no resolution (104, 106). The vestibuloauditory symptoms listed above may recur in 1–13 years after onset (occurs in 22% of patients) (107).
Neurological involvement was reported in 29–56% of cases and usually occurs after eye and ear manifestations (106, 108). CNS involvement may manifest as ischemic stroke (accounting for 2.5–3% of cases reported), encephalitis (5–6%), meningitis (5–22%), encephalopathy, myelopathy, optic nerve disorders, aneurysm, and cerebral venous thrombosis. PNS conditions in Cogan syndrome patients have been reported as peripheral neuropathy (1–12.5%, frequently mononeuritis multiplex), cranial neuropathy (1–10%, mainly cranial nerve II, V, VI, and VII) and myopathy (106).
Conclusion
Neurological involvement is a common complication of PSV (Table 1), and neurologists play an important role in the identification and diagnosis of PSV patients with otherwise unexplained neurological symptoms as their chief complaint. This article summarizes the neurological manifestations of PSV and hopes to improve neuroscientists' understanding of this broad range of diseases.
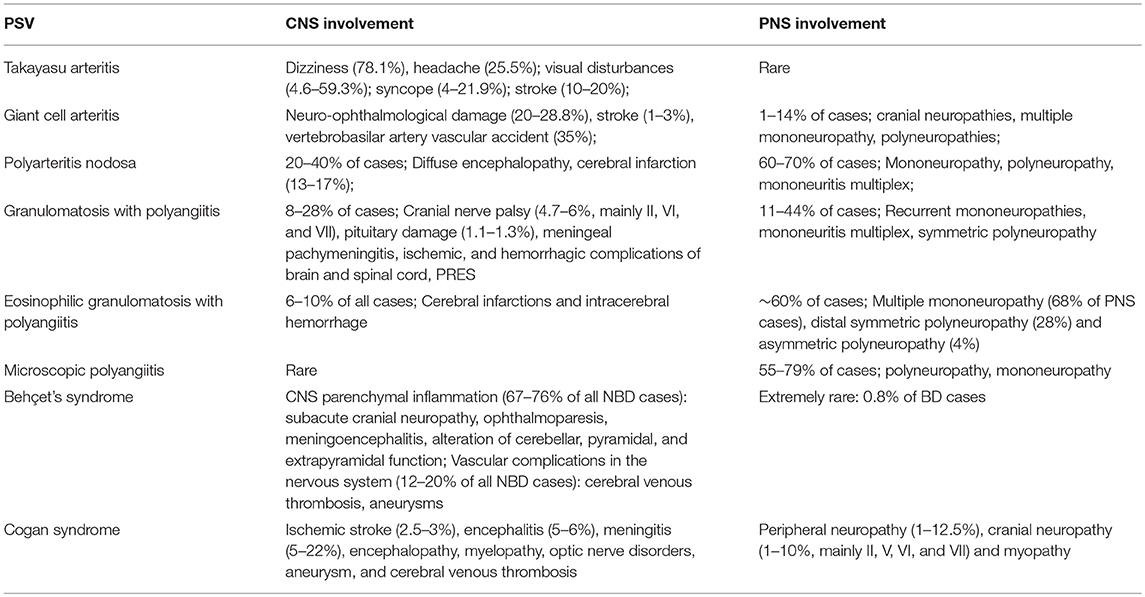
Table 1. Common CNS and PNS involvements of primary systemic vasculitis.
Author Contributions
SZ conceived the article and wrote the manuscript. DY and GT reviewed and edited the manuscript. All authors read and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to the Chongqing Science and Technology Commission, Chongqing Education Commission and the Chongqing Health and Family Planning Commission for funding this project.
References
1. Gonzalez-Gay M, Garcia-Porrua C. Epidemiology of the vasculitides. Rheum Dis Clin North Am. (2001) 27:729–49. doi: 10.1016/S0889-857X(05)70232-5
2. Lane SE, Watts RA, Graham B, Innes NJ, Scott DGI. Are environmental factors important in primary systemic vasculitis? A case-control study. Arthritis Rheum. (2014) 48:814–23. doi: 10.1002/art.10830
3. Mulloy KB. Silica exposure and systemic vasculitis. Environ Health Perspect. (2003) 111:1933–8. doi: 10.1289/ehp.6400
4. Nuyts GD, Van VE, De VA, Daelemans RA, Rorive G, Elseviers MM, et al. Wegener granulomatosis is associated to exposure to silicon compounds: a case-control study. Nephrol Dial Transpl. (1995) 10:1162–5. doi: 10.1093/ndt/10.7.1162
5. Steenland NK, Thun MJ, Ferguson CW, Port FK. Occupational and other exposures associated with male end-stage renal disease: a case/control study. Am J Public Health. (1990) 80:153. doi: 10.2105/AJPH.80.2.153
6. Cuadrado MJ, D'Cruz D, Lloyd M, Mujic F, Khamashta MA, Hughes GRV. Allergic disorders in systemic vasculitis: a case-control study. Br J Rheum. (1994) 33:749. doi: 10.1093/rheumatology/33.8.749
7. Sampaio L, Silva LG, Terroso G, Nadais G, Mariz E, Ventura F. Vasculitic neuropathy. Acta Reumatol Port. (2011) 36:102–9.
8. Bloch DA, Michel BA, Hunder GG, McShane DJ, Arend WP, Calabrese LH, et al. The American College of Rheumatology 1990 criteria for the classification of vasculitis. Patients and methods. Arthritis Rheum. (1990) 33:1068–73. doi: 10.1002/art.1780330803
9. Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. (1994) 37:187–92. doi: 10.1002/art.1780370206
10. Rasmussen N. The 2012 revised international chapel hill consensus conference nomenclature of the vasculitides. Ann Rheum Dis. (2013) 71:16. doi: 10.1136/annrheumdis-2012-eular.1540
11. Pena CE, Pera M, Marcos J, Salas A, Testi A, Garcia MA. Peripheral neurological involvement in primary systemic vasculitis. Ann Rheum Dis. (2015) 74:295–6. doi: 10.1136/annrheumdis-2015-eular.2855
12. Wolf J, Schmitt V, Palm F, Grau AJ, Bergner R. Peripheral neuropathy as initial manifestation of primary systemic vasculitides. J Neurol. (2013) 260:1061–70. doi: 10.1007/s00415-012-6760-7
13. Finsterer J. Systemic and non-systemic vasculitis affecting the peripheral nerves. Acta Neurologica Belgica. (2009) 109:100–13.
14. Kurt S, Alsharabati M, Lu L, Claussen GC, Oh SJ. Asymptomatic vasculitic neuropathy. Muscle Nerve. (2015) 52:34–8. doi: 10.1002/mus.24494
15. Kim HJ, Suh DC, Kim JK, Kim SJ, Lee JH, Choi CG, et al. Correlation of neurological manifestations of Takayasu's arteritis with cerebral angiographic findings. Clin Imaging. (2005) 29:79–85. doi: 10.1016/j.clinimag.2004.04.026
16. Yang L, Zhang H, Jiang X, Song L, Qin F, Zou Y, et al. Clinical features and outcomes of Takayasu arteritis with neurological symptoms in China: a retrospective study. J Rheum. (2015) 42:1846–52. doi: 10.3899/jrheum.150097
17. Duarte MM, Geraldes R, Sousa R, Alarcão J, Costa J. Stroke and transient ischemic attack in takayasu's arteritis: a systematic review and meta-analysis. J Stroke Cerebrovasc Dis. (2016) 25:781–91. doi: 10.1016/j.jstrokecerebrovasdis.2015.12.005
18. Park MC, Lee SW, Park YB, Chung NS, Lee SK. Clinical characteristics and outcomes of Takayasu's arteritis: analysis of 108 patients using standardized criteria for diagnosis, activity assessment, and angiographic classification. Scand J Rheumatol. (2005) 34:284. doi: 10.1080/03009740510026526
19. Couture P, Chazal T, Rosso C, Haroche J, Léger A, Hervier B, et al. Cerebrovascular events in Takayasu arteritis: a multicenter case-controlled study. J Neurol. (2018) 265:757–63. doi: 10.1007/s00415-018-8744-8
20. Takayama K, Nakagawa H, Iwasaki S, Taoka T, Myouchin K, Wada T, et al. Multiple cerebral aneurysms associated with Takayasu arteritis successfully treated with coil embolization. Rad Med Med Imaging Rad Oncol. (2008) 26:33–8. doi: 10.1007/s11604-007-0184-9
21. Camara-Lemarroy CR, Lara-Campos JG, Perez-Contreras E, Rodriguez-Gutierrez R, Galarza-Delgado DA. Takayasu's arteritis and posterior reversible encephalopathy syndrome: a case-based review. Clin Rheumatol. (2013) 32:409–15. doi: 10.1007/s10067-012-2151-9
22. Buttgereit F, Dejaco C, Matteson EL, Dasgupta B. Polymyalgia rheumatica and giant cell arteritis: a systematic review. JAMA. (2016) 315:2442–58. doi: 10.1001/jama.2016.5444
23. Salvarani C, Cimino L, Macchioni P, Consonni D, Cantini F, Bajocchi G, et al. Risk factors for visual loss in an Italian population-based cohort of patients with giant cell arteritis. Arthritis Rheum. (2005) 53:293–7. doi: 10.1002/art.21075
24. Pfadenhauer K, Roesler A, Golling A. The involvement of the peripheral nervous system in biopsy proven active giant cell arteritis. J Neurol. (2007) 254:751–5. doi: 10.1007/s00415-006-0428-0
25. Gonzalez-Gay MA, Vazquez-Rodriguez TR, Gomez-Acebo I, Pego-Reigosa R, Lopez-Diaz MJ, Vazquez-Tri-anes MC, et al. Strokes at time of disease diagnosis in a series of 287 patients with biopsy-proven giant cell arteritis. Medicine. (2009) 88:227. doi: 10.1097/MD.0b013e3181af4518
26. Ungprasert P, Wijarnpreecha K, Koster MJ, Thongprayoon C, Warrington KJ. Cerebrovascular accident in patients with giant cell arteritis: a systematic review and meta-analysis of cohort studies. Semi Arthritis Rheum. (2016) 46:361–6. doi: 10.1016/j.semarthrit.2016.07.005
27. Chazal T, Couture P, Rosso C, Haroche J, Léger A, Hervier B, et al. Cerebrovascular events are associated with lower survival in giant cell arteritis: a case-controlled multicenter study. Joint Bone Spine. (2018) 85:383–5. doi: 10.1016/j.jbspin.2017.05.017
28. Pego-Reigosa R, Garcia-Porrua C, Piã±Eiro A, Dierssen T, Llorca J, Gonzalez-Gay MA. Predictors of cerebrovascular accidents in giant cell arteritis in a defined population. Clini Exp Rheumatol. (2004) 22:13–7.
29. Rossi CM, Di Comite G. The clinical spectrum of the neurological involvement in vasculitides. J Neurol Sci. (2009) 285:13–21. doi: 10.1016/j.jns.2009.05.017
30. Amordorado JC, Llorca J, Garciaporrua C, Costa C, Perezfernandez N, Gonzalezgay MA. Audiovestibular manifestations in giant cell arteritis: a prospective study. Medicine. (2003) 82:13–26. doi: 10.1097/00005792-200301000-00002
31. Minagar A, Fowler M, Harris MK, Jaffe SL. Neurologic presentations of systemic vasculitides. Neurol Clin. (2010) 28:171–84. doi: 10.1016/j.ncl.2009.09.015
32. Lhote F, Cohen P, Guillevin L. Polyarteritis nodosa, microscopic polyangiitis and Churg-Strauss syndrome. Semin Respir Crit Care Med. (1998) 19:27–45. doi: 10.1055/s-2007-1009380
33. Reichart MD, Bogousslavsky J, Janzer RC. Early lacunar strokes complicating polyarteritis nodosa: thrombotic microangiopathy. Neurology. (2000) 54:883–9. doi: 10.1212/WNL.54.4.883
34. Moore PM, Cupps TR. Neurological complications of vasculitis. Ann Neurol. (2010) 14:155–67. doi: 10.1002/ana.410140202
35. Gomes V, Capela C, Rodrigues C, Goncalves F. Polyarteritis nodosa with central and peripheral neurological involvement. Eur J Intern Med. (2013) 24:e118. doi: 10.1016/j.ejim.2013.08.300
36. Guillevin L, Lhote F, Gayraud M, Cohen P, Jarrousse B, Lortholary O, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine. (1996) 75:17–28. doi: 10.1097/00005792-199601000-00003
37. Arai M, Shigeno K, Wada M. [A reversible posterior leukoencephalopathy syndrome in a patient with classical polyarteritis nodosa]. Rinsho shinkeigaku. (1997) 37:64–6.
38. Navinan MR, Subasinghe CJ, Kandeepan T, Kulatunga A. Polyarteritis nodosa complicated by posterior reversible encephalopathy syndrome: a case report. BMC Res Notes. (2014) 7:89. doi: 10.1186/1756-0500-7-89
39. Stanzani L, Fusi L, Gomitoni A, Roncoroni M, Villa P, Grampa G. A case of posterior reversible encephalopathy during polyarteritis nodosa vasculitis. Neurol Sci. (2008) 29:163–7. doi: 10.1007/s10072-008-0929-y
40. Ueno KI, Matsushima A, Hineno A, Fukushima K, Tazawa KI, Matsuda M, et al. Polyarteritis nodosa with central nervous system involvement mimicking relapsingremitting multiple sclerosis. Modern Rheumatol. (2014) 24:525–8. doi: 10.3109/14397595.2013.852849
41. Oomura M, Yamawaki T, Naritomi H, Terai T, Shigeno K. Polyarteritis nodosa in association with subarachnoid hemorrhage. Intern Med. (2006) 45:655–8. doi: 10.2169/internalmedicine.45.1632
42. Ribi C, Cohen P, Pagnoux C, Mahr A, Arène JP, Puéchal X, et al. Treatment of polyarteritis nodosa and microscopic polyangiitis without poor-prognosis factors: a prospective randomized study of one hundred twenty-four patients. Arthritis Rheum. (2010) 62:1186–97. doi: 10.1002/art.27340
43. Song JS, Lim MK, Park BH, Park W. Acute pachymeningitis mimicking subdural hematoma in a patient with polyarteritis nodosa. Rheumatol Int. (2005) 25:637–40. doi: 10.1007/s00296-005-0615-9
44. Rubin F, Tran Khai Hoan N, Bonfils P. Sudden bilateral hearing loss revealing polyarteritis nodosa. Eur Ann Otorhinol Head Neck Dis. (2014) 131:265–6. doi: 10.1016/j.anorl.2014.03.003
45. Vazquez-Romo KA, Rodriguez-Hernandez A, Paczka JA, Nu-o-Suarez MA, Rocha-Mu-oz AD, Zavala-Cerna MG. Optic neuropathy secondary to polyarteritis nodosa, case report, and diagnostic challenges. Front Neurol. (2017) 8:490. doi: 10.3389/fneur.2017.00490
46. Alves NR, Magalhaes CM, Almeida Rde F, Santos RC, Gandolfi L, Pratesi R. Prospective study of Kawasaki disease complications: review of 115 cases. Rev Assoc Med Bras. (1992) 57:295–300.
47. Ishida A, Matsuo S, Kawamura S, Nishikawa T. Subarachnoid hemorrhage due to nonbranching aneurysm of the middle cerebral artery in a young adult with a history of Kawasaki disease. Surg Neurol Int. (2014) 5:5. doi: 10.4103/2152-7806.125285
48. Graf J. Central nervous system disease in antineutrophil cytoplasmic antibodies-associated vasculitis. Rheum Dis Clini North Am. (2017) 43:573–8. doi: 10.1016/j.rdc.2017.06.006
49. Imboden JB. Involvement of the peripheral nervous system in polyarteritis nodosa and antineutrophil cytoplasmic antibodies-associated vasculitis. Rheum Dis Clini North Am. (2017) 43:633–9. doi: 10.1016/j.rdc.2017.06.011
50. De Groot K, Schmidt DK, Arlt AC, Gross WL, Reinhold-Keller E. Standardized neurologic evaluations of 128 patients with Wegener granulomatosis. Arch Neurol. (2001) 58:1215–21. doi: 10.1001/archneur.58.8.1215
51. Nishino H, Rubino FA, DeRemee RA, Swanson JW, Parisi JE. Neurological involvement in Wegener's granulomatosis: an analysis of 324 consecutive patients at the Mayo Clinic. Ann Neurol. (1993) 33:4–9. doi: 10.1002/ana.410330103
52. Choi HA, Lee MJ, Chung CS. Characteristics of hypertrophic pachymeningitis in patients with granulomatosis with polyangiitis. Cephalalgia. (2016) 36:169–70. doi: 10.1177/0333102416670318
53. Stone JH. Limited versus severe Wegener's granulomatosis: baseline data on patients in the Wegener's granulomatosis etanercept trial. Arthritis Rheum. (2003) 48:2299. doi: 10.1002/art.11075
54. Reinholdkeller E, Beuge N, Latza U, De GK, Rudert H, Nölle B, et al. An interdisciplinary approach to the care of patients with Wegener's granulomatosis: long-term outcome in 155 patients. Arthritis Rheum. (2000) 43:1021–32. doi: 10.1002/1529–0131(200005)43:5<1021::AID-ANR10>3.0.CO;2-J
55. Seror R, Mahr A, Ramanoelina J, Pagnoux C, Cohen P, Guillevin L. Central nervous system involvement in wegener granulomatosis. Medicine. (2006) 85:54–65. doi: 10.1097/01.md.0000200166.90373.41
56. Kapoor E, Cartin-Ceba R, Specks U, Leavitt J, Erickson B, Erickson D. Pituitary dysfunction in granulomatosis with polyangiitis: the mayo clinic experience. J Clini Endocrinol Metabol. (2014) 99:3988–94. doi: 10.1210/jc.2014-1962
57. De Parisot A, Puechal X, Langrand C, Raverot G, Gil H, Perard L, et al. Pituitary involvement in granulomatosis with polyangiitis: report of 9 patients and review of the literature. Medicine. (2015) 94:e748. doi: 10.1097/MD.0000000000000748
58. De Luna G, Terrier B, Kaminsky P, Le Quellec A, Maurier F, Solans R, et al. Central nervous system involvement of granulomatosis with polyangiitis: clinical-radiological presentation distinguishes different outcomes. Rheumatology. (2015) 54:424–32. doi: 10.1093/rheumatology/keu336
59. Arteaga-Müller G, Camara-Lemarroy CR, Rizo-Topete L, Guerrero-González E, Sánchez-Martinez C, Cruz-Valdez J. Granulomatosis with polyangiitis and posterior reversible encephalopathy syndrome. J Neurol Sci. (2016) 362:204–5. doi: 10.1016/j.jns.2016.01.048
60. Zabihiyeganeh MO, Jahed AD, Poormoghim HA. Reversible posterior leukoencephalopathy and splenic infarction in a patient with Wegener granulomatosis. Int J Rheum Dis. (2010) 13:232. doi: 10.1111/j.1756-185X.2010.01524.x
61. Ohta T, Sakano T, Shiotsu M, Furue T, Ohtani H, Kinoshita Y, et al. Reversible posterior leukoencephalopathy in a patient with Wegener granulomatosis. Pediatr Nephrol. (2004) 19:442–4. doi: 10.1007/s00467-003-1286-y
62. Pugnet G, Pagnoux C, Bézanahary H, Ly KH, Vidal E, Guillevin L. Progressive multifocal encephalopathy after cyclophosphamide in granulomatosis with polyangiitis (wegener) patients: case report and review of the literature. Clini Exp Rheumatol. (2013) 31(Suppl. 75):S62–4.
63. Peters JE, Gupta V, Saeed IT, Offiah C, Jawad ASM. Severe localised granulomatosis with polyangiitis (Wegener's granulomatosis) manifesting with extensive cranial nerve palsies and cranial diabetes insipidus: a case report and literature review. BMC Neurol. (2018) 18:59. doi: 10.1186/s12883-018-1058-8
64. Cattaneo L, Chierici E, Pavone L, Grasselli C, Manganelli P, Buzio C, et al. Peripheral neuropathy in Wegener's granulomatosis, Churg-Strauss syndrome and microscopic polyangiitis. J Neurol Neurosurg Psychiatr. (2007) 78:1119–23. doi: 10.1136/jnnp.2006.111013
65. Sehgal M, Swanson JW, DeRemee RA, Colby TV. Neurologic manifestations of Churg-Strauss syndrome. Mayo Clini Proc. (1995) 70:337–41. doi: 10.4065/70.4.337
66. Hattori N, Ichimura M, Nagamatsu M, Li M, Yamamoto K, Kumazawa K, et al. Clinicopathological features of Churg-Strauss syndrome-associated neuropathy. Brain. (1999) 122(Pt 3):427–39. doi: 10.1093/brain/122.3.427
67. Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine. (1999) 78:26–37. doi: 10.1097/00005792-199901000-00003
68. Decker ML, Emery DJ, Smyth PS, Lu JQ, Lacson A, Yacyshyn E. Microscopic polyangiitis with spinal cord involvement: a case report and review of the literature. J Stroke Cerebrovasc Dis. (2016) 25:1696–704. doi: 10.1016/j.jstrokecerebrovasdis.2016.01.034
69. Camara-Lemarroy CR, Cruz-Moreno MA, Gamboa-Sarquis RN, Gonzalez-Padilla KA, Tamez-Perez HE, Galarza-Delgado DA. Goodpasture syndrome and posterior reversible encephalopathy syndrome. J Neurol Sci. (2015) 354:135–7. doi: 10.1016/j.jns.2015.05.002
70. Lahmer T, Kuchle C, Schirmer L, Heemann U, Lutz J, Thurmel K. Kidney transplant after preexisting posterior reversible encephalopathy syndrome induced by Goodpasture's syndrome. Exp Clini Transplant. (2012) 10:299–301. doi: 10.6002/ect.2011.0177
71. Ozkok A, Elcioglu OC, Bakan A, Atilgan KG, Alisir S, Odabas AR. Reversible posterior leukoencephalopathy in the course of Goodpasture syndrome. Renal Failure. (2012) 34:254–6. doi: 10.3109/0886022X.2011.647211
72. Cha B, Kim DY, Jang H, Hwang SD, Choi HJ, Kim MJ. Unusual case of posterior reversible encephalopathy syndrome in a patient with anti-glomerular basement membrane antibody glomerulonephritis: a case report and review of the literature. Electrolyte Blood Pressure. (2017) 15:12–6. doi: 10.5049/EBP.2017.15.1.12
73. Gutiérrez-Sánchez MJ, Petkov-Stoyanov V, Martín-Navarro JA. Reversible posterior leukoencephalopathy syndrome in Goodpasture syndrome. Nefrologia. (2012) 32:540–3. doi: 10.3265/Nefrologia.pre2012.Mar.11427
74. Terrier B, Cacoub P. Cryoglobulinemia vasculitis: an update. Curr Opinion Rheumatol. (2013) 25:10–8. doi: 10.1097/BOR.0b013e32835b15f7
75. Terrier B, Krastinova E, Marie I, Launay D, Lacraz A, Belenotti P, et al. Management of noninfectious mixed cryoglobulinemia vasculitis: data from 242 cases included in the CryoVas survey. Blood. (2012) 119:5996–6004. doi: 10.1182/blood-2011-12-396028
76. Ferri C, La Civita L, Cirafisi C, Siciliano G, Longombardo G, Bombardieri S, et al. Peripheral neuropathy in mixed cryoglobulinemia: clinical and electrophysiologic investigations. J Rheumatol. (1992) 19:889–95.
77. Ortiz-Sanjuán F, Calvo- Río V, Loricera J, Mata C, Penagos LM, Alvarez L, et al. Henoch-schönlein purpura: clinical spectrum of the disease in 417 patients from a single center. Arthritis Rheum. (2013) 65:S1122. doi: 10.1002/art.38216
78. Garzoni L, Vanoni F, Rizzi M, Simonetti GD, Simonetti BG, Ramelli GP, et al. Nervous system dysfunction in Henoch-Schönlein syndrome: systematic review of the literature. Rheumatology. (2009) 48:1524–9. doi: 10.1093/rheumatology/kep282
79. Soydemir Ö, Dotan Ak P, Aktan Z, Gözke E. Peripheral neuropathy: rare manifestation in Henoch-Schönlein purpura. Eur J Neurol. (2014) 21:711.
80. De Luna G, Vasco PG, Terrier B, Calleja JL, Fraile G. Henoch Schönlein purpura at diagnosis in 3 adult patients: acute onset with peripheral nervous system dysfunction. Eur J Intern Med. (2013) 24:e116. doi: 10.1016/j.ejim.2013.08.294
81. Fukushima K. Severe intestinal lesions and neuropathy associated with henoch-schonlein purpura. Intern Med. (2013) 52:2009–10. doi: 10.2169/internalmedicine.52.0616
82. Campbell SB, Hawley CM, Staples C. Mononeuritis multiplex complicating Henoch-Schonlein purpura. Aust N Z J Med. (1994) 24:580. doi: 10.1111/j.1445-5994.1994.tb01767.x
83. Fiaux E, Benhamou Y, Cailleux N, Levesque H. A rare case of Henoch-Schönlein purpura with neurological involvement. Intern Med J. (2010) 40:795–6. doi: 10.1111/j.1445-5994.2010.02298.x
84. Karamadoukis L, Ludeman L, Williams AJ. Henoch-Schönlein purpura with intracerebral haemorrhage in an adult patient: a case report. J Med Case Rep. (2008) 2:200. doi: 10.1186/1752-1947-2-200
85. Chuah J, Meaney T. Anterior ischaemic optic neuropathy secondary to Henoch-Schönlein Purpura. Eye. (2005) 19:1028. doi: 10.1038/sj.eye.6701764
86. Fielding RE, Hawkins CP, Hand MF, Heath PD, Davies SJ. Seizures complicating adult Henoch-Schonlein purpura. Nephrol Dialysis Transplant. (1998) 13:761–2. doi: 10.1093/ndt/13.3.759
87. Perez C, Maravi E, Olier J, Guarch R. MR imaging of encephalopathy in adult Henoch-Shonlein purpura. Am J Roentgenol. (2000) 175:922–3. doi: 10.2214/ajr.175.3.1750922a
88. Filosto M, Cavallaro T, Pasolini G, Broglio L, Tentorio M, Cotelli M, et al. Idiopathic hypocomplementemic urticarial vasculitis-linked neuropathy. J Neurol Sci. (2009) 284:179–81. doi: 10.1016/j.jns.2009.03.027
89. Lieberman J, Gephardt G, Calabrese LH. Urticaria, nephritis, and pseudotumor cerebri. Cleve Clin J Med. (1990) 57:197–210. doi: 10.3949/ccjm.57.2.197
90. Koul PA, Wahid A, Shah SU, Koul AN, Saleem SM. Hypocomplementemic urticarial vasculitis and lower cranial nerve palsies. J Assoc Physicians India. (2000) 48:536–7.
91. Al-Araji A, Kidd DP. Neuro-Behçet's disease: epidemiology, clinical characteristics, and management. Lancet Neurol. (2009) 8:192–204. doi: 10.1016/S1474-4422(09)70015-8
92. Akman-Demir G, Serdaroglu P, Tasçi B. Clinical patterns of neurological involvement in Behcet's disease: evaluation of 200 patients. Brain. (1999) 122:2171–81. doi: 10.1093/brain/122.11.2171
93. Siva A, Kantarci OH, Saip S, Altintas A, Hamuryudan V, Islak C, et al. Behçet's disease: diagnostic and prognostic aspects of neurological involvement. J Neurol. (2001) 248:95–103. doi: 10.1007/s004150170242
94. Farah S, Al-Shubaili A, Montaser A, Hussein JM, Malaviya AN, Mukhtar M, et al. Behcet's syndrome: a report of 41 patients with emphasis on neurological manifestations. J Neurol Neurosurg Psychiatr. (1998) 64:382–4. doi: 10.1136/jnnp.64.3.382
95. Davatchi F, Chams-Davatchi C, Shams H, Nadji A, Faezi T, Akhlaghi M, et al. Adult Behcet's disease in Iran: analysis of 6075 patients. Int J Rheum Dis. (2016) 19:95–103. doi: 10.1111/1756-185X.12691
96. Kidd DP. Neurological complications of Behcet's syndrome. J Neurol. (2017) 264:2178–83. doi: 10.1007/s00415-017-8436-9
97. Siva A, Saip S. The spectrum of nervous system involvement in Behçet's syndrome and its differential diagnosis. J Neurol. (2009) 256:513–29. doi: 10.1007/s00415-009-0145-6
98. Kurtuncu M, Aydin BN, Gunduz T, Ala S, Akman-Demir G. Neuro-Behcet's disease: analysis of 430 patients. J Neurol Sci. (2017) 381:121–2. doi: 10.1016/j.jns.2017.08.376
99. Al-Fahad SA, Al-Araji AH. Neuro-Behcet's disease in Iraq: a study of 40 patients. J Neurol Sci. (1999) 170:105–11. doi: 10.1016/S0022-510X(99)00165-3
100. Kidd D, Steuer A, Denman AM, Rudge P. Neurological complications in Behçet's syndrome. Brain. (1999) 122:2183–94. doi: 10.1093/brain/122.11.2183
101. Hirohata S. Histopathology of central nervous system lesions in Behcet's disease. J Neurol Sci. (2008) 267:41–7. doi: 10.1016/j.jns.2007.09.041
102. Kurt EA, Kocer N, Ozguler Y, Ucar D, Uygunoglu U, Islak C, et al. An outcome survey of 100 patients with cerebral venous sinus thrombosis due to Behcet's syndrome followed up at a single, dedicated center. Arthritis Rheumatol. (2016) 68:3966–7. doi: 10.1002/art.39977
103. Kutlu G, Semercioglu S, Ucler S, Erdal A, Inan LE. Epileptic seizures in Neuro-Behcet disease: why some patients develop seizure and others not? Seizure. (2015) 26:32–5. doi: 10.1016/j.seizure.2015.01.013
104. Gluth MB, Baratz KH, Matteson EL, Driscoll CL. Cogan syndrome: a retrospective review of 60 patients throughout a half century. Mayo Clin Proc. (2006) 81:483–8. doi: 10.4065/81.4.483
105. Van DS, Mccoll G, Walter M, Jennens I, Bhathal P, Wicks IP. Prolonged prodrome, systemic vasculitis, and deafness in cogan's syndrome. Ann Rheum Dis. (2001) 60:69–71. doi: 10.1136/ard.60.1.69
106. Antonios N, Silliman S. Cogan syndrome: an analysis of reported neurological manifestations. Neurologist. (2012) 18:55–63. doi: 10.1097/NRL.0b013e31823fa3a0
107. Grasland A, Pouchot J, Hachulla E, Blétry O, Papo T, Vinceneux P. Typical and atypical Cogan's syndrome: 32 cases and review of the literature. Rheumatology. (2004) 43:1007. doi: 10.1093/rheumatology/keh228
Keywords: primary systemic vasculitis, central nervous system, peripheral nervous system, neurological involvement, clinical manifestation
Citation: Zhang S, Yuan D and Tan G (2019) Neurological Involvement in Primary Systemic Vasculitis. Front. Neurol. 10:430. doi: 10.3389/fneur.2019.00430
Received: 09 August 2018; Accepted: 09 April 2019;
Published: 26 April 2019.
Edited by:
Patrick Kwan, Monash University, AustraliaReviewed by:
Ché Serguera, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceUlises Gomez-Pinedo, Instituto de Investigación Sanitaria del Hospital Clínico San Carlos, Spain
Copyright © 2019 Zhang, Yuan and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ge Tan, dGl0b3RAc2luYS5jb20=