 Matthew Benger
Matthew Benger Kshitij Mankad2
Kshitij Mankad2 Christos Proukakis
Christos Proukakis Nicholas D. Mazarakis
Nicholas D. Mazarakis Maria Kinali
Maria Kinali- 1Department of Neurosciences, King's College Hospital, London, United Kingdom
- 2Department of Neuroradiology, Great Ormond Street Hospital, London, United Kingdom
- 3Institute of Neurology, University College London, London, United Kingdom
- 4Centre for Neuroinflammation & Neurodegeneration, Imperial College, London, United Kingdom
- 5Honorary Senior Lecturer in Paediatric Neurology, Imperial College, London, United Kingdom
Mutations in the PARK2 gene have been implicated in the pathogenesis of early-onset Parkinson's disease. We present a case of movement disorder in a 4-year-old child from consanguineous parents and with a family history of Dopamine responsive dystonia, who was diagnosed with early-onset Parkinson's disease based on initial identification of a pathogenic PARK2 mutation. However, the evolution of the child's clinical picture was unusually rapid, with a preponderance of pyramidal rather than extrapyramidal symptoms, leading to re-investigation of the case with further imaging and genetic sequencing. Interestingly, a second homozygous mutation in the FA2H gene, implicated in Hereditary spastic paraplegia, was revealed, appearing to have contributed to the novel phenotype observed, and highlighting a potential interaction between the two mutated genes.
Case Background
A 4-year-old right-handed boy from Qatar presented with a 1-year history of increasingly frequent falls and progressive gait abnormality. Pregnancy, birth, and neonatal development all proceeded unremarkably. He sat independently by 6 months and walked by 13 months, although his parents noted he did so “heavily” and on his tiptoes bilaterally. He was able to jump but not hop, and negotiated staircases by holding onto the banisters. The parents had no concerns regarding handwriting, cognition, behavior, vision, or hearing. Finally, no diurnal variation in the symptoms had been noted.
The patient was one of three children born to healthy first cousins. His 3-year-old brother and 18-month-old sister were both asymptomatic. His father's 35-year-old brother presented with dystonia aged 13 years. Although his genotype was untested, he was given a clinical diagnosis of Dopamine-responsive dystonia (DRD), which remains Levodopa-responsive today.
The patient had already received a number of tests in Qatar. These included an MRI scan 6 months prior to presentation (reported as normal) and genetic sequencing of PARK2, a gene well known to cause early-onset Parkinson's disease (EO-PD) (1). Regarding the latter, PCR amplification of exon 4 of PARK2 failed after several attempts, and subsequent MPLA analysis confirmed a homozygous, pathogenic deletion in exon 4 of the PARK2 gene (p.Ala138Glyfs*7), leading to a diagnosis of EO-PD, with both parents subsequently identified as being carriers of the same mutation (Figure 1). However, the patient underwent two empirical trials of Levodopa without benefit, prompting the parents to seek a second neurological opinion.
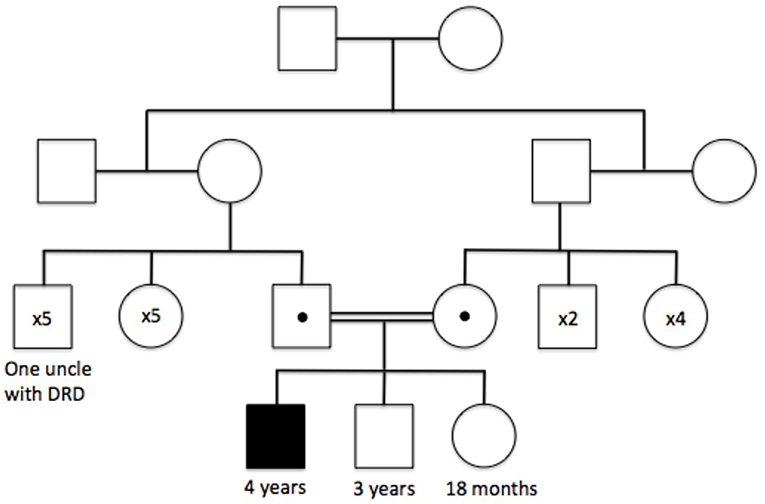
Figure 1. Family tree illustrating the autosomal recessive inheritance of the PARK2 p.Ala138Glyfs*7 mutation, and the clinically affected family members at the time of initial genetic testing.
Examination (Video 1 in Supplementary Material) revealed a markedly paretic gait, with the patient walking, without festination or freezing, on his tiptoes with hips internally rotated in a crouched posture. There was some dystonic posturing of his limbs when lying on the couch. Both lower limbs exhibited hypertonia and hyperreflexia with clonus and upgoing plantars. His upper limbs were less affected, demonstrating subtle rigidity, more pronounced on the left. There was no tremor and no micrographia. There was subtle hypomimia. Cranial nerve examination was normal. The patient followed instructions with minor hesitation and interacted with his siblings normally.
In summary, the clinical picture was predominantly an upper motor neuron-type deficit affecting the patient's lower limbs. In addition to lower limb pyramidal features, there were less pronounced extrapyramidal features including dystonia in the lower limbs, asymmetrical rigidity in the upper limbs, and hypomimia. Hence, this was an early-onset, progressive neurological disorder affecting multiple motor systems. The predominance of pyramidal features, as well as the lack of Levodopa response, argued against this being a disorder driven purely by mutated PARK2.
Diagnostic Investigations
A repeat brain and spine MRI scan was performed under general anesthetic. In contrast to the previous scan, this demonstrated mildly increased T2 signal intensity in the deep and periventricular white matter, predominantly in the peritrigonal region of the lateral ventricles and the occipital region of the cortex, consistent with leukodystrophy. In addition, the repeat MRI demonstrated mild cerebellar volume loss, and bilateral symmetrical iron deposition within the substantiae nigrae and globus pallidi. These radiological changes progressed over a one-year interval (Figure 2). A Dopamine Transporter scan (DaTSCAN) was also carried out at the time of presentation and was reported as normal (Figure 2).
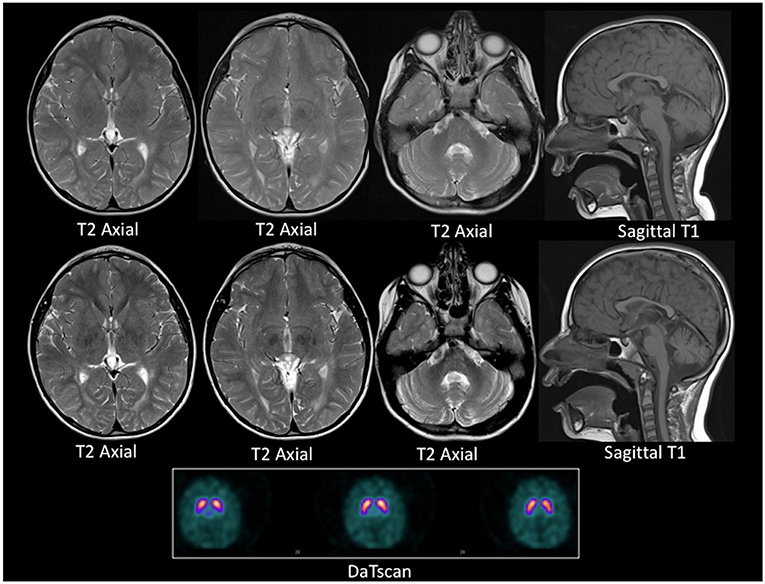
Figure 2. MRI and DaTSCAN images. Top row: Axial T2 weighted images and Sagittal T1 weighted image at age 4 years. Middle row: Axial T2 weighted images Sagittal T1 weighted image at age 5 years. Lower panel: DaTSCAN. All MR images were acquired on a 1.5T scanner. The top and the middle row images show progressive iron deposition in the globus pallidi, more than expected for age, and consistent with neuronal brain iron accumulation. There is global paucity of the cerebral white matter with a thin corpus callosum. There is also cerebellar atrophy between the two time points. The DaTSCAN is normal.
A raft of neurometabolic tests were negative, including CSF studies for neurotransmitters, pyridoxal phosphate and pterin metabolites. Given the patient's spastic paraparetic phenotype, and the above MRI findings, an additional diagnosis of hereditary spastic paraplegia (HSP) was considered.
DNA mutation analyses of a panel of HSP genes revealed a homozygous, glycine to alanine substitution (c.786+1G>A) following exon 5 of Fatty acid 2-hydroxylase (FA2H), a gene responsible for the hydroxylation of myelin galactocerebroside lipid components. In silico analysis of this mutation using Human Splicing Finder software (URL: http://www.umd.be/HSF3/, accessed on 04/04/2019) predicted it would alter an essential “GU” splicing donor sequence of wild type FA2H and should thus be considered pathogenic (2). Mutation of FA2H leads to HSP35 (also known as SPG35), a disorder characterized by childhood-onset spastic paraparesis (±dystonia, ataxia, seizures, progressive cognitive decline) (3, 4) and T2 signal hyperintensities on MRI ± brain iron accumulation (5, 6).
Management
Treatments for HSP are currently limited to those providing symptomatic benefit, alongside genetic counseling for the family. Regular patient follow-up is also crucial, not only to monitor response to symptomatic treatments, but also to identify development of any of the clinically heterogeneous features of HSP35. Furthermore, in this patient's case, whilst the negative DaTSCAN would argue against mutated PARK2-related extrapyramidal features at time of presentation (which were instead more likely to be caused by iron accumulation in the basal ganglia secondary to mutated FA2H), it would be important to continue to monitor for manifestation of mutated PARK2. Indeed, 18 months after the initial presentation, worsening upper limb rigidity was noted, along with bradykinesia, freezing effect, and marked hypomimia, without significant changes in brain iron accumulation on repeat MRI, prompting consideration of a repeat DaTSCAN and levodopa trial at time of writing.
Discussion
Ockham's razor, a frequently cited diagnostic tool, argues that “plurality should never be posited without necessity (7)”. In the case above, the simplest explanation for the patient's symptoms—an altered gait and a propensity to fall—would have pointed to the patient's known recessive PARK2 mutation.
However, there were elements within the history and examination—particularly the pyramidal features and lack of dopamine responsiveness—which could not be “shaved away” for the sake of simplicity. Such features, along with symptom progression, justified the repeat MRI scan, which held the key to diagnosing the additional pathogenic FA2H mutation. The phenotypic overlap demonstrated by this patient in relation to the key clinical features of PARK2-related EO-PD and HSP35 is outlined in Table 1 (9–12).

Table 1. Clinical features of PARK2-associated EO-PO and HSP35 and comparison to the patient studied (8–11).
An additional point of interest remains the genotype of the patient's uncle, who was clinically diagnosed with DSD aged 13 without genotypic confirmation. An intriguing possibility remains that the patient's uncle in fact had homozygous mutation of the PARK2 gene and was demonstrating features of undiagnosed EO-PD (presenting closer to a typical age of onset than in our patient). Certainly, there is considerable phenotypic overlap between DRD and PARK2-related EO-PD (7, 13), alongside the possibility of PARK2 mutations on both maternal and paternal branches of the uncle's family tree due to undocumented consanguinity in previous generations.
At the genetic level, dual homozygous mutations in PARK2 and FA2H have not been reported previously in EO-PD. Both genes play a role in mitochondrial function (a key organelle in the regulation of cellular toxicity and death): the former via a well-established role in optimal mitophagy, the latter via recently discovered links between ceramide metabolism, mitochondrial outer membrane permeability, and electron transport chain activity (14–18). Dual mutation may thus engender additional, or even synergistic, toxicity within nigrostriatal neurons, which might lead to an earlier-than-expected age of symptom onset (typically ~20–40 years) (12, 19). Indeed, whilst the patient's initial DaTSCAN was negative, he went on to develop progressive parkinsonism without corresponding changes in brain iron accumulation on interval MRI suggesting clinical progression of the patient's mutated PARK2-associated phenotype, and prompting consideration of a repeat DaTSCAN and Levodopa trial.
Informed Consent
Written informed consent was obtained from the parents of the participant for the publication of this case report, and from all subjects featured in video material.
Author Contributions
MB: composed manuscript. KM: reported case imaging and selected relevant images formanuscript. CP and NM: aided literature search and edited manuscript. MK: consultant clinician for the case, aided literature search, and edited manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2019.00555/full#supplementary-material
Video 1. Demonstration of patient's gait at time of presentation. Marked lower limb spasticity is noted without festination or freezing. The patient walks on his tiptoes with hips internally rotated.
References
1. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. (1998) 392:605–8. doi: 10.1038/33416
2. Desmet F-O, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. (2009) 37:e67. doi: 10.1093/nar/gkp215
3. Edvardson S, Hama H, Shaag A, Gomori JM, Berger I, Soffer D, et al. Mutations in the fatty acid 2-hydroxylase gene are associated with leukodystrophy with spastic paraparesis and dystonia. Am J Hum Genet. (2008) 83:643–8. doi: 10.1016/j.ajhg.2008.10.010
4. Dick KJ, Eckhardt M, Paisan-Ruiz C, Alshehhi AA, Proukakis C, Sibtain NA, et al. Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35). Hum Mutat. (2010) 31:E1251–60. doi: 10.1002/humu.21205
5. Hourani R, El-Hajj T, Barada WH, Hourani M, Yamout BI. MR imaging findings in autosomal recessive hereditary spastic paraplegia. AJNR Am J Neuroradiol. (2009) 30:936–40. doi: 10.3174/ajnr.A1483
6. Schneider SA, Dusek P, Hardy J, Westenberger A, Jankovic J, Bhatia KP. Genetics and pathophysiology of neurodegeneration with brain iron accumulation (NBIA). Curr Neuropharmacol. (2013) 11:59–79. doi: 10.2174/157015913804999469
7. William of Ockham. Quaestiones et decisiones in quattuor libros Sententiarum Petri Lombardi. Johannes Trechsel. (1495)
8. Potulska-Chromik A, Hoffman-Zacharska D, Łukawska M, Kostera-Pruszczyk A. Dopa-responsive dystonia or early-onset Parkinson disease – Genotype–phenotype correlation. Neurol Neurochir Pol. (2017) 51:1–6. doi: 10.1016/j.pjnns.2016.07.013
9. Dick KJ, Al-Mjeni R, Baskir W, Koul R, Simpson MA, Patton MA, et al. A novel locus for an autosomal recessive hereditary spastic paraplegia (SPG35) maps to 16q21-q23. Neurology. (2008) 71:248. doi: 10.1212/01.wnl.0000319610.29522.8a
10. Massano J, Bhatia KP. Clinical approach to Parkinson's disease: features, diagnosis, and principles of management. Cold Spring Harb Perspect Med. (2012) 2:a008870. doi: 10.1101/cshperspect.a008870
11. Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. (2008) 7:1127–38. doi: 10.1016/S1474-4422(08)70258-8
12. Bruggemann N, Klein C. Parkin Type of early-onset parkinson disease. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, et al, editors. GeneReviews (R). Seattle, WA: University of Washington (1993).
13. Khan NL, Graham E, Critchley P, Schrag AE, Wood NW, Lees AJ, et al. Parkin disease: a phenotypic study of a large case series. Brain J Neurol. (2003) 126(Pt 6):1279–92. doi: 10.1093/brain/awg142
14. Czubowicz K, Strosznajder R. Ceramide in the molecular mechanisms of neuronal cell death. The role of sphingosine-1-phosphate. Mol Neurobiol. (2014) 50:26–37. doi: 10.1007/s12035-013-8606-4
15. Ganesan V, Perera MN, Colombini D, Datskovskiy D, Chadha K, Colombini M. Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis Int J Program Cell Death. (2010) 15:553–62. doi: 10.1007/s10495-009-0449-0
16. Gudz TI, Tserng KY, Hoppel CL. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J Biol Chem. (1997) 272:24154–8. doi: 10.1074/jbc.272.39.24154
17. Kogot-Levin A, Saada A. Ceramide and the mitochondrial respiratory chain. Biochimie. (2014) 100:88–94. doi: 10.1016/j.biochi.2013.07.027
18. Siskind LJ, Kolesnick RN, Colombini M. Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J Biol Chem. (2002) 277:26796–803. doi: 10.1074/jbc.M200754200
Keywords: PARK2 mutation, movement disorder, hereditary spastic paraplegia (HSP), novel phenotype, FA2H gene
Citation: Benger M, Mankad K, Proukakis C, Mazarakis ND and Kinali M (2019) The Interaction of Genetic Mutations in PARK2 and FA2H Causes a Novel Phenotype in a Case of Childhood-Onset Movement Disorder. Front. Neurol. 10:555. doi: 10.3389/fneur.2019.00555
Received: 11 March 2019; Accepted: 09 May 2019;
Published: 29 May 2019.
Edited by:
Ignacio Mata, Cleveland Clinic, United StatesReviewed by:
Manabu Funayama, Juntendo University, JapanJordi Clarimon, Sant Pau Institute for Biomedical Research, Spain
Copyright © 2019 Benger, Mankad, Proukakis, Mazarakis and Kinali. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew Benger, bWF0dGhldy5iZW5nZXJAbmhzLm5ldA==