 Frederike Cosima Oertel
Frederike Cosima Oertel Jana Schließeit
Jana Schließeit Alexander U. Brandt
Alexander U. Brandt Friedemann Paul
Friedemann Paul- 1NeuroCure Clinical Research Center, Berlin Institute of Health, Charité – Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, Berlin, Germany
- 2Experimental and Clinical Research Center, Max-Delbrück-Centrum für Molekulare Medizin and Charité – Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Berlin, Germany
- 3Faculty of Psychology and Neuroscience, Maastricht University, Maastricht, Netherlands
- 4Department of Neurology, University of California, Irvine, Irvine, CA, United States
- 5Department of Neurology, Berlin Institute of Health, Charité – Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, Berlin, Germany
Neuromyelitis optica spectrum disorders (NMOSD) are mostly relapsing autoimmune inflammatory disorders of the central nervous system (CNS) with optic neuritis, myelitis, and brainstem syndromes as clinical hallmarks. With a reported prevalence of up to 70%, cognitive impairment is frequent, but often unrecognized and an insufficiently treated burden of the disease. The most common cognitive dysfunctions are decline in attention and memory performance. Magnetic resonance imaging can be used to access structural correlates of neuropsychological disorders. Cognitive impairment is not only a highly underestimated symptom in patients with NMOSD, but potentially also a clinical correlate of attack-independent changes in NMOSD, which are currently under debate. This article reviews cognitive impairment in NMOSD and discusses associations between structural changes of the CNS and cognitive deficits.
Introduction
Neuromyelitis optica spectrum disorders (NMOSD) are inflammatory autoimmune conditions of the central nervous system (CNS) with a typically relapsing course and a strong female preponderance (1–6). Key clinical features comprise optic neuritis, myelitis, and brainstem syndromes (7–13). Approximately 80% of the patients with NMOSD have pathogenic serum autoantibodies against aquaporin-4 (AQP4), a bidirectional water channel protein predominantly expressed by astrocytes, which is present all over the CNS (7, 14–21). AQP4 appears to not only be important for the internal water balance but its downstream mechanisms also seem to be essential for synaptic plasticity and neuronal functioning due to the involvement of astrocytes, although the exact mechanism of action is still unclear (22). In a subgroup of AQP4 antibody (AQP4-IgG) seronegative NMOSD patients as well as in patients with recurrent optic neuritis and a few patients with multiple sclerosis (MS) an antibody against myelin oligodendrocyte glycoprotein (MOG-IgG) can be detected (23–33). Nowadays, MOG-IgG seropositive patients are mostly assigned to a separate disease entity called MOG-IgG autoimmunity [or MOG encephalomyelitis (MOG-EM)], although they are formally still part of the NMO spectrum (5, 34–36).
In clinical routine, detection of AQP4-specific antibodies in serum allows for discriminating NMOSD from its most common differential diagnosis, MS. The high specificity of AQP4-IgG together with various immunological studies has made clear that NMOSD are not a variant of MS but a separate disease entity (7, 37–39). Subsequently, disease-modifying drugs used in MS, for example interferon-beta, glatiramer acetate, or natalizumab, were found to be ineffective or even harmful for patients with NMOSD (40–45). In contrast, current treatment strategies for patients with NMOSD comprise immunosuppressive therapies with azathioprine or mycophenolate-mofetil and B cell targeting therapies with rituximab (46–54). For most NMOSD patients, these drugs effectively reduce the attack frequency and attack-related accumulation of disability. However, recent studies suggested attack- and lesion-independent “covert” tissue damage in patients with NMOSD, from which clinical implications are not yet entirely clear (55–64) but which presumably contributes to attack-independent symptoms as of which one appears to be cognitive impairment (CI). CI as attack independent symptom was further supported by a study by Saji et al. (65) who tried to proof a permanent interaction between astrocytes and AQP4-IgG which lead finally to dysfunctional synaptic plasticity and hence could be involved in CI in NMOSD AQP4-IgG positive patients. Furthermore, even though CI appears to be a persistent and progressive symptom it still seems to be underrepresented in clinical monitoring and disability scores and often not sufficiently treated (66–70). Over the last years, those neuropsychiatric symptoms in NMOSD came to the forefront, and comparable larger cohorts in observational studies and the application of advanced imaging techniques allowed for the investigation of incidence and structural correlates of these neuropsychological symptoms. This article reviews CI in NMOSD and discusses associations between structural CNS changes and cognitive deficits.
Cognitive Impairment in NMOSD
NMOSD show high rates of comorbidity with other physical and psychological conditions, including CI (67–69, 71). While cognitive dysfunctions have been commonly recognized as a burden in MS, this acknowledgment is still missing in NMOSD and hence studies investigating the link between NMOSD and CI are scarce (72, 73) despite NMOSD patients naming CI as one of their most relevant concerns (74) (see Table 1). In the few studies conducted, patients demonstrate a significant decrease of cognitive abilities and the prevalence of CI in different samples varies between 30 and 70% (71, 75–77). In addition to investigate cognition, each study has used its own assessment method to the end that one common disease specific cognitive test battery for patients with NMOSD is missing (71). Hence, meta-analyses such as the one of Meng et al. are difficult and are usually only able to analyse and make inferences from a small sample of studies (75). Currently, the most commonly used test battery appears to be the Rao's Brief Repeatable Battery of Neuropsychological tests (65, 76, 78). This battery assesses different aspects of cognition for example verbal memory, short and long term memory, processing speed, attention, concentration, and verbal fluency (65).
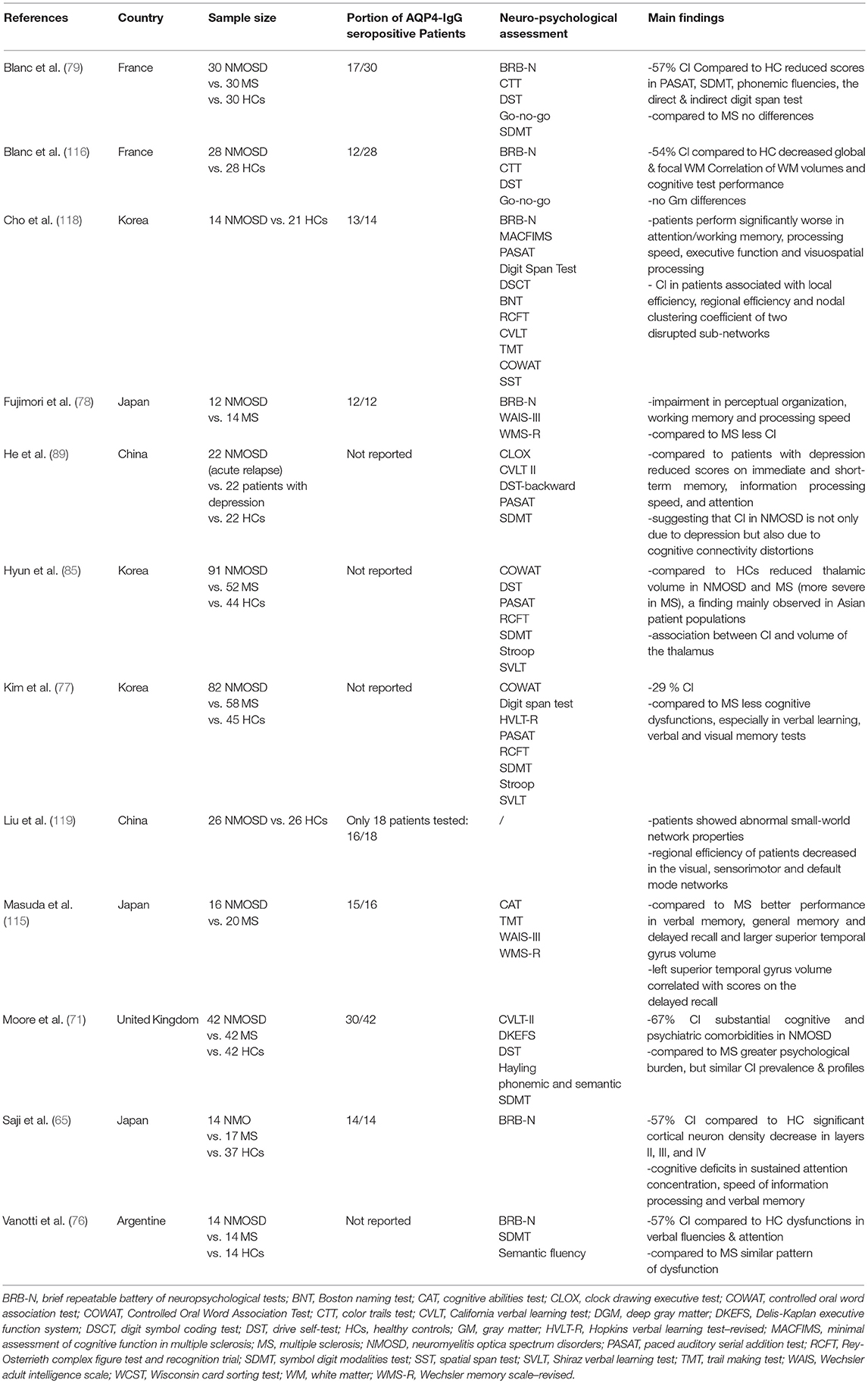
Table 1. The most important original publications on cognitive impairment in NMOSD.
Studies investigating which aspects of cognition are most dysfunctional in patients with NMOSD depict ambiguous results: One of the first studies was conducted by Blanc et al. (79). They found alterations in attention, information processing and verbal fluency (79). The meta-analysis of Meng et al. concluded that patients with NMOSD perform generally worse than healthy controls and that patients are significantly worse in the areas of attention, language, memory, processing speed and executive function (75). Similar findings were made by Saji et al., who found that 57% of patients performed significantly worse in at least three cognitive tests compared to healthy controls (65). Furthermore, they found that deficits are predominantly overt in sustained attention, concentration, verbal memory, and speed of information processing (65). However, verbal fluency on semantic stimulation and spatial reasoning were intact (65). Opposed to these results, Vanotti et al. demonstrated a more pronounced dysfunction in the areas of attention and verbal fluencies (76). The variations across results are not only due to heterogeneity of samples including ethnic background, heterogeneity with regards to antibody status, and other interindividual differences, but also due to differences in assessment, the definition and percentage of CI in samples, correction for depression, as well as analysis of magnetic resonance imaging (MRI) and overall differences in study design (72). In particular, the heterogeneous antibody status and MOG-IgG could be a major confounder, as many studies had mixed samples for example Blanc et al. (17 AQP4-IgG seropositive patients/13 AQP4-IgG seronegative patients who were not tested for MOG-IgG) (79). Other studies for example Vanotti et al. or Liu et al. have not reported antibody status, and hence may have missed a possible association between antibody status and CI (76, 80). Further constraints entail that most of the studies recruited rather small sample sizes, ranging from 12 to 91 patients, and cover only a limited spectrum of ethnic backgrounds, with most studies being from Asian countries (71). Thus, while CI seems to be commonly present in a high percentage of NMOSD patients the specific domain of dysfunctional performance varies greatly between studies and samples. It appears that the most common cognitive dysfunction across all studies is decline in attention and memory performance. Moreover, the question as to the whether NMOSD pathobiology is causative for CI has not been clarified.
AQP4-IgG and Cognitive Impairment
Currently, attack-independent structural changes as part of the pathology in NMOSD are a matter of debate (56–59, 64, 81, 82). The continuous and sometimes deteriorating neuropsychological symptoms might point toward a smoldering disease process in NMOSD independent of clinical attacks, for instance directly caused by the pathogenic antibody AQP4-IgG (56–59). Hence, few studies have tried to investigate the interplay of AQP4-IgG, which usually are a persistent disease factor, and cognitive impairment. Fan et al. investigated the link between AQP4-IgG and cognitive functioning, in particular spatial memory, in mice: They concluded that AQP4-IgG appear to inhibit neuronal plasticity and thus lead to a worsening in memory consolidation and spatial memory, as mice AQP4 knockout mice showed deficits in the acquisition and reversal phase in the Morris water maze (83, 84). This link is further supported by Skucas et al. (86) who found impaired long term potentiation and thus deficits in spatial memory in AQP4 knockout mice (83). While this finding would explain deficits in spatial memory, it provides no explanation for attention deficits, which are often observed in NMOSD patients. An attempt to explain the full extent of poor cognitive functioning in patients with NMOSD was made by Saji et al. who claimed that a unique dynamic between AQP4-IgG and astrocytes is the underlying mechanism belonging to CI (65). According to Saji et al. the unique dynamic exhibited by AQP4-IgG is leading to substantial diffuse cortical neuronal loss throughout the whole brain and hence may lead to neurodegeneration independent of clinical attacks (65). Thus, in contrast to MS where the disease causes demyelinating lesions, AQP4-IgG seropositive NMOSD results in pathological changes of the gray matter especially in the cortical layer II (65). These processes are suggested to include a disruption of glutamate and/or water homeostasis and thus excitotoxicity, release of soluble neurotoxic factors, which may trigger neurodegeneration by diffusing into the gray matter (GM) (65). This pathomechanism would further explain the GM atrophy observed by some studies in patients with AQP4-IgG seropositive NMOSD (80, 85). Other processes possibly induced by downstream mechanisms of AQP4 dysfunction are the activation of the innate immune system and hence activation of microglia as well as other autoimmune properties that could be involved not only in AQP4-IgG seropositive NMOSD but also as response to AQP4-IgG (37, 65, 86, 87). The hypothesis of an immune reaction, leading to pathological downstream mechanisms is further supported by Takeshita et al. (38). They found that, in cell cultures, AQP4-positive astrocytes produced interleukin-6 when exposed to AQP4-IgG. The cytokine interleukin-6 was found to disrupt endothelial cell functioning, which results in impaired blood barrier function (38, 88). On top of that, AQP4 expression seems to play an important role in regulating synaptic plasticity, which might be altered in NMOSD due to AQP4-IgG (88). Although the exact mechanism remains unclear, the existence of CI and other neuropsychological symptoms in AQP4-IgG seropositive NMOSD points toward an attack-independent pathology, potentially induced by the pathogenic antibodies themselves.
Link Between Cognitive Impairment and Depression in NMOSD
Depression is known to also be a common and insufficiently treated symptom in NMOSD: Whereas around 50% of moderately to severely depressed patients with NMOSD receive antidepressant medical treatment, only 50% of treated patients report satisfactory treatment responses (68). Nevertheless, only few studies have investigated the link between depression and CI in patients with NMOSD. On the one hand, studies found a strong association between poor cognitive performance and high levels of depressive symptoms (71, 76). On the other, the study of Blanc et al. reported no association between cognition and depression (79). These opposing results could be explained by small sample sizes, heterogeneous cohorts, and different ethnic backgrounds. Furthermore, as both CI and depression tend to have high prevalence in NMOSD, an association but not causation could be possible (67, 71). On top of that, even if a causal link could be proven, the direction of this association would still be questionable. Especially, since studies focusing on fatigue and quality of life are implying a role of these in depression as well as in cognition (89–91). Therefore, it is advisable to investigate the full spectrum of the disease and its psychological comorbidities instead of only exploring the link between depression and CI and thus eliminating possible confounders.
Association Between Cognitive Impairment and Structural Changes
Numerous studies have described brain tissue alterations in MS [global atrophy, microstructural damage of normal-appearing white matter (WM) and GM)], but studies investigating MRI characteristics in NMOSD are still scarce (92–98). According to the current state of knowledge, up to 80% of AQP4-IgG seropositive NMOSD patients present with cerebral lesions in AQP4-rich sites for example the hypothalamus and periependymal regions and—in contrast to MS—cortical lesions are usually absent (99–105). Also, the few existing MRI studies on MOG-IgG seropositive encephalomyelitis suggest a high similarity with MRI features of AQP4-IgG positive patients with the occasional incidence of characteristic fluffy brainstem lesions (106–108). However, several groups recently reported seizures with cortical MRI involvement on MRI in MOG-IgG seropositive patients which is considered very rare in AQP4-IgG positive NMOSD (109, 110). Whereas, several studies exist describing transsynaptic damage after optic neuritis and myelitis, the existence of global atrophy, and diffuse tissue alterations of WM and GM in AQP4-IgG seropositive NMOSD is still a matter of debate (13, 58, 63, 64, 93, 95, 111, 113, 114).
In order to explain the cognitive dysfunction observed, studies have focused on cortical abnormalities (80, 115). While in MS cognitive dysfunction is linked to cortical lesions, no such correlation can be observed in patients with NMOSD (71, 79, 80, 115). In the study of Liu et al. (80) when comparing 54 NMOSD patients, 28 of which were cognitively preserved and 26 of which had CI, there was no association found between overall brain lesion load and CI (76, 80). Some studies found a correlation between WM and GM atrophy, and CI: Blanc et al. linked focal WM volumes of the brainstem, cerebellum, corticospinal tract, corpus callosum, longitudinal fascicle, and inferior longitudinal fascicle with general CI in NMOSD patients (79). In particular, visual memory, verbal memory, speed of information processing, short term memory and executive functions were found to be impaired (116). Hence, both focal and global WM volume were linked to CI, but no GM atrophy was observed (116). This finding is in line with the work of Finke et al., who observed no changes in deep GM volumes in AQP4-IgG seropositive NMOSD (112). This is further underlined by so far unpublished results from our groups suggesting missing atrophy of the entire thalamic volumes in AQP4-IgG seropositive patients compared to HCs and only selective subnuclei atrophy in attack-related nuclei such as the lateral geniculate nucleus (117). Conflicting with the missing GM and thalamus atrophy was the study conducted by Hyun et al. (85). They described a significant link between volume of the thalamus and CI in patients with NMOSD, with reduced volumes in patients with poorer cognitive performance (85). Nevertheless, the different conclusions about thalamus volume could be due to the fact that Hyun et al. (85) measured their patients after a mean disease duration of 8 years where advanced degeneration has taken place, which potentially could not only be a confounder on its own but also could possibly lead to confounding through depression, advanced disability, and pain. Furthermore, the sample population appears to play an important role when comparing results in respect to GM atrophy, as reduced volume is mainly observed in Asian samples while studies examining a Caucasian sample fail to replicate these results (117). Hence, studies examining cortical volumes in NMOSD should be interpreted carefully.
Further studies investigating structural changes indicate that white matter network alternations could be the underlying reason for cognitive decline in some NMOSD patients (118, 119). One study by Liu et al. investigated the structural connectivity of 26 NMO-patients and 26 sex- and age-matched HCs with help of diffusion tensor tractography (DTI) (119). After performing network analysis, they found alternations in the small-world topology of the white matter structural networks, including abnormal parameters in path length, an increase in small-worldness as well as an increase in normalized clustering. Furthermore, they found an altered global network organization, which is in line with reduced cognitive efficacy observed in NMOSD patients. They suggest that in particular the reduced efficacy of the precuneus (PCUN), a hub belonging to the default-mode network, which is highly involved in cognitive processing, could partially contribute to CI in patients. A similar DTI study investigating white matter networks and cognitive dysfunction in NMOSD was performed by Cho et al. (118). They enrolled 14 NMOSD patients and 21 HCs, and could confirm the finding of Liu et al., that global network strength is decreased (118, 119). Furthermore, they indicated two disrupted sub-networks, each consistent of six hub nodes, including the PCUN. The disrupted networks were significantly linked to poor performance in attention, processing speed and working memory, as well as to visuospatial processing and executive functions. In particular, the local efficiency, regional efficiency and clustering coefficient of these two sub-networks appear to play a role in CI in NMOSD.
Hence, while several structural changes in GM as well as in WM networks seem to occur in NMOSD it appears to be rather difficult to link CI with one particular tissue alternation. On top of that these studies that have investigated the underlying structures of CI face limitations of which a mixed cohort with heterogeneous antibody-status of the patients is one of the most prominent (96).These limitations, in particular the heterogeneous antibody-status, with earlier studies like Blanc et al. (79, 116) reporting a higher seronegative-seropositive ratio than current studies, hamper comparison between studies.
Outlook
Cognitive impairment (CI) appears to be one of the more prominent progressive and attack independent symptoms of NMOSD. Hence, a sensitization of the treating neurologists as well as early and standardized screening tests are therefore necessary to improve the management and treatment of cognitive impairment and other neuropsychiatric symptoms in NMOSD. In the future, adequately powered studies investigating CI, its underlying pathobiological mechanisms as well as longitudinal changes and clinical impact should be a priority of NMOSD research.
With regards to NMOSD-specific pathology, continuous and sometimes deteriorating neuropsychological symptoms might point to covert disease activity in NMOSD independent of clinical attacks. In light of the ongoing discussion on attack-independent structural changes in NMOSD, we should therefore keep in mind that CI might represent a clinical correlate of underlying subtle microstructural CNS changes.
Author Contributions
FO, JS, AB, and FP wrote the manuscript as well as read and approved the final version.
Conflict of Interest Statement
FO was employed of Nocturne, unrelated to this manuscript. AB is founder and holds shares of Motognosis and Nocturne. He is named as inventor on several patent applications describing serum biomarkers for MS, perceptive visual computing for tracking of motor dysfunction and OCT image analysis. FP reports research grants and speaker honoraria from Bayer, Teva, Genzyme, Merck, Novartis, MedImmune and is member of the steering committee of the OCTIMS study (Novartis), all unrelated to this work.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
2. Mori M, Kuwabara S, Paul F. Worldwide prevalence of neuromyelitis optica spectrum disorders. J Neurol Neurosurg Psychiatry. (2018) 89:555–6. doi: 10.1136/jnnp-2017-317566
3. Gold SM, Willing A, Leypoldt F, Paul F, Friese MA. Sex differences in autoimmune disorders of the central nervous system. Semin Immunopathol. (2018) 41:177–88. doi: 10.1007/s00281-018-0723-8
4. Bove R, Elsone L, Alvarez E, Borisow N, Cortez MM, Mateen FJ, et al. Female hormonal exposures and neuromyelitis optica symptom onset in a multicenter study. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e339. doi: 10.1212/NXI.0000000000000339
5. Borisow N, Mori M, Kuwabara S, Scheel M, Paul F. Diagnosis and treatment of NMO spectrum disorder and MOG-encephalomyelitis. Front Neurol. (2018) 9:888. doi: 10.3389/fneur.2018.00888
6. Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflamm. (2012) 9:14. doi: 10.1186/1742-2094-9-14
7. Jarius S, Wildemann B, Paul F. Neuromyelitis optica: clinical features, immunopathogenesis and treatment. Clin Exp Immunol. (2014) 176:149–64. doi: 10.1111/cei.12271
8. Kremer L, Mealy M, Jacob A, Nakashima I, Cabre P, Bigi S, et al. Brainstem manifestations in neuromyelitis optica: a multicenter study of 258 patients. Mult Scler. (2014) 20:843–7. doi: 10.1177/1352458513507822
9. Kitley J, Leite MI, Nakashima I, Waters P, McNeillis B, Brown R, et al. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain. (2012) 135:1834–49. doi: 10.1093/brain/aws109
10. Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology. (1999) 53:1107–14.
11. Pfueller CF, Paul F. Imaging the visual pathway in neuromyelitis optica, imaging the visual pathway in neuromyelitis optica. Mult Scler Int. (2011) 2011:e869814. doi: 10.1155/2011/869814
12. Schmidt F, Zimmermann H, Mikolajczak J, Oertel FC, Pache F, Weinhold M, et al. Severe structural and functional visual system damage leads to profound loss of vision-related quality of life in patients with neuromyelitis optica spectrum disorders. Mult Scler Relat Disord. (2017) 11:45–50. doi: 10.1016/j.msard.2016.11.008
13. Chien C, Scheel M, Schmitz-Hübsch T, Borisow N, Ruprecht K, Bellmann-Strobl J, et al. Spinal cord lesions and atrophy in NMOSD with AQP4-IgG and MOG-IgG associated autoimmunity. Mult Scler. (2018), p. 1–11. doi: 10.1177/1352458518815596
14. Jarius S, Paul F, Franciotta D, Waters P, Zipp F, Hohlfeld R, et al. Mechanisms of Disease: aquaporin-4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol. (2008) 4:202–14. doi: 10.1038/ncpneuro0764
15. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. (2004) 364:2106–12. doi: 10.1016/S0140-6736(04)17551-X
16. Metz I, Beißbarth T, Ellenberger D, Pache F, Stork L, Ringelstein M, et al. Serum peptide reactivities may distinguish neuromyelitis optica subgroups and multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e204. doi: 10.1212/NXI.0000000000000204
17. Zekeridou A, Lennon VA. Aquaporin-4 autoimmunity. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e110. doi: 10.1212/NXI.0000000000000110
18. Hinson SR, Pittock SJ, Lucchinetti CF, Roemer SF, Fryer JP, Kryzer TJ, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. (2007) 69:2221–31. doi: 10.1212/01.WNL.0000289761.64862.ce
19. Waters P, Reindl M, Saiz A, Schanda K, Tuller F, Kral V, et al. Multicentre comparison of a diagnostic assay: aquaporin-4 antibodies in neuromyelitis optica. J Neurol Neurosurg Psychiatry. (2016) 87:1005–15. doi: 10.1136/jnnp-2015-312601
20. Jarius S, Franciotta D, Paul F, Bergamaschi R, Rommer PS, Ruprecht K, et al. Testing for antibodies to human aquaporin-4 by ELISA: sensitivity, specificity, and direct comparison with immunohistochemistry. J Neurol Sci. (2012) 320:32–7. doi: 10.1016/j.jns.2012.06.002
21. Paul F, Jarius S, Aktas O, Bluthner M, Bauer O, Appelhans H, et al. Antibody to aquaporin 4 in the diagnosis of neuromyelitis optica. PLoS Med. (2007) 4:e133. doi: 10.1371/journal.pmed.0040133
22. Szu JI, Binder DK. The role of astrocytic aquaporin-4 in synaptic plasticity and learning and memory. Front Integr Neurosci. (2016) 10:8. doi: 10.3389/fnint.2016.00008
23. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflamm. (2016) 13:279. doi: 10.1186/s12974-016-0717-1
24. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflamm. (2016) 13:280. doi: 10.1186/s12974-016-0718-0
25. Jarius S, Kleiter I, Ruprecht K, Asgari N, Pitarokoili K, Borisow N, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 3: brainstem involvement - frequency, presentation and outcome. J Neuroinflamm. (2016) 13:281. doi: 10.1186/s12974-016-0719-z
26. Pache F, Zimmermann H, Mikolajczak J, Schumacher S, Lacheta A, Oertel FC, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 4: afferent visual system damage after optic neuritis in MOG-IgG-seropositive versus AQP4-IgG-seropositive patients. J Neuroinflamm. (2016) 13:282. doi: 10.1186/s12974-016-0720-6
27. Waters P, Woodhall M, O'Connor KC, Reindl M, Lang B, Sato DK, et al. MOG cell-based assay detects non-MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e89. doi: 10.1212/NXI.0000000000000089
28. Spadaro M, Gerdes LA, Krumbholz M, Ertl-Wagner B, Thaler FS, Schuh E, et al. Autoantibodies to MOG in a distinct subgroup of adult multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e257. doi: 10.1212/NXI.0000000000000257
29. Chalmoukou K, Alexopoulos H, Akrivou S, Stathopoulos P, Reindl M, Dalakas MC. Anti-MOG antibodies are frequently associated with steroid-sensitive recurrent optic neuritis. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e131. doi: 10.1212/NXI.0000000000000131
30. Reindl M, Rostasy K. MOG antibody-associated diseases. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e60. doi: 10.1212/NXI.0000000000000060
31. Hamid SHM, Whittam D, Mutch K, Linaker S, Solomon T, Das K, et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are MOG-IgG positive? A cross sectional study of 132 patients. J Neurol. (2017) 264:2088–94. doi: 10.1007/s00415-017-8596-7
32. Jarius S, Paul F, Aktas O, Asgari N, Dale RC, de Seze J, et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflamm. (2018) 15:134. doi: 10.1186/s12974-018-1144-2
33. Narayan R, Simpson A, Fritsche K, Salama S, Pardo S, Mealy M, et al. MOG antibody disease: a review of MOG antibody seropositive neuromyelitis optica spectrum disorder. Mult Scler Relat Disord. (2018) 25:66–72. doi: 10.1016/j.msard.2018.07.025
34. Sato DK, Callegaro D, Lana-Peixoto MA, Waters PJ, de Jorge FMH, Takahashi T, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. (2014) 82:474–81. doi: 10.1212/WNL.0000000000000101
35. Zamvil SS, Slavin AJ. Does MOG Ig-positive AQP4-seronegative opticospinal inflammatory disease justify a diagnosis of NMO spectrum disorder? Neurol Neuroimmunol Neuroinflamm. (2015) 2:e62. doi: 10.1212/NXI.0000000000000062
36. Hacohen Y, Palace J. Time to separate MOG-Ab-associated disease from AQP4-Ab-positive neuromyelitis optica spectrum disorder. Neurology. (2018) 90:947–8. doi: 10.1212/WNL.0000000000005619
37. Bennett JL, O'Connor KC, Bar-Or A, Zamvil SS, Hemmer B, Tedder TF, et al. B lymphocytes in neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e104. doi: 10.1212/NXI.0000000000000104
38. Takeshita Y, Obermeier B, Cotleur AC, Spampinato SF, Shimizu F, Yamamoto E, et al. Effects of neuromyelitis optica-IgG at the blood-brain barrier in vitro. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e311. doi: 10.1212/NXI.0000000000000311
39. Melamed E, Levy M, Waters PJ, Sato DK, Bennett JL, John GR, et al. Update on biomarkers in neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e134. doi: 10.1212/NXI.0000000000000134
40. Gelfand JM, Cotter J, Klingman J, Huang EJ, Cree BAC. Massive CNS monocytic infiltration at autopsy in an alemtuzumab-treated patient with NMO. Neurol Neuroimmunol Neuroinflamm. (2014) 1:e34. doi: 10.1212/NXI.0000000000000034
41. Stellmann J-P, Krumbholz M, Friede T, Gahlen A, Borisow N, Fischer K, et al. Immunotherapies in neuromyelitis optica spectrum disorder: efficacy and predictors of response. J Neurol Neurosurg Psychiatr. (2017) 88:639–47. doi: 10.1136/jnnp-2017-315603
42. Trebst C, Jarius S, Berthele A, Paul F, Schippling S, Wildemann B, et al. Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol. (2014) 261:1–16. doi: 10.1007/s00415-013-7169-7
43. Ayzenberg I, Schöllhammer J, Hoepner R, Hellwig K, Ringelstein M, Aktas O, et al. Efficacy of glatiramer acetate in neuromyelitis optica spectrum disorder: a multicenter retrospective study. J Neurol. (2016) 263:575–82. doi: 10.1007/s00415-015-7991-1
44. Kleiter I, Hellwig K, Berthele A, Kümpfel T, Linker RA, Hartung H-P, et al. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol. (2012) 69:239–45. doi: 10.1001/archneurol.2011.216
45. Gahlen A, Trampe A-K, Haupeltshofer S, Ringelstein M, Aktas O, Berthele A, et al. Aquaporin-4 antibodies in patients treated with natalizumab for suspected MS. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e363. doi: 10.1212/NXI.0000000000000363
46. Nosadini M, Alper G, Riney CJ, Benson LA, Mohammad SS, Ramanathan S, et al. Rituximab monitoring and redosing in pediatric neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e188. doi: 10.1212/NXI.0000000000000188
47. Valentino P, Marnetto F, Granieri L, Capobianco M, Bertolotto A. Aquaporin-4 antibody titration in NMO patients treated with rituximab: a retrospective study. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e317. doi: 10.1212/NXI.0000000000000317
48. Mealy MA, Wingerchuk DM, Palace J, Greenberg BM, Levy M. Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: multicenter study of treatment efficacy. JAMA Neurol. (2014) 71:324–30. doi: 10.1001/jamaneurol.2013.5699
49. Rommer PS, Dörner T, Freivogel K, Haas J, Kieseier BC, Kümpfel T, et al. Safety and clinical outcomes of rituximab treatment in patients with multiple sclerosis and neuromyelitis optica: experience from a national online registry (GRAID). J Neuroimmune Pharmacol. (2016) 11:1–8. doi: 10.1007/s11481-015-9646-5
50. Paul F. Hope for a rare disease: eculizumab in neuromyelitis optica. Lancet Neurol. (2013) 12:529–31. doi: 10.1016/S1474-4422(13)70089-9
51. Mealy MA, Kim S-H, Schmidt F, López R, Jimenez Arango JA, Paul F, et al. Aquaporin-4 serostatus does not predict response to immunotherapy in neuromyelitis optica spectrum disorders. Mult Scler. (2017) 24:1737–42. doi: 10.1177/1352458517730131
52. Marcinnò A, Marnetto F, Valentino P, Martire S, Balbo A, Drago A, et al. Rituximab-induced hypogammaglobulinemia in patients with neuromyelitis optica spectrum disorders. Neurology. (2018) 5:e498. doi: 10.1212/NXI.0000000000000498
53. Das G, Damotte V, Gelfand JM, Bevan C, Cree BAC, Do L, et al. Rituximab before and during pregnancy: a systematic review, and a case series in MS and NMOSD. Neurology. (2018) 5:e453. doi: 10.1212/NXI.0000000000000453
54. Ellwardt E, Ellwardt L, Bittner S, Zipp F. Monitoring B-cell repopulation after depletion therapy in neurologic patients. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e463. doi: 10.1212/NXI.0000000000000463
55. Liu Y, Duan Y, He Y, Yu C, Wang J, Huang J, et al. A tract-based diffusion study of cerebral white matter in neuromyelitis optica reveals widespread pathological alterations. Mult Scler. (2012) 18:1013–21. doi: 10.1177/1352458511431731
56. Felix CM, Levin MH, Verkman AS. Complement-independent retinal pathology produced by intravitreal injection of neuromyelitis optica immunoglobulin G. J Neuroinflamm. (2016) 13:275. doi: 10.1186/s12974-016-0746-9
57. Jeong IH, Kim HJ, Kim N-H, Jeong KS, Park CY. Subclinical primary retinal pathology in neuromyelitis optica spectrum disorder. J Neurol. (2016) 263:1343–8. doi: 10.1007/s00415-016-8138-8
58. Oertel FC, Kuchling J, Zimmermann H, Chien C, Schmidt F, Knier B, et al. Microstructural visual system changes in AQP4-antibody–seropositive NMOSD. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e334. doi: 10.1212/NXI.0000000000000334
59. Oertel FC, Havla J, Roca-Fernández A, Lizak N, Zimmermann H, Motamedi S, et al. Retinal ganglion cell loss in neuromyelitis optica: a longitudinal study. J Neurol Neurosurg Psychiatry. (2018) 89:1259–65. doi: 10.1136/jnnp-2018-318382
60. Chanson J-B, Lamy J, Rousseau F, Blanc F, Collongues N, Fleury M, et al. White matter volume is decreased in the brain of patients with neuromyelitis optica. Eur J Neurol. (2013) 20:361–7. doi: 10.1111/j.1468-1331.2012.03867.x
61. Yamamura T, Nakashima I. Foveal thinning in neuromyelitis optica: a sign of retinal astrocytopathy? Neurol Neuroimmunol Neuroinflamm. (2017) 4:e347. doi: 10.1212/NXI.0000000000000347
62. Dalmau J. Precision in neuroimmunology. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e345. doi: 10.1212/NXI.0000000000000345
63. Tian D-C, Su L, Fan M, Yang J, Zhang R, Wen P, et al. Bidirectional degeneration in the visual pathway in neuromyelitis optica spectrum disorder (NMOSD). Mult Scler. (2017) 24:1585–93. doi: 10.1177/1352458517727604
64. Ventura RE, Kister I, Chung S, Babb JS, Shepherd TM. Cervical spinal cord atrophy in NMOSD without a history of myelitis or MRI-visible lesions. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e224. doi: 10.1212/NXI.0000000000000224
65. Saji E, Arakawa M, Yanagawa K, Toyoshima Y, Yokoseki A, Okamoto K, et al. Cognitive impairment and cortical degeneration in neuromyelitis optica. Ann Neurol. (2013) 73:65–76. doi: 10.1002/ana.23721
66. Zhao S, Mutch K, Elsone L, Nurmikko T, Jacob A. Neuropathic pain in neuromyelitis optica affects activities of daily living and quality of life. Mult Scler. (2014) 20:1658–61. doi: 10.1177/1352458514522103
67. Chanson J-B, Zéphir H, Collongues N, Outteryck O, Blanc F, Fleury M, et al. Evaluation of health-related quality of life, fatigue and depression in neuromyelitis optica. Eur J Neurol. (2011) 18:836–41. doi: 10.1111/j.1468-1331.2010.03252.x
68. Chavarro VS, Mealy MA, Simpson A, Lacheta A, Pache F, Ruprecht K, et al. Insufficient treatment of severe depression in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e286. doi: 10.1212/NXI.0000000000000286
69. Asseyer S, Schmidt F, Chien C, Scheel M, Ruprecht K, Bellmann-Strobl J, et al. Pain in AQP4-IgG-positive and MOG-IgG-positive neuromyelitis optica spectrum disorders. Mult Scler J Exp Transl Clin. (2018) 4:1–12. doi: 10.1177/2055217318796684
70. Penner I-K, Paul F. Fatigue as a symptom or comorbidity of neurological diseases. Nat Rev Neurol. (2017) 13:662–75. doi: 10.1038/nrneurol.2017.117
71. Moore P, Methley A, Pollard C, Mutch K, Hamid S, Elsone L, et al. Cognitive and psychiatric comorbidities in neuromyelitis optica. J Neurol Sci. (2016) 360:4–9. doi: 10.1016/j.jns.2015.11.031
72. Eizaguirre MB, Alonso R, Vanotti S, Garcea O. Cognitive impairment in neuromyelitis optica spectrum disorders: what do we know? Mult Scler Relat Disord. (2017) 18:225–9. doi: 10.1016/j.msard.2017.10.003
73. Paul F. Pathology and MRI: exploring cognitive impairment in MS. Acta Neurol Scand. (2016) 134:24–33. doi: 10.1111/ane.12649
74. Eaneff S, Wang V, Hanger M, Levy M, Mealy MA, Brandt AU, et al. Patient perspectives on neuromyelitis optica spectrum disorders: data from the PatientsLikeMe online community. Mult Scler Relat Disord. (2017) 17:116–22. doi: 10.1016/j.msard.2017.07.014
75. Meng H, Xu J, Pan C, Cheng J, Hu Y, Hong Y, et al. Cognitive dysfunction in adult patients with neuromyelitis optica: a systematic review and meta-analysis. J Neurol. (2017) 264:1549–58. doi: 10.1007/s00415-016-8345-3
76. Vanotti S, Cores EV, Eizaguirre B, Melamud L, Rey R, Villa A. Cognitive performance of neuromyelitis optica patients: comparison with multiple sclerosis. Arquivos Neuro Psiquiatr. (2013) 71:357–61. doi: 10.1590/0004-282X20130038
77. Kim S-H, Kwak K, Jeong IH, Hyun J-W, Jo H-J, Joung A, et al. Cognitive impairment differs between neuromyelitis optica spectrum disorder and multiple sclerosis. Mult Scler. (2016) 22:1850–8. doi: 10.1177/1352458516636246
78. Fujimori J, Nakashima I, Baba T, Meguro Y, Ogawa R, Fujihara K. Cognitive impairment in neuromyelitis optica spectrum disorders: a comparison of the Wechsler Adult Intelligence Scale-III and the Wechsler Memory Scale Revised with the Rao Brief Repeatable Neuropsychological Battery. eNeurol Sci. (2017) 9:3–7. doi: 10.1016/j.ensci.2017.09.001
79. Blanc F, Zéphir H, Lebrun C, Labauge P, Castelnovo G, Fleury M, et al. Cognitive functions in neuromyelitis optica. Arch Neurol. (2008) 65:84–8. doi: 10.1001/archneurol.2007.16
80. Liu Y, Fu Y, Schoonheim MM, Zhang N, Fan M, Su L, et al. Structural MRI substrates of cognitive impairment in neuromyelitis optica. Neurology. (2015) 85:1491–9. doi: 10.1212/WNL.0000000000002067
81. Bennett J, de Seze J, Lana-Peixoto M, Palace J, Waldman A, Schippling S, et al. Neuromyelitis optica and multiple sclerosis: seeing differences through optical coherence tomography. Mult Scler J. (2015) 21:678–88. doi: 10.1177/1352458514567216
82. Oertel FC, Zimmermann H, Paul F, Brandt AU. Optical coherence tomography in neuromyelitis optica spectrum disorders: potential advantages for individualized monitoring of progression and therapy. EPMA J. (2018) 9:21–33. doi: 10.1007/s13167-017-0123-5
83. Scharfman HE, Binder DK. Aquaporin-4 water channels and synaptic plasticity in the hippocampus. Neurochem Int. (2013) 63:702–11. doi: 10.1016/j.neuint.2013.05.003
84. Fan Y, Liu M, Wu X, Wang F, Ding J, Chen J, et al. Aquaporin-4 promotes memory consolidation in Morris water maze. Brain Struct Funct. (2013) 218:39–50. doi: 10.1007/s00429-011-0373-2
85. Hyun J-W, Park G, Kwak K, Jo H-J, Joung A, Kim J-H, et al. Deep gray matter atrophy in neuromyelitis optica spectrum disorder and multiple sclerosis. Eur J Neurol. (2017) 24:437–45. doi: 10.1111/ene.13224
86. Skucas VA, Mathews IB, Yang J, Cheng Q, Treister A, Duffy AM, et al. Impairment of select forms of spatial memory and neurotrophin-dependent synaptic plasticity by deletion of glial aquaporin-4. J Neurosci. (2011) 31:6392–97. doi: 10.1523/JNEUROSCI.6249-10.2011
87. Rothhammer V, Quintana FJ. Control of autoimmune CNS inflammation by astrocytes. Semin Immunopathol. (2015) 37:625–38. doi: 10.1007/s00281-015-0515-3
88. Hubbard JA, Szu JI, Binder DK. The role of aquaporin-4 in synaptic plasticity, memory and disease. Brain Res Bull. (2018) 136:118–29. doi: 10.1016/j.brainresbull.2017.02.011
89. He D, Chen X, Zhao D, Zhou H. Cognitive function, depression, fatigue, and activities of daily living in patients with neuromyelitis optica after acute relapse. Int J Neurosci. (2011) 121:677–83. doi: 10.3109/00207454.2011.608456
90. Pan J, Zhao P, Cai H, Su L, Wood K, Shi F-D, et al. Hypoxemia, sleep disturbances, and depression correlated with fatigue in neuromyelitis optica spectrum disorder. CNS Neurosci Ther. (2015) 21:599–606. doi: 10.1111/cns.12411
91. Seok JM, Choi M, Cho EB, Lee HL, Kim BJ, Lee KH, et al. Fatigue in patients with neuromyelitis optica spectrum disorder and its impact on quality of life. PLoS ONE. (2017) 12:e0177230. doi: 10.1371/journal.pone.0177230
92. Solomon AJ, Watts R, Dewey BE, Reich DS. MRI evaluation of thalamic volume differentiates MS from common mimics. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e387. doi: 10.1212/NXI.0000000000000387
93. Pache F, Zimmermann H, Finke C, Lacheta A, Papazoglou S, Kuchling J, et al. Brain parenchymal damage in neuromyelitis optica spectrum disorder - A multimodal MRI study. Eur Radiol. (2016) 26:4413–22. doi: 10.1007/s00330-016-4282-x
94. Azevedo CJ, Overton E, Khadka S, Buckley J, Liu S, Sampat M, et al. Early CNS neurodegeneration in radiologically isolated syndrome. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e102. doi: 10.1212/NXI.0000000000000102
95. Raz N, Bick AS, Ben-Hur T, Levin N. Focal demyelinative damage and neighboring white matter integrity: an optic neuritis study. Mult Scler. (2015) 21:562–71. doi: 10.1177/1352458514551452
96. Kremer S, Renard F, Achard S, Lana-Peixoto MA, Palace J, Asgari N, et al. Use of advanced magnetic resonance imaging techniques in neuromyelitis optica spectrum disorder. JAMA Neurol. (2015) 72:815–22. doi: 10.1001/jamaneurol.2015.0248
97. Ramanathan S, Prelog K, Barnes EH, Tantsis EM, Reddel SW, Henderson AP, et al. Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Mult Scler. (2016) 22:470–82. doi: 10.1177/1352458515593406
98. Pawlitzki M, Neumann J, Kaufmann J, Heidel J, Stadler E, Sweeney-Reed C, et al. Loss of corticospinal tract integrity in early MS disease stages. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e399. doi: 10.1212/NXI.0000000000000399
99. Sinnecker T, Schumacher S, Mueller K, Pache F, Dusek P, Harms L, et al. MRI phase changes in multiple sclerosis vs neuromyelitis optica lesions at 7T. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e259. doi: 10.1212/NXI.0000000000000259
100. Kim HJ, Paul F, Lana-Peixoto MA, Tenembaum S, Asgari N, Palace J, et al. MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology. (2015) 84:1165–73. doi: 10.1212/WNL.0000000000001367
101. Kister I, Herbert J, Zhou Y, Ge Y. Ultrahigh-field MR (7 T) imaging of brain lesions in neuromyelitis optica. Mult Scler Int. (2013) 2013:398259. doi: 10.1155/2013/398259
102. Geraldes R, Ciccarelli O, Barkhof F, De Stefano N, Enzinger C, Filippi M, et al. The current role of MRI in differentiating multiple sclerosis from its imaging mimics. Nat Rev Neurol. (2018) 14:199–213. doi: 10.1038/nrneurol.2018.14
103. Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol. (2006) 63:5. doi: 10.1001/archneur.63.7.964
104. Chan KH, Tse CT, Chung CP, Lee RLC, Kwan JSC, Ho PWL, et al. Brain involvement in neuromyelitis optica spectrum disorders. Arch Neurol. (2011) 68:1432–9. doi: 10.1001/archneurol.2011.249
105. Sinnecker T, Dörr J, Pfueller CF, Harms L, Ruprecht K, Jarius S, et al. Distinct lesion morphology at 7-T MRI differentiates neuromyelitis optica from multiple sclerosis. Neurology. (2012) 79:708–14. doi: 10.1212/WNL.0b013e3182648bc8
106. Jurynczyk M, Geraldes R, Probert F, Woodhall MR, Waters P, Tackley G, et al. Distinct brain imaging characteristics of autoantibody-mediated CNS conditions and multiple sclerosis. Brain. (2017) 140:617–27. doi: 10.1093/brain/aww350
107. Jurynczyk M, Tackley G, Kong Y, Geraldes R, Matthews L, Woodhall M, et al. Brain lesion distribution criteria distinguish MS from AQP4-antibody NMOSD and MOG-antibody disease. J Neurol Neurosurg Psychiatry. (2017) 88:132–6. doi: 10.1136/jnnp-2016-314005
108. van Pelt ED, Wong YYM, Ketelslegers IA, Hamann D, Hintzen RQ. Neuromyelitis optica spectrum disorders: comparison of clinical and magnetic resonance imaging characteristics of AQP4-IgG versus MOG-IgG seropositive cases in the Netherlands. Eur J Neurol. (2016) 23:580–7. doi: 10.1111/ene.12898
109. Ogawa R, Nakashima I, Takahashi T, Kaneko K, Akaishi T, Takai Y, et al. MOG antibody–positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e322. doi: 10.1212/NXI.0000000000000322
110. Hamid SHM, Whittam D, Saviour M, Alorainy A, Mutch K, Linaker S, et al. Seizures and encephalitis in myelin oligodendrocyte glycoprotein IgG disease vs aquaporin 4 IgG disease. JAMA Neurol. (2018) 75:65–71. doi: 10.1001/jamaneurol.2017.3196
111. Kuchling J, Brandt AU, Paul F, Scheel M. Diffusion tensor imaging for multilevel assessment of the visual pathway: possibilities for personalized outcome prediction in autoimmune disorders of the central nervous system. EPMA J. (2017) 8:279–94. doi: 10.1007/s13167-017-0102-x
112. Finke C, Heine J, Pache F, Lacheta A, Borisow N, Kuchling J, et al. Normal volumes and microstructural integrity of deep gray matter structures in AQP4+ NMOSD. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e229. doi: 10.1212/NXI.0000000000000229
113. von Glehn F, Jarius S, Cavalcanti Lira RP, Alves Ferreira MC, von Glehn FHR, Costa E Castro SM, et al. Structural brain abnormalities are related to retinal nerve fiber layer thinning and disease duration in neuromyelitis optica spectrum disorders. Mult Scler. (2014) 20:1189–97. doi: 10.1177/1352458513519838
114. Finke C, Zimmermann H, Pache F, Oertel FC, Chavarro VS, Kramarenko Y, et al. Association of visual impairment in neuromyelitis optica spectrum disorder with visual network reorganization. JAMA Neurol. (2018) 75:296–303. doi: 10.1001/jamaneurol.2017.3890
115. Masuda H, Hirano S, Takahashi N, Hatsugano E, Uzawa A, Uchida T, et al. Comparison of cognitive and brain grey matter volume profiles between multiple sclerosis and neuromyelitis optica spectrum disorder. PLoS ONE. (2017) 12:e0184012. doi: 10.1371/journal.pone.0184012
116. Blanc F, Noblet V, Jung B, Rousseau F, Renard F, Bourre B, et al. White matter atrophy and cognitive dysfunctions in neuromyelitis optica. PLoS ONE. (2012) 7:e33878. doi: 10.1371/journal.pone.0033878
117. Papadopoulou A, Oertel FC, Gaetano L, Kuchling J, Zimmermann H, Chien C, et al. Selective, attack-related volume loss of thalamic nuclei in Neuromyelitis Optica Spectrum Disorders. J Neurol Neurosurg Psychiatry. (2019) 1–9. doi: 10.1136/jnnp-2018-320249
118. Cho EB, Han CE, Seo SW, Chin J, Shin J-H, Cho H-J, et al. White matter network disruption and cognitive dysfunction in neuromyelitis optica spectrum disorder. Front Neurol. (2018) 9:1104. doi: 10.3389/fneur.2018.01104
Keywords: neuromyelitis optica spectrum disorders, cognition, neuroinflammation, advanced imaging, MRI
Citation: Oertel FC, Schließeit J, Brandt AU and Paul F (2019) Cognitive Impairment in Neuromyelitis Optica Spectrum Disorders: A Review of Clinical and Neuroradiological Features. Front. Neurol. 10:608. doi: 10.3389/fneur.2019.00608
Received: 08 February 2019; Accepted: 22 May 2019;
Published: 12 June 2019.
Edited by:
Tjalf Ziemssen, Zentrum für Klinische Neurowissenschaften (ZKN), GermanyReviewed by:
Edgar Carnero Contentti, Hospital Alemán, ArgentinaRalf Lürding, University of Regensburg, Germany
Copyright © 2019 Oertel, Schließeit, Brandt and Paul. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Friedemann Paul, ZnJpZWRlbWFubi5wYXVsQGNoYXJpdGUuZGU=
†These authors have contributed equally to this work