 Harald Prüss1,2*
Harald Prüss1,2* Guido Gessner3
Guido Gessner3 Stefan H. Heinemann3
Stefan H. Heinemann3 Franz Rüschendorf4
Franz Rüschendorf4 Ann-Kathrin Ruppert5
Ann-Kathrin Ruppert5 Herbert Schulz5Thomas Sander5Wilhelm Rimpau1
Herbert Schulz5Thomas Sander5Wilhelm Rimpau1- 1Department of Neurology and Experimental Neurology, Charité - Universitätsmedizin Berlin, Berlin, Germany
- 2German Center for Neurodegenerative Diseases (DZNE) Berlin, Bonn, Germany
- 3Department of Biophysics, Center for Molecular Biomedicine, Friedrich Schiller University Jena & Jena University Hospital, Jena, Germany
- 4Max Delbrück Centre for Molecular Medicine, Berlin, Germany
- 5Cologne Center for Genomics, University of Cologne, Cologne, Germany
Mutations in several genes encoding ion channels can cause the long-QT (LQT) syndrome with cardiac arrhythmias, syncope and sudden death. Recently, mutations in some of these genes were also identified to cause epileptic seizures in these patients, and the sudden unexplained death in epilepsy (SUDEP) was considered to be the pathologic overlap between the two clinical conditions. For LQT-associated KCNQ1 mutations, only few investigations reported the coincidence of cardiac dysfunction and epileptic seizures. Clinical, electrophysiological and genetic characterization of a large pedigree (n = 241 family members) with LQT syndrome caused by a 12-base-pair duplication in exon 8 of the KCNQ1 gene duplicating four amino acids in the carboxyterminal KCNQ1 domain (KCNQ1dup12; p.R360_Q361dupQKQR, NM_000218.2, hg19). Electrophysiological recordings revealed no substantial KCNQ1-like currents. The mutation did not exhibit a dominant negative effect on wild-type KCNQ1 channel function. Most likely, the mutant protein was not functionally expressed and thus not incorporated into a heteromeric channel tetramer. Many LQT family members suffered from syncopes or developed sudden death, often after physical activity. Of 26 family members with LQT, seizures were present in 14 (LQTplus seizure trait). Molecular genetic analyses confirmed a causative role of the novel KCNQ1dup12 mutation for the LQT trait and revealed a strong link also with the LQTplus seizure trait. Genome-wide parametric multipoint linkage analyses identified a second strong genetic modifier locus for the LQTplus seizure trait in the chromosomal region 10p14. The linkage results suggest a two-locus inheritance model for the LQTplus seizure trait in which both the KCNQ1dup12 mutation and the 10p14 risk haplotype are necessary for the occurrence of LQT-associated seizures. The data strongly support emerging concepts that KCNQ1 mutations may increase the risk of epilepsy, but additional genetic modifiers are necessary for the clinical manifestation of epileptic seizures.
Introduction
Prolonged QT intervals are the diagnostic hallmark of inherited long-QT (LQT) syndromes which comprise a growing number of mutations in ion channels, with the gene encoding the voltage-gated potassium channel KCNQ1 (Kv7.1) being the most common LQT gene (OMIM #192500: LQT1) (1). LQT syndrome by KCNQ1 mutations has long been considered as a cardiac disease with the risk of ventricular arrhythmias and sudden cardiac death, resulting from prolongation of the cardiac action potential. However, patients with LQT syndrome can also present with epileptic seizures which suggested that the underlying genetic cause might be relevant for both phenotypes (2). In patients with LQT1 syndrome due to KCNQ1 mutations, clinical seizures were consistently observed (3, 4). Indeed, recent research identified the dual expression of KCNQ1 in the heart and in the brain with neurons in forebrain networks and brainstem nuclei expressing the highest levels (5). The same study used a mouse model carrying a human KCNQ1 knock-in mutation which resulted not only in cardiac arrhythmias but also in seizures and sudden unexpected death in epilepsy (SUDEP). The model reflects emerging evidence from clinical cohorts of patients with LQT syndrome in which there is an increased association with epilepsy and seizure-like phenotypes, in particular in the related LQT disease with mutations in the gene encoding the voltage-gated potassium channel KCNH2 (Kv11.1, hERG1) (6, 7).
More than 10 years ago, we have identified a family of Kurdish descent with a novel mutation in the KCNQ1 gene, consisting of a 12-base-pair duplication (KCNQ1dup12) in exon 8 (8). However, it was unclear whether this mutation is causing functional effects, thus plausibly explaining the clinical symptoms. Remarkably, identification of the KCNQ1 mutation in the index patients was antedated by a several years' history of epileptic seizures and treatment with anti-epileptic drugs including carbamazepine. Although suggestive, at that time it was an unsolved question whether KCNQ1 mutations may cause both, cardiac symptoms and epileptic seizures. It was rather contrarily believed that epileptic seizures are only a consequence of cardiac arrhythmias, e.g., torsades de pointes (TdP), a polymorphic ventricular tachycardia potentially leading to cerebral hypoperfusion and seizures (9). For KCNQ1 mutation carriers, only one small family having both, LQT syndrome and epilepsy, has been characterized in detail (10). Thus, further investigations are required to explore pleiotropic effects of KCNQ1 mutations on cardiac symptoms together with epileptic seizures in patients with LQT1 syndrome. Therefore, we markedly extended the characterization of the Kurdish pedigree—one of the largest of a family with LQT syndrome—aiming at the correlation between the KCNQ1dup12 mutation and the clinical manifestation of variable symptoms, in particular syncopes, seizures, and sudden death (11).
Methods
Study Pedigree and Clinical Phenotype
After identification of the index case (IV57) (8), the family was systematically expanded (Supplementary Figure 1), resulting in 241 family members across five generations, for 233 of whom a detailed medical history was available. The pedigree consisted of 117 (48.5%) females and 124 (51.5%) males. The study was approved by the Charité University Hospital Institutional Review Board. All participants gave their written informed consent in accordance with the Declaration of Helsinki.
Determination of the clinical phenotype based on interviews with patients or close relatives, and was performed blinded to the genetic status of the subjects. Characteristic videos demonstrating the differences between syncope and seizures were used to train relatives in better explanation of witnessed events, particularly focusing on duration, aura symptoms, presence of automatisms, focal neurologic signs, and postictal confusion. EEG recordings (which are not the current standard of care for the evaluation of LQT syndrome) were not available for this Kurdish family.
ECG Analysis
Available 12-lead ECG recordings were blindly analyzed by an experienced cardiologist. Measured QT intervals were corrected (QTc) for heart rates between 60 and 80 bpm according to Bazett's formula. For heart rates faster than 80 bpm, Fridericia's formula was used. As repeated ECGs were not available in most cases, LQTS was diagnosed in this study according to the Class IIa recommendation of the European Society of Cardiology (ESC) guidelines (12) by considering QTc times >450 ms in men and >460 ms in women pathologic.
DNA Analyses
Sequence Analyses
DNA samples of 60 family members were extracted from whole blood. Initially, mutation screening of the affected members (IV57, V84) of the index family was performed by Sanger sequencing for an LQT candidate gene panel. Subsequently, whole-exome sequencing was applied for the index patient IV57. Exonic targets were enriched using the SeqCap EZ Human Exome Library v2.0 (Roche, NimbleGen). Sequencing was performed on an Illumina HiSeq2000, using a 100 bp paired-end read protocol.
Genotyping of the KCNQ1 Mutation
Large-scale genotyping of the 12-bp duplication in exon 8 of the KCNQ1 gene (KCNQ1dup12 variation: c.1068_1079delGCAGAAGCAGAGins(GCAGAAGCAGAG)2, NM_000218.2, p.R360_Q361insQKQR, ENSP00000155840, hg19) was carried out by fragment length assay (KCNQ1dup12 variation: genomic reference allele: 128 bp, mutation: 140 bp). Polymerase chain amplification (PCR) of the KCNQ1dup12 variation was performed using flanking fluorescence-labeled forward primer GAGCCTCCTGTCCATTCCTT and the reverse primer ACCGCACCTGAATGAGTGA. Fragment sizes of the biallelic PCR products were separated on ABI 3730 DNA Analyzers and genotypes scored using GENEMAPPER version 3.7 software (Applied Biosystems, Foster City, CA, USA).
Linkage Analyses
Genome-wide linkage mapping was carried out in this multigenerational family including 24 family members of whom 17 members were affected by LQT (Figure 2A) and 12 members also exhibited epileptic seizures (Figure 2B), using 5455 quality-filtered autosomal single nucleotide polymorphisms (SNPs) genotyped by the Illumina HumanLinkage-12 Genotyping BeadChip (Illumina Inc., San Diego, CA, USA). Parametric two-point and multipoint linkage analyses were performed for an affecteds-only classification assuming an autosomal dominant inheritance with a phenocopy rate of 1% and a mutation prevalence of 0.1%. The linkage program MLINK (13) was applied for parametric two-point and MERLIN for parametric multipoint linkage analysis (14). Genotype data quality control and data management was facilitated by the program ALOHOMORA (15). HaploPainter (16) was used for drawing pedigrees with haplotypes based on MERLIN calculations.
Electrophysiology
For this study, expression constructs of KCNQ1 or KCNH2 in pCDNA3 (Invitrogen) and KCNE1 in pEGFP-N1 were used. Mutation KCNQ1dup12 and KCNH2-R878C were introduced by overlap-extension PCR and verified by sequencing. HEK293T cells were maintained with 5% CO2 at 37°C in 45% DMEM + 45% F12 + 10% FCS. Cells were plated on glass coverslips and transfected 24 h later with a constant amount of plasmid DNA encoding wild-type or mutant KCNQ1, or a 1:1 mixture. CD8-encoding DNA (<20% of channel DNA) was cotransfected, allowing identification of transfected cells via CD8-detecting beads (Dynabeads, Life Technologies). For coexpression of KCNE1, an equal amount of DNA encoding C-terminally GFP-tagged KCNE1 was added. In these cases, transfected cells were identified by fluorescence microscopy. Whole-cell patch-clamp measurements were performed with an EPC9 amplifier, operated with PatchMaster software (HEKA Elektronik, Lambrecht, Germany), 2 days after transfection. The bath solution contained (in mM): 146 NaCl, 4 KCl, 2 MgCl2, 2 CaCl2, 10 HEPES (pH 7.4, NaOH). The pipette solution consisted of (in mM): 130 KCl, 2 Mg-ATP, 10 EGTA, 10 HEPES (pH 7.4, KOH). Data were analyzed with FitMaster (HEKA Elektronik) and IgorPro (Wavemetrics, Lake Oswego, OR, USA).
Results
Family Members
The initial diagnosis of LQT syndrome was established in the index patient IV57 (Figure 1A, arrow) when her daughter V84 was diagnosed with a prolonged QT interval after two syncopes. Before that, the index patient had been diagnosed of epilepsy already for more than 20 years. In retrospect, however, several of the “spells” in this patient during adolescence were due to cardiac affection including palpitations, exercise-induced syncope, presyncopal aura, short-lasting unconsciousness with rapid reorientation, most frequently after awakening in the morning. In adulthood, the patient suffered mainly from generalized tonic-clonic seizures (GTCS), prolonged postictal disorientation, enuresis and tongue bite, resulting in treatment with carbamazepine. Treatment with β blockers resulted in complete remission of cardiac symptoms and partial remission of epilepsy (8).

Figure 1. (A) The index patient (arrow) had a long-standing history of seizures. Diagnosis of LQT syndrome was established when her daughter V84 was diagnosed with a prolonged QT interval after two syncopes. (B) The QTc interval (mean ± SEM) was not significantly different between mutation carriers with or without generalized tonic-clonic seizures (GTCS) (P = 0.795, Mann Whitney test).
Following this initial observation, we next analyzed the variable clinical symptoms in one of the largest pedigrees reported of a family with LQT syndrome (Supplementary Figure 1). The expanded pedigree contained 241 family members across five generations. LQT syndrome appeared in the family branches of the founder III24 (Supplementary Figure 1, arrow) with his three marriages (III23, III25, and III27), following an autosomal-dominant segregation pattern. Family history of sudden death was common, 19 children died of SIDS (sudden infant death syndrome) or SUDEP. For example, in one family tree (IV53/IV54) 4 of 8 children died from sudden death at 0–1 years of age.
Aiming primarily at the clinical distinction between symptoms of cardiac syncopes vs. epileptic seizures and due to the lack of available EEGs in this Kurdish pedigree, we interviewed patients and relatives in a standardized way blinded to the genetic status, supported by teaching videos of representative syncope or seizure events. We identified 24 family members with symptoms compatible with syncope, 6 of which passed away at 0–12 years of age. Of the remaining 18, 16 (88.9%) showed prolonged QTc intervals. Clinical assessment revealed 24 family members with a history of GTCS including eight deaths at 0–12 years of age. Two family members had GTCS with normal (V51) or undetermined (V52) QTc intervals, while all remaining 14 patients had GTCS in the context of ECG-confirmed prolonged QTc time (LQTplus seizure trait) (Supplementary Figure 1). However, the length of the QTc interval in mutation carriers was not different between family members with or without GTCS (Figure 1B) (P = 0.795, Mann Whitney test). Also, GTCS frequency and QTc time did not show a significant correlation (P = 0.18, R2 = 0.32, n = 7 family members), collectively suggesting that seizures were not a result of more severe cardiac dysfunction. All 14 patients experienced both types of paroxysmal events, namely those related to cardiac dysfunction (such as with profound pallor at time of spells or exercise-related events) as well as spontaneous epileptic seizures without antecedent cardiac symptoms (e.g., starting during rest and followed by prolonged postictal confusion).
Mutation Screening
Initial Sanger sequencing of the most common LQT genes in the index patient IV57 identified a novel heterozygous 12-bp-duplication in exon 8 of the KCNQ1 gene (KCNQ1dup12; chr11:2606477-2606488(GCAGAAGCAGAG>(GCAGAAGCAGAG)2, hg19) duplicating four amino acids in the carboxyterminal KCNQ1 domain (p.R360_Q361dupQKQR, NM_000218.2, hg19) (8). The KCNQ1dup12 mutation was not detected in gnomAD in 123,136 exomes and 15,496 genomes from unrelated individuals (http://gnomad.broadinstitute.org/). Complementary whole-exome sequencing was carried out in the index patient IV57 affected by the LQTplus seizure trait to search for additional mutations in a panel of 13 genes causing cardiac arrhythmia. We were able to confirm the KCNQ1dup12 mutation and detected a rare heterozygous missense variant in exon 9 of KCNH2, the second most common LQT gene (LQT2: OMIM #613688; dbSNP: rs370393086; chr7:150647022G>A, c.2632C>T, NM_172056.2, p.R878C, CCDS47747.1, hg19). However, protein function prediction did not indicate a substantial functional alteration of the amino acid substitution (p.R878C, ENST00000430723, Polyphen: benign). Electrophysiological characterization of the mutant channels hERG1-R878C upon heterologous expression in HEK293T cells did not reveal functional differences compared to wild-type hERG1 (Supplementary Figure 3). Moreover, this rare KCNH2 missense variant (p.878C) originated from the second wife (III25) of the KCNQ1dup12 mutation founder (III24) and therefore its segregation was restricted to this family branch. Although the rare KCNH2 missense variant was present in the putative LQT phenocopy lacking the KCNQ1dup12 mutation (V85), we did not observe a familial cosegregation with the LQT- or LQTplus seizure trait. Thus, this rare KCNH2 missense variant or other common LQT genes are unlikely to exert a substantial pathogenic effect on the LQT trait in this family.
Strong Linkage Between the KCNQ1dup12 Mutation and Both the LQT Trait and LQTplus Seizure Trait
Genotyping of the KCNQ1dup12 variation in DNA samples of 60 family members detected the always heterozygous KCNQ1dup12 mutation in 24 out of 25 genotyped family members with LQT syndrome and all 14 genotyped family members affected by both LQT syndrome and epileptic seizures (LQTplus seizure trait). One family member (V85) with LQT syndrome did not carry the KCNQ1dup12 mutation and has to be considered as putative phenocopy. Overall, 24 out of 29 KCNQ1dup12 mutation carriers were affected by an LQT syndrome, suggesting an incomplete penetrance of about 80%. Two-point parametric linkage analysis including 68 family members showed significant evidence for linkage between the KCNQ1dup12 mutation and the LQT trait (25 affected individuals) assuming an autosomal dominant inheritance with a phenocopy rate of 1% based on the affected individuals only (maximum LOD score: 5.21 at a recombination fraction (θ) of 0.03, P = 4.83 × 10−7). Likewise, significant evidence for linkage was obtained between the KCNQ1dup12 mutation and the LQTplus seizure trait in 14 affected family members (maximum LOD score: 4.12 at KCNQ1, P = 6.63 × 10−6). To search for other potential LQT loci, we carried out genome-wide parametric multipoint linkage analysis in 24 family members including 17 members with LQT. For an affecteds-only classification and autosomal dominant mode-of-inheritance with a presumed phenocopy rate of 1%, we found a single significant linkage peak at the LQT1/KCNQ1 locus (multipoint LOD score: 3.25, P = 5.47 × 10−5; Figure 2A).
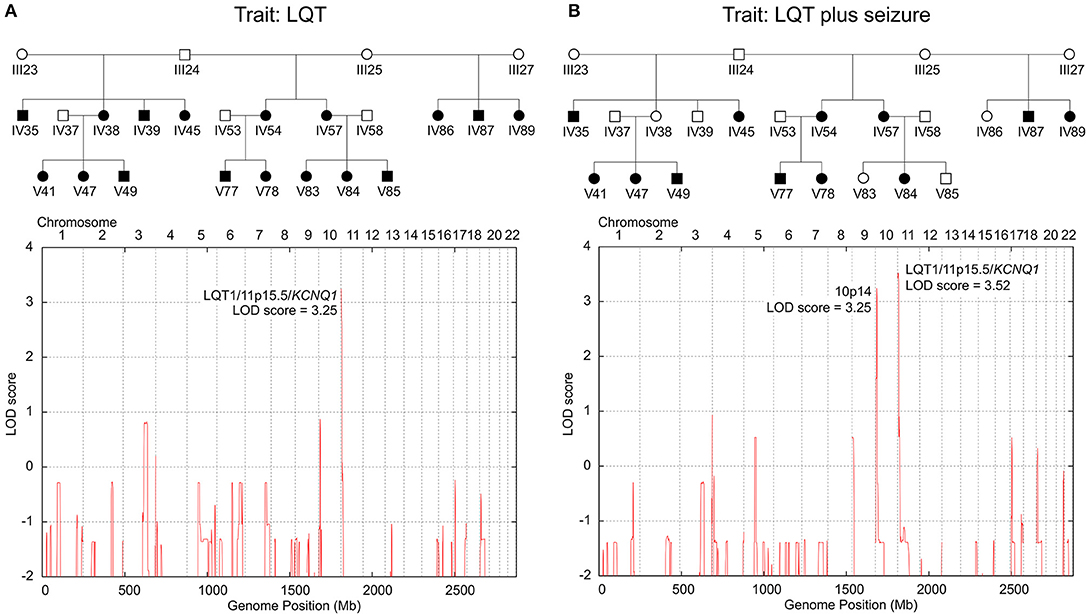
Figure 2. Genome-wide parametric multipoint linkage analyses for two LQT-trait models. (A) Linkage results for the LQT trait only identified a single major LQT-locus at 11p15.5/KCNQ1. (B) Linkage results for the LQT-associated seizure trait revealed cosegregating susceptibility loci at 11p15.5/KCNQ1 and 10p14, suggesting a dominant-dominant two-locus inheritance model. Blackened symbols in the pedigree denote individuals affected by the investigated LQT-trait model (A: LQT only; B: LQT plus epileptic seizure).
Linkage Evidence for an LQT1/KCNQ1 Cosegregating 10p14 Risk Haplotype Suggesting a Two-locus Inheritance Model for the LQTplus Seizure Trait
To examine whether the LQTplus seizure trait may reflect a variable expressivity of the KCNQ1dup12 mutation or whether additional genetic modifier loci are involved, we carried out genome-wide parametric multipoint linkage analyses for an affecteds-only classification using 5455 autosomal SNPs. For the LQTplus seizure trait (12 affected family members), we detected two significant linkage peaks, one predominant peak at the LQT1/KCNQ1 locus in the chromosomal region 11p15.5–p15.4 (multipoint LOD score: 3.52, P = 2.83 × 10−5, Supplementary Figure 4A) and the second linkage peak in the chromosomal region 10p14 (multipoint LOD score = 3.25 at rs1033912, chr10:9592521, P = 5.47 × 10−5, Supplementary Figure 4B) (Figure 2B). The linkage results support a two-locus inheritance model presuming that both the major LQT1/KCNQ1 locus and the modifier locus on chromosome 10p14 are necessary for the coincidental phenotypic expression of LQT and epileptic seizures in the present family. Haplotype analysis of the chromosomal region 10p14 revealed a risk haplotype spanning 3.8 Mb flanked by the SNPs rs2671303 and rs1535976 (chr10:8074577-11850132, hg19) (Supplementary Figure 5). Notably, all 12 family members affected by the LQTplus seizure trait carried both the KCNQ1dup12 mutation and the 10p14 risk haplotype. In addition, two family members (IV38 and IV39) also carried both the KCNQ1dup12 mutation and the 10p14 risk haplotype but exhibited only a severely prolonged LQT interval (>500 ms) without a known history of cardiac syncopes or epileptic seizures. This finding suggests an incomplete penetrance of approximately 80% for the two-locus model.
Electrophysiology
To assess the functional impact of the LQT mutation, the mutation was engineered into a KCNQ1-containing plasmid construct suited for expression in mammalian cells. As shown in Figure 3, KCNQ1dup12-expressing cells exhibited voltage-dependent, partially inactivating outward currents similar to currents observed from non-transfected control cells. Cells transfected with KCNQ1 or an equimolar mixture of wild-type and mutant KCNQ1 DNA exhibited stronger outward currents. Current amplitudes at the end of the depolarizations were determined, normalized to the cell capacitance, and plotted against voltage (Figure 3B; n = 8, each). For positive voltages, current densities of KCNQ1-expressing cells were significantly larger than for KCNQ1dup12-expressing cells or untransfected cells, whereas the latter two were indistinguishable. Upon coexpression of KCNQ1dup12, current densities were slightly but not statistically significantly smaller than that of cells expressing wild-type only. Since the outward currents at the end of the depolarizations are contaminated by endogenous channels, we also analyzed tail currents upon repolarization to −50 mV. KCNQ1-expressing cells exhibited substantial outward tail currents, characteristic for the slow gating of KCNQ1 channels. For KCNQ1dup12 or in untransfected cells, similar currents were barely detectable. Tail current densities from mutant plus wild-type transfected cells were slightly but not statistically significantly smaller than those from cells expressing wild-type only. This finding was reproduced in a second batch of cells with n = 15 cells each (Supplementary Figure 2), suggesting that mutation KCNQ1dup12 does not exhibit a dominant negative effect on KCNQ1 channels. To analyze the channel's voltage dependence, tail current vs. voltage data were fitted with a Boltzmann function. The resulting voltages for half-maximal activation (V0.5) were not significantly different between KCNQ1 (−14 ± 2 mV) and KCNQ1 plus mutant (−14 ± 1 mV, P = 0.76). The corresponding voltages for e-fold change in activation (Ve) were also indistinguishable (9.0 ± 0.5 mV vs. 8.8 ± 0.6 mV, P = 0.81).
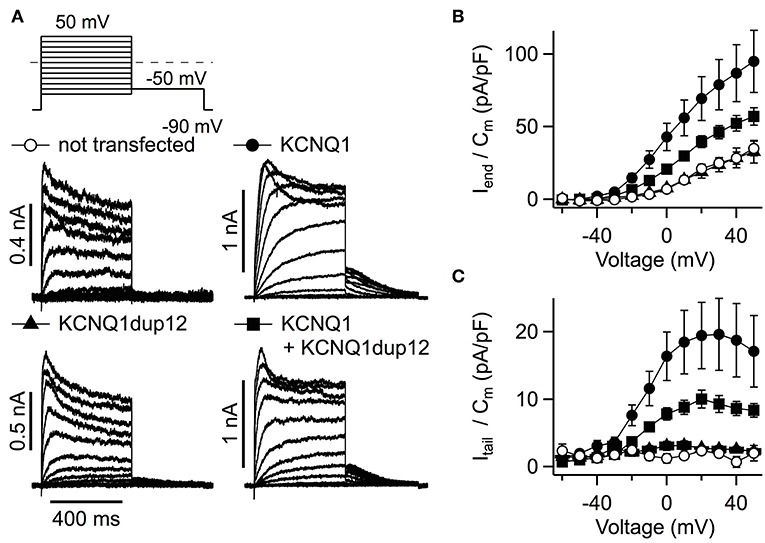
Figure 3. Functional impact of the KCNQ1dup12 mutation on KCNQ1-mediated K+ currents. (A) Superimposed whole-cell current recordings evoked by the pulse protocol shown on top. HEK293T cells were transfected with equal amounts of DNA encoding wild-type or mutant KCNQ1 or a mixture of both, as indicated. Non-transfected cells served as controls. Current amplitude at the end of the depolarization (Iend) and maximal outward tail current (Itail) amplitudes were normalized to the cell capacitance (Cm), averaged and plotted against voltage in (B,C), respectively. Data points are mean ± SEM (n = 8). Straight lines connect data points for clarity.
We also assessed the functional impact of mutation KCNQ1dup12 in heteromeric KCNQ1 + KCNE1 channels, which resemble cardiac IKs. Cells were transfected with equal amounts of KCNQ1 DNA and KCNE1 DNA. KCNE1 was N-terminally fused to GFP allowing identification of transfected cells. Cells transfected with KCNQ1 and KCNE1 exhibited IKs-like currents, characterized by extremely slow activation upon depolarization (Figure 4A). Such types of outward currents were not observed in cells transfected with KCNQ1dup12 plus KCNE1 or KCNE1 alone. The latter exhibited fast activating and partially inactivating currents with indistinguishable current densities determined at the end of the depolarizations (Figure 4B). Upon repolarization to −50 mV, only marginal outward currents were observed. In contrast, KCNQ1-expressing cells exhibited robust outward tail currents. Maximal outward tail current densities (Itail/Cm) are plotted against voltage in Figure 4C. Cotransfection of KCNQ1dup12 did not significantly diminish these current densities. To analyze the voltage dependence of activation, tail current vs. voltage data of those cells clearly differing from those expressing KCNE1 only, were fitted with a Boltzmann function. Resulting V0.5 and Ve data were not significantly different between KCNQ1 plus KCNE1 expressing cells (V0.5: 30.0 ± 3.1 mV and Ve: 13.7 ± 1.0 mV, n = 9) and those cells transfected with KCNQ1dup12 plus KCNQ1 plus KCNE1 (V0.5: 34.7 ± 3.4 mV (P = 0.33) and Ve: 16.2 ± 0.9 mV (P = 0.88, n = 6).
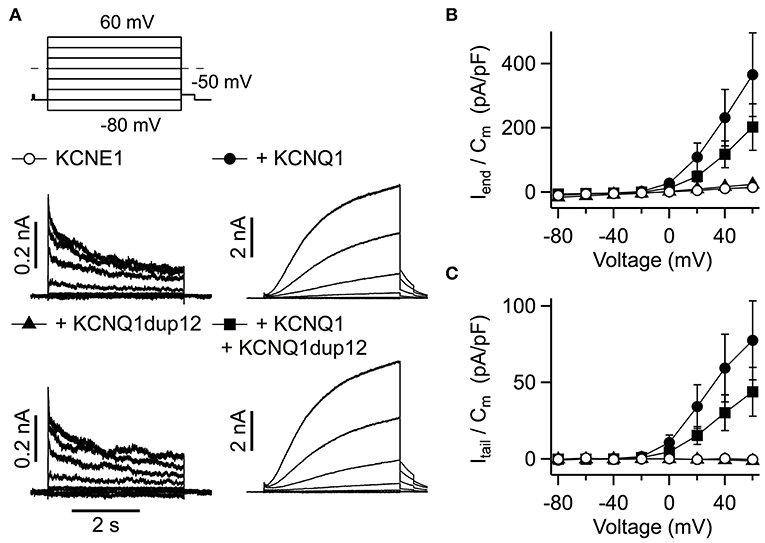
Figure 4. Functional impact of the KCNQ1dup12 mutation on IKs currents. (A) Superimposed whole-cell current recordings evoked by the pulse protocol shown on top. HEK293T cells were transfected with equal amounts of DNA encoding wild-type or mutant KCNQ1 or a mixture of both, as indicated. An equal amount of GFP-tagged KCNE1-encoding DNA was cotransfected. Current amplitude at the end of the depolarization (Iend) and maximal outward tail current (Itail) amplitudes were normalized to the cell capacitance (Cm), averaged and plotted against voltage in (B,C), respectively. Data points are mean ± SEM (n = 15). Straight lines connect data points for clarity.
Discussion
While the clinical overlap between cardiac arrhythmias and epilepsy has been well-established for LQT-associated channelopathies related to the SCN5A and KCNH2 genes (5, 7, 17), and epileptic seizures were found in 12% of patients with LQT syndrome associated with KCNQ1 mutations (4), little is known about the detailed mechanisms and additional genetic risk factors leading to coincidental affection of heart and brain. Experimental murine data support the concept that KCNQ1 mutations can directly lead to seizures, given the protein expression also in the brain (5). A recent study used next-generation sequencing in epilepsy patients with cardiac dysfunction or sudden death to explore the role of already known and novel candidate genes associated with epilepsy and SUDEP (18). Data revealed five new variants including one copy number variant in KCNQ1 (18).
Given that KCNQ1 gene mutations account for up to 48% of congenital LQT syndromes (LQT1, OMIM: #192500) and that our functional analyses confirm a deleterious functional effect of the novel KCNQ1dup12 mutation, our sequence and linkage analyses provide striking evidence that the mutation plays a causative role for the LQT trait in this family. Epileptic seizures were common in the present cohort (LQTplus seizure trait) and frequently appeared independent of cardiac symptoms. Since about 5–10% of LQT patients carry mutations in more than one of the known LQT genes and typically express more severe phenotypes with a younger age at onset (19, 20), we addressed the question whether the LQTplus seizure trait may be explained by variable expressivity of the KCNQ1dup12 mutation or alternatively whether a genetic modifier locus besides KCNQ1 contributes to the coincidence of LQT and seizures. Extensive search for additional mutations in known LQT genes detected a rare heterozygous missense variant of KCNH2 in exon 9. KCNH2 is the second most common LQT gene, and a loss-of-function mutation was associated with epilepsy and sudden death in a family with long QT syndrome (7). However, protein function was not significantly different from wild-type, and the segregation restricted to a small family branch makes a substantial pathogenic effect on the LQT trait in this family highly unlikely.
The manifestation of epileptic seizures also without precedent cardiac arrhythmias in all patients with LQTplus seizures and the absence of family members with epileptic seizures without LQT syndrome implicate the involvement of a genetic modifier. Indeed, genome-wide parametric multipoint linkage analyses revealed a second linkage peak in the chromosomal region 10p14, which was present in all 12 family members exhibiting the LQTplus seizure trait. In turn, KCNQ1 mutation carriers did not have GTCS when lacking the cosegregating 10p14 haplotype. The findings suggest a dominant-dominant two-locus inheritance model in which the major LQT1/KCNQ1 locus at 11p15.5–p15.4 and the modifier locus at 10p14 act together to express the LQTplus seizure trait (Figure 2B; Supplementary Figures 4, 5). The 10p14 risk haplotype encompasses 24 genes, of which the gene encoding the CUG triplet repeat, RNA-binding protein 2 (gene symbol: CELF2, chr10:10838851-11378674, Entrez 10659, hg19) represents a high-ranking candidate gene for the phenotypic expression of LQT-associated seizures. CELF2 is part of CUG-BP, Elav-like family (CELF1-6) and is involved in the regulation of alternative RNA splicing in the brain (21). Mice deficient for Celf4 exhibit a complex seizure disorder that includes both convulsive and non-convulsive seizures resulting from an increased neuronal excitation due to aberrant Nav1.6 (Scn8a) sodium channel activity (22). Likewise, haploinsufficiency of human CELF4 has been implicated to cause an impaired neurodevelopment with febrile seizures in a patient with a truncated CELF4 gene due to a de novo translocation (23). Together, these findings support an involvement of CELF proteins in the regulation of neuronal excitability and epileptogenesis. Notably, whole-exome sequencing in the index patient IV57 did not identify any deleterious coding mutation of the CELF2 gene or other genes located on the risk haplotype on chromosome 10p14. However, mutations in the non-coding sequences of the CELF2 gene may have been missed by targeted sequencing of the CELF2 exons and exon-intron boundaries only.
Recurrent events in the patients of our cohort resulted from two different etiologies. First, they had LQT-related cardiac dysfunctions such as ventricular tachyarrhythmia or syncope. Secondary brain symptoms may be misdiagnosed as epileptic seizures but in fact result from hypoxia-triggered seizures. According to the diagnostic manual of the ILAE's (International League Against Epilepsy) commission on classification and terminology, these paroxysmal events are classified as epilepsy imitators. We reduced the common difficulties in clinical distinction between epileptic seizures and cardiac syncopes by use of representative videos and a detailed history paying particular attention to symptoms which help to distinguish both conditions (such as profound pallor at time of seizures or exercise-related events pointing to cardiac disease).
Second, the family members with LQT-associated seizures exhibited a genetic epilepsy related to the major LQT1/KCNQ1 locus and the cosegregating 10p14 modifier locus, independent of cardiac dysfunction. Thus, the KCNQ1dup12 mutation represented a susceptibility gene not leading to GTCS alone, but caused seizures in combination with the 10p14 risk allele in this LQT1 family. This notion is further supported by clinical observations, including witnessed prolonged grand mal status of 20–30 min in one woman or witnessed GTCS in another by an epileptologist leading to treatment and assessment for epilepsy surgery. It is also supported by equal QTc intervals in mutation carriers with and without GTCS, suggesting that more severe heart disease is not the trigger for seizures.
Electrophysiological recordings with the here described KCNQ1dup12 mutation exhibited no channel activity of the mutant. In the absence or presence of KCNE1 subunits, we did not find substantial KCNQ1- or IKs-like currents types significantly differing in current densities between KCNQ1dup12-transfected cells and their respective controls. This may be explained either by a lack of functional mutant protein, e.g., due to an impaired trafficking, or a complete loss of function, induced by the mutation. In the absence of further evidence for presence of the mutant protein in the plasma membrane, we cannot distinguish between these possibilities. Nevertheless, our data clearly indicate that the mutation does not exhibit a dominant negative effect. Neither with nor without coexpression of KCNE1, expression of the mutant significantly suppressed functional wild-type KCNQ1. Since in both cases coexpression of KCNQ1dup12 did not affect the voltage dependence of the channels, it appears more likely that the mutation impairs functional expression and thus the protein is not incorporated into a heteromeric channel tetramer.
Taken together, we electrophysiologically and genetically characterized a novel KCNQ1 loss-of-function mutation in the largest family with LQT1 syndrome reported so far, demonstrating the causative role of the KCNQ1dup12 mutation for the LQT trait. In addition, we established a genetic two-locus model explaining the coincidental manifestation of LQT-associated epileptic seizures, based on a cosegregation of the KCNQ1dup12 mutation with a strong genetic modifier located in the chromosomal region 10p14. Our present findings provide novel insights into how KCNQ1 mutations in LQT patients can lead to both, cardiac dysfunction with arrhythmias or syncope and increased cerebral excitability with epileptic seizures. They emphasize complex genetic models underlying genetic epilepsies beyond rare single gene effects to explain the remarkably variable phenotypic expressivity across familial epilepsies.
Ethics Statement
The study was approved by the Charité University Hospital Institutional Review Board. All participants gave their written informed consent in accordance with the Declaration of Helsinki.
Author's Note
The authors wish to honor in memory the late Prof. Dieter Janz. His careful attention of patient histories and pioneering genetic studies in epilepsy shaped the present work in all ranks.
Author Contributions
HP, TS, and WR: design and conceptualization of the study. TS, HS, and FR: linkage and haplotype analyses. TS, HS, and A-KR: exome sequencing and variant genotyping. All authors: analysis or interpretation of the data, drafting, and revising the manuscript for intellectual content.
Funding
This work was supported by grants from the European Union Seventh Framework Programs [DESIRE, grant 602531 to TS], the German Research Foundation (DFG) by the EUROCORES Program EuroEPINOMICS [grant SA434/5-1 to TS], the DFG Research Unit FOR 2715 (grant SA434/6-1 to TS), HE2993/8 to SH, as well as the Koeln Fortune Program of the Faculty of Medicine, University of Cologne (to TS and A-KR).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank M. T. Keating (Boston, MA, USA) and M. Koenen (Heidelberg, Germany) for providing plasmids encoding KCNQ1 and KCNE1, respectively. We thank A. Roßner and S. Arend for technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2019.00648/full#supplementary-material
References
1. Goldenberg I, Moss AJ. Long QT syndrome. J Am Coll Cardiol. (2008) 51:2291–300. doi: 10.1016/j.jacc.2008.02.068
2. Hartmann HA, Colom LV, Sutherland ML, Noebels JL. Selective localization of cardiac SCN5A sodium channels in limbic regions of rat brain. Nat Neurosci. (1999) 2:593–5. doi: 10.1038/10147
3. Johnson JN, Hofman N, Haglund CM, Cascino GD, Wilde AA, Ackerman MJ. Identification of a possible pathogenic link between congenital long QT syndrome and epilepsy. Neurology. (2009) 72:224–31. doi: 10.1212/01.wnl.0000335760.02995.ca
4. Auerbach DS, McNitt S, Gross RA, Zareba W, Dirksen RT, Moss AJ. Genetic biomarkers for the risk of seizures in long QT syndrome. Neurology. (2016) 87:1660–8. doi: 10.1212/WNL.0000000000003056
5. Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL, Noebels JL. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med. (2009) 1:2ra6. doi: 10.1126/scitranslmed.3000289
6. Anderson JH, Bos JM, Cascino GD, Ackerman MJ. Prevalence and spectrum of electroencephalogram-identified epileptiform activity among patients with long QT syndrome. Heart Rhythm. (2014) 11:53–7. doi: 10.1016/j.hrthm.2013.10.010
7. Partemi S, Cestele S, Pezzella M, Campuzano O, Paravidino R, Pascali VL, et al. Loss-of-function KCNH2 mutation in a family with long QT syndrome, epilepsy, and sudden death. Epilepsia. (2013) 54:e112–6. doi: 10.1111/epi.12259
8. Rimpau W, Jäger-Roman E, Köpcke C. Epilepsy and long QT-syndrome - case report. Z Epileptol. (2004) 17:103–8. doi: 10.1007/s10309-004-0053-9
9. Zaidi A, Clough P, Cooper P, Scheepers B, Fitzpatrick AP. Misdiagnosis of epilepsy: many seizure-like attacks have a cardiovascular cause. J Am Coll Cardiol. (2000) 36:181–4. doi: 10.1016/S0735-1097(00)00700-2
10. Tiron C, Campuzano O, Perez-Serra A, Mademont I, Coll M, Allegue C, et al. Further evidence of the association between LQT syndrome and epilepsy in a family with KCNQ1 pathogenic variant. Seizure. (2015) 25:65–7. doi: 10.1016/j.seizure.2015.01.003
11. Ackerman MJ. Cardiac channelopathies: it's in the genes. Nat Med. (2004) 10:463–4. doi: 10.1038/nm0504-463
12. Priori SG, Blomstrom-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. (2015) 36:2793–867. doi: 10.1093/eurheartj/ehv316
13. Cottingham RW Jr., Idury RM, Schaffer AA. Faster sequential genetic linkage computations. Am J Hum Genet. (1993) 53:252–63.
14. Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. (2002) 30:97–101. doi: 10.1038/ng786
15. Rüschendorf F, Nurnberg P. ALOHOMORA: a tool for linkage analysis using 10K SNP array data. Bioinformatics. (2005) 21:2123–5. doi: 10.1093/bioinformatics/bti264
16. Thiele H, Nurnberg P. Haplo Painter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. (2005) 21:1730–2. doi: 10.1093/bioinformatics/bth488
17. Parisi P, Oliva A, Coll Vidal M, Partemi S, Campuzano O, Iglesias A, et al. Coexistence of epilepsy and Brugada syndrome in a family with SCN5A mutation. Epilepsy Res. (2013) 105:415–8. doi: 10.1016/j.eplepsyres.2013.02.024
18. Coll M, Striano P, Ferrer-Costa C, Campuzano O, Mates J, Del Olmo B, et al. Targeted next-generation sequencing provides novel clues for associated epilepsy and cardiac conduction disorder/SUDEP. PLoS ONE. (2017) 12:e0189618. doi: 10.1371/journal.pone.0189618
19. Tester DJ, Ackerman MJ. Genetics of long QT syndrome. Methodist Debakey Cardiovasc J. (2014) 10:29–33. doi: 10.14797/mdcj-10-1-29
20. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound mutations: a common cause of severe long-QT syndrome. Circulation. (2004) 109:1834–41. doi: 10.1161/01.CIR.0000125524.34234.13
21. Ladd AN. CUG-BP, Elav-like family (CELF)-mediated alternative splicing regulation in the brain during health and disease. Mol Cell Neurosci. (2013) 56:456–64. doi: 10.1016/j.mcn.2012.12.003
22. Sun W, Wagnon JL, Mahaffey CL, Briese M, Ule J, Frankel WN. Aberrant sodium channel activity in the complex seizure disorder of Celf4 mutant mice. J Physiol. (2013) 591:241–55. doi: 10.1113/jphysiol.2012.240168
Keywords: LQT syndrome, KCNQ1, familial epilepsy, risk haplotype, genetic modifier
Citation: Prüss H, Gessner G, Heinemann SH, Rüschendorf F, Ruppert A-K, Schulz H, Sander T and Rimpau W (2019) Linkage Evidence for a Two-Locus Inheritance of LQT-Associated Seizures in a Multigenerational LQT Family With a Novel KCNQ1 Loss-of-Function Mutation. Front. Neurol. 10:648. doi: 10.3389/fneur.2019.00648
Received: 15 December 2018; Accepted: 03 June 2019;
Published: 25 June 2019.
Edited by:
Edoardo Ferlazzo, Università degli studi Magna Græcia di Catanzaro, ItalyReviewed by:
Pasquale Striano, University of Genoa, ItalyKette D. Valente, University of São Paulo, Brazil
Copyright © 2019 Prüss, Gessner, Heinemann, Rüschendorf, Ruppert, Schulz, Sander and Rimpau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harald Prüss, aGFyYWxkLnBydWVzc0BjaGFyaXRlLmRl