 Siddharth Krishnan
Siddharth Krishnan Catherine B. Lawrence
Catherine B. Lawrence- 1Faculty of Biology, Medicine and Health, Manchester Academic Health Science Centre, Lydia Becker Institute of Immunology and Inflammation, University of Manchester, Manchester, United Kingdom
- 2Manchester Collaborative Centre for Inflammation Research, University of Manchester, Manchester, United Kingdom
- 3Division of Infection, Immunity and Respiratory Medicine, Faculty of Biology, Medicine and Health, Manchester Academic Health Science Centre, University of Manchester, Manchester, United Kingdom
- 4Division of Neuroscience and Experimental Psychology, Faculty of Biology, Medicine and Health, Manchester Academic Health Science Centre, University of Manchester, Manchester, United Kingdom
Infections in the post-acute phase of cerebral ischaemia impede optimal recovery by exacerbating morbidity and mortality. Our review aims to reconcile the increased infection susceptibility of patients post-stroke by consolidating our understanding of compartmentalised alterations to systemic immunity. Mounting evidence has catalogued alterations to numerous immune cell populations but an understanding of the mechanisms of long-range communication between the immune system, nervous system and other organs beyond the involvement of autonomic signalling is lacking. By taking our cues from established and emerging concepts of neuro-immune interactions, immune-mediated inter-organ cross-talk, innate immune training and the role of microbiota-derived signals in central nervous system (CNS) function we will explore mechanisms of how cerebral ischaemia could shape systemic immune function. In this context, we will also discuss a key question: how are immune requirements critical for mediating repair of the ischaemic insult balanced by the need for anti-microbial immunity post-stroke, given that they are mediated by mutually exclusive immune networks? Our reformed understanding of the immune landscape post-stroke and novel mechanisms at play could guide targeted therapeutic interventions and initiate a step-change in the clinical management of these infectious complications post-stroke.
Ischaemic stroke imposes a significant burden on health-care systems and societies across the globe as a consequence of the morbidity and mortality associated with the condition (1, 2). In addition to neurological deficits, infectious complications such as pneumonia and urinary tract infections post-stroke also pose a hurdle to optimal recovery, affecting a significant proportion of patients and exacerbating mortality risk (3–7). Despite significant leaps in our understanding of key players in the immune response post-stroke (8–19) and biomarkers of infection (4, 20, 21), therapy using statins (for immunomodulation), prophylactic beta-blockers (to target sympathetic activation), and antibiotics (to control infection) have proved ineffective in treating infection (22–26). Equally, attempts to improve stroke outcome using neuroprotective therapies to harness the immune system such as dexamethasone, erythropoietin, and many others have been met with limited success in the clinic; in part, due to undesirable effects on systemic immunity. Yet, the question of whether drugs that possess immunomodulatory properties can be purposed for stroke patients remains to be resolved (27). What is clear, however, is the onset of a plethora of immunological alterations involving diverse immune cells and tissues that is essential for both neurological recovery as well as the occurrence of systemic infections through impairments in anti-microbial immunity (13). In this review, we collate previously described alterations to systemic immunity, and delineate how long-range communication between the CNS and the periphery shapes immune control by taking our cues from data that does not examine stroke. We will finally examine whether the cost of tissue repair (of the ischaemic damage) is impairment in anti-microbial immunity and consequently, increased infection susceptibility.
The Systemic Immune Landscape of Stroke
To date, there is a wealth of information surrounding the spectrum of immune alterations that ensue following a stroke in both patients as well as models of experimental stroke. The onset of immunological alterations in the central nervous system (CNS) is sequential; neutrophils are recruited hours after stroke by activated microglia and endothelial cells, followed by monocytes a few days following the insult, whilst T and B cells infiltrate the ischaemic tissue in the succeeding weeks (28–32). This contrasts immune alterations in peripheral tissues where stroke drives alterations to the hepatic cytokine networks as early as 6 h post-stroke whilst other systemic immune effects present 24 h post-stroke, affecting both myeloid and lymphoid populations (12, 14, 17, 33–35). However, the mechanisms by which these rapidly-elicited immune changes morph into immune suppression remain poorly defined.
As well as directly altering immune networks, stroke-driven systemic cytokine signals have also emerged as key players that have been implicated to have divergent effects on outcome following stroke (16, 36, 37). Importantly, hepatic interleukin (IL)-6 and chemokine (C-X-C motif) ligand 1 (CXCL1) drive a rapid and transient inflammatory response (35, 38) whilst primed splenocytes secrete tumour necrosis factor (TNF)-α, interferon (IFN)-γ, chemokine (C-C motif) ligand 2 (CCL2) and IL-2 (39); and granulocyte colony stimulating factor (G-CSF) mobilises monocytes and neutrophils from the bone marrow following experimental stroke (40, 41). Moreover, stroke has also been shown to compromise humoral responses through the induction of hypogammaglobulinaemia via the excretional loss of immunoglobulin (Ig) G and impairments in innate-like B cell responses facilitating IgM loss (12, 42).
Many groups have implicated the loss of lymphocytes (T and natural killer (NK) cells), termed lymphopenia, in circulation as a central feature of stroke-induced immune suppression in patients (43–45), a hallmark also replicated in experimental stroke (14, 45, 46). It is thought that the remaining T lymphocytes are also fundamentally altered, being primed to mount a type-1 response through increased IFN-γ and IL-2 production even years after the ischaemic insult (47). In fact, experimental models of stroke have also demonstrated that CD4+ T cells in Peyer's patches (lymphoid tissue in the small intestine) of mice are primed to secrete increased levels of IL-17 and IFN-γ (15). Similarly, there is also an activation of innate lymphocytes such as invariant natural killer T cells (iNKT) cells in the liver in tandem with a cessation of their patrolling behaviour in the sinusoids (48). Additionally, stroke also leads to the apoptotic loss of splenic marginal zone B cells (12) whilst their loss in the blood and bone marrow is predominantly driven by alterations in lymphopoiesis (49).
As such, even innate immune responses are compromised in stroke patients with impairments in the oxidative burst of neutrophils (34) as well as shifts in the proportions and properties of monocyte populations (9, 17, 50, 51). Specifically, as classical and intermediate monocytes expand within the circulating monocyte pool (50, 51) they accrue deficiencies in their anti-microbial immunity as evidenced by their shedding of CD163, tolerance to endotoxin and their inability to secrete key cytokines such as TNF-α, IL-6, and CCL2. These monocytes, also through their increased secretion of IL-10 and downregulation of human leukocyte antigen-DR isotype (HLA-DR) thus acquire an immune suppressed state (16, 17, 33, 46, 52–54). That said, CD74, the invariant polypeptide chain associated with the HLA complex is upregulated in the peripheral blood mononuclear cells of patients (55). Despite being capable of processing and transporting antigens, monocytes, nevertheless, are not professional antigen-presenting cells (56, 57). Consequently, it remains to be determined if the loss of HLA-DR functionally impacts these monocytes in their ability to prime T cell responses post-stroke. Indeed, murine models of experimental stroke have also implicated impairments in monocyte function with an increase in splenic monocytes; both macrophages and monocytes in the spleen downregulate their expression of major histocompatibility complex II (MHCII) (58, 59). By contrast, the role of non-classical monocytes following stroke is less clear in both patients and experimental models of stroke (60, 61).
The Dynamic Interactions Between the CNS and Systemic Immunity in Stroke
Thus far, we have discussed compartmentalised alterations to systemic immunity post-stroke. In this section, we shall highlight how recruited immune cells interface with locally resident cells and the milieu of cytokines following ischaemic injury in stroke. The identification of meningeal lymphatics in the dural sinuses of the CNS (62, 63) as well as previously undescribed subsets of CNS-resident immune cells such as type 2 innate lymphoid cells (ILC2) (64) has reformed our understanding of immunity in the CNS and its frontiers. It is particularly salient to probe the role of these ILC2s post-stroke given their established function in mediating type 2 immunity (65–69) and the critical role of type 2 immunity in mediating tissue repair (70–73). Essentially, type 2 immunity is an ancient arm of the immune system that is mobilised during infection by multicellular metazoan parasites such as helminths that can drive tissue injury as they develop in the host (70, 74, 75). Characterised by the cytokines IL-3, IL-4, IL-5, IL-9, IL-10, and IL-13, it can be orchestrated by a large repertoire of cells including expanded populations of macrophages, T cell, ILCs, and other sentinels of tissue injury to mediate tissue repair in parallel with parasite expulsion (73, 76, 77). As a result, it becomes important to appreciate the reciprocal regulation that exists between the CNS and systemic immunity in homeostasis (78–80) and how injury to the brain modulates this cross-talk during stroke.
Local cytokine cues facilitate neutrophil recruitment to the ischaemic hemisphere as early as 6 h post-stroke in an intercellular adhesion molecule-1-dependent manner (28, 81). Neutrophils are thought to exacerbate the initial injury through the release of proteases and neutrophil extracellular traps (NETs) that exert neurotoxic effects (82, 83). In particular, the release of matrix metalloproteinase-9, a gelatinase, is thought to compromise the integrity of the blood-brain barrier (BBB) (84, 85) and promote the development of oedema (86). Despite being short-lived, it is thought that neutrophils can take on polarisation states through the acquisition of Ym1 and CD206 expression which has been suggested to be important for their clearance and the resolution of inflammation post-stroke (87).
The role of microglia through their interaction with peripherally-derived leukocytes adds an extra layer of complexity as they are double-edged swords with the capacity to propagate and curtail inflammation, and support neurogenesis (88, 89). Through the acquisition of a classically activated state as a result of astrocytic signals and/or microglial CD8 signalling, they secrete pro-inflammatory cytokines including TNF-α, IL-1α that induce neurotoxic astrocytes (90) and disrupt the integrity of the BBB post-stroke (91). By contrast, they are also capable of limiting neuroinflammation by restraining neutrophil recruitment through the effects of transforming growth factor (TGF)-β on astrocyte-derived CXCL1 (92), limiting neuronal excitotoxicity (93) as well as myelin auto-reactivity (94).
Being one of the first responders to injury or infection (95–97), it comes as no surprise that monocytes are rapidly recruited from the periphery to the ischaemic brain post-stroke (9, 19, 31, 79). Whilst they can infiltrate the brain parenchyma through a breached BBB (98), studies have also demonstrated that the choroid plexus is another means of access to the CNS (95, 99). By employing the CD73 enzyme for extravasation and transmigration, monocytes are thought to infiltrate the CNS via the choroid plexus through interactions between the vascular cell adhesion molecule 1 (VCAM-1) and very late antigen-4 (VLA-4). In the CNS, monocytes are an acute source of pro-inflammatory cytokines that drive neuroinflammation following which the milieu of cytokines directs their differentiation into macrophages with features of alternative activation such as arginase-1 and Ym1 expression to mediate tissue repair (61, 100–103). Nevertheless, the interactions between microglia and monocyte-derived macrophages are indispensable for restraining and resolution of long-term microglial inflammation (61, 104) and promoting neurological recovery in diverse settings of CNS injury (95, 99). Despite their ability to engraft in this niche and mediate repair in contexts of inflammation, studies have shown that these monocyte-derived macrophages maintain a distinct transcriptional identity and cannot replenish microglia (105–107). Although this has been ascribed to the embryonic origins of microglia that are seeded from the yolk sac (108–110), the dynamics of this niche remain unclear in stroke.
Amongst leukocytes recruited to the infarcted brain, T cells are one of the last responders and their dynamics are equally complex with studies suggesting that their functions in situ are influenced by not just the subset but also the route of entry into the CNS (8, 111, 112). It is thought that γδ T cells gain access to the injured brain through the leptomeninges in a C-C chemokine receptor (CCR) type 6-dependent manner (8, 113, 114) meanwhile other T cells can access the CNS through the choroid plexus (112) in addition to migrating across a breached BBB (111). Consequently, γδ T cells exacerbate ischaemic damage through their production of IL-17 (8, 113, 114) whilst CD4+ T cells can take on a type 2 activation state and mediate tissue repair through the production of IL-4 in synergy with macrophages (72, 73, 111).
In conjunction with the previously described alterations to systemic immunity it can thus be observed that stroke initiates a complex cycle of events in the ischaemic brain that shapes immunity and inflammation at distal sites. The notion that inflammation at one site can affect other sites is not a new concept as studies have demonstrated that infection-driven inflammation or antigenic challenges in the lungs are capable of promoting the homing of CD4+ T cells to the gastrointestinal tract where they can drive pathology or protective immunity (115, 116). In this paradigm, it is worth noting that γδ T cells are recruited from the intestinal tract (8) where stroke also modulates inflammation in the Peyer's patches whilst driving shifts in microbial communities (15). It can therefore be observed that by fuelling neuroinflammation, ischaemic damage concurrently shapes immune networks in peripheral tissues such as the blood, bone marrow, and spleen that in turn amplify neuroinflammation and inter-organ cross-talk.
The Reaches of Stroke: Means of Immune Control
Given the diverse range of immune alterations elicited by stroke, it is conceivable that long-range communication mechanisms between the CNS and the peripheral immune system is a prerequisite to mediating these effects. By employing established and emerging immunological data that does not investigate stroke, we will outline plausible mechanisms of how inflammation and injury in the brain in the context of stroke can shape immune networks at distal sites. Thus, we will highlight possible mechanisms that cerebral ischaemia could utilise to effect compartmentalised changes to systemic immunity and add to the concepts of autonomic dysfunction by suggesting avenues of research (Figure 1).
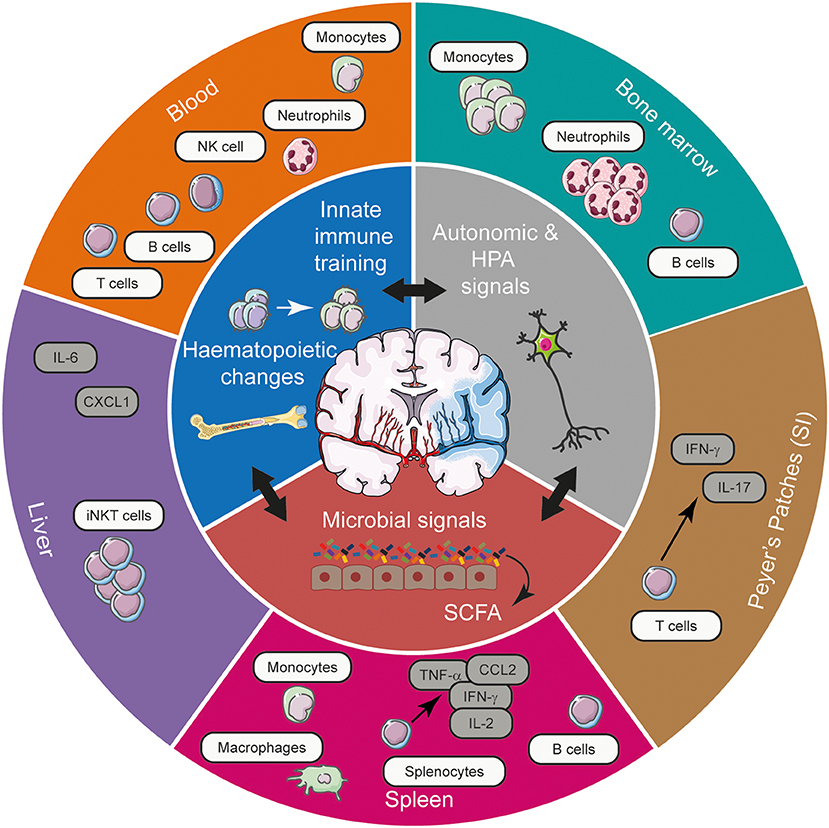
Figure 1. The spectrum of systemic immune alterations post-stroke and the potential mechanisms exploited by stroke to mediate these long-range effects. Stroke, through its effects on autonomic and HPA axis activation, innate immune training, and microbial communities in the gut can gain access to various tissues to shape the systemic immune landscape, affecting cellular, and cytokine networks. Acting in synergy, these means of long-range communication modulate the frequency and functional responsiveness of a plethora of immune cells, tuning the quality of the immune response elicited in various tissues in response to CNS injury. CCL2, chemokine (C-C motif) ligand 2; CXCL1, chemokine (C-X-C motif) ligand 1; IFN-γ, interferon- γ; HPA, hypothalamic-pituitary-adrenal; IL, interleukin; iNKT cell, invariant natural killer T cell; NK cell, natural killer cell; SCFA, short chain fatty acids; SI, small intestine; TNF-α, tumour necrosis factor-α.
Autonomic Signalling
Mounting evidence has ascribed the alterations in systemic immunity to be consequence of increased autonomic signalling, particularly an over activation of the sympathetic nervous system (SNS) through the excessive release of catecholamines, in both patients (43, 117, 118) and experimental stroke (12, 14, 48). Studies have also implicated aberrant cholinergic input (45, 119, 120) and glucocorticoid signalling via the hypothalamic-pituitary-adrenal (HPA) axis (14, 49, 121) in mediating systemic immune dysfunction; again through the excessive release of stress mediators such as cortisol and glucocorticoids into the circulation. In addition to being shaped by circulating catecholamines, many tissues that exhibit altered immune profiles post-stroke such as the gut, lung, spleen, and bone marrow are in fact hard-wired to the SNS (39, 122–125). Mechanistically, it has been posited that neurogenic input from autonomic signals as well as the HPA axis could calibrate systemic immunity by the direct effects of stress mediators on immune cells through adrenergic (126, 127), cholinergic (128, 129) and glucocorticoid receptor signalling (130, 131). Intriguingly, the phenomenon of immunosuppression driven by autonomic and HPA axis-derived signals is not restricted to stroke but is also observed in the broader context of CNS injury such as traumatic brain and spinal cord injury (132–134). Hence, a broader understanding of fundamental neuronal pathways linking the CNS and immune system could guide therapeutic interventions to alleviate infectious complications driven by CNS injury.
Innate Immune Training
Emerging evidence has highlighted the training of innate immune responses by diverse stimuli such as pathogen-associated molecular patterns or cytokines and consequently, the development of immunological memory, a concept traditionally confined to the realms of adaptive immunity (135–144). Given the transient existence of innate cells such as monocytes and neutrophils, it is thought that trained innate immunity is a means to elicit a targeted and enhanced immune reaction to suit host requirements. This has been shown to drive immunometabolic reprogramming via epigenetic rewiring (139, 141) or the replacement of these cells en masse through the generation of functionally poised ones via alterations in haematopoietic output (135, 144). To put this in the context of stroke, recent work has demonstrated that microglial training resulting from repeated administration of bacterial endotoxin is capable of curtailing IL-1β secretion as well as microglial activation (and hence neuroinflammation) following cerebral ischaemia (136). Equally, it is plausible that stroke itself could elicit the training of immune cells in the periphery through its effects on the bone marrow. For example, it has been shown that stroke increases the generation of classical monocytes as a consequence of altered myelopoietic output post-stroke (40). As the bone marrow receives rich sympathetic innervation, it can be envisaged that stroke-driven autonomic signals could tune myelopoiesis to facilitate tissue repair by transducing signals through distinct adrenergic receptor subtypes (125, 145–148). Therefore, the predisposition to repair and replace the entire pool of monocytes and the duration of these changes could impact infection susceptibility as well as the response to a subsequent stroke.
Microbiota-Derived Signals
The interaction of stroke with the commensal microbiota in the gastrointestinal tract is extremely nuanced. Not only does the commensal microbiome play a role in shaping stroke outcome (despite the precise effect being unclear) (8, 15, 18, 149–152) but stroke also mediates shifts in key microbial communities like Firmicutes and Bacteroidetes (153). In homeostasis, the microbiome itself calibrates immunity within the gut microenvironment as well as distal sites through the symbiotic relationship of diverse microbial communities with immune networks by means of microbial metabolites (154–157). In light of data suggesting a detrimental effect of the gut microbiota on stroke outcome (assessed by infarct volume) as well as their role in seeding bacteria that drive pneumonia (8, 149), it therefore becomes difficult to reconcile how commensal bacteria mediate such effects given their indispensable roles in calibrating immunity in homeostasis and injury (158–163). Nevertheless, it is conceivable that the leakiness of the gut barrier induced by stroke (15) could promote the systemic dissemination of pathobionts such as Enterococcus spp., Escherichia coli, and Morganella morganii (149) or result in shifts in key microbial communities that could directly impact systemic immune networks and consequently, stroke outcome (153). Equally, microbial metabolites such as short-chain fatty acids for example, butyrate and propionate are capable of modulating myelopoietic output (increasing the generation of functionally poised monocytes, macrophages and dendritic cells), indicating their ability to train innate immune responses as well (156, 157). In light of these data, it is reasonable to hypothesise that stroke, by shaping gut microbial communities, could also train myeloid responses through the generation of primed progenitors, simultaneously influencing immune networks, shaping both infarct repair and infection susceptibility in distal tissues such as the lungs and bone marrow. However, the precise contributions of stroke in driving shifts in microbial communities and innate immune training remain to be elucidated.
A Balancing act: Ischaemic Injury Repair vs. Anti-Microbial Immunity
Thus far, we have summarised the plethora of effects that stroke has on systemic immunity as well as the means it employs to shape immune function, taking our cues from established and emerging immunological evidence. Taken together, the net effect of the immune alterations elicited by stroke appear to facilitate the repair of the ischaemic insult given that both mononuclear phagocytes as well as T cells appear to adopt an alternatively activated state (14, 31, 61). This is unsurprising as a large body of evidence has identified critical roles for type 2 immunity in mediating tissue repair, a response conserved across vertebrates (74, 75, 164–166). Studies have shown that type 2 cytokines such as IL-4 and IL-13 promote the alternative activation of macrophages via the expression of molecules such as arginase and Ym1 and facilitate collagen fibril assembly (70, 72, 73, 167). However, mononuclear phagocytes with the predisposition to take on an alternatively activated fate remain in circulation and police the immune responses in various tissues but their plasticity, and hence ability, to classically activate and then mediate antimicrobial immunity is questionable. This is due to the fact that the transcriptional and epigenetic landscapes that drive classical and alternative activation, at least in macrophages, are mutually opposing (168). Although tissue-resident macrophages can reversibly polarise in a GATA6-dependent manner (169), whether these principles apply to monocytes and in the context of stroke requires further investigation.
Viewing the paradigm from the vantage point of an immunologist raises some pertinent questions. Is the cost of repairing the ischaemic damage an impairment of antimicrobial defences? Are the various effects on systemic immunity a means to dampen immune responses to antigens that are now exposed to the immune system through a breached BBB following a stroke? Intriguingly, studies have identified that some contexts of stroke (170–174) and other settings of spinal cord (132) and traumatic brain injury (175–177) could also initiate auto-immunity through antibodies to myelin-derived proteins. As a result, immunosuppression post-stroke could be construed as a means to dampen debilitating auto-immunity post-stroke by skewing immunity towards a type 2 phenotype and limiting the splenic B cell pool to constrain humoral and T cell-mediated immunity towards exposed brain antigens. The simultaneous autonomic activation could educate monocytes to take on features of alternative activation in order to augment repair of the injured brain tissue prior to entry though alterations in haematopoiesis in the bone marrow. An inadvertent consequence of this cascade could be increased infection susceptibility due to inadequate plasticity in the polarisation fate acquired during generation. Thus, the systemic availability of poised mononuclear phagocytes could underpin impaired antimicrobial defences in the lungs but it remains to be determined if this is indeed the case.
Concluding Remarks
Data from clinical and pre-clinical paradigms of stroke highlight a diverse range of effects on systemic immunity that can be mediated directly and indirectly by the ischaemic damage. An appreciation of not just the changes themselves but the means employed to elicit them could inform the choice of viable therapies to mitigate infections post-stroke by digressing from traditional choices of antibiotics and beta-blockers. It is therefore imperative we re-examine data from clinical trials of immunotherapy in settings of auto-immunity and cancer to appreciate the remit of immunotherapy. By taking cues from these studies, the clinical trials of immunomodulatory therapies in stroke could provide insight into the fundamental mechanisms at play and more importantly, the suitability of immunotherapies for bolstering neuroprotection and antimicrobial immunity in stroke. This in turn could guide approaches for targeted therapies in the clinical management of neuroprotection and infection to improve outcome following stroke.
Author Contributions
SK conceived the idea for the manuscript. SK and CL wrote and edited the manuscript together.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge the BBSRC grant BB/M025977/1 awarded to Dr. Joanne E. Konkel which supports SK. We would also like to thank Dr. Catriona Cunningham and Flora McClure for critical review of this manuscript and acknowledge Servier Medical Art for templates used in the figure.
References
1. O'Donnell MJ, Chin SL, Rangarajan S, Xavier D, Liu L, Zhang H, et al. Global and regional effects of potentially modifiable risk factors associated with acute stroke in 32 countries (INTERSTROKE): a case-control study. Lancet. (2016) 388:761–75. doi: 10.1016/S0140-6736(16)30506-2
2. Di Carlo A. Human and economic burden of stroke. Age Ageing. (2009) 38:4–5. doi: 10.1093/ageing/afn282
3. Armstrong JR, Mosher BD. Aspiration pneumonia after stroke: intervention and prevention. Neurohospitalist. (2011) 1:85–93. doi: 10.1177/1941875210395775
4. Hoffmann S, Harms H, Ulm L, Nabavi DG, Mackert BM, Schmehl I, et al. Stroke-induced immunodepression and dysphagia independently predict stroke-associated pneumonia - the PREDICT study. J Cereb Blood Flow Metab. (2017) 37:3671–82. doi: 10.1177/0271678X16671964
5. Ovbiagele B, Hills NK, Saver JL, Johnston SC California Acute Stroke Prototype Registry Investigators. Frequency and determinants of pneumonia and urinary tract infection during stroke hospitalization. J Stroke Cerebrovasc Dis. (2006) 15:209–13. doi: 10.1016/j.jstrokecerebrovasdis.2006.05.004
6. Vermeij FH, Scholte op Reimer WJ, de Man P, van Oostenbrugge RJ, Franke CL, de Jong G, et al. Stroke-associated infection is an independent risk factor for poor outcome after acute ischemic stroke: data from the Netherlands Stroke Survey. Cerebrovasc Dis. (2009) 27:465–71. doi: 10.1159/000210093
7. Westendorp WF, Nederkoorn PJ, Vermeij JD, Dijkgraaf MG, van de Beek D. Post-stroke infection: a systematic review and meta-analysis. BMC Neurol. (2011) 11:110. doi: 10.1186/1471-2377-11-110
8. Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal gammadelta T cells. Nat Med. (2016) 22:516–23. doi: 10.1038/nm.4068
9. Chamorro Á, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol. (2012) 8:401–10. doi: 10.1038/nrneurol.2012.98
10. Gelderblom M, Gallizioli M, Ludewig P, Thom V, Arunachalam P, Rissiek B, et al. IL-23 (Interleukin-23)-producing conventional dendritic cells control the detrimental IL-17 (Interleukin-17) response in stroke. Stroke. (2018) 49:155–64. doi: 10.1161/STROKEAHA.117.019101
11. Liesz A, Hagmann S, Zschoche C, Adamek J, Zhou W, Sun L, et al. The spectrum of systemic immune alterations after murine focal ischemia: immunodepression versus immunomodulation. Stroke. (2009) 40:2849–58. doi: 10.1161/STROKEAHA.109.549618
12. McCulloch L, Smith CJ, McColl BW. Adrenergic-mediated loss of splenic marginal zone B cells contributes to infection susceptibility after stroke. Nat Commun. (2017) 8:15051. doi: 10.1038/ncomms16151
13. Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. (2005) 6:775–86. doi: 10.1038/nrn1765
14. Prass K, Meisel C, Höflich C, Braun J, Halle E, Wolf T, et al. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med. (2003) 198:725–36. doi: 10.1084/jem.20021098
15. Singh V, Roth S, Llovera G, Sadler R, Garzetti D, Stecher B, et al. Microbiota dysbiosis controls the neuroinflammatory response after stroke. J Neurosci. (2016) 36:7428–40. doi: 10.1523/JNEUROSCI.1114-16.2016
16. Smith CJ, Emsley HC, Udeh CT, Vail A, Hoadley ME, Rothwell NJ, et al. Interleukin-1 receptor antagonist reverses stroke-associated peripheral immune suppression. Cytokine. (2012) 58:384–9. doi: 10.1016/j.cyto.2012.02.016
17. Urra X, Cervera A, Obach V, Climent N, Planas AM, Chamorro A. Monocytes are major players in the prognosis and risk of infection after acute stroke. Stroke. (2009) 40:1262–8. doi: 10.1161/STROKEAHA.108.532085
18. Winek K, Meisel A, Dirnagl U. Gut microbiota impact on stroke outcome: fad or fact? J Cereb Blood Flow Metab. (2016) 36:891–8. doi: 10.1177/0271678X16636890
19. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. (2011) 17:796–808. doi: 10.1038/nm.2399
20. Hochmeister S, Engel O, Adzemovic MZ, Pekar T, Kendlbacher P, Zeitelhofer M, et al. Lipocalin-2 as an infection-related biomarker to predict clinical outcome in ischemic stroke. PLoS ONE. (2016) 11:e0154797. doi: 10.1371/journal.pone.0154797
21. Warusevitane A, Karunatilake D, Sim J, Smith C, Roffe C. Early diagnosis of pneumonia in severe stroke: clinical features and the diagnostic role of C-reactive protein. PLoS ONE. (2016) 11:e0150269. doi: 10.1371/journal.pone.0150269
22. Lin SP, Long YM, Chen XH. The effects of statins on infections after stroke or transient ischemic attack: a meta-analysis. PLoS ONE. (2015) 10:e0130071. doi: 10.1371/journal.pone.0130071
23. Maier IL, Karch A, Mikolajczyk R, Bähr M, Liman J. Effect of beta-blocker therapy on the risk of infections and death after acute stroke–a historical cohort study. PLoS ONE. (2015) 10:e0116836. doi: 10.1371/journal.pone.0116836
24. Westendorp WF, Vermeij JD, Brouwer MC, Roos YB, Nederkoorn PJ, van de Beek D, et al. Pre-stroke use of beta-blockers does not lower post-stroke infection rate: an exploratory analysis of the preventive antibiotics in stroke study. Cerebrovasc Dis. (2016) 42:506–11. doi: 10.1159/000450926
25. Xi YG, Tian X, Chen WQ, Zhang S, Zhang S, Ren WD, et al. Antibiotic prophylaxis for infections in patients with acute stroke: a systematic review and meta-analysis of randomized controlled trials. Oncotarget. (2017) 8:81075–87. doi: 10.18632/oncotarget.19039
26. Kalra L, Irshad S, Hodsoll J, Simpson M, Gulliford M, Smithard D, et al. Prophylactic antibiotics after acute stroke for reducing pneumonia in patients with dysphagia (STROKE-INF): a prospective, cluster-randomised, open-label, masked endpoint, controlled clinical trial. Lancet. (2015) 386:1835–44. doi: 10.1016/S0140-6736(15)00126-9
27. Smith CJ, Denes A, Tyrrell PJ, Di Napoli M. Phase II anti-inflammatory and immune-modulating drugs for acute ischaemic stroke. Expert Opin Investig Drugs. (2015) 24:623–43. doi: 10.1517/13543784.2015.1020110
28. Perez-de-Puig I, Miró-Mur F, Ferrer-Ferrer M, Gelpi E, Pedragosa J, Justicia C, et al. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol. (2015) 129:239–57. doi: 10.1007/s00401-014-1381-0
29. Doyle KP, Quach LN, Solé M, Axtell RC, Nguyen TV, Soler-Llavina GJ, et al. B-lymphocyte-mediated delayed cognitive impairment following stroke. J Neurosci. (2015) 35:2133–45. doi: 10.1523/JNEUROSCI.4098-14.2015
30. Liesz A, Zhou W, Mracskó É, Karcher S, Bauer H, Schwarting S, et al. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain. (2011) 134(Pt 3):704–20. doi: 10.1093/brain/awr008
31. Miró-Mur F, Pérez-de-Puig I, Ferrer-Ferrer M, Urra X, Justicia C, Chamorro A, et al. Immature monocytes recruited to the ischemic mouse brain differentiate into macrophages with features of alternative activation. Brain Behav Immun. (2016) 53:18–33. doi: 10.1016/j.bbi.2015.08.010
32. Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. (2010) 87:779–89. doi: 10.1189/jlb.1109766
33. Chamorro A, Amaro S, Vargas M, Obach V, Cervera A, Torres F, et al. Interleukin 10, monocytes and increased risk of early infection in ischaemic stroke. J Neurol Neurosurg Psychiatry. (2006) 77:1279–81. doi: 10.1136/jnnp.2006.100800
34. Ruhnau J, Schulze K, Gaida B, Langner S, Kessler C, Bröker B, et al. Stroke alters respiratory burst in neutrophils and monocytes. Stroke. (2014) 45:794–800. doi: 10.1161/STROKEAHA.113.003342
35. Chapman KZ, Dale VQ, Dénes A, Bennett G, Rothwell NJ, Allan SM, et al. A rapid and transient peripheral inflammatory response precedes brain inflammation after experimental stroke. J Cereb Blood Flow Metab. (2009) 29:1764–8. doi: 10.1038/jcbfm.2009.113
36. Emsley HC, Smith CJ, Georgiou RF, Vail A, Hopkins SJ, Rothwell NJ, et al. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry. (2005) 76:1366–72. doi: 10.1136/jnnp.2004.054882
37. Smith CJ, Hulme S, Vail A, Heal C, Parry-Jones AR, Scarth S, et al. SCIL-STROKE (Subcutaneous Interleukin-1 Receptor Antagonist in Ischemic Stroke): a randomized controlled phase 2 trial. Stroke. (2018) 49:1210–6. doi: 10.1161/STROKEAHA.118.020750
38. Campbell SJ, Perry VH, Pitossi FJ, Butchart AG, Chertoff M, Waters S, et al. Central nervous system injury triggers hepatic CC and CXC chemokine expression that is associated with leukocyte mobilization and recruitment to both the central nervous system and the liver. Am J Pathol. (2005) 166:1487–97. doi: 10.1016/S0002-9440(10)62365-6
39. Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. (2006) 26:654–65. doi: 10.1038/sj.jcbfm.9600217
40. Courties G, Herisson F, Sager HB, Heidt T, Ye Y, Wei Y, et al. Ischemic stroke activates hematopoietic bone marrow stem cells. Circ Res. (2015) 116:407–17. doi: 10.1161/CIRCRESAHA.116.305207
41. Denes A, McColl BW, Leow-Dyke SF, Chapman KZ, Humphreys NE, Grencis RK, et al. Experimental stroke-induced changes in the bone marrow reveal complex regulation of leukocyte responses. J Cereb Blood Flow Metab. (2011) 31:1036–50. doi: 10.1038/jcbfm.2010.198
42. Liesz A, Roth S, Zorn M, Sun L, Hofmann K, Veltkamp R. Acquired immunoglobulin G deficiency in stroke patients and experimental brain ischemia. Exp Neurol. (2015) 271:46–52. doi: 10.1016/j.expneurol.2015.04.021
43. Harms H, Reimnitz P, Bohner G, Werich T, Klingebiel R, Meisel C, et al. Influence of stroke localization on autonomic activation, immunodepression, and post-stroke infection. Cerebrovasc Dis. (2011) 32:552–60. doi: 10.1159/000331922
44. Hug A, Dalpke A, Wieczorek N, Giese T, Lorenz A, Auffarth G, et al. Infarct volume is a major determiner of post-stroke immune cell function and susceptibility to infection. Stroke. (2009) 40:3226–32. doi: 10.1161/STROKEAHA.109.557967
45. Liu Q, Jin WN, Liu Y, Shi K, Sun H, Zhang F, et al. Brain ischemia suppresses immunity in the periphery and brain via different neurogenic innervations. Immunity. (2017) 46:474–87. doi: 10.1016/j.immuni.2017.02.015
46. Vogelgesang A, Grunwald U, Langner S, Jack R, Bröker BM, Kessler C, et al. Analysis of lymphocyte subsets in patients with stroke and their influence on infection after stroke. Stroke. (2008) 39:237–41. doi: 10.1161/STROKEAHA.107.493635
47. Theodorou GL, Marousi S, Ellul J, Mougiou A, Theodori E, Mouzaki A, et al. T helper 1 (Th1)/Th2 cytokine expression shift of peripheral blood CD4+ and CD8+ T cells in patients at the post-acute phase of stroke. Clin Exp Immunol. (2008) 152:456–63. doi: 10.1111/j.1365-2249.2008.03650.x
48. Wong CH, Jenne CN, Lee WY, Léger C, Kubes P. Functional innervation of hepatic iNKT cells is immunosuppressive following stroke. Science. (2011) 334:101–5. doi: 10.1126/science.1210301
49. Courties G, Frodermann V, Honold L, Zheng Y, Herisson F, Schloss MJ, et al. Glucocorticoids regulate bone marrow B lymphopoiesis after stroke. Circ Res. (2019) 124:1372–85. doi: 10.1161/CIRCRESAHA.118.314518
50. Kaito M, Araya S, Gondo Y, Fujita M, Minato N, Nakanishi M, et al. Relevance of distinct monocyte subsets to clinical course of ischemic stroke patients. PLoS ONE. (2013) 8:e69409. doi: 10.1371/journal.pone.0069409
51. Grosse GM, Schulz-Schaeffer WJ, Teebken OE, Schuppner R, Dirks M, Worthmann H, et al. Monocyte subsets and related chemokines in carotid artery stenosis and ischemic stroke. Int J Mol Sci. (2016) 17:433. doi: 10.3390/ijms17040433
52. Haeusler KG, Schmidt WU, Föhring F, Meisel C, Helms T, Jungehulsing GJ, et al. Cellular immunodepression preceding infectious complications after acute ischemic stroke in humans. Cerebrovasc Dis. (2008) 25:50–8. doi: 10.1159/000111499
53. Hernández-Jiménez E, Gutierrez-Fernández M, Cubillos-Zapata C, Otero-Ortega L, Rodríguez-Frutos B, Toledano V, et al. Circulating monocytes exhibit an endotoxin tolerance status after acute ischemic stroke: mitochondrial DNA as a putative explanation for poststroke infections. J Immunol. (2017) 198:2038–46. doi: 10.4049/jimmunol.1601594
54. Zhang DP, Yan FL, Xu HQ, Zhu YX, Yin Y, Lu HQ. A decrease of human leucocyte antigen-DR expression on monocytes in peripheral blood predicts stroke-associated infection in critically-ill patients with acute stroke. Eur J Neurol. (2009) 16:498–505. doi: 10.1111/j.1468-1331.2008.02512.x
55. Yang L, Kong Y, Ren H, Li M, Wei CJ, Shi E, et al. Upregulation of CD74 and its potential association with disease severity in subjects with ischemic stroke. Neurochem Int. (2017) 107:148–55. doi: 10.1016/j.neuint.2016.11.007
56. Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. (2013) 39:599–610. doi: 10.1016/j.immuni.2013.08.007
57. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. (2017) 17:349–62. doi: 10.1038/nri.2017.28
58. McCulloch L, Alfieri A, McColl BW. Experimental stroke differentially affects discrete subpopulations of splenic macrophages. Front Immunol. (2018) 9:1108. doi: 10.3389/fimmu.2018.01108
59. Liesz A, Dalpke A, Mracsko E, Antoine DJ, Roth S, Zhou W, et al. DAMP signaling is a key pathway inducing immune modulation after brain injury. J Neurosci. (2015) 35:583–98. doi: 10.1523/JNEUROSCI.2439-14.2015
60. Michaud JP, Pimentel-Coelho PM, Tremblay Y, Rivest S. The impact of Ly6Clow monocytes after cerebral hypoxia-ischemia in adult mice. J Cereb Blood Flow Metab. (2014) 34:e1–9. doi: 10.1038/jcbfm.2014.80
61. Wattananit S, Tornero D, Graubardt N, Memanishvili T, Monni E, Tatarishvili J, et al. Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J Neurosci. (2016) 36:4182–95. doi: 10.1523/JNEUROSCI.4317-15.2016
62. Absinta M, Ha SK, Nair G, Sati P, Luciano NJ, Palisoc M, et al. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. Elife. (2017) 6:e29738. doi: 10.7554/eLife.29738
63. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. (2015) 523:337–41. doi: 10.1038/nature14432
64. Gadani SP, Smirnov I, Smith AT, Overall CC, Kipnis J. Characterization of meningeal type 2 innate lymphocytes and their response to CNS injury. J Exp Med. (2017) 214:285–96. doi: 10.1084/jem.20161982
65. Spencer SP, Wilhelm C, Yang Q, Hall JA, Bouladoux N, Boyd A, et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science. (2014) 343:432–7. doi: 10.1126/science.1247606
66. Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature. (2015) 519:242–6. doi: 10.1038/nature14115
67. Monticelli LA, Buck MD, Flamar AL, Saenz SA, Tait Wojno ED, Yudanin NA, et al. Arginase 1 is an innate lymphoid-cell-intrinsic metabolic checkpoint controlling type 2 inflammation. Nat Immunol. (2016) 17:656–65. doi: 10.1038/ni.3421
68. Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour RE, Nyman J, Dionne D, et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature. (2017) 549:351–6. doi: 10.1038/nature24029
69. Klose CSN, Mahlakõiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature. (2017) 549:282–6. doi: 10.1038/nature23676
70. Allen JE, Sutherland TE. Host protective roles of type 2 immunity: parasite killing and tissue repair, flip sides of the same coin. Semin Immunol. (2014) 26:329–40. doi: 10.1016/j.smim.2014.06.003
71. Minutti CM, Modak RV, Macdonald F, Li F, Smyth DJ, Dorward DA, et al. A macrophage-pericyte axis directs tissue restoration via amphiregulin-induced transforming growth factor beta activation. Immunity. (2019) 50:645–654.e6. doi: 10.1016/j.immuni.2019.01.008
72. Minutti CM, Knipper JA, Allen JE, Zaiss DM. Tissue-specific contribution of macrophages to wound healing. Semin Cell Dev Biol. (2017) 61:3–11. doi: 10.1016/j.semcdb.2016.08.006
73. Minutti CM, Jackson-Jones LH, García-Fojeda B, Knipper JA, Sutherland TE, Logan N, et al. Local amplifiers of IL-4Ralpha-mediated macrophage activation promote repair in lung and liver. Science. (2017) 356:1076–80. doi: 10.1126/science.aaj2067
74. Allen JE, Wynn TA. Evolution of Th2 immunity: a rapid repair response to tissue destructive pathogens. PLoS Pathog. (2011) 7:e1002003. doi: 10.1371/journal.ppat.1002003
75. Skugor S, Glover KA, Nilsen F, Krasnov A. Local and systemic gene expression responses of Atlantic salmon (Salmo salar L.) to infection with the salmon louse (Lepeophtheirus salmonis). BMC Genomics. (2008) 9:498. doi: 10.1186/1471-2164-9-498
76. Jenkins SJ, Ruckerl D, Thomas GD, Hewitson JP, Duncan S, Brombacher F, et al. IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J Exp Med. (2013) 210:2477–91. doi: 10.1084/jem.20121999
77. Allen JE, Maizels RM. Diversity and dialogue in immunity to helminths. Nat Rev Immunol. (2011) 11:375–88. doi: 10.1038/nri2992
78. Kipnis J Multifaceted interactions between adaptive immunity and the central nervous system. Science. (2016) 353:766–71. doi: 10.1126/science.aag2638
79. Prinz M, Priller J. The role of peripheral immune cells in the CNS in steady state and disease. Nat Neurosci. (2017) 20:136–44. doi: 10.1038/nn.4475
80. Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol. (2017) 18:123–31. doi: 10.1038/ni.3666
81. Connolly ES Jr, Winfree CJ, Springer TA, Naka Y, Liao H, Yan SD, et al. Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J Clin Invest. (1996) 97:209–16. doi: 10.1172/JCI118392
82. Allen C, Thornton P, Denes A, McColl BW, Pierozynski A, Monestier M, et al. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J Immunol. (2012) 189:381–92. doi: 10.4049/jimmunol.1200409
83. Stowe AM, Adair-Kirk TL, Gonzales ER, Perez RS, Shah AR, Park TS, et al. Neutrophil elastase and neurovascular injury following focal stroke and reperfusion. Neurobiol Dis. (2009) 35:82–90. doi: 10.1016/j.nbd.2009.04.006
84. McColl BW, Rothwell NJ, Allan SM. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci. (2008) 28:9451–62. doi: 10.1523/JNEUROSCI.2674-08.2008
85. Justicia C, Panés J, Solé S, Cervera A, Deulofeu R, Chamorro A, et al. Neutrophil infiltration increases matrix metalloproteinase-9 in the ischemic brain after occlusion/reperfusion of the middle cerebral artery in rats. J Cereb Blood Flow Metab. (2003) 23:1430–40. doi: 10.1097/01.WCB.0000090680.07515.C8
86. García-Culebras A, Durán-Laforet V, Peña-Martínez C, Ballesteros I, Pradillo JM, Díaz-Guzmán J, et al. Myeloid cells as therapeutic targets in neuroinflammation after stroke: specific roles of neutrophils and neutrophil-platelet interactions. J Cereb Blood Flow Metab. (2018) 38:2150–64. doi: 10.1177/0271678X18795789
87. Cuartero MI, Ballesteros I, Moraga A, Nombela F, Vivancos J, Hamilton JA, et al. N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARgamma agonist rosiglitazone. Stroke. (2013) 44:3498–508. doi: 10.1161/STROKEAHA.113.002470
88. Thored P, Heldmann U, Gomes-Leal W, Gisler R, Darsalia V, Taneera J, et al. Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia. (2009) 57:835–49. doi: 10.1002/glia.20810
89. Neumann J, Gunzer M, Gutzeit HO, Ullrich O, Reymann KG, Dinkel K. Microglia provide neuroprotection after ischemia. FASEB J. (2006) 20:714–6. doi: 10.1096/fj.05-4882fje
90. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
91. Pradillo JM, Denes A, Greenhalgh AD, Boutin H, Drake C, McColl BW, et al. Delayed administration of interleukin-1 receptor antagonist reduces ischemic brain damage and inflammation in comorbid rats. J Cereb Blood Flow Metab. (2012) 32:1810–9. doi: 10.1038/jcbfm.2012.101
92. Li D, Lang W, Zhou C, Wu C, Zhang F, Liu Q, et al. Upregulation of microglial ZEB1 ameliorates brain damage after acute ischemic stroke. Cell Rep. (2018) 22:3574–86. doi: 10.1016/j.celrep.2018.03.011
93. Szalay G, Martinecz B, Lénárt N, Környei Z, Orsolits B, Judák L, et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun. (2016) 7:11499. doi: 10.1038/ncomms11499
94. Römer C, Engel O, Winek K, Hochmeister S, Zhang T, Royl G, et al. Blocking stroke-induced immunodeficiency increases CNS antigen-specific autoreactivity but does not worsen functional outcome after experimental stroke. J Neurosci. (2015) 35:7777–94. doi: 10.1523/JNEUROSCI.1532-14.2015
95. Ge R, Tornero D, Hirota M, Monni E, Laterza C, Lindvall O, et al. Choroid plexus-cerebrospinal fluid route for monocyte-derived macrophages after stroke. J Neuroinflammation. (2017) 14:153. doi: 10.1186/s12974-017-0909-3
96. Leuschner F, Rauch PJ, Ueno T, Gorbatov R, Marinelli B, Lee WW, et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med. (2012) 209:123–37. doi: 10.1084/jem.20111009
97. Strauss-Ayali D, Conrad SM, Mosser DM. Monocyte subpopulations and their differentiation patterns during infection. J Leukoc Biol. (2007) 82:244–52. doi: 10.1189/jlb.0307191
98. McColl BW, Rothwell NJ, Allan SM. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1- and neutrophil-dependent mechanisms. J Neurosci. (2007) 27:4403–12. doi: 10.1523/JNEUROSCI.5376-06.2007
99. Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J, et al. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity. (2013) 38:555–69. doi: 10.1016/j.immuni.2013.02.012
100. Gliem M, Schwaninger M, Jander S. Protective features of peripheral monocytes/macrophages in stroke. Biochim Biophys Acta. (2016) 1862:329–38. doi: 10.1016/j.bbadis.2015.11.004
101. Garcia-Bonilla L, Faraco G, Moore J, Murphy M, Racchumi G, Srinivasan J, et al. Spatio-temporal profile, phenotypic diversity, and fate of recruited monocytes into the post-ischemic brain. J Neuroinflammation. (2016) 13:285. doi: 10.1186/s12974-016-0750-0
102. Ritzel RM, Patel AR, Grenier JM, Crapser J, Verma R, Jellison ER, et al. Functional differences between microglia and monocytes after ischemic stroke. J Neuroinflammation. (2015) 12:106. doi: 10.1186/s12974-015-0329-1
103. Chu HX, Broughton BR, Kim HA, Lee S, Drummond GR, Sobey CG. Evidence that Ly6C(hi) monocytes are protective in acute ischemic stroke by promoting M2 macrophage polarization. Stroke. (2015) 46:1929–37. doi: 10.1161/STROKEAHA.115.009426
104. Greenhalgh AD, Zarruk JG, Healy LM, Baskar Jesudasan SJ, Jhelum P, Salmon CK, et al. Peripherally derived macrophages modulate microglial function to reduce inflammation after CNS injury. PLoS Biol. (2018) 16:e2005264. doi: 10.1371/journal.pbio.2005264
105. Cronk JC, Filiano AJ, Louveau A, Marin I, Marsh R, Ji E, et al. Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J Exp Med. (2018) 215:1627–47. doi: 10.1084/jem.20180247
106. Shemer A, Grozovski J, Tay TL, Tao J, Volaski A, Süß P, et al. Engrafted parenchymal brain macrophages differ from microglia in transcriptome, chromatin landscape and response to challenge. Nat Commun. (2018) 9:5206. doi: 10.1038/s41467-018-07548-5
107. Huang Y, Xu Z, Xiong S, Sun F, Qin G, Hu G, et al. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat Neurosci. (2018) 21:530–40. doi: 10.1038/s41593-018-0090-8
108. Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. (2014) 15:300–12. doi: 10.1038/nrn3722
109. Prinz M, Erny D, Hagemeyer N. Ontogeny and homeostasis of CNS myeloid cells. Nat Immunol. (2017) 18:385–92. doi: 10.1038/ni.3703
110. Goldmann T, Wieghofer P, Jordão MJ, Prutek F, Hagemeyer N, Frenzel K, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. (2016) 17:797–805. doi: 10.1038/ni.3423
111. Brait VH, Arumugam TV, Drummond GR, Sobey CG. Importance of T lymphocytes in brain injury, immunodeficiency, and recovery after cerebral ischemia. J Cereb Blood Flow Metab. (2012) 32:598–611. doi: 10.1038/jcbfm.2012.6
112. Llovera G, Benakis C, Enzmann G, Cai R, Arzberger T, Ghasemigharagoz A, et al. The choroid plexus is a key cerebral invasion route for T cells after stroke. Acta Neuropathol. (2017) 134:851–68. doi: 10.1007/s00401-017-1758-y
113. Arunachalam P, Ludewig P, Melich P, Arumugam TV, Gerloff C, Prinz I, et al. CCR6 (CC Chemokine Receptor 6) is essential for the migration of detrimental natural interleukin-17-producing gammadelta T cells in stroke. Stroke. (2017) 48:1957–65. doi: 10.1161/STROKEAHA.117.016753
114. Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, et al. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med. (2009) 15:946–50. doi: 10.1038/nm.1999
115. Wang J, Li F, Wei H, Lian ZX, Sun R, Tian Z. Respiratory influenza virus infection induces intestinal immune injury via microbiota-mediated Th17 cell-dependent inflammation. J Exp Med. (2014) 211:2397–410. doi: 10.1084/jem.20140625
116. Ruane D, Brane L, Reis BS, Cheong C, Poles J, Do Y, et al. Lung dendritic cells induce migration of protective T cells to the gastrointestinal tract. J Exp Med. (2013) 210:1871–88. doi: 10.1084/jem.20122762
117. Liesz A, Rüger H, Purrucker J, Zorn M, Dalpke A, Möhlenbruch M, et al. Stress mediators and immune dysfunction in patients with acute cerebrovascular diseases. PLoS ONE. (2013) 8:e74839. doi: 10.1371/journal.pone.0074839
118. Chamorro A, Amaro S, Vargas M, Obach V, Cervera A, Gómez-Choco M, et al. Catecholamines, infection, and death in acute ischemic stroke. J Neurol Sci. (2007) 252:29–35. doi: 10.1016/j.jns.2006.10.001
119. Lafargue M, Xu L, Carlès M, Serve E, Anjum N, Iles KE, et al. Stroke-induced activation of the alpha7 nicotinic receptor increases Pseudomonas aeruginosa lung injury. FASEB J. (2012) 26:2919–29. doi: 10.1096/fj.11-197384
120. Engel O, Akyüz L, da Costa Goncalves AC, Winek K, Dames C, Thielke M, et al. Cholinergic pathway suppresses pulmonary innate immunity facilitating pneumonia after stroke. Stroke. (2015) 46:3232–40. doi: 10.1161/STROKEAHA.115.008989
121. Mracsko E, Liesz A, Karcher S, Zorn M, Bari F, Veltkamp R. Differential effects of sympathetic nervous system and hypothalamic-pituitary-adrenal axis on systemic immune cells after severe experimental stroke. Brain Behav Immun. (2014) 41:200–9. doi: 10.1016/j.bbi.2014.05.015
122. Veiga-Fernandes H, Mucida D. Neuro-immune interactions at barrier surfaces. Cell. (2016) 165:801–11. doi: 10.1016/j.cell.2016.04.041
123. Matteoli G, Boeckxstaens GE. The vagal innervation of the gut and immune homeostasis. Gut. (2013) 62:1214–22. doi: 10.1136/gutjnl-2012-302550
124. Chamorro A, Urra X, Planas AM. Infection after acute ischemic stroke: a manifestation of brain-induced immunodepression. Stroke. (2007) 38:1097–103. doi: 10.1161/01.STR.0000258346.68966.9d
125. Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. (2006) 124:407–21. doi: 10.1016/j.cell.2005.10.041
126. Pirzgalska RM, Seixas E, Seidman JS, Link VM, Sánchez NM, Mahú I, et al. Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat Med. (2017) 23:1309–18. doi: 10.1038/nm.4422
127. Shaked I, Hanna RN, Shaked H, Chodaczek G, Nowyhed HN, Tweet G, et al. Transcription factor Nr4a1 couples sympathetic and inflammatory cues in CNS-recruited macrophages to limit neuroinflammation. Nat Immunol. (2015) 16:1228–34. doi: 10.1038/ni.3321
128. Pavlov VA, Parrish WR, Rosas-Ballina M, Ochani M, Puerta M, Ochani K, et al. Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain Behav Immun. (2009) 23:41–5. doi: 10.1016/j.bbi.2008.06.011
129. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. (2000) 405:458–62. doi: 10.1038/35013070
130. Varga G, Ehrchen J, Tsianakas A, Tenbrock K, Rattenholl A, Seeliger S, et al. Glucocorticoids induce an activated, anti-inflammatory monocyte subset in mice that resembles myeloid-derived suppressor cells. J Leukoc Biol. (2008) 84:644–50. doi: 10.1189/jlb.1107768
131. van de Garde MD, Martinez FO, Melgert BN, Hylkema MN, Jonkers RE, Hamann J. Chronic exposure to glucocorticoids shapes gene expression and modulates innate and adaptive activation pathways in macrophages with distinct changes in leukocyte attraction. J Immunol. (2014) 192:1196–208. doi: 10.4049/jimmunol.1302138
132. Held KS, Lane TE. Spinal cord injury, immunodepression, and antigenic challenge. Semin Immunol. (2014) 26:415–20. doi: 10.1016/j.smim.2014.03.003
133. Kraft P, Drechsler C, Schuhmann MK, Gunreben I, Kleinschnitz C. Characterization of peripheral immune cell subsets in patients with acute and chronic cerebrovascular disease: a case-control study. Int J Mol Sci. (2015) 16:25433–49. doi: 10.3390/ijms161025433
134. Schwulst SJ, Trahanas DM, Saber R, Perlman H. Traumatic brain injury-induced alterations in peripheral immunity. J Trauma Acute Care Surg. (2013) 75:780–8. doi: 10.1097/TA.0b013e318299616a
135. Askenase MH, Han SJ, Byrd AL, Morais da Fonseca D, Bouladoux N, Wilhelm C, et al. Bone-marrow-resident NK cells prime monocytes for regulatory function during infection. Immunity. (2015) 42:1130–42. doi: 10.1016/j.immuni.2015.05.011
136. Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. (2018) 556:332–8. doi: 10.1038/s41586-018-0023-4
137. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden CDCC, Li Y, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell. (2018) 172:135–146.e9. doi: 10.1016/j.cell.2017.11.025
138. van der Meer JW, Joosten LA, Riksen N, Netea MG. Trained immunity: a smart way to enhance innate immune defence. Mol Immunol. (2015) 68:40–4. doi: 10.1016/j.molimm.2015.06.019
139. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. (2018) 172:162–175.e14. doi: 10.1016/j.cell.2017.12.013
140. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: a program of innate immune memory in health and disease. Science. (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
141. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. (2012) 12:223–32. doi: 10.1016/j.chom.2012.06.006
142. Netea MG, van Crevel R. BCG-induced protection: effects on innate immune memory. Semin Immunol. (2014) 26:512–7. doi: 10.1016/j.smim.2014.09.006
143. Netea MG, van der Meer JW. Trained immunity: an ancient way of remembering. Cell Host Microbe. (2017) 21:297–300. doi: 10.1016/j.chom.2017.02.003
144. Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. (2018) 172:147–161.e12. doi: 10.1016/j.cell.2017.11.034
145. Dénes A, Boldogkoi Z, Uhereczky G, Hornyák A, Rusvai M, Palkovits M, et al. Central autonomic control of the bone marrow: multisynaptic tract tracing by recombinant pseudorabies virus. Neuroscience. (2005) 134:947–63. doi: 10.1016/j.neuroscience.2005.03.060
146. Méndez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. (2008) 452:442–7. doi: 10.1038/nature06685
147. Vasamsetti SB, Florentin J, Coppin E, Stiekema LCA, Zheng KH, Nisar MU, et al. Sympathetic neuronal activation triggers myeloid progenitor proliferation and differentiation. Immunity. (2018) 49:93–106.e7. doi: 10.1016/j.immuni.2018.05.004
148. Maryanovich M, Zahalka AH, Pierce H, Pinho S, Nakahara F, Asada N, et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat Med. (2018) 24:782–91. doi: 10.1038/s41591-018-0030-x
149. Stanley D, Mason LJ, Mackin KE, Srikhanta YN, Lyras D, Prakash MD, et al. Translocation and dissemination of commensal bacteria in post-stroke infection. Nat Med. (2016) 22:1277–84. doi: 10.1038/nm.4194
150. Wen SW, Wong CHY. An unexplored brain-gut microbiota axis in stroke. Gut Microbes. (2017) 8:601–6. doi: 10.1080/19490976.2017.1344809
151. Winek K, Dirnagl U, Meisel A. The gut microbiome as therapeutic target in central nervous system diseases: implications for stroke. Neurotherapeutics. (2016) 13:762–74. doi: 10.1007/s13311-016-0475-x
152. Winek K, Engel O, Koduah P, Heimesaat MM, Fischer A, Bereswill S, et al. Depletion of cultivatable gut microbiota by broad-spectrum antibiotic pretreatment worsens outcome after murine stroke. Stroke. (2016) 47:1354–63. doi: 10.1161/STROKEAHA.115.011800
153. Spychala MS, Venna VR, Jandzinski M, Doran SJ, Durgan DJ, Ganesh BP, et al. Age-related changes in the gut microbiota influence systemic inflammation and stroke outcome. Ann Neurol. (2018) 84:23–36. doi: 10.1002/ana.25250
154. Thion MS, Low D, Silvin A, Chen J, Grisel P, Schulte-Schrepping J, et al. Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell. (2018) 172:500–516.e16. doi: 10.1016/j.cell.2017.11.042
155. Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. (2014) 157:121–41. doi: 10.1016/j.cell.2014.03.011
156. Trompette A, Gollwitzer ES, Pattaroni C, Lopez-Mejia IC, Riva E, Pernot J, et al. Dietary fiber confers protection against flu by shaping Ly6c(-) patrolling monocyte hematopoiesis and CD8(+) T cell metabolism. Immunity. (2018) 48:992–1005.e8. doi: 10.1016/j.immuni.2018.04.022
157. Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. (2014) 20:159–66. doi: 10.1038/nm.3444
158. Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. (2014) 343:1249288. doi: 10.1126/science.1249288
159. Naik S, Bouladoux N, Linehan JL, Han SJ, Harrison OJ, Wilhelm C, et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature. (2015) 520:104–8. doi: 10.1038/nature14052
160. Harrison OJ, Linehan JL, Shih HY, Bouladoux N, Han SJ, Smelkinson M, et al. Commensal-specific T cell plasticity promotes rapid tissue adaptation to injury. Science. (2019) 363:eaat6280. doi: 10.1126/science.aat6280
161. Belkaid Y, Bouladoux N, Hand TW. Effector and memory T cell responses to commensal bacteria. Trends Immunol. (2013) 34:299–306. doi: 10.1016/j.it.2013.03.003
162. Belkaid Y, Naik S. Compartmentalized and systemic control of tissue immunity by commensals. Nat Immunol. (2013) 14:646–53. doi: 10.1038/ni.2604
163. Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature. (2013) 498:113–7. doi: 10.1038/nature12240
164. Tapia VS, Herrera-Rojas M, Larrain J. JAK-STAT pathway activation in response to spinal cord injury in regenerative and non-regenerative stages of Xenopus laevis. Regeneration. (2017) 4:21–35. doi: 10.1002/reg2.74
165. Secombes CJ, Zou J. Evolution of interferons and interferon receptors. Front Immunol. (2017) 8:209. doi: 10.3389/fimmu.2017.00209
166. Gause WC, Wynn TA, Allen JE. Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nat Rev Immunol. (2013) 13:607–14. doi: 10.1038/nri3476
167. Knipper JA, Willenborg S, Brinckmann J, Bloch W, Maaß T, Wagener R, et al. Interleukin-4 receptor alpha signaling in myeloid cells controls collagen fibril assembly in skin repair. Immunity. (2015) 43:803–16. doi: 10.1016/j.immuni.2015.09.005
168. Piccolo V, Curina A, Genua M, Ghisletti S, Simonatto M, Sabò A, et al. Opposing macrophage polarization programs show extensive epigenomic and transcriptional cross-talk. Nat Immunol. (2017) 18:530–40. doi: 10.1038/ni.3710
169. Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell. (2014) 157:832–44. doi: 10.1016/j.cell.2014.04.016
170. Jin WN, Gonzales R, Feng Y, Wood K, Chai Z, Dong JF, et al. Brain ischemia induces diversified neuroantigen-specific T-cell responses that exacerbate brain injury. Stroke. (2018) 49:1471–8. doi: 10.1161/STROKEAHA.118.020203
171. Becker KJ, Tanzi P, Zierath D, Buckwalter MS. Antibodies to myelin basic protein are associated with cognitive decline after stroke. J Neuroimmunol. (2016) 295–296:9–11. doi: 10.1016/j.jneuroim.2016.04.001
172. Doyle KP, Buckwalter MS. Does B lymphocyte-mediated autoimmunity contribute to post-stroke dementia? Brain Behav Immun. (2017) 64:1–8. doi: 10.1016/j.bbi.2016.08.009
173. Gee JM, Kalil A, Thullbery M, Becker KJ. Induction of immunologic tolerance to myelin basic protein prevents central nervous system autoimmunity and improves outcome after stroke. Stroke. (2008) 39:1575–82. doi: 10.1161/STROKEAHA.107.501486
174. Urra X, Miró F, Chamorro A, Planas AM. Antigen-specific immune reactions to ischemic stroke. Front Cell Neurosci. (2014) 8:278. doi: 10.3389/fncel.2014.00278
175. Zhang Z, Zoltewicz JS, Mondello S, Newsom KJ, Yang Z, Yang B, et al. Human traumatic brain injury induces autoantibody response against glial fibrillary acidic protein and its breakdown products. PLoS ONE. (2014) 9:e92698. doi: 10.1371/journal.pone.0092698
176. Javidi E, Magnus T. Autoimmunity after ischemic stroke and brain injury. Front Immunol. (2019) 10:686. doi: 10.3389/fimmu.2019.00686
Keywords: cerebral ischaemia, post-stroke infection, systemic immunity, innate immune training, neuroimmunology
Citation: Krishnan S and Lawrence CB (2019) Old Dog New Tricks; Revisiting How Stroke Modulates the Systemic Immune Landscape. Front. Neurol. 10:718. doi: 10.3389/fneur.2019.00718
Received: 29 March 2019; Accepted: 18 June 2019;
Published: 02 July 2019.
Edited by:
Johannes Boltze, University of Warwick, United KingdomReviewed by:
Adam Denes, Institute of Experimental Medicine (MTA), HungaryBrad A. Sutherland, University of Tasmania, Australia
Renée Jade Turner, University of Adelaide, Australia
Copyright © 2019 Krishnan and Lawrence. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Siddharth Krishnan, c2lkZGhhcnRoLmtyaXNobmFuQG1hbmNoZXN0ZXIuYWMudWs=