 Jun Ma
Jun Ma Lin Wang1
Lin Wang1 Xinhua Wan
Xinhua Wan- 1Department of Neurology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences, Beijing, China
- 2Department of Geriatrics, Qilu Hospital of Shandong University, Jinan, China
Background: Dystonia is a movement disorder with high clinical and genetic heterogeneity. Recently mutations in lysine-specific histone methyltransferase 2B (KMT2B) gene have been reported to be associated with early-onset progressive dystonia.
Methods: We performed whole-exome sequencings (WES) in a cohort of early-onset dystonia patients from China. Bioinformatics analysis and cosegregation testings were conducted to select candidate causal variants. The effects of identified variants were classified according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines.
Results: Three novel KMT2B variants were identified, including p.Q1359* in patient 1, p.R1487AfsTer7 in patient 2, and p.R152W in patient 3. Among these variants, the nonsense variant p.Q1359* and the frameshift variant p.R1487AfsTer7 showed high pathogenicity and were rated as pathogenic according to the ACMG guideline. Regarding the phenotypes of these two patients with pathogenic variants, patient 2 showed the similar presentation as reported whereas patient 1 seemly harbored the atypical presentations, including later onset age, atypical sites of onset and milder degree of dystonia.
Conclusions: We further report three dystonia patients with novel variants in KMT2B and expand the spectrums of genotype and phenotype of KMT2B.
Introduction
Dystonia is known as a group of clinically and etiologically heterogeneous disorders characterized by sustained or intermittent muscle contractions causing abnormal, often repetitive, movements, postures, or both (1). Genetically defective components play an important role in the genesis of dystonia. With the advent of next-generation sequencing (NGS) technology, an expanding spectrum of dystonia associated genes have been identified (2). In 2016, two groups independently reported mutations in a newly identified gene, lysine-specific histone methyltransferase 2B (KMT2B) in patients with early-onset generalized dystonia (3, 4). Till now, more than 40 patients with different KMT2B variants have been reported, among which only one is from China (3–13). In this study, we screened KMT2B in a cohort of early-onset Chinese dystonia patients by whole-exome sequencing to expand the current knowledge on this gene.
Methods
Subjects
This study was carried out in a cohort of 52 unrelated dystonia patients (29 females, 23 males) from the Movement Disorders Clinic in the Department of Neurology, Peking Union Medical College Hospital in China. Detailed demographic and clinical characteristics of all recruited patients were summarized in the Table S1. All these patients were required to be diagnosed as dystonia with onset of dystonia before 26 years old (the cut-off age of early-onset defined by Chinese dystonia guideline). The mean age at onset was 15.7 years ranging from 1 to 26. Among these patients, 18 had focal dystonia, 10 had segmental dystonia, 12 had multifocal dystonia, 11 had generalized dystonia, and 1 had paroxysmal dystonia. Patients who were suspected of acquired etiologies were excluded. Most patients (43/52, 82.7%) were categorized as isolated dystonia, whereas nine patients manifested with additional symptoms including myoclonus (2/9), cognitive impairment (3/9), parkinsonism (3/9), chorea(1/9), epilepsy (1/9), and impaired vision (1/9). The study was approved by the ethics committee of Peking Union Medical College Hospital. Written informed consents were obtained from all patients or their legal guardians.
Genetic Analysis
Genomic DNAs were extracted from peripheral blood samples of all the patients. After exclusion of the two most common genes (TOR1A, THAP1) for dystonia by Sanger sequencing, whole-exome sequencings (WES) were carried out in all patients. The detailed methods for WES were shown in Supplementary Materials. Briefly, variants were called, aligned, annotated and filtered. Only missense, nonsense, splice-site, in-frame insertion/deletion and frameshift variants with population minor allele frequency (MAF) <1% in public databases of normal human variation were selected for further assessment. The referenced public databases included 1000 Genomes Project (http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/), Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org/) and genome Aggregation Database (genomAD, http://gnomad-old.broadinstitute.org/). Pathogenicity prediction were performed by SIFT (http://sift.jcvi.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), MutationTaster (http://www.mutationtaster.org), and Combined Annotation Dependent Depletion (CADD; http://cadd.gs.washington.edu). Potential causal variants were further verified by Sanger sequencing and tested for cosegregation in their available family members. Then these variants were tested in 100 Chinese unrelated healthy individuals by Sanger sequencing. The clinical effects of identified variants were classified according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines (14).
Results
In this study, three novel KMT2B (NM_014727.2) variants were detected, including one nonsense variant c.4075C>T (p.Q1359*) in patient 1, one frameshift variant c.4458delC (p.R1487AfsTer7) in patient 2, and one missense variant c.454C>T (p.R152W) in patient 3 (Table 1). The clinical features of affected patients were summarized in Table 2.
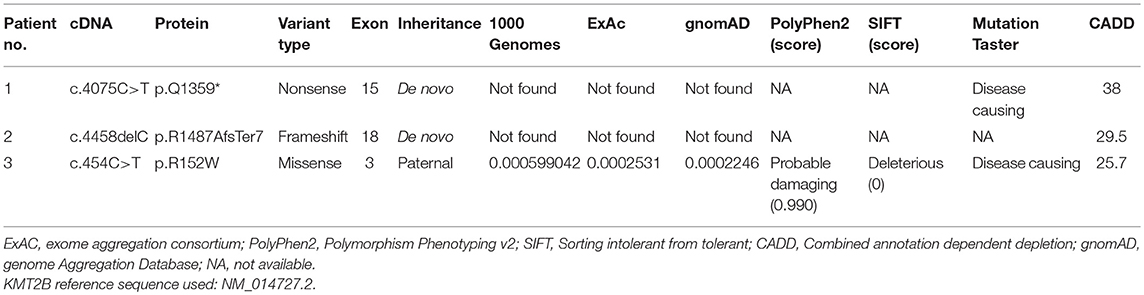
Table 1. Summary of KMT2B novel variants identified in three probands with dystonia.
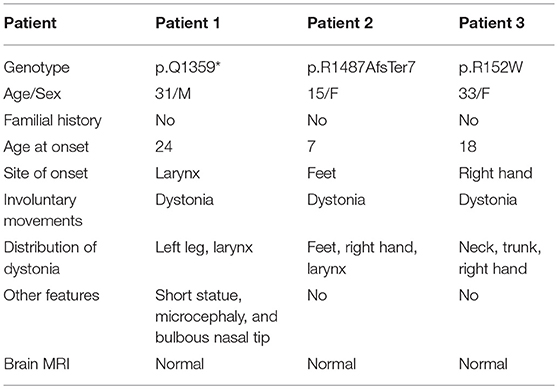
Table 2. Clinical findings of patients with KMT2B variants in this study.
Patient 1
Patient 1 was a 31 year-old man who was the only child born to his healthy non-consanguineous parents. Family history and delivery history of the patient were unremarkable. At the age of 24, he first developed slurred speech. The symptom progressed slowly. Mild dystonia of left leg were later noticed when he was admitted to our hospital (Video S1). Also, short statue, microcephaly and bulbous nasal tip were present in this patient. However, there was no evidence of additional neurological features such as developmental delay, intellectual disability and seizure. Brain magnetic resonance imaging (MRI) was unremarkable from the age of 24 to 31. Administration of medication, including levodopa, baclofen and benzhexol, did not show any clinical benefit. Whole-exome sequencing (WES) was performed and detected a novel heterozygous stop-gain variant c.4075C>T (p.Q1359*) in KMT2B. The variant was absent in dbSNP, ExAc, 1000 Genomes, and gnomAD. It was predicted to “disease causing” by MutationTaster and scored 38 by CADD. Segregation analysis revealed that the stop-gain variant was absent in his parents which indicated it was de novo (Table 1; Figure 1).
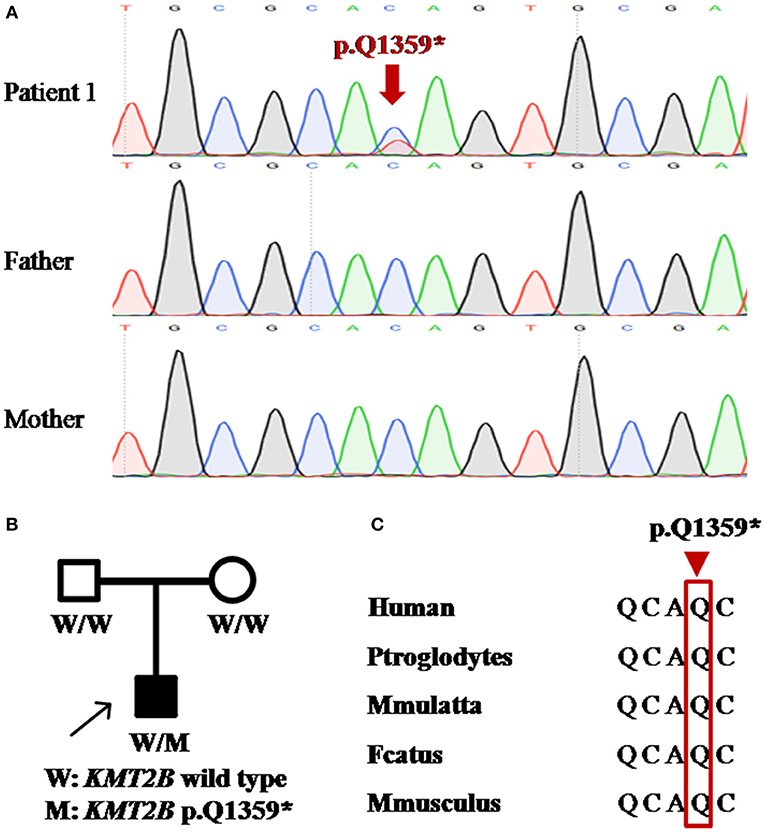
Figure 1. Detection of KMT2B nonsense mutation p.Q1359* in patient 1. (A) DNA sequencing chromatograms of portions of KMT2B gene nonsense mutation (p.Q1359*) (red arrowed). (B) Pedigree chart of patient 1. (A,B) demonstrated the de novo status of the nonsense mutation. (C) Conservation across multiple species at position 1359 (red rectangle).
Patient 2
Patient 2 was a 15 year-old girl from a non-consanguineous family with negative family history. She firstly experienced abnormal gait with dystonia posturing in her feet at the age of 7 years. During the following years, she developed slurred speech, followed by abnormal posture of right hand, leading to handwriting difficulty (Video S2). The severity of these symptoms mildly progressed, especially the severe dysphonia. Neuroimaging of brain showed no obvious abnormalities. Medical interventions including levodopa were of little effect. The testings of gene GCH1, TOR1A, and THAP1 did not detect any pathogenic variants. WES was then performed and uncovered a heterozygous KMT2B frameshift variant c.4458delC (p.R1487AfsTer7), which was never reported before. This variant was absent in dbSNP, ExAc, 1000 Genomes and gnomAD, and predicted a score of 29.5 by CADD. Segregation analysis demonstrated the de novo status of the variant (Table 1; Figure 2).
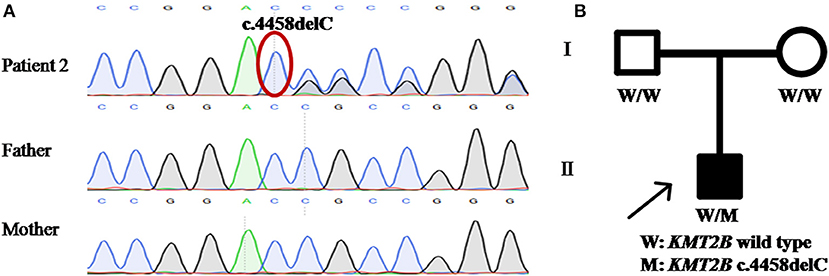
Figure 2. Detection of KMT2B frameshift variant p.R1487AfsTer7 in patient 2. (A) DNA sequencing chromatograms of portions of KMT2B gene frameshift variant p.R1487AfsTer7 (red circle). (B) Pedigree chart of patient 2. (A,B) demonstrated the de novo status of the frameshift variant.
Patient 3
Patient 3 was a 33 year-old woman, who first developed right-handed writer's cramp at 18 years old. The symptom was improved after the treatment of botulinum toxin A injection. At 28 years old, a remarkable spasmodic torticollis was developed and progressively deteriorated in the following years. On current examination, in addition to the demonstrated cervical dystonia, abnormal posture of trunk was also noted (Video S3). In this patient, exome sequencing uncovered a novel heterozygous missense variant c.454C>T (p.R152W) in KMT2B, which was predicted to be damaging by SIFT, PolyPhen2, and MutationTaster. The CADD score was 25.7. The variant was rare in ExAc, 1000 Genomes and gnomAD (MAF 0.02531%, 0.0599042%, and 0.02246%, respectively). Segregation analysis revealed that the variant was inherited from her father who did not show any abnormal symptoms at current examination (Table 1; Figure S1).
In addition, several variants in other dystonia related genes were also identified, including GCH1 c.638_641del in patient 4, PINK1 c.1474C>T and c.938C>T in patient 7, SGCE c.304C>T in patient 26, VPS13A c.7867C>T in patient 23, and ANO3 c.970A>G in patient 40.
Discussion
KMT2B is a large gene (NM_014727.2: 37 exons, 8,148 bp), encoding a lysine methlytransferase specifically responsible for the methylation of histone H3 at lysine 4 (H3K4), an important epigenetic modification associated with active gene transcription (4). Its mutations have been recently reported to cause early-onset generalized dystonia (3, 4). Up to now, more than 40 patients with different KMT2B variants have been reported, including cases with interstitial microdeletion encompassing the entire gene, as well as cases with pathogenic variants (frameshift, in-frame deletion, splice-site, nonsense and missense variants) (3–11). For the majority of patients, KMT2B variants were confirmed as de novo, but autosomal dominant inheritance with reduced penetrance was also reported (4).
In this study, we found one nonsense mutation, one frameshift mutation and one missense mutation in three dystonia patients, among which c.4075C>T (p.Q1359*) was present in patient 1, c.4458delC (p.R1487AfsTer7) in patient 2, and c.454C>T (p.R152W) in patient 3. According to the criteria published by the ACMG (14), the variant c.4075C>T (p.Q1359*) in patient 1 was rated as “pathogenic” because this variant was a null variant in KMT2B [very strong pathogenic criterion (PVS)], proven to be de novo in origin [strong pathogenic criterion 2 (PS2)], absent from population databases [moderate pathogenic criterion 2 (PM2)], and predicted to be deleterious by multiple computational methods [supporting pathogenic criterion 3 (PP3)]. Of note, this nonsense variant occurred within the functionally important PHD domain (4). Similar to above, the frameshift variant c.4458delC in patient 2 should also be categorized to be “pathogenic,” because it belonged to PVS, PS2, PM2, and PP3. In contrast, the missense variant c.454C>T in patient 3 was of uncertain significance because it only met the criteria PP3 (In silico analysis supporting a deleterious effect) according to the consensus of ACMG. The variant was not located in any functional domains of KMT2B protein. Notably, co-segregations of this patient were difficult to judge because of the possible incomplete penetrance of KMT2B. Consequently, further functional verification of this missense variant may be helpful.
Regarding the phenotypes, patients with KMT2B variants present a relatively similar disease course progressively evolving from childhood-onset lower-limb dystonia into generalized dystonia with prominent bulbar and craniocervical involvement (3, 4). In our study, between the patients with pathogenic KMT2B variants, patient 2 showed the similar presentations as reported whereas patient 1 seemly harbored the atypical presentations. Firstly, patient 1 had a later onset age (24 years) than the typical KMT2B-mutated patients (5.8 years in the original report) (3, 4). Secondly, the site of onset with larynx was also atypical because lower-limb dystonia was the initial symptom in the majority patients. What's more, as the core feature, the symptoms of dystonia in this patient were seemly much milder than previously reported patients. As reported, in addition to dystonia, other features were present to varying degrees in the majority of patients, including developmental delay, intellectual disability, oculomotor disturbances, microcephaly, and dysmorphic features (such as elongated face, bulbous nasal tip, and short stature) (4). In our study, patient 1 showed short statue, microcephaly and bulbous nasal tip in addition to dystonia, whereas patient 2 presented with isolated dystonia. Finally, symmetrical hypointensity of the globus pallidi on brain MRI were observed in some reported cases (4). However, no abnormal neuroimaging findings were showed in our patients. In addition, patient 3 also presented with atypical manifestations, including a later onset age (18 years old), atypical site of onset (right hand) and milder degree of dystonia. However, the meaning of the phenotype is unclear concerning the uncertainty of the pathogenicity of R152W.
With the development of sequencing technology, more and more atypical cases with KMT2B mutations have been reported, such as paroxysmal cervical dystonia, or isolated oromandibular dystonia, or global development delay without any evidence of dystonia (4, 9). Thus, clinically heterogeneous phenotypes bring great challenge on precise diagnosis of dystonia. This study suggests that performing WES on affected individuals is an effective method for mapping genes of patients with possible genetic cause.
In conclusion, we report three dystonia patients with novel variants in KMT2B and broaden the spectrums of genotype and phenotype of KMT2B. Among these variants, the novel nonsense variant c.4075C>T (p.Q1359*) and the frameshift variant c.4458delC (p.R1487AfsTer7) show high pathogenicity whereas the missense variant need to further verify.
Data Availability
The sequencing data in our manuscript has been uploaded to SRA (Sequence Read Archive) of NCBI. The SRA accession is PRJNA549023. It will be accessible with the following link after the indicated release date: https://www.ncbi.nlm.nih.gov/sra/PRJNA549023.
Ethics Statement
The study was approved by the ethics committee of Peking Union Medical College Hospital. Written informed consents were obtained from all patients or their legal guardians in accordance with the Declaration of Helsinki.
Author Contributions
JM: conception of the work, data acquisition, statistical analysis, and writing of the first draft. XW: design and organization of the work, manuscript review, and critique. LW, YY, and SL: data acquisition, manuscript review, and critique.
Funding
This study was supported by National Key Research and Development Program of China (2018YFC1314700).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank all the study participants.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2019.00729/full#supplementary-material
References
1. Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord. (2013) 28:863–73. doi: 10.1002/mds.25475
2. Lohmann K, Klein C. Update on the genetics of dystonia. Curr Neurol Neurosci Rep. (2017) 17:26. doi: 10.1007/s11910-017-0735-0
3. Zech M, Boesch S, Maier EM, Borggraefe I, Vill K, Laccone F, et al. Haploinsufficiency of KMT2B, encoding the lysine-specific histone methyltransferase 2B, results in early-onset generalized dystonia. Am J Hum Genet. (2016) 99:1377–87. doi: 10.1016/j.ajhg.2016.10.010
4. Meyer E, Carss KJ, Rankin J, Nichols JM, Grozeva D, Joseph AP, et al. Mutations in the histone methyltransferase gene KMT2B cause complex early-onset dystonia. Nat Genet. (2017) 49:223–37. doi: 10.1038/ng.3740
5. Lange LM, Tunc S, Tennstedt S, Munchau A, Klein C, Assmann B, et al. A novel, in-frame KMT2B deletion in a patient with apparently isolated, generalized dystonia. Mov Disord. (2017) 32:1495–7. doi: 10.1002/mds.27137
6. Zech M, Jech R, Havrankova P, Fecikova A, Berutti R, Urgosik D, et al. KMT2B rare missense variants in generalized dystonia. Mov Disord. (2017) 32:1087–91. doi: 10.1002/mds.27026
7. Zech M, Jech R, Wagner M, Mantel T, Boesch S, Nocker M, et al. Molecular diversity of combined and complex dystonia: insights from diagnostic exome sequencing. Neurogenetics. (2017) 18:195–205. doi: 10.1007/s10048-017-0521-9
8. Baizabal-Carvallo JF, Alonso-Juarez M. Generalized dystonia associated with mutation in the histone methyltransferase gene KMT2B (DYT28) and white matter abnormalities. Parkinsonism Relat Disord. (2018) 49:116–7. doi: 10.1016/j.parkreldis.2018.01.016
9. Faundes V, Newman WG, Bernardini L, Canham N, Clayton-Smith J, Dallapiccola B, et al. Histone lysine methylases and demethylases in the landscape of human developmental disorders. Am J Hum Genet. (2018) 102:175–87. doi: 10.1016/j.ajhg.2017.11.013
10. Kawarai T, Miyamoto R, Nakagawa E, Koichihara R, Sakamoto T, Mure H, et al. Phenotype variability and allelic heterogeneity in KMT2B-associated disease. Parkinsonism Relat Disord. (2018) 52:55–61. doi: 10.1016/j.parkreldis.2018.03.022
11. Zhou XY, Wu JJ, Sun YM. An atypical case of early-onset dystonia with a novel missense variant in KMT2B. Parkinsonism Relat Disord. (2018). doi: 10.1016/j.parkreldis.2018.09.020. [Epub ahead of print].
12. Bras A, Ribeiro JA, Sobral F, Moreira F, Morgadinho A, Januario C. Early-onset oromandibular-laryngeal dystonia and Charlot gait: new phenotype of DYT-KMT2B. Neurology. (2019) 92:919. doi: 10.1212/WNL.0000000000007469
13. Klein C, Baumann H, Olschewski L, Hanssen H, Munchau A, Ferbert A, et al. De-novo KMT2B mutation in a consanguineous family: 15-Year follow-up of an Afghan dystonia patient. Parkinsonism Relat Disord. (2019). doi: 10.1016/j.parkreldis.2019.03.018. [Epub ahead of print].
14. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics andGenomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
Keywords: dystonia, whole-exome sequencing, KMT2B, novel, Chinese
Citation: Ma J, Wang L, Yang Y, Li S and Wan X (2019) Identification of Novel KMT2B Variants in Chinese Dystonia Patients via Whole-Exome Sequencing. Front. Neurol. 10:729. doi: 10.3389/fneur.2019.00729
Received: 30 January 2019; Accepted: 19 June 2019;
Published: 04 July 2019.
Edited by:
Antonio Pisani, University of Rome Tor Vergata, ItalyReviewed by:
Niccolo Mencacci, Northwestern University, United StatesYih-Ru Wu, Chang Gung Memorial Hospital, Taiwan
Copyright © 2019 Ma, Wang, Yang, Li and Wan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinhua Wan, ZHJ4aHdhbkAxNjMuY29t