 Parham Habibzadeh1,2
Parham Habibzadeh1,2 Soroor Inaloo3Mohammad Silawi1Hassan Dastsooz1,4Mohammad Ali Farazi Fard1Forough Sadeghipour1Zahra Faghihi1Mohaddeseh Rezaeian1
Soroor Inaloo3Mohammad Silawi1Hassan Dastsooz1,4Mohammad Ali Farazi Fard1Forough Sadeghipour1Zahra Faghihi1Mohaddeseh Rezaeian1 Majid Yavarian1Johann Böhm5
Majid Yavarian1Johann Böhm5 Mohammad Ali Faghihi1,6*
Mohammad Ali Faghihi1,6*- 1Persian BayanGene Research and Training Center, Shiraz University of Medical Sciences, Shiraz, Iran
- 2Student Research Committee, Shiraz University of Medical Sciences, Shiraz, Iran
- 3Neonatal Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
- 4Italian Institute for Genomic Medicine (IIGM), University of Turin, Turin, Italy
- 5Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), Inserm, CNRS, Université de Strasbourg, Illkirch, France
- 6Center for Therapeutic Innovation, Department of Psychiatry and Behavioral Sciences, University of Miami Miller School of Medicine, Miami, FL, United States
Mitochondrial complex III deficiency nuclear type 2 is an autosomal-recessive disorder caused by mutations in TTC19 gene. TTC19 is involved in the preservation of mitochondrial complex III, which is responsible for transfer of electrons from reduced coenzyme Q to cytochrome C and thus, contributes to the formation of electrochemical potential and subsequent ATP generation. Mutations in TTC19 have been found to be associated with a wide range of neurological and psychological manifestations. Herein, we report on a 15-year-old boy born from first-degree cousin parents, who initially presented with psychiatric symptoms. He subsequently developed progressive ataxia, spastic paraparesis with involvement of caudate bodies and lentiform nuclei with cerebellar atrophy. Eventually, the patient developed gastrointestinal involvement. Using whole-exome sequencing (WES), we identified a novel homozygous frameshift mutation in the TTC19 gene in the patient (NM_017775.3, c.581delG: p.Arg194Asnfs*16). Advanced genetic sequencing technologies developed in recent years have not only facilitated identification of novel disease genes, but also allowed revelations about novel phenotypes associated with mutations in the genes already linked with other clinical features. Our findings expanded the clinical features of TTC19 mutation to potentially include gastrointestinal involvement. Further functional studies are needed to elucidate the underlying pathophysiological mechanisms.
Background
Mitochondrial respiratory chain (MRC), consisting of five enzymatic complexes embedded in the inner mitochondrial membrane, has an important role in providing cells with ATP. Mutations in the nuclear or mitochondrial DNA affecting MRC could result in a wide array of disorders with various neurological and non-neurological presentations (1). Complex III, consisting of 11 subunits, is responsible for the transfer of electrons from reduced coenzyme Q to cytochrome C and thus, contributes to the formation of electrochemical potential ultimately leading to the production of ATP (2).
Mitochondrial complex III deficiency, nuclear type 2 (OMIM: 615157) is an autosomal-recessive disorder caused by mutations in TTC19 gene. TTC19 (Entrez Gene: 54902; OMIM: 613814) is involved in the preservation of complex III function by allowing turnover of Rieske protein through removal of the N-terminal proteolytic fragment of the protein (3). Mutations in TTC19 have been shown to be linked with rapidly progressive neurological impairment (2), spinocerebellar ataxia (4–6), progressive psychosis (4), Leigh syndrome (7), developmental delay and regression in childhood (2, 8–10), bilateral cherry red spots, and failure to thrive (10). Clinical, neuroimaging, and biochemical findings in patients with pathogenic mutations in TTC19 gene are summarized in Table 1.
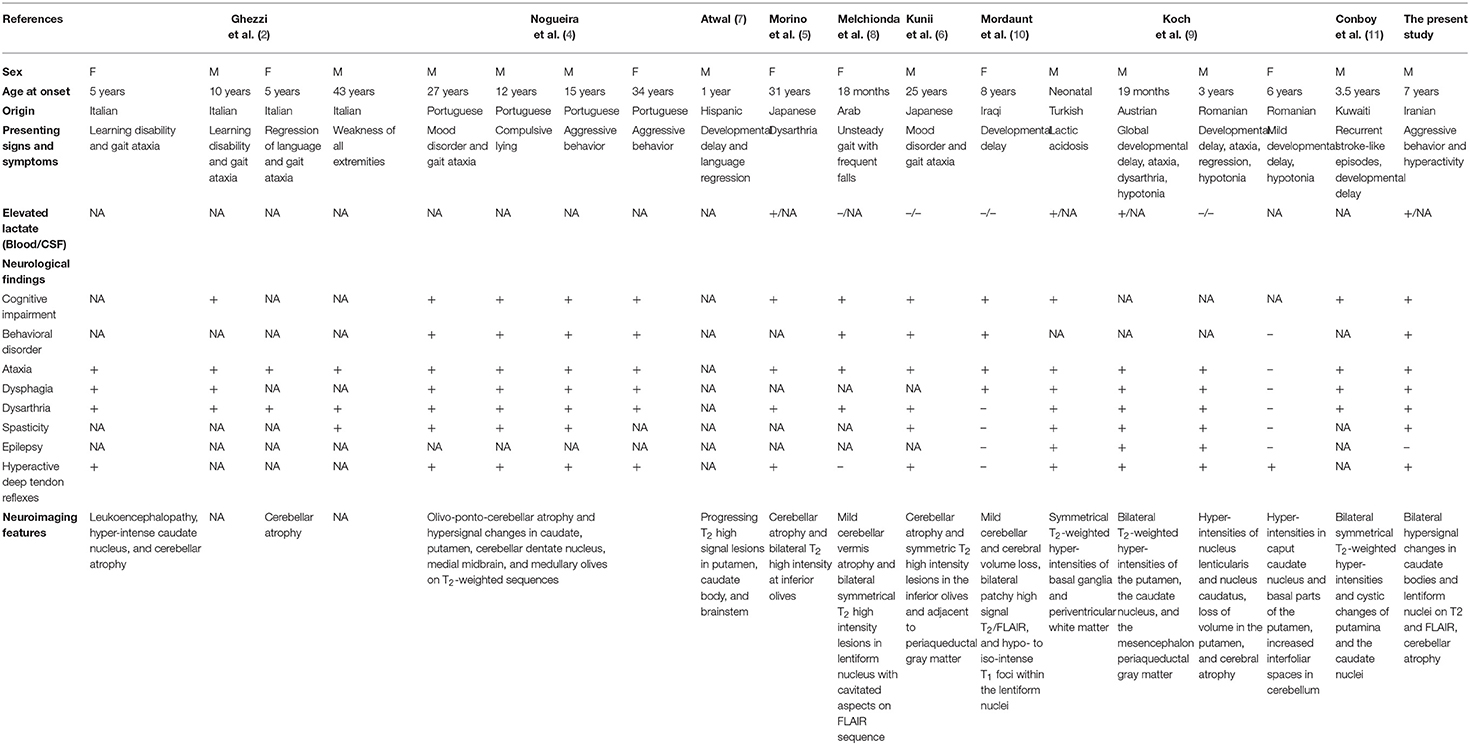
Table 1. Clinical, neuroimaging, and biochemical findings in patients with TTC19 mutations.
Herein, we report on a 15-year-old boy presenting with psychiatric symptoms, progressive ataxia, spastic paraparesis, bilious vomiting, and constipation with a novel homozygous frameshift mutation in TTC19.
Case Presentation
The patient is a 15-year-old boy, who is the first and only child of consanguineous healthy parents who were first cousins. He initially presented at the age of 7 years with psychiatric symptoms including aggressive behavior and hyperactivity for which he was under treatment with methylphenidate (Ritalin), risperidone, olanzapine, and biperiden. The patient had normal psychomotor development until the age of 13 years, when he began to develop speech difficulty. Between the age of 13 and 14, he started to experience gait disturbance and difficulty walking, which progressed in the following year and made the patient wheelchair-bound. On physical examination, the patient was emaciated and cachectic; weighing 45 kg with a height of 165 cm, his body mass index (BMI) was 16.5 kg/m2. Neurological examination was significant for bilateral hyperactive deep tendon reflexes, severe ataxia, tremor, horizontal nystagmus, and spasticity, which was more pronounced in the lower extremities. The patient made limited eye contact and appeared to have intellectual impairment. In addition, musculoskeletal examination was notable for pes cavus.
Laboratory results including serum electrolytes, plasma ammonia, liver function test, blood amino-acid analysis, and cerebrospinal fluid examination were all normal. However, his blood lactate level was elevated to 27 mg/dL (reference range: 4.5–19.8 mg/dL). Magnetic resonance imaging (MRI) of the brain showed hypersignal changes bilaterally in caudate bodies and lentiform nuclei on T2 and fluid attenuated inversion recovery imaging (FLAIR). The lesions appeared as hypodensities in computed tomography (CT). In addition, cerebellar atrophy was detected (Figure 1).
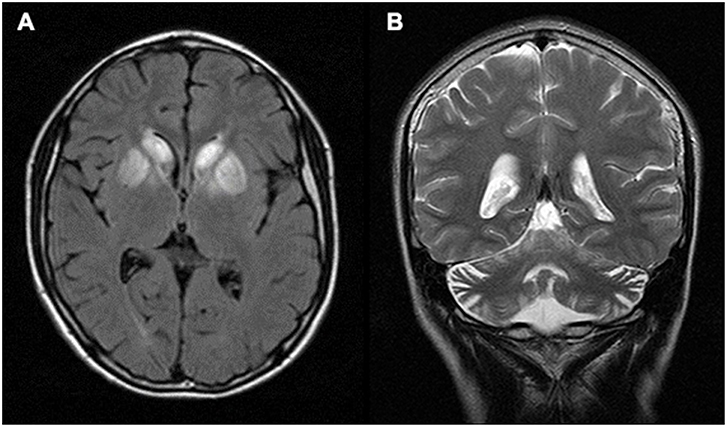
Figure 1. Brain MRI. (A) Axial FLAIR image of the patient showing hyper-signal changes bilaterally in caudate bodies and lentiform nuclei. (B) Coronal T2 image showing cerebellar atrophy.
The patient's condition deteriorated significantly in subsequent months, leading to severe cognitive impairment and mutism. In addition, the patient developed dysphagia, bilious vomiting, and constipation. Upper endoscopy revealed erythematous lesions distributed in the entire esophagus. Furthermore, gastric mucosa in the fundus, body, and antrum was hyperemic with multiple erosions. These lesions were also seen in the bulb and the second part of duodenum. A percutaneous endoscopic gastrostomy (PEG) tube was inserted due to feeding problems.
Total genomic DNA was extracted from the patient's blood sample using QIAamp DNA Blood Mini kit (Qiagen, Germany). Subsequently, whole-exome sequencing (WES) was performed using Illumina NextSeq500 instrument. Variants with an allele frequency of more than 0.005 in gnomAD, EXAC, and our in-house database were excluded. In addition, synonymous and non-coding variants were filtered. Subsequently, considering the autosomal-recessive pattern of inheritance, homozygous, and compound heterozygous variants were analyzed (Table S1). Finally, correlation of the patient's clinical findings with phenotypes associated with the genes harboring identified genetic variations revealed that our patient was homozygous for a previously undescribed frameshift deletion mutation in TTC19 gene (NM_017775.3: c.581delG: p.Arg194Asnfs*16). This novel mutation along with other previously reported pathogenic variants are represented in Figure 2. Sanger sequencing of exon six of the gene was also performed in the patient and his parents, using the following forward (5′-ATTCACAGTTGGCTCATCACTC-3′) and reverse (5′-AGATGTTGTGTGCCCCACTA-3′) primers. It was confirmed that both parents were heterozygous for the mutation, and that the proband was homozygous for this mutation (Figure 3).
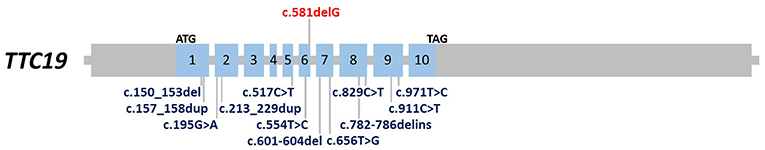
Figure 2. Schematic representation of TTC19 gene (GenBank accession no. NM_017775.3). The blue boxes indicate exons. Novel (shown in red) and previously reported mutations are shown.
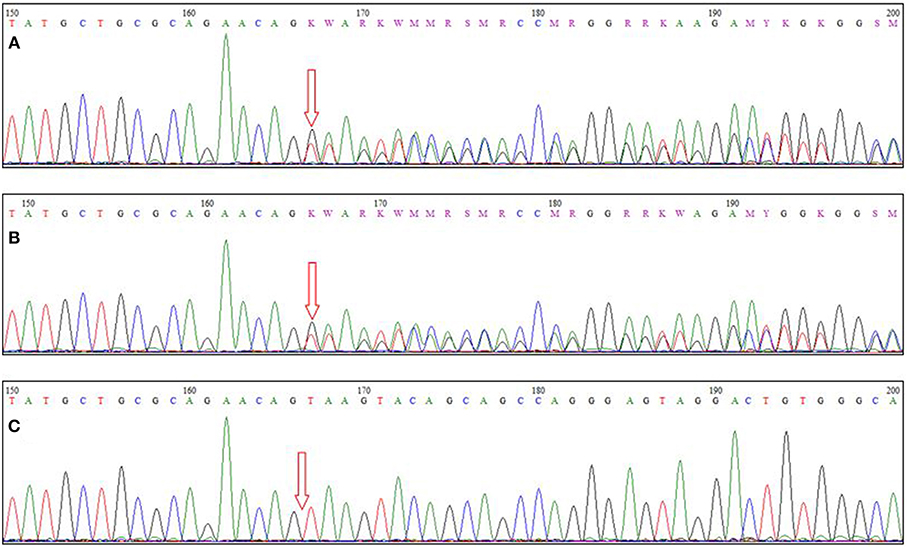
Figure 3. Sequence chromatograms of (A,B) parents and (C) the patient. The arrow indicates the site of the causative mutation.
Written informed consent was obtained from the patient's parents. This study was conducted in accordance with ethical standards of the declaration of Helsinki.
Discussion and Conclusion
Mitochondrial disorders are clinically and genetically heterogeneous, greatly hindering the diagnosis of these disorders. Recent advances made in genetic technologies, allowing sequencing and in-depth investigation of the genome, has substantially improved the diagnosis of mitochondrial diseases. Autosomal-recessive cerebellar ataxias (ARCA) are a diverse group of neurodegenerative disorders characterized by movement incoordination and unsteadiness (12). A growing number of defects in biological pathways such as deficiency of DNA repair, defects in lipoprotein assembly, chaperone dysfunction, and mitochondrial defects can lead to ARCA (12). However, in almost half of the patients the genetic cause remains elusive (13). Herein, we reported a mitochondrial ARCA in a 15-year-old patient who presented with progressive ataxia, spastic paraparesis, psychiatric, and gastrointestinal symptoms with a novel frameshift deletion in TTC19 gene. Protein-protein interaction analysis revealed an interaction of TTC19 with many genes, including ZFYVE26, which has been identified as the cause of autosomal-recessive spastic paraplegia 15 (14, 15). Furthermore, animal models have highlighted the importance of TTC19 gene. TTC19-null adult Drosophilia melanogaster exhibited reduced lifespan, low fertility, adult-onset motor impairment, and abnormal optomotor function (2).
The neurological and psychiatric manifestations of the patient described here coincides well with the manifestations reported earlier; progressive signs and symptoms of basal ganglia and cerebellar dysfunction such as ataxia, dysarthria, and spasticity were observed. Furthermore, psychiatric manifestations are often seen in dominant types of cerebellar ataxia, such as spinocerebellar ataxia 7 and 8; they are generally uncommon in those with ARCA (16, 17). However, it seems that patients with mutations in TTC19 gene are more likely to present psychiatric manifestations, as described in our patient (4, 6).
Patients with mitochondrial disorders are frequently reported to have gastrointestinal symptoms. These symptoms might be the predominant presentation in disorders like mitochondrial neurogastrointestinal encephalomyopathy (MNGIE), whereas in other disorders like Leigh syndrome, gastrointestinal symptoms are usually less prominent compared with neurological presentations (18). Despite the fact that dysphagia is reported in previous reports, constipation and bilious vomiting have not hitherto been reported in the literature. This might be due to intestinal pseudo-obstruction, which has also been reported in other mitochondrial disorders (19, 20).
Advanced genetic sequencing technologies developed in recent years, such as WES, have not only facilitated identification of novel disease genes, but also allowed revelations about novel phenotypes associated with mutations in the genes already linked with other clinical features. Our findings expanded the clinical features of TTC19 mutation to potentially include gastrointestinal involvement. Further functional studies are however needed to shed light over the underlying pathophysiological mechanisms.
Data Availability
All data are available from the corresponding author on request.
Ethics Statement
The Ethics Committee of the Persian BayanGene Research and Training Center approved the study protocol. The parents signed a written informed consent to participate in this study. Written informed consent was obtained from the parents of the patient for the publication of this case report.
Author Contributions
MAF conceived and designed the study, collected, assembled, and interpreted NGS data. PH and SI clinically evaluated the patient. PH drafted the manuscript. SI, MAF, MY, and JB revised the manuscript. PH and JB did the bioinformatics analysis. MS, HD, MAFF, FS, ZF, and MR did the genetic studies.
Funding
This study was supported by the NIMAD research grant (940714) awarded to MAF.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the family members for participating in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2019.00944/full#supplementary-material
References
1. Dimauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. (2003) 348:2656–68. doi: 10.1056/NEJMra022567
2. Ghezzi D, Arzuffi P, Zordan M, Da Re C, Lamperti C, Benna C, et al. Mutations in TTC19 cause mitochondrial complex III deficiency and neurological impairment in humans and flies. Nat Genet. (2011) 43:259–63. doi: 10.1038/ng.761
3. Bottani E, Cerutti R, Harbour ME, Ravaglia S, Dogan SA, Giordano C, et al. TTC19 plays a husbandry role on UQCRFS1 turnover in the biogenesis of mitochondrial respiratory complex III. Mol Cell. (2017) 67:96–105.e104. doi: 10.1016/j.molcel.2017.06.001
4. Nogueira C, Barros J, Sa MJ, Azevedo L, Taipa R, Torraco A, et al. Novel TTC19 mutation in a family with severe psychiatric manifestations and complex III deficiency. Neurogenetics. (2013) 14:153–60. doi: 10.1007/s10048-013-0361-1
5. Morino H, Miyamoto R, Ohnishi S, Maruyama H, Kawakami H. Exome sequencing reveals a novel TTC19 mutation in an autosomal recessive spinocerebellar ataxia patient. BMC Neurol. (2014) 14:5. doi: 10.1186/1471-2377-14-5
6. Kunii M, Doi H, Higashiyama Y, Kugimoto C, Ueda N, Hirata J, et al. A Japanese case of cerebellar ataxia, spastic paraparesis and deep sensory impairment associated with a novel homozygous TTC19 mutation. J Hum Genet. (2015) 60:187–91. doi: 10.1038/jhg.2015.7
7. Atwal PS. Mutations in the complex III assembly factor tetratricopeptide 19 gene TTC19 are a rare cause of leigh syndrome. JIMD Rep. (2014) 14:43–5. doi: 10.1007/8904_2013_282
8. Melchionda L, Damseh NS, Abu Libdeh BY, Nasca A, Elpeleg O, Zanolini A, et al. A novel mutation in TTC19 associated with isolated complex III deficiency, cerebellar hypoplasia, and bilateral basal ganglia lesions. Front Genet. (2014) 5:397. doi: 10.3389/fgene.2014.00397
9. Koch J, Freisinger P, Feichtinger RG, Zimmermann FA, Rauscher C, Wagentristl HP, et al. Mutations in TTC19: expanding the molecular, clinical and biochemical phenotype. Orphanet J Rare Dis. (2015) 10:40. doi: 10.1186/s13023-015-0254-5
10. Mordaunt DA, Jolley A, Balasubramaniam S, Thorburn DR, Mountford HS, Compton AG, et al. Phenotypic variation of TTC19-deficient mitochondrial complex III deficiency: a case report and literature review. Am J Med Genet A. (2015) 167:1330–6. doi: 10.1002/ajmg.a.36968
11. Conboy E, Selcen D, Brodsky M, Gavrilova R, Ho ML. Novel homozygous variant in TTC19 causing mitochondrial complex III deficiency with recurrent stroke-like episodes: expanding the phenotype. Semin Pediatr Neurol. (2018) 26:16–20. doi: 10.1016/j.spen.2018.04.003
12. Anheim M, Tranchant C, Koenig M. The autosomal recessive cerebellar ataxias. N Engl J Med. (2012) 366:636–46. doi: 10.1056/NEJMra1006610
13. Pandolfo M, Manto M. Cerebellar and afferent ataxias. Continuum. (2013) 19:1312–43. doi: 10.1212/01.CON.0000436158.39285.22
14. Hanein S, Martin E, Boukhris A, Byrne P, Goizet C, Hamri A, et al. Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including Kjellin syndrome. Am J Hum Genet. (2008) 82:992–1002. doi: 10.1016/j.ajhg.2008.03.004
15. Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, et al. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. (2009) 37:D412–416. doi: 10.1093/nar/gkn760
16. Embirucu EK, Martyn ML, Schlesinger D, Kok F. Autosomal recessive ataxias: 20 types, and counting. Arq Neuropsiquiatr. (2009) 67:1143–56. doi: 10.1590/S0004-282X2009000600036
17. Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. (2010) 9:885–94. doi: 10.1016/S1474-4422(10)70183-6
18. Rahman S. Gastrointestinal and hepatic manifestations of mitochondrial disorders. J Inherit Metab Dis. (2013) 36:659–73. doi: 10.1007/s10545-013-9614-2
19. Blondon H, Polivka M, Joly F, Flourie B, Mikol J, Messing B. Digestive smooth muscle mitochondrial myopathy in patients with mitochondrial-neuro-gastro-intestinal encephalomyopathy (MNGIE). Gastroenterol Clin Biol. (2005) 29:773–8. doi: 10.1016/S0399-8320(05)86346-8
Keywords: mitochondrial diseases, mitochondrial encephalomyopathies, TTC19, mitochondrial complex III deficiency, neurodegenerative diseases
Citation: Habibzadeh P, Inaloo S, Silawi M, Dastsooz H, Farazi Fard MA, Sadeghipour F, Faghihi Z, Rezaeian M, Yavarian M, Böhm J and Faghihi MA (2019) A Novel TTC19 Mutation in a Patient With Neurological, Psychological, and Gastrointestinal Impairment. Front. Neurol. 10:944. doi: 10.3389/fneur.2019.00944
Received: 05 April 2019; Accepted: 15 August 2019;
Published: 04 September 2019.
Edited by:
Jie Shen, Brigham and Women's Hospital and Harvard Medical School, United StatesReviewed by:
Filippo M. Santorelli, Fondazione Stella Maris (IRCCS), ItalyGeorgia Xiromerisiou, University of Thessaly, Greece
Copyright © 2019 Habibzadeh, Inaloo, Silawi, Dastsooz, Farazi Fard, Sadeghipour, Faghihi, Rezaeian, Yavarian, Böhm and Faghihi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohammad Ali Faghihi, bWZhZ2hpaGlAbWVkLm1pYW1pLmVkdQ==