 Ali Shoeibi
Ali Shoeibi Nahid Olfati
Nahid Olfati Irene Litvan
Irene Litvan- 1Department of Neurology, Faculty of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran
- 2UC San Diego Department of Neurosciences, Parkinson and Other Movement Disorder Center, La Jolla, CA, United States
Progressive supranuclear palsy (PSP) is a four-repeat tau proteinopathy. Abnormal tau deposition is not unique for PSP and is the basic pathologic finding in some other neurodegenerative disorders such as Alzheimer's disease (AD), age-related tauopathy, frontotemporal degeneration, corticobasal degeneration, and chronic traumatic encephalopathy. While AD research has mostly been focused on amyloid beta pathology until recently, PSP as a prototype of a primary tauopathy with high clinical-pathologic correlation and a rapid course is a crucial candidate for tau therapeutic research. Several novel approaches to slow disease progression are being developed. It is expected that the benefits of translational research in this disease will extend beyond the PSP population. This article reviews advances in the diagnosis, epidemiology, pathology, hypothesized etiopathogenesis, and biomarkers and disease-modifying therapeutic approaches of PSP that is leading it to become a frontrunner in translation.
Introduction
Progressive supranuclear palsy (PSP) is a primary tauopathy that is playing an increasingly important role in the field. Better understanding of PSP clinicopathological correlations and pathogenesis has led to a revision of the diagnosis and search for new biomarkers and disease-modifying therapeutic approaches. Because of the rapidly progressive nature of the disease, PSP is an excellent candidate both for pre-clinical, animal model studies of tauopathies, and development of novel therapeutics that could be translated into the clinic. This article reviews the advances in the diagnosis, epidemiology, pathology, hypothesized etiopathogenesis, and biomarkers and disease-modifying therapeutic approaches of PSP that is leading it to become a frontrunner in translation. A summary of the progress in PSP research in the last 25 years is depicted in Figure 1 (1–27).
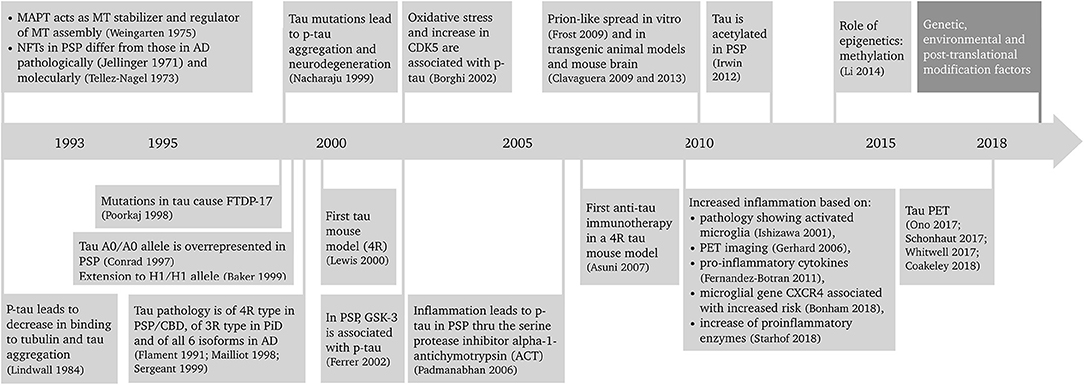
Figure 1. Twenty-five years of progress in progressive supranuclear palsy research. AD, Alzheimer's disease; CBD, corticobasal degeneration; CDK5, cyclin-dependent kinase 5; CXCR4, chemokine receptor type 4; FTDP-17, frontotemporal dementia with parkinsonism linked to chromosome 17; GSK-3, glycogen synthase kinase 3; MAPT, microtubule associated protein tau gene; MT, microtubule; NFT, neurofibrillary tangle; p-tau, phosphorylated tau; PET, positron emission tomography; PiD, Pick's disease; PSP, progressive supranuclear palsy; 3R, tau protein with 3 repeat domains; 4R, tau protein with 4 repeat domains.
Diagnosis
PSP was described as a clinicopathologic entity in 1964 by Steele et al. (28) who described nine patients with characteristic steady progressive vertical supranuclear gaze palsy, pseudobulbar palsy, cognitive impairment, retrocollis and axial rigidity. These features differentiated it from Parkinson's disease (PD) and encephalitic and vascular parkinsonism. They also mapped the neuronal degeneration characterized by neuronal cell loss, neurofibrillary tangles and gliosis in certain nuclei of the rostral midbrain, basal ganglia, and cerebellum (28).
Ten years later PSP was characterized as the prototype of subcortical dementias in view of its significant executive dysfunction and absence of cortical features (29). Early recurrent falls and slowing of vertical saccades were recognized later and were considered as the distinctive features of the National Institute of Neurological Disorders and Stroke and Society for PSP (NINDS-SPSP) possible clinical diagnostic inclusion criteria published in 1996 (30). The probable NINDS-SPSP criteria require both severe postural instability with falls within the first year of symptom onset and vertical supranuclear gaze palsy. These set of diagnostic criteria require lack of features excluding PSP such as cortical dementia resembling Alzheimer's disease (AD), autonomic disturbances or limb cerebellar features resembling multiple system atrophy, hallucinations, and delusions resembling dementia with Lewy bodies, lateralized cortical/motor features resembling corticobasal syndrome, oculomasticatory myoclonus characteristic of Whipple's disease and magnetic resonance imaging (MRI) abnormalities suggesting other disorders such as vascular parkinsonism, etc.
Pure akinesia with gait freezing was the first atypical PSP presentation that was reported by Japanese researchers in a number of pathologically diagnosed PSP cases in 1987 (31–33). Other PSP phenotypic presentations were later described in pathologically confirmed PSP patients (34), recognizing the PSP clinical heterogeneity. The original constellation of findings is now called PSP-Richardson syndrome (PSP-RS) or classical PSP (30).
The International Parkinson and Movement Disorder Society (MDS) has recently standardized the definition of several PSP phenotypic presentations (35). The new criteria (MDS-PSP) (35) classify the core PSP clinical features into four domains: ocular motor dysfunction, postural instability, akinesia, and cognitive dysfunction, which are classified into three levels of certainty (Table 1). Combinations of these features in different stages of the disease (especially early stages) determine the phenotype of PSP (35). In view of the considerable overlap between phenotypes (36), a set of four rules has recently been proposed by the MDS PSP study group to guide assignment of a unique phenotypical diagnosis to those patients who fulfill criteria for multiple phenotypes (37). However, it seems that still more refinement of the criteria is needed (38).
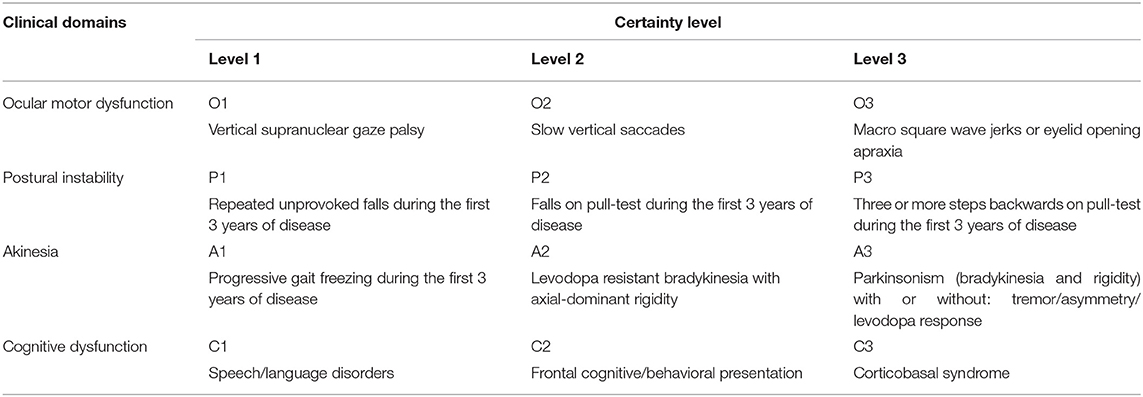
Table 1. Defining clinical features of the PSP phenotypes based on MDS-PSP criteria.
PSP-Parkinsonism (PSP-P) is a retrospective diagnosis made after patients presenting with usually asymmetric parkinsonism with or without resting tremor or levodopa response resembling PD later develop typical PSP-RS features including postural instability and/or vertical supranuclear gaze palsy (34, 39). Williams et al. first defined PSP-P based on these features (34, 39), but the MDS criteria added an axial dominant parkinsonism (A2 in Table 1) as a more specific, though less sensitive, presentation for PSP-P (40).
PSP-progressive gait freezing (PSP-PGF) was originally described in Japan (32), and is currently defined as predominant transient motor blocks or start hesitations in the first 3 years of symptom onset which is unresponsive to levodopa and is unaccompanied with rigidity, tremor, or dementia early in disease course (35). Postural instability, falls and eye movement disturbances are late features of this variant and some patients never develop vertical supranuclear gaze palsy. Prominent axial and neck rigidity without limb rigidity are distinctive features for PSP-PGF. Conspicuous frontal subcortical dementia and levodopa responsive parkinsonism are not usual features (41). Patients with this phenotype, as well as the PSP-P phenotype have a longer survival than those with the PSP-RS.
A frontal behavioral/cognitive syndrome is the presenting feature of PSP in a small subset of patients (PSP-F) (42, 43). This phenotype is characterized by severe apathy, disinhibition, compulsive behavior and loss of insight. Typical PSP-RS features develop years later in the course of the disease. The MDS-PSP criteria defines frontal behavioral/cognitive syndrome as having two out of five basic cognitive features: apathy, bradyphrenia, dysexecutive syndrome, reduced phonemic verbal fluency, and socially inappropriate behaviors (impulsivity, disinhibition, or perseveration).
PSP-non-fluent, agrammatic primary progressive aphasia/progressive apraxia of speech (PSP-nfaPPA/AOS), or the speech/language variant (PSP-SL) is another cortical PSP variant (44) that is believed to have high specificity to represent a probable underlying 4R tauopathy pathology, along with the corticobasal syndrome (CBS) (35). PSP-CBS presents with a combination of cortical (ideomotor orobuccal or limb apraxia, parietal sensory dysfunction, and alien limb phenomenon) and movement (non-levodopa responsive akinesia, rigidity, and stimulus sensitive myoclonus) abnormalities. The standardized MDS definition requires presence of at least one feature of each, cortical or movement-related, categories. Sometimes these patients are clinically indistinguishable from patients with CBD-CBS, but the early presence of supranuclear vertical gaze palsy/slow vertical saccade or postural instability may favor the PSP-CBS diagnosis (45–48).
In the search for an early diagnosis of prodromal PSP, the MDS designates two variants: PSP-ocular motor (PSP-OM), defined as vertical supranuclear gaze palsy (O1), and PSP-postural instability (PSP-PI), defined as falls or postural instability on the pull test (P1 or P2). Both exclude other PSP clinical findings.
There are other rare variants described, which have yet to be standardized: PSP-primary lateral sclerosis (PSP-PLS) (49–51) and PSP-cerebellar ataxia (PSP-C) (52–54), but these are rare and have a very low predictive accuracy for PSP pathology (35).
Overall, the mean disease duration for PSP patient is about 6–8 years, with the shortest duration for PSP-RS (55). The main predictors of a short survival are the PSP-RS variant, early presence of falls, cognitive disorders, and dysphagia (56). Pneumonia and sepsis are considered as the leading causes of death in PSP patients (57).
Epidemiology
PSP is the most common atypical parkinsonian disorder. It was considered as having an approximate prevalence of 5–6 per 100,000 (58, 59). However, higher PSP prevalence has been reported from Japan, Switzerland, and the United Kingdom (55, 60, 61). This higher prevalence could be related to the aging of population, increased general awareness of the condition, inclusion of various disease phenotypes and also the fact that recruitment occurred within a government-based program in Japan that provides support for rare disorders such as PSP. The fact that in some non-selected community-based brain autopsy series (62–64) 3–5% of cases with no or minimal clinical findings have showed PSP pathologies suggests that the true prevalence of PSP is probably much higher. PSP prevalence increases with age and shows no gender predominance (58, 59).
The cause of PSP is still unknown. The intriguing explanation of exposures to mitochondrial toxins as probable cause of PSP and the PSP-like Guadeloupean parkinsonism (65), as well as a cluster of PSP cases in an industrial region (66), suggests the role of environmental/toxic exposures in the PSP pathogenesis. A recent large incidence-based case-control study of 284 PSP and 284 matched controls (ENGENE-PSP) (67) showed that PSP is associated with lower education and exposure to well-water also supports those findings. The association of PSP with lower education is in accordance with previous studies (68, 69). The association with well-water consumption suggests pesticide exposure. Though pesticide, organic solvents, and metal exposures, assessed by an industrial hygienist and toxicologist, as well as living in or close to a rural area were significantly associated with PSP in univariate analyses, these factors were, however, not statistically significant in the multivariate analysis (67), which could be in part related to the relatively small study size. Note the possible contributory role of metal exposure found in the PSP cluster in Northern France (66) and the association between firearm use in veterans and PSP found in the ENGENE-PSP case-control study (70).
The epidemiological studies that assessed the PSP environmental risk factors are summarized in Table 2 (66–74). Available evidence collectively suggests a possible role of low education, metal exposure and consumption of well-water. It could be hypothesized that metal exposure and possible exposure to pesticides through well-water in people who work in those fields who may have low education could lead to increase in oxidative injury that in turn could lead to tau aggregation in susceptible individuals. However, it remains unclear why low education remains as an independent factor in the multivariate analyses. More research is needed to further identify the specific agent(s) responsible and possible mechanisms that would explain these observations (Table 2). Unfortunately only one of those studies had a relatively large sample to identify risk factors (67).
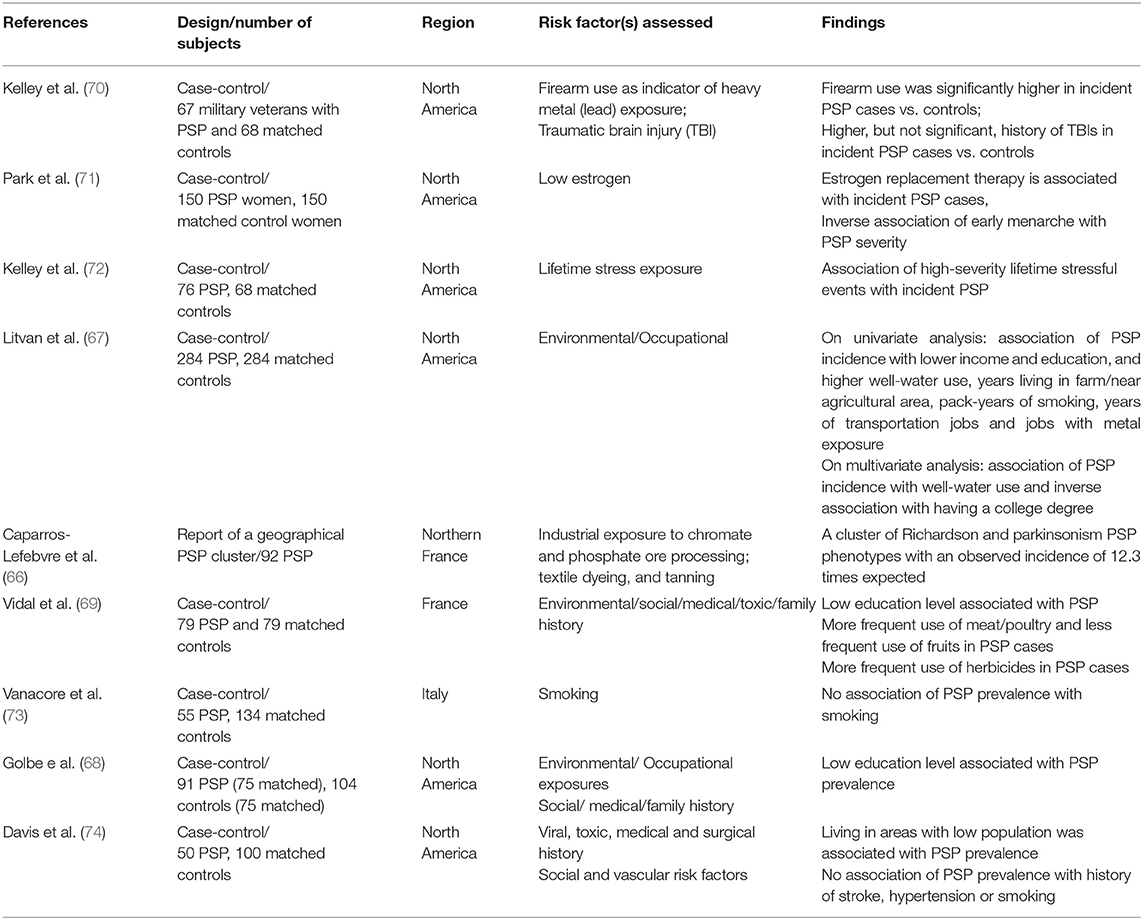
Table 2. Environmental epidemiological risk factors studies in PSP.
Pathology
Olszewski reported neurofibrillary tangles (NFTs) and gliosis in the basal ganglia (mainly globus pallidus and subthalamic nucleus), brainstem structures (predominantly superior colliculi, substantia nigra, periaqueductal gray matter, and pontine tegmentum), and cerebellar dentate nucleus in their cases (28). Pathological criteria of PSP, however, were developed about 30 years later (75) and subsequently revised and validated in 1996 (76) providing the basis for PSP research till present. It defines definite PSP as high density of NFTs and neuropil threads in at least three of these following areas: pallidum, subthalamic nucleus, substantia nigra, or pons. These changes should be accompanied by a pathology of low to high density in at least three of the following areas: striatum, medulla, oculomotor complex, or dentate nucleus. Fulfillment of these criteria in a patient with PSP-compatible history, after exclusion of ischemic and degenerative lesions diagnostic of other disorders, will define definite PSP. However, the tufted astrocyte which is characteristic of the disease was described a few years later by Komori et al. (77).
The establishment of the presence (78, 79) and central role of the tau protein in NFTs (8, 80, 81) and recognition of the six tau isoforms (82) was the key to the later identification of PSP as a 4R-tauopathy. This implies an over representation of the 4-repeat domain containing tau (4R-tau) isoforms relative to the 3-repeat containing ones (3R-tau) in its pathological tau aggregates (7, 83). This is in contrast to what occurs in healthy subjects that have both, 3- and 4-repeat tau isoforms in equal proportions. In PSP, hyperphosphorylated 4R-tau assembles into 13–14 nm straight filaments (84) that aggregate to form dense perikaryal “globose” NFTs in neurons and characteristic glial inclusions named “tufted astrocytes” (85, 86). It is hypothesized that PSP tau pathology in PSP-RS starts in the pallido-luyso-nigral areas and then spreads to the pontine nuclei, other basal ganglia structures, cerebellar dentate nucleus as well as frontal and parietal cortices. The various phenotypic presentations are a consequence of the pathology in different brain areas (87, 88).
In corticobasal degeneration, another 4-repeat tauopathy, neuronal NFTs are more disperse and less argyrophilic than in PSP and astrocytic plaques are the typical lesions because the aggregated tau is mainly located in cell processes, leaving cell soma almost devoid of aggregates. In contrast, PSP's tufted astrocytes are laden with tau fibrillar deposits at soma, with propagation to the cell processes (86, 89).
In contrast to these 4R tauopathies, tau pathology in AD mainly includes bundles of filaments that are composed of both 3R and 4R tau isoforms (90). These filaments are arranged in an antiparallel helical pattern as opposed to the straight filaments of PSP (91). AD tau aggregates mainly in neurons in special areas of the brain starting from transentorhinal regions with subsequent spread to the neocortical association areas (92).
Genetics of PSP
Tau is a microtubule associated protein coded by the MAPT (microtubule associated protein tau) gene located on chromosome 17q21. MAPT contains 16 exons. Alternate splicing of exons 2, 3 (inclusion of either 0, 1, or 2 near N-terminal inserts) and 10 [inclusion or exclusion of repeat-domain number 2 (R2)] produces all six tau isoforms (82) (Figure 2). Tau is extensively expressed in the brain and is mostly located in axons. It is believed traditionally that tau acts as a microtubule (MT) stabilizer and regulator of MTs assembly (94). Further studies showed that tau can regulate fast axonal transport (95) and probably plays a role in stabilizing nuclear DNA (96). To perform its functions, tau bears an intrinsically disordered conformation, meaning that it lacks a stable folded structure and this feature allows its interaction with a wide range of regulatory molecules (97, 98). Each tau isoform consists of 4 domains; an acidic N-terminus region containing a phosphatase-activation domain (PAD), a proline-rich domain, a MT binding region (with 3 or 4 repeat domains), and a neutral highly conserved C-terminal domain. In its monomeric water-soluble state, tau takes a paperclip conformation (99) that keeps the phosphatase-activation domain unexposed. The proline-rich domain includes much of the tau's potentially phosphorylatable regulatory sites, and the C-terminus region contains residues that probably prevent tau aggregation (100, 101).
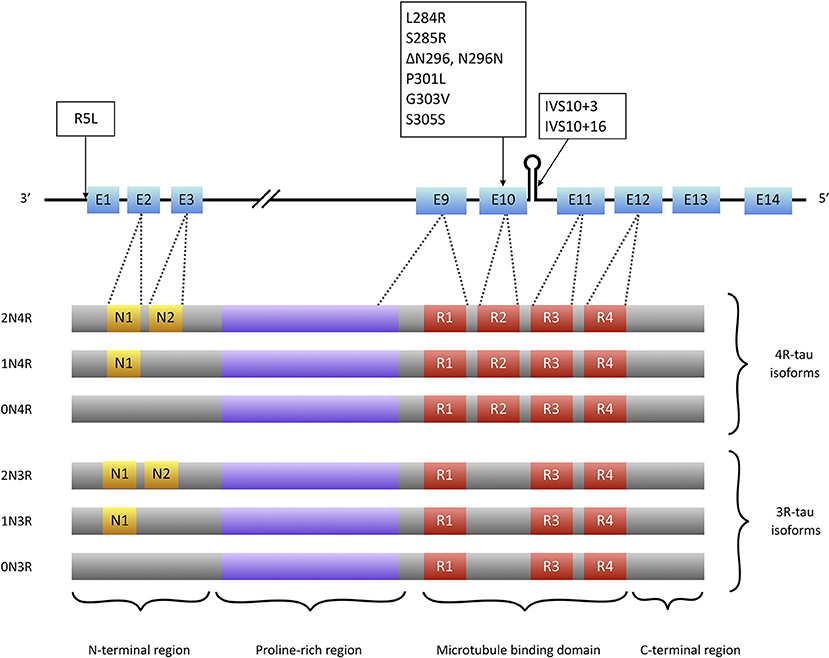
Figure 2. Tau isoforms and conformations. Six tau isoforms result from alternate splicing of exons 2, 3, and 10 (E2, E3, and E10). Tau mutations that present with PSP-like phenotypes (boxes) are mainly located at exon 10 or its splice site. There is only one mutation (R5L) located outside exon 10 that causes a PSP phenotype with brainstem 4R-tau aggregates but also 3R-tau-containing aggregates in cortical areas (93).
The human MAPT gene is affected by a large inversion polymorphism that generates a region of linkage disequilibrium defined by two extended haplotypes, H1 and H2. The H1 haplotype is more frequent in PSP patients compared to the general population (9, 102). In fact, the H1 haplotype is present in 95% of the PSP patients compared to 75% of the general populations with an estimated odds ratio of 5.5, which indicates that H1 haplotype risk for PSP is the same to that of apolipoprotein E (APOE) ε3/ε4 risk allele for AD (103). A common variant of this haplotype, H1c sub-haplotype, has been shown to be accountable for the associated risk of the H1 haplotype with PSP and a number of other neurodegenerative disorders (104–106). A single nucleotide polymorphism (SNP) in the H1c background, rs242557, carries the major associated risk of this haplotype in PSP and CBD (107, 108). More recently three other H1 sub-haplotypes have been proposed to be associated with PSP including H1d, H1g, and H1o (109). The exact mechanism of pathogenesis of the H1 haplotype is not yet known, however there are some clues to confirm or denote to its role: (1) the PSP-like clinical phenotype has been observed in patients with familial tauopathy (frontotemporal degeneration with parkinsonism linked to chromosome 17, FTDP-17) who have an H1/H1 genotype in contrast to the frontal dementia-predominant phenotype in those with H1/H2 genotype (110), (2) increase in translation of total tau with a higher 4R:3R-tau ratio has been shown to be related to H1c sub-haplotype (111), (3) epigenetic studies have shown differential methylation pattern at this haplotype in PSP patients vs. controls (21, 112, 113), and (4) recently, increased plasma tau levels have been found to be associated with the H1c sub-haplotype (114). A MAPT variant, p.A152T, has also been found as a risk factor for PSP and other tauopathies (115).
Genome-wide association studies (GWAS) also found PSP-associated SNPs in other genes including STX6, EIF2AK3, and MOBP (104, 107). STX6 encodes the protein syntaxin 6 which is involved in protein trafficking through the endoplasmic reticulum (116). However, it is not yet known how this altered function would affect the tau metabolism in neurons and glia. (116) EIF2AK3 encodes an RNA-like pancreatic endoplasmic reticulum kinase (PERK), a regulator of the unfolded protein response (UPR) of this organelle. UPR is triggered when the endoplasmic reticulum is overloaded by unfolded proteins, and causes a reduction in overall protein translation and enhances autophagy (117). UPR is activated in regions of PSP brains involved by tau pathology (118). Studies on cultured neurons derived from PSP patients also showed that tauopathy-associated PERK alleles produce a functionally impaired kinase that is associated with neuronal damage due to endoplasmic reticulum stress (119). Product of the gene MOBP is a central nervous system (CNS) myelin structural protein highly expressed in the involved brain regions in PSP (120). Two recent GWAS studies with meta analyses (121, 122) revealed additional SNPs significantly associated with PSP inside or near three other genes: SLCO1A2, DUSP10, and RUNX2. SLCO1A2 codes for a solute carrier organic anion transporter that is highly expressed in areas commonly involved in PSP. A SNP found in an intergenic region near DUSP10 possibly has influence on tau hyperphosphorylation (122). RUNX2 codes for a transcription factor with an effect on differentiation of osteoblasts but its role in PSP is not yet known (121). These two large studies confirmed findings of previous studies except for an SNP in EIF2AK3 that did not reach genome-wide significance in one of these studies (121). Further studies are needed to confirm the latter findings and their implication in the PSP pathogenesis.
Etiopathogenesis
It is not yet known which inciting event(s) trigger dysregulation of the tau protein and which tau abnormality precedes others in the sporadic PSP cases. The exact role of recent epidemiological studies described above need to be further studied (Table 2). A notion of possible contribution of tau “toxic gain of function” as the basic pathogenic underpinning of tauopathies was challenged after a study by SantaCruz et al. (123) who showed that switching off the abnormal expression of tau in a transgenic tauopathy mouse model stopped the neurodegeneration process and improved the cognitive function despite continued deposition of NFTs (124). Similar results were reported in other studies (125, 126). These findings indicate that the process of NFT formation could be dissociated from neurodegeneration and open a window for further discussion on the probability of existence of other more toxic soluble tau species that account for neurodegeneration (127, 128).
Abnormal Post-translational Modifications
After translation, tau undergoes numerous regulatory post-translational modifications (PTMs) including phosphorylation, acetylation, methylation, truncation, among others. These PTMs can result in changes in tau conformation and in its affinity to MTs as well as its propensity to form aggregates (129). In the adult human brain, phosphorylation and dephosphorylation of tau by different kinases and phosphatases at various epitopes regulate the tau function, binding to microtubules and other membrane or nucleic acid partners, and axonal transport (95, 130). Tau contains 85 phosphorylatable epitopes (including serine, threonine, and tyrosine residues) but only 10 epitopes are phosphorylated in the normal brain compared to 16 epitopes in the PSP brains (131, 132). Abnormal tau phosphorylation is associated with a range of disturbances including: (1) tau detachment from MTs and impaired axonal transport (by unmasking of the PAD) (95, 101, 133, 134), (2) tau aggregation (especially through phosphorylation at the C-terminal region) (135), (3) redistribution of tau from axons to cell soma and dendritic processes causing impaired synaptic function and plasticity accompanied by α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) and N-methyl-D-aspartate (NMDA) receptor rearrangements (136), and (4) impaired tau degradation by the proteasome via impairing its recognition by chaperons (137).
Considering the profound role of tau hyperphosphorylation in mediating various pathogenic processes leading to neurodegeneration in PSP, efforts have been mainly directed to elucidate the role of tau kinases and phosphatases. Tau is the substrate for a large number of kinases including proline-directed kinases, particularly glycogen synthase kinase 3 (GSK3) and cyclin-dependent kinase 5 (CDK5), many non-proline directed serine, and threonine kinases such as calcium/calmodulin-dependent protein kinase II, microtubule affinity-regulating kinases, cAMP-dependent protein kinase A) and tyrosine kinases (Fyn, Src, Abl) (101). GSK3 can phosphorylate almost half of tau's phosphorylatable sites and its abnormal activation has been shown in PSP as well as CBD and AD brains and is believed to be associated with pathologic tau hyperphosphorylation and aggregation (15). GSK3 is involved in a signaling pathway mediated by exposed PAD to trigger kinesin-bound cargo delivery. Abnormal exposure of PAD probably results in GSK3 activation and leads to cargo detachment from the MT as well as tau hyperphosphorylation and aggregation (95, 134) (Figure 3). However, the GSK3 inhibitors (including lithium, sodium valproate, and tideglusib) recently evaluated in clinical therapeutic trials of PSP patients failed to slow disease progression (138, 139). Other kinases and phosphatases are under evaluation (140). However, it is not yet known whether tau hyperphosphorylation is a cause for or a consequence of tau aggregation.
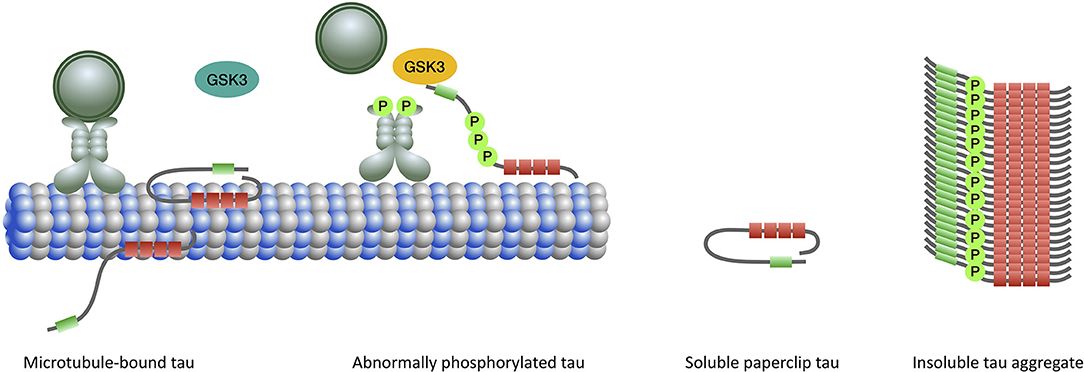
Figure 3. When tau binds to microtubules the N-terminal half of the protein projects away. The extreme N-terminal end of tau contains a phosphatase-activation domain (PAD) (green) that has a role in the regulation of cargo delivery. In the normal paperclip conformation PAD is not exposed, preventing it from triggering the phosphatase-kinase cascade (PKC) and detachment of cargo from microtubule. Activation of PAD and PKC normally occurs at the site of cargo delivery. Abnormal tau phosphorylation (by priming kinases) leads to persistent exposure and activation of PAD and triggering of the PKC which involves overactivation of GSK3β (yellow). Impairment of the fast axonal transport subsequently ensues. In the aggregated form, the microtubule binding domains constitute the core of the filament with the N- and C-terminal regions forming a fuzzy coat around it.
In addition, in PSP there is physiological and pathological acetylation on various lysine residues of the tau molecule (20). Acetylation of various residues provide stabilization of MT-bound tau and regulation of tau phosphorylation, aggregation, degradation, subcellular redistribution, truncation and tau liquid-liquid phase separation (20, 141–146). Acetylation at K280, which is located in the repeat domain 2 of the 4R tau, is a well-known pathological mechanism of acetylation in PSP and other tauopathies (20). However, further research is needed to reveal the acetylation profile of tau in PSP and evaluate its potential implication as a novel therapeutic strategy.
The addition of O-linked N-acetylglucosamine (O-GlcNAc) to the tau protein occurs physiologically and is believed to prevent tau hyperphosphorylation (147, 148). Although reduction of the enzyme involved in tau O-GlcNAcylation was shown in AD brains (149), there are no in vivo or in vitro studies in PSP.
Tau truncation can change its folding and this probably has effects on tau clearance leading to tau aggregation or other various toxic effects (150). Tau fragments enter the cerebrospinal fluid (CSF) and are being used as potential disease markers in many neurodegenerative diseases (151) including PSP. Other tau PTMs including nitration, ubiquitination, sumoylation, methylation, isomerization, and deamidation have mostly been studied in AD, but their role in PSP need to be clarified (101). A detailed profile of sequences and patterns of PSP tau PTMs in neurons and glia could increase our understanding of tau pathogenesis and provide new therapeutic targets.
Mitochondrial Dysfunction
Several lines of evidence indicate the possible role of mitochondrial dysfunction, oxidative injury and defects of energy metabolism in PSP. Based on epidemiological and experimental studies, exposure to herbal neurotoxins containing mitochondrial complex I [CI, NADH:ubiquinone oxidoreductase (152)] inhibitors (mainly fruits and tea made from the Annonaceae family) is a risk factor for a PSP-like parkinsonian disorder with brainstem-predominant 4R-tau inclusions in the French West Indies (153–156). In addition, studies on PSP hybrid cell lines containing mitochondrial DNA showed CI hypofunction (157). PSP patients' brain positron emission tomography (PET) and phosphorous magnetic resonance spectroscopy provide further evidence of energy metabolism failure (158–160). Moreover, PSP brains studies show lipid peroxidation deficits and evidence of oxidative injury (161–163). Although these studies show possible energy metabolism defects in PSP, the exact biochemical basis for the mitochondrial dysfunction is not yet understood. Two recent small clinical trials of the coenzyme Q10 mitochondrial CI enhancer showed marginal or no benefit (164, 165).
Neuroinflammation
Both direct postmortem examination of brain tissue and in vivo ligand-based PET studies show activation of brain macrophages and microglia in the PSP involved brain areas (13, 16, 19). Higher levels of proinflammatory cytokine transcripts, especially interleukin-1β, have also been reported in PSP brains (19) and a recent GWAS showed that the microglial gene CXCR4, is associated with increased risk of PSP and PD (26).
Other studies showed the role of the proinflammatory 5-lipoxygenase enzyme in PSP (26, 166). Despite all evidence for the role of neuroinflammation in the PSP pathogenesis, a PSP case-control study did not find any association between prior use of non-steroidal anti-inflammatory agents and PSP, its disease severity, or the age of symptom onset (167). However, the sample was not large enough to evaluate only anti-inflammatories that cross the blood-brain barrier.
Prion-Like Tau Spread
Several studies show that in PSP abnormally phosphorylated tau fibrils act like self-propagating strains and produce pathogenic “seeds” that are transferable to neighboring cells and are capable of inducing tau aggregates in connecting neurons and glia following neural networks (168–171). However, it is not yet known which mechanisms in the cell-to-cell tau spread are the most relevant (exosomes, release and uptake, tunneling nanotubes, unconventional secretion, or other mechanisms) (172–175). Moreover, although studies are indicative of short-segment filamentous tau species as probable seeds (176, 177), the specific pathogenic PSP tau seeds and their conformation are still unknown. Detailed structure of the core of tau filaments derived from AD, Pick disease and chronic traumatic encephalopathy brains have recently been studied using cryo-electron microscopy (178–180). These studies provide interesting information about specific tau folding in AD, Pick's disease and chronic traumatic encephalopathy. Similar studies are being performed in PSP and corticobasal degeneration that will likely provide clues into the underlying tau pathogenic process, differentiate these disorders and equally important allow the modeling and development of new therapies based on the protein conformation.
Diagnostic Biomarkers
There are no reliable biomarkers for the antemortem diagnosis of PSP. The diagnosis is currently based on clinical criteria. It is particularly challenging to differentiate PSP from a wide range of parkinsonian and dementing disorders during the first few years of disease in the absence of postural instability and ophthalmoparesis (181). Epidemiological studies show a lag of four or more years between the presentation of the first PSP symptom until the correct diagnosis (55, 58). Fortunately, due to increased PSP awareness this diagnosis lag is decreasing. Considering the lack of optimal biomarkers and the wide range of PSP phenotypic presentations that also overlap with other neurodegenerative proteinopathies PSP still remains underdiagnosed. Hopefully, ongoing studies focused on the characterization of PSP-specific biomarkers will soon identify accurate diagnostic and outcome biomarkers that would allow the conduction of therapeutic trials at earlier stages.
Structural Brain Imaging
Conventional brain imaging may show atrophy of midbrain and superior cerebellar peduncle out of proportion to that of the pons and middle cerebellar peduncle. Several imaging indices have been suggested as reliable markers of PSP-RS (182). However, imaging studies on pathologically confirmed PSP cases are limited and other conditions, especially CBD with clinical presentations of the Richardson syndrome, may have false positive indices (183). Therefore, the value of the MRI studies in increasing the certainty of the underlying pathologic diagnosis is unclear. The magnetic resonance parkinsonism index (MRPI) developed by Quattrone et al. (184) as the product of the ratios of pons to midbrain area (Pa/Ma) and middle to superior cerebellar peduncles diameter (MCPd/SCPd) showed a high sensitivity and specificity to differentiate PSP from PD and parkinsonism-predominant multiple system atrophy (MSA-P), another atypical parkinsonian disorder (184–187). A new variant of MRPI, named MRPI 2.0, that in addition incorporates the measurement of the third ventricle diameter (188) seems even more promising. MRPI 2.0 is defined as the MRPI ratio multiplied by the ratio of the third ventricle width of the frontal horn (Figure 4). However, pathologically confirmation in independent samples is still lacking.
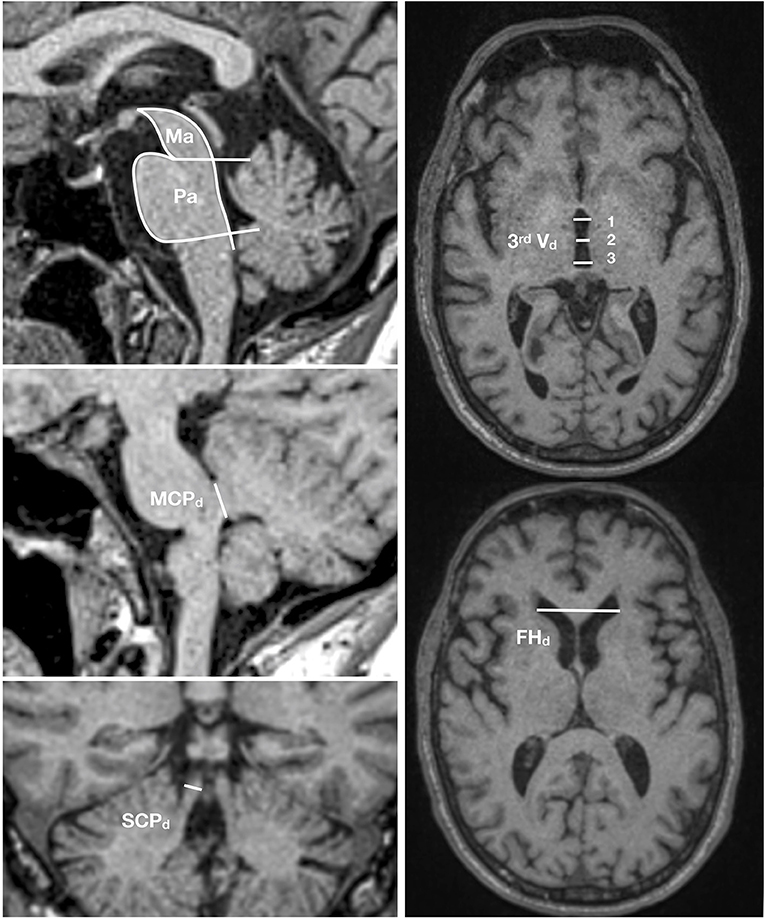
Figure 4. MRPI 2.0 index. This index is the product of the ratios of pons to midbrain area (Pa/Ma), width of middle to superior cerebellar peduncles (MCPd/SCPd), and average third ventricle diameter (measured at three points) to the maximal frontal horn diameter [(3rd Vd 1+2+3/3)/FHd]. MRIPI 2.0 = (Pa/Ma) × (MCPd/SCPd) × (average 3rd Vd/FHd).
Other structural imaging techniques such as voxel based morphometry, volumetry, diffusion weighted, and diffusion tensor imaging and combination of various measurements have been suggested to detect earlier stages of degeneration [for a review see (189)]. Lack of pathological confirmation in most studies as well as inherent limitations and confounders of these modalities are major shortcomings for these studies (190, 191). A number of small pathologically confirmed studies proposed that midbrain atrophy can differentiate PSP from other parkinsonian syndromes (192, 193). A larger pathologically confirmed study, using 3 dimensional MRI volumetry of the combination of midbrain, parietal white matter, temporal gray matter, brainstem, frontal white matter and pons, showed that this measure can reliably differentiate PSP from CBD and controls. However, a later study that differentiated typical and variant PSP phenotypes in their sample of 24 pathologically confirmed PSP showed that midbrain atrophy is associated with typical PSP phenotype (with underlying PSP or CBD pathology) but cannot differentiate PSP pathology presenting with variant phenotypes (183). Free water imaging (194) and diffusion kurtosis imaging (195) have recently been used to address parts of the limitations of structural imaging studies and have showed promising results. An increase in free water was found in several brain areas in PSP patients including basal ganglia, thalamus, midbrain, substantia nigra, cerebellar peduncles, dentate nucleus, cerebellar vermis and lobules V and VI, and corpus callosum. This pattern was in contrast to PD cases who had increased free water only in substantia nigra. MSA patients also had more restricted pattern than PSP cases (i.e., dentate nucleus, subthalamic nucleus, and corpus callosum did not show increased free water in MSA patients). Similarly, changes in free-water-corrected fractional anisotropy values were more pervasive in PSP (increased in putamen, caudate, thalamus, and vermis and decreased in the superior cerebellar peduncle and corpus callosum) than MSA (increased in putamen and caudate) and PD (no significant difference compared to controls) cases. These findings indicate that free water imaging might be used to differentiate various parkinsonian syndromes, however, replication of these findings in pathology proven samples is needed. These findings are in accordance with the results of the previous studies applying diffusion weighted and diffusion tensor imaging to parkinsonian patients (196–198) which in turn reflect the pattern of pathological involvement assessed by voxel based morphometry (199, 200). There are scarce studies focusing on differentiation between PSP phenotypes (201–204) but the results have been contradictory and inconclusive.
Functional Imaging
Various functional imaging techniques have been used in parkinsonian syndromes including magnetic resonance spectroscopy, dopamine transporter imaging, task-free functional MRI, and FDG-PET (182). These techniques are not specific for the underling pathology and do not differentiate between neurodegenerative and non-degenerative processes.
Recently, tau PET imaging makes it possible to detect the distribution and severity of specific forms of tau pathology (182, 205). [11C]PBB3 (phenyl/pyridinyl-butadienyl-benzothiazole/benzothiazolium family), [18F]AV-1451 [aka [18F]flortaucipir, pyrido-indole family], and [18F]THK5351 (arylquinoline family) are the first generation of tau PET tracers tested in clinical studies (205). Unfortunately, [11C]PBB3 has off-target binding to white matter, venous structures and β amyloid (206) and [18F]AV-1451 has off-target binding to monoamine oxidase (MAO)-A, choroid plexus and mineralized or melanin containing structures (207). However, despite these off-target binding, studies on PSP patients showed that [18F]AV-1451 also binds to the PSP-specific subcortical areas with tau pathology, including dentate nucleus, thalamus, midbrain, pallidum and striatum (24, 25, 208, 209), which makes it a favorable tracer in studies of PSP. The pattern has been correlated with postmortem tau pathology in a few patients (24). In fact, [18F]AV-1451 has been reported to be highly sensitive and specific in differentiating PSP from PD (24). Nonetheless, off target binding precludes an early diagnosis because ligand binding is found in the same areas in normal controls except for the dentate nucleus (210–212). On the other hand, because these tracers were developed to detect 3R/4R AD pathology, the PET tau signal is lower in PSP, CBD and in patients carrying MAPT mutations with probable 4R tau pathology compared to those with AD (213). It remains controversial whether the tracer uptake associates with disease severity (208, 211). Despite the fact that postmortem PSP brain autoradiographic studies showed weak binding of this tracer to PSP pathology (207, 214), clinical studies show tau binding in PSP (24, 25, 208, 209). In fact, a recent study, evaluating disease progression in clinically diagnosed PSP-R patients followed up for 12 months using [18F]AV-1451 and midbrain volume on 3 Tesla MRI (215) showed that MRI midbrain atrophy correlated better with clinical disease progression than to [18F]AV-1451 uptake.
A recent study showed a correlation between PSP patients' clinical severity with the [18F]THK5351 signal in 11 PSP patients (216), however, there are concerns about its possible binding to TDP-43 pathology (217).
Second generation tracers including [18F]PM-PBB3, [18F]GTP-1 (ClinicalTrials.gov NCT02640092), [18F]PI-2620 (218, 219), [18F]MK-6240 (220), [18F]R06958948 (221), and [18F]JNJ64349311 (222) have generally showed less off-target binding especially at choroid plexus and MAO enzymes in AD and PSP. However, further validation in different samples including older patients are necessary. It is hoped that ongoing ligand studies identify 4R tau specific ligands. Detailed information of the PSP tau fibrillar structure once available will probably be helpful in designing more specific PSP tau ligands (223).
CSF and Blood Biomarkers
Although high levels of tau oligomers in CSF have been well-incorporated into diagnostic workup of AD (224), tau oligomer measurements in PSP have not yet shown reliable and consistent pattern except for decreased level of total tau and phospho-tau compared to AD and healthy controls (225). It has been hypothesized that measurement of specific truncated forms or PSP-specific epitopes of tau released by degenerating cells in PSP are needed to show real amount of CSF tau in PSP. However, newer ELISAs with antibodies directed to mid- and N-terminal portions of tau showed the same results of lower tau levels in CSF (225). There are studies reporting a reverse association of CSF phospho-tau with disease severity in PSP (226).
Recently a protein amplification technique called real-time quaking-induced conversion (RT-QuIC) (227) or protein misfolding cyclic amplification (PMCA) assay (228), has been applied successfully in identification of small amounts of misfolded proteins in body tissue/fluid samples. RT-QuiC in CSF was recently used to discriminate PiD from other neurodegenerative disorders and healthy controls (229). The 3R-tau filaments of PiD, but not filaments from AD or FTD, seeded recombinant 3R tau monomers. 3R-tau RT-QuIC differentiated PiD from other disorders with high sensitivity and specificity. These results await replication in larger samples. The same method was used to differentiate AD tau seeds from disease controls including cases of 4R (PSP and CBD), 3R (PiD) or 3R+4R (chronic traumatic encephalopathy, primary age-related tauopathy) tauopathies (230). Results showed that AD-RT-QuIC assay can differentiate AD from 3R and 4R tauopathies with high sensitivity and specificity. These techniques are also under evaluation for possible application in identification of 4R-tau PSP seeds. However, at present, these techniques are not quantitative and do not allow to measure the disease severity.
Higher levels of neurofilament light chain (NfL) have been found in atypical parkinsonisms compared to PD (231). NfL is an unspecific marker of axonal loss in central and peripheral nervous system and studies on mouse models of tauopathy, AD, and α-synucleinopathy have showed that its blood and CSF levels are associated with progression and severity of neurodegeneration (232). NfL has been related to disease severity in PSP and changes in its level have been measurable in time-span of clinical trials, suggesting it could be a marker of disease progression in therapeutic trials (226, 233, 234). However, NfL is not suitable for PSP diagnosis since it can be high and associated with disease severity in other disorders such as vascular dementia and frontotemporal degeneration (235, 236). Various combinations of above-mentioned biomarkers have been proposed for the differential diagnosis of PSP, but replication studies are needed (226, 237, 238).
Disease-modifying Therapeutic Approaches
Various disease modifying approaches are now under evaluation based on recent advances in the understanding of PSP pathogenesis (Table 3). Tideglusib, a GSK3-β inhibitor, was among the first disease-modifying agents evaluated in a large double-blind placebo-controlled clinical trial of PSP patients (138). Tideglusib failed to show any clinical effect as other GSK3-β inhibitors did in smaller trials (139). Davunetide, a neuroprotective and microtubule stabilizer, was evaluated with the same negative results (233). However, other phosphorylation inhibitors, microtubule stabilizers and neuroprotective agents are still under evaluation (140). Mitochondrial function enhancing nutrients including coenzyme Q10 have also been tested in clinical trials of PSP patients with no apparent benefit (140, 165).
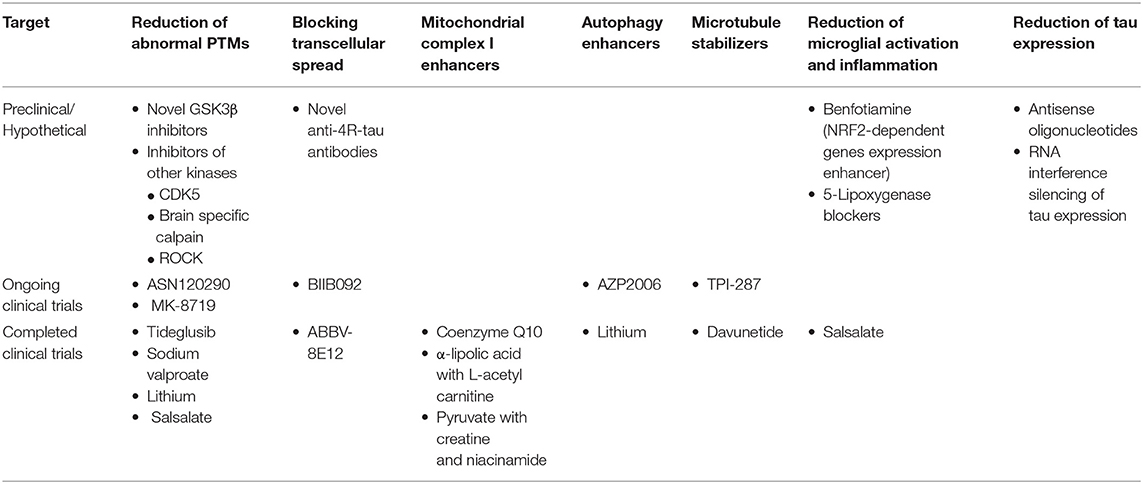
Table 3. Disease modifying therapeutic approaches for PSP based on their target etiopathogenic process.
Anti-tau antibodies are the most promising potential therapeutic strategies that are currently in clinical phase evaluation for PSP. Two humanized antibodies directed to different epitopes of extracellular tau, ABBV-8E12, and BIIB092, entered phase II with the hope to prevent the spread of tau pathology. ABBV-8E12 is a humanized antibody against extracellular fibrillar tau antibody designed to slow down the cell-to-cell spread of tau pathology. BIIB092 is directed to an N-terminally truncated form of extracellular tau. These agents showed no significant adverse events in the phase I trials and are being well-tolerated in ongoing phase II studies (140, 239). Unfortunately, ABBV-8E12 trial was recently discontinued due to lack of benefit.
MAPT gene silencing using antisense oligonucleotides or RNA interference are other promising future therapeutic strategies for tauopathies. IONIS-MAPTRx (BIIB080) is the only antisense oligonucleotide directed to the MAPT gene expression. It is currently under clinical evaluation in mild AD in a phase I/II study (ClinicalTrials.govNCT03186989) and is planned to enter phase II/III in FTLD patients.
Conclusion
PSP is a pathological entity with a wide range of presenting clinical features. It may present with symptoms similar to other neurodegenerative disorders including other atypical parkinsonisms, PD, frontotemporal lobar degeneration, and AD. Recent advances in terms of phenotypic and pathologic characterization, genetics, and molecular imaging have greatly increased our understanding of this unique disorder and have provided clues for the development of disease modifying treatments. Further knowledge about the mechanisms involved in its development, pathological alteration at the level of genes, RNA, tau protein regulation and new insights into the structural details of 4R tau fibrils and seeds, will pave the way for novel therapeutic approaches. Development of 4R tau PET ligands and accurate measures of 4R-tau in blood are of utmost importance for an early diagnosis and measure of disease progression in new therapeutic trials.
Our better understanding of the etiopathogenesis is being translated into experimental therapeutic trials with anti-tau antibodies (240, 241) and a number of other therapeutic modalities, including antisense oligonucleotides (242), tau post-translational modifiers (138), neuroprotective (233), and anti-inflammatory drugs (243). Hopefully soon these new approaches will also translate into clinical practice.
Disclosure
IL was supported by the National Institutes of Health grants: 5P50AG005131-33, 2R01AG038791-06A, U01NS090259, 1U54 NS 092089, U01NS100610, U01NS80818, R25NS098999, P20GM109025; Parkinson Study Group, Michael J Fox Foundation, Lewy Body Association, AVID Pharmaceuticals, Abbvie, Biogen and Roche. She was member of a Lundbeck Advisory Board and participated in a symposium organized by Sunovion. She receives her salary from the University of California, San Diego. She is Chief Editor of Frontiers in Neurology.
AS receives his salary from Mashhad University of Medical Sciences.
NO receives her salary from Mashhad University of Medical Sciences.
Author Contributions
AS and NO prepared the primary draft of the manuscript and figures. IL supervised the prepration of the primary draft, reviewed the manuscript draft, and improved the text. All authors approved the final draft.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AD, Alzheimer's disease; APOE, apolipoprotein E; CBD, corticobasal degeneration; CBD-CBS, corticobasal degeneration with corticobasal syndrome phenotype; CI, mitochondrial complex I; CNS, central nervous system; CSF, cerebrospinal fluid; ENGENE-PSP, environmental genetic PSP risk factor study; FTDP-17, frontotemporal degeneration with parkinsonism linked to chromosome 17; FTLD, frontotemporal lobar degeneration; GSK3, glycogen synthase kinase 3; GWAS, Genome-wide association study; MAO-A, monoamine oxidase A; MAPT, microtubule associated protein tau gene; MDS-PSP, International Parkinson and Movement Disorders Society PSP study group criteria; MRI, magnetic resonance imaging; MRPI, Magnetic resonance parkinsonism index; MSA-P, parkinsonism-predominant multiple system atrophy (); MT, microtubule; NfL, neurofilament light chain; NFTs, neurofibrillary tangles; NINDS-SPSP, National Institute of Neurological Disorders and Stroke and Society for PSP; O-GlcNAc, O-linked N-acetylglucosamine; PAD, phosphatase-activation domain; PD, Parkinson's disease; PERK, pancreatic endoplasmic reticulum kinase; PET, positron emission tomography; PiD, Pick's disease; PMCA, protein misfolding cyclic amplification; PSP, Progressive supranuclear palsy; PSP-C, PSP- cerebellar ataxia; PSP-CBS, PSP- corticobasal syndrome; PSP-F, PSP- behavioral variant of frontotemporal dementia; PSP-nfaPPA/AOS, PSP-non-fluent, agrammatic primary progressive aphasia/progressive apraxia of speech; PSP-OM, PSP-ocular motor; PSP-P, PSP-Parkinsonism; PSP-PGF, PSP- progressive gait freezing; PSP-PI, PSP-postural instability; PSP-PLS, PSP- primary lateral sclerosis; PSP-RS, PSP-Richardson syndrome; PSP-SL, PSP- speech language variant; PTMs, post-translational modifications; R2, repeat domain number 2 of the tau gene; RT-QuIC, real-time quaking-induced conversion; SNPs, single nucleotide polymorphisms; UPR, unfolded protein response; 3R-tau, tau containing 3 repeat domains; 4R-tau, tau containing 4 repeat domains.
References
1. Jellinger K. Progressive supranuclear palsy (subcortical argyrophilic dystrophy). Acta Neuropathol. (1971) 19:347–52. doi: 10.1007/BF00692156
2. Tellez-Nagel I, Wisniewski HM. Ultrastructure of neurofibrillary tangles in Steele-Richardson-Olszewski syndrome. Arch Neurol. (1973) 29:324–7. doi: 10.1001/archneur.1973.00490290064007
3. Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA. (1975) 72:1858–62. doi: 10.1073/pnas.72.5.1858
4. Lindwall G, Cole RD. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem. (1984) 259:5301–5.
5. Flament S, Delacourte A, Verny M, Hauw JJ, Javoy-Agid F. Abnormal Tau proteins in progressive supranuclear palsy. Similarities and differences with the neurofibrillary degeneration of the Alzheimer type. Acta Neuropathol. (1991) 81:591–6. doi: 10.1007/BF00296367
6. Conrad C, Andreadis A, Trojanowski JQ, Dickson DW, Kang D, Chen X, et al. Genetic evidence for the involvement of tau in progressive supranuclear palsy. Ann Neurol. (1997) 41:277–81. doi: 10.1002/ana.410410222
7. Mailliot C, Sergeant N, Bussiere T, Caillet-Boudin ML, Delacourte A, Buee L. Phosphorylation of specific sets of tau isoforms reflects different neurofibrillary degeneration processes. FEBS Lett. (1998) 433:201–4. doi: 10.1016/S0014-5793(98)00910-7
8. Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. (1998) 43:815–25. doi: 10.1002/ana.410430617
9. Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, et al. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. (1999) 8:711–5. doi: 10.1093/hmg/8.4.711
10. Nacharaju P, Lewis J, Easson C, Yen S, Hackett J, Hutton M, et al. Accelerated filament formation from tau protein with specific FTDP-17 missense mutations. FEBS Lett. (1999) 447:195–9. doi: 10.1016/S0014-5793(99)00294-X
11. Sergeant N, Wattez A, Delacourte A. Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: tau pathologies with exclusively “exon 10” isoforms. J Neurochem. (1999) 72:1243–9. doi: 10.1046/j.1471-4159.1999.0721243.x
12. Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. (2000) 25:402–5. doi: 10.1038/78078
13. Ishizawa K, Dickson DW. Microglial activation parallels system degeneration in progressive supranuclear palsy and corticobasal degeneration. J Neuropathol Exp Neurol. (2001) 60:647–57. doi: 10.1093/jnen/60.6.647
14. Borghi R, Giliberto L, Assini A, Delacourte A, Perry G, Smith MA, et al. Increase of cdk5 is related to neurofibrillary pathology in progressive supranuclear palsy. Neurology. (2002) 58:589–92. doi: 10.1212/WNL.58.4.589
15. Ferrer I, Barrachina M, Puig B. Glycogen synthase kinase-3 is associated with neuronal and glial hyperphosphorylated tau deposits in Alzheimer's disease, Pick's disease, progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol. (2002) 104:583–91. doi: 10.1007/s00401-002-0587-8
16. Gerhard A, Trender-Gerhard I, Turkheimer F, Quinn NP, Bhatia KP, Brooks DJ. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in progressive supranuclear palsy. Mov Disord. (2006) 21:89–93. doi: 10.1002/mds.20668
17. Padmanabhan J, Levy M, Dickson DW, Potter H. Alpha1-antichymotrypsin, an inflammatory protein overexpressed in Alzheimer's disease brain, induces tau phosphorylation in neurons. Brain. (2006) 129(Pt 11):3020–34. doi: 10.1093/brain/awl255
18. Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. (2007) 27:9115–29. doi: 10.1523/JNEUROSCI.2361-07.2007
19. Fernandez-Botran R, Ahmed Z, Crespo FA, Gatenbee C, Gonzalez J, Dickson DW, et al. Cytokine expression and microglial activation in progressive supranuclear palsy. Parkinsonism Relat Disord. (2011) 17:683–8. doi: 10.1016/j.parkreldis.2011.06.007
20. Irwin DJ, Cohen TJ, Grossman M, Arnold SE, Xie SX, Lee VM, et al. Acetylated tau, a novel pathological signature in Alzheimer's disease and other tauopathies. Brain. (2012) 135(Pt 3):807–18. doi: 10.1093/brain/aws013
21. Li Y, Chen JA, Sears RL, Gao F, Klein ED, Karydas A, et al. An epigenetic signature in peripheral blood associated with the haplotype on 17q21.31, a risk factor for neurodegenerative tauopathy. PLoS Genet. (2014) 10:e1004211. doi: 10.1371/journal.pgen.1004211
22. Coakeley S, Cho SS, Koshimori Y, Rusjan P, Harris M, Ghadery C, et al. Positron emission tomography imaging of tau pathology in progressive supranuclear palsy. J Cereb Blood Flow Metab. (2017) 37:3150–60. doi: 10.1177/0271678X16683695
23. Ono M, Sahara N, Kumata K, Ji B, Ni R, Koga S, et al. Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain. (2017) 140:764–80. doi: 10.1093/brain/aww339
24. Schonhaut DR, McMillan CT, Spina S, Dickerson BC, Siderowf A, Devous MD Sr, et al. (18) F-flortaucipir tau positron emission tomography distinguishes established progressive supranuclear palsy from controls and Parkinson disease: a multicenter study. Ann Neurol. (2017) 82:622–34. doi: 10.1002/ana.25060
25. Whitwell JL, Lowe VJ, Tosakulwong N, Weigand SD, Senjem ML, Schwarz CG, et al. [(18) F]AV-1451 tau positron emission tomography in progressive supranuclear palsy. Mov Disord. (2017) 32:124–33. doi: 10.1002/mds.26834
26. Bonham LW, Karch CM, Fan CC, Tan C, Geier EG, Wang Y, et al. CXCR4 involvement in neurodegenerative diseases. Transl Psychiatry. (2018) 8:73. doi: 10.1038/s41398-017-0049-7
27. Starhof C, Winge K, Heegaard NHH, Skogstrand K, Friis S, Hejl A. Cerebrospinal fluid pro-inflammatory cytokines differentiate parkinsonian syndromes. J Neuroinflammation. (2018) 15:305. doi: 10.1186/s12974-018-1339-6
28. Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol. (1964) 10:333–59. doi: 10.1001/archneur.1964.00460160003001
29. Albert ML, Feldman RG, Willis AL. The 'subcortical dementia' of progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. (1974) 37:121–30. doi: 10.1136/jnnp.37.2.121
30. Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. (1996) 47:1–9. doi: 10.1212/WNL.47.1.1
31. Homma Y, Takahashi H, Takeda S, Ikuta F. [An autopsy case of progressive supranuclear palsy showing “pure akinesia without rigidity and tremor and with no effect by L-dopa therapy”]. No To Shinkei. (1987) 39:183–7.
32. Imai H, Narabayashi H, Sakata E. “Pure akinesia” and the later added supranuclear ophthalmoplegia. Adv Neurol. (1987) 45:207–12.
33. Matsuo H, Takashima H, Kishikawa M, Kinoshita I, Mori M, Tsujihata M, et al. Pure akinesia: an atypical manifestation of progressive supranuclear palsy. J Neurol Neuosurg Psychiatry. (1991) 54:397–400. doi: 10.1136/jnnp.54.5.397
34. Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L, et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson's syndrome and PSP-parkinsonism. Brain. (2005) 128(Pt 6):1247–58. doi: 10.1093/brain/awh488
35. Hoglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord. (2017) 32:853–64. doi: 10.1002/mds.26987
36. Ali F, Botha H, Whitwell JL, Josephs KA. Utility of the movement disorders society criteria for progressive supranuclear palsy in clinical practice: clinical utility of PSP criteria. Mov Disord Clin Pract. (2019) 6:436–9. doi: 10.1002/mdc3.12807
37. Grimm MJ, Respondek G, Stamelou M, Arzberger T, Ferguson L, Gelpi E, et al. How to apply the movement disorder society criteria for diagnosis of progressive supranuclear palsy. Mov Disord. (2019) 34:1228–32. doi: 10.1002/mds.27666
38. Shoeibi A, Litvan I, Juncos JL, Riley DE, Bordelon Y, Standaert D, et al. Are the International Parkinson disease and Movement Disorder Society progressive supranuclear palsy (IPMDS-PSP) diagnostic criteria accurate enough to differentiate common PSP phenotypes? Parkinsonism Relat Disord (2019). doi: 10.1016/j.parkreldis.2019.10.012
39. Williams DR, Lees AJ. What features improve the accuracy of the clinical diagnosis of progressive supranuclear palsy-parkinsonism (PSP-P)? Mov Disord. (2010) 25:357–62. doi: 10.1002/mds.22977
40. Respondek G, Kurz C, Arzberger T, Compta Y, Englund E, Ferguson LW, et al. Which ante mortem clinical features predict progressive supranuclear palsy pathology? Mov Disord. (2017) 32:995–1005. doi: 10.1002/mds.27034
41. Williams DR, Holton JL, Strand K, Revesz T, Lees AJ. Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Mov Disord. (2007) 22:2235–41. doi: 10.1002/mds.21698
42. Han HJ, Kim H, Park J-H, Shin H-W, Kim GU, Kim DS, et al. Behavioral changes as the earliest clinical manifestation of progressive supranuclear palsy. J Clin Neurol. (2010) 6:148–51. doi: 10.3988/jcn.2010.6.3.148
43. Hassan A, Parisi JE, Josephs KA. Autopsy-proven progressive supranuclear palsy presenting as behavioral variant frontotemporal dementia. Neurocase. (2012) 18:478–88. doi: 10.1080/13554794.2011.627345
44. Josephs KA, Duffy JR. Apraxia of speech and nonfluent aphasia: a new clinical marker for corticobasal degeneration and progressive supranuclear palsy. Curr Opin Neurol. (2008) 21:688–92. doi: 10.1097/WCO.0b013e3283168ddd
45. Tsuboi Y, Josephs KA, Boeve BF, Litvan I, Caselli RJ, Caviness JN, et al. Increased tau burden in the cortices of progressive supranuclear palsy presenting with corticobasal syndrome. Mov Disord. (2005) 20:982–8. doi: 10.1002/mds.20478
46. Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR, et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology. (2006) 66:41–8. doi: 10.1212/01.wnl.0000191307.69661.c3
47. Ling H, Silva R, Massey L, Courtney R, Hondhamuni G, Bajaj N, et al. Characteristics of progressive supranuclear palsy presenting with corticobasal syndrome: a cortical variant. Neuropathol Appl Neurobiol. (2014) 40:149–63. doi: 10.1111/nan.12037
48. Eusebio A, Koric L, Felician O, Guedj E, Ceccaldi M, Azulay JP. Progressive supranuclear palsy and corticobasal degeneration: diagnostic challenges and clinicopathological considerations. Rev Neurol. (2016) 172:488–502. doi: 10.1016/j.neurol.2016.07.009
49. Lopez G, Bayulkem K, Hallett M. Progressive supranuclear palsy (PSP): Richardson syndrome and other PSP variants. Acta Neurol Scand. (2016) 134:242–9. doi: 10.1111/ane.12546
50. Pringle C, Hudson A, Munoz D, Kiernan J, Brown W, Ebers G. Primary lateral sclerosis: clinical features, neuropathology and diagnostic criteria. Brain. (1992) 115:495–520. doi: 10.1093/brain/115.2.495
51. Nagao S, Yokota O, Nanba R, Takata H, Haraguchi T, Ishizu H, et al. Progressive supranuclear palsy presenting as primary lateral sclerosis but lacking parkinsonism, gaze palsy, aphasia, or dementia. J Neurol Sci. (2012) 323:147–53. doi: 10.1016/j.jns.2012.09.005
52. Kanazawa M, Shimohata T, Toyoshima Y, Tada M, Kakita A, Morita T, et al. Cerebellar involvement in progressive supranuclear palsy: a clinicopathological study. Mov Disord. (2009) 24:1312–8. doi: 10.1002/mds.22583
53. Koga S, Aoki N, Uitti RJ, Van Gerpen JA, Cheshire WP, Josephs KA, et al. When DLB, PD, and PSP masquerade as MSA An autopsy study of 134 patients. Neurology. (2015) 85:404–12. doi: 10.1212/WNL.0000000000001807
54. Koga S, Josephs KA, Ogaki K, Labbé C, Uitti RJ, Graff-Radford N, et al. Cerebellar ataxia in progressive supranuclear palsy: an autopsy study of PSP-C. Mov Disord. (2016) 31:653–62. doi: 10.1002/mds.26499
55. Coyle-Gilchrist IT, Dick KM, Patterson K, Vazquez Rodriquez P, Wehmann E, Wilcox A, et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology. (2016) 86:1736–43. doi: 10.1212/WNL.0000000000002638
56. Glasmacher SA, Leigh PN, Saha RA. Predictors of survival in progressive supranuclear palsy and multiple system atrophy: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. (2017) 88:402–11. doi: 10.1136/jnnp-2016-314956
57. Moscovich M, Boschetti G, Moro A, Teive HA, Hassan A, Munhoz RP. Death certificate data and causes of death in patients with parkinsonism. Parkins Relat Disord. (2017) 41:99–103. doi: 10.1016/j.parkreldis.2017.05.022
58. Nath U, Ben-Shlomo Y, Thomson RG, Morris HR, Wood NW, Lees AJ, et al. The prevalence of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) in the UK. Brain. (2001) 124(Pt 7):1438–49. doi: 10.1093/brain/124.7.1438
59. Golbe LI. The epidemiology of progressive supranuclear palsy. Handb Clin Neurol. (2008) 89:457–9. doi: 10.1016/S0072-9752(07)01242-0
60. Takigawa H, Kitayama M, Wada-Isoe K, Kowa H, Nakashima K. Prevalence of progressive supranuclear palsy in Yonago: change throughout a decade. Brain Behav. (2016) 6:e00557. doi: 10.1002/brb3.557
61. Fleury V, Brindel P, Nicastro N, Burkhard PR. Descriptive epidemiology of parkinsonism in the Canton of Geneva, Switzerland. Parkins Relat Disord. (2018) 54:30–9. doi: 10.1016/j.parkreldis.2018.03.030
62. Dugger BN, Hentz JG, Adler CH, Sabbagh MN, Shill HA, Jacobson S, et al. Clinicopathological outcomes of prospectively followed normal elderly brain bank volunteers. J Neuropathol Exp Neurol. (2014) 73:244–52. doi: 10.1097/NEN.0000000000000046
63. Nogami A, Yamazaki M, Saito Y, Hatsuta H, Sakiyama Y, Takao M, et al. Early stage of progressive supranuclear palsy: a neuropathological study of 324 consecutive autopsy cases. J Nippon Med Sch. (2015) 82:266–73. doi: 10.1272/jnms.82.266
64. Yoshida K, Hata Y, Kinoshita K, Takashima S, Tanaka K, Nishida N. Incipient progressive supranuclear palsy is more common than expected and may comprise clinicopathological subtypes: a forensic autopsy series. Acta Neuropathol. (2017) 133:809–23. doi: 10.1007/s00401-016-1665-7
65. Lannuzel A, Ho glinger GU, Champy P, Michel PP, Hirsch EC, Ruberg M. Is atypical parkinsonism in the Caribbean caused by the consumption of Annonacae? J Neural Transm. (2006) 70(Suppl.):153–7. doi: 10.1007/978-3-211-45295-0_24
66. Caparros-Lefebvre D, Golbe LI, Deramecourt V, Maurage CA, Huin V, Buee-Scherrer V, et al. A geographical cluster of progressive supranuclear palsy in northern France. Neurology. (2015) 85:1293–300. doi: 10.1212/WNL.0000000000001997
67. Litvan I, Lees PS, Cunningham CR, Rai SN, Cambon AC, Standaert DG, et al. Environmental and occupational risk factors for progressive supranuclear palsy: case-control study. Mov Disord. (2016) 31:644–52. doi: 10.1002/mds.26512
68. Golbe LI, Rubin RS, Cody RP, Belsh JM, Duvoisin RC, Grosmann C, et al. Follow-up study of risk factors in progressive supranuclear palsy. Neurology. (1996) 47:148–54. doi: 10.1212/WNL.47.1.148
69. Vidal JS, Vidailhet M, Derkinderen P, de Gaillarbois TD, Tzourio C, Alperovitch A. Risk factors for progressive supranuclear palsy: a case-control study in France. J Neurol Neurosurg Psychiatry. (2009) 80:1271–4. doi: 10.1136/jnnp.2008.149849
70. Kelley KD, Checkoway H, Hall DA, Reich SG, Cunningham C, Litvan I. Traumatic brain injury and firearm use and risk of progressive supranuclear palsy among veterans. Front Neurol. (2018) 9:474. doi: 10.3389/fneur.2018.00474
71. Park HK, Ilango S, Charriez CM, Checkoway H, Riley D, Standaert DG, et al. Lifetime exposure to estrogen and progressive supranuclear palsy: environmental and Genetic PSP study. Mov Disord. (2018) 33:468–72. doi: 10.1002/mds.27336
72. Kelley KD, Peavy G, Edland S, Rogers W, Riley DE, Bordelon Y, et al. The role of stress as a risk factor for progressive supranuclear palsy. J Parkinsons Dis. (2017) 7:377–83. doi: 10.3233/JPD-160945
73. Vanacore N, Bonifati V, Fabbrini G, Colosimo C, Marconi R, Nicholl D, et al. Smoking habits in multiple system atrophy and progressive supranuclear palsy. European Study Group on Atypical Parkinsonisms. Neurology. (2000) 54:114–9. doi: 10.1212/WNL.54.1.114
74. Davis PH, Golbe LI, Duvoisin RC, Schoenberg BS. Risk factors for progressive supranuclear palsy. Neurology. (1988) 38:1546–52. doi: 10.1212/WNL.38.10.1546
75. Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, et al. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology. (1994) 44:2015–9. doi: 10.1212/WNL.44.11.2015
76. Litvan I, Hauw JJ, Bartko JJ, Lantos PL, Daniel SE, Horoupian DS, et al. Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol. (1996) 55:97–105. doi: 10.1097/00005072-199601000-00010
77. Komori T, Arai N, Oda M, Nakayama H, Mori H, Yagishita S, et al. Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. (1998) 96:401–8. doi: 10.1007/s004010050911
78. Brion JP, Passareiro H, Nunez J, Flament-Durand J. Immunological detection of tau protein in neurofibrillary tangles of Alzheimer's disease. Arch Biol. (1985) 96:229–35.
79. Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci USA. (1988) 85:4051–5. doi: 10.1073/pnas.85.11.4051
80. Foster NL, Wilhelmsen K, Sima AA, Jones MZ, D'Amato CJ, Gilman S. Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Conference Participants. Ann Neurol. (1997) 41:706–15. doi: 10.1002/ana.410410606
81. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. (1998) 393:702–5. doi: 10.1038/31508
82. Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. (1989) 3:519–26. doi: 10.1016/0896-6273(89)90210-9
83. Schoch KM, DeVos SL, Miller RL, Chun SJ, Norrbom M, Wozniak DF, et al. Increased 4R-tau induces pathological changes in a human-tau mouse model. Neuron. (2016) 90:941–7. doi: 10.1016/j.neuron.2016.04.042
84. Arima K. Ultrastructural characteristics of tau filaments in tauopathies: immuno-electron microscopic demonstration of tau filaments in tauopathies. Neuropathology. (2006) 26:475–83. doi: 10.1111/j.1440-1789.2006.00669.x
85. Nishimura M, Namba Y, Ikeda K, Oda M. Glial fibrillary tangles with straight tubules in the brains of patients with progressive supranuclear palsy. Neurosci Lett. (1992) 143:35–8. doi: 10.1016/0304-3940(92)90227-X
86. Dickson DW, Rademakers R, Hutton ML. Progressive supranuclear palsy: pathology and genetics. Brain Pathol. (2007) 17:74–82. doi: 10.1111/j.1750-3639.2007.00054.x
87. Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol. (2009) 8:270–9. doi: 10.1016/S1474-4422(09)70042-0
88. Armstrong MJ. Progressive Supranuclear Palsy: an Update. Curr Neurol Neurosci Rep. (2018) 18:12. doi: 10.1007/s11910-018-0819-5
89. Dickson DW. Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol. (1999) 246(Suppl. 2):II6–15. doi: 10.1007/BF03161076
90. Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. (1992) 8:159–68. doi: 10.1016/0896-6273(92)90117-V
91. Kidd M. Paired helical filaments in electron microscopy of Alzheimer's disease. Nature. (1963) 197:192–3. doi: 10.1038/197192b0
92. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. (1991) 82:239–59. doi: 10.1007/BF00308809
93. Poorkaj P, Muma NA, Zhukareva V, Cochran EJ, Shannon KM, Hurtig H, et al. An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol. (2002) 52:511–6. doi: 10.1002/ana.10340
94. Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell. (1992) 3:1141–54. doi: 10.1091/mbc.3.10.1141
95. Kanaan NM, Morfini G, Pigino G, LaPointe NE, Andreadis A, Song Y, et al. Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. Neurobiol Aging. (2012) 33:826.e815–30. doi: 10.1016/j.neurobiolaging.2011.06.006
96. Wei Y, Qu MH, Wang XS, Chen L, Wang DL, Liu Y, et al. Binding to the minor groove of the double-strand, tau protein prevents DNA from damage by peroxidation. PLoS ONE. (2008) 3:e2600. doi: 10.1371/journal.pone.0002600
97. Schwalbe M, Ozenne V, Bibow S, Jaremko M, Jaremko L, Gajda M, et al. Predictive atomic resolution descriptions of intrinsically disordered hTau40 and alpha-synuclein in solution from NMR and small angle scattering. Structure. (2014) 22:238–49. doi: 10.1016/j.str.2013.10.020
98. Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. (2015) 16:18–29. doi: 10.1038/nrm3920
99. Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. (2006) 45:2283–93. doi: 10.1021/bi0521543
100. Wang Y, Garg S, Mandelkow EM, Mandelkow E. Proteolytic processing of tau. Biochem Soc Trans. (2010) 38:955–61. doi: 10.1042/BST0380955
101. Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. (2017) 133:665–704. doi: 10.1007/s00401-017-1707-9
102. Stefansson H, Helgason A, Thorleifsson G, Steinthorsdottir V, Masson G, Barnard J, et al. A common inversion under selection in Europeans. Nat Genet. (2005) 37:129–37. doi: 10.1038/ng1508
103. Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. (1997) 278:1349–56. doi: 10.1001/jama.278.16.1349
104. Pittman AM, Myers AJ, Abou-Sleiman P, Fung HC, Kaleem M, Marlowe L, et al. Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J Med Genet. (2005) 42:837–46. doi: 10.1136/jmg.2005.031377
105. Pittman AM, Fung HC, de Silva R. Untangling the tau gene association with neurodegenerative disorders. Hum Mol Genet. (2006) 15:R188–95. doi: 10.1093/hmg/ddl190
106. Zhang CC, Zhu JX, Wan Y, Tan L, Wang HF, Yu JT, et al. Meta-analysis of the association between variants in MAPT and neurodegenerative diseases. Oncotarget. (2017) 8:44994–007. doi: 10.18632/oncotarget.16690
107. Hoglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. (2011) 43:699–705. doi: 10.1038/ng.859
108. Kouri N, Ross OA, Dombroski B, Younkin CS, Serie DJ, Soto-Ortolaza A, et al. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun. (2015) 6:7247. doi: 10.1038/ncomms8247
109. Heckman MG, Brennan RR, Labbe C, Soto AI, Koga S, DeTure MA, et al. Association of MAPT subhaplotypes with risk of progressive supranuclear palsy and severity of tau pathology. JAMA Neurol. (2019) 76:710–7. doi: 10.1001/jamaneurol.2019.0250
110. Baba Y, Tsuboi Y, Baker MC, Uitti RJ, Hutton ML, Dickson DW, et al. The effect of tau genotype on clinical features in FTDP-17. Parkinsonism Relat Disord. (2005) 11:205–8. doi: 10.1016/j.parkreldis.2005.01.003
111. Myers AJ, Pittman AM, Zhao AS, Rohrer K, Kaleem M, Marlowe L, et al. The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol Dis. (2007) 25:561–70. doi: 10.1016/j.nbd.2006.10.018
112. Allen M, Burgess JD, Ballard T, Serie D, Wang X, Younkin CS, et al. Gene expression, methylation and neuropathology correlations at progressive supranuclear palsy risk loci. Acta Neuropathol. (2016) 132:197–211. doi: 10.1007/s00401-016-1576-7
113. Huin V, Deramecourt V, Caparros-Lefebvre D, Maurage CA, Duyckaerts C, Kovari E, et al. The MAPT gene is differentially methylated in the progressive supranuclear palsy brain. Mov Disord. (2016) 31:1883–90. doi: 10.1002/mds.26820
114. Chen J, Yu JT, Wojta K, Wang HF, Zetterberg H, Blennow K, et al. Genome-wide association study identifies MAPT locus influencing human plasma tau levels. Neurology. (2017) 88:669–76. doi: 10.1212/WNL.0000000000003615
115. Coppola G, Chinnathambi S, Lee JJ, Dombroski BA, Baker MC, Soto-Ortolaza AI, et al. Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer's diseases. Hum Mol Genet. (2012) 21:3500–12. doi: 10.1093/hmg/dds161
116. Jung JJ, Inamdar SM, Tiwari A, Choudhury A. Regulation of intracellular membrane trafficking and cell dynamics by syntaxin-6. Biosci Rep. (2012) 32:383–91. doi: 10.1042/BSR20120006
117. Nijholt DA, de Graaf TR, van Haastert ES, Oliveira AO, Berkers CR, Zwart R, et al. Endoplasmic reticulum stress activates autophagy but not the proteasome in neuronal cells: implications for Alzheimer's disease. Cell Death Differ. (2011) 18:1071–81. doi: 10.1038/cdd.2010.176
118. Stutzbach LD, Xie SX, Naj AC, Albin R, Gilman S, Group PSPGS, et al. The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer's disease. Acta Neuropathol Commun. (2013) 1:31. doi: 10.1186/2051-5960-1-31
119. Yuan SH, Hiramatsu N, Liu Q, Sun XV, Lenh D, Chan P, et al. Tauopathy-Associated PERK alleles are functional hypomorphs that increase neuronal vulnerability to ER stress. Hum Mol Genet. (2018) 28:584–97. doi: 10.1093/hmg/ddy297
120. Montague P, McCallion AS, Davies RW, Griffiths IR. Myelin-associated oligodendrocytic basic protein: a family of abundant CNS myelin proteins in search of a function. Dev Neurosci. (2006) 28:479–87. doi: 10.1159/000095110
121. Chen JA, Chen Z, Won H, Huang AY, Lowe JK, Wojta K, et al. Joint genome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. Mol Neurodegener. (2018) 13:41. doi: 10.1186/s13024-018-0270-8
122. Sanchez-Contreras MY, Kouri N, Cook CN, Serie DJ, Heckman MG, Finch NA, et al. Replication of progressive supranuclear palsy genome-wide association study identifies SLCO1A2 and DUSP10 as new susceptibility loci. Mol Neurodegenerat. (2018) 13:37. doi: 10.1186/s13024-018-0267-3
123. Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. (2005) 309:476–81. doi: 10.1126/science.1113694
124. Trojanowski JQ, Lee VM. Pathological tau: a loss of normal function or a gain in toxicity? Nat Neurosci. (2005) 8:1136–7. doi: 10.1038/nn0905-1136
125. Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, et al. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. (2001) 293:711–4. doi: 10.1126/science.1062382
126. Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, et al. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci. (2011) 31:2511–25. doi: 10.1523/JNEUROSCI.5245-10.2011
127. Congdon EE, Duff KE. Is tau aggregation toxic or protective? J Alzheimers Dis. (2008) 14:453–7. doi: 10.3233/JAD-2008-14415
128. Cowan CM, Mudher A. Are tau aggregates toxic or protective in tauopathies? Front Neurol. (2013) 4:114. doi: 10.3389/fneur.2013.00114
129. Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. (2016) 17:5–21. doi: 10.1038/nrn.2015.1
130. Kanaan NM, Himmelstein DS, Ward SM, Combs B, Binder LI. Chapter 56 - tau protein: biology and pathobiology. In: LeDoux MS, editors. Movement Disorders. 2nd ed. Boston, MA: Academic Press (2015). p. 857–74. doi: 10.1016/B978-0-12-405195-9.00056-1
131. Hanger DP, Byers HL, Wray S, Leung KY, Saxton MJ, Seereeram A, et al. Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J Biol Chem. (2007) 282:23645–54. doi: 10.1074/jbc.M703269200
132. Wray S, Saxton M, Anderton BH, Hanger DP. Direct analysis of tau from PSP brain identifies new phosphorylation sites and a major fragment of N-terminally cleaved tau containing four microtubule-binding repeats. J Neurochem. (2008) 105:2343–52. doi: 10.1111/j.1471-4159.2008.05321.x
133. Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. (2009) 15:112–9. doi: 10.1016/j.molmed.2009.01.003
134. Kanaan NM, Morfini GA, LaPointe NE, Pigino GF, Patterson KR, Song Y, et al. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J Neurosci. (2011) 31:9858–68. doi: 10.1523/JNEUROSCI.0560-11.2011
135. Liu F, Li B, Tung EJ, Grundke-Iqbal I, Iqbal K, Gong CX. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur J Neurosci. (2007) 26:3429–36. doi: 10.1111/j.1460-9568.2007.05955.x
136. Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. (2010) 68:1067–81. doi: 10.1016/j.neuron.2010.11.030
137. Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. (2007) 117:648–58. doi: 10.1172/JCI29715
138. Tolosa E, Litvan I, Hoglinger GU, Burn D, Lees A, Andres MV, et al. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov Disord. (2014) 29:470–8. doi: 10.1002/mds.25824
139. Leclair-Visonneau L, Rouaud T, Debilly B, Durif F, Houeto JL, Kreisler A, et al. Randomized placebo-controlled trial of sodium valproate in progressive supranuclear palsy. Clin Neurol Neurosurg. (2016) 146:35–9. doi: 10.1016/j.clineuro.2016.04.021
140. Shoeibi A, Olfati N, Litvan I. Preclinical, phase I, and phase II investigational clinical trials for treatment of progressive supranuclear palsy. Expert Opin Investig Drugs. (2018) 27:349–61. doi: 10.1080/13543784.2018.1460356
141. Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. (2010) 67:953–66. doi: 10.1016/j.neuron.2010.08.044
142. Cook C, Carlomagno Y, Gendron TF, Dunmore J, Scheffel K, Stetler C, et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum Mol Genet. (2014) 23:104–16. doi: 10.1093/hmg/ddt402
143. Cohen TJ, Constance BH, Hwang AW, James M, Yuan CX. Intrinsic tau acetylation is coupled to auto-proteolytic tau fragmentation. PLoS ONE. (2016) 11:e0158470. doi: 10.1371/journal.pone.0158470
144. Tracy TE, Sohn PD, Minami SS, Wang C, Min SW, Li Y, et al. Acetylated tau obstructs KIBRA-mediated signaling in synaptic plasticity and promotes tauopathy-related memory loss. Neuron. (2016) 90:245–60. doi: 10.1016/j.neuron.2016.03.005
145. Ferreon JC, Jain A, Choi KJ, Tsoi PS, MacKenzie KR, Jung SY, et al. Acetylation disfavors tau phase separation. Int J Mol Sci. (2018) 19:E1360. doi: 10.3390/ijms19051360
146. Haj-Yahya M, Lashuel HA. Protein semisynthesis provides access to tau disease-associated Post-translational Modifications (PTMs) and paves the way to deciphering the tau PTM code in health and diseased states. J Am Chem Soc. (2018) 140:6611–21. doi: 10.1021/jacs.8b02668
147. Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc Natl Acad Sci USA. (2004) 101:10804–9. doi: 10.1073/pnas.0400348101
148. Smet-Nocca C, Broncel M, Wieruszeski JM, Tokarski C, Hanoulle X, Leroy A, et al. Identification of O-GlcNAc sites within peptides of the Tau protein and their impact on phosphorylation. Mol Biosyst. (2011) 7:1420–9. doi: 10.1039/c0mb00337a
149. Liu F, Shi J, Tanimukai H, Gu J, Gu J, Grundke-Iqbal I, et al. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer's disease. Brain. (2009) 132(Pt 7):1820–32. doi: 10.1093/brain/awp099
150. Quinn JP, Corbett NJ, Kellett KAB, Hooper NM. Tau proteolysis in the pathogenesis of tauopathies: neurotoxic fragments and novel biomarkers. J Alzheimers Dis. (2018) 63:13–33. doi: 10.3233/JAD-170959
151. Jabbari E, Zetterberg H, Morris HR. Tracking and predicting disease progression in progressive supranuclear palsy: CSF and blood biomarkers. J Neurol Neurosurg Psychiatry. (2017) 88:883–8. doi: 10.1136/jnnp-2017-315857
152. Mimaki M, Wang X, McKenzie M, Thorburn DR, Ryan MT. Understanding mitochondrial complex I assembly in health and disease. Biochim Biophys Acta. (2012) 1817:851–62. doi: 10.1016/j.bbabio.2011.08.010
153. Kotake Y, Yoshida M, Ogawa M, Tasaki Y, Hirobe M, Ohta S. Chronic administration of 1-benzyl-1,2,3,4-tetrahydroisoquinoline, an endogenous amine in the brain, induces parkinsonism in a primate. Neurosci Lett. (1996) 217:69–71. doi: 10.1016/0304-3940(96)13065-2
154. Kawai H, Makino Y, Hirobe M, Ohta S. Novel endogenous 1,2,3,4-tetrahydroisoquinoline derivatives: uptake by dopamine transporter and activity to induce parkinsonism. J Neurochem. (1998) 70:745–51. doi: 10.1046/j.1471-4159.1998.70020745.x
155. Caparros-Lefebvre D, Elbaz A. Possible relation of atypical parkinsonism in the French West Indies with consumption of tropical plants: a case-control study. Caribbean Parkinsonism Study Group. Lancet. (1999) 354:281–6. doi: 10.1016/S0140-6736(98)10166-6
156. Caparros-Lefebvre D, Sergeant N, Lees A, Camuzat A, Daniel S, Lannuzel A, et al. Guadeloupean parkinsonism: a cluster of progressive supranuclear palsy-like tauopathy. Brain. (2002) 125(Pt 4):801–11. doi: 10.1093/brain/awf086
157. Swerdlow RH, Golbe LI, Parks JK, Cassarino DS, Binder DR, Grawey AE, et al. Mitochondrial dysfunction in cybrid lines expressing mitochondrial genes from patients with progressive supranuclear palsy. J Neurochem. (2000) 75:1681–4. doi: 10.1046/j.1471-4159.2000.0751681.x
158. Blin J, Baron JC, Dubois B, Pillon B, Cambon H, Cambier J, et al. Positron emission tomography study in progressive supranuclear palsy. Brain hypometabolic pattern and clinicometabolic correlations. Arch Neurol. (1990) 47:747–52. doi: 10.1001/archneur.1990.00530070035009
159. Martinelli P, Scaglione C, Lodi R, Iotti S, Barbiroli B. Deficit of brain and skeletal muscle bioenergetics in progressive supranuclear palsy shown in vivo by phosphorus magnetic resonance spectroscopy. Mov Disord. (2000) 15:889–93. doi: 10.1002/1531-8257(200009)15:5<889::AID-MDS1020>3.0.CO;2-T
160. Stamelou M, Pilatus U, Reuss A, Magerkurth J, Eggert KM, Knake S, et al. In vivo evidence for cerebral depletion in high-energy phosphates in progressive supranuclear palsy. J Cereb Blood Flow Metab. (2009) 29:861–70. doi: 10.1038/jcbfm.2009.2
161. Albers DS, Augood SJ, Martin DM, Standaert DG, Vonsattel JP, Beal MF. Evidence for oxidative stress in the subthalamic nucleus in progressive supranuclear palsy. J Neurochem. (1999) 73:881–4. doi: 10.1046/j.1471-4159.1999.0730881.x
162. Albers DS, Augood SJ, Park LC, Browne SE, Martin DM, Adamson J, et al. Frontal lobe dysfunction in progressive supranuclear palsy: evidence for oxidative stress and mitochondrial impairment. J Neurochem. (2000) 74:878–81. doi: 10.1046/j.1471-4159.2000.740878.x
163. Odetti P, Garibaldi S, Norese R, Angelini G, Marinelli L, Valentini S, et al. Lipoperoxidation is selectively involved in progressive supranuclear palsy. J Neuropathol Exp Neurol. (2000) 59:393–7. doi: 10.1093/jnen/59.5.393
164. Stamelou M, Reuss A, Pilatus U, Magerkurth J, Niklowitz P, Eggert KM, et al. Short-Term Effects of Coenzyme Q10 in Progressive Supranuclear Palsy: a Randomized, Placebo-Controlled Trial. Mov Disord. (2008) 23:942–9. doi: 10.1002/mds.22023
165. Apetauerova D, Scala SA, Hamill RW, Simon DK, Pathak S, Ruthazer R, et al. CoQ10 in progressive supranuclear palsy: a randomized, placebo-controlled, double-blind trial. Neurol Neuroimmunol Neuroinflamm. (2016) 3:e266. doi: 10.1212/NXI.0000000000000266
166. Giannopoulos PF, Chu J, Sperow M, Li JG, Yu WH, Kirby LG, et al. Pharmacologic inhibition of 5-lipoxygenase improves memory, rescues synaptic dysfunction, and ameliorates tau pathology in a transgenic model of tauopathy. Biol Psychiatry. (2015) 78:693–701. doi: 10.1016/j.biopsych.2015.01.015
167. Marras C, Cunningham CR, Hou J, Proudfoot J, Standaert DG, Juncos J, et al. Anti-inflammatory drug use and progressive supranuclear palsy. Parkins Relat Disord. (2018) 48:89–92. doi: 10.1016/j.parkreldis.2017.11.346
168. Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. (2009) 11:909–13. doi: 10.1038/ncb1901
169. Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. (2011) 286:15317–31. doi: 10.1074/jbc.M110.209296
170. Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA. (2013) 110:9535–40. doi: 10.1073/pnas.1301175110
171. Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's-like tauopathy. J Neurosci. (2013) 33:1024–37. doi: 10.1523/JNEUROSCI.2642-12.2013
172. Vella LJ, Sharples RA, Nisbet RM, Cappai R, Hill AF. The role of exosomes in the processing of proteins associated with neurodegenerative diseases. Eur Biophys J. (2008) 37:323–32. doi: 10.1007/s00249-007-0246-z
173. Holmes BB, Diamond MI. Prion-like properties of Tau protein: the importance of extracellular Tau as a therapeutic target. J Biol Chem. (2014) 289:19855–61. doi: 10.1074/jbc.R114.549295
174. Abounit S, Wu JW, Duff K, Victoria GS, Zurzolo C. Tunneling nanotubes: a possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion. (2016) 10:344–51. doi: 10.1080/19336896.2016.1223003
175. Katsinelos T, Zeitler M, Dimou E, Karakatsani A, Muller HM, Nachman E, et al. Unconventional secretion mediates the trans-cellular spreading of tau. Cell Rep. (2018) 23:2039–55. doi: 10.1016/j.celrep.2018.04.056
176. Falcon B, Cavallini A, Angers R, Glover S, Murray TK, Barnham L, et al. Conformation determines the seeding potencies of native and recombinant Tau aggregates. J Biol Chem. (2015) 290:1049–65. doi: 10.1074/jbc.M114.589309
177. Jackson SJ, Kerridge C, Cooper J, Cavallini A, Falcon B, Cella CV, et al. Short fibrils constitute the major species of seed-competent tau in the brains of mice transgenic for human P301S Tau. J Neurosci. (2016) 36:762–72. doi: 10.1523/JNEUROSCI.3542-15.2016
178. Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, et al. Cryo-EM structures of tau filaments from Alzheimer's disease. Nature. (2017) 547:185–90. doi: 10.1038/nature23002
179. Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, et al. Structures of filaments from Pick's disease reveal a novel tau protein fold. Nature. (2018) 561:137–40. doi: 10.1038/s41586-018-0454-y
180. Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature. (2019) 568:420–3. doi: 10.1038/s41586-019-1026-5
181. Litvan I. Atypical Parkinsonian Disorders: Clinical and Research Aspects. 1 ed. Totowa, N.J.: Humana Press (2005). doi: 10.1385/1-59259-834-X:001
182. Whitwell JL, Hoglinger GU, Antonini A, Bordelon Y, Boxer AL, Colosimo C, et al. Radiological biomarkers for diagnosis in PSP: where are we and where do we need to be? Mov Disord. (2017) 32:955–71. doi: 10.1002/mds.27038
183. Whitwell JL, Jack CR Jr, Parisi JE, Gunter JL, Weigand SD, Boeve BF, et al. Midbrain atrophy is not a biomarker of progressive supranuclear palsy pathology. Eur J Neurol. (2013) 20:1417–22. doi: 10.1111/ene.12212
184. Quattrone A, Nicoletti G, Messina D, Fera F, Condino F, Pugliese P, et al. MR imaging index for differentiation of progressive supranuclear palsy from Parkinson disease and the Parkinson variant of multiple system atrophy. Radiology. (2008) 246:214–21. doi: 10.1148/radiol.2453061703
185. Morelli M, Arabia G, Novellino F, Salsone M, Giofre L, Condino F, et al. MRI measurements predict PSP in unclassifiable parkinsonisms: a cohort study. Neurology. (2011) 77:1042–7. doi: 10.1212/WNL.0b013e31822e55d0
186. Zanigni S, Calandra-Buonaura G, Manners DN, Testa C, Gibertoni D, Evangelisti S, et al. Accuracy of MR markers for differentiating Progressive Supranuclear Palsy from Parkinson's disease. Neuroimage Clin. (2016) 11:736–42. doi: 10.1016/j.nicl.2016.05.016
187. Nigro S, Arabia G, Antonini A, Weis L, Marcante A, Tessitore A, et al. Magnetic Resonance Parkinsonism Index: diagnostic accuracy of a fully automated algorithm in comparison with the manual measurement in a large Italian multicentre study in patients with progressive supranuclear palsy. Eur Radiol. (2017) 27:2665–75. doi: 10.1007/s00330-016-4622-x
188. Quattrone A, Morelli M, Nigro S, Quattrone A, Vescio B, Arabia G, et al. A new MR imaging index for differentiation of progressive supranuclear palsy-parkinsonism from Parkinson's disease. Parkins Relat Disord. (2018) 54:3–8. doi: 10.1016/j.parkreldis.2018.07.016
189. Stezin A, Lenka A, Jhunjhunwala K, Saini J, Pal PK. Advanced structural neuroimaging in progressive supranuclear palsy: where do we stand? Parkins Relat Disord. (2017) 36:19–32. doi: 10.1016/j.parkreldis.2016.12.023
190. Tuch DS, Reese TG, Wiegell MR, Makris N, Belliveau JW, Wedeen VJ. High angular resolution diffusion imaging reveals intravoxel white matter fiber heterogeneity. Magn Reson Med. (2002) 48:577–82. doi: 10.1002/mrm.10268
191. Meijer FJ, van Rumund A, Tuladhar AM, Aerts MB, Titulaer I, Esselink RA, et al. Conventional 3T brain MRI and diffusion tensor imaging in the diagnostic workup of early stage parkinsonism. Neuroradiology. (2015) 57:655–69. doi: 10.1007/s00234-015-1515-7
192. Massey LA, Micallef C, Paviour DC, O'Sullivan SS, Ling H, Williams DR, et al. Conventional magnetic resonance imaging in confirmed progressive supranuclear palsy and multiple system atrophy. Mov Disord. (2012) 27:1754–62. doi: 10.1002/mds.24968
193. Massey LA, Jager HR, Paviour DC, O'Sullivan SS, Ling H, Williams DR, et al. The midbrain to pons ratio: a simple and specific MRI sign of progressive supranuclear palsy. Neurology. (2013) 80:1856–61. doi: 10.1212/WNL.0b013e318292a2d2
194. Planetta PJ, Ofori E, Pasternak O, Burciu RG, Shukla P, DeSimone JC, et al. Free-water imaging in Parkinson's disease and atypical parkinsonism. Brain. (2016) 139(Pt 2):495–508. doi: 10.1093/brain/awv361
195. Ito K, Ohtsuka C, Yoshioka K, Kameda H, Yokosawa S, Sato R, et al. Differential diagnosis of parkinsonism by a combined use of diffusion kurtosis imaging and quantitative susceptibility mapping. Neuroradiology. (2017) 59:759–69. doi: 10.1007/s00234-017-1870-7
196. Blain CR, Barker GJ, Jarosz JM, Coyle NA, Landau S, Brown RG, et al. Measuring brain stem and cerebellar damage in parkinsonian syndromes using diffusion tensor MRI. Neurology. (2006) 67:2199–205. doi: 10.1212/01.wnl.0000249307.59950.f8
197. Nicoletti G, Tonon C, Lodi R, Condino F, Manners D, Malucelli E, et al. Apparent diffusion coefficient of the superior cerebellar peduncle differentiates progressive supranuclear palsy from Parkinson's disease. Mov Disord. (2008) 23:2370–6. doi: 10.1002/mds.22279
198. Rizzo G, Martinelli P, Manners D, Scaglione C, Tonon C, Cortelli P, et al. Diffusion-weighted brain imaging study of patients with clinical diagnosis of corticobasal degeneration, progressive supranuclear palsy and Parkinson's disease. Brain. (2008) 131(Pt 10):2690–700. doi: 10.1093/brain/awn195
199. Padovani A, Borroni B, Brambati SM, Agosti C, Broli M, Alonso R, et al. Diffusion tensor imaging and voxel based morphometry study in early progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. (2006) 77:457–63. doi: 10.1136/jnnp.2005.075713
200. Josephs KA, Whitwell JL, Dickson DW, Boeve BF, Knopman DS, Petersen RC, et al. Voxel-based morphometry in autopsy proven PSP and CBD. Neurobiol Aging. (2008) 29:280–289. doi: 10.1016/j.neurobiolaging.2006.09.019
201. Agosta F, Pievani M, Svetel M, Jecmenica Lukic M, Copetti M, Tomic A, et al. Diffusion tensor MRI contributes to differentiate Richardson's syndrome from PSP-parkinsonism. Neurobiol Aging. (2012) 33:2817–26. doi: 10.1016/j.neurobiolaging.2012.02.002
202. Saini J, Bagepally BS, Sandhya M, Pasha SA, Yadav R, Pal PK. In vivo evaluation of white matter pathology in patients of progressive supranuclear palsy using TBSS. Neuroradiology. (2012) 54:771–80. doi: 10.1007/s00234-011-0983-7
203. Hong JY, Yun HJ, Sunwoo MK, Ham JH, Lee JM, Sohn YH, et al. Comparison of regional brain atrophy and cognitive impairment between pure akinesia with gait freezing and Richardson's syndrome. Front Aging Neurosci. (2015) 7:180. doi: 10.3389/fnagi.2015.00180
204. Quattrone A, Morelli M, Williams DR, Vescio B, Arabia G, Nigro S, et al. MR parkinsonism index predicts vertical supranuclear gaze palsy in patients with PSP-parkinsonism. Neurology. (2016) 87:1266–73. doi: 10.1212/WNL.0000000000003125
205. Saint-Aubert L, Lemoine L, Chiotis K, Leuzy A, Rodriguez-Vieitez E, Nordberg A. Tau PET imaging: present and future directions. Mol Neurodegener. (2017) 12:19. doi: 10.1186/s13024-017-0162-3
206. Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. (2013) 79:1094–108. doi: 10.1016/j.neuron.2013.07.037
207. Lowe VJ, Curran G, Fang P, Liesinger AM, Josephs KA, Parisi JE, et al. An autoradiographic evaluation of AV-1451 Tau PET in dementia. Acta Neuropathol Commun. (2016) 4:58. doi: 10.1186/s40478-016-0315-6
208. Cho H, Choi JY, Hwang MS, Lee SH, Ryu YH, Lee MS, et al. Subcortical (18) F-AV-1451 binding patterns in progressive supranuclear palsy. Mov Disord. (2017) 32:134–40. doi: 10.1002/mds.26844
209. Passamonti L, Vazquez Rodriguez P, Hong YT, Allinson KS, Williamson D, Borchert RJ, et al. 18F-AV-1451 positron emission tomography in Alzheimer's disease and progressive supranuclear palsy. Brain. (2017) 140:781–91. doi: 10.1093/brain/aww340
210. Smith R, Schain M, Nilsson C, Strandberg O, Olsson T, Hagerstrom D, et al. Increased basal ganglia binding of (18) F-AV-1451 in patients with progressive supranuclear palsy. Mov Disord. (2017) 32:108–14. doi: 10.1002/mds.26813
211. Smith R, Scholl M, Honer M, Nilsson CF, Englund E, Hansson O. Tau neuropathology correlates with FDG-PET, but not AV-1451-PET, in progressive supranuclear palsy. Acta Neuropathol. (2017) 133:149–51. doi: 10.1007/s00401-016-1650-1
212. Whitwell JL, Ahlskog JE, Tosakulwong N, Senjem ML, Spychalla AJ, Petersen RC, et al. Pittsburgh Compound B and AV-1451 positron emission tomography assessment of molecular pathologies of Alzheimer's disease in progressive supranuclear palsy. Parkins Relat Disord. (2018) 48:3–9. doi: 10.1016/j.parkreldis.2017.12.016
213. Jones DT, Knopman DS, Graff-Radford J, Syrjanen JA, Senjem ML, Schwarz CG, et al. In vivo (18)F-AV-1451 tau PET signal in MAPT mutation carriers varies by expected tau isoforms. Neurology. (2018) 90:e947–54. doi: 10.1212/WNL.0000000000005117
214. Marquie M, Normandin MD, Vanderburg CR, Costantino IM, Bien EA, Rycyna LG, et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol. (2015) 78:787–800. doi: 10.1002/ana.24517
215. Whitwell JL, Tosakulwong N, Schwarz CG, Botha H, Senjem ML, Spychalla AJ, et al. MRI outperforms [18F]AV-1451 PET as a longitudinal biomarker in progressive supranuclear palsy. Mov Disord. (2018) 34:105–13. doi: 10.1002/mds.27546
216. Brendel M, Schonecker S, Hoglinger G, Lindner S, Havla J, Blautzik J, et al. [(18)F]-THK5351 PET correlates with topology and symptom severity in progressive supranuclear palsy. Front Aging Neurosci. (2017) 9:440. doi: 10.3389/fnagi.2017.00440
217. Lee H, Seo S, Lee SY, Jeong HJ, Woo SH, Lee KM, et al. [18F]-THK5351 PET imaging in patients with semantic variant primary progressive aphasia. Alzheimer Dis Assoc Disord. (2018) 32:62–9. doi: 10.1097/WAD.0000000000000216
218. Barret O, Seibyl J, Stephens A, Madonia J, Alagille D, Mueller A, et al. First in Human Characterization of PI-2620, a Next Generation PET Tracer for Assessing Tau in Alzheimer's Disease and Other Tauopathies. (2017) Vienna: Alzheimer's & Parkinson's Diseases Congress.
219. Stephens A, Seibyl J, Mueller A, Barret O, Berndt M, Madonia J, et al. Characterization of Novel PET Tracers for the Assessment of Tau Pathology in Alzheimer's Disease and Other Tauopathies. (2017) Vienna: Alzheimer's & Parkinson's Diseases Congress.
220. Lohith TG, Bennacef I, Vandenberghe R, Vandenbulcke M, Salinas-Valenzuela C, Declercq R, et al. First-in-human brain imaging of Alzheimer dementia patients and elderly controls with (18)F-MK-6240, a PET tracer targeting neurofibrillary tangle pathology. J Nucl Med. (2018) 60:107–14. doi: 10.2967/jnumed.118.208215
221. Wong DF, Comley RA, Kuwabara H, Rosenberg PB, Resnick SM, Ostrowitzki S, et al. Characterization of 3 novel tau radiopharmaceuticals, (11)C-RO-963, (11)C-RO-643, and (18)F-RO-948, in healthy controls and in Alzheimer subjects. J Nucl Med. (2018) 59:1869–76. doi: 10.2967/jnumed.118.209916
222. Declercq L, Rombouts F, Koole M, Fierens K, Marien J, Langlois X, et al. Preclinical evaluation of (18)F-JNJ64349311, a novel PET tracer for Tau imaging. J Nucl Med. (2017) 58:975–81. doi: 10.2967/jnumed.116.185199
223. Goedert M, Yamaguchi Y, Mishra SK, Higuchi M, Sahara N. Tau filaments and the development of positron emission tomography tracers. Front Neurol. (2018) 9:70. doi: 10.3389/fneur.2018.00070
224. Olsson B, Lautner R, Andreasson U, Ohrfelt A, Portelius E, Bjerke M, et al. CSF and blood biomarkers for the diagnosis of Alzheimer's disease: a systematic review and meta-analysis. Lancet Neurol. (2016) 15:673–84. doi: 10.1016/S1474-4422(16)00070-3
225. Wagshal D, Sankaranarayanan S, Guss V, Hall T, Berisha F, Lobach I, et al. Divergent CSF tau alterations in two common tauopathies: Alzheimer's disease and progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. (2015) 86:244–50. doi: 10.1136/jnnp-2014-308004
226. Rojas JC, Bang J, Lobach IV, Tsai RM, Rabinovici GD, Miller BL, et al. CSF neurofilament light chain and phosphorylated tau 181 predict disease progression in PSP. Neurology. (2018) 90:e273–81. doi: 10.1212/WNL.0000000000004859
227. Wilham JM, Orru CD, Bessen RA, Atarashi R, Sano K, Race B, et al. Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog. (2010) 6:e1001217. doi: 10.1371/journal.ppat.1001217
228. Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. (2001) 411:810–3. doi: 10.1038/35081095
229. Saijo E, Ghetti B, Zanusso G, Oblak A, Furman JL, Diamond MI, et al. Ultrasensitive and selective detection of 3-repeat tau seeding activity in Pick disease brain and cerebrospinal fluid. Acta Neuropathol. (2017) 133:751–65. doi: 10.1007/s00401-017-1692-z
230. Kraus A, Saijo E, Metrick MA II, Newell K, Sigurdson CJ, Zanusso G, et al. Seeding selectivity and ultrasensitive detection of tau aggregate conformers of Alzheimer disease. Acta Neuropathol. (2018) 137:585–98. doi: 10.1007/s00401-018-1947-3
231. Hansson O, Janelidze S, Hall S, Magdalinou N, Lees AJ, Andreasson U, et al. Blood-based NfL: a biomarker for differential diagnosis of parkinsonian disorder. Neurology. (2017) 88:930–7. doi: 10.1212/WNL.0000000000003680
232. Bacioglu M, Maia LF, Preische O, Schelle J, Apel A, Kaeser SA, et al. Neurofilament light chain in blood and CSF as marker of disease progression in mouse models and in neurodegenerative diseases. Neuron. (2016) 91:56–66. doi: 10.1016/j.neuron.2016.05.018
233. Boxer AL, Lang AE, Grossman M, Knopman DS, Miller BL, Schneider LS, et al. Davunetide in patients with progressive supranuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol. (2014) 13:676–85. doi: 10.1016/S1474-4422(14)70088-2
234. Rojas JC, Karydas A, Bang J, Tsai RM, Blennow K, Liman V, et al. Plasma neurofilament light chain predicts progression in progressive supranuclear palsy. Ann Clin Transl Neurol. (2016) 3:216–25. doi: 10.1002/acn3.290
235. Scherling CS, Hall T, Berisha F, Klepac K, Karydas A, Coppola G, et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol. (2014) 75:116–26. doi: 10.1002/ana.24052
236. Skillback T, Farahmand B, Bartlett JW, Rosen C, Mattsson N, Nagga K, et al. CSF neurofilament light differs in neurodegenerative diseases and predicts severity and survival. Neurology. (2014) 83:1945–53. doi: 10.1212/WNL.0000000000001015
237. Borroni B, Malinverno M, Gardoni F, Grassi M, Parnetti L, Agosti C, et al. A combination of CSF tau ratio and midsaggital midbrain-to-pons atrophy for the early diagnosis of progressive supranuclear palsy. J Alzheimers Dis. (2010) 22:195–203. doi: 10.3233/JAD-2010-100333
238. Magdalinou NK, Paterson RW, Schott JM, Fox NC, Mummery C, Blennow K, et al. A panel of nine cerebrospinal fluid biomarkers may identify patients with atypical parkinsonian syndromes. J Neurol Neurosurg Psychiatry. (2015) 86:1240–7. doi: 10.1136/jnnp-2014-309562
239. Abstracts of the 2018 International Congress of Parkinson's disease and movement disorders((R)). Mov Disord. (2018) 33(Suppl. 2):S1–929. doi: 10.1002/mds.116
240. West T, Braunstein JB, Fogelman I, Boxer AL, Hu H, Verghese PB, et al. Safety, tolerability and pharmacokinetics of ABBV-8E12, a humanized monoclonal antibody, in phase 1, single ascending dose, placebo-controlled study in subjects with progressive supranuclear palsy [abstract]. In: Clinical Trials on Alzheimer's Disease (CTAD) 2016. San Diego, CA (2016).
241. Boxer A, Qureshi I, Grundman M, Tirucherai GS, Bechtold C, Ahlijanian M, et al. Multiple ascending dose study of the Tau-directed monoclonal antibody BIIB092 in patients with progressive supranuclear palsy [abstract]. In: American Academy of Neurology Annual Meeting 2018. Los Angeles, CA (2018).
242. Lane RM, Kordasiewicz HB, Smith AE, Mignon L, Miller T, Narayanan P, et al. Rationale for and development of IONIS-MAPTRx, the First Tau-lowering Antisense oligonucleotide, in patients with Mild AD [abstract]. In: 142nd Annual Meeting of The American Neurological Association. San Diego, CA (2017). doi: 10.1016/j.jalz.2017.07.242
Keywords: progressive supranuclear palsy, tauopathy, translational research, epidemiology, etiopathogenesis, biomarker
Citation: Shoeibi A, Olfati N and Litvan I (2019) Frontrunner in Translation: Progressive Supranuclear Palsy. Front. Neurol. 10:1125. doi: 10.3389/fneur.2019.01125
Received: 26 August 2019; Accepted: 08 October 2019;
Published: 22 October 2019.
Edited by:
Antonio Pisani, University of Rome Tor Vergata, ItalyReviewed by:
Giovanni Rizzo, University of Bologna, ItalyMarian L. Dale, Oregon Health & Science University, United States
Copyright © 2019 Shoeibi, Olfati and Litvan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irene Litvan, aWxpdHZhbkB1Y3NkLmVkdQ==