 Ewelina Palasz1
Ewelina Palasz1 Wiktor Niewiadomski2
Wiktor Niewiadomski2 Anna Gasiorowska1,2
Anna Gasiorowska1,2 Adrianna Wysocka1
Adrianna Wysocka1 Anna Stepniewska2
Anna Stepniewska2 Grazyna Niewiadomska1*
Grazyna Niewiadomska1*- 1Neurobiology Center, Nencki Institute of Experimental Biology, Polish Academy of Science, Warsaw, Poland
- 2Department of Applied Physiology, Mossakowski Medical Research Centre, Polish Academy of Sciences, Warsaw, Poland
Parkinson's disease (PD) is manifested by progressive motor, autonomic, and cognitive disturbances. Dopamine (DA) synthesizing neurons in the substantia nigra (SN) degenerate, causing a decline in DA level in the striatum that leads to the characteristic movement disorders. A disease-modifying therapy to arrest PD progression remains unattainable with current pharmacotherapies, most of which cause severe side effects and lose their efficacy with time. For this reason, there is a need to seek new therapies supporting the pharmacological treatment of PD. Motor therapy is recommended for pharmacologically treated PD patients as it alleviates the symptoms. Molecular mechanisms behind the beneficial effects of motor therapy are unknown, nor is it known whether such therapy may be neuroprotective in PD patients. Due to obvious limitations, human studies are unlikely to answer these questions; therefore, the use of animal models of PD seems indispensable. Motor therapy in animal models of PD characterized by the loss of dopaminergic neurons has neuroprotective and neuroregenerative effects, and the completeness of neuronal protection may depend on (i) degree of neuronal loss, (ii) duration and intensity of exercise, and (iii) time elapsed between insult and commencing of training. As the physical activity is neuroprotective for dopaminergic neurons, the question arises what is the mechanism of this protective action. A current hypothesis assumes a central role of neurotrophic factors in the neuroprotection of dopaminergic neurons, even though it is still not clear whether increased DA level in the nigrostriatal axis results from neurogenesis of dopaminergic neurons in the SN, recovery of the phenotype of dopaminergic neurons, increased sprouting of the residual dopaminergic axons in the striatum, or generation of local striatal neurons from inhibitory interneurons. In the present review, we discuss studies describing the influence of physical exercise on the PD-like changes manifested in animal models of the disease and focus our interest on the current state of knowledge on the mechanism of neuroprotection induced by physical activity as a supportive therapy in PD.
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disorder. Due to the frequency of occurrence, it is a serious medical, social, and economic problem. Currently, no proven neuroprotective or disease-modifying treatment is available for PD. Several agents can be used to treat the motor symptoms as well as non-motor symptoms, such as depression, fatigue, sleep disorders, and wakefulness, associated with dopamine (DA) deficiency.
Although pharmacological therapy is the current gold standard in the treatment of PD, recent clinical trials have shown that physical activity alleviates and slows down the development of movement impairments, reduces depression and anxiety, and improves mood state, cognitive function, and sleep quality (1, 2). Recent studies have shown that regular physical activity, such as strength training, walking, flexibility, balance, and aerobic training or dance, adjusted to the severity of the disease and to the current PD patient's state of health, is able to enhance brain plasticity, which plays a key role in improving motor and cognitive functions (3).
Physical activity has been found beneficial for persons with PD; however, there is still no answer as to which type, frequency, or intensity of physical exercise is the most effective in relieving Parkinsonian symptoms. The lack of standardized terminology, protocols, interventions, and outcome measures limits the comparison of data and makes them difficult to interpret.
Aerobic training, aimed mainly to increase cardiovascular capacity, has a beneficial effect on PD patients. Reuter et al. (4) study showed that flexibility and relaxation program, walking, and Nordic walking reduced the pain and improved the quality of life of all patients. Nordic walking proved superior in improving postural stability and gait, and other researchers (5–8) confirmed improvement of movement parameters by this kind of walking. On the other hand, Bello et al. (9) demonstrated the superiority of training on the treadmill compared to overground walking in improving gait and body balance. In yet another study, treadmill training was also found to improve speed, cadence, stride length, and distance walked (10).
A common symptom of PD is muscle weakness. The strength (resistance) training increases muscle mass and bone mineral density, sustains body balance, and thus improves the quality of life of PD individuals. Scandalis et al. (11) showed improved gait function in patients with mild to moderate PD subjected to resistance training. Hirsch et al. (12) confirmed a lower fall risk and a longer independent life of PD individuals in response to resistance and balance exercises.
Improvement in motor function can be seen as specific effect of physical training. However, such activity also preserves or improves cognitive function in PD patients, which suggests training to act as a disease-modifying factor. Physical exercise has been shown to improve performance in verbal fluency tests and to reduce spatial working memory errors in cognitive tests. Cruise et al. (13) showed that a combination of strength and cardiovascular training improved executive function in the course of PD. Tabak et al. (14) and Nocera et al. (15) in small case studies observed improvement in executive function in PD patients after aerobic exercises.
Although the collective evidence supports physical activity as a measure for PD prevention, the mechanism underlying the diminished risk of PD development in physically active persons is still not fully understood (16). Based on animal studies, several mechanisms are believed to explain the effects of physical activity as an adjuvant therapy against PD: increased synthesis and release of neurotrophins (17–20), restoration of the equilibrium between the level and interactions of neurotransmitters (21, 22), increased resistance to oxidative stress (23), reduced inflammatory process in the brain (21, 24, 25), and enhanced synaptogenesis (26), angiogenesis (27), and neurogenesis (28, 29) (Figure 1). In the present review, we aimed to describe the influence of physical exercise on PD symptoms in animal models of the disease and report the current state of knowledge concerning the mechanism underlying the neuroprotective effects of physical activity as supportive therapy in PD.
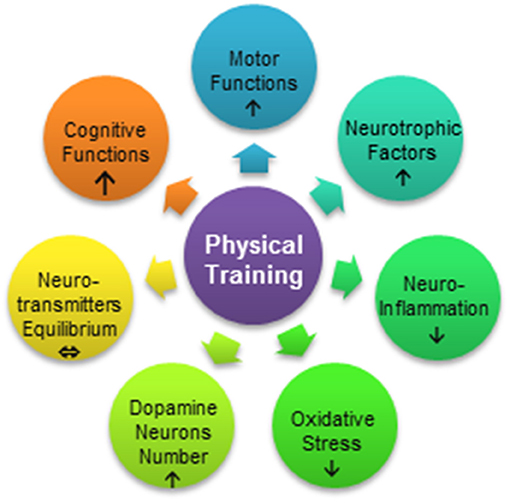
Figure 1. Processes influenced by exercise in Parkinson's disease. Physical training can improve motor function, reduce neuroinflammation, and increase resistance to oxidative stress or reduce the stress level. Motor therapy results in mobilization of neurotrophic factors, protection of dopamine neurons, and restoration of the equilibrium between neurotransmitters such as dopamine and glutamate. Physical exercise may also lead to the enhancement of cognitive functions.
Exercise Induces Recovery of Motor Function and Neuroprotection in Animal Models of PD
Motor Performance
Many studies in animal models of PD showed that various forms of physical activity, differing in type of effort, duration, intensity, and starting point with respect to neurotoxic insult, were neuroprotective and suppressed processes involved in PD pathology (30–32).
The two commonly used toxins leading to the degeneration of nigral dopaminergic neurons and, in consequence, to DA depletion in the striatum are 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 6-hydroxydopamine [6-OHDA]. The most commonly adopted forms of physical activity in animals are the running wheel, performed voluntarily, and forced treadmill training. There are data proving a reduction of motor performance after administration of PD-inducing neurotoxins (30–33) and a protective effect of physical activity (19, 34–36) against behavioral impairments caused by these neurotoxins in animals. However, the detection and experimental quantification of mouse motor impairments that truthfully mimic the symptoms of PD patients has proved difficult. To detect reliably behavioral deficits using standard mouse motor tests such as the rotarod, inverted grid test, and general locomotor activity monitoring in open field, very large doses of MPTP must be used and behavioral alterations often appear to be transient (32, 37, 38). Movement disorders are only slight in mice performing simple motor tasks, even if the loss of dopaminergic neurons in SN after exposure to MPTP ranges from 60 to 70% (39–41), and this may explain why some researchers report no detectable motor deterioration. Nevertheless, it has been shown that physical activity has both protective (42, 43) and restorative (22, 44, 45) effects on motor parameters such as velocity (46), locomotor activity (42), or body balance and coordination (34, 43).
Several studies report that physical exercise led to motor function improvement in rotarod test in a mouse model of PD (47–51). The MPTP-treated groups that completed a treadmill exercise regimen achieved significantly higher maximal velocity than those in the MPTP sedentary group, although there are data showing no effect of MPTP on the rotarod performance (32, 52). A few reports show that mice treated with MPTP have reduced locomotion; however, there are also studies that report no such changes (53–55) as well as reports in which a restorative effect of endurance exercise on locomotion measured in the open field was noticed (42).
Movement in the upside-down position seems to be a task so complex that it should reveal impairments caused by the loss of dopaminergic neurons; therefore, the inverted grid test was introduced by Tillerson et al. (32). During this test, mouse movement on the underside of the horizontal grid is recorded. Tillerson et al. (32, 56) detected sustained behavioral deficits in MPTP-treated mice up to 28 days post-injection, and these deficits were inversely correlated with striatal DA content and expression of the dopamine transporter (DAT), vesicular monoamine transporter 2 (VMAT2), and tyrosine hydroxylase (TH). The inverted grid test was then used by other researchers (57–63), but none of them demonstrated any correlation between motor performance and the degree of dopaminergic neuron loss. Interestingly, in the study that provides an in-depth analysis of many motor parameters (64), no adverse effect of MPTP treatment was noticed.
The detection and experimental quantification of motor impairments in rodents that truthfully mimic the anomalies of Parkinsonian patients proved difficult either due to inadequate behavioral tests or—very likely—because motor impairment takes another form in these animals. So, at present, despite the known ability of physical exercise to promote motoric improvement, our knowledge regarding the most beneficial form, duration, intensity, and frequency of exercise is still insufficient.
Protection of Dopaminergic Neurons
Depending on when physical training is commenced with respect to neurotoxic insult, one may examine (i) the preventive role of exercise against PD once such insult follows training; (ii) the modification of the course of disease, when the chronic treatment is paralleled with training; and (iii) the neuroregenerative effect of exercise when physical training is applied post-insult.
The neuroprotective role of exercise against the loss of dopaminergic neurons was studied by Gerecke et al. (65), where voluntary physical activity on the running wheel was applied before acute administration of MPTP. The authors determined the critical duration of voluntary exercise necessary for neuroprotection of dopaminergic neurons. They found that 1 month of training provided no neuroprotection, 2 months of training conferred partial protection, and only the first 3 months of training prevented any loss of substantia nigra pars compacta (SNpc) dopaminergic neurons by subsequent neurotoxin treatment. However, beside duration of the training, the distance covered also turned out to be essential. It was found that the longer distance run on the wheel, the lesser loss of DA neuron in the 3-month training group, so only mice that had run the longest distance were completely protected against MPTP toxicity.
The effects of ongoing training on the progression of PD were modeled with daily treadmill running during chronic neurotoxin treatment realized with 10 injections of MPTP spread over 5 weeks. Such chronic MPTP treatment leads to permanent, lasting at least 6 months, neurological deficits resembling, though incompletely, PD. This is unlike acute and subacute MPTP treatments, after which neurological deficits and behavioral changes soon wane spontaneously (65).
Ahmad et al. (66) using this chronic mouse model proved that 10- and 18-week-long treadmill training, started 1 week before commencing MPTP treatment, reduced loss of dopaminergic neurons in the ventral tegmental area (VTA). It should be noted, though, that this model encompasses both parallel application of neurotoxin and physical training as well as post-insult training. Somewhat surprisingly, the number of DA neurons was greater following 18 weeks of exercise training, than 10, which may prove that either new DA neurons appeared and/or some DA neurons that lost their phenotype regained it during subsequent 8 weeks.
Pothakos et al. (42), using the same model, started treadmill training 1 week before MPTP treatment, continued it during intoxication lasting 5 weeks, and over the subsequent 8–12 weeks. They did not observe either the recovery of striatal DA or preservation of TH positive neurons in the SNpc in exercising MPTP-treated mice. It may be important that different doses of MPTP have been used in these studies−12.5 vs. 25 mg/kg of MPTP per injection. The importance of dose was confirmed by Lau et al. (19). In the same chronic model of PD with 18-week treadmill training, which was started 1 week before MPTP intoxication, they observed nearly complete preservation of TH immunopositive neurons in the SNpc and the level of striatal DA comparable to that in control mice. The importance of MPTP dosing was reflected in the loss of 55% of TH positive neurons observed in the study of Lau et al. (19) who used 15 mg MPTP/kg/injection and a 72% loss in the study of Pothakos et al. (42) who applied 25 mg/kg/injection.
Two studies that used the protocol described above delivered contrasting results: Pothakos et al. (42) did not observe either the recovery of striatal DA or preservation of TH positive neurons in the SNpc in exercising MPTP-treated mice, whereas Lau et al. (19) found nearly complete preservation of TH immunopositive neurons in the SNpc and the level of striatal DA comparable to that in control mice. The possible explanation of this discrepancy may be the dose of MPTP, which, in Pothakos et al. study, is twice as big as in Lau et al. study. The difference in MPTP dose was reflected in the 72% loss of nigral DA neurons in the former vs. 55% in the latter study.
Studies on the beneficial effects of exertion applied after neurotoxin application show varying results: from no effect to partial or complete preservation of the number of TH-positive nigral neurons. Aerobic training lasting 4 weeks and started after 5 weeks of treatment with 25 mg MPTP/kg/injection slightly raised nigrostriatal TH and DA levels as compared with sedentary MPTP-injected mice (67). Kintz et al. (22) found that training lasting 37 days, which started 5 days after acute dosing with MPTP, did not return the level of striatal DA reduced by the neurotoxin. Zhao et al. (68) found that the number of nigral dopaminergic neurons returned almost to that observed in control mice after 4 weeks of vibration training following 1 week of MPTP treatment and was also significantly greater than that in sedentary MPTP-treated animals. This was accompanied by similar changes in the striatal DA level. In another study (69), it was found that 8 weeks of progressive treadmill that started 2 weeks after chronic high-dose (25 mg/kg/injection) MPTP administration caused nearly complete restoration of TH-positive neurons in the SNpc, as well as similar recovery of TH and DAT in MPTP-treated exercising mice.
Increased number of dopaminergic neurons observed in neurotoxin-treated animals after physical exercise could be the result of better protection of neurons or neurogenesis. In a study performed by Jang et al. (45), mice were intraperitoneally injected with 25 mg/kg MPTP daily for 1 week. Training on the treadmill lasted 6 weeks and was applied 4 weeks after the last MPTP dose. This training induced neurogenesis in MPTP-treated mice, evidenced by an increased number of bromodeoxyuridine (BrdU)-positive neurons, and attenuated the loss of dopaminergic neurons as manifested by higher levels of TH and DAT. The authors also showed enhanced autophagy, exemplified by changes in autophagy-related protein levels (e.g., microtubule-associated protein 1A/1B-light chain 3—MAP1LC3, nucleoporin p62—p62, Beclin 1, Beclin 2 (Bcl-2)/adenovirus E1B-19 kDa interacting protein—BNIP3, lysosome-associated membrane protein 2—LAMP2, cathepsin L and transcription factor EB—TFEB), and augmented antioxidant capability (e.g., increased level of superoxide dismutase-1—SOD-1, catalase, glutathione peroxidase 1—GPX1, heme oxygenase-1—HO-1, and DJ1) as compared with the results obtained in the MPTP group with no treadmill training.
Also, in a rat model of PD induced by 6-OHDA injections, significant preservation of TH-immunopositive neurons in the SNpc and fibers in the striatum was observed after 4 weeks of treadmill training applied 24 h post-neurotoxin insult (70).
The neuroprotective potency even of a late (i.e., post-neurotoxic insult) physical training was recently illustrated by our study (71), where training of the same duration and intensity attenuated neuronal loss to the same degree, both when commenced before or after chronic MPTP treatment.
Although most reports prove that physical activity positively affects motor functions (19, 34, 44, 47–50, 72) and increases the level of DA in the striatum (19, 73), there are also studies reporting the lack of effects of physical exercise on these parameters (22, 52, 53, 55). For example, Gerecke et al. (65) have found that, even though 3 months of training completely protected dopaminergic neurons against MPTP-induced neurotoxicity, the level of DA and its metabolites in the striatum was significantly lower compared to controls. In order to account for these discrepant findings, the authors hypothesize that uptake of 1-methyl-4-phenylpyridinium (MPP+) by DAT generates the formation of hydrogen peroxide and nitrosylated proteins. These compounds damage the dopaminergic terminals but not the cell body. It is also possible that MPP+ alone lowers the ability of dopaminergic neurons to transport DA into striatal terminals leading to decreased DA level.
Regulation of Dopaminergic and Glutamatergic Transmission via Physical Activity
Degeneration of dopaminergic neurons in the SNpc and striatal loss of axonal terminals are key pathological features of PD in humans (74–76) and in toxin-induced animal models (19, 42, 71, 77). DA deficiency in PD also leads to loss of dendritic spines within the striatum, which results in motor impairments (78). Two different types of dopaminergic activity can be observed in the striatum: a phasic, brief high-amplitude increase of DA release that acts at the synaptic space through the low affinity D1 receptor (D1R) and a tonic DA release of low amplitude that acts through the high-affinity D2 receptors (D2R) located in the extrasynaptic space. The striatum is functionally subdivided into ventral and dorsal areas, which participate in different aspects of motor control (74). The dorsal striatum is mainly composed of two subpopulations of medium spiny neurons (MSNs): DA D1 receptor-expressing MSNs that constitute the striatonigral or direct pathway (dMSNs) and DA D2 receptor-expressing MSNs that constitute the striatopallidal or indirect pathway (iMSNs). It has been suggested that each pathway has a different role in motor control, with dMSNs being involved in the main aspects of motor control, including motor program selection and activation, and iMSNs in selection and activation of a context-specific motor programs based on the integration of motivational/emotional signaling with sensory-motor inputs (79).
Toy et al. (77) have found a decrease in dendritic spine density in both D1R and D2R containing MSNs after acute MPTP administration and an increase in dendritic spine density and arborization in MSNs of both pathways after 30 days of intensive treadmill exercise. Recovery of dendritic spines was associated with increased expression of post-synaptic density protein 95 (PSD-95) and of presynaptic synaptophysin, leading to increased synaptogenesis in the dorsolateral striatum and improvement in motor performance (77).
The deficiency of DA leads to structural and functional changes in MSNs. These changes usually entail increased glutamatergic projection and hyper-excitability of the indirect pathway (D2R-iMSNs) (80). The prolonged elevated level of glutamate in the intercellular space results in longer depolarization and in disturbances of ionic homeostasis that elicits excitotoxicity and, in consequence, cell death. The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic-acid receptor (AMPAR), a fast-acting ionotropic glutamate receptor, plays a critical role in these processes. AMPARs are tetrameric channels composed of various combinations of four glutamate receptor subunits, GluA1–GluA4. In particular, the presence or absence of GluA2 decides about important electrophysiological channel properties, including calcium (Ca2+) permeability. Reduced level of GluA2 in AMPAR channels may lead to increased permeability for calcium ions.
Alterations in AMPARs expression have also been noted in animal models of PD characterized by a reduced level of DA. Kintz et al. (22) have noticed that MPTP treatment alone or combined with exercise evoked changes in AMPAR level limited to the D2R-iMSNs striatopallidal pathway. They observed an increase in the level of AMPARs lacking GluA2 in MPTP-treated mice. Increased expression of the GluA2 subunit of the AMPAR in animals that started to exercise after acute MPTP administration suggested the restoration of normal AMPAR subunit expression. Enhanced expression of GluA2-lacking AMPAR possibly potentiates glutamatergic signaling, which leads to hyperexcitability of the striatopallidal projection pathway observed as a consequence of DA depletion (81). Exercise increased the presence of GluA2 subunit in AMPARs in MPTP-treated mice. This could decrease Ca2+ influx and diminish glutamatergic drive leading to reduced glutamatergic projection and thus increased survival of dopaminergic neurons.
The hypothesis of increased glutamatergic projection in the course of PD and reduced glutamatergic tonus in response to physical activity has been strengthened by Scone et al. (21), who showed that while levels of vesicular glutamate transporter 1 (VGLUT1) and glutamate transporter-1 (GLT-1) were elevated after MPTP administration, the level of these transporters was decreased following physical activity, restoring glutamate homeostasis in treated mice. Fisher et al. (44) analyzed the neuroprotective effect of 30 days of treadmill training on proportions between dopaminergic and glutamatergic projection in C57 BL/6J mice acutely administered with MPTP. The treadmill training was started 4 days after MPTP or saline treatment. The MPTP exercise group demonstrated significantly reduced DAT immunoreactivity, higher expression of D2R mRNA, and a significant decrease in the density of glutamate terminals compared to the MPTP sedentary group. An increase in the density of nerve terminal glutamate immunolabeling, characteristic for MPTP lesioning, may, therefore, reflect a decrease in the extracellular levels of striatal glutamate. Consequently, the effect of exercise in an MPTP-lesioned brain may be the increased release of glutamate at the synapse, which may alter DA receptor subtype expression or/and function of medium spiny neurons.
Exercise Mobilizes Neurotrophic Factors
Post-mortem examinations of PD brain in human and animal models demonstrate a decreased level of neurotrophic factors (NTFs) in the nigrostriatal pathway (82–84), although this reduction could be partially due to the loss of SNpc dopaminergic neurons, which specifically express one of the NTFs, the brain-derived neurotrophic factor (BDNF). Decreased efficiency of neurotrophic factors, such as BDNF, nerve growth factor (NGF), neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4) in PD may be connected with a reduced level of cyclic nucleotides. Such reduction leads to dysregulation of transcription of NFT encoding genes, mediated by the cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB) (85–87).
NTFs are endogenous proteins that promote differentiation, maintenance, function, and plasticity of the nervous system and allow neurons to survive and repair after injury (88). Therefore, NTFs may serve as potential therapeutic agents in the treatment of neurodegenerative diseases including PD. For example, BDNF, mesencephalic astrocyte-derived neurotrophic factor (MANF), glial cell line-derived neurotrophic factor (GDNF), and cerebral dopamine neurotrophic factor (CDNF) have been shown to be neuroprotective and neurorestorative toward damaged dopaminergic neurons in cell cultures and in various PD animal models (89). In vivo, NTFs induce survival of nigrostriatal DA cell bodies and fibers improving motor performance compromised in parkinsonian animals.
The best known trophic factors that protect dopaminergic neurons against oxidative stress are BDNF and GDNF. Initially, the potential therapeutic role of BDNF and GDNF in dopaminergic neurons was found in in vitro studies (31, 89, 90). BDNF is widely and abundantly expressed in the brain and significantly involved in several aspects of neuronal development and maturation, plasticity, and recovery mechanisms (91, 92). BDNF also supports the survival of several types of neurons, including mesencephalic dopaminergic, septal cholinergic, and striatal gamma-aminobutyric-acid-releasing (GABAergic) neurons. Binding of BDNF to the high-affinity tropomyosin-related kinase B (TrkB) receptor leads to phosphorylation of TrkB and activation of the three essential downstream intracellular signaling cascades within neuronal somata: phospholipase C-γ (PLCγ), phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT), and mitogen-activated protein kinase/extracellular signal-related kinase (MAPK/ERK) pathways. Furthermore, BDNF may activate the CREB transcription factor and CREB-binding protein (CBP) and regulate expression of genes encoding proteins involved in stress resistance and cell survival and even neural plasticity (93).
GDNF, in turn, is believed to be the most important neurotrophic factor in the nigrostriatal dopaminergic system and therefore is considered to have therapeutic potential for neuroprotective and regenerative interventions in PD (88). GDNF promotes the survival and maturation of dopaminergic neurons and increases their high-affinity DA uptake (89). It exerts similar effects on motor, adrenergic, parasympathetic, enteric, and somatic sensory neurons (94). GDNF binds with high affinity to glycosylphosphatidylinositol-linked receptor α1 (GFRα1), which is highly expressed in midbrain dopaminergic neurons when measured at mRNA and protein levels. The complex of GDNF and GFRα1, in turn, recruits a transmembrane rearranged during transfection (Ret) receptor and triggers downstream signaling, which controls mitochondrial morphology and complex I activity in dopaminergic neurons (94). GDNF, after binding to its receptor, activates signaling pathways leading to stimulation of dopaminergic neuron excitability, inhibition of DAT activity, and stimulation of TH phosphorylation (95).
There is increasing evidence that NTFs are critical for exercise-induced neuroprotection. The study of Lau et al. (19) has shown that the exercise-induced recovery of cell number and motor behavior in the chronic MPTP mouse model of PD was associated with an improved mitochondrial function and an increase in the brain region-specific levels of BDNF and GDNF. Furthermore, Zhao et al. (68) demonstrated that in MPTP mice, 4 weeks of vibration training almost completely restored dopaminergic neurons in the SN, lost due to MPTP treatment, and DA levels in the striatum, and significantly increased the level of BDNF in the striatum. The authors postulated that long-lasting vibration training could protect dopaminergic neurons from MPTP-induced damage probably by upregulating BDNF. In turn, Gerecke et al. (20), using BDNF+/− mice, showed that exercise was not able to protect dopaminergic neurons from MPTP-induced neurodegeneration.
In another study by Tajiri et al. (70), daily forced treadmill training was used in the 6-OHDA acute rat model of PD. In the 6-OHDA training group, behavioral recovery in the cylinder test and a significant decrease in the number of amphetamine-induced rotations were observed. This was accompanied by the preservation of dopaminergic fibers in the striatum and neurons in the SNpc. In addition, BrdU/doublecortin (Dcx) co-staining revealed enhanced proliferation and migration of neural progenitor cells from the subventricular zone (SVZ) toward the injection site. At the same time, BDNF and GDNF levels were upregulated in the striatum. Physical training in animal models of PD, beside neuroprotection of the dopaminergic system, may enhance neurogenesis and progenitor cell migration through upregulating BDNF-TrkB and GDNF-Ret signaling, which can, in turn, stimulate certain signaling cascades, including those that activate the PI3K/Akt pathway or the extracellular signal-regulated kinases 1 and 2 (ERK1/2) cascade. The PI3K/Akt signaling pathway transmits anti-apoptotic signals stemming from neurotrophins. This pathway activates transcription factors such as CREB and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), which trigger the expression of genes involved in cell survival, such as Bcl-2 and other inhibitors of apoptosis. It also suppresses the expression of pro-apoptotic genes, such as the Bcl-2-associated death promoter (BAD) and Forkhead box (FOX) transcription factors (96). Similarly, the ERK1/2 signaling cascade induces anti-apoptotic genes such as those encoding Bcl-2 and the transcription factor CREB. ERK1/2 also mediates neuritic outgrowth induced by neurotrophic factors (97) and phosphorylation of TH, which increases its enzymatic activity, thus enhancing DA synthesis (98).
Additionally, physical activity, through the induction of neurotrophin signaling pathways, affects synaptic plasticity in MPTP mice. In the study of Zhu et al. (99), the authors have demonstrated that long-term potentiation (LTP) impairment in the MPTP model was due to the decrease in hippocampal BDNF level. Both induction of endogenous BDNF synthesis by memantine or application of exogenous BDNF restored the LTP deficit in the MPTP model. In addition to the effects of memantine on LTP, MPTP-enhanced long-term depression (LTD) was also reversed by memantine administration. These data suggest that compounds that can activate BDNF synthesis have the potential to protect memory in PD.
The evidence that exercise, through regulation of growth factors, secures successful brain function was demonstrated in several other studies (100–104). Also in our study (71), the results suggest that 10 weeks of training on the treadmill, no matter if started before or after PD induction, have a protective effect against dopaminergic neuron degeneration in the chronic MPTP mouse model of PD. Neuroprotective properties of exercise were most likely associated with increased expression of endogenous NTFs and reduced inflammation in the brain. Although the actual sequence of events needs to be discerned, it is likely that NTFs protect neurons against degeneration and in this way prevent the inflammatory response. However, the other way is also possible, i.e., that the strength of inflammation is regulated by the neurotrophin signaling pathways irrespective of neurodegeneration. It is likely that both ways operate when training accompanies neurotoxic assault and also when it is applied after such assault. In summary, these studies underscore the view that physical activity remains neuroprotective even during the advanced stage of PD and therefore provide strong support for starting and continuing physical activity at any point of the disease.
Physical Training Modulates Neuroinflammatory Mechanisms
Although the death of dopaminergic neurons is a well-characterized pathological feature of PD, the primary cause of the disease is still not clear. Since the first observations of reactive microglia and astrocytes in post-mortem brain samples of PD patients were made (105, 106), numerous studies have proposed a role of glial cells in the neuropathology of PD (107–109). Furthermore, infiltration of cells of the peripheral immune system into the brain is also likely to play a role in the pathomechanism of PD (110, 111). It is not clear whether inflammation is a primary cause of PD or a secondary event, that is, the consequence of neuronal death. Short-lasting inflammatory reactions may induce NTFs (112, 113) and may protect neurons against reactive oxygen species (ROS) (114) and glutamate neuronal hyperexcitability (115) but chronic inflammation usually leads to degeneration and neuronal damage (113, 116, 117).
Neuroinflammation means activation of microglia and reactive astrocytes, which, in turn, produce cytokines, chemokines, reactive oxygen, and nitrogen species, secondary messengers, prostaglandins, and protein complement cascades (118). Elevated levels of interleukin-6 (IL-6) acting as pro-inflammatory cytokine, tumor necrosis factor alpha (TNF-α), interleukin-1 beta (IL-1β), and nitric oxide synthase (NOS), found in the SN, putamen, as well as in the cerebrospinal fluid (CSF) and serum of PD patients suggest that, in PD, glial cells acquire a pro-inflammatory phenotype (119).
Both the pro-inflammatory cytokines and their receptors play a role in the pathogenesis of PD. A glial reaction in the brain involving astrocytes and microglia have been described in several toxin-induced animal models of PD (21, 46, 120, 121). Loss of dopaminergic neurons is associated with activated microglia (122–124), which, in turn, activates astrocytes. Increasing evidence suggests that the CD200 protein and its receptor, CD200R, play a critical role during microglia activation. Deficits in the CD200–CD200R pathway exacerbate microglial activation and degeneration of dopaminergic neurons (125).
Liddelow et al. (126) have shown in in vitro and in vivo studies that activated microglia stimulate, in turn, astrocytes by secreting interleukin-1 alpha (IL-1α), TNFα, and complement component 1q (C1q). Lofrumento et al. (109) have observed a significant increase in IL-1β, TNF-α, and IL-6 mRNA expression level and an increase in both mRNA and protein levels of their respective receptors IL-1RI (IL-1 receptor type I), TNF-αRI, and IL-6Rα in the SN of MPTP-treated animals in comparison with untreated mice.
Astrogliosis is characterized mainly by an increase in the number and size of astrocytes and increased expression of the glial fibrillary acidic protein (GFAP). Due to pro-inflammatory activation, astrocytes lose the ability to promote neuronal survival, neurite outgrowth, synaptogenesis, and phagocytosis (127). Astrogliosis has been confirmed during PD (35, 128). Sconce et al. (21) have shown an increase in GFAP immunoreactivity and a reduced ratio of phosphorylated to non-phosphorylated component 3 of the nuclear factor of activated T-cells (NFATc3) within the SN in MPTP mice.
The neuroprotective effect of physical activity has been linked to the prevention and modulation of the inflammatory process (129). Recent evidence suggests that physical activity, in addition to modifying the cardiovascular, muscular, and hormonal systems, also affects the immune system, which can lead to the overall anti-inflammatory effect in the whole body (130), including attenuation of the inflammatory processes in the brain in the course of neurodegenerative diseases.
Sconce et al. (21) have reported that running wheel exercise affects behavioral deficits, as well as dopaminergic, glutamatergic, and inflammatory biomarkers in a progressive MPTP mouse model of PD. The authors noticed recovery of motor abilities, increased VMAT2 expression, decreased glycosylated-DAT expression, reduced levels of VGLUT1 and GLT-1, and lower levels of the inflammatory marker, component 3 of the nuclear factor of activated T-cells (NFATc3), and of the astrocytic marker, GFAP, in MPTP/exercised mice as compared to MPTP mice without exercise. The anti-inflammatory effect of different types of physical activity has been verified by Goes et al. (24). The authors found that 4-week swimming training alleviated cognitive and motor impairment, and prevented both the increase of ROS and IL-1β levels and inhibition of glutathione peroxidase (GPx) glutathione S-transferase (GST) and glutathione reductase (GR) activities. This training also restored DA, 3,4-dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) activity levels in the striatum of mice administered with 6-OHDA (24).
Recent studies have suggested that interaction between alpha-synuclein (α-syn) and toll-like receptor 2 (TLR2) may play a critical role in the onset of neuroinflammation (131). Jang et al. (132) observed in mice that 8-week treadmill training started after completion of chronic MPTP treatment eliminated motor coordination deficits, increased nigrostriatal TH level, and diminished expression of α-syn in the striatum. This, in consequence, led to downregulation of TLR2 signaling molecules such as myeloid differentiation primary response 88 (MyD88), tumor necrosis factor receptor-associated factor 6 (TRAF6), and phosphorylated transforming growth factor-β-activated protein kinase 1 (p-TAK1). What was interesting, physical activity significantly decreased GFAP level and increased the level of phosphorylated NFATc3, thus reducing the susceptibility of dopaminergic neurons to apoptosis.
In another study (71), the immunohistochemical staining and ELISA assay against GFAP showed an increased number of astrocytes and elevated GFAP concentration in SNpc and VTA in MPTP sedentary mice. On the other hand, treadmill training caused reduced GFAP concentration in the MPTP-trained group, comparable to the concentration of GFAP observed in the control groups. Furthermore, staining against CD11b and ionized calcium-binding adapter molecule 1 (Iba1), the markers of microglia, also showed higher intensity in the SNpc and VTA of MPTP mice without treadmill training compared with results obtained in controls and the MPTP group with treadmill training (71).
Both subtypes of glial cells, astrocytes and microglia, may be activated in two different ways, resulting in a pro-inflammatory (classical M1 activation) or anti-inflammatory (alternative M2 activation) formation of response. In the latter case, stimulated microglia show increased expression of cytokines recognized as an anti-inflammatory, such as interleukin-10 (IL-10), transforming growth factor beta (TGFβ), insulin growth factor 1 (IGF-1), NGF, and BDNF (133). Astrocytes, similarly to microglia, secrete anti-inflammatory compounds, among them neurotrophic factors such as GDNF, BDNF, and MANF, which revive injured dopaminergic neurons and uphold their survival (134, 135).
It is likely that physical training applied in MPTP mice not only reduces M1-type pro-inflammatory glial activation but also promotes M2-type neuroprotective activation of microglia. The increased synthesis of trophic factors observed after prolonged physical training could favor the neuroprotective pathway of microglia activation. In consequence, dopaminergic neurons protected by neurotrophins do not degenerate and thus do not send signals that mobilize the pro-inflammatory response and proliferation of glial cells. Training may reduce the reactivity of microglia and mitigate inflammation by altering multiple metabolic and transcriptional processes (136). For instance, it may inhibit the activity of glycogen synthase kinase 3 (GSK-3), which is a major regulator of the balance between the pro- and anti-inflammatory mediators in immune cells, including microglia (137). GSK-3 stimulates activated microglia to release pro-inflammatory cytokines such as IL-1β, IL-6, and TNFα, and inhibits the release of anti-inflammatory cytokines such as IL-10 (138–140). The way the exercise affects GSK-3 activity may rely on the activation of some extracellular factors, including those mediated by BDNF (141, 142) and GDNF (143, 144), which are known to inhibit GSK-3.
Physical Training Attenuates Mitochondrial Dysfunction and Oxidative Stress
It has long been recognized that oxidative stress may be important in the etiology of a variety of late-onset neurodegenerative diseases including PD (145–147). Oxidative stress is connected with the production of ROS, which show strong oxidizing properties. Although ROS at physiological concentrations play a pivotal role in several cellular signaling pathways such as cell cycle regulation, phagocytosis, and enzyme activation, excessive generation of ROS leads to several harmful effects including damage to DNA, lipids, and proteins (148).
In the PD brain, the nigral polyunsaturated free fatty acid level is reduced, while the nigral level of lipid peroxidation markers (malondialdehyde and 4-hydroxynonenal) is elevated (149). Also, protein oxidative damage is evidenced by the presence of protein carbonyls (150). Some results suggest that reactive nitrogen species contribute to PD etiology due to their role played in nitration and nitrosylation of certain proteins (151). Oxidative stress leading to cell death may occur in the SNpc because of (i) augmented production of H2O2 due to increased DA turnover, (ii) reduced brain capacity to eliminate H2O2 because of glutathione (GSH) deficiency, or (iii) promotion of •OH formation caused by elevated level of the reactive iron (152, 153). Synthesis of neuronal GSH requires delivery of GSH precursors, which mainly arise from extracellular cleavage of astrocyte-derived GSH. Lower nigral levels of GSH have been found in post-mortem PD patient brains when compared with age-matched controls. Diminished GSH release from astrocytes may be caused by depletion of reduced GSH in astrocytes, which in turn is caused by increased oxidant production by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) (154).
Mitochondria are a key point for programmed cell death (PCD) where apoptosis may be triggered by several factors produced as a result of intracellular stress, such as ROS and calcium ions (Ca2+) overload (155). It has been reported that, due to oxidative stress, there are more deletions in mitochondrial DNA of the surviving nigral dopaminergic neurons of PD patients (156, 157). The hypothesis of mitochondrial dysfunction in PD was strengthened after the discovery of mutations in several genes encoding mitochondrial proteins that give rise to a familial form of PD (e.g., PINK1 and PARK2, DJ-1) (158).
The involvement of mitochondrial dysfunction and mitochondrial ROS production has been demonstrated also in experimental animal models using either toxin (MPTP) or genetically altered mice. The post-mortem examination of SNpc in PD patients showed a significant increase in the H-subunit and a significant reduction in the L-subunit of ferritin, which was associated with increased iron concentration, ROS overproduction, and neuronal death (159). A reduction in complex I activity has been demonstrated in the SN, lymphocytes, and platelets of PD patients (160). Moreover, increased oxidative stress has been detected in both the rotenone and MPTP-induced toxin models and in genetic models of PD (160, 161). Studies in animals have indicated that MPP+, the active metabolite of MPTP, and rotenone inhibit ATP synthesis by blocking electron transport. This blockade is due to binding to complex I. MPP+, similarly to rotenone, produces superoxide anions in electron transport particles, which supports the view that MPP+ is primarily a mitochondrial toxin (162). The neurotoxic effects of MPP+ can be effectively prevented by antioxidants, indicating that the neurotoxicity of this compound is due to oxidative stress (163).
Many studies demonstrate a positive effect of physical exercise on inhibition of oxidative stress (47, 69, 164–172). Using a chronic mouse model of PD, Patki, and Lau (173) revealed that 18-week treadmill training inhibited cytochrome c release, elevated levels of p53, and upregulated the expression of mitochondrial transcription factor A (THAM) and of peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α), which are known to be associated with mitochondrial dysfunction and loss of dopaminergic neurons.
Koo et al. (69, 174) demonstrated, in a MPTP/probenecid-induced mouse model of PD, that treadmill training improved mitochondrial function and promoted autophagy via the sirtuin-1 (SIRT1) signaling pathway causing α-syn level to decrease. This subsequently attenuated the loss of dopaminergic neurons due to reducing apoptotic cell death mediated by α-syn. Most importantly, training increased expression of SIRT1 that, in turn, enhanced mitochondrial biogenesis and reduced oxidative stress by activating PGC-1α. Furthermore, activation of SIRT1-activated microtubule-associated protein 1 light chain 3 (MAP1LC3) and, in consequence, promoted autophagic clearance of α-syn. Improved mitochondrial function and augmented autophagy caused by physical training were accompanied by ameliorated motor abilities in mice chronically treated with MPTP. Koo et al. (174) have also demonstrated that training on the treadmill, applied after MPTP administration and lasting 8 weeks, reduced the loss of dopaminergic neurons, altered the level of proteins involved in apoptosis (increased Bcl-2 and decreased Bcl-2-associated X protein-Bax and caspase-3), increased expression of core proteins of the translocase of the outer mitochondrial membrane (TOM) complex, which is part of the mitochondrial import machinery (MIM), and increased the level of mitochondrial electron transport chain proteins (cyclooxygenase—COX-I, COX-IV, and mitochondrial 70 kDa heat shock protein—mtHSP70).
In the Al-Jarrah et al. (175) study, the authors have shown a positive impact of exercise training on the concentration of nitric oxide in the striatum of a chronic MPTP mouse model of PD. The 4-week training on the treadmill started after MPTP injections significantly decreased the level of neuronal nitric oxide synthase (nNOS) and inducible form of NOS (iNOS) in animals subjected to MPTP administration and treadmill training compared with the MPTP sedentary group. Four weeks of treadmill training improved the motor skills, evaluated through analyzing gait on the CatWalk, in a unilateral 6-OHDA rat model of PD (167, 168). This training also improved dopaminergic neuron viability, recovered mitochondrial function, and attenuated oxidative stress in PD rats. It was suggested that this phenomenon may be associated with improved mitochondrial turnover, that is, mitochondrial fusion, fission, and clearance, giving rise to increased quantities of mitochondria. It was demonstrated that exercise prevented 6-OHDA-induced loss of TH immunolabeling and activated the nuclear factor (erythroid-derived 2)-like 2 (Nrf2)–antioxidant response element (ARE) axis (i.e., ARE-dependent transcription) in the nigrostriatal pathway in C57BL/6 mice (176). The Nrf2-ARE is a major cellular defense mechanism against oxidative stress, which regulates the expression of antioxidant enzymes, such as gamma-glutamylcysteine ligase (γGCL) and HO-1. The Nrf2-ARE signaling pathway is strongly involved in neuroprotection and anti-inflammatory response. Treadmill training also stimulated mitochondrial biogenesis in the striatum of animals that were more resistant to the oxidant, 6-OHDA, and a nitric oxide donor, namely, (±)-S-nitroso-N-acetylpenicillamine. Similar results were obtained in the study of Tsou et al. (172) who showed that exercise enhanced the nigrostriatal Nrf2-mediated antioxidant defense capacity to protect dopaminergic neurons against MPP+-induced toxicity in mice.
The study, designed to investigate the potential neuroprotective effect of swimming training in a mouse model of PD induced by 6-OHDA (24), demonstrated that a 4-week-long training attenuated the following features associated with PD: increased number of falls in the rotarod test, impairment of long-term memory in the object recognition test, depressive-like behavior in the tail suspension test, and, at the same time, increase in ROS and IL-1β levels, inhibition of GPx activity, increase in GR and GST activities, and decrease in DA, HVA, and DOPAC levels.
Altogether, the findings presented in the above sections indicate that the beneficial effects induced by exercise, when neurodegenerative diseases, such as PD, are concerned, are mostly due to attenuation of oxidative stress and inflammation and elevation of NTFs.
Conclusions
Epidemiological data support the notion of beneficial effects of physical activity on preventing the risk, development, and progression of PD. Due to the fact that the effectiveness of pharmacological treatment decreases with the development of the disease and because properly selected physical training does not cause side effects, the physical activity should be recommended, as a supportive therapy, to patients suffering from PD. The physical activity not only reduces motor impairments but also improves cognitive functions.
Studies in animal models of PD indicate that physical activity may prevent the loss of, protect, or restore dopaminergic neurons probably by activating signaling cascades triggered by the increased availability of neurotrophins (Figure 2). Furthermore, neurotrophins can stabilize intracellular calcium concentration, induce antioxidant enzyme expression, and suppress the release of proinflammatory cytokines. Neurotrophic factors also provide important extracellular signals regulating neurogenesis in the adult brain. Finally, exercise improves motor circuitry through alterations in DA and glutamate neurotransmission.
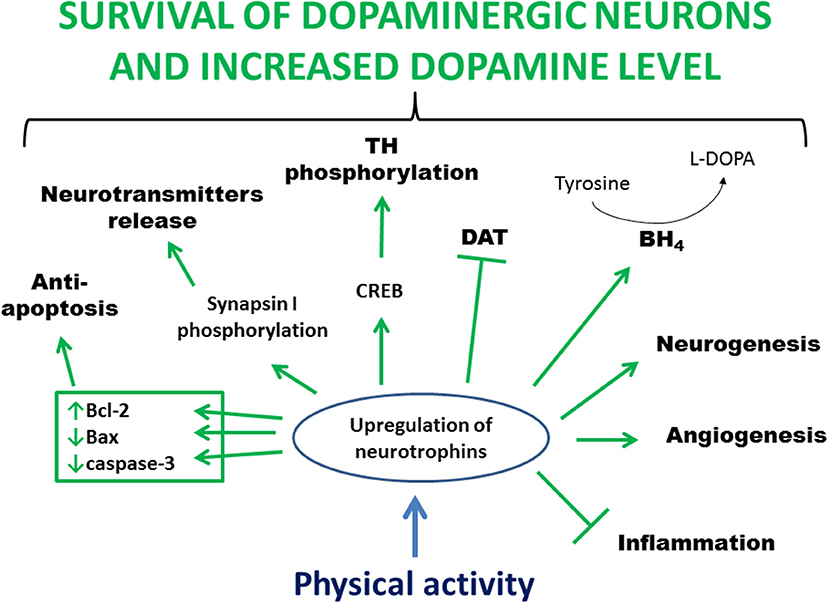
Figure 2. Main processes involved in exercise-induced and neurotrophin-mediated increased viability of dopaminergic neurons. Physical activity leads to an increased level of neurotrophins in the brain. This upregulation stimulates anti-apoptotic proteins, mediates clustering and release of synaptic vesicles, activates CREB leading to phosphorylation and activation of TH, increases BH4 level allowing conversion of tyrosine to L-DOPA, activates a pathway that inhibits DAT activity, increases blood flow and angiogenesis, enhances neurogenesis, and reduces inflammation. In summary, all these processes lead to greater survival of dopaminergic neurons and increased level of dopamine. Bcl-2, B-cell lymphoma 2; BH4, tetrahydrobiopterin; CREB, cAMP response-element binding protein; DAT, dopamine transporter; L-DOPA, L-3,4-dihydroxyphenylalanine; TH, tyrosine hydroxylase.
However, animal models utilizing neurotoxins do not completely reproduce clinical symptoms and pathologies of PD in humans. Therefore, when MPTP is administered to animals, we are talking about inducing parkinsonism, not systemic PD. MPTP animal models are most often used to study the pathogenesis and progression of the disease and the effectiveness of drugs. Among the disadvantages of the animal model of PD caused by MPTP administration, the short-term and acute nature of pathological changes is mentioned. This disadvantage is remedied by the method of administration of neurotoxin used in chronic models of induction of parkinsonism in which the loss of dopaminergic neurons persists for at least several months. Another disadvantage is the difficulty or even impossibility to reproduce the characteristic movement disorders seen in humans. Lack of these disorders can be caused by a different organization of movement control by the nervous system in human and in mice. In animals, motor control is not so highly hierarchical and there is lesser contribution of the cerebral cortex and subcortical structures to locomotion than in humans.
It is likely that neurotoxin-based animal models of PD are adequate experimental models to study aspects of the beneficial effects of physical exertion, concerning changes at the tissue and cellular level and may be well-used to investigate such phenomena as neuroprotection, induction of trophic factors and neurogenesis, alleviation of inflammation, reduction of oxidative stress, and improvement of mitochondrial function. On the other hand, these models seems not so useful in studying the effect of exercise on alleviating movement disorders, because the results of the latest studies indicate that the loss of motor function after administration of neurotoxins is insignificant and difficult to capture/notice in the behavioral tests used.
It is possible that the outline, as presented in this review, of mechanisms by which physical activity confers neuroprotection is valid. However, more research is needed to confirm these mechanisms and elucidate their details. There is also a question what is the optimal type, intensity, and duration of the exercise when it should be performed to be maximally effective.
Specifically, as the majority of data supporting the ability of physical activity to protect DA neurons stems from neurotoxin PD models, the question arises whether this property of physical activity would persist in other animal models of PD. With a positive answer to this question, the next issue to be cleared would be whether there are common neuroprotective mechanisms. The existence of such common mechanisms will be strongly supportive to continue physical activity by PD patients during the whole course of illness as a disease-modifying factor.
Author Contributions
EP, WN, and GN contributed equally to this work and wrote the main body of the paper. AG wrote the Introduction section. AW and AS made the figures and create the references. All authors reviewed the final manuscript.
Funding
This work was funded by NCN grant 2014/15/B/NZ4/05041 and TauRx Therapeutics/WisTa Laboratories Ltd., Singapore, and by funds from the Nencki Institute of Experimental Biology.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Muhlack S, Welnic J, Woitalla D, Müller T. Exercise improves efficacy of levodopa in patients with Parkinson's disease. Mov Disord. (2007) 22:427–30. doi: 10.1002/mds.21346
2. Fayyaz M, Jaffery SS, Anwer F, Zil-E-Ali A, Anjum I. The effect of physical activity in Parkinson's disease: a mini-review. Cureus. (2018) 10:e2995. doi: 10.7759/cureus.2995
3. Petzinger G, Fisher B, McEwen S, Beeler S, Walsh J, Jakowec MENT. Exercise-enhanced neuroplasticity targeting motor and cognitive circuitry in Parkinson's disease. Lancet Neurol. (2013) 12:716–26. doi: 10.1016/S1474-4422(13)70123-6
4. Reuter I, Mehnert S, Leone P, Kaps M, Oechsner M, Engelhardt M. Effects of a flexibility and relaxation programme, walking, and nordic walking on Parkinson's disease. J Aging Res. (2011) 2011:1–18. doi: 10.4061/2011/232473
5. Szefler-Derela J, Suszynski K, Doroniewicz I, Kowalczyk A, Opala G, Kwiek SJ. Nordic walking in Parkinson disease rehabilitation. Ann Acad Medicae Silesiensis. (2014) 68:361–7.
6. Ebersbach G, Ebersbach A, Gandor F, Wegner B, Wissel J, Kupsch A. Impact of physical exercise on reaction time in patients with Parkinson's disease—data from the Berlin BIG study. Arch Phys Med Rehabil. (2014) 95:996–9. doi: 10.1016/j.apmr.2013.10.020
7. Cugusi L, Solla P, Serpe R, Carzedda T, Piras L, Oggianu M, et al. Effects of a nordic walking program on motor and non-motor symptoms, functional performance and body composition in patients with Parkinson's disease. NeuroRehabilitation. (2015) 37:245–54. doi: 10.3233/NRE-151257
8. Monteiro EP, Franzoni LT, Cubillos DM, de Oliveira Fagundes A, Carvalho AR, Oliveira HB, et al. Effects of Nordic walking training on functional parameters in Parkinson's disease: a randomized controlled clinical trial. Scand J Med Sci Sports. (2017) 27:351–8. doi: 10.1111/sms.12652
9. Bello O, Sanchez JA, Lopez-Alonso V, Márquez G, Morenilla L, Castro X, et al. The effects of treadmill or overground walking training program on gait in Parkinson's disease. Gait Posture. (2013) 38:590–5. doi: 10.1016/j.gaitpost.2013.02.005
10. Nadeau A, Pourcher E, Corbeil P. Effects of 24 wk of treadmill training on gait performance in Parkinson's disease. Med Sci Sport Exerc. (2014) 46:645–55. doi: 10.1249/MSS.0000000000000144
11. Scandalis TA, Bosak A, Berliner JC, Helman LL, Wells MR. Resistance training and gait function in patients with Parkinson's disease. Am J Phys Med Rehabil. (2001) 80:38–43. doi: 10.1097/00002060-200101000-00011
12. Hirsch MA, Toole T, Maitland CG, Rider RA. The effects of balance training and high-intensity resistance training on persons with idiopathic Parkinson's disease. Arch Phys Med Rehabil. (2003) 84:1109–17. doi: 10.1016/S0003-9993(03)00046-7
13. Cruise KE, Bucks RS, Loftus AM, Newton RU, Pegoraro R, Thomas MG. Exercise and Parkinson's: benefits for cognition and quality of life. Acta Neurol Scand. (2011) 123:13–9. doi: 10.1111/j.1600-0404.2010.01338.x
14. Tabak R, Aquije G, Fisher BE. Aerobic exercise to improve executive function in Parkinson disease. J Neurol Phys Ther. (2013) 37:58–64. doi: 10.1097/NPT.0b013e31829219bc
15. Nocera JR, Altmann LJP, Sapienza C, Okun MS, Hass CJ. Can exercise improve language and cognition in Parkinson's disease? A case report. Neurocase. (2010) 16:301–6. doi: 10.1080/13554790903559663
16. LaHue SC, Comella CL, Tanner CM. The best medicine? The influence of physical activity and inactivity on Parkinson's disease. Mov Disord. (2016) 31:1444–54. doi: 10.1002/mds.26728
17. Phillips C, Baktir MA, Srivatsan M, Salehi A. Neuroprotective effects of physical activity on the brain: a closer look at trophic factor signaling. Front Cell Neurosci. (2014) 8:170. doi: 10.3389/fncel.2014.00170
18. Sleiman SF, Henry J, Al-Haddad R, El Hayek L, Abou Haidar E, Stringer T, et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body β-hydroxybutyrate. Elife. (2016) 5:e15092 doi: 10.7554/eLife.15092
19. Lau Y-S, Patki G, Das-Panja K, Le W-D, Ahmad SO. Neuroprotective effects and mechanisms of exercise in a chronic mouse model of Parkinson's disease with moderate neurodegeneration. Eur J Neurosci. (2011) 33:1264–74. doi: 10.1111/j.1460-9568.2011.07626.x
20. Gerecke KM, Jiao Y, Pagala V, Smeyne RJ. Exercise does not protect against MPTP-induced neurotoxicity in BDNF happloinsufficent mice. PLoS ONE. (2012) 7:1–11. doi: 10.1371/journal.pone.0043250
21. Sconce MD, Churchill MJ, Greene RE, Meshul CK. Intervention with exercise restores motor deficits but not nigrostriatal loss in a progressive MPTP mouse model of Parkinson's disease. Neuroscience. (2015) 299:156–74. doi: 10.1016/j.neuroscience.2015.04.069
22. Kintz N, Petzinger GM, Akopian G, Ptasnik S, Williams C, Jakowec MW, et al. Exercise modifies α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor expression in striatopallidal neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mouse. J Neurosci Res. (2013) 91:1492–507. doi: 10.1002/jnr.23260
23. Chalimoniuk M, Jagsz S, Sadowska-Krepa E, Chrapusta SJ, Klapcinska B, Langfort J. Diversity of endurance training effects on antioxidant defenses and oxidative damage in different brain regions of adolescent male rats. J Physiol Pharmacol. (2015) 66:539–47.
24. Goes ATR, Souza LC, Filho CB, Del Fabbro L, De Gomes MG, Boeira SP, et al. Neuroprotective effects of swimming training in a mouse model of Parkinson's disease induced by 6-hydroxydopamine. Neuroscience. (2014) 256:61–71. doi: 10.1016/j.neuroscience.2013.09.042
25. Real CC, Garcia PC, Britto LRG. Treadmill exercise prevents increase of neuroinflammation markers involved in the dopaminergic damage of the 6-OHDA Parkinson's disease model. J Mol Neurosci. (2017) 63:36–49. doi: 10.1007/s12031-017-0955-4
26. Ambrogini P, Lattanzi D, Ciuffoli S, Betti M, Fanelli M, Cuppini R. Physical exercise and environment exploration affect synaptogenesis in adult-generated neurons in the rat dentate gyrus: possible role of BDNF. Brain Res. (2013) 1534:1–12. doi: 10.1016/j.brainres.2013.08.023
27. Lopez-Lopez C, LeRoith D, Torres-Aleman I. Insulin-like growth factor I is required for vessel remodeling in the adult brain. Proc Natl Acad Sci USA. (2004) 101:9833–8. doi: 10.1073/pnas.0400337101
28. Van der Borght K, Kóbor-Nyakas DÉ, Klauke K, Eggen BJL, Nyakas C, Van der Zee EA, et al. Physical exercise leads to rapid adaptations in hippocampal vasculature: Temporal dynamics and relationship to cell proliferation and neurogenesis. Hippocampus. (2009) 19:928–36. doi: 10.1002/hipo.20545
29. van Praag H, Shubert T, Zhao C, FH G. Exercise enhances learning and hippocampal neurogenesis in aged mice. J Neurosci. (2005) 25:8680–5. doi: 10.1523/JNEUROSCI.1731-05.2005
30. Fredriksson A, Archer T. MPTP-induced behavioural and biochemical deficits: a parametric analysis. J Neural Transm Park Dis Dement Sect. (1994) 7:123–32. doi: 10.1007/BF02260967
31. Rozas G, López-Martín E, Guerra MJ, Labandeira-García JL. The overall rod performance test in the MPTP-treated-mouse model of Parkinsonism. J Neurosci Methods. (1998) 83:165–75. doi: 10.1016/S0165-0270(98)00078-8
32. Tillerson JL, Caudle WM, Reverón ME, Miller GW. Detection of behavioral impairments correlated to neurochemical deficits in mice treated with moderate doses of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Exp Neurol. (2002) 178:80–90. doi: 10.1006/exnr.2002.8021
33. Chung YC, Baek JY, Kim SR, Ko HW, Bok E, Shin W-H, et al. Capsaicin prevents degeneration of dopamine neurons by inhibiting glial activation and oxidative stress in the MPTP model of Parkinson's disease. Exp Mol Med. (2017) 49:e298. doi: 10.1038/emm.2016.159
34. Wu SY, Wang TF, Yu L, Jen CJ, Chuang JI, Wu FS, et al. Running exercise protects the substantia nigra dopaminergic neurons against inflammation-induced degeneration via the activation of BDNF signaling pathway. Brain Behav Immun. (2011) 25:135–46. doi: 10.1016/j.bbi.2010.09.006
35. Hsueh SC, Chen KY, Lai JH, Wu CC, Yu YW, Luo Y, et al. Voluntary physical exercise improves subsequent motor and cognitive impairments in a rat model of Parkinson's disease. Int J Mol Sci. (2018) 19:508. doi: 10.3390/ijms19020508
36. Rafie F, Shabazi M, Sheikh M, Naghdi N, Sheibani V. Effects of voluntary exercise on motor function in Parkinson's disease model of rats. Ann Appl Sport Sci. (2017) 5:81–6. doi: 10.18869/acadpub.aassjournal.5.2.81
37. Heikkila RE, Sonsalla PK. The MPTP-treated mouse as a model of Parkinsonism: how good is it? Neurochem Int. (1992) 20:299–303. doi: 10.1016/0197-0186(92)90256-Q
38. Sundström E, Fredriksson A, Archer T. Chronic neurochemical and behavioral changes in MPTP-lesioned C57BL/6 mice: a model for Parkinson's disease. Brain. (1990) 28:181–8. doi: 10.1016/0006-8993(90)91656-2
39. Bové J, Perier C. Neurotoxin-based models of Parkinson's disease. Neuroscience. (2012) 211:51–76. doi: 10.1016/j.neuroscience.2011.10.057
40. Blandini F, Armentero M-T. Animal models of Parkinson's disease. FEBS J. (2012) 279:1156–66. doi: 10.1111/j.1742-4658.2012.08491.x
41. Bazzu G, Calia G, Puggioni G, Spissu Y, Rocchitta G, Debetto P, et al. alpha-Synuclein- and MPTP-generated rodent models of Parkinson's disease and the study of extracellular striatal dopamine dynamics: a microdialysis approach. CNS Neurol Disord Drug Targets. (2010) 9:482–90. doi: 10.2174/187152710791556177
42. Pothakos K, Kurz MJ, Lau Y-S. Restorative effect of endurance exercise on behavioral deficits in the chronic mouse model of Parkinson's disease with severe neurodegeneration. BMC Neurosci. (2009) 10:6. doi: 10.1186/1471-2202-10-6
43. Tuon T, Valvassori SS, Lopes-Borges J, Luciano T, Trom CB, Silva LA, et al. Physical training exerts neuroprotective effects in the regulation of neurochemical factors in an animal model of Parkinson's disease. Neuroscience. (2012) 227:305–12. doi: 10.1016/j.neuroscience.2012.09.063
44. Fisher BE, Petzinger GM, Nixon K, Hogg E, Bremmer S, Meshul CK, et al. Exercise-induced behavioral recovery and neuroplasticity in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mouse basal ganglia. J Neurosci Res. (2004) 77:378–90. doi: 10.1002/jnr.20162
45. Jang Y, Kwon I, Song W, Cosio-Lima LM, Lee Y. Endurance exercise mediates neuroprotection against MPTP-mediated Parkinson's disease via enhanced neurogenesis, antioxidant capacity, and autophagy. Neuroscience. (2018) 379:292–301. doi: 10.1016/j.neuroscience.2018.03.015
46. Chen YH, Kuo TT, Kao JH, Huang EYK, Hsieh TH, Chou YC, et al. Exercise ameliorates motor deficits and improves dopaminergic functions in the rat hemi-Parkinson's model. Sci Rep. (2018) 8:3973. doi: 10.1038/s41598-018-22462-y
47. Chen Y, Zhang Y, Li L, Hölscher C. Neuroprotective effects of geniposide in the MPTP mouse model of Parkinson's disease. Eur J Pharmacol. (2015) 768:21–7. doi: 10.1016/j.ejphar.2015.09.029
48. Lu M, Su C, Qiao C, Bian Y, Ding J, Hu G. Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of Parkinson's disease via autophagy and mitochondrial ROS clearance. Int J Neuropsychopharmacol. (2016) 19:pyw047. doi: 10.1093/ijnp/pyw047
49. Liu J, Huang D, Xu J, Tong J, Wang Z, Huang L, et al. Tiagabine protects dopaminergic neurons against neurotoxins by inhibiting microglial activation. Sci Rep. (2015) 5:15720. doi: 10.1038/srep15720
50. Klein C, Rasinska J, Empl L, Sparenberg M, Poshtiban A, Hain EG, et al. Physical exercise counteracts MPTP-induced changes in neural precursor cell proliferation in the hippocampus and restores spatial learning but not memory performance in the water maze. Behav Brain Res. (2016) 307:227–38. doi: 10.1016/j.bbr.2016.02.040
51. Sung Y-H, Kim S-C, Hong H-P, Park C-Y, Shin M-S, Kim C-J, et al. Treadmill exercise ameliorates dopaminergic neuronal loss through suppressing microglial activation in Parkinson's disease mice. Life Sci. (2012) 91:1309–16. doi: 10.1016/j.lfs.2012.10.003
52. Lau YS, Fung YK, Trobough KL, Cashman JR, Wilson JA. Depletion of striatal dopamine by the N-oxide of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Neurotoxicology. (1991) 12:189–99.
53. Meredith GE, Rademacher DJ. MPTP mouse models of Parkinson's disease: an update. J Park Dis. (2012) 1:19–33. doi: 10.3233/JPD-2011-11023
54. Zhang Q, Heng Y, Mou Z, Huang J, Yuan Y, Chen N. Reassessment of subacute MPTP-treated mice as animal model of Parkinson's disease. Acta Pharmacol Sin. (2017) 38:1317–28. doi: 10.1038/aps.2017.49
55. Schamne MG, Mack JM, Moretti M, Matheus FC, Walz R, Lanfumey L, et al. The gender-biased effects of intranasal MPTP administration on anhedonic- and depressive-like behaviors in C57BL/6 mice: the role of neurotrophic factors. Neurotox Res. (2018) 34:808–19. doi: 10.1007/s12640-018-9912-4
56. Tillerson JL, Caudle WM, Reverón ME, Miller GW. Exercise induces behavioral recovery and attenuates neurochemical deficits in rodent models of Parkinson's disease. Neuroscience. (2003) 119:899–911. doi: 10.1016/S0306-4522(03)00096-4
57. Fleming SM. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human -synuclein. J Neurosci. (2004) 24:9434–40. doi: 10.1523/JNEUROSCI.3080-04.2004
58. Caudle WM, Richardson JR, Wang MZ, Taylor TN, Guillot TS, McCormack AL, et al. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci. (2007) 27:8138–48. doi: 10.1523/JNEUROSCI.0319-07.2007
59. Manning-Bo AB, Caudle WM, Perez XA, Reaney SH, Paletzki R, Isla MZ, et al. Increased vulnerability of nigrostriatal terminals in DJ-1-deficient mice is mediated by the dopamine transporter. Neurobiol Dis. (2007) 27:141–50. doi: 10.1016/j.nbd.2007.03.014
60. Price DA, Martinez AA, Seillier A, Koek W, Acosta Y, Fernandez E, et al. WIN55,212-2, a cannabinoid receptor agonist, protects against nigrostriatal cell loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Eur J Neurosci. (2009) 29:2177–86. doi: 10.1111/j.1460-9568.2009.06764.x
61. Kim ST, Son HJ, Choi JH, Ji IJ, Hwang O. Vertical grid test and modified horizontal grid test are sensitive methods for evaluating motor dysfunctions in the MPTP mouse model of Parkinson's disease. Brain Res. (2010) 1306:176–83. doi: 10.1016/j.brainres.2009.09.103
62. Salama M, ElDakroory SAEA, ElTantawy D, Ghanem AA, Elghaffar HA, Elhusseiny M, ElHak SG. Regenerative effects of umbilical cord matrix cells (UCMCs) in a rodent model of rotenone neurotoxicity. Environ Toxicol Pharmacol. (2012) 34:338–44. doi: 10.1016/j.etap.2012.05.005
63. Korecka JA, Eggers R, Swaab DF, Bossers K, Verhaagen J. Modeling early Parkinson's disease pathology with chronic low dose MPTP treatment. Restor Neurol Neurosci. (2013) 31:155–67. doi: 10.3233/RNN-110222
64. Niewiadomski W, Palasz E, Skupinska M, Zylinski M, Steczkowska M, Gasiorowska A, et al. TracMouse: a computer aided movement analysis script for the mouse inverted horizontal grid test. Sci Rep. (2016) 6:39331. doi: 10.1038/srep39331
65. Gerecke KM, Jiao Y, Pani A, Pagala V, Smeyne RJ. Exercise protects against MPTP-induced neurotoxicity in mice. Brain Res. (2010) 1341:72–83. doi: 10.1016/j.brainres.2010.01.053
66. Ahmad SO, Park JH, Stenho-Bittel L, Lau YS. Effects of endurance exercise on ventral tegmental area neurons in the chronic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and probenecid-treated mice. Neurosci Lett. (2009) 450:102–5. doi: 10.1016/j.neulet.2008.11.065
67. Al-Jarrah M, Pothakos K, Novikova L, Smirnova IV, Kurz MJ, Stehno-Bittel L, et al. Endurance exercise promotes cardiorespiratory rehabilitation without neurorestoration in the chronic mouse model of Parkinsonism with severe neurodegeneration. Neuroscience. (2007) 149:28–37. doi: 10.1016/j.neuroscience.2007.07.038
68. Zhao L, He LX, Huang SN, Gong LJ, Li L, Lv YY, et al. Protection of dopamine neurons by vibration training and up-regulation of brain-derived neurotrophic factor in a MPTP mouse model of Parkinson's disease. Physiol Res. (2014) 63:649–57.
69. Koo J-H, Cho J-Y. Treadmill exercise attenuates α-synuclein levels by promoting mitochondrial function and autophagy possibly via SIRT1 in the chronic MPTP/P-induced mouse model of Parkinson's disease. Neurotox Res. (2017) 32:473–86. doi: 10.1007/s12640-017-9770-5
70. Tajiri N, Yasuhara T, Shingo T, Kondo A, Yuan W, Kadota T, et al. Exercise exerts neuroprotective effects on Parkinson's disease model of rats. Brain Res. (2010) 1310:200–7. doi: 10.1016/j.brainres.2009.10.075
71. Palasz E, Niewiadomski W, Gasiorowska A, Mietelska-Porowska A, Niewiadomska G. Neuroplasticity and neuroprotective effect of treadmill training in the chronic mouse model of Parkinson's disease. Neural Plast. (2019) 2019:1–14. doi: 10.1155/2019/8215017
72. Shin M-S, Jeong H-Y, An D-I, Lee H-Y, Sung Y-H. Treadmill exercise facilitates synaptic plasticity on dopaminergic neurons and fibers in the mouse model with Parkinson's disease. Neurosci Lett. (2016) 621:28–33. doi: 10.1016/j.neulet.2016.04.015
73. Zhang L, Feng Y, Ji W, Liu J, Liu K. Effect of voluntary wheel running on striatal dopamine level and neurocognitive behaviors after molar loss in rats. Behav Neurol. (2017) 2017:1–6. doi: 10.1155/2017/6137071
74. Brichta L, Greengard P. Molecular determinants of selective dopaminergic vulnerability in Parkinson's disease: an update. Front Neuroanat. (2014) 8:152. doi: 10.3389/fnana.2014.00152
75. Olanow CW, Agid Y, Mizuno Y, Albanese A, Bonucelli U, Damier P, et al. Levodopa in the treatment of Parkinson's disease: current controversies. Mov Disord. (2004) 19:997–1005. doi: 10.1002/mds.20243
76. Burke RE, O'Malley K. Axon degeneration in Parkinson's disease. Exp Neurol. (2013) 246:72–83. doi: 10.1016/j.expneurol.2012.01.011
77. Toy WA, Petzinger GM, Leyshon BJ, Akopian GK, Walsh JP, Hoffman MV, et al. Treadmill exercise reverses dendritic spine loss in direct and indirect striatal medium spiny neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson's disease. Neurobiol Dis. (2014) 63:201–9. doi: 10.1016/j.nbd.2013.11.017
78. Pisani A, Centonze D, Bernardi G, Calabresi P. Striatal synaptic plasticity: implications for motor learning and Parkinson's disease. Mov Disord. (2005) 20:395–402. doi: 10.1002/mds.20394
79. Yager LM, Garcia AF, Wunsch AM, Ferguson SM. The ins and outs of the striatum: role in drug addiction. Neuroscience. (2015) 301:529–41. doi: 10.1016/j.neuroscience.2015.06.033
80. Petzinger GM, Holschneider DP, Fisher BE, McEwen S, Kintz N, Halliday M, et al. The effects of exercise on dopamine neurotransmission in Parkinson's disease: targeting neuroplasticity to modulate basal ganglia circuitry. Brain Plast. (2015) 1:29–39. doi: 10.3233/BPL-150021
81. Hernandez-Echeagaray E, Starling AJ, Cepeda C, Levine MS. Modulation of AMPA currents by D2 dopamine receptors in striatal medium-sized spiny neurons: are dendrites necessary? Eur J Neurosci. (2004) 19:2455–63. doi: 10.1111/j.0953-816X.2004.03344.x
82. Chauhan NB, Siegel GJ, Lee JM. Depletion of glial cell line-derived neurotrophic factor in substantia nigra neurons of Parkinson's disease brain. J Chem Neuroanat. (2001) 21:277–88. doi: 10.1016/S0891-0618(01)00115-6
83. Siegel GJ, Chauhan NB, Service N, Hines E, Hospital VA, Rm F. Neurotrophic factors in Alzheimer's and Parkinson's disease brain. Brain Res Rev. (2000) 33:199–227. doi: 10.1016/S0165-0173(00)00030-8
84. Parain K, Murer M, Yan Q, Faucheux B, Agid Y, Hirsch E, et al. Reduced expression of brain derived neurotrophic factor protein in Parkinson's disease substantia nigra. Neuroreport. (1999) 10:557–61. doi: 10.1097/00001756-199902250-00021
85. Sharma S, Deshmukh R. Vinpocetine attenuates MPTP-induced motor deficit and biochemical abnormalities in Wistar rats. Neuroscience. (2015) 286:393–403. doi: 10.1016/j.neuroscience.2014.12.008
86. Schwenkgrub J, Zaremba M, Joniec-Maciejak I, Cudna A, Mirowska-Guzel D, Kurkowska-Jastrzebska I. The phosphodiesterase inhibitor, ibudilast, attenuates neuroinflammation in the MPTP model of Parkinson's disease. PLoS ONE. (2017) 12:e0182019. doi: 10.1371/journal.pone.0182019
87. Sharma S, Kumar K, Deshmukh R, Sharma PL. Phosphodiesterases: regulators of cyclic nucleotide signals and novel molecular target for movement disorders. Eur J Pharmacol. (2013) 714:486–97. doi: 10.1016/j.ejphar.2013.06.038
88. Aron L, Klein R. Repairing the parkinsonian brain with neurotrophic factors. Trends Neurosci. (2011) 34:88–100. doi: 10.1016/j.tins.2010.11.001
89. Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. (1993) 260:1130–2. doi: 10.1126/science.8493557
90. Hyman C, Hofer M, Barde Y-A, Juhasz M, Yancopoulos GD, Squinto SP, et al. BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature. (1991) 350:230–2. doi: 10.1038/350230a0
91. Tome D, Fonseca C, Campos F, Baltazar G. Role of neurotrophic factors in Parkinson's disease. Curr Pharm Des. (2017) 23:809–38. doi: 10.2174/1381612822666161208120422
92. Mohammadi A, Amooeian VG, Rashidi E. Dysfunction in brain-derived neurotrophic factor signaling pathway and susceptibility to Schizophrenia, Parkinson's and Alzheimer's diseases. Curr Gene Ther. (2018) 18:45–63. doi: 10.2174/1566523218666180302163029
93. Bathina S, Das UN. Brain-derived neurotrophic factor and its clinical implications. Arch Med Sci. (2015) 6:1164–78. doi: 10.5114/aoms.2015.56342
94. Kramer ER, Liss B. GDNF-Ret signaling in midbrain dopaminergic neurons and its implication for Parkinson disease. FEBS Lett. (2015) 589:3760–72. doi: 10.1016/j.febslet.2015.11.006
95. Tenenbaum L, Humbert-Claude M. Glial cell line-derived neurotrophic factor gene delivery in Parkinson's disease: a delicate balance between neuroprotection, trophic effects, and unwanted compensatory mechanisms. Front Neuroanat. (2017) 11:29. doi: 10.3389/fnana.2017.00029
96. Lee E-R, Kim J-Y, Kang Y-J, Ahn J-Y, Kim J-H, Kim B-W, et al. Interplay between PI3K/Akt and MAPK signaling pathways in DNA-damaging drug-induced apoptosis. Biochim Biophys Acta. (2006) 1763:958–68. doi: 10.1016/j.bbamcr.2006.06.006
97. García-Martínez JM, Pérez-Navarro E, Gavaldà N, Alberch J. Glial cell line-derived neurotrophic factor promotes the arborization of cultured striatal neurons through the p42/p44 mitogen-activated protein kinase pathway. J Neurosci Res. (2006) 83:68–79. doi: 10.1002/jnr.20713
98. Lindgren N, Leak RK, Carlson KM, Smith AD, Zigmond MJ. Activation of the extracellular signal-regulated kinases 1 and 2 by glial cell line-derived neurotrophic factor and its relation to neuroprotection in a mouse model of Parkinson's disease. J Neurosci Res. (2008) 86:2039–49. doi: 10.1002/jnr.21641
99. Zhu G, Li J, He L, Wang X, Hong X. MPTP-induced changes in hippocampal synaptic plasticity and memory are prevented by memantine through the BDNF-TrkB pathway. Br J Pharmacol. (2015) 172:2354–68. doi: 10.1111/bph.13061
100. van Praag H, Christie BR, Sejnowski TJ, Gage FH. Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci USA. (1999) 96:13427–31. doi: 10.1073/pnas.96.23.13427
101. Yasuhara T, Hara K, Maki M, Matsukawa N, Fujino H, Date I, et al. Lack of exercise, via hindlimb suspension, impedes endogenous neurogenesis. Neuroscience. (2007) 149:182–91. doi: 10.1016/j.neuroscience.2007.07.045
102. Yi SS, Hwang IK, Yoo K-Y, Park OK, Yu J, Yan B, et al. Effects of treadmill exercise on cell proliferation and differentiation in the subgranular zone of the dentate gyrus in a rat model of type II diabetes. Neurochem Res. (2009) 34:1039–46. doi: 10.1007/s11064-008-9870-y
103. Wu C-W, Chen Y-C, Yu L, Chen H, Jen CJ, Huang A-M, et al. Treadmill exercise counteracts the suppressive effects of peripheral lipopolysaccharide on hippocampal neurogenesis and learning and memory. J Neurochem. (2007) 103:2471–81. doi: 10.1111/j.1471-4159.2007.04987.x
104. Smith AD, Zigmond MJ. Can the brain be protected through exercise? Lessons from an animal model of parkinsonism. Exp Neurol. (2003) 184:31–9. doi: 10.1016/j.expneurol.2003.08.017
105. Teismann P, Schulz JB. Cellular pathology of Parkinson's disease: astrocytes, microglia and inflammation. Cell Tissue Res. (2004) 318:149–61. doi: 10.1007/s00441-004-0944-0
106. McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, McGeer EG. Microglia in degenerative neurological disease. Glia. (1993) 7:84–92. doi: 10.1002/glia.440070114
107. Dehmer T, Lindenau J, Haid S, Dichgans J, Schulz JB. Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. J Neurochem. (2000) 74:2213–6. doi: 10.1046/j.1471-4159.2000.0742213.x
108. Kohutnicka M, Lewandowska E, Kurkowska-Jastrzebska I, Członkowski A, Członkowska A. Microglial and astrocytic involvement in a murine model of Parkinson's disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Immunopharmacology. (1998) 39:167–80. doi: 10.1016/S0162-3109(98)00022-8
109. Lofrumento DD, Saponaro C, Cianciulli A, De Nuccio F, Mitolo V, Nicolardi G, et al. MPTP-induced neuroinflammation increases the expression of pro-inflammatory cytokines and their receptors in mouse brain. Neuroimmunomodulation. (2011) 18:79–88. doi: 10.1159/000320027
110. Kurkowska-Jastrzebska I, Wronska A, Kohutnicka M, Członkowski A, Członkowska A. The inflammatory reaction following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxication in mouse. Exp Neurol. (1999) 156:50–61. doi: 10.1006/exnr.1998.6993
111. Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. (2008) 119:182–92. doi: 10.1172/JCI36470
112. Baquet ZC. Brain-derived neurotrophic factor is required for the establishment of the proper number of dopaminergic neurons in the substantia nigra pars compacta. J Neurosci. (2005) 25:6251–9. doi: 10.1523/JNEUROSCI.4601-04.2005
113. Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol Ser A. (2014) 69:S4–9. doi: 10.1093/gerona/glu057
114. Benarroch EE. Neuron-astrocyte interactions: partnership for normal function and disease in the central nervous system. Mayo Clin Proc. (2005) 80:1326–38. doi: 10.4065/80.10.1326
115. Pita-Almenar JD, Zou S, Colbert CM, Eskin A. Relationship between increase in astrocytic GLT-1 glutamate transport and late-LTP. Learn Mem. (2012) 19:615–26. doi: 10.1101/lm.023259.111
116. Walker KA. Inflammation and neurodegeneration: chronicity matters. Aging. (2018) 11:3–4. doi: 10.18632/aging.101704
117. McGeer PL. Inflammation and the degenerative diseases of aging. Ann N Y Acad Sci USA. (2004) 1035:104–16. doi: 10.1196/annals.1332.007
118. Vivekanantham S, Shah S, Dewji R, Dewji A, Khatri C, Ologunde R. Neuroinflammation in Parkinson's disease: role in neurodegeneration and tissue repair. Int J Neurosci. (2015) 125:717–25. doi: 10.3109/00207454.2014.982795
119. Carta AR, Mulas G, Bortolanza M, Duarte T, Pillai E, Fisone G, et al. l-DOPA-induced dyskinesia and neuroinflammation: do microglia and astrocytes play a role? Eur J Neurosci. (2017) 45:73–91. doi: 10.1111/ejn.13482
120. Haggerty T, Credle J, Rodriguez O, Wills J, Oaks AW, Masliah E, et al. Hyperphosphorylated Tau in an α-synuclein-overexpressing transgenic model of Parkinson's disease. Eur J Neurosci. (2011) 33:1598–610. doi: 10.1111/j.1460-9568.2011.07660.x
121. Rodrigues RW, Gomide VC, Chadi G. Astroglial and microglial reaction after a partial nigrostriatal degeneration induced by the striatal injection of different doses of 6-hydroxydopamine. Int J Neurosci. (2001) 109:91–126. doi: 10.3109/00207450108986528
122. Liu J, Wang MW, Gu P, Ma QY, Wang YY, Geng Y, et al. Microglial activation and age-related dopaminergic neurodegeneration in MPTP-treated SAMP8 mice. Brain Res. (2010) 1345:213–20. doi: 10.1016/j.brainres.2010.05.043
123. Sawada H, Hishida R, Hirata Y, Ono K, Suzuki H, Muramatsu S, et al. Activated microglia affect the nigro-striatal dopamine neurons differently in neonatal and aged mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Neurosci Res. (2007) 85:1752–61. doi: 10.1002/jnr.21241
124. Bian M juan, Li L mei, Yu M, Fei J, Huang F. Elevated interleukin-1β induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine aggravating dopaminergic neurodegeneration in old male mice. Brain Res. (2009) 1302:256–64. doi: 10.1016/j.brainres.2009.07.030
125. Wang XJ, Zhang S, Yan ZQ, Zhao YX, Zhou HY, Wang Y, et al. Impaired CD200-CD200R-mediated microglia silencing enhances midbrain dopaminergic neurodegeneration: Roles of aging, superoxide, NADPH oxidase, and p38 MAPK. Free Radic Biol Med. (2011) 50:1094–106. doi: 10.1016/j.freeradbiomed.2011.01.032
126. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactinve astroctes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
127. Neal M, Richardson JR. Epigenetic regulation of astrocyte function in neuroinflammation and neurodegeneration. Biochim Biophys Acta. (2018) 1864:432–43. doi: 10.1016/j.bbadis.2017.11.004
128. Al-Jarrah MD, Jamous M. Effect of endurance exercise training on the expression of GFAP, S100B, and NSE in the striatum of chronic/progressive mouse model of Parkinson's disease. NeuroRehabilitation. (2011) 28:359–63. doi: 10.3233/NRE-2011-0664
129. Cotman CW, Berchtold NC, Christie L-A. Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci. (2007) 30:464–72. doi: 10.1016/j.tins.2007.06.011
130. Gleeson M, Bishop NC, Stensel DJ, Lindley MR, Mastana SS, Nimmo MA. The anti-inflammatory effects of exercise: mechanisms and implications for the prevention and treatment of disease. Nat Rev Immunol. (2011) 11:607–10. doi: 10.1038/nri3041
131. Dimatelis JJ, Hendricks S, Hsieh J, Vlok NM, Bugarith K, Daniels WMU, et al. Exercise partly reverses the effect of maternal separation on hippocampal proteins in 6-hydroxydopamine-lesioned rat brain. Exp Physiol. (2013) 98:233–44. doi: 10.1113/expphysiol.2012.066720
132. Jang Y, Koo JH, Kwon I, Kang EB, Um HS, Soya H, et al. Neuroprotective effects of endurance exercise against neuroinflammation in MPTP-induced Parkinson's disease mice. Brain Res. (2017) 1655:186–93. doi: 10.1016/j.brainres.2016.10.029
133. Łabuzek K, Skrudlik E, Gabryel B, Okopien B. Anti-inflammatory microglial cell function in the light of the latest scientific research. Ann Acad Medicae Silesiensis. (2015) 69:99–110. doi: 10.18794/aams/32608
134. McGeer PL, McGeer EG. Glial reactions in Parkinson's disease. Mov Disord. (2008) 23:474–83. doi: 10.1002/mds.21751
135. Saavedra A, Baltazar G, Duarte EP. Interleukin-1β mediates GDNF up-regulation upon dopaminergic injury in ventral midbrain cell cultures. Neurobiol Dis. (2007) 25:92–104. doi: 10.1016/j.nbd.2006.08.019
136. Cobianchi S, Arbat-Plana A, Lopez-Alvarez VM, Navarro X. Neuroprotective effects of exercise treatments after injury: the dual role of neurotrophic factors. Curr Neuropharmacol. (2017) 15:495–518. doi: 10.2174/1570159X14666160330105132
137. Yuskaitis CJ, Jope RS. Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell Signal. (2009) 21:264–73. doi: 10.1016/j.cellsig.2008.10.014
138. Maixner DW, Weng H-R. The role of glycogen synthase kinase 3 beta in neuroinflammation and pain. J Pharm Pharmacol. (2013) 1:001. doi: 10.13188/2327-204X.1000001
139. Wang M-J, Huang H-Y, Chen W-F, Chang H-F, Kuo J-S. Glycogen synthase kinase-3β inactivation inhibits tumor necrosis factor-α production in microglia by modulating nuclear factor κB and MLK3/JNK signaling cascades. J Neuroinflammation. (2010) 7:99. doi: 10.1186/1742-2094-7-99
140. Green HF, Nolan YM. GSK-3 mediates the release of IL-1β, TNF-α and IL-10 from cortical glia. Neurochem Int. (2012) 61:666–71. doi: 10.1016/j.neuint.2012.07.003
141. Yang J-W, Ma W, Luo T, Wang D-Y, Lu J-J, Li X-T, et al. BDNF promotes human neural stem cell growth via GSK-3 β -mediated crosstalk with the wnt/ β -catenin signaling pathway. Growth Factors. (2016) 34:19–32. doi: 10.3109/08977194.2016.1157791
142. Yang J, Ru J, Ma W, Gao Y, Liang Z, Liu J, Guo J, et al. BDNF promotes the growth of human neurons through crosstalk with the Wnt/β-catenin signaling pathway via GSK-3β. Neuropeptides. (2015) 54:35–46. doi: 10.1016/j.npep.2015.08.005
143. Yue P, Gao L, Wang X, Ding X, Teng J. Intranasal administration of GDNF protects against neural apoptosis in a rat model of Parkinson's disease through PI3K/Akt/GSK3β pathway. Neurochem Res. (2017) 42:1366–74. doi: 10.1007/s11064-017-2184-1
144. Zhou Y, Zhu J, Lv Y, Song C, Ding J, Xiao M, et al. Kir6.2 deficiency promotes mesencephalic neural precursor cell differentiation via regulating miR-133b/GDNF in a Parkinson's disease mouse model. Mol Neurobiol. (2018) 55:8550–62. doi: 10.1007/s12035-018-1005-0
145. Chen X, Guo C, Kong J. Oxidative stress in neurodegenerative diseases. Neural Regen Res. (2012) 7:376–85. doi: 10.3969/j.issn.1673-5374.2012.05.009
146. Liu Z, Zhou T, Ziegler AC, Dimitrion P, Zuo L. Oxidative stress in neurodegenerative diseases: from molecular mechanisms to clinical applications. Oxid Med Cell Longev. (2017) 2017:1–11. doi: 10.1155/2017/2525967
147. Thanan R, Oikawa S, Hiraku Y, Ohnishi S, Ma N, Pinlaor S, et al. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int J Mol Sci. (2014) 16:193–217. doi: 10.3390/ijms16010193
148. Manoharan S, Guillemin GJ, Abiramasundari RS, Essa MM, Akbar M, Akbar MD. Oxid Med Cell Longev. (2016) 2016:8590578. doi: 10.1155/2016/8590578
149. Dalfó E, Portero-Otín M, Ayala V, Martínez A, Pamplona R, Ferrer I. Evidence of oxidative stress in the neocortex in incidental lewy body disease. J Neuropathol Exp Neurol. (2005) 64:816–30. doi: 10.1097/01.jnen.0000179050.54522.5a
150. Beal MF. Oxidatively modified proteins in aging and disease. Free Radic Biol Med. (2002) 32:797–803. doi: 10.1016/S0891-5849(02)00780-3
151. Brown GC, Borutaite V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim Biophys Acta. (2004) 1658:44–9. doi: 10.1016/j.bbabio.2004.03.016
152. Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson's disease. J Parkinsons Dis. (2013) 3:461–91. doi: 10.3233/JPD-130230
153. Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. (2009) 7:65–74. doi: 10.2174/157015909787602823
154. Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem. (2003) 384:505–16. doi: 10.1515/BC.2003.059
155. Galluzzi L, Blomgren K, Kroemer G. Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci. (2009) 10:481–94. doi: 10.1038/nrn2665
156. Seet RCS, Lee C-YJ, Lim ECH, Tan JJH, Quek AML, Chong W-L, et al. Oxidative damage in Parkinson disease: measurement using accurate biomarkers. Free Radic Biol Med. (2010) 48:560–6. doi: 10.1016/j.freeradbiomed.2009.11.026
157. Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. (2006) 38:515–7. doi: 10.1038/ng1769
158. Koziorowski D, Hoffman-Zacharska D, Sławek J, Jamrozik Z, Janik P, Potulska-Chromik A, et al. Incidence of mutations in the PARK2, PINK1, PARK7 genes in Polish early-onset Parkinson disease patients. Neurol Neurochir Pol. (2013) 47:319–24. doi: 10.5114/ninp.2013.36756
159. Friedman A, Arosio P, Finazzi D, Koziorowski D, Galazka-Friedman J. Ferritin as an important player in neurodegeneration. Park Relat Disord. (2011) 17:423–30. doi: 10.1016/j.parkreldis.2011.03.016
160. Schapira AH V. Mitochondrial dysfunction in neurodegenerative diseases. Neurochem Res. (2008) 33:2502–9. doi: 10.1007/s11064-008-9855-x
161. Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, et al. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. (2009) 33:627–38. doi: 10.1016/j.molcel.2009.02.013
162. Lotharius J, O'Malley KL. The Parkinsonism-inducing Drug 1-Methyl-4-phenylpyridinium triggers intracellular dopamine oxidation. J Biol Chem. (2000) 275:38581–8. doi: 10.1074/jbc.M005385200
163. González-Polo R, Soler G, Rodriguezmartin A, Moran J, Fuentes J. Protection against MPP+ neurotoxicity in cerebellar granule cells by antioxidants. Cell Biol Int. (2004) 28:373–80. doi: 10.1016/j.cellbi.2004.03.005
164. Wi S, Lee JW, Kim M, Park C-H, Cho S-R. An enriched environment ameliorates oxidative stress and olfactory dysfunction in Parkinson's disease with α-synucleinopathy. Cell Transplant. (2018) 27:831–9. doi: 10.1177/0963689717742662
165. Elokda A, DiFrancisco-Donoghue J, Lamberg EM, Werner WG. Effects of exercise induced oxidative stress on glutathione levels in Parkinson's disease on and off medication. J Neurol. (2010) 257:1648–53. doi: 10.1007/s00415-010-5584-6
166. Finaud J, Lac G, Filaire E. Oxidative Stress: relationship with exercise and training. Sport Med. (2006) 36:327–58. doi: 10.2165/00007256-200636040-00004
167. Chuang C-S, Chang J-C, Cheng F-C, Liu K-H, Su H-L, Liu C-S. Modulation of mitochondrial dynamics by treadmill training to improve gait and mitochondrial deficiency in a rat model of Parkinson's disease. Life Sci. (2017) 191:236–44. doi: 10.1016/j.lfs.2017.10.003
168. Costa RO da, Gadelha-Filho CVJ, Costa AEM da, Feitosa ML, Araújo DP de, Lucena JD de, et al. The treadmill exercise protects against dopaminergic neuron loss and brain oxidative stress in Parkinsonian rats. Oxid Med Cell Longev. (2017) 2017:1–10. doi: 10.1155/2017/2138169
169. Alkadhi KA. Exercise as a positive modulator of brain function. Mol Neurobiol. (2018) 55:3112–30. doi: 10.1007/s12035-017-0516-4
170. Archer T, Kostrzewa RM. Exercise and nutritional benefits in PD: rodent models and clinical settings. Curr Top Behav Neurosci. (2016) 29:333–51. doi: 10.1007/7854_2015_409
171. Monteiro-Junior RS, Cevada T, Oliveira BRR, Lattari E, Portugal EMM, Carvalho A, et al. We need to move more: neurobiological hypotheses of physical exercise as a treatment for Parkinson's disease. Med Hypotheses. (2015) 85:537–41. doi: 10.1016/j.mehy.2015.07.011
172. Tsou Y-H, Shih C-T, Ching C-H, Huang J-Y, Jen CJ, Yu L, et al. Treadmill exercise activates Nrf2 antioxidant system to protect the nigrostriatal dopaminergic neurons from MPP+ toxicity. Exp Neurol. (2015) 263:50–62. doi: 10.1016/j.expneurol.2014.09.021
173. Patki G, Lau YS. Impact of exercise on mitochondrial transcription factor expression and damage in the striatum of a chronic mouse model of Parkinson's disease. Neurosci Lett. (2011) 505:268–72. doi: 10.1016/j.neulet.2011.10.036
174. Koo J-H, Cho J-Y, Lee U-B. Treadmill exercise alleviates motor deficits and improves mitochondrial import machinery in an MPTP-induced mouse model of Parkinson's disease. Exp Gerontol. (2017) 89:20–9. doi: 10.1016/j.exger.2017.01.001
175. Al-Jarrah M, Obaidat H, Bataineh Z, Walton L, Al-Khateeb A. Endurance exercise training protects against the upregulation of nitric oxide in the striatum of MPTP/probenecid mouse model of Parkinson's disease. NeuroRehabilitation. (2013) 32:141–7. doi: 10.3233/NRE-130831
176. Aguiar AS, Duzzioni M, Remor AP, Tristão FSM, Matheus FC, Raisman-Vozari R, et al. Moderate-intensity physical exercise protects against experimental 6-hydroxydopamine-induced hemiparkinsonism through Nrf2-antioxidant response element pathway. Neurochem Res. (2016) 41:64–72. doi: 10.1007/s11064-015-1709-8
Keywords: Parkinson's disease, physical activity, neurotrophic factors, neuroplasticity, dopaminergic system
Citation: Palasz E, Niewiadomski W, Gasiorowska A, Wysocka A, Stepniewska A and Niewiadomska G (2019) Exercise-Induced Neuroprotection and Recovery of Motor Function in Animal Models of Parkinson's Disease. Front. Neurol. 10:1143. doi: 10.3389/fneur.2019.01143
Received: 30 May 2019; Accepted: 11 October 2019;
Published: 01 November 2019.
Edited by:
Ali Keshavarzian, Rush University, United StatesReviewed by:
Angel Sesar, Complejo Hospitalario Universitario de Santiago, SpainPedro Ribeiro, Federal University of Rio de Janeiro, Brazil
Copyright © 2019 Palasz, Niewiadomski, Gasiorowska, Wysocka, Stepniewska and Niewiadomska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Grazyna Niewiadomska, Zy5uaWV3aWFkb21za2FAbmVuY2tpLmdvdi5wbA==