 Karl B. Alstadhaug
Karl B. Alstadhaug Anna P. Andreou
Anna P. Andreou- 1Nordland Hospital Trust, Bodø, Norway
- 2Institute of Clinical Medicine, The Arctic University of Norway, Tromsø, Norway
- 3Headache Research, Wolfson CARD, Institute of Psychiatry, Psychology and Neuroscience, King's College London, London, United Kingdom
- 4The Headache Centre, Guy's and St Thomas', NHS Foundation Trust, London, United Kingdom
Background: The actions of caffeine as an antagonist of adenosine receptors have been extensively studied, and there is no doubt that both daily and sporadic dietary consumption of caffeine has substantial biological effects on the nervous system. Caffeine influences headaches, the migraine syndrome in particular, but how is unclear.
Materials and Methods: This is a narrative review based on selected articles from an extensive literature search. The aim of this study is to elucidate and discuss how caffeine may affect the migraine syndrome and discuss the potential pathophysiological pathways involved.
Results: Whether caffeine has any significant analgesic and/or prophylactic effect in migraine remains elusive. Neither is it clear whether caffeine withdrawal is an important trigger for migraine. However, withdrawal after chronic exposure of caffeine may cause migraine-like headache and a syndrome similar to that experienced in the prodromal phase of migraine. Sensory hypersensitivity however, does not seem to be a part of the caffeine withdrawal syndrome. Whether it is among migraineurs is unknown. From a modern viewpoint, the traditional vascular explanation of the withdrawal headache is too simplistic and partly not conceivable. Peripheral mechanisms can hardly explain prodromal symptoms and non-headache withdrawal symptoms. Several lines of evidence point at the hypothalamus as a locus where pivotal actions take place.
Conclusion: In general, chronic consumption of caffeine seems to increase the burden of migraine, but a protective effect as an acute treatment or in severely affected patients cannot be excluded. Future clinical trials should explore the relationship between caffeine withdrawal and migraine, and investigate the effects of long-term elimination.
“I am not acquainted with any agents which equal these substances (coffee and tea, aa), in the power of removing headache without leaving inconvenient results. And as their physiological action is so purely cerebral, restoring the intellectual faculties, and ministering to the sensations of well-being, as well as lessening any sad emotions, we have here an adequate presumption, were any required, that this headache is seated in the nerves, which are immediately related with the molecular action of the brain.”
John Addington Symonds, the Goulstonian lecture for 1858 (1)
Introduction
It is well-know that caffeine can stimulate wakefulness, increase concentration and decrease the sensation of fatigue (2), but how does it affect one of the most common human agonies (3), headaches? Caffeine is commonly used as analgesic adjuvant for the acute treatment of pain. However, despite the flattering description of the efficacy above, the general analgesic effect of caffeine seems at best modest (4). Besides, chronic consumption of it may have a flip side, withdrawal may cause caffeine withdrawal (5, 6), a syndrome including symptoms such as drowsiness, headache, mood-changes, difficulty focusing, nausea and muscle pain/stiffness (Box 1). Even small amounts of caffeine have been shown to suppress this (5). Headache can occur independently of the other symptoms (7), and Caffeine-withdrawal headache (Box 2), properly described in the 1940s (8, 9), is recognized as an own diagnostic entity by the International Classification of Headache Disorders (ICHD-3) (10). Results from both experimental and clinical studies indicate a high rate of caffeine withdrawal in the modern society, that may even be underestimated (5). The real world extent and clinical (physiological and psychological) importance are not well-known (11). The dual effects of caffeine in headaches, relieving on one side and triggering on the other side, make caffeine a very interesting substance in headache pathophysiology research. Still, the prevailing theory of the withdrawal headache is basically a rebound vasodilation due to caffeine's vasoconstrictive effect (12), at large a too simplistic theory that is not in conformity with modern views of headache pathophysiology (13). The caffeine withdrawal syndrome, which includes symptoms suggestive of the prodromal phase of migraine, is hardly of peripheral origin. Based on the current established knowledge on migraine pathophysiology, this narrative review aims to explore how caffeine, which has profound biological effect as an adenosine receptor antagonist (14), may influence pathways involved in headaches, with a particular focus in migraine.
Box 1. Diagnostic criteria for Caffeine-withdrawal according to the DSM-5.
A. Prolonged daily use of caffeine.
B. Abrupt cessation of caffeine use, or reduction in the amount of caffeine used, followed within 24 h by 3 or more of the following symptoms:
a) Headache.
b) Marked fatigue or drowsiness.
c) Dysphoric or depressed mood, or irritability.
d) Difficulty concentrating.
e) Symptoms of nausea, vomiting, or muscle pain/stiffness.
C. Clinically significant distress or impairment in social, occupational, or other important areas of functioning.
D. Not due to the direct physiological effects of a general medical condition and are not better accounted for by another mental disorder.
Box 2. Diagnostic criteria for Caffeine-withdrawal headache according to the ICHD-3.
A. Headache fulfilling criterion C.
B. Caffeine consumption of >200 mg/d for > 2 weeks, which has been interrupted or delayed.
C. Evidence of causation demonstrated by both of the following:
1. Headache has developed within 24 h after last caffeine intake
2. Either or both of the following:
a) Headache is relieved within 1 h by intake of caffeine 100 mg.
b) Headache has resolved within 7 days after caffeine withdrawal.
D. Not better accounted for by another ICHD-3 diagnosis.
Materials and Methods
The article is based on unsystematic searches in PubMed with terms like “caffeine and headache,” “caffeine withdrawal,” “adenosine and headache,” with more, and on own knowledge of older and recent literature on migraine. A discretionary selection of publications was made.
Results and Discussion
Caffeine and Adenosine
Caffeine is a major constituent of coffee and tea, but also naturally occur in guarana, cola nuts, cocoa, and several other plants (2). Soft drinks, energy drinks, and dietary supplements are also important sources, in particular among the younger population (15). The typical level in an ordinary cup of coffee varies between 50 and 100 mg (2). After oral ingestion, caffeine is rapidly and completely absorbed (99%), with peak plasma concentration reached usually within an hour (16, 17). Caffeine passes through all biological membranes, including the blood-brain barrier, and is distributed in all body fluids (18). It is metabolized by the cytochrome P450 system, the isoenzyme CYP1A2 responsible for 90% of caffeine clearance, and in adults it has a typical half-life of 5 h (19). Numerous factors, including a broad and variable genetic basis (20, 21), modify caffeine clearance.
The actions of caffeine in doses relevant to human consumption (50 to several 100 milligrams) are through antagonism of G protein-coupled purinergic (P1) receptors, more specifically the adenosine receptors (ARs), preferentially the A1R and A2AR (18, 22, 23). The human genes encoding for these two receptors are ADORA1 and ADORA2, respectively. Single nucleotide polymorphisms (SNPs) in ADORA2 appear to play a role in an individual's subjective response to caffeine (24–26).
Adenosine is found in every cell in the form of adenosine 5′-diphosphate (ADP) or adenosine 5′-triphosphate (ATP). It is continuously formed by breakdown of ATP and the physiological effects of adenosine are directly related to the metabolic activity (27). It modulates the activity of numerous cells, including mast cells, smooth muscle cells, platelets, and neurons (28). In the nervous system, adenosine appears to play an important role in modulating brain neurotransmitter release, locomotion, reward, sleep/wakefulness, cognition, and analgesia (14). Adenosine is neither stored nor released as a classical neurotransmitter, but may exert marked effects on neuronal excitability through G protein-coupled adenosine receptors (AR) A1, A2A, A2B, and A3 in both the peripheral and the central nervous system. The A1R is virtually found everywhere in the brain. In general it mediates tonic inhibition, especially through inhibition of glutamate release from presynaptic nerve endings (27). The A2AR is concentrated in dopamine-rich regions (18), especially on striato-pallidal GABAergic neurons (23), where it exerts excitatory effects on neurons and contributes to inhibition of motor activity (29). The A2AR actions are complicated by the fact that it co-localize and form heteromeres with dopamine-, cannabinoid-, and glutamate receptors (23). The roles of A2BR and A3R are not as well understood, but it appears that adenosine has a lower affinity for these receptors compared to A1R and A2AR receptors. Their activation is more likely to happen under conditions of hypoxia during which there is increased adenosine availability (30, 31).
The Caffeine's Effects Are in General Opposite to the Effects of Adenosine
The brain levels of adenosine in cats and rodents in resting physiological condition have been estimated to 30–200 nM/L, concentrations sufficient to activate A1, A2A and possibly A3 receptors if numerous on the cells (30, 31), but in most tissues the adenosine signaling is not very prominent (14). Even low concentrations of caffeine, such as 1–10 μM achieved after consumption of a single cup of coffee, result in significant antagonism of adenosine A1 and A2A receptors and may result in increased alertness (32).
Sleep and Arousal
An important function of adenosine in the CNS is its involvement in the sleep/arousal system. Adenosine has sleep-promoting effects (33). During sustained and prolonged wakefulness the extracellular adenosine accumulates in the basal forebrain cholinergic region, and it declines slowly during recovery sleep (34). This rise of adenosine reduces the cortical activity by direct A1R modulation of the corticopetal system, the major extrathalamic relay of the reticular ascending system (RAS) to the cortex, and indirectly via A2AR-modulation of the hypothalamus (35). It has been shown, for instance, that infusion of a specific A2AR agonist into the subarachnoidal space, inducing NREM sleep, will cause increased activity in the ventrolateral-preoptic (VLPO) area of the anterior hypothalamus and a reduced activity of the tuberomammilary nuclei (TMN) in the posterior hypothalamus as seen by increased number of Fos-positive neurons (36). Caffeine-induced wakefulness depends on adenosine A2A receptors (37). It seems that blocking of A2AR in nucleus accumbens inhibits the GABAergic output to the lateral hypothalamus, the TMN and the locus coeruleus (LC), causing activation and the major arousal effect of caffeine (38). Genetic knockout models have shown that both A1 and A2A receptors are involved in mediating the sleep-promoting properties of adenosine in the brain (39). Moreover, the arousal effects of caffeine seen in wild-type animals are blunted in ADORA2A-knockout mice (38). It is therefore conceivable that adenosine receptor ligands could be used as normal cognitive enhancers or sleep promoters.
Pain
Experimental data indicate that caffeine at doses between 25 and 100 mg/kg have intrinsic antinociceptive effects (40). To extrapolate data derived from animal experiments to humans, it is generally assumed that giving 10 mg/kg caffeine to a rat corresponds to giving 3.5 mg/kg (about two to three cups of coffee) to a 70 kg human (18), although such an assumption may be premature given the higher metabolic rate of rodents and hence the shorter half-life of caffeine in their system. Based on this assumption, caffeine at doses around 600–1,200 mg may be needed to achieve anti-nociception in caffeine-naive humans. As an adjuvant caffeine (≥65 mg) can potentiate the analgesic properties of other medications (4, 41) by 40% (42), and there is a possibility that caffeine alone in such low doses might have intrinsic analgesic properties for some types of pain, such as headache. Ward et al. employed a double-blind placebo-controlled crossover design to assess whether caffeine alone (60 and 130 mg) has independent analgesic effects on non-migrainous headaches, and found equivalent effects to acetaminophen (43). Caffeine, and combination analgesics with caffeine may be used in tension-type headache, but frequent use is not recommended due to the risk of developing medication-overuse headache (41). It is also used in hypnic headache (44) and post-lumbar puncture headache (45), but probably not to the analgesic effect per se. In hypnic headache the effect of caffeine is claimed to go “beyond the usual analgesic effects observed in other headache disorders” (44). Hypothalamic effects, as described in the next section, may play a role. The effect on post-lumbar puncture headache, which has “not conclusive evidence” according to a Cochrane report from 2013 (45), has been attributed non-analgesic mechanisms, including adenosine-mediated vasoconstriction (46, 47).
Analgesic properties of caffeine may be hard to reconcile with analgesic effects of adenosine. When the concentrations of adenosine increase during stressful conditions, including noxious stimulation, it may reduce pain (48), and adenosine receptor agonists produce antinociception in a variety of pain models (49). Mice lacking A1R show signs of increased anxiety and hyperalgesia, and the antinociceptive effects of adenosine seen in wild-type mice (50) cannot be shown (48). A1R is present on peripheral sensory nerve terminals and in lamina II of the spinal cord, and it has been suggested that peripheral anti-nociception achieved by activation of A1R occurs via blocked release of endogenous calciton gene-related peptide (CGRP) (51) and substance P (52). Under inflammatory conditions, experimental data indicate that the analgesic effect is by reducing hypersensitivity through a central mechanism (49, 53). This is in accordance with human studies, showing that intrathecal injection of adenosine does not cause antinociception to acute thermal or chemical stimuli, but reduce allodynia from intradermal capsaicin injection (53). Adenosine may also increase pain mediated by A2A receptors (28). Experimental data indicate that there are pro-nociceptive A2ARs on the peripheral nerve terminals (28, 54), and that blocking of these may cause antinociception (40). Caffeine in doses normally consumed by humans can probably act as an analgesic, partly through blockade of A2A receptor (40). Central dopaminergic mechanisms may also be involved. Like other analgesics (55), caffeine increases dopamine release (18), probably via inhibition of A2AR (56). Notably, A2A receptor antagonists did not affect dural meningeal vasodilatation caused by CGRP in a rat model (57). The recent marketing of CGRP receptor antibodies and the development of CGRP antagonists has been considered a major breakthrough in the migraine field (58). Despite the fact that A2AR does not appear to be located in the spinal cord, a specific inhibition of the activity of the intermediolateral cell column (the sympathetic system) was shown by Brooks et al., indicating that the A2AR may be located on presynaptic inhibitory terminals of descending fibers from higher brain centers (59).
It is clear that both adenosine and blockade of adenosine by caffeine may cause anti-nociception. Since the nanomolar affinities of adenosine for A1R and A2AR are almost the same, this indicates a fine balanced modulation of the pain processing (30), making it very difficult to predict the net effects of caffeine on nociception in humans.
Caffeine Overuse and Withdrawal
Caffeine causes increased well-being in small to moderate doses and its overuse has the potential to cause physical dependence (60). The caffeine dependence syndrome has been recognized by the World Health Organization as a behavioral disorders due to frequent use of caffeine (61). Considerable evidence suggests that this is due to enhanced dopaminergic activity, especially via blocking of A2AR causing increased dopamine release in the ventral striatum (nucleus accumbens) (26). However, there is a compensatory up-regulation of the adenosine system, causing increased functional sensitivity to adenosine during withdrawal (5, 10). These molecular changes appear to increase functional sensitivity to adenosine during caffeine abstinence, and play an important role in the behavioral and physiological effects produced by caffeine withdrawal (5, 10). In humans, caffeine withdrawal following chronic consumption may give rise to a time-limited syndrome comprising of headache, drowsiness, mood-changes, difficulty focusing, nausea, and muscle pain/stiffness (Box 1) (5, 6, 62). Consequently, caffeine consumption may be maintained to avoid withdrawal symptoms. Caffeine-withdrawal headache is a headache developing within 24 h after regular consumption of caffeine in excess of 200 mg/day for more than 2 weeks, which has been interrupted. According to the ICHD3 classification, this type of headache resolves spontaneously within 7 days in the absence of further consumption (10).
Further, repeated exposure to caffeine may lead to rapid development of tolerance, preferentially to the A1-blocking effect, and in some cases it even may result in opposite effects than expected (14). By drinking three to four cups of coffee regularly around 50% A1 and A2A receptor occupancy can be achieved for several hours, and many of the actions of caffeine are due to this AR blockade (18).
The Migraine Syndrome
Migraine is a disorder characterized by recurrent attacks of head pain associated with hypersensitivity to sensory stimuli (10) and when full-blown it involves different phases (63). (a) A prodromal or premonitory phase hours prior to the onset of the headache with a broad range of symptoms (Table 1), which patients can reliably recognize, and thus predict the occurrence of a headache, (b) Transient neurological symptoms, known as migraine aura (typically visual alterations), just before the actual headache starts (in migraine with aura patients), (c) An intense headache, typically involving only one site of the head, accompanied by nausea, sensitivity to light, noise and smells, (d) The postdrome phase following the resolution of the headache and characterized mainly by fatigue and inability to concentrate (66). Understanding the mechanisms involved in the transition from a headache-free to the headache state is crucial in understanding the underlying cause of headaches and the development abortive drugs. In many primary headache disorders, but especially migraine (67, 68), several external factors have been reported to trigger this transition; stress, bright light and lack of sleep are probably the most commonly reported (69). The periodicity of migraine attacks strongly indicates involvement of internal clock mechanisms in its pathophysiology (70).
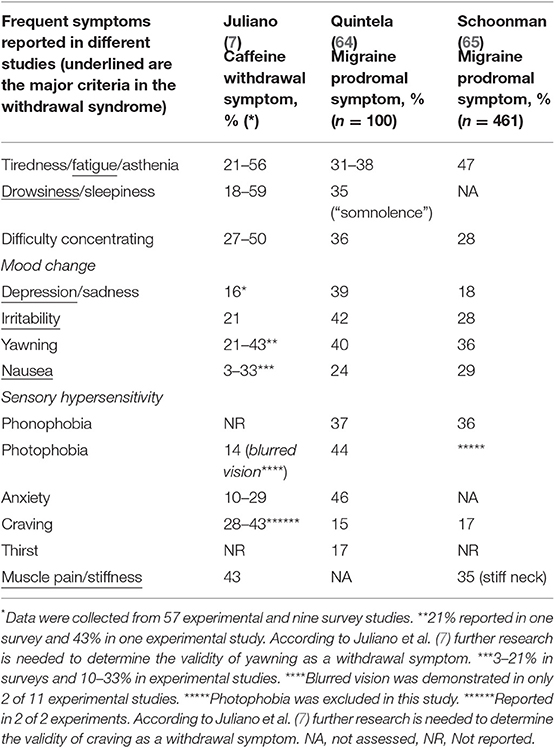
Table 1. Caffeine withdrawal symptoms and migraine prodromal symptoms.
Does the Migraine Prodromal Syndrome and Caffeine-Withdrawal Syndrome Share the Same Symptoms?
The prodromal, or premonitory, phase of migraine is usually defined as the period 2–48 h prior to aura or migraine headache with symptoms (Table 1) indicating an attack. Thirty to 90% of migraine patients report such a phase (71). In a prospective electronic diary study, Giffin et al. found that yawning, increased emotionality and concentration difficulties (difficulty reading and speaking) were the most reliable predictors (72). In another study by Schoonman et al., where patients had to choose among 12 specific premonitory symptoms, the most frequently reported were fatigue, phonophobia and yawning. Increased emotionality and concentration difficulties were reported by almost one third (10). Kelman found that tiredness, mood change and gastrointestinal symptoms (nausea) were the most frequent reported symptoms, and that yawning was rarely reported (69). However, there were specific questions about the former categories, but none about the latter symptom. In a prospective survey of 100 unselected patients, Quintela et al. (64) found that anxiety, phonophobia, irritability, unhappiness and yawning were the most common reported prodromal symptoms, whereas asthenia, tiredness, somnolence and concentration difficulties were the most common symptoms reported in the postdromal phase. Due to different methodologies used to identify symptoms, it may be difficult to compare results from different studies, but interestingly, the most prevalent symptoms reported are much the same as the symptoms of the caffeine-withdrawal syndrome. In Table 1 findings from the very good and comprehensive review about caffeine withdrawal symptoms made by Juliano et al. (7) are compared with findings from two of the mentioned surveys. These data strongly suggest that the same or similar pathophysiological pathways may be involved in both the prodromal phase of migraine and the caffeine withdrawal syndrome. However, sensory hypersensitivity, a cardinal migraineous feature (10), does not seem to be part of the caffeine withdrawal syndrome. This may nevertheless be due to an underlying thalamocortical dysrhythmia (73), that appears to be specific for migraine. The overlap of symptoms may also be due to the fact that caffeine withdrawal may act as a trigger of a migraine attack in migraine patients?
Is the Caffeine Withdrawal Headache Similar to Migraine?
Headache as a caffeine withdrawal symptom has been frequently studied (7), and in some case reports and experimental studies it has been characterized. However, premorbid primary headache syndromes, even in otherwise solid papers (74), are seldom reported. It seems though that subjects with a history of frequent headaches (75) are at increased risk, and that migraineurs and subjects who experience withdrawal headache share some comorbidity such as major depression and anxiety (5). To “produce experimental headaches which would be more physiological than experimental histamine and nitrite headaches,” Dreisbach in 1940 gave a number of non-habitual coffee drinkers capsules of 10–12 grains (650–780 mg) of caffeine daily for 1 week and then withdrew the treatment. On the day of withdrawal, almost all developed a moderate to severe headache. In migraineurs, “typical migraine syndromes” ensued (8). Unfortunately, the report does not give sufficient data for valid interpretation. Three years later, however, he and a colleague reported 38 similar trials in 24 persons of whom 5 had migraine. In 21 of the trials the subjects experienced their worst headache ever, in 11 it was definite but not severe, and in 6 there was slight or no headache. All of the migraineours experienced caffeine withdrawal headache “quite different from the migraine syndrome.” It is reason to question this statement. The headache was accompanied by nausea in 4 and vomiting in 1. In 4 subjects the headache was consistently accompanied by serous rhinorrhea. This may indicate cranial autonomic symptoms, frequently seen in migraine (76). No information about other accompanying symptoms was given. It was argued that 2 subjects had headache localized to the opposite side of the head from their usual migraine. In one subject, who used to have left-sided frontal migraine, it occurred bilaterally and was localized in the occipital region. Our clinical experience, however, confirms that is not unlikely for migraine to switch sides or to be bilateral rather than unilateral. Scotomas, regularly experienced by 3 migraine patients, did not occur. Scotoma, is a symptom usually described in migraine with aura patients and to date no headache-induced substance has been found to consistently trigger migraine aura. Further, two of the migraineurs had migraine attacks on the height of the caffeine stimulation, and in one of these subjects this reoccurred in a second trial. In these trials, and in later studies the headache is described to typically evolve gradually (60, 77, 78), being diffuse (79), throbbing (9), severe (60, 80), intensified with exercise (77), and Valsalva manoeuver (9) and having a mean duration for 2–3 days (81).
Despite being claimed that caffeine-related headache has a non-migrainous clinical presentation (82), the description of caffeine withdrawal headache given in the literature is not very different from migraine. With the high prevalence of migraine (83), and the lack of information on pre-existing headache in many studies taken into consideration, there seems to be a clear shortcoming in the knowledge when it comes to separating caffeine withdrawal as a migraine trigger from caffeine withdrawal headache per se.
Does Caffeine Cause Headaches?
Caffeine itself is rarely reported as a trigger of migraine (68), but as noticed by Driesbach it probably has the potential (9). As an important cause of medication-overuse headache (84) caffeine has been strongly incriminated, partly due to the withdrawal syndrome encouraging patients to continue their overuse (85–87). In a double blinded, randomized, placebo-controlled, 12 week crossover study of 45 healthy subjects who habitually consumed 4 to 6 cups of coffee a day (81),withdrawal caused headaches during the first or second day in 19 (42%) (81). The ratio of subjects reporting headaches during caffeine weeks and during non-caffeine weeks was 1. However, what is obvious, but not commented by the authors, is that when the withdrawal weeks (week 1 and 7) are removed the ratio increases to 7.7. Subjects also reported improved sleep during the non-caffeine weeks.
Caffeine-overuse may even cause chronic headache in children (15). Hering-Hanit and Gadoth reported 36 children with chronic headache, none with a prior history of migraine, who consumed excessive amounts of caffeine in the form of cola drinks (in average ~190 mg caffeine/d), and of whom 33 completely recovered after gradual withdrawal. Three children continued to have infrequent migraine without aura after withdrawal (15). In general, most chronic daily headaches in children appear to be migraine related (88). Caffeine has also been a suspected risk factor for episodic migraine evolving into chronic migraine, which in essence is defined as headache on ≥15 d/month for >3 months (66). A strong positive correlation between caffeine consumption and both episodic (89) and chronic migraine (90) has been found. In a retrospective population-based study, Scher et al. found that dietary and medicinal caffeine consumption appears to be only a modest risk factor for developing chronic daily headache, including chronic migraine (91). Overall, no relationship between current caffeine consumption and headache was found, neither was that found in two further studies (92, 93). However, in the cross-sectional population-based study by Boardman et al., headache sufferers were more than twice as likely to be heavy caffeine consumers compared to non-sufferers (92). In a Norwegian large-scale cross-sectional population-based study only a small association between high consumption of caffeine (>540 mg/d) and infrequent migraine was found (94). In this study chronic headache was more prevalent among individuals with low caffeine consumption (mean of 125 mg/d).
In general, chronic consumption of caffeine seems to be associated with increased migraine burden. Either chronic caffeine increases the risk of migraine per se, or in contrast, has beneficial effects in patients severely affected by migraine. Whether long-term elimination of caffeine diet will reduce the migraine burden has not been studied previously.
Pathophysiological Aspects
In 1990, Welch et al. substantiated the concept of migraine as a state of central neuronal hyperexcitability (95). Later it has become clear that some sort of cortical dysexcitability may even be present between attack (73). The mechanisms underlying the abnormal regulation of cortical function and the cyclic features of migraine remain largely unknown, but older, largely intuition based theories of hypothalamic dysfunction as the cause of periodicity of attacks have in recent years had a renaissance, especially after brain imaging evidence of hypothalamic activity both in the prodromal (96) and the pain phase (97) of migraine. There is accumulating indirect evidence for a pivotal role of hypothalamus in many primary headache disorders (98–102). The discovery of a mutation in a clock-gene (CK1δ) causing so called familiar advanced sleep phase syndrome was strongly linked to migraine both clinically and experimentally (103). Mice engineered to carry the mutation exhibit lower threshold for cortical spreading depression (CSD), the phenomenon underlying aura (see beneath), and increased sensitivity to noxious stimuli after nitro-glycerine treatment. A clear relationship between sleep and migraine exists (104), and caffeine can certainly disrupt sleep (105). Disrupted sleep-patterns predispose, and sleep per se protects against migraine (100). The risk of getting a migraine attack is low during sleep, but spikes in the morning, especially when associated with insomnia (106), and peaks in the afternoon probably due to work-related stress (107). Menstrual migraine and weekend headaches are also clear examples of the periodicity. The higher prevalence of migraine on weekend mornings reported was attributed to caffeine withdrawal by Couturier et al. (108).
Adenosine Can Cause Headache
It is now accepted that the pain during a migraine attack is perceived to be felt on intracranial structures, such as, the dura mater and intracranial vasculature (109, 110). Activation of the ophthalmic division (V1) of the trigeminal nerve is regarded primarily responsible for causing the pain in primary headache disorders (102). Upon activation of the trigeminal fibers, several neuropeptides such as substance P, neurokinin A, calcitonin gene-related peptide (CGRP), pituitary adenylate cyclase activating peptide (PACAP) and nitric oxide (NO) are released into the innervated tissue (dura mater and meningeal vessels) with the potential to cause neurogenic inflammation. Migraine has been attributed to such a local sterile meningeal inflammation (111), possibly involving release of histamine from mast cells (112). Activation of the trigeminal fibers is referred to as “trigeminovascular activation” and is considered the key event in causing headache. Accumulating data support a pivotal role of CGRP in migraine (58), and when given intravenously a delayed migraine-like headache will be induced in a large fraction of migraineurs but not in controls (113). CGRP causes only a modest, around 10%, vasodilation which is unlikely to activate perivascular trigeminal afferents (114). In general, the former “vascular theory” of migraine has been abandoned by several experiments showing that vasodilatation of neither extracranial- or intracranial/meningeal arteries is neither necessary nor sufficient to cause migraine headache (113). However, vasodilatation may worsen pain in an already sensitized pain network. The trigeminal fibers, that carry the sensory information from the intracranial structures, project on second-order neurons within the trigeminocervical complex (TCC; trigeminal nucleus caudalis, C1 and C2 spinal levels). These neurons give rise to the main ascending trigeminothalamic pathway that carries sensory information to third order neurons in the thalamus, before processing the information to higher cortical areas (115).
As with other recognized experimental triggers of migraine (116), such as CGRP, it is hard to conceive how adenosine itself, not passing the blood-brain barrier readily (14), could act centrally to elicit a migraine cascade. Further, adenosine has been ascribed peripheral anti-nociceptive effects by blocking the release of CGRP (51). Nevertheless, increased levels of adenosine in the blood during migraine attacks have been reported (117), and headaches are often reported when adenosine is used intravenously in cardiology (118). The A2A agonist regadenoson does also induce headache frequently (119). The drug dipyramidole, which increases the level of adenosine by inhibiting adenosine re-uptake, can cause headache as an adverse effect in one third of the patients. It has been also reported to increase the migraine frequency in migraine patients (120) and to elicit migraine in an experimental setting (121). In addition, intravenous application of adenosine can trigger migraine attacks (122). Based on this, antagonizing adenosine by caffeine may theoretically have an abortive effect in migraine. The problem is, as mentioned earlier, that chronic caffeine consumption in humans seems to increase the migraine burden. Supported by experimental findings (123, 124), an increased vascular tone due to up-regulation of adenosine receptors and compensatory increased levels of plasma adenosine have been suggested mechanisms underlying this risk (125, 126). Increased cerebral blood velocities have been measured after caffeine withdrawal (12), and there is no doubt that caffeine has the opposite effect causing a decrease in cerebral blood flow due to central vasoconstriction. However, the neuronal-vascular coupling is complex and in a functional magnetic resonance imaging study (fMRI), reduced cerebral perfusion induced by caffeine was independent of previous caffeine consumption (127). In a study of the rat middle meningeal artery, caffeine was found to reverse the relaxing effect of adenosine, mainly mediated by blocking of A2AR (128). Whether vasoconstriction significantly contributes to the effects of caffeine in withdrawal headache remains unclear since vasodilation hardly is a primary cause of the headache. As mentioned earlier, blocking of A2AR does not seem to affect neurogenic vasodilation (57). Further, discrepancy between different studies has been observed. In an experimental cat migraine model, the A1R agonist GR79236 had a dose-dependent inhibitory effect on trigeminovascular nociceptive transmission (129). This effect was explored in humans. In 12 healthy female volunteers the adenosine A1 receptor agonist GR79236 was shown to inhibit trigeminal nociception, as measured by the blink reflex (130). Further studies in pigs did not show any effect on capsaicin-induced CGRP release (131), and in a multicenter evaluation of the adenosine agonist GR79236X in patients with dental pain after third molar extraction, no efficacy was shown when compared to placebo (132).
Caffeine Has the Ability to Induce Cortical Excitability, and May Even Predispose for CSD?
Traditionally, the migraine aura has been considered a distinctive phase, but probably it is a consequence of the same or parallel mechanisms that triggers the pain (133, 134). It is believed that the migraine aura itself is caused by so called cortical spreading depression (CSD) (135). CSD is a wave of neuronal depolarisation linked with depressed neuronal activity and blood flow changes (136). In animals, CSD can quite readily be induced by focal cortical stimulation for example by applying K+ (137). Despite being accepted by most experts as a plausible mechanism of migraine aura and even headache in migraine without aura (“silent spreading depression”) it has been difficult to prove that CSD actually is the underlying mechanism in humans. It also remains enigmatic how CSD could be triggered in patients during migraine. So called calcium waves in astrocyte networks is a speculative (138), but alluring mechanism that could offer an alternative explanation to the classical CSD. Recent advances support the idea that astrocytes could play an important role in spreading depression initiation (139). ATP-receptors (P2 receptors) are both necessary and sufficient for propagation of calcium waves (140), and thus has the potential to initiate and sustain a heighten state of neuronal excitability. Caffeine may possibly modulate this susceptibility (141). An A2A receptor gene haplotype has been reported to be associated with migraine with aura (142), but the findings should be reproduced. There appears to be only one published case where intravenous adenosine precipitated migraine aura (122). Activation of A1R has been shown to increase K+ conductance and thus hyperpolarize CNS neurons (50). It is thus more plausible that acute caffeine could increase the susceptibility to CSD, and pre-published reports supported that (143, 144). However, caffeine exposure did not affect the susceptibility to CSD in a recent study of mice (82). Curiously, CSD can induce yawning in rats (145).
Yawning Indicates Hypothalamic Alterations
It has been demonstrated that activating the A1R on TMN-neurons increases NREM sleep (146), and that blocking them on hypocretinergic neurons of the lateral hypothalamus increases wakefulness (147). Both neurotransmitter systems have been suggested to play an important role in migraine (148, 149). The TMN of the posterior hypothalamus has been suggested to play a role in the initial phases of a migraine attack and to be responsible for the morning occurring migraine attacks (148, 150). During drowsiness and normal recovery sleep the firing from the histaminergic neurons are reduced or absent, but during wakening and arousal they fire, allegedly the most wake-selective firing pattern identified to date (151). Adenosine may well have a protective effect against migraine during sleep, but during non-recovery sleep and wakefulness disrupted homeostasis may cause increased histaminergic firing predisposing for headaches. It has been postulated that yawning is the manifestation of a switch in brain states from “default mode” to an “attentional mode” by increasing clearance of adenosine (152). It remains to be proven in experimental models that increased histaminergic firing sensitizes the TCC.
Yawning may also be indicative of an individual's inability to properly maintain thermal brain homeostasis (153). If yawning occurs without being associated with tiredness, it may perhaps indicate a thermoregulatory dysfunction. In the study of Schoonman et al. of the premonitory symptoms of migraine there was no correlation between “sleep problems” and yawning (Spearman's rank correlation of 0.024) (65). Further, Jacome described 3 migraineurs with compulsive yawning as a prodromal symptom, independent of fatigue and drowsiness (154). In a recent cross-sectional study, 45.4% of 339 migraineurs reported repetitive yawning during migraine attacks (155). Sleepiness was significantly more often reported in patients with yawning compared to those who did not yawn during their migraine attacks.
Thermoregulation and sleep are interrelated. It is well-known that yawning has a clear circadian pattern parallel to the rise in body and brain temperature, normally occurring most often before sleep onset and after waking (153). A hypothesis that migraine attacks serve to restore the brain temperature has recently been put forward (156). In general, the neurons of the brain are very sensitive to variations in the temperature (157). Short visual stimulation of the rat invokes a rise in temperature over the visual cortex (158), and during prolonged (4 min) visual stimulation in man, an increase in regional cerebral blood flow caused an average decrease in temperature by 0.2°C (159). Histamine has been shown to mainly excite heat-sensitive neurons in the anterior hypothalamus, causing hypothermia. In contrast to adenosine (160), caffeine increase body temperature parallel to arousal during circadian misalignment in humans (161). Injecting neuropeptide orexin-A into the rat PVN elicits a cortical arousal response followed by yawning (162), and injecting it into the ventromedial hypothalamus it induces hyperthermic reactions (163).
Based on the fact that migraineurs show a lower threshold for central dopamine receptor activation than normal subjects (164), and that exogenously administered dopamine receptor agonists may produce some symptoms experienced in the prodromal phase of migraine such as drowsiness and yawning, dopamine may play an important role in migraine pathophysiology. This theory is consistent with the idea that caffeine withdrawal symptoms are due to increased sensitivity of adenosine, causing increased drowsiness (due to increased disinhibition of VLPO sleep-active neurons reducing histaminergic tone) and excessive yawning due to increased dopaminergic tone. It has been shown that injecting dopamine (D2) agonists into the paraventricular nucleus of the hypothalamus (PVN) of rats, increases local nitric oxide (NO) production and thereby activates central oxytocinergic neurotransmission, inducing yawning (165). However, microinjection of other substances into the PVN, such as histamine (166) and nitroglycerine (167) also induces yawning. As distinct from dopamine agonists, that seldom induces headache (168), both donors of NO and histamine are established triggers in pharmacological models of migraine (116). Glyceryl trinitrate has even been shown to both induce prodromal symptoms of migraine (169) and activate the hypothalamus (96).
Caffeine Can Alter the Circadian System and Neuronal Excitability
It has been proposed that the hypersensitivity to light during migraine may be exerted by intrinsically photosensitive retinal ganglion cells (ipRGCs) through a pathway that modulates the activity of dura-sensitive thalamocortical neurons (170). The ipRGCs entrain the circadian rhythms and influence sleep/wakefulness (171). Adenosine, through A1R inhibition of glutamate release, seems to fine-tune the circadian system through gating of both photic and non-photic input to the superior biological clock, the suprachiasmatic nucleus (27). Giving an A1R agonist during midday, phase-shift mimicking the effect of a 3 h sleep deprivation procedure may be achieved in hamsters (172). Caffeine increases light responsiveness of the mouse circadian pacemaker (173), and it has been shown that chronic exposure to caffeine interferes with the ability of the SCN to entrain normally to light, and that it potentiates phase-delays (174). Further, it has been claimed that glasses that filter out blue light can reduce the frequency of migraine attacks with short periods of usage (175), and in an experimental study light stimulation with the peak wavelength of ipRGC induced migraine attacks more frequently than extensive-wavelength (176). Based on this, it may be speculated that caffeine interferes with the ability of the biological clock to entrain to light, causing increased excitability in pathways involved in migraine headache, including the dura-sensitive thalamocortical neurons.
The pharmacology of both the TCC and the thalamus provides interesting insights into migraine pathophysiology, as they are prominent sites of action of migraine specific medication-triptans (177), of clinically active preventives (178, 179) and of other potential anti-migraine compounds (180). The modulation of sensory transmission in the thalamus, assumes further significance as it has been shown not only to be a pivotal area for the development of sensory hypersensitivity to light during migraine (170), but could also participate in the development of hypersensitivity to noise (181), and non-cranial allodynia that is frequently seen in migraine patients (182). Experimental data show that presynaptic adenosine A1 receptors on thalamo-cortical neurons can mediate reduced cortical excitability directly (183). When interacting with serotonin, adenosine may also modulate thalamic sensory gating during sleep (184). If the caffeine withdrawal syndrome is due to increased sensitivity to adenosine, this may explain why sensory hypersensitivity is not a symptom of caffeine withdrawal. It is tempting to speculate that caffeine can increase the sensory hypersensitivity that accompanies migraine headache.
Both the TCC and the thalamus have reciprocal direct or indirect connections with multiple brainstem, midbrain and cortical nuclei that control the excitability of the ascending trigeminothalamic pathway (185). These brain nuclei make up the descending pain modulatory system (115), which is a powerful regulator of pain-related activity along the ascending trigeminothalamic pathway. Disruption of normal endogenous descending modulatory tone may play a critical role in primary headache disorders, but what really alters the excitability of the ascending trigeminothalamic pathway in a manner that a migraine attack develops in susceptible individuals, remains to be revealed. As mentioned earlier, central dopaminergic mechanisms (via A2AR) may be involved. In an experimental headache model, stimulation of the dopaminergic A11 significantly inhibited peri-MMA dural and noxious pinch evoked firing of neurons in the TCC, an effect that was blocked by a D2 receptor antagonist (186). Whether the A11 neurons have adenosine receptors, and whether caffeine induces release of dopamine from the A11 nucleus, is not known.
Conclusion
The current opinion is that caffeine both can relieve and trigger headaches. It has to be clarified whether caffeine withdrawal triggers or merely resembles the migraine syndrome. The nature of the caffeine withdrawal syndrome needs to be better understood. In assessing the clinical effects of caffeine withdrawal, there is a chance that a triggered migraine syndrome is interpreted as part of the caffeine withdrawal syndrome, explaining an overlap between these two. If it triggers migraine it offers a good human migraine model. A clinical trial thoroughly evaluating the withdrawal syndrome specifically in subjects with migraine should be performed. Furthermore, whether long-term elimination of caffeine diet will reduce or increase the burden of migraine should also be evaluated.
There is compelling evidence that adenosine may trigger headaches, but how this poor blood-brain-barrier penetrating substance can trigger central mechanisms when given intravenously remains enigmatic. Chronically blocking adenosine receptors by habitually drinking coffee seems to increase the burden of migraine, and it is tempting to believe that this causes an increased sensitivity to adenosine, evident when caffeine is withdrawn. Caffeine withdrawal and migraine prodromal symptoms are definitely caused by alterations in the CNS. Looking beyond the peripheral effects, central adenosine mechanisms should be explored in experimental headache models. The link between adenosine, the circadian system, sleep, and pain points at the posterior hypothalamus as a locus in quo. The effects of caffeine on the TMN and the A11 may offer novel insight, and the A2AR seems to be of particular interest. Two positron emission tomography (PET) A2AR-ligands have been developed, and this may render human in vivo imaging studies possible (14). Future research should also confirm and investigate a role of receptor genes like ADORA2A in migraine and caffeine withdrawal.
Author Contributions
AA conceived the manuscript. KA wrote it with support from AA.
Funding
This research received grant from Helse Nord (project number SFP1207-14).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Symonds JA. Gulstonian lectures for 1858. Headache. Medical Times and Gazette. (London) (1858). p. 37.
2. Heckman MA, Weil J, Gonzalez de Mejia E. Caffeine (1, 3, 7-trimethylxanthine) in foods: a comprehensive review on consumption, functionality, safety, and regulatory matters. J Food Sci. (2010) 75:R77–87. doi: 10.1111/j.1750-3841.2010.01561.x
3. Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. (2012) 380:2163–96. doi: 10.1016/S0140-6736(12)61729-2
4. Derry CJ, Derry S, Moore RA. Caffeine as an analgesic adjuvant for acute pain in adults. Cochrane Database Syst Rev. (2012) 3:CD009281. doi: 10.1002/14651858.CD009281.pub2
5. Griffiths RR, Reissig CJ. Substance Abuse: Caffeine Use Disorders. Psychiatry. John Wiley & Sons, Ltd. (2008). 1019–40 p. doi: 10.1002/9780470515167.ch56
6. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-5. Washington, DC: American Psychiatric Association (2013).
7. Juliano LM, Griffiths RR. A critical review of caffeine withdrawal: empirical validation of symptoms and signs, incidence, severity, and associated features. Psychopharmacology. (2004) 176:1–29. doi: 10.1007/s00213-004-2000-x
8. The Scientific Proceedings of The American Society For Pharmacology And Experimental Therapeutics, Incorporated. Thirty-First Annual Meeting, New Orleans, Louisiana, March 13–16, 1940. J Pharmacol Exp Ther. (1940) 69:273–310.
10. Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition. Cephalalgia. (2018) 38:1–211. doi: 10.1177/0333102417738202
11. Addicott MA. Caffeine use disorder: a review of the evidence and future implications. Curr Addict Rep. (2014) 1:186–92. doi: 10.1007/s40429-014-0024-9
12. Couturier EG, Laman DM, van Duijn MA, van Duijn H. Influence of caffeine and caffeine withdrawal on headache and cerebral blood flow velocities. Cephalalgia. (1997) 17:188–90. doi: 10.1046/j.1468-2982.1997.1703188.x
13. Charles A. The pathophysiology of migraine: implications for clinical management. Lancet Neurol. (2018) 17:174–82. doi: 10.1016/S1474-4422(17)30435-0
14. Chen JF, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets–what are the challenges? Nat Rev Drug Discov. (2013) 12:265–86. doi: 10.1038/nrd3955
15. Hering-Hanit R, Gadoth N. Caffeine-induced headache in children and adolescents. Cephalalgia. (2003) 23:332–5. doi: 10.1046/j.1468-2982.2003.00576.x
16. Blanchard J, Sawers SJ. The absolute bioavailability of caffeine in man. Eur J Clin Pharmacol. (1983) 24:93–8. doi: 10.1007/BF00613933
17. Lang R, Dieminger N, Beusch A, Lee YM, Dunkel A, Suess B, et al. Bioappearance and pharmacokinetics of bioactives upon coffee consumption. Anal Bioanal Chem. (2013) 405:8487–503. doi: 10.1007/s00216-013-7288-0
18. Fredholm BB, Battig K, Holmen J, Nehlig A, Zvartau EE. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev. (1999) 51:83–133.
19. Denaro CP, Brown CR, Jacob P III, Benowitz NL. Effects of caffeine with repeated dosing. Eur J Clin Pharmacol. (1991) 40:273–8. doi: 10.1007/BF00315208
20. Yang A, Palmer AA, de Wit H. Genetics of caffeine consumption and responses to caffeine. Psychopharmacology. (2010) 211:245–57. doi: 10.1007/s00213-010-1900-1
21. Landolt HP. “No thanks, coffee keeps me awake”: individual caffeine sensitivity depends on ADORA2A genotype. Sleep. (2012) 35:899–900. doi: 10.5665/sleep.1942
22. Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov. (2006) 5:247–64. doi: 10.1038/nrd1983
23. de Lera Ruiz M, Lim YH, Zheng J. Adenosine A2A receptor as a drug discovery target. J Med Chem. (2014) 57:3623–50. doi: 10.1021/jm4011669
24. Rogers PJ, Hohoff C, Heatherley SV, Mullings EL, Maxfield PJ, Evershed RP, et al. Association of the anxiogenic and alerting effects of caffeine with ADORA2A and ADORA1 polymorphisms and habitual level of caffeine consumption. Neuropsychopharmacology. (2010) 35:1973–83. doi: 10.1038/npp.2010.71
25. Hohoff C, Garibotto V, Elmenhorst D, Baffa A, Kroll T, Hoffmann A, et al. Association of adenosine receptor gene polymorphisms and in vivo adenosine A1 receptor binding in the human brain. Neuropsychopharmacology. (2014) 39:2989–99. doi: 10.1038/npp.2014.150
26. Ferre S, O'Brien MC. Alcohol and caffeine: the perfect storm. J Caffeine Res. (2011) 1:153–62. doi: 10.1089/jcr.2011.0017
27. Ruby CL, O'Connor KM, Ayers-Ringler J, Choi DS. Adenosine and glutamate in neuroglial interaction: implications for circadian disorders and alcoholism. Adv Neurobiol. (2014) 11:103–19. doi: 10.1007/978-3-319-08894-5_6
28. Ledent C, Vaugeois JM, Schiffmann SN, Pedrazzini T, El Yacoubi M, Vanderhaeghen JJ, et al. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature. (1997) 388:674–8. doi: 10.1038/41771
29. Aoyama S, Kase H, Borrelli E. Rescue of locomotor impairment in dopamine D2 receptor-deficient mice by an adenosine A2A receptor antagonist. J Neurosci. (2000) 20:5848–52. doi: 10.1523/JNEUROSCI.20-15-05848.2000
30. Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International union of pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. (2001) 53:527–52.
31. Fredholm BB. Adenosine–a physiological or pathophysiological agent? J Mol Med. (2014) 92:201–6. doi: 10.1007/s00109-013-1101-6
32. Fredholm BB, Battig K, Holmen J, Nehlig A, Zvartau EE. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev. (1999) 51:83–133.
33. Basheer R, Strecker RE, Thakkar MM, McCarley RW. Adenosine and sleep-wake regulation. Prog Neurobiol. (2004) 73:379–96. doi: 10.1016/j.pneurobio.2004.06.004
34. Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW. Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science. (1997) 276:1265–8. doi: 10.1126/science.276.5316.1265
35. Ferre S. Role of the central ascending neurotransmitter systems in the psychostimulant effects of caffeine. J Alzheimers Dis. (2010) 20(Suppl. 1):S35–49. doi: 10.3233/JAD-2010-1400
36. Scammell TE, Gerashchenko DY, Mochizuki T, McCarthy MT, Estabrooke IV, Sears CA, et al. An adenosine A2a agonist increases sleep and induces Fos in ventrolateral preoptic neurons. Neuroscience. (2001) 107:653–63. doi: 10.1016/S0306-4522(01)00383-9
37. Huang ZL, Qu WM, Eguchi N, Chen JF, Schwarzschild MA, Fredholm BB, et al. Adenosine A2A, but not A1, receptors mediate the arousal effect of caffeine. Nat Neurosci. (2005) 8:858–9. doi: 10.1038/nn1491
38. Lazarus M, Shen HY, Cherasse Y, Qu WM, Huang ZL, Bass CE, et al. Arousal effect of caffeine depends on adenosine A2A receptors in the shell of the nucleus accumbens. J Neurosci. (2011) 31:10067–75. doi: 10.1523/JNEUROSCI.6730-10.2011
39. Huang ZL, Urade Y, Hayaishi O. The role of adenosine in the regulation of sleep. Curr Top Med Chem. (2011) 11:1047–57. doi: 10.2174/156802611795347654
41. Bendtsen L, Evers S, Linde M, Mitsikostas DD, Sandrini G, Schoenen J. EFNS guideline on the treatment of tension-type headache - report of an EFNS task force. Eur J Neurol. (2010) 17:1318–25. doi: 10.1111/j.1468-1331.2010.03070.x
42. Zhang WY. A benefit-risk assessment of caffeine as an analgesic adjuvant. Drug Saf. (2001) 24:1127–42. doi: 10.2165/00002018-200124150-00004
43. Ward N, Whitney C, Avery D, Dunner D. The analgesic effects of caffeine in headache. Pain. (1991) 44:151–5. doi: 10.1016/0304-3959(91)90129-L
44. Holle D, Obermann M. Hypnic headache and caffeine. Expert Rev Neurother. (2012) 12:1125–32. doi: 10.1586/ern.12.100
45. Basurto Ona X, Uriona Tuma SM, Martinez Garcia L, Sola I, Bonfill Cosp X. Drug therapy for preventing post-dural puncture headache. Cochrane Database Syst Rev. (2013) 2:CD001792. doi: 10.1002/14651858.CD001792.pub3
46. Camann WR, Murray RS, Mushlin PS, Lambert DH. Effects of oral caffeine on postdural puncture headache. A double-blind, placebo-controlled trial. Anesth Analg. (1990) 70:181–4. doi: 10.1213/00000539-199002000-00009
47. Moyer JH, Tashnek AB, Miller SI, Snyder H, Bowman RO. The effect of theophylline with ethylenediamine (aminophylline) and caffeine on cerebral hemodynamics and cerebrospinal fluid pressure in patients with hypertensive headaches. Am J Med Sci. (1952) 224:377–85. doi: 10.1097/00000441-195210000-00002
48. Johansson B, Halldner L, Dunwiddie TV, Masino SA, Poelchen W, Gimenez-Llort L, et al. Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc Natl Acad Sci USA. (2001) 98:9407–12. doi: 10.1073/pnas.161292398
49. Li X, Conklin D, Pan HL, Eisenach JC. Allosteric adenosine receptor modulation reduces hypersensitivity following peripheral inflammation by a central mechanism. J Pharm Exp Ther. (2003) 305:950–5. doi: 10.1124/jpet.102.047951
50. Sawynok J. Adenosine receptor activation and nociception. Eur J Pharmacol. (1998) 347:1–11. doi: 10.1016/S0014-2999(97)01605-1
51. Carruthers AM, Sellers LA, Jenkins DW, Jarvie EM, Feniuk W, Humphrey PP. Adenosine A(1) receptor-mediated inhibition of protein kinase A-induced calcitonin gene-related peptide release from rat trigeminal neurons. Mol Pharmacol. (2001) 59:1533–41. doi: 10.1124/mol.59.6.1533
52. Sjolund KF, Sollevi A, Segerdahl M, Lundeberg T. Intrathecal adenosine analog administration reduces substance P in cerebrospinal fluid along with behavioral effects that suggest antinociception in rats. Anesth Analg. (1997) 85:627–32. doi: 10.1213/00000539-199709000-00025
53. Eisenach JC, Hood DD, Curry R. Preliminary efficacy assessment of intrathecal injection of an American formulation of adenosine in humans. Anesthesiology. (2002) 96:29–34. doi: 10.1097/00000542-200201000-00011
54. Ferre S, Diamond I, Goldberg SR, Yao L, Hourani SM, Huang ZL, et al. Adenosine A2A receptors in ventral striatum, hypothalamus and nociceptive circuitry implications for drug addiction, sleep and pain. Prog Neurobiol. (2007) 83:332–47. doi: 10.1016/j.pneurobio.2007.04.002
55. Jarcho JM, Mayer EA, Jiang ZK, Feier NA, London ED. Pain, affective symptoms, and cognitive deficits in patients with cerebral dopamine dysfunction. Pain. (2012) 153:744–54. doi: 10.1016/j.pain.2012.01.002
56. Ferre S, Fuxe K, von Euler G, Johansson B, Fredholm BB. Adenosine-dopamine interactions in the brain. Neuroscience. (1992) 51:501–12. doi: 10.1016/0306-4522(92)90291-9
57. Haanes KA, Labastida-Ramirez A, Chan KY, de Vries R, Shook B, Jackson P, et al. Characterization of the trigeminovascular actions of several adenosine A2A receptor antagonists in an in vivo rat model of migraine. J Headache Pain. (2018) 19:41. doi: 10.1186/s10194-018-0867-x
58. Edvinsson L, Haanes KA, Warfvinge K, Krause DN. CGRP as the target of new migraine therapies - successful translation from bench to clinic. Nat Rev Neurol. (2018) 14:338–50. doi: 10.1038/s41582-018-0003-1
59. Brooke RE, Deuchars J, Deuchars SA. Input-specific modulation of neurotransmitter release in the lateral horn of the spinal cord via adenosine receptors. J Neurosci. (2004) 24:127–37. doi: 10.1523/JNEUROSCI.4591-03.2004
60. Griffiths RR, Evans SM, Heishman SJ, Preston KL, Sannerud CA, Wolf B, et al. Low-dose caffeine physical dependence in humans. J Pharmacol Exp Ther. (1990) 255:1123–32.
61. WHO. The ICD-10 Classification of Mental and Behavioural Disorders: Clinical Descriptions and Diagnostic Guidelines. Geneva (1992).
62. Juliano LM, Huntley ED, Harrell PT, Westerman AT. Development of the caffeine withdrawal symptom questionnaire: caffeine withdrawal symptoms cluster into 7 factors. Drug Alcohol Depend. (2012) 124:229–34. doi: 10.1016/j.drugalcdep.2012.01.009
63. Blau JN. Resolution of migraine attacks: sleep and the recovery phase. J Neurol Neurosurg Psychiatry. (1982) 45:223–6. doi: 10.1136/jnnp.45.3.223
64. Quintela E, Castillo J, Munoz P, Pascual J. Premonitory and resolution symptoms in migraine: a prospective study in 100 unselected patients. Cephalalgia. (2006) 26:1051–60. doi: 10.1111/j.1468-2982.2006.01157.x
65. Schoonman GG, Evers DJ, Terwindt GM, van Dijk JG, Ferrari MD. The prevalence of premonitory symptoms in migraine: a questionnaire study in 461 patients. Cephalalgia. (2006) 26:1209–13. doi: 10.1111/j.1468-2982.2006.01195.x
66. Headache Classification Committee of the International Headache Society (IHS). The international classification of headache disorders, 3rd edition (beta version). Cephalalgia. (2013) 33:629–808. doi: 10.1177/0333102413485658
68. Peroutka SJ. What turns on a migraine? A systematic review of migraine precipitating factors. Curr Pain Headache Rep. (2014) 18:454. doi: 10.1007/s11916-014-0454-z
69. Kelman L. The triggers or precipitants of the acute migraine attack. Cephalalgia. (2007) 27:394–402. doi: 10.1111/j.1468-2982.2007.01303.x
70. Alstadhaug K. Periodicity of migraine. In: Clarke LB, editor. Migraine Disorders Research Trends. New York, NY: Nova Science (2007). p. 107–44.
71. Becker WJ. The premonitory phase of migraine and migraine management. Cephalalgia. (2013) 33:1117–21. doi: 10.1177/0333102412437390
72. Giffin NJ, Ruggiero L, Lipton RB, Silberstein SD, Tvedskov JF, Olesen J, et al. Premonitory symptoms in migraine: an electronic diary study. Neurology. (2003) 60:935–40. doi: 10.1212/01.WNL.0000052998.58526.A9
73. Coppola G, Ambrosini A, Di Clemente L, Magis D, Fumal A, Gerard P, et al. Interictal abnormalities of gamma band activity in visual evoked responses in migraine: an indication of thalamocortical dysrhythmia? Cephalalgia. (2007) 27:1360–7. doi: 10.1111/j.1468-2982.2007.01466.x
74. Silverman K, Evans SM, Strain EC, Griffiths RR. Withdrawal syndrome after the double-blind cessation of caffeine consumption. N Engl J Med. (1992) 327:1109–14. doi: 10.1056/NEJM199210153271601
75. Weber JG, Klindworth JT, Arnold JJ, Danielson DR, Ereth MH. Prophylactic intravenous administration of caffeine and recovery after ambulatory surgical procedures. Mayo Clinic Proc. (1997) 72:621–6. doi: 10.1016/S0025-6196(11)63567-2
76. Barbanti P, Fabbrini G, Pesare M, Vanacore N, Cerbo R. Unilateral cranial autonomic symptoms in migraine. Cephalalgia. (2002) 22:256–9. doi: 10.1046/j.1468-2982.2002.00358.x
77. Greden JF, Victor BS, Fontaine P, Lubetsky M. Caffeine-withdrawal headache: a clinical profile. Psychosomatics. (1980) 21:411–3, 417–8. doi: 10.1016/S0033-3182(80)73670-8
78. Roller L. Caffeinism: subjective quantitative aspect of withdrawal syndrome. Med J Aust. (1981) 1:146. doi: 10.5694/j.1326-5377.1981.tb135407.x
79. Lader M, Cardwell C, Shine P, Scott N. Caffeine withdrawal symptoms and rate of metabolism. J Psychopharmacol. (1996) 10:110–8. doi: 10.1177/026988119601000205
80. Griffiths RR, Woodson PP. Reinforcing properties of caffeine: studies in humans and laboratory animals. Pharmacol Biochem Behav. (1988) 29:419–27. doi: 10.1016/0091-3057(88)90180-3
81. van Dusseldorp M, Katan MB. Headache caused by caffeine withdrawal among moderate coffee drinkers switched from ordinary to decaffeinated coffee: a 12 week double blind trial. BMJ. (1990) 300:1558–9. doi: 10.1136/bmj.300.6739.1558
82. Yalcin N, Chen SP, Yu ES, Liu TT, Yen JC, Atalay YB, et al. Caffeine does not affect susceptibility to cortical spreading depolarization in mice. J Cereb Blood Flow Metab. (2019) 39:740–50. doi: 10.1177/0271678X18768955
83. Leonardi M, Steiner TJ, Scher AT, Lipton RB. The global burden of migraine: measuring disability in headache disorders with WHO's Classification of Functioning, Disability and Health (ICF). J Headache Pain. (2005) 6:429–40. doi: 10.1007/s10194-005-0252-4
84. Alstadhaug KB, Ofte HK, Kristoffersen ES. Preventing and treating medication overuse headache. Pain Rep. (2017) 2:e612. doi: 10.1097/PR9.0000000000000612
85. Diener HC, Limmroth V. Medication-overuse headache: a worldwide problem. Lancet Neurol. (2004) 3:475–83. doi: 10.1016/S1474-4422(04)00824-5
86. Dong Z, Chen X, Steiner TJ, Hou L, Di H, He M, et al. Medication-overuse headache in China: Clinical profile, and an evaluation of the ICHD-3 beta diagnostic criteria. Cephalalgia. (2014) 35:644–51. doi: 10.1177/0333102414552533
87. Scher AI, Lipton RB, Stewart WF, Bigal M. Patterns of medication use by chronic and episodic headache sufferers in the general population: results from the frequent headache epidemiology study. Cephalalgia. (2010) 30:321–8. doi: 10.1111/j.1468-2982.2009.01913.x
88. Hershey AD. Chronic daily headaches in children. Expert Opin Pharmacother. (2003) 4:485–91. doi: 10.1517/14656566.4.4.485
89. Takeshima T, Ishizaki K, Fukuhara Y, Ijiri T, Kusumi M, Wakutani Y, et al. Population-based door-to-door survey of migraine in Japan: the Daisen study. Headache. (2004) 44:8–19. doi: 10.1111/j.1526-4610.2004.04004.x
90. Bigal ME, Sheftell FD, Rapoport AM, Tepper SJ, Lipton RB. Chronic daily headache: identification of factors associated with induction and transformation. Headache. (2002) 42:575–81. doi: 10.1046/j.1526-4610.2002.02143.x
91. Scher AI, Stewart WF, Lipton RB. Caffeine as a risk factor for chronic daily headache: a population-based study. Neurology. (2004) 63:2022–7. doi: 10.1212/01.WNL.0000145760.37852.ED
92. Boardman HF, Thomas E, Millson DS, Croft PR. Psychological, sleep, lifestyle, and comorbid associations with headache. Headache. (2005) 45:657–69. doi: 10.1111/j.1526-4610.2005.05133.x
93. Wiendels NJ, Knuistingh Neven A, Rosendaal FR, Spinhoven P, Zitman FG, Assendelft WJ, et al. Chronic frequent headache in the general population: prevalence and associated factors. Cephalalgia. (2006) 26:1434–42. doi: 10.1111/j.1468-2982.2006.01210.x
94. Hagen K, Thoresen K, Stovner LJ, Zwart JA. High dietary caffeine consumption is associated with a modest increase in headache prevalence: results from the Head-HUNT Study. J headache Pain. (2009) 10:153–9. doi: 10.1007/s10194-009-0114-6
95. Welch KM, D'Andrea G, Tepley N, Barkley G, Ramadan NM. The concept of migraine as a state of central neuronal hyperexcitability. Neurol Clin. (1990) 8:817–28. doi: 10.1016/S0733-8619(18)30319-0
96. Maniyar FH, Sprenger T, Monteith T, Schankin C, Goadsby PJ. Brain activations in the premonitory phase of nitroglycerin-triggered migraine attacks. Brain. (2014) 137(Pt 1):232–41. doi: 10.1093/brain/awt320
97. Denuelle M, Fabre N, Payoux P, Chollet F, Geraud G. Hypothalamic activation in spontaneous migraine attacks. Headache. (2007) 47:1418–26. doi: 10.1111/j.1526-4610.2007.00776.x
98. Holle D, Naegel S, Obermann M. Pathophysiology of hypnic headache. Cephalalgia. (2014) 34:806–12. doi: 10.1177/0333102414535996
99. Holle D, Obermann M. Cluster headache and the hypothalamus: causal relationship or epiphenomenon? Expert Rev Neurother. (2011) 11:1255–63. doi: 10.1586/ern.11.115
100. Alstadhaug KB. Migraine and the hypothalamus. Cephalalgia. (2009) 29:809–17. doi: 10.1111/j.1468-2982.2008.01814.x
101. Holle D, Katsarava Z, Obermann M. The hypothalamus: specific or nonspecific role in the pathophysiology of trigeminal autonomic cephalalgias? Curr Pain Headache Rep. (2011) 15:101–7. doi: 10.1007/s11916-010-0166-y
102. Noseda R, Burstein R. Migraine pathophysiology: anatomy of the trigeminovascular pathway and associated neurological symptoms, cortical spreading depression, sensitization, and modulation of pain. Pain. (2013) 154(Suppl. 1):S44–53. doi: 10.1016/j.pain.2013.07.021
103. Brennan KC, Bates EA, Shapiro RE, Zyuzin J, Hallows WC, Huang Y, et al. Casein kinase idelta mutations in familial migraine and advanced sleep phase. Sci Transl Med. (2013) 5:183ra56. doi: 10.1126/scitranslmed.3005784
104. Holland PR. Headache and sleep: shared pathophysiological mechanisms. Cephalalgia. (2014) 34:725–44. doi: 10.1177/0333102414541687
105. Wesensten NJ. Legitimacy of concerns about caffeine and energy drink consumption. Nutr Rev. (2014) 72(Suppl 1):78–86. doi: 10.1111/nure.12146
106. Alstadhaug K, Salvesen R, Bekkelund S. Insomnia and circadian variation of attacks in episodic migraine. Headache. (2007) 47:1184–8. doi: 10.1111/j.1526-4610.2007.00858.x
107. Alstadhaug K, Salvesen R, Bekkelund S. 24-hour distribution of migraine attacks. Headache. (2008) 48:95–100. doi: 10.1111/j.1526-4610.2007.00779.x
108. Couturier EG, Hering R, Steiner TJ. Weekend attacks in migraine patients: caused by caffeine withdrawal? Cephalalgia. (1992) 12:99–100. doi: 10.1046/j.1468-2982.1992.1202099.x
109. Ray BS, Wolff HG. Experimental studies on headache. Pain sensitive structures of the head and their significance in headache. Arch Surg. (1940) 41:813–56. doi: 10.1001/archsurg.1940.01210040002001
110. Kemp WJ III, Tubbs RS, Cohen-Gadol AA. The innervation of the cranial dura mater: neurosurgical case correlates and a review of the literature. World Neurosurg. (2012) 78:505–10. doi: 10.1016/j.wneu.2011.10.045
111. Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med. (2002) 8:136–42. doi: 10.1038/nm0202-136
112. Levy D, Kainz V, Burstein R, Strassman AM. Mast cell degranulation distinctly activates trigemino-cervical and lumbosacral pain pathways and elicits widespread tactile pain hypersensitivity. Brain Behav Immun. (2012) 26:311–7. doi: 10.1016/j.bbi.2011.09.016
113. Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Ann Rev Physiol. (2013) 75:365–91. doi: 10.1146/annurev-physiol-030212-183717
114. Brennan KC, Charles A. An update on the blood vessel in migraine. Curr Opin Neurol. (2010) 23:266–74. doi: 10.1097/WCO.0b013e32833821c1
115. Fields H. State-dependent opioid control of pain. Nat Rev Neurosci. (2004) 5:565–75. doi: 10.1038/nrn1431
116. Ashina M, Hansen JM, Olesen J. Pearls and pitfalls in human pharmacological models of migraine: 30 years' experience. Cephalalgia. (2013) 33:540–53. doi: 10.1177/0333102412475234
117. Guieu R, Devaux C, Henry H, Bechis G, Pouget J, Mallet D, et al. Adenosine and migraine. Can J Neurol Sci. (1998) 25:55–8. doi: 10.1017/S0317167100033497
118. Delacretaz E. Clinical practice. Supraventricular tachycardia. N Engl J Med. (2006) 354:1039–51. doi: 10.1056/NEJMcp051145
119. Nguyen KL, Bandettini WP, Shanbhag S, Leung SW, Wilson JR, Arai AE. Safety and tolerability of regadenoson CMR. Eur Heart J Cardiovasc Imaging. (2014) 15:753–60. doi: 10.1093/ehjci/jet278
121. Kruuse C, Lassen LH, Iversen HK, Oestergaard S, Olesen J. Dipyridamole may induce migraine in patients with migraine without aura. Cephalalgia. (2006) 26:925–33. doi: 10.1111/j.1468-2982.2006.01137.x
122. Brown SG, Waterer GW. Migraine precipitated by adenosine. Med J Aust. (1995) 162:389, 391. doi: 10.5694/j.1326-5377.1995.tb139957.x
123. Fredholm BB. Adenosine actions and adenosine receptors after 1 week treatment with caffeine. Acta Physiol Scand. (1982) 115:283–6. doi: 10.1111/j.1748-1716.1982.tb07078.x
124. Varani K, Portaluppi F, Merighi S, Ongini E, Belardinelli L, Borea PA. Caffeine alters A2A adenosine receptors and their function in human platelets. Circulation. (1999) 99:2499–502. doi: 10.1161/01.CIR.99.19.2499
125. Shapiro RE. Caffeine and headaches. Neurol Sci. (2007) 28(Suppl. 2):S179–83. doi: 10.1007/s10072-007-0773-5
126. Shapiro RE. Caffeine and headaches. Curr Pain Headache Rep. (2008) 12:311–5. doi: 10.1007/s11916-008-0052-z
127. Laurienti PJ, Field AS, Burdette JH, Maldjian JA, Yen YF, Moody DM. Relationship between caffeine-induced changes in resting cerebral perfusion and blood oxygenation level-dependent signal. AJNR Am J Neuroradiol. (2003) 24:1607–11.
128. Haanes KA, Edvinsson L. Expression and characterization of purinergic receptors in rat middle meningeal artery-potential role in migraine. PLoS ONE. (2014) 9:e108782. doi: 10.1371/journal.pone.0108782
129. Goadsby PJ, Hoskin KL, Storer RJ, Edvinsson L, Connor HE. Adenosine A1 receptor agonists inhibit trigeminovascular nociceptive transmission. Brain. (2002) 125(Pt 6):1392–401. doi: 10.1093/brain/awf141
130. Giffin NJ, Kowacs F, Libri V, Williams P, Goadsby PJ, Kaube H. Effect of the adenosine A1 receptor agonist GR79236 on trigeminal nociception with blink reflex recordings in healthy human subjects. Cephalalgia. (2003) 23:287–92. doi: 10.1046/j.1468-2982.2003.00511.x
131. Arulmani U, Heiligers JP, Centurion D, Garrelds IM, Villalon CM, Saxena PR. Lack of effect of the adenosine A1 receptor agonist, GR79236, on capsaicin-induced CGRP release in anaesthetized pigs. Cephalalgia. (2005) 25:1082–90. doi: 10.1111/j.1468-2982.2005.00967.x
132. Sneyd JR, Langton JA, Allan LG, Peacock JE, Rowbotham DJ. Multicentre evaluation of the adenosine agonist GR79236X in patients with dental pain after third molar extraction. Br J Anaesth. (2007) 98:672–6. doi: 10.1093/bja/aem075
133. Hansen JM, Lipton RB, Dodick DW, Silberstein SD, Saper JR, Aurora SK, et al. Migraine headache is present in the aura phase: a prospective study. Neurology. (2012) 79:2044–9. doi: 10.1212/WNL.0b013e3182749eed
134. Akcali D, Sayin A, Sara Y, Bolay H. Does single cortical spreading depression elicit pain behaviour in freely moving rats? Cephalalgia. (2010) 30:1195–206. doi: 10.1177/0333102409360828
135. Olesen J. Regional cerebral blood flow and oxygen metabolism during migraine with and without aura. Cephalalgia. (1998) 18:2–4. doi: 10.1046/j.1468-2982.1998.1801001-3.x
136. Leão AA. Spreading depression of activity in cerebral cortex. J Neurophysiol. (1944) 7:359–90. doi: 10.1152/jn.1944.7.6.359
137. Andreou AP, Summ O, Charbit AR, Romero-Reyes M, Goadsby PJ. Animal models of headache: from bedside to bench and back to bedside. Expert Rev Neurother. (2010) 10:389–411. doi: 10.1586/ern.10.16
138. Charles A, Brennan KC. The neurobiology of migraine. Handb Clin Neurol. (2010) 97:99–108. doi: 10.1016/S0072-9752(10)97007-3
139. Cozzolino O, Marchese M, Trovato F, Pracucci E, Ratto GM, Buzzi MG, et al. Understanding spreading depression from headache to sudden unexpected death. Front Neurol. (2018) 9:19. doi: 10.3389/fneur.2018.00019
140. Gallagher CJ, Salter MW. Differential properties of astrocyte calcium waves mediated by P2Y1 and P2Y2 receptors. J Neurosci. (2003) 23:6728–39. doi: 10.1523/JNEUROSCI.23-17-06728.2003
141. Stefanello N, Spanevello RM, Passamonti S, Porciuncula L, Bonan CD, Olabiyi AA, et al. Coffee, caffeine, chlorogenic acid, and the purinergic system. Food Chem Toxicol. (2019) 123:298–313. doi: 10.1016/j.fct.2018.10.005
142. Hohoff C, Marziniak M, Lesch KP, Deckert J, Sommer C, Mossner R. An adenosine A2A receptor gene haplotype is associated with migraine with aura. Cephalalgia. (2007) 27:177–81. doi: 10.1111/j.1468-2982.2007.01254.x
143. Keselman I, Zyuzin J, Charles A. Modulation of Migraine-Related Cortical Excitability by Adenosine and its Receptors. Headache Congress International Headache Society and American Headache Society, Boston, MA (2013).
144. Yalcin N, Yu E, Atalay Y, Ayata C, Eikermann-Haerter K. The migraine trigger caffeine increases susceptibility to spreading depolarization. In: 56th Annual Scientific Meeting. Los Angeles, CA: American Headache Society (2014).
145. Huston J. Yawning and penile erection induced in rats by cortical spreading depression. Nature. (1971) 232:274–5. doi: 10.1038/232274a0
146. Oishi Y, Huang ZL, Fredholm BB, Urade Y, Hayaishi O. Adenosine in the tuberomammillary nucleus inhibits the histaminergic system via A1 receptors and promotes non-rapid eye movement sleep. Proc Natl Acad Sci USA. (2008) 105:19992–7. doi: 10.1073/pnas.0810926105
147. Thakkar MM, Engemann SC, Walsh KM, Sahota PK. Adenosine and the homeostatic control of sleep: effects of A1 receptor blockade in the perifornical lateral hypothalamus on sleep-wakefulness. Neuroscience. (2008) 153:875–80. doi: 10.1016/j.neuroscience.2008.01.017
148. Alstadhaug KB. Histamine in migraine and brain. Headache. (2014) 54:246–59. doi: 10.1111/head.12293
149. Rainero I, De Martino P, Pinessi L. Hypocretins and primary headaches: neurobiology and clinical implications. Expert Rev Neurother. (2008) 8:409–16. doi: 10.1586/14737175.8.3.409
150. Burstein R, Jakubowski M. Unitary hypothesis for multiple triggers of the pain and strain of migraine. J Comp Neurol. (2005) 493:9–14. doi: 10.1002/cne.20688
151. Westerink BH, Cremers TI, De Vries JB, Liefers H, Tran N, De Boer P. Evidence for activation of histamine H3 autoreceptors during handling stress in the prefrontal cortex of the rat. Synapse. (2002) 43:238–43. doi: 10.1002/syn.10043
152. Walusinski O. How yawning switches the default-mode network to the attentional network by activating the cerebrospinal fluid flow. Clin Anat. (2014) 27:201–9. doi: 10.1002/ca.22280
153. Gallup AC, Eldakar OT. The thermoregulatory theory of yawning: what we know from over 5 years of research. Front Neurosci. (2012) 6:188. doi: 10.3389/fnins.2012.00188
154. Jacome DE. Compulsive yawning as migraine premonitory symptom. Cephalalgia. (2001) 21:623–5. doi: 10.1046/j.1468-2982.2001.00214.x
155. Guven B, Guven H, Comoglu SS. Migraine and Yawning. Headache. (2018) 58:210–6. doi: 10.1111/head.13195
156. Horvath C. Alterations in brain temperatures as a possible cause of migraine headache. Med Hypotheses. (2014) 82:529–34. doi: 10.1016/j.mehy.2014.02.004
157. Busto R, Dietrich WD, Globus MY, Valdes I, Scheinberg P, Ginsberg MD. Small differences in intraischemic brain temperature critically determine the extent of ischemic neuronal injury. J Cereb Blood Flow Metab. (1987) 7:729–38. doi: 10.1038/jcbfm.1987.127
158. Shevelev IA. Functional imaging of the brain by infrared radiation (thermoencephaloscopy). Prog Neurobiol. (1998) 56:269–305. doi: 10.1016/S0301-0082(98)00038-0
159. Yablonskiy DA, Ackerman JJ, Raichle ME. Coupling between changes in human brain temperature and oxidative metabolism during prolonged visual stimulation. Proc Natl Acad Sci USA. (2000) 97:7603–8. doi: 10.1073/pnas.97.13.7603
160. Bennet DW, Drury AN. Further observations relating to the physiological activity of adenine compounds. J Physiol. (1931) 72:288–320. doi: 10.1113/jphysiol.1931.sp002775
161. McHill AW, Smith BJ, Wright KP Jr. Effects of caffeine on skin and core temperatures, alertness, and recovery sleep during circadian misalignment. J Biol Rhythms. (2014) 29:131–43. doi: 10.1177/0748730414523078
162. Sato-Suzuki I, Kita I, Seki Y, Oguri M, Arita H. Cortical arousal induced by microinjection of orexins into the paraventricular nucleus of the rat. Behav Brain Res. (2002) 128:169–77. doi: 10.1016/S0166-4328(01)00307-2
163. Monda M, Viggiano AN, Viggiano A, Viggiano E, Lanza A, De Luca V. Hyperthermic reactions induced by orexin A: role of the ventromedial hypothalamus. Eur J Neurosci. (2005) 22:1169–75. doi: 10.1111/j.1460-9568.2005.04309.x
164. Blin O, Azulay JP, Masson G, Aubrespy G, Serratrice G. Apomorphine-induced yawning in migraine patients: enhanced responsiveness. Clin Neuropharmacol. (1991) 14:91–5. doi: 10.1097/00002826-199102000-00008
165. Sanna F, Succu S, Melis MR, Argiolas A. Dopamine agonist-induced penile erection and yawning: differential role of D(2)-like receptor subtypes and correlation with nitric oxide production in the paraventricular nucleus of the hypothalamus of male rats. Behav Brain Res. (2012) 230:355–64. doi: 10.1016/j.bbr.2012.02.033
166. Seki Y, Sato-Suzuki I, Kita I, Oguri M, Arita H. Yawning/cortical activation induced by microinjection of histamine into the paraventricular nucleus of the rat. Behav Brain Res. (2002) 134:75–82. doi: 10.1016/S0166-4328(01)00454-5
167. Melis MR, Stancampiano R, Lai C, Argiolas A. Nitroglycerin-induced penile erection and yawning in male rats: mechanism of action in the brain. Brain Res Bull. (1995) 36:527–31. doi: 10.1016/0361-9230(94)00236-T
168. Cerbo R, Barbanti P, Buzzi MG, Fabbrini G, Brusa L, Roberti C, et al. Dopamine hypersensitivity in migraine: role of the apomorphine test. Clin Neuropharm. (1997) 20:36–41. doi: 10.1097/00002826-199702000-00004
169. Afridi SK, Kaube H, Goadsby PJ. Glyceryl trinitrate triggers premonitory symptoms in migraineurs. Pain. (2004) 110:675–80. doi: 10.1016/j.pain.2004.05.007
170. Noseda R, Kainz V, Jakubowski M, Gooley JJ, Saper CB, Digre K, et al. A neural mechanism for exacerbation of headache by light. Nat Neurosci. (2010) 13:239–45. doi: 10.1038/nn.2475
171. Sodhi P, Hartwick AT. Adenosine modulates light responses of rat retinal ganglion cell photoreceptors througha cAMP-mediated pathway. J Physiol. (2014) 592(Pt 19):4201–20. doi: 10.1113/jphysiol.2014.276220
172. Antle MC, Steen NM, Mistlberger RE. Adenosine and caffeine modulate circadian rhythms in the Syrian hamster. Neuroreport. (2001) 12:2901–5. doi: 10.1097/00001756-200109170-00029
173. van Diepen HC, Lucassen EA, Yasenkov R, Groenen I, Ijzerman AP, Meijer JH, et al. Caffeine increases light responsiveness of the mouse circadian pacemaker. Eur J Neurosci. (2014) 40:3504–11. doi: 10.1111/ejn.12715
174. Verbanes NM, Zisk CF, Marinos LN, Dietzel JD, Maziarz CM, Ruby CL. Caffeine potentiates circadian photic phase-resetting and delays light entrained onset in mice. In: Society of Neuroscience's 44th Annual Meeting; November 15–19. Washington, DC (2014).
175. Good PA, Taylor RH, Mortimer MJ. The use of tinted glasses in childhood migraine. Headache. (1991) 31:533–6. doi: 10.1111/j.1526-4610.1991.hed3108533.x
176. Tatsumoto M, Eda T, Ishikawa T, Ayama M, Hirata K. Light of Intrinsically Photosensitive Retinal Ganglion Cell (ipRGC) Causing Migraine Headache Exacerbation. Headache Congress International Headache Society and American Headache Society, Boston, MA (2013).
177. Shields KG, Goadsby PJ. Serotonin receptors modulate trigeminovascular responses in ventroposteromedial nucleus of thalamus: a migraine target? Neurobiol Dis. (2006) 23:491–501. doi: 10.1016/j.nbd.2006.04.003
178. Andreou AP, Goadsby PJ. Topiramate in the treatment of migraine: a kainate (glutamate) receptor antagonist within the trigeminothalamic pathway. Cephalalgia. (2011) 31:1343–58. doi: 10.1177/0333102411418259
179. Andreou AP, Shields KG, Goadsby PJ. GABA and valproate modulate trigeminovascular nociceptive transmission in the thalamus. Neurobiol Dis. (2010) 37:314–23. doi: 10.1016/j.nbd.2009.10.007
180. Andreou AP, Goadsby PJ. Therapeutic potential of novel glutamate receptor antagonists in migraine. Expert Opin Investig Drugs. (2009) 18:789–803. doi: 10.1517/13543780902913792
181. Filippov IV, Williams WC, Krebs AA, Pugachev KS. Dynamics of infraslow potentials in the primary auditory cortex: component analysis and contribution of specific thalamic-cortical and non-specific brainstem-cortical influences. Brain Res. (2008) 1219:66–77. doi: 10.1016/j.brainres.2008.04.011
182. Burstein R, Jakubowski M, Garcia-Nicas E, Kainz V, Bajwa Z, Hargreaves R, et al. Thalamic sensitization transforms localized pain into widespread allodynia. Ann Neurol. (2010) 68:81–91. doi: 10.1002/ana.21994
183. Fontanez DE, Porter JT. Adenosine A1 receptors decrease thalamic excitation of inhibitory and excitatory neurons in the barrel cortex. Neuroscience. (2006) 137:1177–84. doi: 10.1016/j.neuroscience.2005.10.022
184. Yang YC, Hu CC, Huang CS, Chou PY. Thalamic synaptic transmission of sensory information modulated by synergistic interaction of adenosine and serotonin. J Neurochem. (2014) 128:852–63. doi: 10.1111/jnc.12499
185. Akerman S, Holland PR, Goadsby PJ. Diencephalic and brainstem mechanisms in migraine. Nat Rev Neurosci. (2011) 12:570–84. doi: 10.1038/nrn3057
Keywords: headache, caffeine, adenosine, dopaminergic, histaminergic, circadian, yawning, withdrawal
Citation: Alstadhaug KB and Andreou AP (2019) Caffeine and Primary (Migraine) Headaches—Friend or Foe? Front. Neurol. 10:1275. doi: 10.3389/fneur.2019.01275
Received: 17 September 2019; Accepted: 18 November 2019;
Published: 03 December 2019.
Edited by:
Uwe Reuter, Charité Medical University of Berlin, GermanyReviewed by:
Mark Braschinsky, University of Tartu, EstoniaAnish Bahra, National Hospital for Neurology and Neurosurgery (NHNN), United Kingdom
Copyright © 2019 Alstadhaug and Andreou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karl B. Alstadhaug, S2FybC5iam9ybmFyLmFsc3RhZGhhdWdAbmxzaC5ubw==