 Eun Hye Oh1
Eun Hye Oh1 Jin-Hong Shin1
Jin-Hong Shin1 Hyang-Sook Kim1Jae Wook Cho1
Hyang-Sook Kim1Jae Wook Cho1 Seo Young Choi2
Seo Young Choi2 Kwang-Dong Choi2Je-Keun Rhee3
Kwang-Dong Choi2Je-Keun Rhee3 Seowhang Lee4
Seowhang Lee4 Changwook Lee4
Changwook Lee4 Jae-Hwan Choi1*
Jae-Hwan Choi1*- 1Department of Neurology, Research Institute for Convergence of Biomedical Science and Technology, Pusan National University School of Medicine, Pusan National University Yangsan Hospital, Yangsan, South Korea
- 2Department of Neurology, Pusan National University Hospital, Pusan National University School of Medicine and Biomedical Research Institute, Busan, South Korea
- 3School of Systems Biomedical Science, Soongsil University, Seoul, South Korea
- 4Department of Biological Sciences, School of Life Sciences, Ulsan National Institute of Sciences and Technology, Ulsan, South Korea
Objectives: The cause of Meniere's disease (MD) is unclear but likely involves genetic and environmental factors. The aim of this study was to investigate the genetic basis underlying MD by screening putative candidate genes for MD.
Methods: Sixty-eight patients who met the diagnostic criteria for MD of the Barany Society were included. We performed targeted gene sequencing using next generation sequencing (NGS) panel composed of 45 MD-associated genes. We identified the rare variants causing non-synonymous amino acid changes, stop codons, and insertions/deletions in the coding regions, and excluded the common variants with minor allele frequency >0.01 in public databases. The pathogenicity of the identified variants was analyzed by various predictive tools and protein structural modeling.
Results: The average read depth for the targeted regions was 1446.3-fold, and 99.4% of the targeted regions were covered by 20 or more reads, achieving the high quality of the sequencing. After variant filtering, annotation, and interpretation, we identified a total of 15 rare heterozygous variants in 12 (17.6%) sporadic patients. Among them, four variants were detected in familial MD genes (DTNA, FAM136A, DPT), and the remaining 11 in MD-associated genes (PTPN22, NFKB1, CXCL10, TLR2, MTHFR, SLC44A2, NOS3, NOTCH2). Three patients had the variants in two or more genes. All variants were not detected in our healthy controls (n = 100). No significant differences were observed between patients with and without a genetic variant in terms of sex, mean age of onset, bilaterality, the type of MD, and hearing threshold at diagnosis.
Conclusions: Our study identified rare variants of putative candidate genes in some of MD patients. The genes were related to the formation of inner ear structures, the immune-associated process, or systemic hemostasis derangement, suggesting the multiple genetic predispositions in the development of MD.
Introduction
Meniere's disease (MD) is a clinical syndrome that consists of episodes of spontaneous vertigo usually associated with unilateral fluctuating sensorineural hearing loss (SNHL), tinnitus and aural fullness (1). Almost 150 years have elapsed since Prosper Meniere first described the clinical entity, but unfortunately, the exact pathophysiology of this condition is yet to be fully understood. Histopathological studies in human temporal bones have found endolymphatic hydrops (EH) in most patients with MD, but the origin of the EH is unknown (2).
MD is considered a multifactorial disorder associated with the various factors including anatomical abnormalities, infections, allergens, and autoimmune disorders. Most MD cases are sporadic, but familial MD is observed in 8–10% of sporadic cases, suggesting genetic susceptibility to the development of MD (3–5). To search a candidate gene associated with MD, many approaches including linkage analysis, case-control study or the sequencing of selected genes have been previously used (6–10). A variety of common single nucleotide polymorphisms (SNPs) have been meaningfully detected in patients with MD (11–35) (Supplementary Table 1). Most of them were the genes related to the inflammation or regulating the ionic composition and water transport of the inner ear. However, the effect size of these common variants was small (odds ratio < 2). Furthermore, a common variant could not reflect the entire heritability of MD. Recent studies using whole-exome sequencing (WES) for Spanish families with MD have identified probably pathogenic rare variants in candidate genes including FAM136A, DTNA, PRKCB, DPT, and SEMA3D (36–38). Since these genes encode proteins that may be relevant to the formation or maintaining of inner ear structures, the identified rare variants are expected to account for the genetic contribution of MD, but further replicative studies in distinct populations are needed. Thus, the aim of this study was to explore the previously proposed MD-associated genes using targeted NGS to investigate the genetic basis underlying MD.
Materials and Methods
Subjects
We recruited 68 unrelated patients with definite MD who visited a tertiary dizziness clinic from 2015 to 2018. The diagnosis of definite MD was made based on the criteria established by the Classification Committee of the Barany Society (1). All patients met the following criteria: (1) Two or more spontaneous episodes of vertigo, each lasting 20 min to 12 h, (2) Audiometrically documented low- to medium-frequency sensorineural hearing loss in the affected ear on at least one occasion before, during, or after one of the episodes of vertigo, (3) Fluctuating aural symptoms (hearing, tinnitus, or earfullness) in the affected ear, (4) Not better accounted for by another vestibular diagnosis. A brain MRI was performed to rule out any neurological lesions. The patients included 38 males and 30 females with age ranging from 28 to 89 years (mean age 60.2 ± 12.0 years). The mean age of onset was 57.5 ± 11.3 years. Most patients (n = 63, 93%) had a unilateral MD. Six had at least one family member with a history of MD-like symptoms. According to the phenotype, 18 (26%) were classified as delayed MD based on a previous history of sensorineural hearing loss (months or years) before the onset of vertigo episodes, while the others (n = 50) showed classic MD phenotype (39).
All experiments followed the tenets of the Declaration of Helsinki, and informed consents were obtained after the nature and possible consequences of this study had been explained to the participants. This study was approved by the institutional review boards of Pusan National University Yangsan Hospital.
Targeted Next-Generation Sequencing
Targeted genes were collected from the literature review. The keywords “Meniere's disease” and “gene” were used to search the MD-associated genes in PubMed, resulting in 101 papers when this study was initiated (August, 2017). After excluding the genes showing no correlation with MD, we selected 45 genes used for targeted NGS (Supplementary Table 1). The selected genes were largely classified into two categories as follow: (1) “familial MD gene,” the pathogenic genes for familial MD identified by high-throughput sequencing (36–38); (2) “MD-associated gene,” the candidate genes contributing to the development of MD demonstrated by association study or network-based study (11–35, 40). The MD gene panel was designed by the Suredesign webtool (Agilent) to cover the exons and 20 bp in the flanking regions.
Genomic DNA was extracted from the blood sample of all patients. For the generation of standard exome capture libraries, we used the Agilent SureSelect Target Enrichment protocol for Illumina paired-end sequencing library (ver. B.3, June 2015) with 1 μg input DNA. The quantification of DNA and the DNA quality was measured by PicoGreen and Nanodrop. The qualified genomic DNA sample was randomly fragmented by Covaris followed by adapter ligation, purification, hybridization, and PCR. Captured libraries were subjected to Agilent 2100 Bioanalyzer to estimate the quality and were loaded on to the Illumina HiSeq2500 (San Diego, USA) according to the manufacturer's instructions. Raw image files were processed by HCS1.4.8 for base-calling with default parameters and the sequences of each individual were generated as 100 bp paired-end reads. Sequence reads were aligned to the human reference genome sequence (GRCh37.3, hg19) using the Burrows-Wheeler Aligner (BWA, version 0.7.12). PCR duplicate reads were marked and removed with Picard tools (version 1.92). Genome Analysis Toolkit (GATK, version 2.3-9) was used for indel realignment and base recalibration. Variation annotation and interpretation analysis were performed using SnpEff (version 4.2).
Identification of Rare or Novel Variants
To identify the possible pathogenic variants, we first filtered out synonymous and non-coding variants, and extracted the variants causing non-synonymous amino acid changes, stop codons, in-frame insertions/deletions in coding regions, or changes to splice site sequences in exon/intron boundaries. Then, common variants with a minor allele frequency (MAF) > 0.01 that represented in dbSNP147, the Exome Aggregation Consortium (ExAC), gnomAD, 1,000 Genomes Project, and NHLBI GO Exome Sequencing Project (ESP) 6500 were excluded. The pathogenicity of the non-synonymous variants was analyzed using Sorting Intolerant From Tolerant (SIFT), Likelihood Ratio Test (LRT), Polyphen2, MutationTaster, Functional Analysis Through Hidden Markov Models (FATHMM), and Combined Annotation Dependent Depletion (CADD) phred score. The higher CADD phred scores indicate that a variant is more likely to have deleterious effects (41). The strength of ectopic splicing sites created by intronic variants was evaluated by the Human Splicing Finder program. All rare and novel variants were annotated for previously reported disease-causing variants using the Human Gene Mutation Database. All variants were confirmed by Sanger DNA sequencing, and were screened in 100 normal controls.
Protein Structural Modeling
The structural modeling of missense variants of PTPN22 (PDB accession code: 2P6X), MTHFR (PDB accession code: 6FCX), NOTCH2 (PDB accession code: 2OO4) and CXCL10 (PDB accession code: 1O80) were generated using I-TASSER server. The confidence scores (C-scores) indicating significance of threading template alignments and the convergence parameters of modeling simulation were calculated for each predicted model to estimate the quality of model (42). The calculated C-scores of PTPN22, MTHFR, NOTCH2, CXCL10 were 1.61, 0.07, 0.36, 0.34, respectively. The best model was selected based on the template modeling score (TM-score) and the root mean square deviation (RMSD) value indicating proper topology of the mutant model compared to the PDB template. The optimization of predicted models was performed by PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.), and each mutant model was aligned with that of wild type to compare three-dimensional structures.
Statistical Analyses
To investigate the association between the genetic variants and disease manifestation, we calculated the odds ratio for each variant using Fisher's exact test from the allele frequency of ExAC and gnomAD database as controls (total population and East Asian population). P-values were corrected for multiple testing by the total amount of variants found for each gene following Bonferroni approach. We also compared the clinical characteristics between patients with and without rare variants. Continuous variables (mean age, age of onset, and hearing threshold at diagnosis) were compared using the Mann-Whitney test, and categorical variables (sex, bilaterality, the type of MD, and the presence of family history) using Fisher's exact test. A statistical analysis was performed using SPSS software (SPSS Inc., Chicago, IL, USA) and a p < 0.05 was considered as significant.
Results
The average read depth for the targeted regions was 1446.3-fold, and 99.4% of the targeted regions were covered by 20 or more reads, achieving the high quality of the sequencing (Supplementary Table 2). After variant filtering, annotation, and interpretation, we identified a total of 15 rare heterozygous variants (two novel and 13 rare variants) in 12 (17.6%) sporadic patients (Table 1). Among them, four variants were detected in familial MD gene, and the remaining 11 in MD-associated gene. Three patients (P-29, P-60, P-66) had the variants in two or more genes. All variants were not detected in our healthy controls (n = 100).
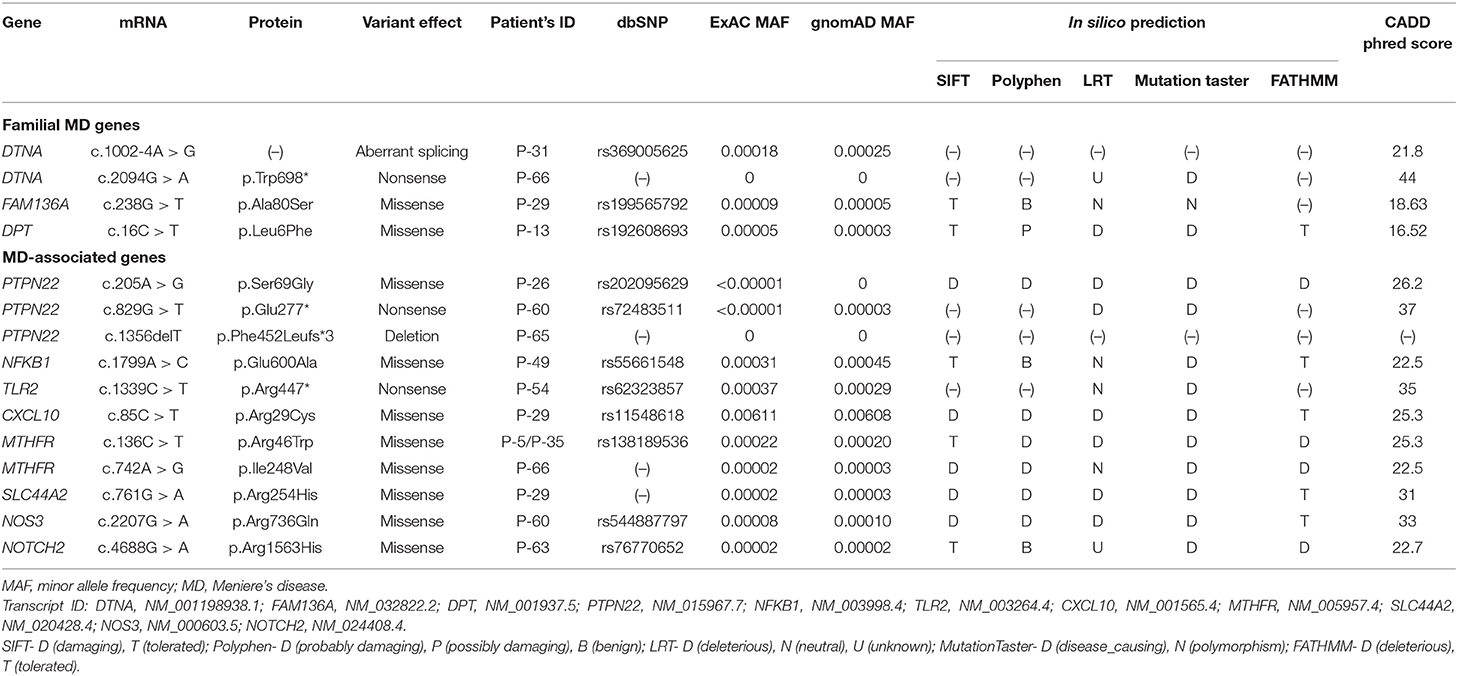
Table 1. Identified rare variants in putative candidate genes associated with Meniere's disease.
Variants in Familial MD Genes
We identified one novel and three rare heterozygous variants in three familial MD genes. Two variants were detected in DTNA. One was a rare variant affecting splice acceptor site located in intron 8 (P-31: c.1002-4A > G, Figure 1A). Human Splicing Finder predicted that this variant will alter the splice site and make new splice sites resulting in three bases longer on exon 9. Another was a nonsense variant resulting in a premature stop codon (P-66: c.2094G > A, p.W698X, Figure 1B). It was absent in all public database, and predicted to be deleterious by MutationTaster and CADD scores. The other rare variants were detected in FAM136A (P-29: c.238G > T, p.A80S, Figure 1C) and DPT (P-13: c.16C > T, p.L6F, Figure 1D). Both were a missense variant which a single nucleotide change results in a codon that codes for a different amino acid. Of them, the variant in DPT changed highly conserved amino acid and was predicted as protein-damaging potential by three prediction tools (Polyphen2, LRT, MutationTaster).
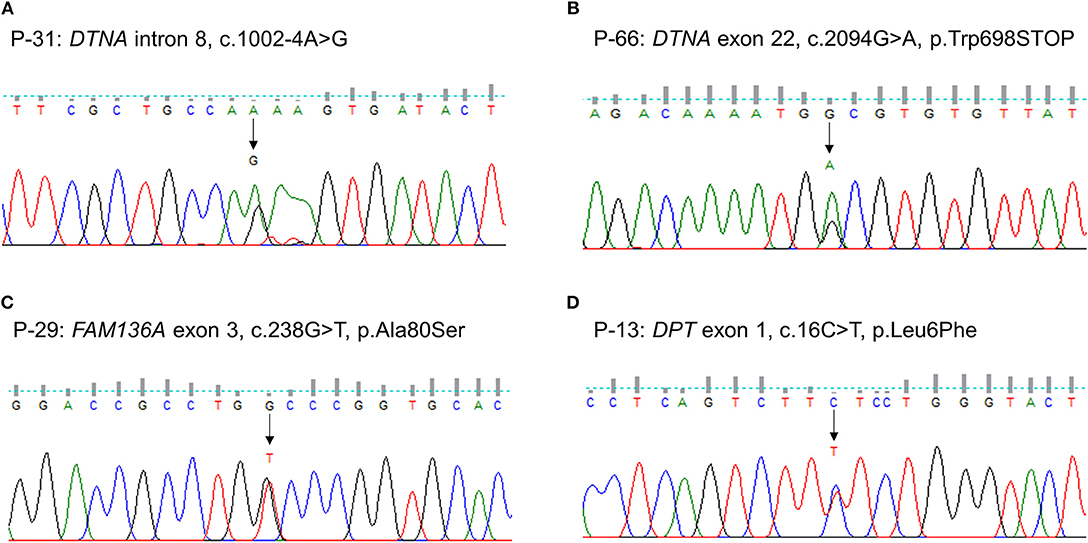
Figure 1. Sequencing results of the rare variants in familial MD genes identified by targeted next-generation sequencing. The chromatograms show two rare heterozygous variants in DTNA: (A) one is a splice site variant at the 4th nucleotide of the splice acceptor site in intron 8 (c.1002-4A > G, P-31); (B) another is a novel nonsense variant in exon 22 causing a premature stop codon (c.2094G > A, p.W698X, P-66). The others are a heterozygous missense rare variant in exon 3 of FAM136A causing the substitution of alanine by serine at position 80 (c.238G > T, p.A80S, P-29) (C) and in exon 1 of DPT changing the highly conserved leucine by phenylalanine at position 6 (c.16C > T, p.L6F, P-13) (D).
Variants in MD-Associated Genes
We identified one novel and 10 rare heterozygous variants in eight MD-associated genes. Interestingly, possibly pathogenic variants of PTPN22 were detected in three unrelated patients. Two were truncated variants caused by premature stop codon (P-60: c.829G > T, p.E277X, Figure 2A) or single base deletion (P-65: c.1356delT, p.F452Lfs*3, Figure 2B). The nonsense variant was predicted to be deleterious by LRT, MutationTaster and CADD score. The remaining one was a missense variant with protein-damaging potential predicted by all prediction tools (P-26: c.205A > G, p.S69G, Figure 2C). Protein structural modeling predicted that p.S69G variant may disrupt the positions of surrounding residues (p.F68, p.N87, p.K90, and p.Q267) by steric hindrance effects, and eventually may affect the enzymatic activity of PTPN22 (Figure 2D).
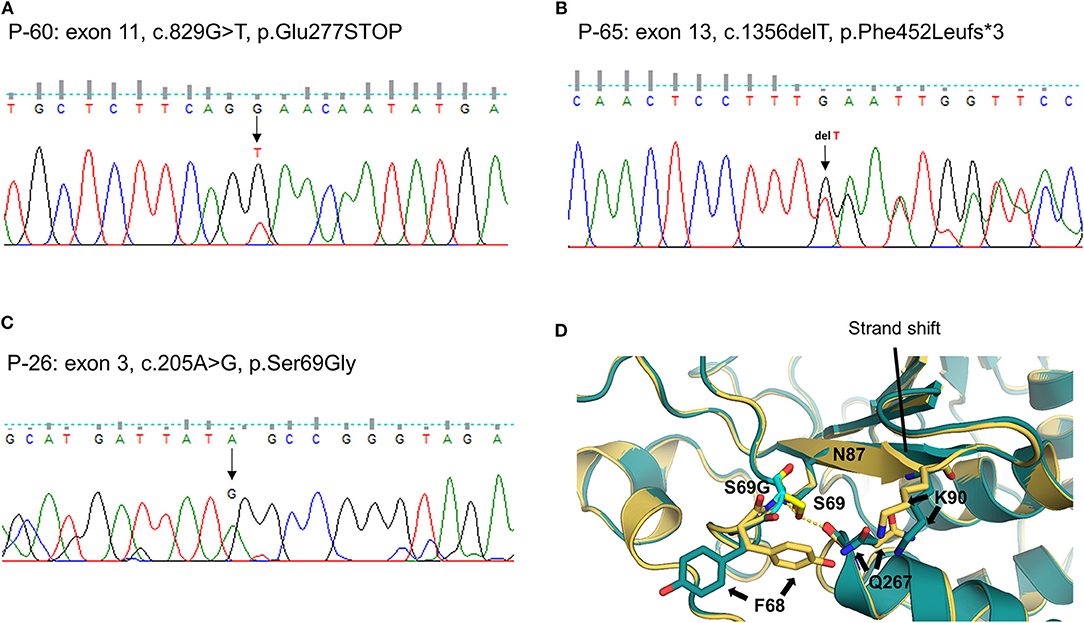
Figure 2. Sequencing results and protein structural modeling of the rare variants in PTPN22 identified by targeted next-generation sequencing. The chromatograms show three rare heterozygous variants: (A) one is a nonsense variant in exon 11 resulting in a premature stop codon (c.829G > T, p.E277X, P-60); (B) another is single base deletion in exon 13 that leads to frameshift and premature stop codon 3 amino acids downstream (c.1356delT, p.F452Lfs*3, P-65); (C) the other is a missense rare variant in exon 3 causing the substitution of highly conserved serine by glycine at position 69 (c.205A > G, p.S69G, P-26). Protein structural modeling predicts that p.S69G variant may affect the enzyme activity of PTPN22 by a disruption of hydrogen bonds network between p.S69 and p.N87 residues, and the steric hindrance effect resulting from the rotation of p.F68, p.Q267, and p.K90 residues (D), yellow: wild type PTPN22, teal: p.S69G mutant PTPN22).
We also detected two missense variants of MTHFR in three unrelated patients (P-5 and P-35: c.136C > T, p.R46W, Figure 3A; P-66: c.742A > G, p.I248V, Figure 3B). They were considered likely pathogenic by four of the prediction tools. Protein structural modeling revealed that p.R46W variant may cause breakage of salt bridges with surrounding residues (p.D188, p.D191, and p.D223), resulting in destabilizing the β-strand (β5and β6 Figure 3C). On the other hand, p.I248V variant showed no discernible differences when the secondary structure as well as surrounding residue orientation were analyzed.
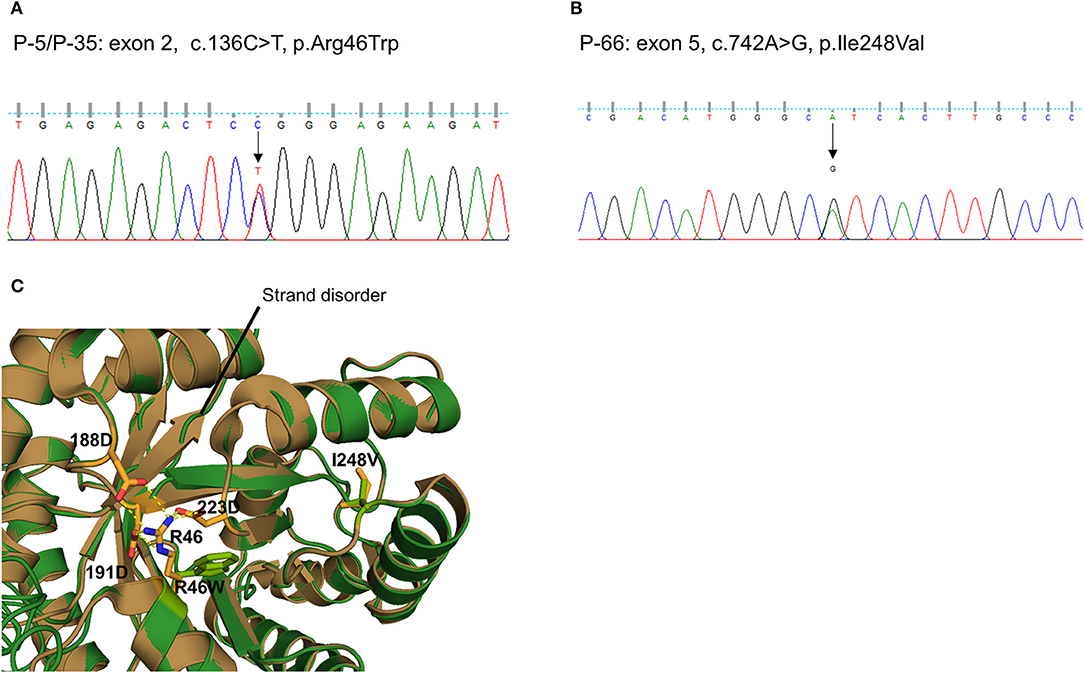
Figure 3. Sequencing results and protein structural modeling of the rare variants in MTHFR identified by targeted next-generation sequencing. The chromatograms show two rare heterozygous variants: (A) one is a missense rare variant in exon 2 causing the substitution of highly conserved arginine by tryptophan at position 46 (c.136C > T, p.R46W, P-5/P-35); (B) another is also a missense rare variant in exon 5 causing the substitution of highly conserved isoleucine by valine at position 248 (c.742A > G, p.I248V, P-66). (C) Protein structural modeling shows that p.R46W variant may cause breakage of salt bridges with side chains of p.D188, p.D223, and p.D191 residues. This leads to destabilize the central β-sheet which is essential for reductase activity. On the other hands, p.I248V variant reveals no discernible differences when secondary structure as well as surrounding residue orientation are analyzed (brown: wild type MTHFR, green: p.R46W/p.I248V mutant MTHFR).
The other five missense variants had protein-damaging potential predicted by at least one prediction tool: SLC44A2 (P-29: c.761G > A, p.R254H, Figure 4A), NOS3 (P-60: c.2207G > A, p.R736Q, Figure 4B), NFKB1 (P-49: c.1799A > C, p.E600A, Figure 4C), NOTCH2 (P-63: c.4688G > A, p.R1563H, Figure 4E), and CXCL10 (P-29: c.85C > T, p.R29C, Figure 4F). Protein structural modeling predicted that the p.R1563H variant in NOTCH2 may destabilize the Lin-12/Notch repeat (LNR) domain by impairing a hydrogen bond network with surrounding residues (p.D1511, p.F1513, and p.D1515), while the p.R29C variant in CXCL10 may cause no conformational change in overall structure. The remaining one was a nonsense variant of TLR2 (P-54: c.1339C > T, p.R447X, Figure 4D), which was predicted to be deleterious by MutationTaster and CADD score.
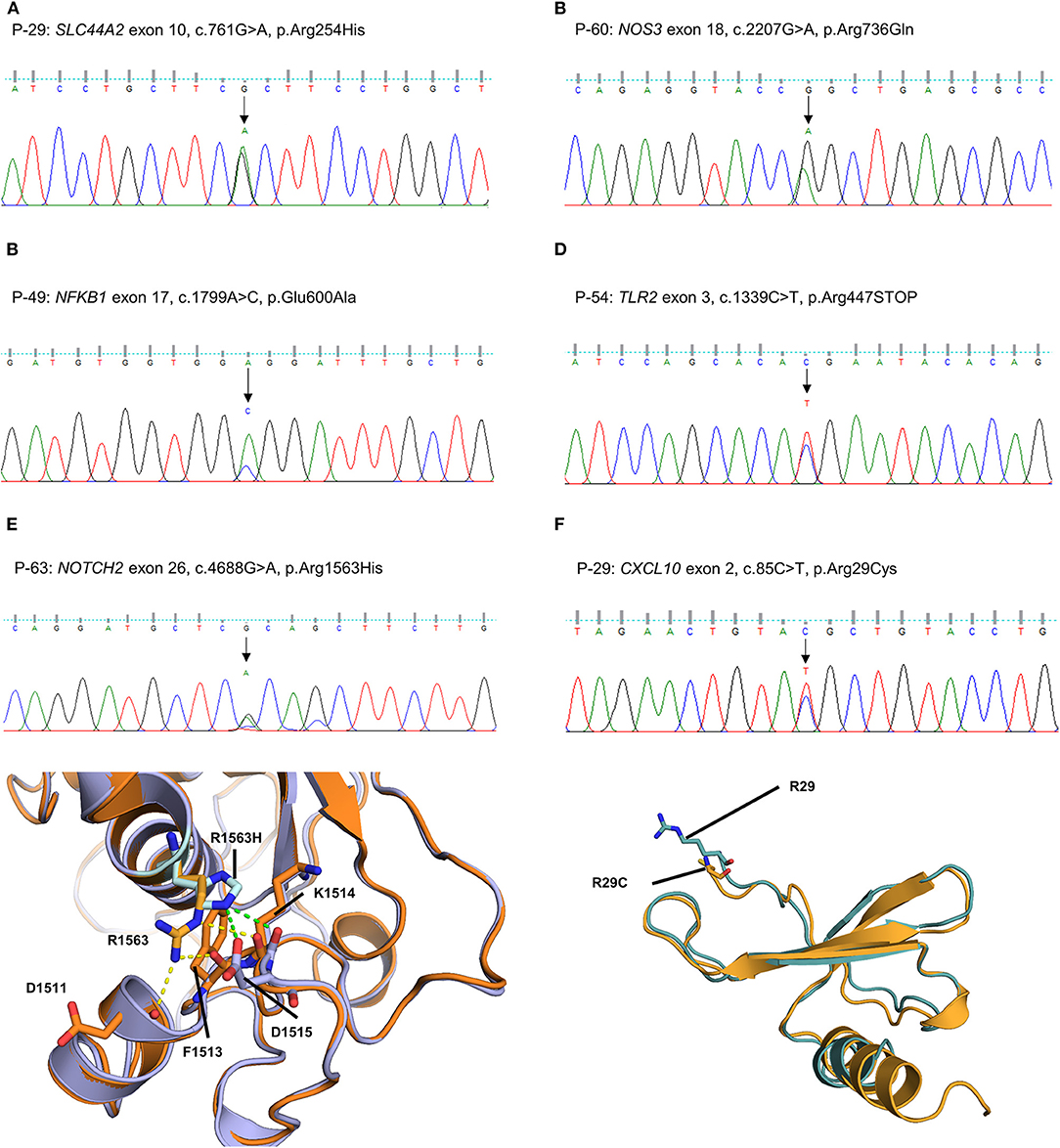
Figure 4. Sequencing results and protein structural modeling of the rare variants in other MD-associated genes identified by targeted next-generation sequencing. (A–D) The chromatograms show a heterozygous missense rare variant in SLC44A2 (exon 10, c.761G > A, p.R254H, P-29), NOS3 (exon 18, c.2207G > A, p.R736Q, P-60), and NFKB1 (exon 17, c.1799A > C, p.E600A, P-49), and a nonsense variant in exon 3 of TLR2 causing premature stop codon (c.1339C > T, p.R447X, P-54). (E) P-63 has a heterozygous missense variant in exon 26 of NOTCH2 causing the substitution of highly conserved arginine by histidine at position 1563 (c.4688G > A, p.R1563H). In protein structural modeling, p.R1563H variant may result in loss of interaction with p.D1511 and p.F1513 residues, and form new hydrogen bond network with main chain of p.K1514 residue and side chain of p.D1515 (green dash line). These impaired interactions make p.D1515 residue to lose ability to interact with a calcium ion, and eventually destabilize the Lin-12/Notch repeat domain required for regulating the NOTCH2 receptor (orange: wild type NOTCH2, purple: p.R1563H mutant NOTCH2). (F) P-29 had a heterozygous missense rare variant in exon 2 of CXCL10 causing the substitution of highly conserved arginine by cystein at position 85 (c.85C > T, p.R29C). However, there is no conformational change in overall structure because the R29 residue is located at the N-terminal end and not involved in interaction with neighboring residues (teal: wild type CXCL10, orange: p.R29C mutant CXCL10).
Copy number variation (CNV) analysis using CNVkit from targeted DNA sequencing data did not reveal any pathogenic CNV in each patient.
Association Analysis of a Genetic Variant
When using the allele frequency of total population from ExAC and gnomAD database, 14 of the 15 rare variants were significantly associated with MD (unadjusted p-value), but eight of them showed significant association after correcting for multiple testing (Bonferroni) in both databases (Table 2). The genes included DTNA, PTPN22, MTHFR, SLC44A2, and NOTCH2. In particular, all rare variants of PTPN22 and MTHFR were significantly associated with MD even after correcting for multiple testing. Compared to the allele frequency of East Asians, 10 variants were significantly associated with MD (unadjusted p-value) in both databases, but no significant associations were observed in all variants after correcting for multiple testing (Table 2).
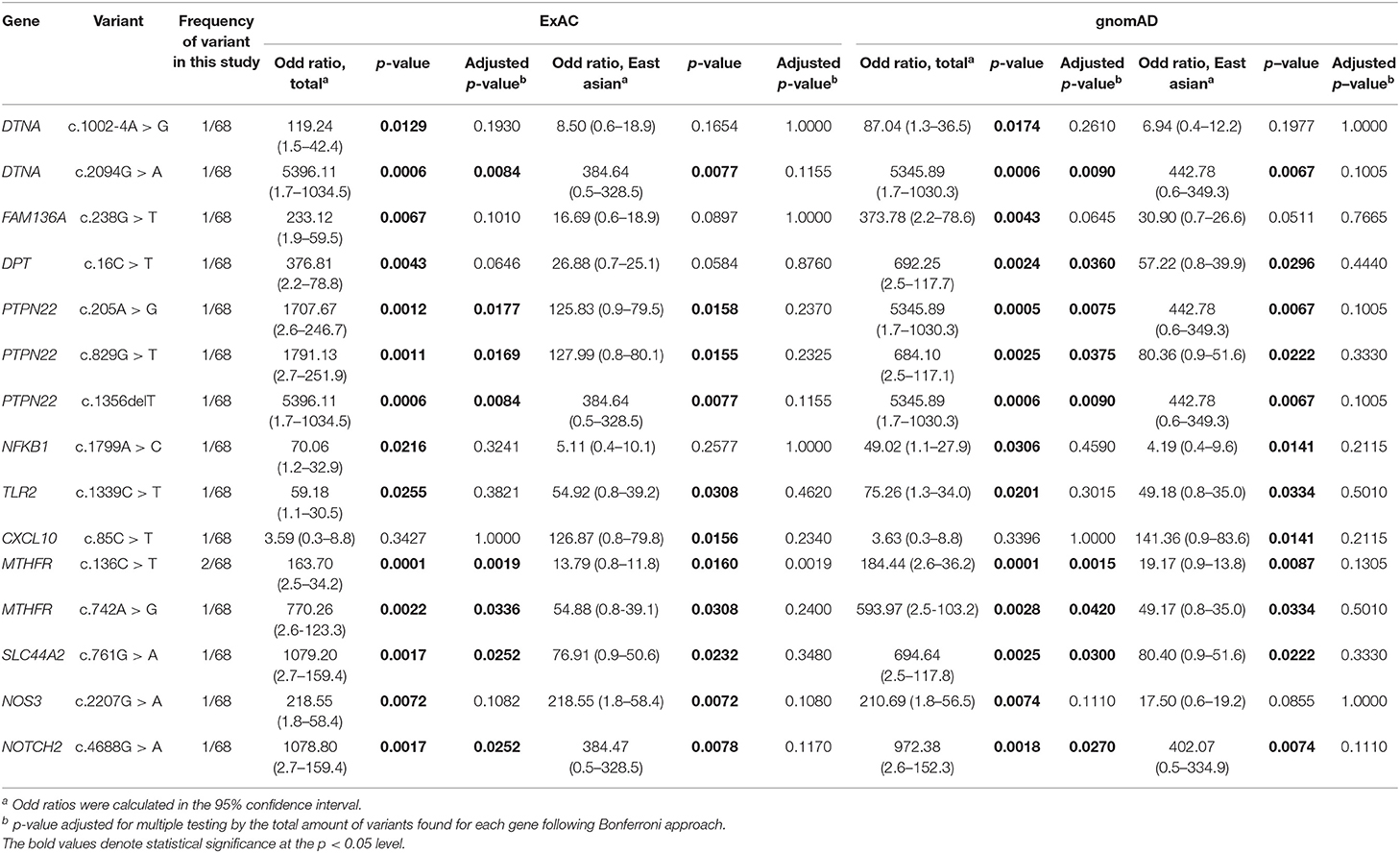
Table 2. Genetic association analysis results for rare variants.
Phenotypes of Patients With a Genetic Variant
The mean age of onset in patients with a novel/rare variant was 53 ± 11.7 years, which showed a tendency of early-onset disease rather than the patients without a genetic variant, but this was not significantly different (vs. 58.4 ± 11.1 years, p = 0.133, Table 3). Also, no significant differences were observed between the two groups in terms of sex, bilaterality, the type of MD, and hearing threshold at diagnosis (Table 3). Three patients with variants in two or more genes did not have distinct clinical features compared to those without variants.
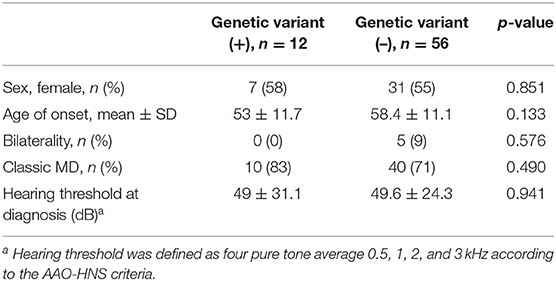
Table 3. Phenotypic differences between patients with and without a genetic variant.
Discussion
To the best of our knowledge, this is the first study on extensive genetic screening with regard to putative candidate MD genes. By using targeted NGS, we identified 15 rare heterozygous variants in 11 candidate MD genes, which some of them had the pathogenic potential by in silico prediction or protein structural modeling. Our results highlight the genetic landscape of MD.
Over the last 20 years, many candidate genes have been proposed for MD based on the so-called “common disease-common variants” paradigm that prevailed in complex disease genetic studies (8–10). It was thought that the common variants may lead to genetic susceptibility to complex polygenic disease (43). By this approach, some SNPs associated with MD have been identified in the genes related to the inflammation or regulating the ionic composition and water transport of the inner ear (11–35). However, most of the SNPs had small effect size with an odds ratio <2, and several variants were located in the non-coding regions of the genome. In addition, none of them would be replicated among a different ethnic group (26, 44, 45). These suggest that a common genetic variant cannot explain the entire heritability of MD and another investigation may be needed (46).
The MD has strong familial aggregation, and most of these families show an autosomal dominant mode of inheritance with incomplete penetrance (3–5). Thus, the optimal methods identifying novel genes may be a NGS technique or a massive parallel sequencing targeting for familial MD. Recent advances in molecular diagnostics have made it possible to explore the entire genome of individuals. By using WES, a Spanish group has identified novel or rare variants in the FAM136A, DTNA, PRKCB, DPT, and SEMA3D in several families with MD (36–38). These proteins were expressed in the neurosensorial epithelium of the crista ampullaris of the rat by immunohistochemistry. This suggests that they may play a significant role in the formation or maintaining of inner ear structures, and the identified rare variants are expected to contribute to the development of EH by the altered protein functions. In this study, we identified some rare variants of FAM136A, DTNA, and DPT genes in sporadic cases. Especially, one variant in DTNA (c.2094G > A) had highly pathogenic potential by truncating the protein. The DTNA encodes α-dystrobrevin (DB), a structural protein of the dystrophin-associated protein complex (36). The absence of α-DB resulted in abnormal brain capillary permeability, progressively escalating brain edema in the mouse model (47). Also, it was expressed in the vestibular system at early stages of development in mice, suggesting a relevant role in the maturation of the vestibular system (48). Although their pathogenicity should be confirmed by functional study, the rare variants in familial MD genes may have large effects on phenotype by impairing protein function, and gene-environment interactions may be strongest for rare alleles (49–51).
In addition, we detected some rare variants in the immune-associated genes including PTPN22, NFKB1, TLR2, and CXCL10. There have been growing evidences on the role of autoimmunity and immunological mechanisms in the development of MD as follows: (1) The increased prevalence of autoimmune disease among MD patients, (2) The elevated levels of autoantibodies and immunocomplexes in MD patients, (3) The association of MD with HLA-types and genetic polymorphisms, and (4) The positive response to steroid (52–54). Recent studies have found that basal levels of proinflammatory cytokines were increased in some patients with MD (55, 56). Gene expression study using mRNA also demonstrated that immune-related genes such as GSTM1, TMEM176A, and TMEM176B were highly expressed in MD patients compared to normal controls (57). Through our study, the PTPN22 may be of particular interest to support the immune-associated process in MD. The identified rare variants had a pathogenic potential by truncating protein or inducing structural instability. The PTPN22 encodes lymphoid tyrosine phosphatase (LYP), which is a strong negative regulator of T cell activation, and is expressed in various immunocytes including T- and B-cells (58). It has been found to be associated with autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus, and type 1 diabetes (59–61). A previous study also found significant association between putative functional SNP (rs2476601) and bilateral MD in the Spanish population (31). The NFKB1 encodes a transcription factor that regulates inflammation and immune responses, and has been linked to a number of inflammatory diseases, such as autoimmune arthritis, asthma, and glomerulonephritis (15). Some allelic variants in NFKB1 are known to modify the hearing outcome in patients with MD and unilateral SNHL (15). The TLR2 is a member of the Toll-like receptor family, which plays a fundamental role in pathogen recognition and activation of innate immunity (40). The CXCL10 encodes interferon gamma-induced protein 10 (IP-10), a small cytokine belonging to the CXC chemokine family (40). IP-10 is an important mediator of the inflammatory response to interferons, and has been reported to contribute to immune mediated apoptosis in the ear, inducing human presbycusis (62). All these things support the possible autoimmune etiology or immunological mechanisms in the development of MD.
For several decades, a vascular theory has been proposed as the mechanism of MD's attacks (63). The classic attacks are characterized by acute loss of vestibular response and low-tone hearing followed by an apparent recovery over hours. These unique characteristics may be explained by the differential sensitivity of inner ear tissues to transient ischemia and the excitotoxic cascade lasting several hours by ischemia/reperfusion injury (63). Most animal models demonstrated that both the endolymphatic hydrops and the impaired perfusion pressure were needed to induce MD's attacks (64, 65). Many studies have also documented an association between MD and presumed vascular disorders including migraine, sickle cell disease, Behcet's disease, and branch retinal artery occlusion (66–71). Among the identified genes in this study, several genes including MTHFR, NOS3, SLC44A2, and NOTCH2 are associated with prothrombotic risk factors and various vascular disorders (72–75). The MTHFR encodes methylenetetrahydrofolate reductase which catalyzes the conversion of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate, a co-substrate for homocystein remethylation to methionine (29). Specific polymorphisms in MTHFR (rs1801131, rs1801133) have been proposed as predisposing inherited vascular risk factors in the development of MD and sudden SNHL (29, 76, 77). The NOS3 encodes a nitric oxide synthase 3, which is responsible for the generation of nitric oxide (NO), a vasodilator in the vascular endothelium (13). NOS3 is also located in the inner and outer hair cells, and a polymorphism of NOS3 (rs1799983) was significantly associated with the risk of sudden SNHL and MD (13). The SLC44A2 encodes a choline transporter-like protein 2 (CTL2) which plays a role in choline transport or uptake with CTL1 (32). Two polymorphisms in SLC44A2 (rs2288904, rs9797861) were linked to venous thrombosis, coronary artery disease, and stroke, and rs2288904 was associated with severity of MD (32). Furthermore, in vivo binding of anti-SLC44A2 antibody induced hearing loss in mice and guinea pigs (78, 79). Thus, their genetic deficiency may cause the increased permeability of vascular endothelial cells, oxidative stress response, and increased vascular resistance, resulting in thrombosis and disturbance of micro-circulation in the inner ear (32).
Although the diagnostic criteria established by the Classification Committee of the Barany Society may improve the accuracy in clinical diagnosis of MD, the phenotypic heterogeneity is observed and some patients may have co-morbid conditions with MD, such as migraine or autoimmune disorders (39). Recent study suggests that an enrichment of rare variants in hearing loss genes such as GJB2, SLC26A, or USH1G may contribute to explain these phenotypic heterogeneities (80). Our patients in this study also showed a broad phenotypic spectrum regarding the onset age, clinical subgroups, and hearing threshold at diagnosis, and some had an accumulation of rare variants in two or more MD genes. Although we confined our gene panel to putative candidate genes associated with MD, our results also suggest the additive effect of several rare variants in the variable expressivity of MD phenotype. Alternatively, the burden of copy number variation can help understand phenotypic heterogeneity (81). Further studies may be needed to confirm these theories.
This study has potential limitations. Despite the extensive genetic screening using NGS, more than 80% of our patients did not have any likely pathogenic variant in putative candidate MD genes. This low detection rate may be due to a high proportion of sporadic cases in our study. Or, the unknown genes or additional factors including epigenetic and environmental modification are likely to contribute to the development of MD (46). We also did not perform functional study determining pathogenicity of our variants. Indeed, several variants in our study were predicted to be benign by in silico prediction tools, or affect only some isoforms of a gene. Despite the rarity and putative pathogenicity of the variants, establishing the pathogenicity may be difficult without a functional study, especially in sporadic cases. With regard to variants of familial MD gene, we could not determine if they were de novo mutations because of parental death. Finally, our panel did not include the GSTM1 or histamine H4 receptor genes, which have recently been suggested as a possible candidate gene of MD (57, 82). All of these should be considered when interpreting our results.
In conclusion, we identified rare variants of putative candidate genes in some of MD patients. The identified genes were related to the formation of inner ear structures, the immune-associated process, or systemic hemostasis derangement, suggesting the multiple genetic predispositions in the development of MD. Since there are still many MD patients without genetic variants, further assessments for the candidate genes will be needed.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
All experiments followed the tenets of the Declaration of Helsinki, and informed consents were obtained after the nature and possible consequences of this study had been explained to the participants. This study was approved by the institutional review boards of Pusan National University Yangsan Hospital.
Author Contributions
EO analyzed and interpreted the data and wrote the manuscript. J-HS and H-SK perfomed the experiment. SC, K-DC, J-KR, and JC analyzed and interpreted the data. SL and CL interpreted the data and revised the manuscript. J-HC contributed to the design and conceptualization of the study, and revised the manuscript.
Funding
This research was supported by Research Institute for Convergence of Biomedical Science and Technology Grant (30-2018-016), Pusan National University Yangsan Hospital.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2019.01424/full#supplementary-material
References
1. Lopez-Escamez JA, Carey J, Chung WH, Goebel JA, Magnusson M, Mandala M, et al. Diagnostic criteria for Meniere's disease. J Vestib Res. (2015) 25:1–7. doi: 10.3233/VES-150549
2. Rauch SD, Merchant SN, Thedinger BA. Meniere's syndrome and endolymphatic hydrops: double-blind temporal bone study. Ann Otol Rhinol Laryngol. (1989) 98:873–83. doi: 10.1177/000348948909801108
3. Requena T, Espinosa-Sanchez JM, Cabrera S, Trinidad G, Soto-Varela A, Santos-Perez S, et al. Familial clustering and genetic heterogeneity in Meniere's disease. Clin Genet. (2014) 85:245–52. doi: 10.1111/cge.12150
4. Morrison AW, Bailey ME, Morrison GA. Familial Meniere's disease: clinical and genetic aspects. J Laryngol Otol. (2009) 123:29–37. doi: 10.1017/S0022215108002788
5. Lee JM, Kim MJ, Jung J, Kim HJ, Seo YJ, Kim SH. Genetic aspects and clinical characteristics of familial Meniere's disease in a South Korean population. Laryngoscope. (2015) 125:2175–80. doi: 10.1002/lary.25207
6. Klar J, Frykholm C, Friberg U, Dahl N. A Meniere's disease gene linked to chromosome 12p12.3. Am J Med Genet B Neuropsychiatr Genet. (2006) 141b:463–7. doi: 10.1002/ajmg.b.30347
7. Arweiler-Harbeck D, Horsthemke B, Jahnke K, Hennies HC. Genetic aspects of familial Meniere's disease. Otol Neurotol. (2011) 32:695–700. doi: 10.1097/MAO.0b013e318216074a
8. Frejo L, Giegling I, Teggi R, Lopez-Escamez JA, Rujescu D. Genetics of vestibular disorders: pathophysiological insights. J Neurol. (2016) 263 (Suppl. 1):S45–53. doi: 10.1007/s00415-015-7988-9
9. Roman-Naranjo P, Gallego-Martinez A, Lopez Escamez JA. Genetics of vestibular syndromes. Curr Opin Neurol. (2018) 31:105–10. doi: 10.1097/WCO.0000000000000519
10. Requena T, Espinosa-Sanchez JM, Lopez-Escamez JA. Genetics of dizziness: cerebellar and vestibular disorders. Curr Opin Neurol. (2014) 27:98–104. doi: 10.1097/WCO.0000000000000053
11. Teggi R, Lanzani C, Zagato L, Delli Carpini S, Manunta P, Bianchi G, et al. Gly460Trp alpha-adducin mutation as a possible mechanism leading to endolymphatic hydrops in Meniere's syndrome. Otol Neurotol. (2008) 29:824–8. doi: 10.1097/MAO.0b013e318180a4b1
12. Nishio N, Teranishi M, Uchida Y, Sugiura S, Ando F, Shimokata H, et al. Polymorphisms in genes encoding aquaporins 4 and 5 and estrogen receptor α in patients with Ménière's disease and sudden sensorineural hearing loss. Life Sci. (2013) 92:541–6. doi: 10.1016/j.lfs.2013.01.019
13. Teranishi M, Uchida Y, Nishio N, Kato K, Otake H, Yoshida T, et al. Polymorphisms in genes involved in the free-radical process in patients with sudden sensorineural hearing loss and Ménière's disease. Free Radic Res. (2013) 47:498–506. doi: 10.3109/10715762.2013.793319
14. Yazdani N, Mojbafan M, Taleba M, Amiri P, Nejadian F, Amoli MM. Sex-specific association of RANTES gene−403 variant in Meniere's disease. Eur Arch Otorhinolaryngol. (2015) 272:2221–5. doi: 10.1007/s00405-014-3151-y
15. Cabrera S, Sanchez E, Requena T, Martinez-Bueno M, Benitez J, Perez N, et al. Intronic variants in the NFKB1 gene may influence hearing forecast in patients with unilateral sensorineural hearing loss in Meniere's disease. PLoS ONE. (2014) 9:e112171. doi: 10.1371/journal.pone.0112171
16. Teggi R, Colombo B, Trimarchi M, Bianco M, Manfredi A, Corti A. Altered chromogranin a circulating levels in Meniere's Disease. Dis Markers. (2015) 2015:643420. doi: 10.1155/2015/643420
17. Vrabec JT, Liu L, Li B, Leal SM. Sequence variants in host cell factor C1 are associated with Meniere's disease. Otol Neurotol. (2008) 29:561–6. doi: 10.1097/MAO.0b013e318168d23b
18. Khorsandi MT, Amoli MM, Borghei H, Emami H, Amiri P, Amirzargar A, et al. Associations between HLA-C alleles and definite Meniere's Disease. Iranian J Allergy Asthma Immunol. (2011) 10:119–22. doi: 10.02/ijaai.119122
19. Melchiorri L, Martini A, Rizzo R, Berto A, Adinolfi E, Baricordi OR. Human leukocyte antigen-A, -B, -C and -DR alleles and soluble human leukocyte antigen class I serum level in Meniere's disease. Acta Otolaryngol Suppl. (2002) 548:26–9. doi: 10.1080/00016480260094938
20. Meng X, Lian N, Yang Z, Liu C, Tang L. An association study of histocompatibility leukocyte antigen-class II with Meniere's disease. Zhonghua Er Bi Yan Hou Ke Za Zhi. (2001) 36:25–7.
21. Lopez-Escamez JA, Vilchez JR, Soto-Varela A, Santos-Perez S, Perez-Garrigues H, Aran I, et al. HLA-DRB1*1101 allele may be associated with bilateral Meniere's disease in southern European population. Otol Neurotol. (2007) 28:891–5. doi: 10.1097/MAO.0b013e3180dca1cc
22. Koo JW, Oh SH, Chang SO, Park MH, Lim MJ, Yoo TJ, et al. Association of HLA-DR and type II collagen autoimmunity with Meniere's disease. Tissue Antigens. (2003) 61:99–103. doi: 10.1034/j.1399-0039.2003.610112.x
23. Kawaguchi S, Hagiwara A, Suzuki M. Polymorphic analysis of the heat-shock protein 70 gene (HSPA1A) in Ménière's disease. Acta Otolaryngol. (2008) 128:1173–7. doi: 10.1080/00016480801901675
24. Furuta T, Teranishi M, Uchida Y, Nishio N, Kato K, Otake H, et al. Association of interleukin-1 gene polymorphisms with sudden sensorineural hearing loss and Ménière's disease. Int J Immunogenet. (2011) 38:249–54. doi: 10.1111/j.1744-313X.2011.01004.x
25. Doi K, Sato T, Kuramasu T, Hibino H, Kitahara T, Horii A, et al. Ménière's disease is associated with single nucleotide polymorphisms in the human potassium channel genes, KCNE1 and KCNE3. ORL J Otorhinolaryngol Relat Spec. (2005) 67:289–93. doi: 10.1159/000089410
26. Hietikko E, Kotimäki J, Okuloff A, Sorri M, Männikkö M. A replication study on proposed candidate genes in Ménière's disease, and a review of the current status of genetic studies. Int J Audiol. (2012) 51:841–5. doi: 10.3109/14992027.2012.705900
27. Gazquez I, Moreno A, Aran I, Soto-Varela A, Santos S, Perez-Garrigues H, et al. MICA-STR A.4 is associated with slower hearing loss progression in patients with Meniere's disease. Otol Neurotol. (2012) 33:223–9. doi: 10.1097/MAO.0b013e31824296c8
28. Yazdani N, Khorsandi A, Shtiani MT, Zarandy MM, Mohammadi SJ, Ghazavi H, et al. Association between MIF gene variation and Meniere's disease. Int J Immunogenet. (2013) 40:488–91. doi: 10.1111/iji.12058
29. Huang Y, Teranishi M, Uchida Y, Nishio N, Kato K, Otake H, et al. Association between polymorphisms in genes encoding methylenetetrahydrofolate reductase and the risk of Ménière's disease. J Neurogenet. (2013) 27:5–10. doi: 10.3109/01677063.2013.770510
30. Lopez-Escamez JA, Moreno A, Bernal M, Perez-Garrigues H, Santos-Perez S, Soto-Varela A, et al. Poly(ADP-ribose) polymerase-1 (PARP-1) spanning promoter region confer protection to bilateral Meniere's disease. Acta Otolaryngol. (2009) 129:1222–5. doi: 10.3109/00016480802684080
31. Lopez-Escamez JA, Saenz-Lopez P, Acosta L, Moreno A, Gazquez I, Perez-Garrigues H, et al. Association of a functional polymorphism of PTPN22 encoding a lymphoid protein phosphatase in bilateral Meniere's disease. Laryngoscope. (2010) 120:103–7. doi: 10.1002/lary.20650
32. Nair TS, Kommareddi PK, Galano MM, Miller DM, Kakaraparthi BN, Telian SA, et al. SLC44A2 single nucleotide polymorphisms, isoforms, and expression: association with severity of Meniere's disease? Genomics. (2016) 108:201–8. doi: 10.1016/j.ygeno.2016.11.002
33. Requena T, Gazquez I, Moreno A, Batuecas A, Aran I, Soto-Varela A, et al. Allelic variants in TLR10 gene may influence bilateral affectation and clinical course of Meniere's disease. Immunogenetics. (2013) 65:345–55. doi: 10.1007/s00251-013-0683-z
34. Greco A, Gallo A, Fusconi M, Marinelli C, Macri GF, De Vincentiis M. Meniere's disease might be an autoimmune condition? Autoimmunity Reviews. (2012) 11:731–8. doi: 10.1016/j.autrev.2012.01.004
35. Teggi R, Zagato L, Delli Carpini S, Citterio L, Cassandro C, Albera R, et al. Genetics of ion homeostasis in Ménière's Disease. Eur Arch Otorhinolaryngol. (2017) 274:757–63. doi: 10.1007/s00405-016-4375-9
36. Requena T, Cabrera S, Martin-Sierra C, Price SD, Lysakowski A, Lopez-Escamez JA. Identification of two novel mutations in FAM136A and DTNA genes in autosomal-dominant familial Meniere's disease. Hum Mol Genet. (2015) 24:1119–26. doi: 10.1093/hmg/ddu524
37. Martín-Sierra C, Requena T, Frejo L, Price SD, Gallego-Martinez A, Batuecas-Caletrio A, et al. A novel missense variant in PRKCB segregates low-frequency hearing loss in an autosomal dominant family with Meniere's disease. Hum Mol Genet. (2016) 25:3407–15. doi: 10.1093/hmg/ddw183
38. Martín-Sierra C, Gallego-Martinez A, Requena T, Frejo L, Batuecas-Caletrío A, Lopez-Escamez JA. Variable expressivity and genetic heterogeneity involving DPT and SEMA3D genes in autosomal dominant familial Meniere's disease. Eur J Hum Genet. (2017) 25:200–7. doi: 10.1038/ejhg.2016.154
39. Frejo L, Martin-Sanz E, Teggi R, Trinidad G, Soto-Varela A, Santos-Perez S, et al. Extended phenotype and clinical subgroups in unilateral Meniere disease: a cross-sectional study with cluster analysis. Clin Otolaryngol. (2017) 42:1172–80. doi: 10.1111/coa.12844
40. Li L, Wang Y, An L, Kong X, Huang T. A network-based method using a random walk with restart algorithm and screening tests to identify novel genes associated with Menière's disease. PLoS ONE. (2017) 12:e0182592. doi: 10.1371/journal.pone.0182592
41. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. (2019) 47:D886–94. doi: 10.1093/nar/gky1016
42. Yang J, Zhang Y. Protein structure and function prediction using I-TASSER. Curr Protoc Bioinform. (2015) 52:1–15. doi: 10.1002/0471250953.bi0508s52
43. Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. (2001) 17:502–10. doi: 10.1016/S0168-9525(01)02410-6
44. Li YJ, Jin ZG, Xu XR. Variants in the KCNE1 or KCNE3 gene and risk of Meniere's disease: a meta-analysis. J Vestib Res. (2016) 25:211–8. doi: 10.3233/VES-160569
45. Campbell CA, Della Santina CC, Meyer NC, Smith NB, Myrie OA, Stone EM, et al. Polymorphisms in KCNE1 or KCNE3 are not associated with Meniere disease in the Caucasian population. Am J Med Genet A. (2010) 152a:67–74. doi: 10.1002/ajmg.a.33114
46. Gazquez I, Lopez-Escamez JA. Genetics of recurrent vertigo and vestibular disorders. Curr Genomics. (2011) 12:443–50. doi: 10.2174/138920211797248600
47. Lien CF, Mohanta SK, Frontczak-Baniewicz M, Swinny JD, Zablocka B, Górecki DC. Absence of glial α-dystrobrevin causes abnormalities of the blood-brain barrier and progressive brain edema. J Biol Chem. (2012) 287:41374–85. doi: 10.1074/jbc.M112.400044
48. Lien CF, Vlachouli C, Blake DJ, Simons JP, Górecki DC. Differential spatio-temporal expression of alpha-dystrobrevin-1 during mouse development. Gene Expr Patterns. (2004) 4:583–93. doi: 10.1016/j.modgep.2004.01.015
49. Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. (2010) 11:446–50. doi: 10.1038/nrg2809
50. Lettre G. Rare and low-frequency variants in human common diseases and other complex traits. J Med Genet. (2014) 51:705–14. doi: 10.1136/jmedgenet-2014-102437
51. Saint Pierre A, Génin E. How important are rare variants in common disease? Brief Funct Genomics. (2014) 13:353–61. doi: 10.1093/bfgp/elu025
52. Tomoda K, Suzuka Y, Iwai H, Yamashita T, Kumazawa T. Menière's disease and autoimmunity: clinical study and survey. Acta Otolaryngol Suppl. (1993) 500:31–4. doi: 10.3109/00016489309126174
53. Filipo R, Mancini P, Nostro G. Ménière's disease and autoimmunity. Ann N Y Acad Sci. (1997) 830:299–305. doi: 10.1111/j.1749-6632.1997.tb51900.x
54. Kim SH, Kim JY, Lee HJ, Gi M, Kim BG, Choi JY. Autoimmunity as a candidate for the etiopathogenesis of Meniere's disease: detection of autoimmune reactions and diagnostic biomarker candidate. PLoS ONE. (2014) 9:e111039. doi: 10.1371/journal.pone.0111039
55. Frejo L, Gallego-Martinez A, Requena T, Martin-Sanz E, Amor-Dorado JC, Soto-Varela A, et al. Proinflammatory cytokines and response to molds in mononuclear cells of patients with Meniere disease. Sci Rep. (2018) 8:5974. doi: 10.1038/s41598-018-23911-4
56. Flook M, Frejo L, Gallego-Martinez A, Martin-Sanz E, Rossi-Izquierdo M, Amor-Dorado JC, et al. Differential proinflammatory signature in vestibular migraine and meniere disease. Front Immunol. (2019) 10:1229. doi: 10.3389/fimmu.2019.01229
57. Sun Y, Zhang D, Sun G, Lv Y, Li Y, Li X, et al. RNA-sequencing study of peripheral blood mononuclear cells in sporadic Ménière's disease patients: possible contribution of immunologic dysfunction to the development of this disorder. Clin Exp Immunol. (2018) 192:33–45. doi: 10.1111/cei.13083
58. Mustelin T, Alonso A, Bottini N, Huynh H, Rahmouni S, Nika K, et al. Protein tyrosine phosphatases in T cell physiology. Mol Immunol. (2004) 41:687–700. doi: 10.1016/j.molimm.2004.04.015
59. Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. (2004) 75:330–7. doi: 10.1086/422827
60. de Lima SC, Adelino JE, Crovella S, de Azevedo Silva J, Sandrin-Garcia P. PTPN22 1858C > T polymorphism and susceptibility to systemic lupus erythematosus: a meta-analysis update. Autoimmunity. (2017) 50:428–34. doi: 10.1080/08916934.2017.1385774
61. Lee YH, Rho YH, Choi SJ, Ji JD, Song GG, Nath SK, et al. The PTPN22 C1858T functional polymorphism and autoimmune diseases–a meta-analysis. Rheumatology. (2007) 46:49–56. doi: 10.1093/rheumatology/kel170
62. Dong Y, Li M, Liu P, Song H, Zhao Y, Shi J. Genes involved in immunity and apoptosis are associated with human presbycusis based on microarray analysis. Acta Otolaryngol. (2014) 134:601–8. doi: 10.3109/00016489.2014.880795
63. Foster CA, Breeze RE. The Meniere attack: an ischemia/reperfusion disorder of inner ear sensory tissues. Med Hypotheses. (2013) 81:1108–15. doi: 10.1016/j.mehy.2013.10.015
64. Takumida M, Akagi N, Anniko M. A new animal model for Meniere's disease. Acta Otolaryngol. (2008) 128:263–71. doi: 10.1080/00016480701497436
65. Kimura R. Experimental endolymphatic hydrops. In: Harris JP, editor. Meniere's Disease. The Hague: Kugler publications; 1999. p. 115–24.
66. Lee H, Lopez I, Ishiyama A, Baloh R. Can migraine damage the inner ear? Arch Neurol. (2000) 57:1631–4. doi: 10.1001/archneur.57.11.1631
67. Lee H, Whitman GT, Lim JG, Yi SD, Cho YW, Ying S, et al. Hearing symptoms in migrainous infarction. Arch Neurol. (2003) 60:113–6. doi: 10.1001/archneur.60.1.113
68. Marcus RE, Lee YM. Inner ear disorders in a family with sickle cell thalassemia. Arch Otolaryngol. (1976) 102:703–5.
69. Schwanitz HJ, Knop J, Bonsmann G. Behcet disease with inner ear involvement. Zeitschrift fur Hautkrankheiten. (1984) 59:1173–4.
70. Brama I, Fainaru M. Inner ear involvement in Behcet's disease. Arch Otolaryngol. (1980) 106:215–7. doi: 10.1001/archotol.1980.00790280023004
71. Recupero SM, Abdolrahimzadeh R, Occhiuto S, Tripodi A. G, Branch retinal artery occlusion in a young woman with Meniere's disease. Ophthalmologica. (1998) 212:77–9.
72. Liew SC, Gupta ED. Methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism: epidemiology, metabolism and the associated diseases. Eur J Med Genet. (2015) 58:1–10. doi: 10.1016/j.ejmg.2014.10.004
73. Tilburg J, Adili R, Nair TS, Hawley ME, Tuk DC, Jackson M, et al. Characterization of hemostasis in mice lacking the novel thrombosis susceptibility gene Slc44a2. Thromb Res. (2018) 171:155–9. doi: 10.1016/j.thromres.2018.09.057
74. Oliveira-Paula GH, Lacchini R, Tanus-Santos JE. Clinical and pharmacogenetic impact of endothelial nitric oxide synthase polymorphisms on cardiovascular diseases. Nitric Oxide. (2017) 63:39–51. doi: 10.1016/j.niox.2016.08.004
75. Rasheed MA, Kantoush N, Abd El-Ghaffar N, Farouk H, Kamel S, Ibrahim AA, et al. Expression of JAZF1, ABCC8, KCNJ11and Notch2 genes and vitamin D receptor polymorphisms in type 2 diabetes, and their association with microvascular complications. Ther Adv Endocrinol Metab. (2017) 8:97–108. doi: 10.1177/2042018817708910
76. Shu J, Yin S, Tan AZ, He M. Association between the methylenetetrahydrofolate reductase gene C677T polymorphism and sudden sensorineural hearing loss: a meta-analysis. Eur Arch Otorhinolaryngol. (2015) 272:2267–74. doi: 10.1007/s00405-014-3198-9
77. Uchida Y, Sugiura S, Ando F, Shimokata H, Nakashima T. Association of the C677T polymorphism in the methylenetetrahydrofolate reductase gene with sudden sensorineural hearing loss. Laryngoscope. (2010) 120:791–5. doi: 10.1002/lary.20809
78. Nair TS, Prieskorn DM, Miller JM, Dolan DF, Raphael Y, Carey TE. KHRI-3 monoclonal antibody-induced damage to the inner ear: antibody staining of nascent scars. Hear Res. (1999) 129:50–60. doi: 10.1016/S0378-5955(98)00220-2
79. Nair TS, Raphael Y, Dolan DF, Parrett TJ, Perlman LS, Brahmbhatt VR, et al. Monoclonal antibody induced hearing loss. Hear Res. (1995) 83:101–13. doi: 10.1016/0378-5955(94)00194-U
80. Gallego-Martinez A, Requena T, Roman-Naranjo P, Lopez-Escamez JA. Excess of rare missense variants in hearing loss genes in sporadic meniere disease. Front Genet. (2019) 10:76. doi: 10.3389/fgene.2019.00076
81. Girirajan S1, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med. (2012) 367:1321–31. doi: 10.1056/NEJMoa1200395
Keywords: Meniere's disease, endolymphatic hydrops, gene, whole-exome sequencing, rare variant
Citation: Oh EH, Shin J-H, Kim H-S, Cho JW, Choi SY, Choi K-D, Rhee J-K, Lee S, Lee C and Choi J-H (2020) Rare Variants of Putative Candidate Genes Associated With Sporadic Meniere's Disease in East Asian Population. Front. Neurol. 10:1424. doi: 10.3389/fneur.2019.01424
Received: 06 November 2019; Accepted: 31 December 2019;
Published: 22 January 2020.
Edited by:
Jose Antonio Lopez-Escamez, Andalusian Autonomous Government of Genomics and Oncological Research (GENYO), SpainReviewed by:
Teresa Requena, University of Edinburgh, United KingdomBarbara Vona, University of Tübingen, Germany
Copyright © 2020 Oh, Shin, Kim, Cho, Choi, Choi, Rhee, Lee, Lee and Choi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jae-Hwan Choi, cmFjaGVsYm9sYW5AaGFubWFpbC5uZXQ=