 Samya Chakravorty1,2,3,4*
Samya Chakravorty1,2,3,4* Babi Ramesh Reddy Nallamilli5
Babi Ramesh Reddy Nallamilli5 Satish Vasant Khadilkar6,7,8
Satish Vasant Khadilkar6,7,8 Madhu Bala Singla6,7,8
Madhu Bala Singla6,7,8 Ashish Bhutada6
Ashish Bhutada6 Rashna Dastur9
Rashna Dastur9 Pradnya Satish Gaitonde9
Pradnya Satish Gaitonde9 Laura E Rufibach10
Laura E Rufibach10 Logan Gloster1,4Madhuri Hegde4,5*
Logan Gloster1,4Madhuri Hegde4,5*- 1Emory University Department of Pediatrics, Atlanta, GA, United States
- 2Emory University Department of Human Genetics, Atlanta, GA, United States
- 3Division of Neurosciences, Children's Healthcare of Atlanta, Atlanta, GA, United States
- 4School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA, United States
- 5PerkinElmer Genomics, Global Laboratory Services, Waltham, MA, United States
- 6Department of Neurology, Bombay Hospital, Mumbai, India
- 7Department of Neurology, Sir J J Group of Hospitals, Grant Medical College, Mumbai, India
- 8Bombay Hospital Institute of Medical Sciences, Mumbai, India
- 9Centre for Advanced Molecular Diagnostics in Neuromuscular Disorders (CAMDND), Mumbai, India
- 10Jain Foundation, Seattle, WA, United States
Objective: Inherited myopathies comprise more than 200 different individually rare disease-subtypes, but when combined together they have a high prevalence of 1 in 6,000 individuals across the world. Our goal was to determine for the first time the clinical- and gene-variant spectrum of genetic myopathies in a substantial cohort study of the Indian subcontinent.
Methods: In this cohort study, we performed the first large clinical exome sequencing (ES) study with phenotype correlation on 207 clinically well-characterized inherited myopathy-suspected patients from the Indian subcontinent with diverse ethnicities.
Results: Clinical-correlation driven definitive molecular diagnosis was established in 49% (101 cases; 95% CI, 42–56%) of patients with the major contributing pathogenicity in either of three genes, GNE (28%; GNE-myopathy), DYSF (25%; Dysferlinopathy), and CAPN3 (19%; Calpainopathy). We identified 65 variant alleles comprising 37 unique variants in these three major genes. Seventy-eight percent of the DYSF patients were homozygous for the detected pathogenic variant, suggesting the need for carrier-testing for autosomal-recessive disorders like Dysferlinopathy that are common in India. We describe the observed clinical spectrum of myopathies including uncommon and rare subtypes in India: Sarcoglycanopathies (SGCA/B/D/G), Collagenopathy (COL6A1/2/3), Anoctaminopathy (ANO5), telethoninopathy (TCAP), Pompe-disease (GAA), Myoadenylate-deaminase-deficiency-myopathy (AMPD1), myotilinopathy (MYOT), laminopathy (LMNA), HSP40-proteinopathy (DNAJB6), Emery-Dreifuss-muscular-dystrophy (EMD), Filaminopathy (FLNC), TRIM32-proteinopathy (TRIM32), POMT1-proteinopathy (POMT1), and Merosin-deficiency-congenital-muscular-dystrophy-type-1 (LAMA2). Thirteen patients harbored pathogenic variants in >1 gene and had unusual clinical features suggesting a possible role of synergistic-heterozygosity/digenic-contribution to disease presentation and progression.
Conclusions: Application of clinically correlated ES to myopathy diagnosis has improved our understanding of the clinical and genetic spectrum of different subtypes and their overlaps in Indian patients. This, in turn, will enhance the global gene-variant-disease databases by including data from developing countries/continents for more efficient clinically driven molecular diagnostics.
Introduction
To date, >250 genes are associated with various inherited neuromuscular disorders (NMDs) (1–4) comprising a broad heterogeneous group of genetic myopathies. However, significant numbers of affected patients remain without a definitive molecular diagnosis due to novel gene or multiple gene associations with disease (5, 6). The autosomal-dominant forms represent <10% of all genetic myopathies (6–8) and are usually associated with milder clinical presentation and adult onset. For the more common autosomal-recessive forms, multiple monogenic subtypes have been identified including those that make up the limb-girdle muscular dystrophies (LGMDs) (5, 9). Definitive molecular diagnosis is a prerequisite for patient participation in clinical-trials or precision medicine treatment and management. Hence, it is important to understand the genetic etiologies and subtype prevalence in different populations to enhance global variant databases for more efficient and accurate gene-variant classification and diagnosis.
Several studies reported the use of next-generation-sequencing (NGS) for molecular diagnosis in small/large genetic myopathy patient-cohorts in different countries (6, 10–12). But the application of exome sequencing (ES) for a large cohort of myopathy patients from the Indian subcontinent has not been reported previously. Similar to the rising awareness of the importance of African genomes or exomes in global public health and enhancing public databases, it is important to study the populations of the Indian subcontinent and Asia as a whole comprising of the largest proportion of the world population that offers diverse ethnic and genetic backgrounds (13–16). Moreover, the sociocultural belief systems and traditions have led to community marriages, consanguinity, and intra-communal exogamy in sections of the Indian population. Here, we performed the first large-scale ES study on 207 clinically well-characterized inherited myopathy-suspected patients of diverse ethnicities from the Indian subcontinent to identify the causative gene and disease-subtypes. Our results yield interesting observations on Indian myopathy clinical and gene-variant landscape and approaches to both clinical and genetic diagnosis in muscular dystrophy and myopathy in general.
Materials and Methods
Standard Protocol Approvals, Registrations, and Patient Consents
Written, informed consent was obtained from all individuals or minors' parent or legal guardian or next of kin for the publication of any potentially identifiable images or data included in this article. Written informed consents were obtained from all participants of the study for all procedures according to approved ethics committee Institutional Review Board protocol. All participants (including parents or legal guardians in case of minor patients) were provided pre- and post-test routine genetic counseling.
Patients and Study Design
In an effort to understand the genetic basis of myopathies, over a 3 year period (2016–2019) a total of 207 patients with clinical suspicion of an inherited myopathy were recruited (see Questionnaires in Supplementary Material) based on the following inclusion and exclusion criteria. Inclusion criteria: Patients with a suspected clinical diagnosis of genetic myopathy. Exclusion Criteria: 1. Patients with confirmed diagnosis of Duchenne muscular dystrophy (DMD), facioscapulohumeral muscular dystrophy (FSHD), myotonic dystrophy (DM1 and DM2), mitochondrial myopathy, and acquired myopathies. DMD, myotonic dystrophies, and FSHD were genetically diagnosed. Mitochondrial myopathies were diagnosed based on muscle biopsy immunohistochemistry and/or genetic diagnosis. We used modified Gomori trichrome, cytochrome oxidase (COX), and nicotinamide adenine dinucleotide (NADH) stains for the demonstration of mitochondrial dysfunction, supported by electron microscopy as needed. There was no age restriction and the patients were recruited from across the Indian subcontinent at a single tertiary care hospital at Mumbai, India.
Clinical Evaluation
All patients underwent comprehensive clinical evaluation. Clinical history including age of onset, initial symptoms, region in which weakness started first, and functional status of the patient was collected whenever available for all patients recruited in the study. Pattern of weakness including upper limbs or lower limbs or both, proximal or distal or both, symmetrical or asymmetrical, and differential weakness was determined. Detailed pedigree analysis was performed. Available affected family members were examined. Detailed history regarding the origins and ancestry and marriage customs practiced in the specific ethnic community was documented. Patients underwent detailed clinical examination with focus on the motor system. Hypertrophy, atrophy of muscles was determined from inspection. Neck flexors, extensors, and other total 16 pairs of muscles (trapezius, deltoid, serratus anterior, biceps, brachioradialis, triceps, wrist flexors and extensors, ileopsoas, hip adductors, abductors, extensors, quadriceps, hamstrings, tibialis anterior, gastrocnemius-soleus) were examined as per the MRC grading system of 0 to 5 (17). Small muscles of hands, feet, and paraspinal muscle were also examined. Serum creatine kinase was determined at the time of presentation prior to electromyography. Electromyography and nerve conduction studies were performed in all patients. Electrocardiography was done in all patients and echocardiography was done in selected symptomatic patients.
Muscle Biopsy Procedure
Muscle biopsy procedure was performed for only the consenting patients as per routine methodology. Muscle biopsy was performed from clinically affected muscle with power between MRC grades 3 to 4. Biopsy specimens were snap frozen in chilled isopentane for frozen sections and later taken in formalin for paraffin embedding. Routine assessment of morphological changes was carried out on frozen and paraffin sections. Biopsy specimens from those patients who consented for the procedure were grouped into dystrophic, myopathic, dystrophic with inflammation, myositis, mixed (neurogenic and myopathic) changes, and some no diagnosis. Histochemical studies were done using ATPase and NADH-TR (nicotinamide adenine dinucleotide [NAD] + hydrogen [H] and tetrazolium reductase) stains. Frozen sections of 6 microns were stained with dystrophin 1 (C terminus-mouse monoclonal Dy8\6C5), dystrophin 2 (mid-rod domain mouse monoclonal Dy4\6D3), dysferlin (mouse monoclonal NCL-Hamlet-2), and α (Ad1\20A6), β (Bsarc\5B1), γ (35 DAG\21B5), and δ (Dsarc 3\12C1) sarcoglycan antibodies (Novocastra labs), using peroxidase method and standard controls.
Molecular Diagnosis by Exome Sequencing
Exome sequencing was performed in a Clinical Laboratory Improvement Amendments and College of American Pathologists (CLIA-CAP)-certified laboratory. Peripheral intravenous blood was collected from enrolled patients into 10-ml commercial tubes containing ethylene-diamine tetraacetic acid (EDTA). These samples were shipped immediately overnight on ice to our laboratory for further analysis. DNA was extracted from whole blood using the Qiagen genomic DNA extraction kit according to the manufacturer's instructions. We performed exome sequencing using an Agilent V5Plus exome capture kit to identify disease causative variants. Sequencing was performed using the Illumina HiSeq 2500 sequencing system with 100-basepair (bp) paired-end reads (similar to bidirectional Sanger sequencing), with an average coverage of 100X in the target region. The target region includes the exon and 10 bp of flanking intronic region. NextGENe software mapped the DNA sequence reads to the published human genome build UCSC hg19 reference sequence. Primary data analysis was also performed using Illumina DRAGEN Bio-IT Platform v.2.03. Secondary and tertiary data analysis was also performed using PerkinElmer's internal ODIN v.1.01 software for SNVs and Biodiscovery's NxClinical v.4.3 or Illumina DRAGEN Bio-IT Platform v.2.03 for copy number variant (CNV) and absence of heterozygosity (AOH). Reads were aligned and filtered as described previously (18, 19). In short, in the filtering process, variants in genes that are not relevant to the patient's clinical phenotypes were filtered in the clinical setting using population-wide minor allele frequency, predicted effect on protein function or splicing, literature evidence, and disease-variant databases such as the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk), ClinVar (http://www.clinvar.com), Online Mendelian Inheritance of Man (https://www.ncbi.nlm.nih.gov/omim), genome Aggregation Database (gnomAD: https://gnomad.broadinstitute.org/), and using our internal Emory Genetics Lab (EGL) EmVClass database (http://www.egl-eurofins.com/emvclass/emvclass.php). Variants that do not meet QC metrics, such as those with poor coverage (<20×), were considered less likely to be real, treated as false positives, and therefore filtered. Variants with a minor allele frequency of >0.01 are polymorphisms by definition and less likely to be pathogenic. Silent changes and intronic variants beyond the consensus splice donor/acceptor sequences or beyond the 10 bp intronic flanking sequences are less likely to be pathogenic and were filtered in the analysis unless a known clinically significant rare deep-intronic variant is identified. Rare single nucleotide variants (SNVs) meeting internal quality assessment guidelines are confirmed by Sanger sequence analysis.
Molecular diagnosis was performed by classifying variants with clinical data correlation to identify most likely causal disease gene and as per American College of Medical Genetics and Genomics (ACMG) guidelines (20) and with clinical data correlation. Diagnostic yield was calculated based on definitive diagnosis of patients harboring pathogenic or likely pathogenic variants in autosome-recessive/dominant, and X-linked myopathy genes. Prevalence of each identified myopathy subtype identified by our clinical exome sequencing diagnostic program was established and compared. When rare reportable variants in our study were not found in NIH ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), nature of variant, literature evidence, and correlation with established specific clinical symptoms of the patient's myopathy was used to identify most likely causal gene and to classify the variant, keeping in mind ACMG guidelines. When no literature evidence was found, the variant was classified based on nature of variant such as non-sense variant or frameshift variant causing a premature stop codon or a stop-loss or a large deletion or duplication in genes known to affect by loss of function modality, as well as correlation with established specific clinical symptoms of the patient's myopathy. High-confidence rare variants were taken as novel when not found in public databases and included in the analysis to interpret relevant disease type causality with clinical correlation. Patient cases with variants of uncertain significances (VUSs) were not included in the molecular diagnostic yield calculations.
Results
Molecular Diagnosis of Recruited Patients
A total of 207 unrelated myopathy-suspected patients were recruited in the current study from diverse regions of India with different ethnicities and religion, as well as varied social and marriage customs. Molecular diagnosis was established in 49% (101/207; 95% CI, 42–56%) of the patients with the majority having pathogenic variants identified in one of the following genes, GNE (31%; 31/101), DYSF (27%; 27/101), or CAPN3 (19%; 19/101), indicating that these genes are the major contributors to genetic myopathy in India (Figure 1). Autosomal-recessive forms were much more frequent constituting 95/101 (94%) of the genetically diagnosed patients. Uncommon autosomal/X-linked recessive-subtypes identified were: 5 sarcoglycanopathies (LGMD R3–R6; 5%), 3 anoctaminopathies (LGMD R12 anoctamin-related; 3%), 2 myoadenylate-deaminase-deficiency-myopathies (2%), 2 Pompe disease (2%), 2 telethoninopathies (LGMDR7 telethonin-related; 2%), 1 X-linked Emery-Dreifuss-muscular-dystrophy (EDMD1 emerin-related; 1%), 1 Merosin-deficiency-congenital-muscular-dystrophy-type 1 (MDC1 or LGMDR23 laminin α2-related; 1%), 1 POMT1-proteinopathy (LGMDR11 POMT1-related; 1%). Autosomal-dominant-subtypes were less common with 3 laminopathies (EDMD2; 3%), 1 myotilinopathy (Myofibrillar Myopathy; 1%), and 1 HSP40-proteinopathy (LGMDD1 DNAJB6-related; 1%). Fourteen patients had no reportable variants and were truly negative for this study.
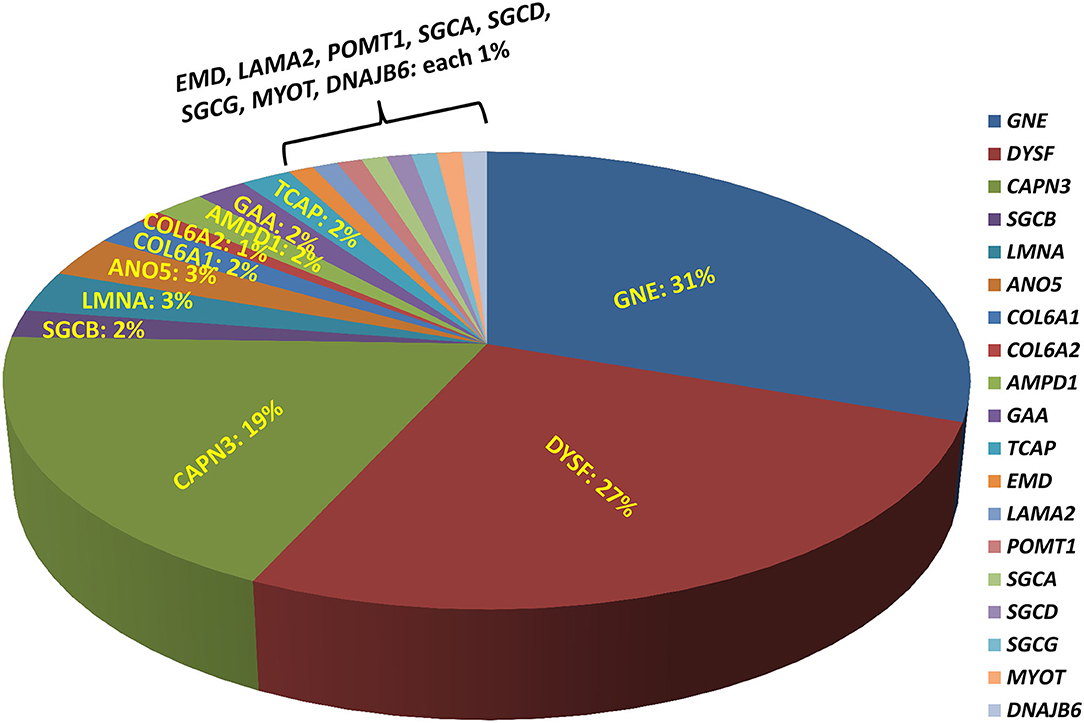
Figure 1. Molecular diagnosis with different genetic myopathy subtypes. Molecular diagnosis using exome sequencing has been established in 49% of the patients with the majority having pathogenic variants identified in one of the following genes: GNE (31%), DYSF (27%), CAPN3 (19%), indicating that these genes are likely the major contributors to genetic myopathy-like phenotype.
We identified 65 variant alleles comprised of 37 unique variants in the three major genes (GNE, DYSF, CAPN3). A total of 14 unique variants were identified in GNE (Figure 2A). The present study confirmed the absence of a mutational hot-spot region in GNE in the Indian patients. The majority of the variants detected in GNE (12) were missense variants located throughout the GNE gene, suggesting an important role of missense changes in causing GNE-myopathy in people of Indian subcontinent origin (Table 1). The most common pathogenic variants c.2179G>A (p/V727M) and c.1760T>C/c.1853T>C (p.I587T/p.I618T) were detected in most of the GNE patients (Figure 2D) with extensive homozygosity observed for the c.1760T>C (p.I587T) common pathogenic variant. Only one non-sense variant c.385C>T (p.R129X), one frameshift variant c.397_398dupAT, and no splice site variants were identified in GNE.
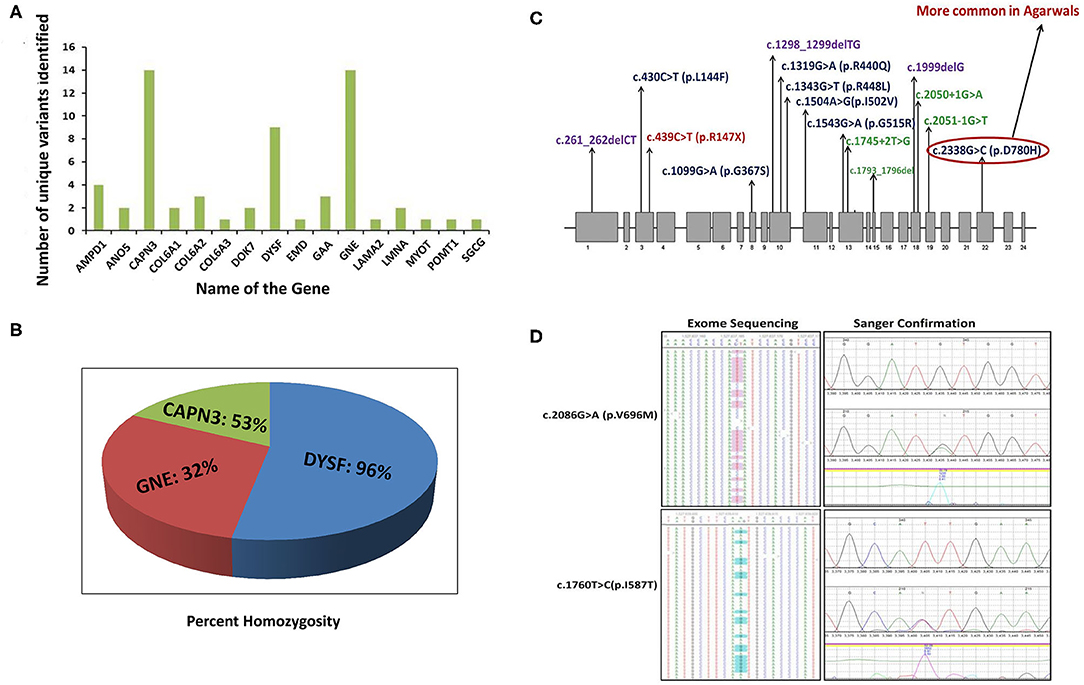
Figure 2. Unique pathogenic variants and percent homozygosity identified in major myopathy genes. (A) Number of unique pathogenic variants identified in major myopathy genes. (B) The percentage of homozygosity observed in the major myopathy subtypes. Among the three major myopathy subtypes, 96% of the limb-girdle muscular dystrophy R2 (LGMDR2) patients caused by variants in DYSF gene were identified with homozygous variants. (C) Mutation spectrum of CAPN3 gene: CAPN3 variants identified in the current study are distributed throughout the gene. There is no specific mutation hot spot region but 3 variants are located in exon 10 indicating it as more unstable region prone to genetic variation. (D) Two common variants in GNE gene: The two common variants in GNE gene were detected in exome sequencing with high confidence and also further confirmed by Sanger sequencing. Left side panel indicates the variants confirmation by exome sequencing. Right side panel indicates the variants confirmation by Sanger sequencing.

Table 1. Molecular and Clinical aspects of CAPN3, DYSF, and GNE patients.
CAPN3 and DYSF are the next major contributing genes in Indian genetic myopathy patients. Calpainopathy (CAPN3, LGMDR1) represents the most frequent LGMD-subtype worldwide (6, 77, 78) but was the second most common myopathy found in our cohort. No specific mutational hot spot was identified, and all 14 identified unique variants in 9 patients were distributed throughout the CAPN3 gene (Figure 2C). Homozygosity was observed in 4 out of 9 CAPN3 patients (45%) (Figures 2A,B).
The 9 unique variants identified in 9 DYSF patients were also distributed throughout the DYSF gene. Interestingly, we found that 96% (26/27) of the individuals with a dysferlinopathy molecular diagnosis were homozygous for the detected pathogenic variant (Figure 2B), explaining why autosomal recessive disorders like dysferlinopathy are the more common form of inherited myopathy found in India.
In addition, the two Pompe genetically diagnosed Indian patients did not have either any homozygous GAA variants or the leaky splice-variant c.-32-13T>G that are more common in adult-onset Pompe in many other populations.
Clinical Characteristics of Genetically-Diagnosed Patients
Pictorial examples (Figure 3) and details of clinical features of common subtypes (Tables 1, 2) and uncommon subtypes (Tables 3, 4) of myopathies identified in our Indian cohort are provided. The mean age of onset was 22.23 years (birth−60 years). Cardiac involvement was observed in a total of 6 patients: one each for AMPD1, GAA, DYSF, LMNA, and two GNE-myopathies. Patient #2 (AMPD1) and 38 (LMNA: Laminopathy or EDMD) showed atrioventricular conduction blocks, with AMPD1 patient showing 30% ejection fraction. Patient #47 (DYSF) showed proximo-distal weakness and mild sub-clinical cardiomyopathy (ejection fraction 50%) resembling few reports of miyoshi myopathy (126, 127). Patient #73 (GAA: Pompe disease) had dilated cardiomyopathy with 32% ejection fraction and also showed respiratory involvement with breathlessness on exertion. Patient #89 and 90 (GNE-myopathies) showed both sub-clinical cardiomyopathy evidenced by mild reduction of ejection fraction on the 2D echocardiography. Among the 6 patients, only patient #73 (Pompe disease) showed breathlessness on exertion. Data on pulmonary function tests on any case were not available. Available muscle biopsy results indicated that 66% (31 out of 47) of the patients had dystrophic or myopathic changes with/without inflammation. A total of 4 patients were non-ambulatory and required wheelchair assistance (Tables 1–4 and Supplementary Table 1).
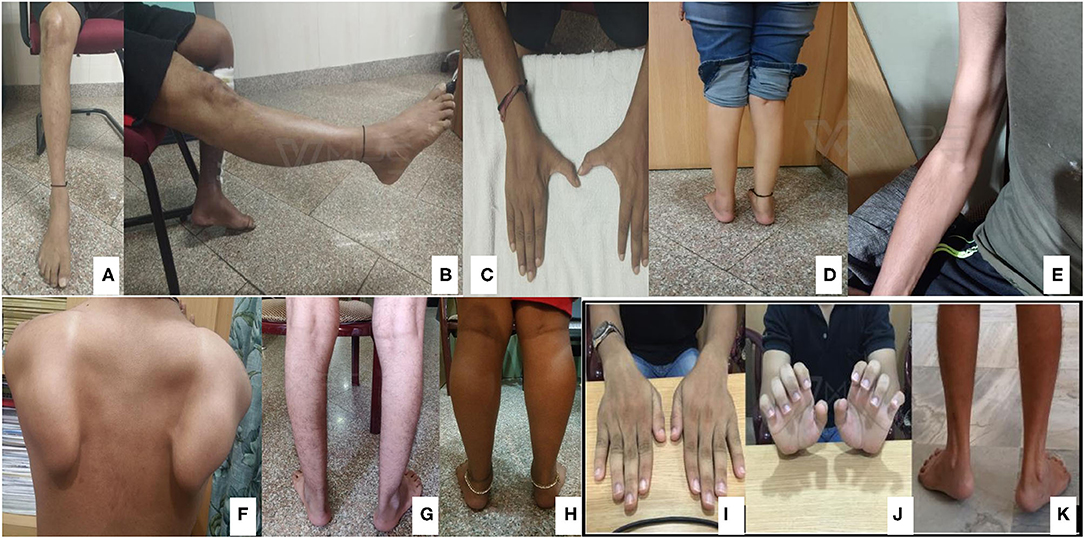
Figure 3. Classical features of common subtypes identified. GNE myopathy (A–C): Tibialis anterior (TA) muscle wasting, normal quadriceps, and first dorsal interossei (FDI) muscle wasting identified, respectively. Dysferlinopathy (D,E): Gastrocnemius wasting and biceps lump. Calpainopathy (F,G): Scapular winging and ankle contracture. Sarcoglycanopathy (H): Calf hypertrophy. Collagenopathy (I–K): Long finger flexor and ankle contractures.
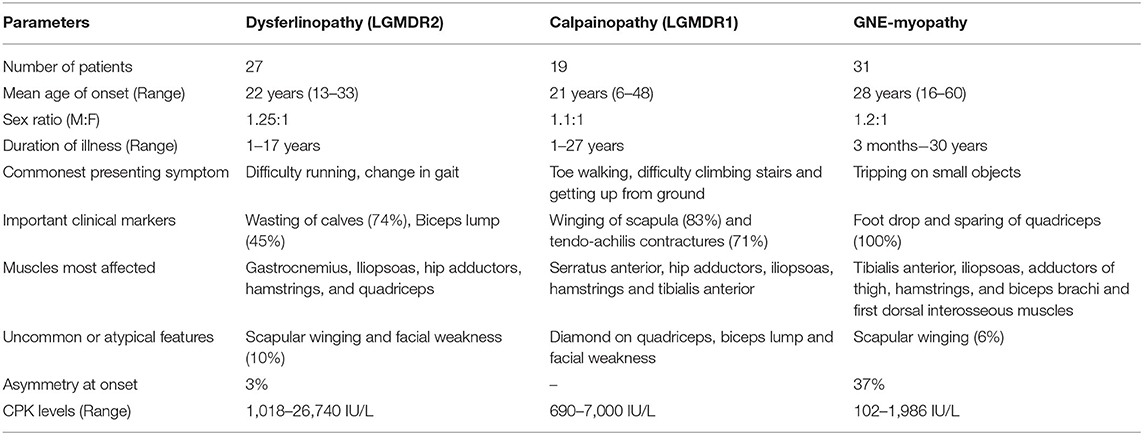
Table 2. Clinical features of common subtypes.
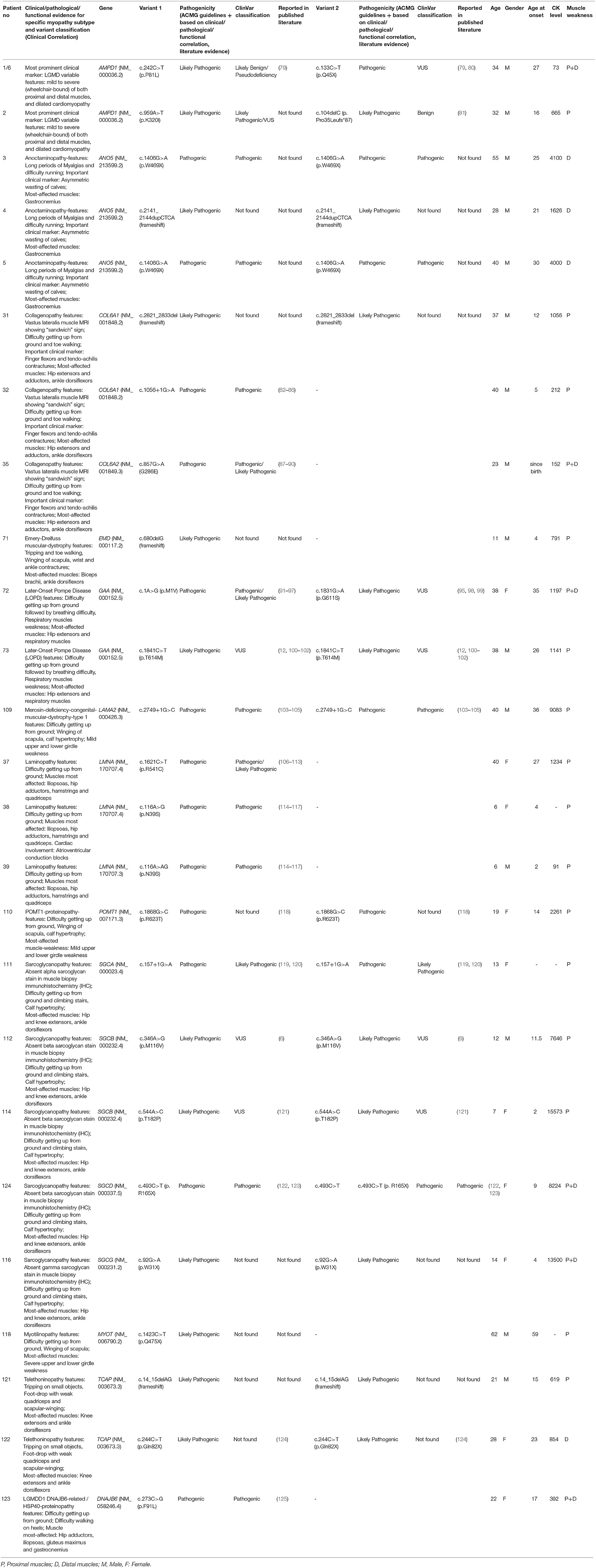
Table 3. Uncommon subtypes identified in Indian patients.
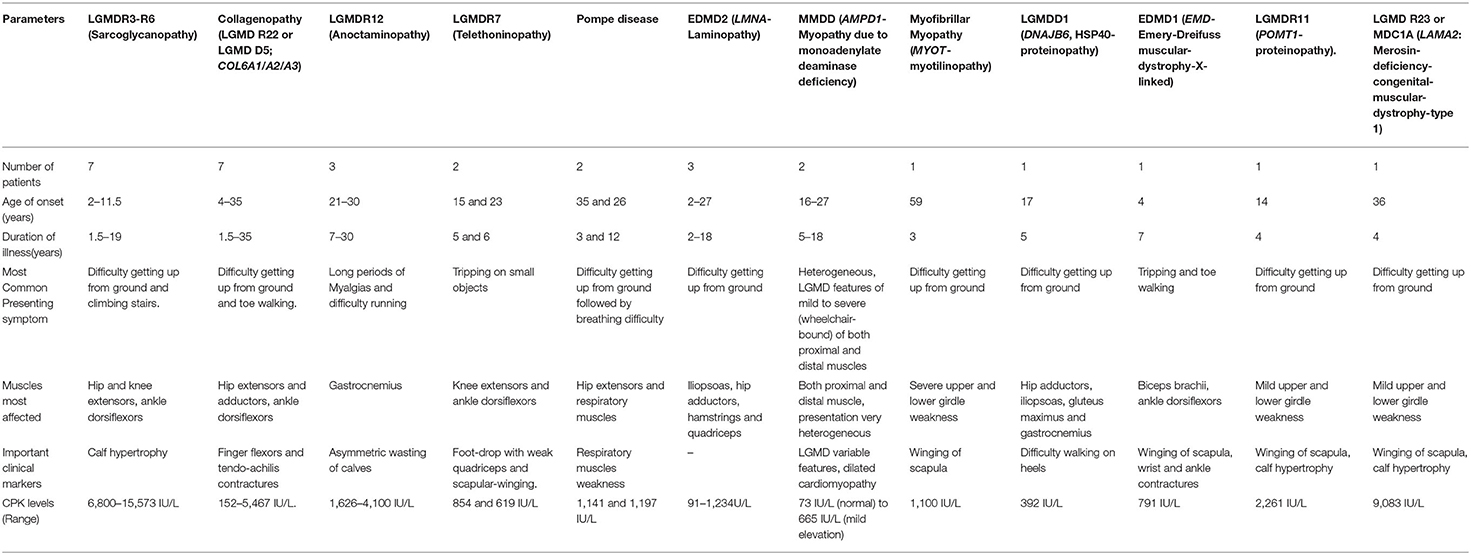
Table 4. Clinical features of uncommon subtypes.
Common Subtypes
GNE myopathy, dysferlinopathy, and calpainopathy were identified as the three most common subtypes of inherited myopathies. Normal range of creatine kinase (CK) in this study was 30–160 IU/L. The details of the pattern of clinical weakness and uncommon clinical features of these three groups are described below.
GNE-Myopathy
The onset-age varied (16–60 years) with the mean at 28 years in 17 females and 14 males. Total duration of illness was between 3 months and 30 years with lower limb onset in all except one with upper limb involvement simultaneously. Asymmetry at onset was seen in 13 patients. Creatine kinase (CK) levels ranged between 102 and 1,986 IU/L. Two patients had loss of ambulation with long duration of illness (17 and 13 years).
Pattern of Weakness
Most patients had conventional presentation with weakness affecting the tibialis-anterior (TA) maximally along with sparing of the quadriceps. With advancing duration of the disease, patients also showed a degree of weakness in the iliopsoas, adductors of thigh, hamstrings in lower-limbs and biceps, and first dorsal interosseous muscles in the upper-limbs (denoted as D+P in Table 2).
Rare Clinical Features
Two patients had severe affection of proximal-muscles of the upper-limbs with winging, later into the illness (Table 2).
Dysferlinopathy
Mean onset age was 22 years (13–33 years) in 15 males and 12 females. Duration of illness ranged from 1 to 17 years. Family history was positive in 6 and history of consanguinity was elicited in 6 patients. Weakness began in the lower limbs of all patients: proximo-distal in 7 patients, only proximal in 21 patients, and only distal in the remaining 3 patients. Biceps lump was seen in 14 patients. Loss of ambulation developed in one patient 10 years into the illness. CK levels highly varied and were between 1,018 and 26,740 IU/L.
Pattern of Weakness
Most patients had proximal-onset of lower-limbs-weakness with maximum affection of iliopsoas, hip adductors, hamstrings, quadriceps, and gastrocnemius muscles. These patients had difficulty in walking on toes, wasting of calves, and high CK-levels (Table 2).
Rare Clinical Features
Scapular winging and facial weakness was seen in 3 patients, in advanced stages of the disease. Asymmetry of weakness was seen in only 1 patient. This unusual patient had one pathogenic variant in the ANO5 gene along with a homozygous pathogenic variant in DYSF. Interestingly, this patient (patient #50 in Table 5) showed a phenotype common to both dysferlinopathy and anoctaminopathy (high CK, proximal and distal muscle weakness, asymmetrical weakness) and features unusual to both types (facial weakness). Presence of inflammation in muscle biopsy and absence of Dysferlin staining were the manifestations of dysferlinopathy at the histological level.
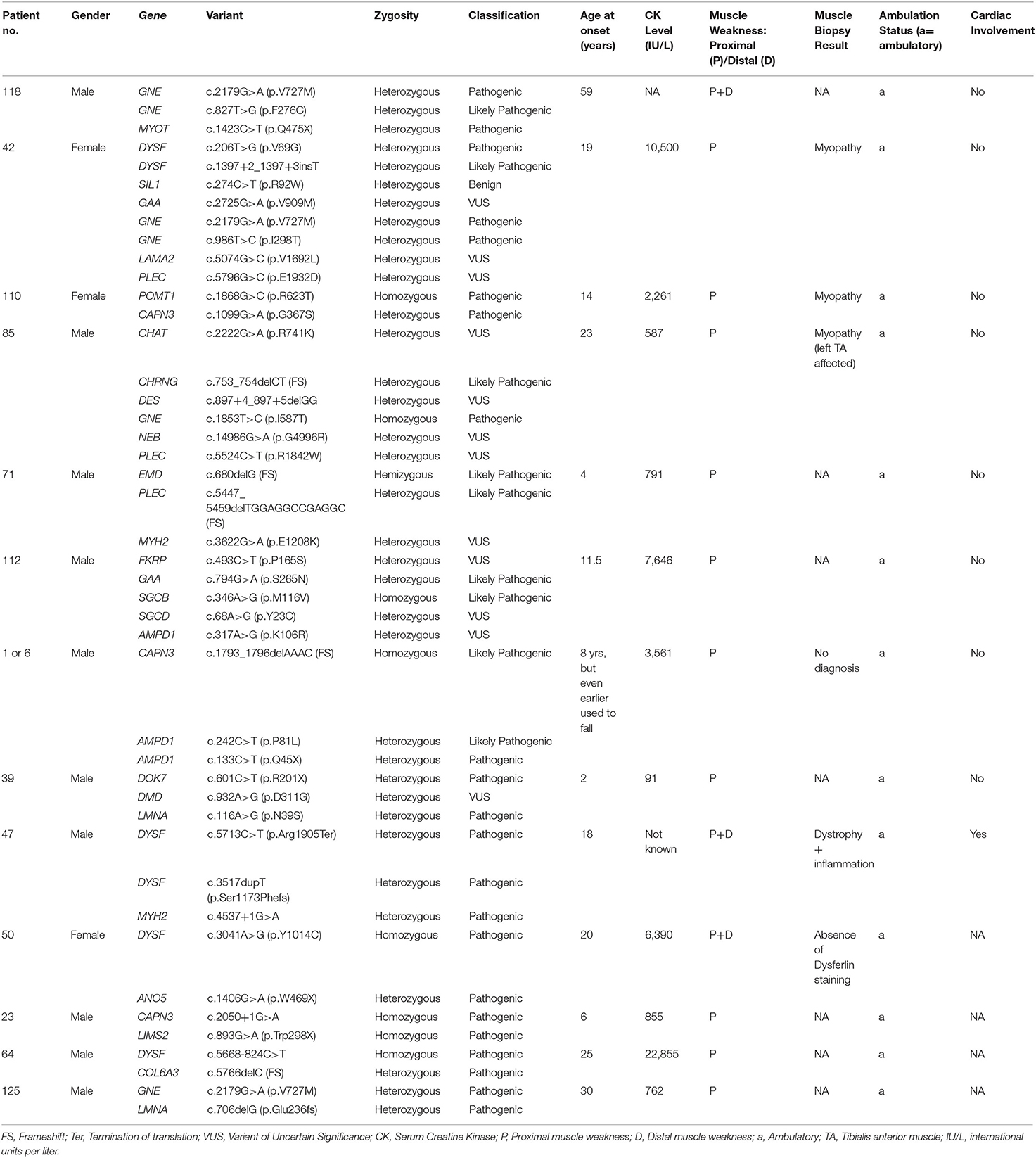
Table 5. Clinical symptoms and genotype of patients with pathogenic variants in more than one gene.
Calpainopathy
Mean onset age was 21 years (6–48 years) in 10 males and 9 females. Duration of illness ranged from 1 to 27 years. Family history was positive in 3 and history of consanguinity was present in 5 patients. Lower limb onset of weakness was seen in 21 patients, upper limbs in 2, and upper and lower limbs were simultaneously affected in 1 patient. The weakness was proximal in 18, proximo-distal in 5, and distal in 1 patient. Winging was present in 15/19 and tendo-achilis contractures were seen in 12/19 patients. Loss of ambulation developed in one patient 27 years into the illness. CK levels ranged from 690 to 7,000 IU/L.
Pattern of Weakness
Most patients had the conventional proximal weakness onset in the lower-limbs. Maximum weakness was seen in the hip adductors, iliopsoas, hamstrings, tibialis anterior (TA), and serratus anterior muscles. Tendoachillis contractures and toe-walking was common.
Rare Clinical Features
One patient had diamond on quadriceps and biceps lump, which is commonly described in dysfelinopathy. One other patient had mild facial weakness.
Clinical Features of Genetically Diagnosed Uncommon Subtypes in India
The clinical features of the rare and uncommon genetically diagnosed subtypes found in this study have been provided in Table 4. These individual groups were small consisting of few or a single patient each and were comprised of sarcoglycanopathies (SGCA/SGCB/SGCD/SGCG), collagenopathies (COL6A1/COL6A2/COL6A3), anoctaminopathy (ANO5), telethoninopathy (TCAP), Pompe disease (GAA), myoadenylate deaminase deficiency myopathy (AMPD1), myotilinopathy (MYOT), laminopathy (LMNA), HSP 40 proteinopathy (DNAJB6), Emery Dreifuss muscular dystrophy (EMD), POMT1 proteinopathy (POMT1), and merosin deficient congenital muscular dystrophy (LAMA2). Interestingly, myoadenylate deaminase deficiency myopathy due to AMPD1 pathogenic variants was only identified in 2 patients out of 207 Indian patient cohort (~1%) suggesting lower prevalence among individuals in the Indian subcontinent area and Asians in general. These 2 patients were 32 and 34 years old and showed heterogeneous symptoms. One had mild limb girdle weakness with slightly elevated CK-levels (665 IU/L). The second patient was wheelchair bound and had a long history with severe weakness of both proximal and distal muscles, cardiomyopathy but with normal CK levels (73 IU/L). Along with having compound heterozygous pathogenic AMPD1 variants (c.242C>T and c.133C>T), the patient with the severe phenotype also harbored homozygous CAPN3 pathogenic variant (c.1793_1796delAAAC; patient # 1 or 6), which could be associated with higher clinical severity. These AMPD1 cases suggest more heterogeneity in the nature of muscle weakness and clinical overlap of AMPD1-associated myopathy with limb-girdle features in the Indian population.
We also identified 21 patients carrying VUSs in CAPN3, DYSF, GNE, COL6A1, COL6A2, COL6A3, FLNC, SGCB, TRIM32 genes that has strong clinical correlation with the gene and corresponding myopathy subtype of interest but without further information reclassify them as likely pathogenic or pathogenic (Table 6). These cases are not included in the diagnosed patients in our cohort. Further segregation and functional studies are needed to confirm these VUSs' pathogenicity.
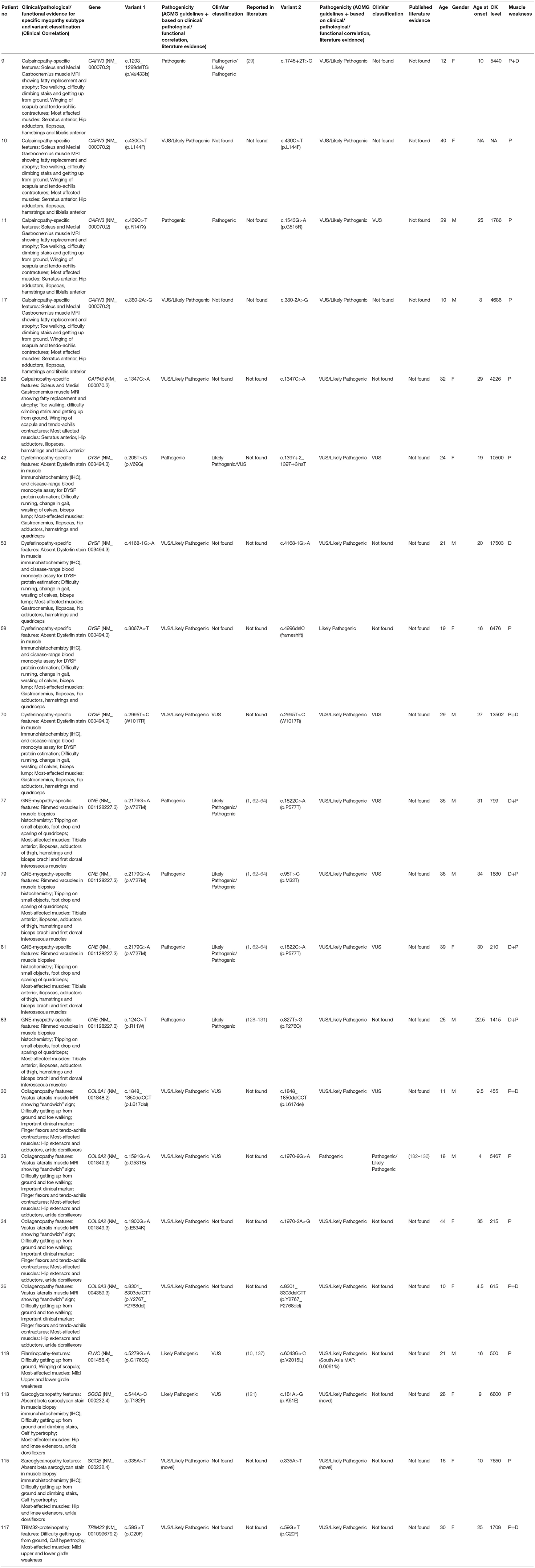
Table 6. Molecular and Clinical aspects of patients (not included in diagnostic yield) with Variants of Uncertain Significance (VUSs) or Likely Pathogenic but with Clinical Correlation with Gene of interest.
Multigenic Myopathies: A Novel Disease Mechanism
Interestingly, 13 patients were identified with pathogenic variants in more than one myopathy genes (Table 5), all showing unusual presentations. We describe below such cases of multigenic unusual presentations identified in this study.
Patient #118 with one pathogenic variant each in the GNE (OMIM #603824) and myotilin (MYOT; OMIM #604103) genes started with disease symptoms very late, beginning at 59 years of age. His phenotype largely resembled GNE-myopathy with foot-drop and quadriceps-sparing. However, disease onset was both proximal and distal, which also could resemble both subtypes. By the time foot-drop started, difficulty in rising from the ground and low chairs had already developed. In this patient, both the genetic abnormalities could be contributing to the proximo-distal presentation resembling both autosomal-dominant myotilinopathy (138) as well as autosomal-recessive GNE-myopathy that has been reported to show early onset of proximal weakness mimicking LGMD especially with hamstring weakness (139). Muscle biopsy staining showed rimmed vacuoles, a common feature of both GNE-myopathy and MYOT-related myofibrillar myopathy. Myotilin immunostaining could have helped further but was not available.
Patient #42 had pathogenic variants in the GNE gene and she additionally harbored one likely pathogenic variant in the dysferlin (DYSF) gene. Her phenotype was strongly suggestive of a GNE-myopathy but had onset of proximal muscle weakness and presented a high CK level (1050 IU/L), which are a very well-known presentation of dysferlinopathy suggesting blended phenotype. GNE-myopathy patients are known to have proximal weakness only in later stages of illness. Her muscle biopsy confirmed rimmed vacuoles but immunocytochemistry for dysferlin was not available, resembling GNE-myopathy more than dysferlinopathy.
Patient #110 showed the most unusual symptoms: quadriceps-sparing and foot drop resembling GNE-myopathy, but with early and prominent development of scapular-winging and calf-hypertrophy. Interestingly, a heterozygous pathogenic variant in CAPN3 (OMIM #114240) and homozygous pathogenic variant in POMT1 (OMIM #607423) were present without any GNE variant. While the CAPN3 variant can explain adductor, posterior thigh weakness, and scapular winging, POMT1 variants are known to be associated with hypertrophic calves (140) in limb-girdle muscular dystrophy (LGMDR11). Quadriceps-sparing and prominent foot-drop are unusual in the demonstrated genetic abnormalities.
Another patient (patient #50) with a homozygous pathogenic variant in DYSF and a heterozygous pathogenic variant in ANO5 showed phenotypes of dysferlinopathy and anoctaminopathy that are representative of both subtypes (20 years onset age, calf atrophy, CK of 6,390 IU/L, combination of proximal and distal weakness, presence of inflammation in muscle-biopsy, asymmetry of muscle weakness, weakness in dorsiflexors of ankle joints, partial foot drop) and features unusual to both (facial weakness).
We identified one patient clinically diagnosed with LGMDR2 but with an unexpected severe progression from onset age at 18 years to difficulty in standing by 24 years of age. This patient had pathogenic variants in both DYSF and MYH2 genes (patient #47). Heterozygous loss of MYH2 coupled with complete loss of DYSF could be contributing to the rapid disease progression, and severe phenotype of both proximal and distal muscle weakness along with cardiac-involvement observed in this individual.
Patient #23, a 19 year-old boy, showed homozygous pathogenic variants in both CAPN3 and LIMS2 genes. He suffered from progressive weakness of hip and shoulder girdles. The phenotype consisted of scapular winging, lordosis, severe weakness of hip adductor muscles, and mild tendo-achillis contractures, resembling calpainopathies. He also had mild calf hypertrophy. While transient or persistent calf hypertrophy has been documented in calpainopathies (141), it is also a feature of the LIMS2-patients. He did not have the triangular tongue or cardiomyopathy seen in LIMS2-patients.
Patient #125 had one pathogenic variant each in both LMNA and GNE genes with three generations being affected. He had limb-girdle pattern of weakness with slow progression along with calf hypertrophy and subclinical cardiac involvement in the form of atrioventricular conduction block. His CK level was mildly raised (762 IU/L). All these features are in favor of autosomal-dominant laminopathy. His examination did not reveal any features suggestive of GNE-myopathy but since GNE variants can be segregated either recessively or dominantly, longitudinal patient and family natural history is being followed through.
Similar multigenic combinations of pathogenic variants were detected for GNE, ANO5, MYOT, CAPN3, LIMS2, COL6A3, POMT1, CHRNG, EMD, PLEC, GAA, SGCB, DMD, DOK7, and LMNA genes in a total of 13 individuals (Table 5).
Discussion
Most of the inherited myopathies impose difficulties with physical activity, walking, poor quality of life, and ultimately cause a heavy burden on both the affected individuals and their families. Identifying the correct diagnosis of these inherited myopathies can aid in disease management, treatment, and family planning. Clinical diagnosis is based on the distribution of predominant muscle weakness, inheritance mode, and associated symptoms but is often highly elusive due to the overlap in clinical presentation. Therefore, molecular diagnosis is necessary to confirm the identifying disease-causative gene. This study provides for the first time molecular diagnosis and clinical information on a fairly large cohort size of genetic myopathies in patients of diverse ethnic backgrounds seen in the Indian subcontinent.
This cohort includes a large variety of inherited myopathies including GNE-myopathy, varieties of LGMDs, collagenopathies, metabolic myopathies, and related others that have a significant phenotypic overlap. GNE, DYSF, and CAPN3 are the three major genetic contributors to these myopathies in the Indian subcontinent. Even though we recruited patients from across the Indian subcontinent, as the evaluation was performed in a major single center hospital, there could be limitations to the interpretation of the prevalence. With upcoming or ongoing interventional or natural history study clinical trials on these myopathy subtypes (https://clinicaltrials.gov/ct2/results?recrs=ab&cond=GNE+Myopathy+&term=&cntry=&state=&city=&dist=, https://www.sarepta.com/science/gene-therapy-engine), this study will enable cohorts from the Indian subcontinent to be included in patient registries that, in turn, will enhance the clinical trials by including different populations with varied ethnicities as well as better monitoring of trial efficacies. Our diagnostic yield of 49% is considerably high with careful clinical correlation of the gene and rare variant identified. Another reason for a high diagnostic yield in an Indian subcontinent cohort is the high prevalence of consanguinity leading to greater homozygosity as found in all the three major contributing genes: GNE, DYSF, and CAPN3 (Figure 2B). Moreover, our careful clinical pre-screening with inclusion criteria of genetic myopathies and excluding DMD, FSHD, DM1, DM2, mitochondrial myopathies, and acquired myopathies enabled a higher diagnostic yield and further suggests the importance of phenotype correlation of clinical genetic testing results. The clinical features of the three major genetic myopathies followed those mentioned in the literature to a large extent such as GNE myopathy patients with weakness of the tibialis anterior muscles with sparing of quadriceps, dysferlinopathy patients having gastrocnemius weakness and proximal weakness, and calpainopathies having winging of scapulae and hip girdle weakness, mainly of adductor muscles. In addition, GNE-myopathy patients in our cohort also had proximal weakness, possibly representing advanced stages of the disease. The clinical features of the common and uncommon types are summarized in Tables 2, 4, respectively.
Molecular findings related to GNE-myopathy patients identified in this study have been reported recently (65) with the most common pathogenic variant in our cohort being the c.1760T>C/c.1853T>C (p.I587T/p.I618T). All patients identified with this variant had their ancestry originating in Rajasthan, a province in the north-west part of India, and all belonged to the Maheshwari and Jain ethnic communities of Rajasthan. This variant has recently been described as the most common variant in Roma of Europe, with an ethnic founder effect (66, 67). Our results and previous studies support the current view that Rajasthan in India is the origin of Roma ethnic group of Europe (65). Moreover, p.V727M is potentially a founder variant in Indian subcontinent since it was seen with high prevalence in our study and previous studies (142–144), and hence this variant is likely present in carriers at higher rates in the north-western areas of Indian subcontinent. This suggests the need for family tracing and carrier testing for inherited rare disorders such as GNE-myopathy for places where traditionally rural consanguineous marriages are prevalent.
Calpainopathy (LGMDR1) was considered a strict autosomal-recessive LGMD-subtype for many years, but patients carrying specific single pathogenic deletion variants in the CAPN3 gene are reported recently showing dominant disease segregation (6, 145), which was not identified in Indian patients in the current study.
Limb-girdle muscular dystrophy type 2B (LGMDR2) and Miyoshi myopathy (MM) caused by variants in the dysferlin gene, DYSF (146), are the two major clinical types of dysferlinopathy (147), characterized by proximal muscle-weakness, difficulty in running and climbing stairs, and increased fatigue (148). The higher (96%) homozygosity rate of pathogenic variant in DYSF gene explains why autosomal recessive disorders like dysferlinopathy are more common in the Indian subcontinent. DYSF gene is much larger than some of the other genes such as GNE and CAPN3, the other two most prevalent myopathy forms in this study. Thus, DYSF likely harbors more variants and therefore has a higher chance of homozygosity. Moreover, due to the sociocultural customs of consanguinity in some parts of the Indian subcontinent, our study strongly supports the immediate need for offering genetic counseling and carrier screening tests for genetic disorders in India and other countries where endogamous and consanguineous marriages are commonplace.
In this cohort many other more rare genetic myopathies were seen (Tables 3, 4 and Supplementary Table 1). Out of these, sarcoglycanopathies and collagenopathies were seen more frequently than other myopathies. The phenotypes of most myopathies were comparable to literature descriptions. There were some unusual clinical features and heterogeneity as well, such as that in AMPD1 associated myopathy where ranges from mild symptoms to severe limb-girdle features progressing fast to wheel-chair assistance and dilated cardiomyopathy were observed. Only 2 AMPD1-associated myopathy cases were identified in our Indian subcontinent cohort suggesting lower prevalence in Asia similar to what was shown before in the Japanese population (149), compared to being one of the most common genetic myopathies among Caucasians (1 in 50–100 individuals in general population) (150). The AMPD1 case with severe clinical symptoms also harbored homozygous pathogenic truncated variant in CAPN3, which may result in the greater disease severity. Further studies are needed to resolve the gene contributions and for better understanding of AMPD1's clinical significance. The diagnosis of myoadenylate deaminase deficiency is challenging given about 2% of muscle biopsies may have enzyme deficiency without clinical correlation (151–153). Deep phenotyping, immunohistochemical studies, western blot analysis, or muscle imaging as well as functional studies are necessary to verify this group of myopathies.
The interesting finding of 13 patients with pathogenic variants in more than one myopathy gene (Table 5) and showing unusual presentations suggest a possible role of synergistic-heterozygosity and digenic contribution to unusual myopathies, similar to our recent finding in the US LGMD population (6). Multigenic combinations of pathogenic variants were detected involving 15 genes, and 13 patients in this study suggest a high prevalence of synergistic heterozygosity in genetic myopathies. In some cases, the phenotype exhibited features of both the genes (e.g., GNE and MYOT, patient #118; DYSF and ANO5, patient #50, in Table 5), and in some patients phenotype favored one gene over the other (e.g., patient #125, Table 5). In some other cases, there were clinical features unexplained by the identified genetic variants such as patient #110 (Table 5) showing GNE-myopathy features without any GNE variant but pathogenic variants in POMT1 and CAPN3. Thus, when faced with an atypical phenotype of inherited myopathy, the possibility of pathogenic variants in more than one myopathic gene exists, and clinical exome or genome sequencing should be considered. Moreover, the result of genetic testing in such multigenic cases need to be interpreted cautiously and muscle immunostaining should be considered. For multigenic myopathies, some cases may be due to a genetically unidentified or a novel gene, especially those with heterozygous VUS in genes known to cause only recessive diseases. With widespread use of extensive panels, focused or whole-exome, and genome-sequencing, more such instances will be unearthed, helping further understanding of the pathophysiology and expressions of the genetic abnormalities. Functional omics platforms such as RNA sequencing, proteomics, and metabolomics (154, 155) of the muscle tissue and segregation studies are needed in these unusual cohorts to understand the variable expressions of the genes at the effector organ and the phenotypic variability. We propose to carry out this work in the future, which is a current limitation of this study.
True negative findings for the 14 patients with myopathy clinical symptoms suggest the need for whole genome sequencing (WGS) in the future to discover new myopathy causal genes harboring variants such as large deletions or duplication or deep-intronic variants that may have been missed with ES. A study limitation was that even though our inclusion criteria did not have any age restriction for subject recruitment, the neuromuscular clinic caters mainly to the adult patients and hence pediatric cases were underrepresented in this study. Though this is a single center study, patients were referred to the center from all over the Indian subcontinent with diverse ethnicities and religious origins. Another limitation of our study was the lack of segregation analysis of compound heterozygous variant combinations identified in patients of our cohort. This is due to difficulty in the current setting in the region based on from where the patients travel and come to the clinic and unavailability of parents or children of the patients. Segregation study is needed that will further enhance our understanding of the variant distribution in the population and for better family planning.
There is a paucity of population-based genetic testing in the Indian subcontinent in public databases that may lead to insufficiency of minor allele frequency, and lack of reports of similar myopathy patient(s) harboring the variant or unavailable functional data is a limitation in this study. Yet, this also points toward the immediate need and importance of studies such as ours on genetic testing with clinical correlation on large Asian cohorts, either disease-specific or general healthy population.
Finally, to our knowledge, the current study is the first comprehensive clinical-exome sequencing effort on a large genetic myopathy cohort in the Indian subcontinent that has enhanced our understanding of the spectrum of gene-variant-myopathy-subtype landscape in India yielding a high diagnostic rate. In this genomic era, studies such as this in different developing countries and continents on specific disease cohorts with diverse ethnicities will enable enhancement of the repertoire in the global genomic databases such as ExAc (http://exac.broadinstitute.org/), gnomAD (https://gnomad.broadinstitute.org/), and also country-specific large databases such as the UK100,000 Genome project (https://www.genomicsengland.co.uk/about-genomics-england/the-100000-genomes-project/), NIH All of US Genomics program (https://allofus.nih.gov/), and others for faster and more accurate variant classification, faster enhanced diagnostics, and the understanding of genotype-phenotype correlations.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found here: https://doi.org/10.5061/dryad.tmpg4f4w6 and in European Nucleotide Archive (ENA) under study accession number PRJEB40370.
Ethics Statement
The studies involving human participants were reviewed and approved by the Emory University Institutional Review Board (IRB). The patients/participants provided their written informed consent to participate in this study. Written, informed consent was obtained from all individuals or minors' parent or legal guardian or next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
SC and MH had full access to all data in this study, take responsibility for the integrity of the data and the accuracy of the data analysis, designed, conceptualized, and oversaw the entire study. Acquisition, analysis, or interpretation of data was done by SC, BN, SK, MS, AB, RD, PG, LR, and LG. Drafting of the manuscript was done by SC, SK, and MS. Statistical analysis was performed by SC, BN, SK, RD, PG, and LR. SC obtained funding. SC, MH, and SK supervised the entire study. Critical revision of the manuscript for important intellectual content was done by all authors.
Funding
This work was supported by the Jain Foundation Focused Research Grant, Muscular Dystrophy Association Grant (MDA578400), and Sanofi-Genzyme Investigator Sponsored Studies Grant (SGZ-2017-11829) to SC. BN and MH are employees of Perkin Elmer Genomics. MH is also an adjunct professor of Georgia Institute of Technology.
Conflict of Interest
SC reports grants from Muscular Dystrophy Association and Jain Foundation during the conduct of the study. MH reports that she is the VP and CSO of Global Lab Services, PerkinElmer, Inc. that does diagnostic testing and has equity in the company without any conflict of interest.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the patients and their families for study participation, and the funding sources. Part of this work was available online as a preprint (156). We thank Dr. Arunkanth Ankala of Emory University Department of Human Genetics, Mrs. Khanjan K. Gandhi of Emory University Winship Cancer Center, and Dr. Lora Bean of PerkinElmer Genomics for assisting with data management.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2020.559327/full#supplementary-material
References
1. Chakravorty S, Berger K, Arafat D, Nallamilli BRR, Subramanian HP, Joseph S, et al. Clinical utility of RNA sequencing to resolve unusual GNE myopathy with a novel promoter deletion. Muscle Nerve. (2019) 60:98–103. doi: 10.1002/mus.26486
2. Benarroch L, Bonne G, Rivier F, Hamroun D. The 2020 version of the gene table of neuromuscular disorders (nuclear genome). Neuromusc Disord. (2019) 29:980–1018. doi: 10.1016/j.nmd.2019.10.010
3. Bonne G, Rivier F, Hamroun D. The 2019 version of the gene table of neuromuscular disorders (nuclear genome). Neuromusc Disord. (2018) 28:1031–63. doi: 10.1016/j.nmd.2018.09.006
4. Bonne G, Rivier F, Hamroun D. The 2018 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromusc Disord. (2017) 27:1152–83. doi: 10.1016/j.nmd.2017.10.005
5. Nigro V, Savarese M. Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myol. (2014) 33:1–12.
6. Nallamilli BRR, Chakravorty S, Kesari A, Tanner A, Ankala A, Schneider T, et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol. (2018) 5:1574–87. doi: 10.1002/acn3.649
7. Guglieri M, Magri F, D'Angelo MG, Prelle A, Morandi L, Rodolico C, et al. Clinical, molecular, and protein correlations in a large sample of genetically diagnosed Italian limb girdle muscular dystrophy patients. Hum Mutat. (2008) 29:258–66. doi: 10.1002/humu.20642
8. Rahimov F, Kunkel LM. The cell biology of disease: cellular and molecular mechanisms underlying muscular dystrophy. J Cell Biol. (2013) 201:499–510. doi: 10.1083/jcb.201212142
9. Mahmood OA, Jiang XM. Limb-girdle muscular dystrophies: where next after six decades from the first proposal (Review). Mol Med Rep. (2014) 9:1515–32. doi: 10.3892/mmr.2014.2048
10. Yu M, Zheng Y, Jin S, Gang Q, Wang Q, Yu P, et al. Mutational spectrum of Chinese LGMD patients by targeted next-generation sequencing. PLoS ONE. (2017) 12:e0175343. doi: 10.1371/journal.pone.0175343
11. Savarese M, Di Fruscio G, Torella A, Fiorillo C, Magri F, Fanin M, et al. The genetic basis of undiagnosed muscular dystrophies and myopathies: results from 504 patients. Neurology. (2016) 87:71–6. doi: 10.1212/WNL.0000000000002800
12. Reddy HM, Cho KA, Lek M, Estrella E, Valkanas E, Jones MD, et al. The sensitivity of exome sequencing in identifying pathogenic mutations for LGMD in the United States. J Hum Genet. (2017) 62:243–52. doi: 10.1038/jhg.2016.116
13. Majumder PP, Basu A. A genomic view of the peopling and population structure of India. Cold Spring Harb Perspect Biol. (2014) 7:a008540. doi: 10.1101/cshperspect.a008540
14. Majumder PP. The human genetic history of South Asia. Curr Biol. (2010) 20:R184–7. doi: 10.1016/j.cub.2009.11.053
15. Moorjani P, Patterson N, Loh PR, Lipson M, Kisfali P, Melegh BI, et al. Reconstructing Roma history from genome-wide data. PLoS ONE. (2013) 8:e58633. doi: 10.1371/journal.pone.0058633
16. Krithika S, Maji S, Vasulu TS. A microsatellite study to disentangle the ambiguity of linguistic, geographic, ethnic and genetic influences on tribes of India to get a better clarity of the antiquity and peopling of South Asia. Am J Phys Anthropol. (2009) 139:533–46. doi: 10.1002/ajpa.21018
17. Mendell JR, Florence J. Manual muscle testing. Muscle Nerve. (1990) 13:S16–20. doi: 10.1002/mus.880131307
18. Jones MA, Bhide S, Chin E, Ng BG, Rhodenizer D, Zhang VW, et al. Targeted polymerase chain reaction-based enrichment and next generation sequencing for diagnostic testing of congenital disorders of glycosylation. Genet Med. (2011) 13:921–32. doi: 10.1097/GIM.0b013e318226fbf2
19. Chakravorty S, Ankala A, Hegde MR. Implementation of exome sequencing assay. In: Netto GJ, Kaul KL, editors. Genomic Applications in Pathology. Cham: Springer International Publishing (2019). p. 261–81.
20. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
21. de Morrée A, Lutje Hulsik D, Impagliazzo A, van Haagen HHHBM, de Galan P, van Remoortere A, et al. Calpain 3 is a rapid-action, unidirectional proteolytic switch central to muscle remodeling. PLoS ONE. (2010) 5:e11940. doi: 10.1371/journal.pone.0011940
22. Fanin M, Fulizio L, Nascimbeni AC, Spinazzi M, Piluso G, Ventriglia VM, et al. Molecular diagnosis in LGMD2A: mutation analysis or protein testing? Hum Mutat. (2004) 24:52–62. doi: 10.1002/humu.20058
23. Groen EJ, Charlton R, Barresi R, Anderson LV, Eagle M, Hudson J, et al. Analysis of the UK diagnostic strategy for limb girdle muscular dystrophy 2A. Brain. (2007) 130(Pt. 12):3237–49. doi: 10.1093/brain/awm259
24. Shirafuji T, Otsuka Y, Kobessho H, Minami N, Hayashi Y, Nishino I, et al. [Case of LGMD2A (calpainopathy) clinically presenting as Miyoshi distal myopathy]. Clin Neurol. (2008) 48:651–5. doi: 10.5692/clinicalneurol.48.651
25. Sáenz A, Leturcq F, Cobo AM, Poza JJ, Ferrer X, Otaegui D, et al. LGMD2A: genotype-phenotype correlations based on a large mutational survey on the calpain 3 gene. Brain. (2005) 128(Pt. 4):732–42. doi: 10.1093/brain/awh408
26. Richard I, Roudaut C, Saenz A, Pogue R, Grimbergen JE, Anderson LV, et al. Calpainopathy-a survey of mutations and polymorphisms. Am J Hum Genet. (1999) 64:1524–40. doi: 10.1086/302426
27. Khadilkar SV, Chaudhari CR, Dastur RS, Gaitonde PS, Yadav JG. Limb-girdle muscular dystrophy in the agarwals: utility of founder mutations in CAPN3 gene. Ann Indian Acad Neurol. (2016) 19:108–11. doi: 10.4103/0972-2327.175435
28. Ankala A, Kohn JN, Dastur R, Gaitonde P, Khadilkar SV, Hegde MR. Ancestral founder mutations in calpain-3 in the Indian Agarwal community: historical, clinical, and molecular perspective. Muscle Nerve. (2013) 47:931–7. doi: 10.1002/mus.23763
29. Todorova A, Kress W, Mueller C. Novel mutations in the calpain 3 gene in Germany. Clin Genet. (2005) 67:356–8. doi: 10.1111/j.1399-0004.2005.00398.x
30. Nilsson MI, Macneil LG, Kitaoka Y, Alqarni F, Suri R, Akhtar M, et al. Redox state and mitochondrial respiratory chain function in skeletal muscle of LGMD2A patients. PLoS ONE. (2014) 9:e102549. doi: 10.1371/journal.pone.0102549
31. Piluso G, Politano L, Aurino S, Fanin M, Ricci E, Ventriglia VM, et al. Extensive scanning of the calpain-3 gene broadens the spectrum of LGMD2A phenotypes. J Med Genet. (2005) 42:686–93. doi: 10.1136/jmg.2004.028738
32. Krahn M, Lopez de Munain A, Streichenberger N, Bernard R, Pécheux C, Testard H, et al. CAPN3 mutations in patients with idiopathic eosinophilic myositis. Ann Neurol. (2006) 59:905–11. doi: 10.1002/ana.20833
33. Krahn M, Pécheux C, Chapon F, Béroud C, Drouin-Garraud V, Laforet P, et al. Transcriptional explorations of CAPN3 identify novel splicing mutations, a large-sized genomic deletion and evidence for messenger RNA decay. Clin Genet. (2007) 72:582–92. doi: 10.1111/j.1399-0004.2007.00906.x
34. Blázquez L, Azpitarte M, Sáenz A, Goicoechea M, Otaegui D, Ferrer X, et al. Characterization of novel CAPN3 isoforms in white blood cells: an alternative approach for limb-girdle muscular dystrophy 2A diagnosis. Neurogenetics. (2008) 9:173–82. doi: 10.1007/s10048-008-0129-1
35. Bartoli M, Desvignes JP, Nicolas L, Martin K. Exome sequencing as a second-tier diagnostic approach for clinically suspected dysferlinopathy patients. Muscle Nerve. (2014) 50:1007–10. doi: 10.1002/mus.24344
36. Krahn M, Goicoechea M, Hanisch F, Groen E, Bartoli M, Pécheux C, et al. Eosinophilic infiltration related to CAPN3 mutations: a pathophysiological component of primary calpainopathy? Clin Genet. (2011) 80:398–402. doi: 10.1111/j.1399-0004.2010.01620.x
37. Balci B, Aurino S, Haliloglu G, Talim B, Erdem S, Akcören Z, et al. Calpain-3 mutations in Turkey. Eur J Pediatr. (2006) 165:293–8. doi: 10.1007/s00431-005-0046-3
38. Stehlikova K, Skalova D, Zidkova J, Mrazova L, Vondracek P, Mazanec R, et al. Autosomal recessive limb-girdle muscular dystrophies in the czech republic. BMC Neurol. (2014) 14:154. doi: 10.1186/s12883-014-0154-7
39. Krahn M, Bernard R, Pecheux C, Hammouda el H, Eymard B, Lopez de Munain A, et al. Screening of the CAPN3 gene in patients with possible LGMD2A. Clin Genet. (2006) 69:444–9. doi: 10.1111/j.1399-0004.2006.00603.x
40. Xi J, Blandin G, Lu J, Luo S, Zhu W, Béroud C, et al. Clinical heterogeneity and a high proportion of novel mutations in a Chinese cohort of patients with dysferlinopathy. Neurol India. (2014) 62:635–9. doi: 10.4103/0028-3886.149386
41. Krahn M, Béroud C, Labelle V, Nguyen K, Bernard R, Bassez G, et al. Analysis of the DYSF mutational spectrum in a large cohort of patients. Hum Mutat. (2009) 30:E345–75. doi: 10.1002/humu.20910
42. Aoki M, Liu J, Richard I, Bashir R, Britton S, Keers SM, et al. Genomic organization of the dysferlin gene and novel mutations in Miyoshi myopathy. Neurology. (2001) 57:271–8. doi: 10.1212/WNL.57.2.271
43. Jin SQ, Yu M, Zhang W, Lyu H, Yuan Y, Wang ZX. Dysferlin gene mutation spectrum in a large cohort of chinese patients with dysferlinopathy. Chin Med J. (2016) 129:2287–93. doi: 10.4103/0366-6999.190671
44. Huang Y, Verheesen P, Roussis A, Frankhuizen W, Ginjaar I, Haldane F, et al. Protein studies in dysferlinopathy patients using llama-derived antibody fragments selected by phage display. Eur J Hum Genet. (2005) 13:721–30. doi: 10.1038/sj.ejhg.5201414
45. Rosales XQ, Gastier-Foster JM, Lewis S, Vinod M, Thrush DL, Astbury C, et al. Novel diagnostic features of dysferlinopathies. Muscle Nerve. (2010) 42:14–21. doi: 10.1002/mus.21650
46. Nguyen K, Bassez G, Krahn M, Bernard R, Laforêt P, Labelle V, et al. Phenotypic study in 40 patients with dysferlin gene mutations: high frequency of atypical phenotypes. Arch Neurol. (2007) 64:1176–82. doi: 10.1001/archneur.64.8.1176
47. Vincent AE, Rosa HS, Alston CL, Grady JP, Rygiel KA, Rocha MC, et al. Dysferlin mutations and mitochondrial dysfunction. Neuromusc Disord. (2016) 26:782–8. doi: 10.1016/j.nmd.2016.08.008
48. Middel V, Zhou L, Takamiya M, Beil T, Shahid M, Roostalu U, et al. Dysferlin-mediated phosphatidylserine sorting engages macrophages in sarcolemma repair. Nat Commun. (2016) 7:12875. doi: 10.1038/ncomms12875
49. Szymanska S, Rokicki D, Karkucinska-Wieckowska A, Szymanska-Debinska T, Ciara E, Ploski R, et al. Case report of an adolescent girl with limb-girdle muscular dystrophy type 2B - the usefulness of muscle protein immunostaining in the diagnosis of dysferlinopathies. Folia Neuropathol. (2014) 52:452–6. doi: 10.5114/fn.2014.47847
50. Gallardo E, de Luna N, Diaz-Manera J, Rojas-García R, Gonzalez-Quereda L, Flix B, et al. Comparison of dysferlin expression in human skeletal muscle with that in monocytes for the diagnosis of dysferlin myopathy. PLoS ONE. (2011) 6:e29061. doi: 10.1371/journal.pone.0029061
51. Turan S, Farruggio AP, Srifa W, Day JW, Calos MP. Precise correction of disease mutations in induced pluripotent stem cells derived from patients with limb girdle muscular dystrophy. Mol Ther. (2016) 24:685–96. doi: 10.1038/mt.2016.40
52. Stehlíková K, Skálová D, Zídková J, Haberlová J, Vohánka S, Mazanec R, et al. Muscular dystrophies and myopathies: the spectrum of mutated genes in the czech republic. Clin Genet. (2017) 91:463–9. doi: 10.1111/cge.12839
53. Klinge L, Aboumousa A, Eagle M, Hudson J, Sarkozy A, Vita G, et al. New aspects on patients affected by dysferlin deficient muscular dystrophy. J Neurol Neurosurg Psychiatry. (2010) 81:946–53. doi: 10.1136/jnnp.2009.178038
54. Vilchez JJ, Gallano P, Gallardo E, Lasa A, Rojas-García R, Freixas A, et al. Identification of a novel founder mutation in the DYSF gene causing clinical variability in the Spanish population. Arch Neurol. (2005) 62:1256–9. doi: 10.1001/archneur.62.8.1256
55. Bean LJ, Tinker SW, da Silva C, Hegde MR. Free the data: one laboratory's approach to knowledge-based genomic variant classification and preparation for EMR integration of genomic data. Hum Mutat. (2013) 34:1183–8. doi: 10.1002/humu.22364
56. Park YE, Kim HS, Lee CH, Nam TS, Choi YC, Kim DS. Two common mutations (p.Gln832X and c.663+1G>C) account for about a third of the DYSF mutations in Korean patients with dysferlinopathy. Neuromusc Disord. (2012) 22:505–10. doi: 10.1016/j.nmd.2011.12.007
57. Lo HP, Cooper ST, Evesson FJ, Seto JT, Chiotis M, Tay V, et al. Limb-girdle muscular dystrophy: diagnostic evaluation, frequency and clues to pathogenesis. Neuromusc Disord. (2008) 18:34–44. doi: 10.1016/j.nmd.2007.08.009
58. Harris E, Bladen CL, Mayhew A, James M, Bettinson K, Moore U, et al. The clinical outcome study for dysferlinopathy: an international multicenter study. Neurol Genet. (2016) 2:e89. doi: 10.1212/NXG.0000000000000089
59. Dastur RS, Gaitonde PS, Kachwala M, Nallamilli BRR, Ankala A, Khadilkar SV, et al. Detection of dysferlin gene pathogenic variants in the Indian population in patients predicted to have a dysferlinopathy using a blood-based monocyte assay and clinical algorithm: a model for accurate and cost-effective diagnosis. Ann Indian Acad Neurol. (2017) 20:302–8. doi: 10.4103/aian.AIAN_129_17
60. Dominov JA, Uyan O, McKenna-Yasek D, Nallamilli BRR, Kergourlay V, Bartoli M, et al. Correction of pseudoexon splicing caused by a novel intronic dysferlin mutation. Ann Clin Transl Neurol. (2019) 6:642–54. doi: 10.1002/acn3.738
61. Winckler PB, da Silva AMS, Coimbra-Neto AR, Carvalho E, Cavalcanti EBU, Sobreira CFR, et al. Clinicogenetic lessons from 370 patients with autosomal recessive limb-girdle muscular dystrophy. Clin Genet. (2019) 96:341–53. doi: 10.1111/cge.13597
62. Carrillo N, Malicdan MC, Huizing M. GNE myopathy. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews(®) [Internet]. Seattle, WA: University of Washington (1993–2020).
63. Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. (2001) 29:83–7. doi: 10.1038/ng718
64. Argov Z, Eisenberg I, Grabov-Nardini G, Sadeh M, Wirguin I, Soffer D, et al. Hereditary inclusion body myopathy: the middle eastern genetic cluster. Neurology. (2003) 60:1519–23. doi: 10.1212/01.WNL.0000061617.71839.42
65. Khadilkar SV, Nallamilli BRR, Bhutada A, Hegde M, Gandhi K, Faldu HD, et al. A report on GNE myopathy: individuals of Rajasthan ancestry share the Roma gene. J Neurol Sci. (2017) 375:239–40. doi: 10.1016/j.jns.2017.02.005
66. Chamova T, Guergueltcheva V, Gospodinova M, Krause S, Cirak S, Kaprelyan A, et al. GNE myopathy in Roma patients homozygous for the p.I618T founder mutation. Neuromusc Disord. (2015) 25:713–8. doi: 10.1016/j.nmd.2015.07.004
67. Celeste FV, Vilboux T, Ciccone C, de Dios JK, Malicdan MC, Leoyklang P, et al. Mutation update for GNE gene variants associated with GNE myopathy. Hum Mutat. (2014) 35:915–26. doi: 10.1002/humu.22583
68. Cho A, Hayashi YK, Monma K, Oya Y, Noguchi S, Nonaka I, et al. Mutation profile of the GNE gene in Japanese patients with distal myopathy with rimmed vacuoles (GNE myopathy). J Neurol Neurosurg Psychiatry. (2014) 85:914–7. doi: 10.1136/jnnp-2013-305587
69. Chaouch A, Brennan KM, Hudson J, Longman C, McConville J, Morrison PJ, et al. Two recurrent mutations are associated with GNE myopathy in the North of Britain. J Neurol Neurosurg Psychiatry. (2014) 85:1359–65. doi: 10.1136/jnnp-2013-306314
70. Cerino M, Gorokhova S, Béhin A, Urtizberea JA, Kergourlay V, Salvo E, et al. Novel pathogenic variants in a french cohort widen the mutational spectrum of GNE myopathy. J Neuromusc Dis. (2015) 2:131–6. doi: 10.3233/JND-150074
71. Saechao C, Valles-Ayoub Y, Esfandiarifard S, Haghighatgoo A, No D, Shook S, et al. Novel GNE mutations in hereditary inclusion body myopathy patients of non-middle eastern descent. Genet Test Mol Biomark. (2010) 14:157–62. doi: 10.1089/gtmb.2009.0157
72. de Dios JK, Shrader JA, Joe GO, McClean JC, Williams K, Evers R, et al. Atypical presentation of GNE myopathy with asymmetric hand weakness. Neuromusc Disord. (2014) 24:1063–7. doi: 10.1016/j.nmd.2014.07.006
73. Park YE, Kim DS, Choi YC, Shin JH. Progression of GNE myopathy based on the patient-reported outcome. J Clin Neurol. (2019) 15:275–84. doi: 10.3988/jcn.2019.15.3.275
74. Chen Y, Xi J, Zhu W, Lin J, Luo S, Yue D, et al. GNE myopathy in Chinese population: hotspot and novel mutations. J Hum Genet. (2019) 64:11–6. doi: 10.1038/s10038-018-0525-9
75. Zhao J, Wang Z, Hong D, Lv H, Zhang W, Chen J, et al. Mutational spectrum and clinical features in 35 unrelated mainland Chinese patients with GNE myopathy. J Neurol Sci. (2015) 354:21–6. doi: 10.1016/j.jns.2015.04.028
76. No D, Valles-Ayoub Y, Carbajo R, Khokher Z, Sandoval L, Stein B, et al. Novel GNE mutations in autosomal recessive hereditary inclusion body myopathy patients. Genet Test Mol Biomark. (2013) 17:376–82. doi: 10.1089/gtmb.2012.0408
77. Fanin M, Nascimbeni AC, Fulizio L, Angelini C. The frequency of limb girdle muscular dystrophy 2A in northeastern Italy. Neuromusc Disord. (2005) 15:218–24. doi: 10.1016/j.nmd.2004.11.003
78. Pathak P, Sharma MC, Sarkar C, Jha P, Suri V, Mohd H, et al. Limb girdle muscular dystrophy type 2A in India: a study based on semi-quantitative protein analysis, with clinical and histopathological correlation. Neurol India. (2010) 58:549–54. doi: 10.4103/0028-3886.68675
79. Morisaki T, Gross M, Morisaki H, Pongratz D, Zöllner N, Holmes EW. Molecular basis of AMP deaminase deficiency in skeletal muscle. Proc Natl Acad Sci USA. (1992) 89:6457–61. doi: 10.1073/pnas.89.14.6457
80. Castro-Gago M, Gómez-Lado C, Pérez-Gay L, Eirís-Puñal J, Martínez EP, García-Consuegra I, et al. Primary adenosine monophosphate (AMP) deaminase deficiency in a hypotonic infant. J Child Neurol. (2011) 26:734–7. doi: 10.1177/0883073810390367
81. Johnston JJ, Lewis KL, Ng D, Singh LN, Wynter J, Brewer C, et al. Individualized iterative phenotyping for genome-wide analysis of loss-of-function mutations. Am J Hum Genet. (2015) 96:913–25. doi: 10.1016/j.ajhg.2015.04.013
82. Lucioli S, Giusti B, Mercuri E, Vanegas OC, Lucarini L, Pietroni V, et al. Detection of common and private mutations in the COL6A1 gene of patients with Bethlem myopathy. Neurology. (2005) 64:1931–7. doi: 10.1212/01.WNL.0000163990.00057.66
83. Trempe JF, Sauvé V, Grenier K, Seirafi M, Tang MY, Ménade M, et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science. (2013) 340:1451–5. doi: 10.1126/science.1237908
84. Lamandé SR, Shields KA, Kornberg AJ, Shield LK, Bateman JF. Bethlem myopathy and engineered collagen VI triple helical deletions prevent intracellular multimer assembly and protein secretion. J Biol Chem. (1999) 274:21817–22. doi: 10.1074/jbc.274.31.21817
85. Pan TC, Zhang RZ, Sudano DG, Marie SK, Bönnemann CG, Chu ML. New molecular mechanism for Ullrich congenital muscular dystrophy: a heterozygous in-frame deletion in the COL6A1 gene causes a severe phenotype. Am J Hum Genet. (2003) 73:355–69. doi: 10.1086/377107
86. Baker NL, Mörgelin M, Pace RA, Peat RA, Adams NE, Gardner RJ, et al. Molecular consequences of dominant Bethlem myopathy collagen VI mutations. Ann Neurol. (2007) 62:390–405. doi: 10.1002/ana.21213
87. Fan Y, Liu A, Wei C, Yang H, Chang X, Wang S, et al. Genetic and clinical findings in a Chinese cohort of patients with collagen VI-related myopathies. Clin Genet. (2018) 93:1159–71. doi: 10.1111/cge.13230
88. Lampe AK, Dunn DM, von Niederhausern AC, Hamil C, Aoyagi A, Laval SH, et al. Automated genomic sequence analysis of the three collagen VI genes: applications to Ullrich congenital muscular dystrophy and Bethlem myopathy. J Med Genet. (2005) 42:108–20. doi: 10.1136/jmg.2004.023754
89. Butterfield RJ, Foley AR, Dastgir J, Asman S, Dunn DM, Zou Y, et al. Position of glycine substitutions in the triple helix of COL6A1, COL6A2, and COL6A3 is correlated with severity and mode of inheritance in collagen VI myopathies. Hum Mutat. (2013) 34:1558–67. doi: 10.1002/humu.22429
90. Long CG, Braswell E, Zhu D, Apigo J, Baum J, Brodsky B. Characterization of collagen-like peptides containing interruptions in the repeating Gly-X-Y sequence. Biochemistry. (1993) 32:11688–95. doi: 10.1021/bi00094a027
91. Parini R, De Lorenzo P, Dardis A, Burlina A, Cassio A, Cavarzere P, et al. Long term clinical history of an Italian cohort of infantile onset Pompe disease treated with enzyme replacement therapy. Orphanet J Rare Dis. (2018) 13:32. doi: 10.1186/s13023-018-0771-0
92. Fukuhara Y, Fuji N, Yamazaki N, Hirakiyama A, Kamioka T, Seo JH, et al. A molecular analysis of the GAA gene and clinical spectrum in 38 patients with Pompe disease in Japan. Mol Genet Metab Rep. (2018) 14:3–9. doi: 10.1016/j.ymgmr.2017.10.009
93. Pittis MG, Donnarumma M, Montalvo AL, Dominissini S, Kroos M, Rosano C, et al. Molecular and functional characterization of eight novel GAA mutations in Italian infants with Pompe disease. Hum Mutat. (2008) 29:E27–36. doi: 10.1002/humu.20753
94. Remiche G, Ronchi D, Magri F, Lamperti C, Bordoni A, Moggio M, et al. Extended phenotype description and new molecular findings in late onset glycogen storage disease type II: a northern Italy population study and review of the literature. J Neurol. (2014) 261:83–97. doi: 10.1007/s00415-013-7137-2
95. Bali DS, Goldstein JL, Banugaria S, Dai J, Mackey J, Rehder C, et al. Predicting Cross-Reactive Immunological Material (CRIM) status in Pompe disease using GAA mutations: lessons learned from 10 years of clinical laboratory testing experience. Am J Med Genet C Semin Med Genet. (2012) 160c:40–9. doi: 10.1002/ajmg.c.31319
96. Hermans MM, van Leenen D, Kroos MA, Beesley CE, Van Der Ploeg AT, Sakuraba H, et al. Twenty-two novel mutations in the lysosomal alpha-glucosidase gene (GAA) underscore the genotype-phenotype correlation in glycogen storage disease type II. Hum Mutat. (2004) 23:47–56. doi: 10.1002/humu.10286
97. Löscher WN, Huemer M, Stulnig TM, Simschitz P, Iglseder S, Eggers C, et al. Pompe disease in Austria: clinical, genetic and epidemiological aspects. J Neurol. (2018) 265:159–64. doi: 10.1007/s00415-017-8686-6
98. Turaça LT, de Faria DO, Kyosen SO, Teixeira VD, Motta FL, Pessoa JG, et al. Novel GAA mutations in patients with Pompe disease. Gene. (2015) 561:124–31. doi: 10.1016/j.gene.2015.02.023
99. Fu L, Qiu W, Yu Y, Guo Y, Zhao P, Zhang X, et al. Clinical and molecular genetic study of infantile-onset Pompe disease in Chinese patients: identification of 6 novel mutations. Gene. (2014) 535:53–9. doi: 10.1016/j.gene.2013.10.066
100. Beltran Papsdorf TB, Howard JF Jr, Chahin N. Pearls & Oysters: clues to the diagnosis of adult-onset acid maltase deficiency. Neurology. (2014) 82:e73–5. doi: 10.1212/WNL.0000000000000163
101. Bali DS, Tolun AA, Goldstein JL, Dai J, Kishnani PS. Molecular analysis and protein processing in late-onset Pompe disease patients with low levels of acid α-glucosidase activity. Muscle Nerve. (2011) 43:665–70. doi: 10.1002/mus.21933
102. Liao HC, Chiang CC, Niu DM, Wang CH, Kao SM, Tsai FJ, et al. Detecting multiple lysosomal storage diseases by tandem mass spectrometry–a National Newborn Screening Program in Taiwan. Clin Chim Acta Int J Clin Chem. (2014) 431:80–6. doi: 10.1016/j.cca.2014.01.030
103. Oliveira J, Santos R, Soares-Silva I, Jorge P, Vieira E, Oliveira ME, et al. LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin Genet. (2008) 74:502–12. doi: 10.1111/j.1399-0004.2008.01068.x
104. Rajakulendran S, Parton M, Holton JL, Hanna MG. Clinical and pathological heterogeneity in late-onset partial merosin deficiency. Muscle Nerve. (2011) 44:590–3. doi: 10.1002/mus.22196
105. Beytía Mde L, Dekomien G, Hoffjan S, Haug V, Anastasopoulos C, Kirschner J. High creatine kinase levels and white matter changes: clinical and genetic spectrum of congenital muscular dystrophies with laminin alpha-2 deficiency. Mol Cell Probes. (2014) 28:118–22. doi: 10.1016/j.mcp.2013.11.002
106. Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. (2014) 16:601–8. doi: 10.1038/gim.2013.204
107. Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, et al. Reassessment of mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. (2017) 19:192–203. doi: 10.1038/gim.2016.90
108. Forissier JE, Bonne G, Bouchier C, Duboscq-Bidot L, Richard P, Wisnewski C, et al. Apical left ventricular aneurysm without atrio-ventricular block due to a lamin A/C gene mutation. Eur J Heart Fail. (2005) 5:821–5. doi: 10.1016/s1388-9842(03)00149-1
109. Hookana E, Junttila MJ, Särkioja T, Sormunen R, Niemelä M, Raatikainen MJ, et al. Cardiac arrest and left ventricular fibrosis in a finnish family with the lamin A/C mutation. J Cardiovasc Electrophysiol. (2008) 19:743–7. doi: 10.1111/j.1540-8167.2007.01017.x
110. Forissier JF, Bonne G, Bouchier C, Duboscq-Bidot L, Richard P, Wisnewski C, et al. Apical left ventricular aneurysm without atrio-ventricular block due to a lamin A/C gene mutation. Eur J Heart Fail. (2003) 5:821–5. doi: 10.1016/S1388-9842(03)00149-1
111. Lakdawala NK, Funke BH, Baxter S, Cirino AL, Roberts AE, Judge DP, et al. Genetic testing for dilated cardiomyopathy in clinical practice. J Cardiac Fail. (2012) 18:296–303. doi: 10.1016/j.cardfail.2012.01.013
112. Dialynas G, Flannery KM, Zirbel LN, Nagy PL, Mathews KD, Moore SA, et al. LMNA variants cause cytoplasmic distribution of nuclear pore proteins in Drosophila and human muscle. Hum Mol Genet. (2012) 21:1544–56. doi: 10.1093/hmg/ddr592
113. Dittmer TA, Sahni N, Kubben N, Hill DE, Vidal M, Burgess RC, et al. Systematic identification of pathological lamin A interactors. Mol Biol Cell. (2014) 25:1493–510. doi: 10.1091/mbc.e14-02-0733
114. Benedetti S, Menditto I, Degano M, Rodolico C, Merlini L, D'Amico A, et al. Phenotypic clustering of lamin A/C mutations in neuromuscular patients. Neurology. (2007) 69:1285–92. doi: 10.1212/01.wnl.0000261254.87181.80
115. Pasqualin LM, Reed UC, Costa TV, Quedas E, Albuquerque MA, Resende MB, et al. Congenital muscular dystrophy with dropped head linked to the LMNA gene in a Brazilian cohort. Pediatr Neurol. (2014) 50:400–6. doi: 10.1016/j.pediatrneurol.2013.11.010
116. Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. (2011) 32:557–63. doi: 10.1002/humu.21438
117. Quijano-Roy S, Mbieleu B, Bönnemann CG, Jeannet PY, Colomer J, Clarke NF, et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol. (2008) 64:177–86. doi: 10.1002/ana.21417
118. Godfrey C, Clement E, Mein R, Brockington M, Smith J, Talim B, et al. Refining genotype–phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. (2007) 130:2725–35. doi: 10.1093/brain/awm212
119. Carrié A, Piccolo F, Leturcq F, de Toma C, Azibi K, Beldjord C, et al. Mutational diversity and hot spots in the alpha-sarcoglycan gene in autosomal recessive muscular dystrophy (LGMD2D). J Med Genet. (1997) 34:470–5. doi: 10.1136/jmg.34.6.470
120. van der Kooi AJ, Frankhuizen WS, Barth PG, Howeler CJ, Padberg GW, Spaans F, et al. Limb-girdle muscular dystrophy in the Netherlands: gene defect identified in half the families. Neurology. (2007) 68:2125–8. doi: 10.1212/01.wnl.0000264853.40735.3b
121. Duggan DJ, Gorospe JR, Fanin M, Hoffman EP, Angelini C. Mutations in the sarcoglycan genes in patients with myopathy. N Engl J Med. (1997) 336:618–24. doi: 10.1056/NEJM199702273360904
122. Khadilkar SV, Singh RK, Hegde M, Urtizberea A, Love DR, Chong B. Spectrum of mutations in sarcoglycan genes in the Mumbai region of western India: high prevalence of 525del T. Neurol India. (2009) 57:406–10. doi: 10.4103/0028-3886.55603
123. Duggan DJ, Manchester D, Stears KP, Mathews DJ, Hart C, Hoffman EP. Mutations in the delta-sarcoglycan gene are a rare cause of autosomal recessive limb-girdle muscular dystrophy (LGMD2). Neurogenetics. (1997) 1:49–58. doi: 10.1007/s100480050008
124. Barresi R, Morris C, Hudson J, Curtis E, Pickthall C, Bushby K, et al. Conserved expression of truncated telethonin in a patient with limb-girdle muscular dystrophy 2G. Neuromusc Disord. (2015) 25:349–52. doi: 10.1016/j.nmd.2014.12.006
125. Ruggieri A, Brancati F, Zanotti S, Maggi L, Pasanisi MB, Saredi S, et al. Complete loss of the DNAJB6 G/F domain and novel missense mutations cause distal-onset DNAJB6 myopathy. Acta Neuropathol Commun. (2015) 3:44. doi: 10.1186/s40478-015-0224-0
126. Wenzel K, Geier C, Qadri F, Hubner N, Schulz H, Erdmann B, et al. Dysfunction of dysferlin-deficient hearts. J Mol Med. (2007) 85:1203–14. doi: 10.1007/s00109-007-0253-7
127. Choi ER, Park SJ, Choe YH, Ryu DR, Chang SA, Choi JO, et al. Early detection of cardiac involvement in Miyoshi myopathy: 2D strain echocardiography and late gadolinium enhancement cardiovascular magnetic resonance. J Cardiovasc Magn Reson. (2010) 12:31. doi: 10.1186/1532-429X-12-31
128. Huizing M, Rakocevic G, Sparks SE, Mamali I, Shatunov A, Goldfarb L, et al. Hypoglycosylation of alpha-dystroglycan in patients with hereditary IBM due to GNE mutations. Mol Genet Metab. (2004) 81:196–202. doi: 10.1016/j.ymgme.2003.11.012
129. Weihl CC, Baloh RH, Lee Y, Chou TF, Pittman SK, Lopate G, et al. Targeted sequencing and identification of genetic variants in sporadic inclusion body myositis. Neuromusc Disord. (2015) 25:289–96. doi: 10.1016/j.nmd.2014.12.009
130. Krause S, Aleo A, Hinderlich S, Merlini L, Tournev I, Walter MC, et al. GNE protein expression and subcellular distribution are unaltered in HIBM. Neurology. (2007) 69:655–9. doi: 10.1212/01.wnl.0000267426.97138.fd
131. Penner J, Mantey LR, Elgavish S, Ghaderi D, Cirak S, Berger M, et al. Influence of UDP-GlcNAc 2-epimerase/ManNAc kinase mutant proteins on hereditary inclusion body myopathy. Biochemistry. (2006) 45:2968–77. doi: 10.1021/bi0522504
132. Deconinck N, Richard P, Allamand V, Behin A, Lafôret P, Ferreiro A, et al. Bethlem myopathy: long-term follow-up identifies COL6 mutations predicting severe clinical evolution. J Neurol Neurosurg Psychiatry. (2015) 86:1337–46. doi: 10.1136/jnnp-2013-307245
133. Deconinck N, Dion E, Ben Yaou R, Ferreiro A, Eymard B, Briñas L, et al. Differentiating emery-dreifuss muscular dystrophy and collagen VI-related myopathies using a specific CT scanner pattern. Neuromusc Disord. (2010) 20:517–23. doi: 10.1016/j.nmd.2010.04.009
134. Martoni E, Urciuolo A, Sabatelli P, Fabris M, Bovolenta M, Neri M, et al. Identification and characterization of novel collagen VI non-canonical splicing mutations causing Ullrich congenital muscular dystrophy. Hum Mutat. (2009) 30:E662–72. doi: 10.1002/humu.21022
135. Foley AR, Hu Y, Zou Y, Yang M, Medne L, Leach M, et al. Large genomic deletions: a novel cause of Ullrich congenital muscular dystrophy. Ann Neurol. (2011) 69:206–11. doi: 10.1002/ana.22283
136. Briñas L, Richard P, Quijano-Roy S, Gartioux C, Ledeuil C, Lacène E, et al. Early onset collagen VI myopathies: genetic and clinical correlations. Ann Neurol. (2010) 68:511–20. doi: 10.1002/ana.22087
137. Cui H, Wang J, Zhang C, Wu G, Zhu C, Tang B, et al. Mutation profile of FLNC gene and its prognostic relevance in patients with hypertrophic cardiomyopathy. Mol Genet Genomic Med. (2018) 6:1104–13. doi: 10.1002/mgg3.488
138. Olivé M, Goldfarb LG, Shatunov A, Fischer D, Ferrer I. Myotilinopathy: refining the clinical and myopathological phenotype. Brain. (2005) 128(Pt. 10):2315–26. doi: 10.1093/brain/awh576
139. Park YE, Kim HS, Choi ES, Shin JH, Kim SY, Son EH, et al. Limb-girdle phenotype is frequent in patients with myopathy associated with GNE mutations. J Neurol Sci. (2012) 321:77–81. doi: 10.1016/j.jns.2012.07.061
140. Balci B, Uyanik G, Dincer P, Gross C, Willer T, Talim B, et al. An autosomal recessive limb girdle muscular dystrophy (LGMD2) with mild mental retardation is allelic to Walker-Warburg syndrome (WWS) caused by a mutation in the POMT1 gene. Neuromusc Disord. (2005) 15:271–5. doi: 10.1016/j.nmd.2005.01.013
141. Fardeau M, Hillaire D, Mignard C, Feingold N, Feingold J, Mignard D, et al. Juvenile limb-girdle muscular dystrophy: clinical, histopathological and genetic data from a small community living in the reunion Island. Brain. (1996) 119:295–308. doi: 10.1093/brain/119.1.295
142. Nishino I, Carrillo-Carrasco N, Argov Z. GNE myopathy: current update and future therapy. J Neurol Neurosurg Psychiatry. (2015) 86:385–92. doi: 10.1136/jnnp-2013-307051
143. Nalini A, Gayathri N, Nishino I, Hayashi YK. GNE myopathy in India. Neurol India. (2013) 61:371–4. doi: 10.4103/0028-3886.117609
144. Argov Z, Mitrani Rosenbaum S. GNE myopathy: two clusters with history and several founder mutations. J Neuromusc Dis. (2015) 2:S73–S6. doi: 10.3233/JND-150087
145. Vissing J, Barresi R, Witting N, Van Ghelue M, Gammelgaard L, Bindoff LA, et al. A heterozygous 21-bp deletion in CAPN3 causes dominantly inherited limb girdle muscular dystrophy. Brain. (2016) 139(Pt. 8):2154–63. doi: 10.1093/brain/aww133
146. Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. (1998) 20:31–6. doi: 10.1038/1682
147. Urtizberea JA, Bassez G, Leturcq F, Nguyen K, Krahn M, Levy N. Dysferlinopathies. Neurol India. (2008) 56:289–97. doi: 10.4103/0028-3886.43447
148. Prelle A, Sciacco M, Tancredi L, Fagiolari G, Comi GP, Ciscato P, et al. Clinical, morphological and immunological evaluation of six patients with dysferlin deficiency. Acta Neuropathol. (2003) 105:537–42. doi: 10.1007/s00401-002-0654-1
149. Morisaki H, Higuchi I, Abe M, Osame M, Morisaki T. First missense mutations (R388W and R425H) of AMPD1 accompanied with myopathy found in a Japanese patient. Hum Mutat. (2000) 16:467–72. doi: 10.1002/1098-1004(200012)16:6<467::AID-HUMU3>3.0.CO;2-V
150. Gross M, Rotzer E, Kolle P, Mortier W, Reichmann H, Goebel HH, et al. A G468-T AMPD1 mutant allele contributes to the high incidence of myoadenylate deaminase deficiency in the caucasian population. Neuromusc Disord. (2002) 12:558–65. doi: 10.1016/S0960-8966(02)00008-1
151. Norman B, Glenmark B, Jansson E. Muscle AMP deaminase deficiency in 2% of a healthy population. Muscle Nerve. (1995) 18:239–41. doi: 10.1002/mus.880180216
152. Verzijl HT, van Engelen BG, Luyten JA, Steenbergen GC, van den Heuvel LP, ter Laak HJ, et al. Genetic characteristics of myoadenylate deaminase deficiency. Ann Neurol. (1998) 44:140–3. doi: 10.1002/ana.410440124
153. Norman B, Mahnke-Zizelman DK, Vallis A, Sabina RL. Genetic and other determinants of AMP deaminase activity in healthy adult skeletal muscle. J Appl Physiol. (1998) 85:1273–8. doi: 10.1152/jappl.1998.85.4.1273
154. Chakravorty S, Hegde M. Inferring the effect of genomic variation in the new era of genomics. Hum Mutat. (2018) 39:756–73. doi: 10.1002/humu.23427
155. Vladutiu GD. Heterozygosity: an expanding role in proteomics. Mol Genet Metab. (2001) 74:51–63. doi: 10.1006/mgme.2001.3240
Keywords: inherited myopathies, LGMD, exome sequencing, next generation sequencing, molecular diagnostics, India, subcontinent
Citation: Chakravorty S, Nallamilli BRR, Khadilkar SV, Singla MB, Bhutada A, Dastur R, Gaitonde PS, Rufibach LE, Gloster L and Hegde M (2020) Clinical and Genomic Evaluation of 207 Genetic Myopathies in the Indian Subcontinent. Front. Neurol. 11:559327. doi: 10.3389/fneur.2020.559327
Received: 05 May 2020; Accepted: 23 September 2020;
Published: 05 November 2020.
Edited by:
Edoardo Malfatti, INSERM U1179 Handicap neuromusculaire : Physiopathologie, Biothérapie et Pharmacologie appliquées (END-ICAP), FranceReviewed by:
Monkol Lek, Yale University, United StatesLuisa Politano, University of Campania Luigi Vanvitelli, Italy
Teerin Liewluck, Mayo Clinic, United States
Copyright © 2020 Chakravorty, Nallamilli, Khadilkar, Singla, Bhutada, Dastur, Gaitonde, Rufibach, Gloster and Hegde. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samya Chakravorty, c2FteWEuY2hha3Jhdm9ydHlAZW1vcnkuZWR1; Madhuri Hegde, bWFkaHVyaS5oZWdkZUBwZXJraW5lbG1lci5jb20=