 Juan Darío Ortigoza-Escobar
Juan Darío Ortigoza-Escobar- Movement Disorders Unit, Institut de Recerca Sant Joan de Déu, CIBERER-ISCIII and European Reference Network for Rare Neurological Diseases (ERN-RND), Barcelona, Spain
Inherited metabolic diseases or inborn errors of metabolism frequently manifest with both hyperkinetic (dystonia, chorea, myoclonus, ataxia, tremor, etc.) and hypokinetic (rigid-akinetic syndrome) movement disorders. The diagnosis of these diseases is in many cases difficult, because the same movement disorder can be caused by several diseases. Through a literature review, two hundred and thirty one inborn errors of metabolism presenting with movement disorders have been identified. Fifty-one percent of these diseases exhibits two or more movement disorders, of which ataxia and dystonia are the most frequent. Taking into account the wide range of these disorders, a methodical evaluation system needs to be stablished. This work proposes a six-step diagnostic algorithm for the identification of inborn errors of metabolism presenting with movement disorders comprising red flags, characterization of the movement disorders phenotype (type of movement disorder, age and nature of onset, distribution and temporal pattern) and other neurological and non-neurological signs, minimal biochemical investigation to diagnose treatable diseases, radiological patterns, genetic testing and ultimately, symptomatic, and disease-specific treatment. As a strong action, it is emphasized not to miss any treatable inborn error of metabolism through the algorithm.
Introduction
Inborn errors of metabolism (IEMs) are defined as any condition that leads to a disturbance of a metabolic pathway, irrespective of whether it is associated with abnormalities in biochemical laboratory tests. Indeed, IEMs include not only enzymes or transporters deficiencies or superactivities, but also chaperons and transcription factors abnormalities. Recently, a current nosology by Ferreira et al., defines 1,015 IEMs in 130 groups (1).
IEMs encompass a large group of single gene disorders that can affect all organs and lead to a variety of symptoms (2). Most of the IEMs are multisystem diseases with neurological (intellectual disability/developmental delay, cognitive regression, hypotonia, spasticity, neuropathy, vision and hearing impairment, encephalopathy/stroke, epilepsy, etc.) and non-neurological (failure to thrive, vomiting, hepatomegaly, splenomegaly, renal tubular acidosis, nephrolithiasis, etc.) manifestations. Commonly initial symptoms and signs of IEMs are somewhat nonspecific (3). Movement disorders (MD) are among the most usual neurological symptoms in children with IEMs and account for a significant part of the morbidity and mortality. MD manifest in IEMs that cause diffuse CNS or selective basal ganglia involvement. As it is known, the basal ganglia participate in the control of voluntary movement and on the other hand this brain area is especially vulnerable to certain IEMs as metal storage defects, energy metabolism and lysosomal storages disorders (4, 5).
A central problem is the large number and variety of IEMs with MD and the poor recognition of these disorders that make it challenging for the pediatric neurologist to decide on the initial evaluation with the consequent delay in diagnosis and timely treatment. This work herein facilitates an update information with new IEMs that present MD, suggests red flags and diagnostics clues for suspecting IEMs, proposes the minimum biochemical studies as stated in each MD and the differential diagnoses according to the neuroradiological findings and provides evidence on symptomatic or disease specific-treatment through a six-step algorithm.
Overview of Movement Disorders in Inborn Errors of Metabolism
Based on the distinct pathway involved, IEMs can be categorized as described by the nosology of Ferreira et al. into the following groups (1): (1) Disorders of nitrogen-containing compounds, (2) Disorders of vitamins, cofactors, metals, and minerals, (3) Disorders of carbohydrates, (4) Mitochondrial disorders of energy metabolism, (5) Disorders of lipids, (6) Disorders of Tetrapyrroles, (7) Storage disorders, (8) Disorders of peroxisome and oxalate, and (9) Congenital disorders of glycosylation. This accurate and updated nosology will be applied in this review, in which a total of 231 IEMs presenting with MD will be included. The search strategy, the selection criteria and the data extraction applied are described in Supplementary File 1. The IEMs presenting with MD appear in Supplementary Table 1.
Whereas, MD are classified into two main categories, hyperkinetic, and hypokinetic movements. Hyperkinetic MD are unwanted or excess movements which include dystonia, choreoathetosis, tremor, myoclonus, tics, and stereotypies. Definition and classification of pediatric hyperkinetic movements were described by Sanger et al. (6), although; this classification did not include ataxia. Hypokinetic movements are called hypokinetic-rigid syndrome or Parkinsonism (7). Both hyperkinetic and hypokinetic MD are crucial clinical findings with significant implications for diagnosis and treatment.
MD in the context of IEMs are generally a combination of different MD, therefore a MD oftentimes does not predict the type of IEM. The 51% of IEMs of this review presents 2 or more MD. In this group there are well-known diseases namely neurotransmitters disorders (8), cerebrotendinous xanthomatosis (9), Wilson disease (10), POLG deficiency (11), Niemann Pick disease type C (12), and Glucose transporter 1 deficiency (13) that can exhibit virtually any MD and that must always be incorporated in the differential diagnosis on either child with MD. It should be noted that many of these referred IEMs are also treatable.
Rarely, there is an isolated MD, commonly mild ataxia or chorea, as part of a more diffuse clinical picture. Furthermore, the MD can evolve over a period of time in the same patient, by the progress in the severity of the main MD [e.g., dystonia in PANK2-Pantothenate kinase-associated neurodegeneration (14), FA2H-Fatty acid hydroxylase-associated neurodegeneration (15), SLC39A14 deficiency (16), TIMM8A-Deafness-Dystonia-Optic Neuronopathy Syndrome (17), or ataxia in COQ8A deficiency (18), MVK-mevalonate kinase deficiency (19), SNX14 deficiency (20), and COG5-CDG (21) or by presenting new further MDs], in particular HRS in relation to neurodegeneration [e.g., Leigh syndrome (22), CLN1 (23), PLA2G6-associated neurodegeneration (24), WDR45-associated neurodegeneration (25), Tay Sachs or Sandhoff disease (26), and L-2-hydroxyglutarate Dehydrogenase deficiency (27)]. Ataxia and dystonia together represent >50% of the cases of MD, with myoclonus and hypokinetic rigid syndrome being the least frequent MD. The percentages of the different MD in the IEMs are detailed in Figure 1.
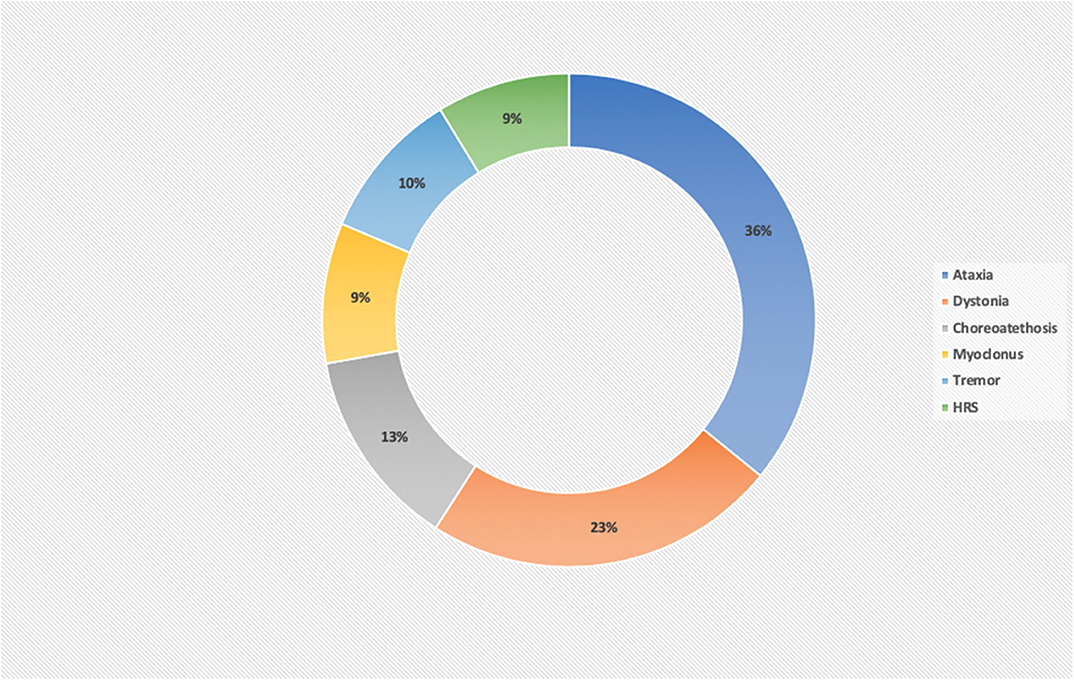
Figure 1. Percentage of movement disorders in a series of 231 inborn error of metabolism disorders. Ataxia and dystonia are the most frequent MDs in IEMs, comprising about 60% of the disorders. HRS and myoclonus are the least frequent MD and for this reason, the most useful MD in guiding the diagnosis of a definite IEM.
Several studies have shown that IEMs account for less than a quarter of diagnoses in children with MD, for this reason timely recognition of these underlying IEMs often allows disease-specific treatments and the best possible outcome for the affected child (3, 28). The percentage of diagnosis reached depends on the population included and on the biochemical or genetics analysis conducted. Diagnosis rate is lower in children with acute symptoms and substantially higher in children with chronic pathology. In this review, six studies investigating IEMs presenting with MD in 909 children and adults have been found (3, 29–33). The groups examined were very heterogeneous while other study exclusively included patients with dystonia (29). When all movement disorders were included, dystonia and ataxia were the most frequent MD. Most of the studies applied targeted next generation sequencing panels obtaining a diagnostic yield ranged between 11 and 51%. Nevertheless, among three other studies in children with acute pathology presenting to the pediatric emergency department, the percentage of children with IEMs was between two and eight percent of all cases. These cases included IEMs as various as ceroidolipofuscinosis, mucopolisacaridosis, pantothenate kinase-associated neurodegeneration, glutaric aciduria type 1, MELAS, and Leigh syndrome (34–36). The full-description of all these studies is displayed in Supplementary Table 2.
Regrettably, there are only a few studies that make a comprehensive description of the MD in the IEMs and most of the knowledge is based on individual cases or series of very few patients. Quite apart from the fact that many of these report do not provide in-depth characterization of the MD. Some studies that include large series of patients with IEMs and MD are: Tetrahydrobiopterin (BH4) deficiencies (37), cobalamin-related remethylation disorders (38), SERAC1 deficiency (39), mitochondrial disorders (22), PLA2G6-associated neurodegeneration (24), lysosomal storage disorders (40), cerebrotendinous xanthomatosis (9), and congenital disorders of glycosylation (41). IEMs with MD as a primary or prominent feature and treatable causes should always be in mind. These groups of IEMs are listed in Table 1.
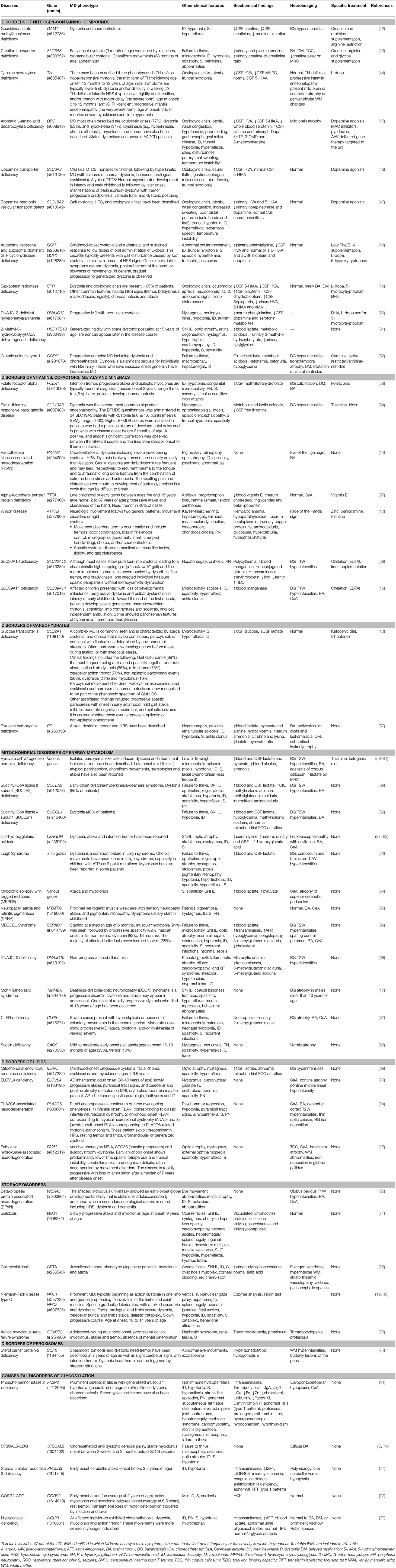
Table 1. Movement disorders and neurological/non-neurological characterization, biochemical findings, neuroimaging, and disease-specific treatment in IEMs with MD as a prominent feature.
IEMs should repeatedly be considered, even in patients in whom an acquired cause of MD is assumed (e.g., infantile cerebral palsy). In a review by Leach et al. they identified 67 treatable IEMs that mimicked infantile cerebral palsy. Seventy-four percent of these IEMs have a specific treatment or a treatment that allows stabilizing/preventative effects. Fifty-seven percent of these IEMs are detected with straightforward metabolic/biochemical tests (80).
With regards to previous diagnostic algorithms of IEMs and MD, a simple scheme for adult patients was proposed by Sedel et al. that demonstrated a diagnostic approach based first on the clinical course of symptoms and the brain MRI (4). This very simple scheme allows the exclusion of the most frequent etiologies speedily whilst it cannot be applied altogether to childhood-onset MD because the diseases differ from adults.
One as well the other, IEM and MD impact intensively children's quality of life (QOL). In the study by Eggink et al., it was found that increasing severity and lower adaptative abilities of MD positively correlated with lower (QOL) scores (2). For this reason, adequate treatment can better ameliorate the quality of life of these children.
Approach to Child With Suspected IEM and MD
In this work, a six-step diagnostic algorithm was drafted and represented in Figure 2. The algorithm is based on simple steps that are listed below. The algorithm helps decide whether to consider a metabolic cause in a patient with MD. It also guides in the characterization of the MD, considering the characteristics that will most help guide a defined IEMs. It presents a list of neurological and non-neurological symptoms to take into account to establish the differential diagnosis of IEMs. Dependent on each MD, it proposes a list of minimal biochemical studies and also a list of IEMs to consider in fulfillment of the neuroradiological findings. Finally, a summary of the available treatments of both the IEMs and the MD is made. Once more, it is very important to note that the algorithm encourages thinking about treatable causes across different steps of the process.
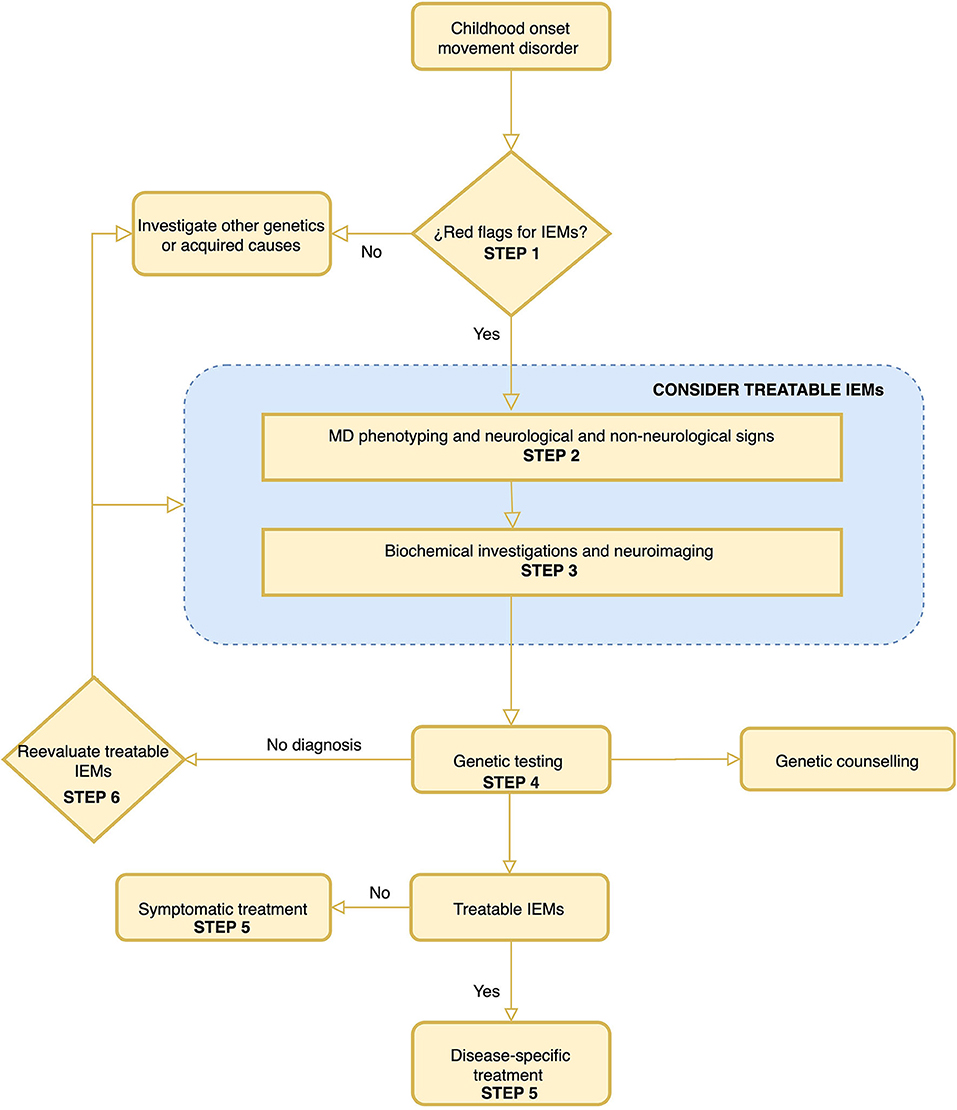
Figure 2. A proposed algorithm for the diagnosis of inborn error of metabolism in childhood-onset movement disorders. The most important steps of this algorithm are step 1 “Red Flags for IEMs” and step 6 “Rethink of treatable IEMs.” It is important to highlight that treatable IEMs should always be taken into consideration. The rationalization of the different steps is developed in the text of the article.
Step 1. When to Suspect an IEM in MD?
IEMs should be included in the differential diagnosis of all children with MD, even though there are several signs and symptoms that should indicate that a particular MD may be produced by an IEM. Some of these signs and symptoms are (3, 4, 81):
1) Diffuse clinical picture with several neurological and non-neurological signs,
2) Combination of different MD (ataxia plus dystonia, dystonia plus parkinsonism, chorea plus dystonia and myoclonus with any other MD or neurological or non-neurological signs, other than seizures),
3) Acute or subacute onset, remarkably if the onset is associated with encephalopathy/coma or if onset is precipitated by a concurrent febrile illness, starvation, physical exhaustion, or after a high-protein ingestion,
4) Insidious onset in a patient with multiple previous non-systemic manifestations,
5) Consanguinity and/or recessive or X-linked inheritance pattern,
6) Distinct neuroradiological findings for instance basal ganglia abnormalities, white matter involvement (hypomyelination/leukoencephalopathy) or cerebellar atrophy,
7) Atypical or progressively abnormal MD that fails to respond to standard treatment, and
8) MD that are not explained by classical etiologies (e.g., structural brain lesion, infectious/parainfectious or autoimmune disorders, toxic or drug induced MD and other well-known genetic or neurodegenerative MD, etc.).
Step 2. Characterize the MD Phenotype and Other Neurological and Non-neurological Signs
Neurological Signs
Movement Disorder
Phenomenology of MD
Not surprisingly this is the most significant step in the algorithm and also the one that can offer the most difficulty. The characterization of the MD must be in line with the definitions of Sanger et al., and reinforce in the own experience of the physician (6). As far as possible it is of the utmost importance to have also the assessment of an expert in pediatric MD. In a straightforward manner, it should be determined:
1. If the child presents unwanted excess movement (hyperkinetic movements) will be classified taking into account the definitions for dystonia, chorea, myoclonus, tremor, and stereotypies fairly detailed by Sanger et al. (6). The definition of ataxia is not included in the latter; accordingly, this definition will be used:
a. Ataxia: consists of the decomposition of movement due to breakdown of normal coordinated execution of a voluntary movement. Dysmetria, dysdiadochokinesia, intentional tremor, hypotonia, wide-based gait, or inability to tandem walk are frequently associated signs of ataxia. Early onset cerebellar ataxia consists in a large group of patients with rare disorders, manifesting symptomatic cerebellar ataxia before the age of 25 years (82).
It is worth noting that tics are not habitually associated with IEMs, hence have not been include in this article, despite the fact that few cases of patients with tics have been reported in the following IEMs: Lesch-Nyhan disease (83), juvenile neuronal ceroid-lipofuscinosis (84), pantothenate kinase-associated neurodegeneration (85), Wilson disease (86), POLG disease (87), mannosidosis (88), COASY-associated neurodegeneration (89), and Maple Syrup Urine Disease (90).
2. If the child presents decreased (hypokinesia) and slow (bradykinesia) movements, rigidity or rest tremor. These are all signs of parkinsonism or hypokinetic rigid syndrome. These signs commonly do not appear all at the same time in a child, therefore the syndrome is routinely “incomplete” (7). The diagnosis of HRS greatly narrow possible IEMs. In this instance, is extremely helpful to observe the recommendations made by Garcia-Cazorla et al. to separate etiologies on the basis of age at onset (7). HRS starting at very young age (<2 years) can guide us to the diagnosis of monoamine neurotransmitter defects, mitochondrial diseases or Neurodegeneration with Brain Iron Accumulation disorders. On the contrary, HRS starting in children older than 2 year of age, can guide us in addition to those formerly mentioned in the following disorders: Wilson disease (10), GLUT-1 deficiency (13), Niemann-Pick type C (12), Gaucher disease (27), manganese accumulation disorders (16, 56), glutaric aciduria type 1 (52), Celia's encephalopathy (91), gangliosidosis (26), and cerebrotendinous xanthomatosis (9). Although it should be noted that HRS generally appears predominantly in the initial stages of the monoamine neurotransmitter defects or mitochondrial disorders, emerging later in the remaining IEMs.
By virtue of the response to treatment, it is important to highlight a very important group of treatable IEMs frequently presenting with HRS: the monoamine neurotransmitter disorders. The clinical phenotypes of these IEMs are predominantly neurological and the symptoms are similar to other childhood neurological disorders, such as cerebral palsy, hypoxic ischemic encephalopathy, or epileptic encephalopathies, as a result the monoamine neurotransmitter disorders are not recognized and are often misdiagnosed. Marked diurnal variation of motor symptoms (axial hypotonia and gait disturbances) and MD (dystonia, HRS, dyskinesia/chorea, tremor, and less commonly myoclonus) are often prominent along with autonomic dysfunction (sweating, hypersalivation, nasal congestion, and temperature dysregulation). Detailed MD phenotype, other clinical features as well as biochemical findings and neuroimaging of monoamine neurotransmitter disorders appears in Table 1. Prompt diagnosis is fundamental, considering that many respond to treatment and that adequate therapy is curative in some disorders. Conversely, the remarkable response to L-dopa of any childhood-onset undiagnosed MD should also guide us to this group of IEMs (8).
Commonly a child affected by an IEM present more than one type of MD. To follow the proposed six-step algorithm, we should take into account all the MD that we can identify regardless the fact that we will always take into more consideration: (1) the most prominent MD in the patient or (2) the least frequent (more specific) MD in all cases. In this sense, diseases that show myoclonus or HRS are less numerous, limiting for the best our diagnostic options. A list of treatable IEMs according to the MD they present appears in Supplementary Table 3.
MD Age at Onset and Type of Onset
In some cases, the MD begins earlier in the prenatal or the neonatal period. With respect to infants and children, there are plenty IEMs presenting with MD that can debut at these group of ages, consequently the isolated age of onset criteria will not be useful to differentiate between the different IEMs. Although, there are some IEMs presenting MD frequently before 2 years of age. Many MD in IEMs have an onset in adulthood regardless numerous cases with non-specific non-neurological symptoms as early as childhood or adolescence (Table 2).
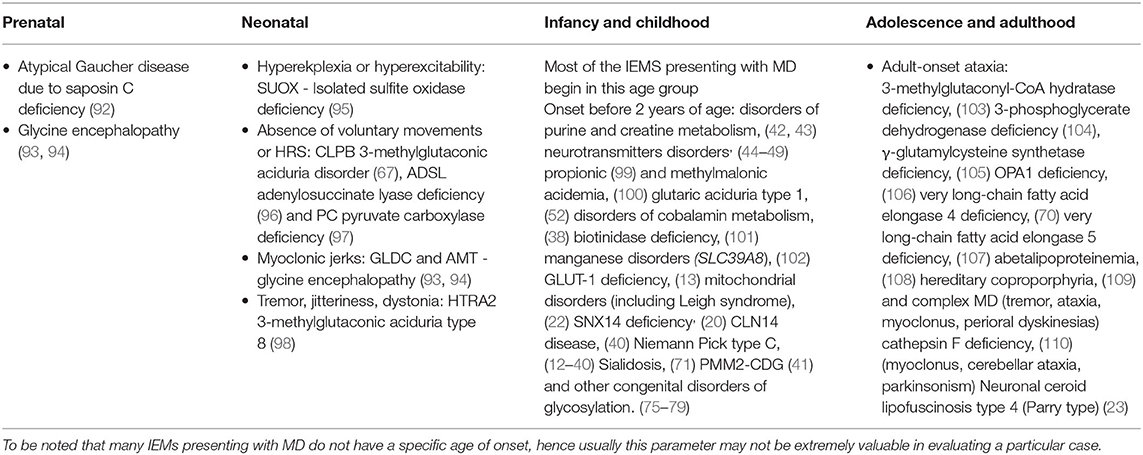
Table 2. IEMs presenting with MD by age at onset.
MD that appear acutely, associated or not with encephalopathy, are generally caused by diseases that are considered medical emergencies. In cases where the onset of MD coincides with a metabolic decompensation, there is several times an acute injury to the basal ganglia or the cerebral cortex, in most cases with accompanying encephalopathy/coma, as is often the case in Leigh syndrome (22), glutaric aciduria type 1 (52), propionic acidemia (99), and other organic acidurias or mitochondrial disorders. Injury to the basal ganglia can also occur with the appearance of MD delayed as in the β-ketothiolase deficiency (111). In the remaining IEMs, there is very often no clear onset of MD disorder.
In a large number of these diseases there is an identifiable trigger as for example: fever [ataxia: Maple Syrup Urine Disease (112), episodic ataxia type 6 (113), milder phenotypes of glycine encephalopathy (93, 94), and GORS2-CDG (78), dystonia: β-ketothiolase deficiency (111) and mitochondrial short-chain enoyl-CoA hydratase 1 deficiency (114)], fasting or fever [(complex MD: GLUT1 deficiency (13)], exercise [(ataxia or dystonia: mitochondrial short-chain enoyl-CoA hydratase 1 deficiency (114), pyruvate dehydrogenase complex deficiency (58–61)] and lead exposure [ataxia: hereditary coproporphyria (109)]. In other cases, the trigger is regularly more subtle as stressful situations and dystonic head tremor in sterol carrier protein-2 deficiency (74) or else minor febrile illnesses and brief episodes of ataxia in citrullinemia (115).
Notwithstanding, status dystonicus is a rare and life-threatening MD emergency. It has been described in the following IEMs: Wilson disease (116), Pantothenate kinase-associated neurodegeneration (117–120), aromatic l-amino acid decarboxylase (AADC) deficiency (45) and SLC6A3 dopamine transporter deficiency syndrome (121). Management is adjusted to the characteristics and complications of each patient. The use of dystonia action plans can facilitate intervention or avoid progression to dystonic state. One of these plans have been describe in detail by Allen et al. (122). If the dystonic refractory state persists despite drug therapy, early consideration of intrathecal baclofen or deep brain stimulation is required.
Finally, involuntary MD can in addition appear during treatment, for example tremor with the administration of vitamin B12.
MD Distribution
Focal MDs in IEMs are rare and are generally associated with asymmetric radiologic injury. Multifocal or segmental MDs have been described in: dystonia: PMM2-CDG (41), myoclonus: Pyridoxamine 5′-phosphate oxidase deficiency (123) and hemiplegic episodes: episodic ataxia type 6 (113) and MELAS (124). In the remaining IEMs, the distribution of the MD is usually generalized. In this context, the MD distribution is not very useful to guide the diagnosis.
On the contrary, in certain patients, the distribution can be very useful, as in the case of oromandibular dystonia noted in just a few of IEMs: cerebral creatine deficiency (43), GCDH1 dopa-responsive dystonia (48), PLA2G6-associated neurodegeneration (24), COASY-associated neurodegeneration (125), CP-aceruloplasminemia (126), fucosidosis (127), hypermanganesemia due to SLC39A14 (16), CLN3 (128), GM1 gangliosidosis (26), TIMM8A-Deafness-Dystonia-Optic Neuronopathy Syndrome (17), and biotin-thiamine responsive basal ganglia disease (54).
In a similar manner, dystonic head tremor observed in AFG3L2-spinocerebellar ataxia type 28 (129), GBA2-Autosomal recessive spastic paraplegia type 46 (130), PEX6-Peroxin 6 deficiency (131), PEX16- Peroxin 16 deficiency (132), Niemann Pick type C disease (12), NAXE-NAD(P)HX epimerase deficiency (133), TTPA-vitamin E deficiency (55), and SCP2-Sterol carrier protein-2 deficiency (74) gives an excellent clue for the diagnosis of the IEMs.
MD Temporal Pattern
In the majority of IEMs, as expected, MD presented persistently and continuously, due to this circumstances, the temporary pattern does not usually provide diagnostic benefits. In contrast, it is very useful in a restricted group of IEMs presenting with paroxysmal or intermittent MD (Table 3) as well as the monoamine neurotransmitter disorders whereby there is a very characteristic diurnal fluctuation of symptoms: motor symptoms, including MD, become more prominent in the evening and improve after sleep.
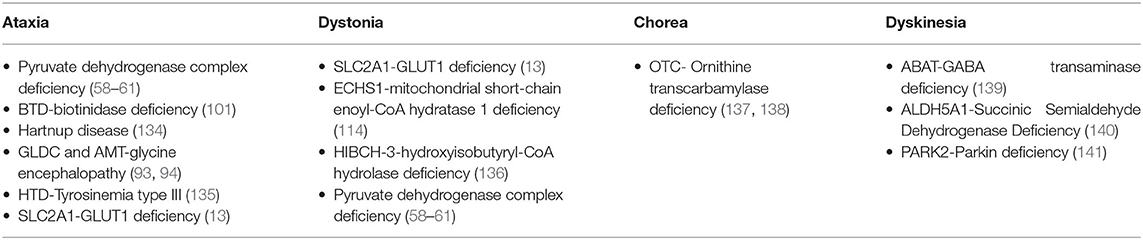
Table 3. IEMs presenting with paroxysmal MD.
Other Neurological Signs
In the second step, the neurological evaluation is completed with other signs and symptoms that can guide the diagnosis. The following signs and symptoms will be taken into account:
a. Intellectual disability or developmental delay, cognitive regression, autistic features, self-injurious behavior
b. Tone muscle and osteotendinous reflexes: hypotonia, pyramidal signs or spasticity and hypo/hyperreflexia
c. Episodic encephalopathy/coma
d. Seizures or epileptic encephalopathy
e. Microcephaly/Macrocephaly
f. Myopathy, contractures, polyneuropathy or pes cavus
g. Optic atrophy, nystagmus, ptosis, oculogyric crises, oculomotor apraxia, ectopia lentis, glaucoma, strabismus, retinitis pigmentosa, retinal degeneration, chorioretinal dystrophy, cherry red-spot, supranuclear gaze palsy, ophthalmoplegia, and cataracts
h. Sensorineural hearing loss (SNHL).
It is imperative to emphasize that the ophthalmological exploration can find numerous signs, which are ordinarily more or less specific and can exceedingly help guide the diagnosis of the child.
Non-neurological Signs
Thus, far as possible these non-neurological signs and symptoms will be considered during the evaluation of a patient with IEMs:
a. Dysmorphic or coarse facies
b. Intrauterine growth retardation (IUGR), hidrops fetalis, postnatal failure to thrive, or short stature
c. Vomiting, hepatomegaly, cirrhosis, splenomegaly, intestinal pseudo-obstruction, and pancreatitis
d. Cardiomyopathy and long QT syndrome
f. Frequent infections,
g. Renal tubular acidosis, renal failure, nephrolithiasis, nephrotic syndrome, and glomerulosclerosis
h. Other: gout, hair abnormalities (trichorrhexis nodosa), episodic hypothermia, skin (blonde hair, light-sensitive dermatitis, eczema, seborrheic dermatitis, alopecia, xanthelasmata, xanthomas, cutaneous leiomyomata, etc.), delayed teething, apneas, and skeletal abnormalities (scoliosis, osteoporosis, chondrocalcinosis, dysostosis multiplex etc.), etc.
The phenotype of the MD, as well as the neurological and non-neurological signs of each IEMs appears in the Supplementary Table 1. Some of the non-neurological symptoms are very specific to certain IEMS and may further assist in the diagnostic process.
Step 3. Biochemical Investigation and Neuroimaging
Eighty-eight percent of the 231 IEMs with MD can be detected through biochemical tests in blood, urine, feces, fibroblasts or CSF. Nevertheless, the diagnosis in the remaining IEMs will only be accomplish with the genetic testing. The tests to request are heterogeneous and will vary in line with the assumed MD and the interest in excluding certain diseases. First-tier and second-tier testing have been considered for each MD in order to prioritize the diagnosis of treatable diseases (see Table 4). In the second-tier testing, more specialized or less common biochemical analysis (enzymatic activities, porphyrins, etc.) have been included. However, some tests of the second-tier should be considered at the beginning of the diagnostic process if the suspicion of a particular disease is very high. As expected, this list of biochemical tests is broader for patients with ataxia, the most frequent MD in patients with IEMs and the one with the most possible etiologies whereas the list become more restricted for patients with myoclonus and hypokinetic-rigid syndrome.
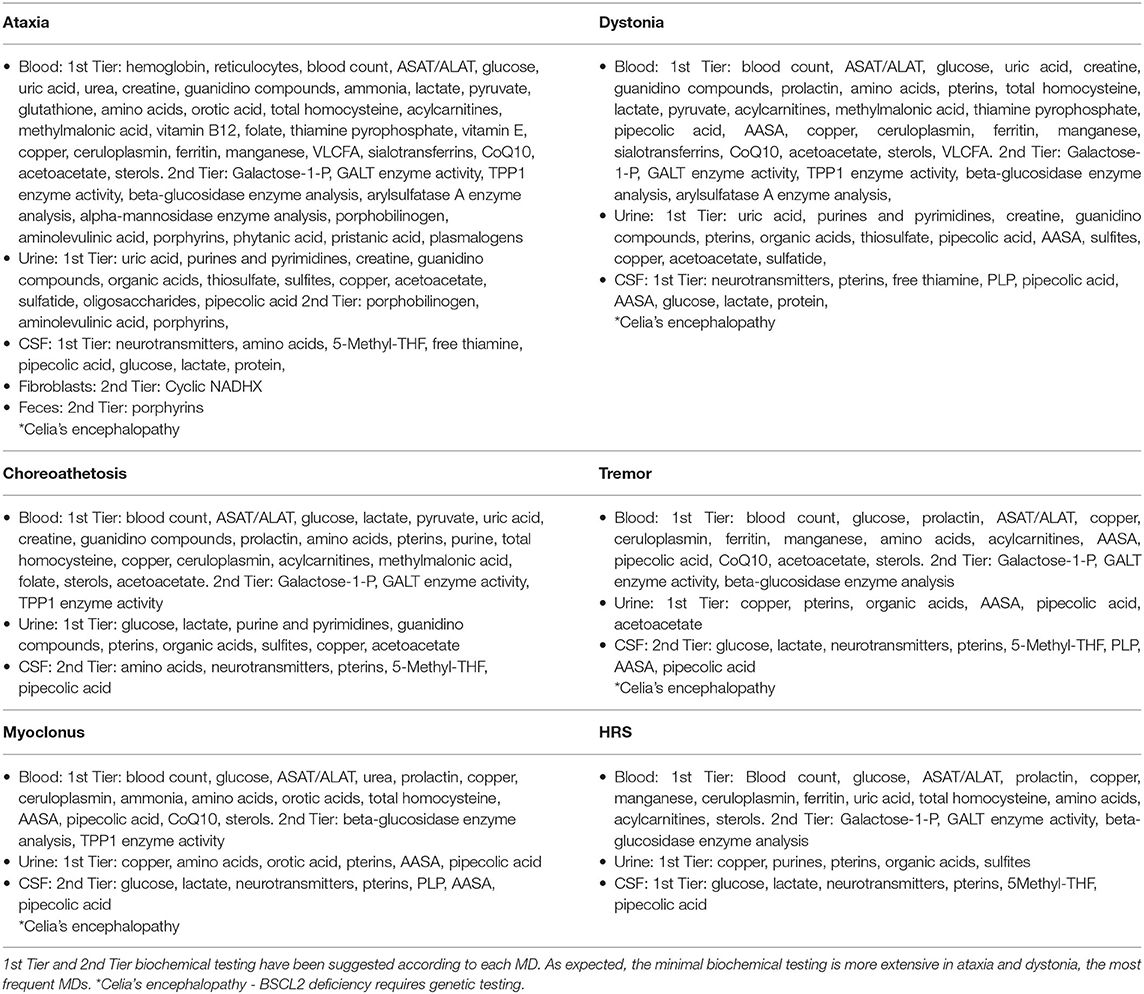
Table 4. Minimal biochemical testing to diagnose treatable IEMs based on the particular MD.
Subsequent confirmation of a definite disease may be done through additional biochemical or genetic tests. In many countries where there is difficulty in performing biochemical tests, genetic testing should be done sooner rather than later. A vital aspect in the biochemical study of a patient is to know at all times the diseases that have been ruled out with the requested tests and, therefore, to make a more thought-out plan for each patient. An alternative and sometimes controversial approach is the early performance of the lumbar puncture in a patient with MD. Lumbar puncture is regularly included in the initial evaluation of all children with MD, first and foremost when there is no speedy availability of genetic tests. This approach is correct unless there is a differential diagnostic hypothesis in which a lumbar puncture is not considered necessary. Thus, analysis of cerebrospinal fluid in IEMs presenting with MD is essential and should be considered as a 1st Tier biochemical investigation, especially in cases of ataxia, dystonia, or HRS, taking into account the need for the precise diagnosis of certain IEMs (monoamine neurotransmitter disorders, GLUT-1 deficiency, etc.) with the consequent establishment of a specific treatment (8, 13).
In this section it is necessary to comment on newborn screening programs since many patients are diagnosed now even before presenting neurological or non-neurological symptoms. The main purpose of newborn screening programs is to diagnose genetic disorders early, allowing treatment to begin before symptoms appear. The quantification of amino acids and acylcarnitines in dried blood spots (DBS) by MS/MS allows the simultaneous detection of more than 30 metabolic disorders, including those associated with amino acid, organic acid, and fatty acid metabolism. Other disorders detected using different techniques, e.g., galactosemia, biotinidase deficiency, Pompe disease, and mucopolysaccharidosis type I, are now included in some NBS programs. In a study by Navarrete et al., diagnosis of an inborn error of metabolism were confirmed in fifty-nine percent of patients tested using a personalized exome sequencing panel (142).
Along with these baseline biochemical testing, neuroimage can identify patterns of treatable inborn errors of metabolism and helps to exclude other secondary causes of MD guiding further testing. Consequently, neuroimage should be considered part of the initial diagnostic evaluation (81). In chronic MD it is essential to make a correct assessment of the neuroimaging, which generally gives many clues to the specific diagnosis. In the IEMs that cause MD, a combination of cerebral atrophy, cerebellar atrophy, white matter alteration (hypomyelination, demyelination, nonspecific lesions, or decreased volume) and basal ganglia involvement (hyperintensity, metal deposit, etc.) can be observed. Additionally, less frequently injuries in other areas like the brain stem, corpus callosum or the spinal cord or metabolic can be detected.
In the study by Cordeiro et al., basal ganglia or gray matter abnormalities with or without white matter changes were present in sixty seven percent of the patients with IEMs (3). It is of the utmost importance to keep in mind that many IEMs can present normal neuroimaging in the initial stages and exclusively progress to cerebral or cerebellar atrophy in advanced stages of the disease. Therefore, it is necessary to be careful with the interpretation of a normal brain MRI. Spectroscopy can be useful in some cases: Creatine transporter deficiency (43), SLC13A5-Epileptic encephalopathy, early infantile (143), 25, Pyruvate dehydrogenase complex deficiency (58–61), thiamine pyrophosphokinase deficiency, biotin-thiamine-responsive basal ganglia disease, mitochondrial thiamine pyrophosphate transporter deficiency (54), and many other mitochondrial disorders (Figure 4). Detection of intracerebral or basal ganglia calcification is also suggestive of certain IEMs (Table 5, Figure 3): Dihydropteridine reductase deficiency (37), folate receptor alpha deficiency, dihydrofolate reductase deficiency, hereditary folate malabsorption (144), phenylketonuria (144), Krabbe disease (145) and Aicardi-Goutières syndrome (146). Figure 3 and Figure 4 present the radiological pattern and MR Spectroscopy of some IEMs and Table 5 shows a list of IEMs with disease-specific treatment classified according to the neuroradiological pattern.
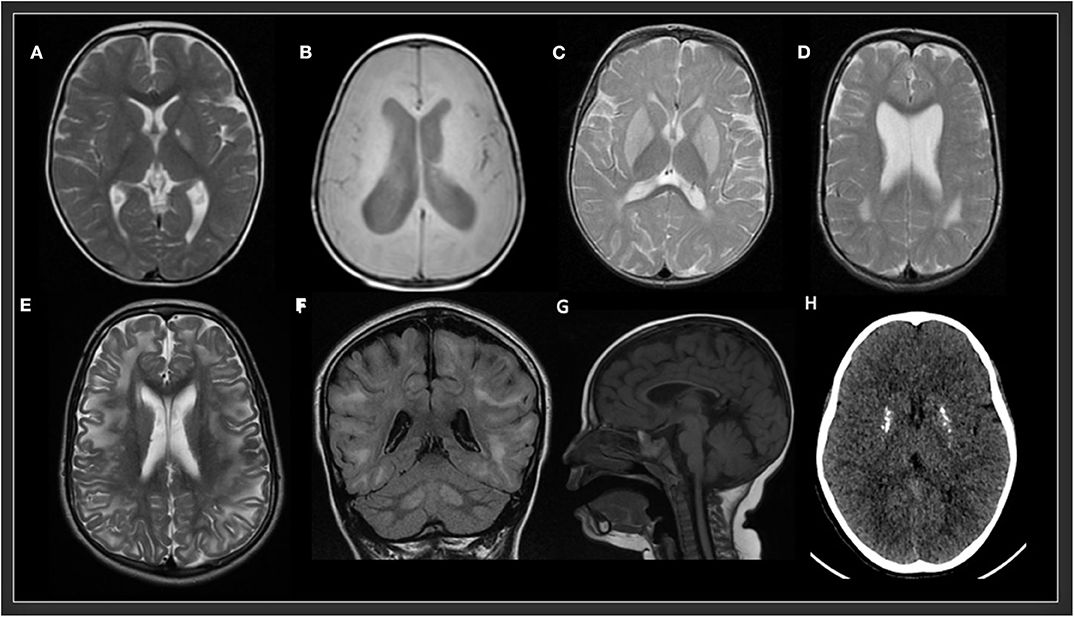
Figure 3. Radiological patterns in selected inborn errors of metabolism. (A) Mitochondrial short-chain enoyl-CoA hydratase 1 deficiency (ECHS1). MRI (T2W) showing bilateral symmetric signal hyperintensity in globus pallidus and a small cavitation in the left globus pallidus. (B) Type 3 Gaucher disease (GBA). MRI (T1W) showing decrease volume of white matter and hydrocephalus. (C) Glutaric aciduria type 1 (GCDH). MRI (T2 FSE) showing bilateral symmetric signal hyperintensity in putamen and globus pallidus and posterior periventricular white matter abnormalities. (D) Rhizomelic chondrodysplasia punctata, type 1 (PEX7). MRI (T2) showing bilateral symmetric posterior periventricular white matter hyperintensity and ventriculomegaly. (E,F) L-2-hydroxyglutaric aciduria (L2HGA). MRI (T2 and FLAIR) showing bilateral diffuse cerebral white matter and dentate nuclei hyperintensities. (G) Methylmalonic aciduria and homocystinuria, cblC type (MMACHC). MRI (T1) showing a very thin corpus callosum. (H) Aicardi-Goutières syndrome 2 (RNASEH2B). CT showing multiple calcifications in basal ganglia.
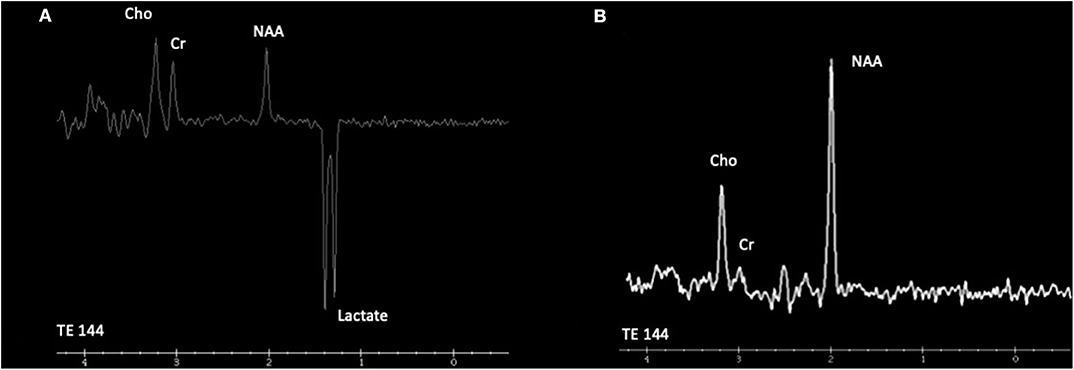
Figure 4. Cranial proton magnetic resonance spectroscopy (MRS). (A) Leigh syndrome (NDUFS4). Magnetic resonance spectroscopy using intermediate echo time (144 milliseconds) at the right caudate nuclei reveals the presence of an obvious inverted doublet of lactate at 1.33 ppm. (B) Cerebral creatine deficiency syndrome 1 (SLC6A8). Magnetic resonance spectroscopy using intermediate echo time (144 milliseconds) at the posterior periventricular white matter level reveals low creatine peak at 3.0 ppm. Cho, choline; Cr, creatine; NAA, N-acetyl aspartate.
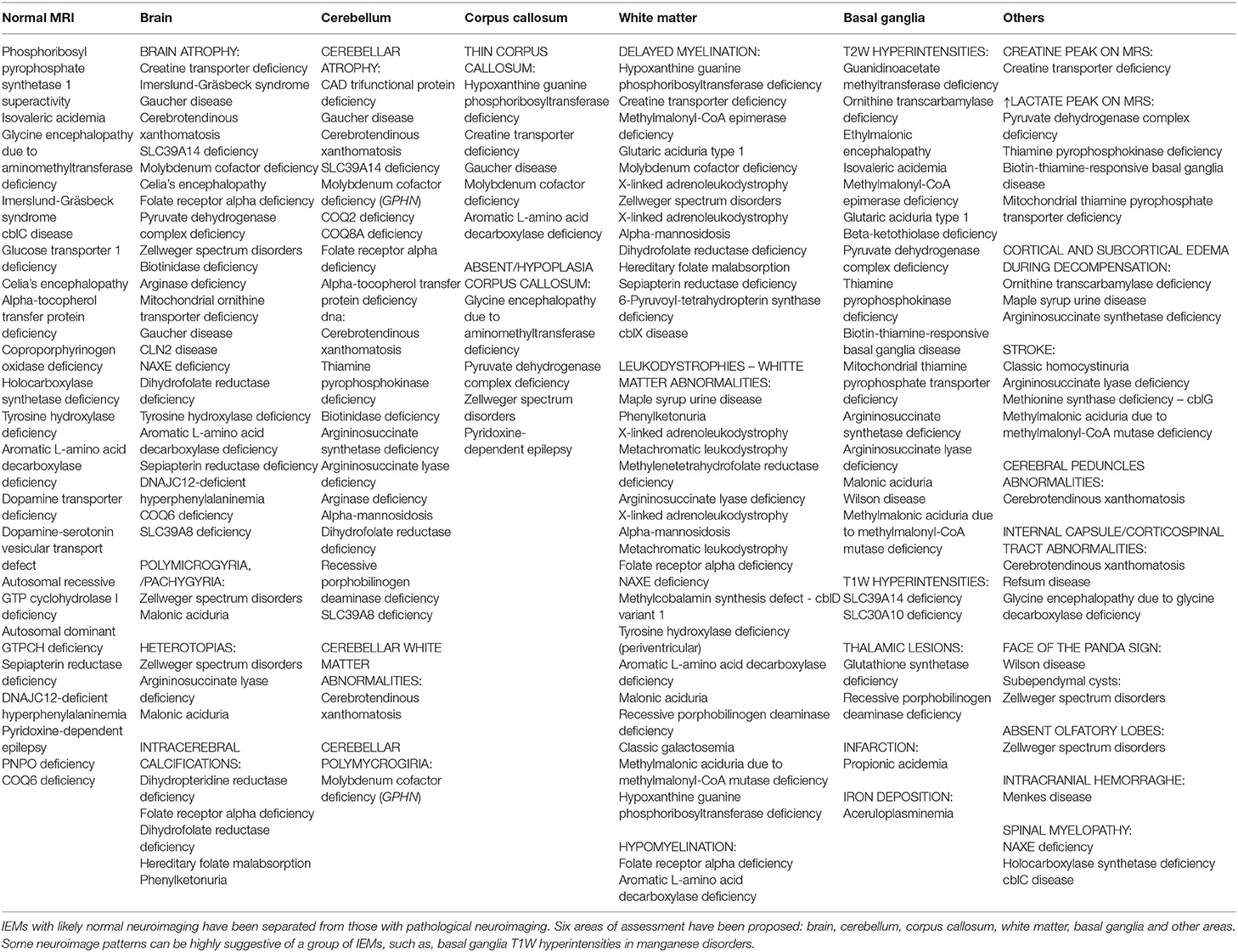
Table 5. Radiological findings in IEMs with MD.
Step 4. Genetic Investigation in IEMs
Genetic analysis is currently performed as a definitive test to confirm a diagnosis and to identify the exact gene involved. The genetic testing strategy will depend on the clinical setting and the available resources. Diagnostic yield of karyotype, chromosomal microarray or single gene sequencing (unless there is a very clear biochemical biomarker) is in many cases very low and therefore should not be indicated as the first option. In the case of IEMs and MD, the recommended testing would be targeted next generation sequencing panels or whole exome/genome sequencing (81). The diagnostic yield of the different reported studies ranged between eleven and fifty one percent (see Supplementary Table 2). In a study by Cordeiro et al., targeted next-generation sequencing panels and whole exome sequencing increased diagnostic rate to more than 40%. In the same study, a genetic diagnosis provided either disease-specific or symptomatic therapy in thirty eight percent of the patients with a genetic diagnosis, highlighting the importance of genetic investigations to confirm underlying genetic cause in patients with pediatric MD. Early detection of treatable IEM allows for referral to expert centers and timely interventions to prevent disease progression and to improve neurological functioning. Additionally, detection of IEM for which no treatment currently exists allows for genetic counseling of the affected families (3).
Step 5. Treatment of Pediatric Metabolic Movement Disorders
MDs are a major medical problem. Treatment of children with IEMs-associated MD may include both symptomatic treatment of MDs and disease-specific management of the underlying IEM. Pharmacologic treatments for IEMs may comprise specialized dietary modifications, cofactor, vitamins, or supplements addition, enzyme replacement therapy, hematopoietic stem cell transplantation, and gene therapy (81). Successful treatment and better outcome frequent take place with early recognition of an underlying treatable IEM, therefore there should be an increased consciousness of these IEMs (147). A complete list of the specific treatments of each IEMs appears in Supplementary Table 1. Regrettably, only thirty-eight percent of the IEMs included in this article have a disease-specific treatment. Although, over the last decades, new treatment approaches have changed the scope of IEMs from a group of rare, untreatable, and often fatal disorders to an important cause of potentially treatable diseases.
By contrast, when disease modification therapies are not possible, symptomatic therapy may be the only treatment that is available. Symptomatic therapy of MD requires multidisciplinary approach ranging from avoiding identifiable triggers, pharmacologic interventions, physiotherapy, to intrathecal baclofen pump, deep brain stimulation and gene therapy (3). Koy et al. has reviewed prodigiously the management of MD including dose range and guidelines for drugs commonly used to treat pediatric MD (148). The reader may refer to this review for more information. Even with treatment, MD hardly resolve completely and can cause life-long disability. Nevertheless, symptomatic treatment may improve the functional abilities and quality of life, correct orthopedic complications or relieve pain of children with generalized dystonia.
Recently, Mohammad et al., outlined a treatment approach for children with MD, including IEMs disorders. In this approach, phenomenology of MD, associated comorbidities (in particular intellectual disability and psychiatric disorders), etiology (and natural history of the disorder), level of functional impairment, and risk of a definite therapy are determined sequentially before making a treatment decision. As in any other MD, the administration of quality of life scales or scales for the specific MD is recommended, in order to have a useful measure of the effect of the treatment. The risk of a given treatment should always be considered before starting, since the adverse effects of the treatment may be worse than the MD itself. Providentially, side effects are transient and reversible for most treatments (149).
Keeping up to date with the emergence of new gene therapies is essential. Gene therapy trials using viral vectors results in improvement of MD and motor function in early clinical studies of AADC deficiency (150, 151).
Kojima et al., conducted an open-label phase 1/2 study including six patients whom received adeno-associated virus vector harboring DDC via bilateral intraputaminal infusions. Five of these patients presented a severe phenotype while the remaining patient presented a moderate phenotype. At follow-up for up to 2 years, motor function improved markedly in all patients. They were able to stand with support to run or ride a bicycle. The treatment was more effective in the younger patients. Cognitive and verbal functions were also improved in the patient with the moderate phenotype. Clinical signs such as dystonia and oculogyric seizures disappeared or decreased markedly. As an adverse effect, transient chorea was observed (150). Tseng et al., demonstrated that there was an improvement in the microstructural integrity of white matter tracts using Brain diffusion tensor imaging (DTI) 1 year after AADC gene therapy (151).
In the same way, enzyme-replacement therapy for CNL2 have resulted in significant less decline in motor and language function than that in historical controls although with serious adverse events comprising failure of the intraventricular device and device-related infections (152).
For many patients, medical management alone is insufficient. Hence, many of these drug-refractory patients experience significant symptomatic improvement after baclofen intrathecal infusion pump or deep brain stimulation (DBS) following an appropriate selection process. Intrathecal baclofen pump has been indicated in several patients with spasticity or severe dystonia and progressive IEMs, in some cases for palliative purposes, for example: Niemann Pick disease type C (153), Sjögren-Larsson syndrome (154), Metachromatic leukodystrophy (155), and Gaucher disease type 2 (156). Regarding DBS, Supplementary Table 4 describes the role and evidence of DBS in selected IEMs and childhood-onset MD. Twenty-five articles were included with fifty-four patients who underwent DBS surgery. The mean age of surgery was 12.2 ± 4.6 years (range: 1–18 y) and the most frequent targets were internal globus pallidus (19 studies) and subthalamic nucleus (5 studies) (157–181). In summary, DBS can be recommended for treatment of dystonia in pantothenate kinase associated neurodegeneration or Lesch–Nyhan syndrome (182). Patients with pantothenate kinase associated neurodegeneration repeatedly respond well to DBS even if there may not be sustained at long-term effect because of ongoing neurodegeneration in the pallidum. While DBS appears effective in Lesch–Nyhan syndrome patients both in reducing dystonia and self-mutilating behavior. No beneficial effect was observed in the following IEMs: X-linked adrenoleukodystrophy, methylmalonic aciduria, mitochondrial disorder and glutaric aciduria type 1.
Step 6. Reevaluate Treatable IEMs
IEMs presenting with prominent MD can be noticed in Table 1. It is of fundamental importance to reevaluate these treatable IEMs and ensure that they have been completely discarded, both biochemically and genetically. This is a process that must be performed consistently in all patients without diagnosis. Finally, other genetic or acquired causes that could explain the movement disorders will have to be investigated.
Future Directions
Further collaborative investigations are needed in which an elaborated description of the MD is made, including the largest cohorts possible. It would also be beneficial if more research was done on the application of intrathecal baclofen pump and deep brain stimulation in IEMs. The future is very promising, both in new diagnostic techniques in particular metabolomics and in new gene therapies. Metabolomics can be described as a complete and simultaneous analysis of small molecules in a biological sample. The aims of this analysis are to improve the diagnosis, understand the factors that contribute to the progression and susceptibility of a certain disease, and monitor the long-term metabolic effects of therapies. The scope and usefulness of this approach in diagnosing IEMs is expected to drastically change in the near future and it is very likely that they will be included in upcoming diagnostic algorithms (183).
Conclusion
This review proposes a straightforward six-step algorithm to IEMs presenting with MD in childhood. The major components of this algorithm include the initial suspicion on an IEM, the characterization of the MD phenotype and other neurological and non-neurological signs, biochemical, genetic and imaging testing based on the type of MD. Finally, an update of the treatable IEMs and the therapies available for the MD is made. The application of this algorithm will potentially allow clinicians to avoid blind and poorly efficacious explorations or biological test.
Author Contributions
JDO-E wrote and reviewed the final manuscript and provide data for all tables and figures.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2020.582160/full#supplementary-material
References
1. Ferreira C, van Karnebeek C, Vockley J, Blau N. A proposed nosology of inborn errors of metabolism. Genet Med. (2019) 21:102–6. doi: 10.1038/s41436-018-0022-8
2. Eggink H, Kuiper A, Peall K, Contarino M, Bosh A, Post B, et al. Rare inborn errors of metabolism with movement disorders: a case study to evaluate the impact upon quality of life and adaptive functioning. Orphanet J Rare Dis. (2014) 9:177. doi: 10.1186/s13023-014-0177-6
3. Cordeiro D, Bullivant G, Siriwardena K, Evans A, Kovayashi J, Cohn R, et al. Genetic landscape of pediatric movement disorders and management implications. Neurol Genet. (2018) 4:e265. doi: 10.1212/NXG.0000000000000265
4. Sedel F, Saudubray JM, Roze E, Agid Y, Vidailhet M. Movement disorders and inborn errors of metabolism in adults: a diagnostic approach. J Inherit Metab Dis. (2008) 31:308–18. doi: 10.1007/s10545-008-0854-5
5. Ferreira C, Hoffmann G, Blau N. Clinical and biochemical footprints of inherited metabolic diseases. I. Movement disorders. Mol Genet Metab. (2019) 127:28–30. doi: 10.1016/j.ymgme.2019.03.007
6. Sanger T, Chen D, Fehlings D, Hallett M, Lang A, Mink J, et al. Definition and classification of hyperkinetic movements in childhood. Mov Disord. (2010) 25:1538–49. doi: 10.1002/mds.23088
7. Garcia-Cazorla A, Ortez C, Pérez-Dueñas B, Serrano M, Pineda M, Campistol J, et al. Hypokinetic-rigid syndrome in children and inborn errors of metabolism. Eur J Paediatr Neurol. (2011) 15:295–302. doi: 10.1016/j.ejpn.2011.04.013
8. Ng J, Papandreou A, Heales S, Kurian M. Monoamine neurotransmitter disorders–Clinical advances and future perspectives. Nat Rev Neurol. (2015) 11:567–84. doi: 10.1038/nrneurol.2015.172
9. Stelten B, van de Warrenburg B, Wevers R, Verrips A. Movement disorders in cerebrotendinous xanthomatosis. Parkinsonism Relat Disord. (2019) 58:12–6. doi: 10.1016/j.parkreldis.2018.07.006
10. Weiss KH. Wilson Disease. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (1999). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1512/
11. Cohen BH, Chinnery PF, Copeland WC. POLG-Related Disorders. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (2010). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK26471/
12. Patterson M. Niemann-pick disease type C. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2000). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1296/
13. Wang D, Pascual JM, De Vivo D. Glucose transporter type 1 deficiency syndrome. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2002). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1430/
14. Gregory A, Hayflick SJ. Pantothenate kinase-associated neurodegeneration. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2002). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1490/
15. Rattay T, Lindig T, Baets J, Smets K, Deconick T, Söhn A, et al. FAHN/SPG35: a Narrow phenotypic spectrum across disease classifications. Brain. (2019) 142:1561–72. doi: 10.1093/brain/awz102
16. Tuschl K, Meyer E, Valdivia L, Zhao N, Dadswell C, Abdul-Sada A, et al. Mutations in sLC39A14 disrupt manganese homeostasis and cause childhood-Onset parkinsonism-Dystonia. Nat Commun. (2016) 7:11601. doi: 10.1038/ncomms11601
17. Tranebjærg L. Deafness-dystonia-optic neuronopathy syndrome. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2003). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1216/
18. Terraciano A, Renaldo F, Zanni G, D'Amico A, Pastore A, Barresi S, et al. The use of muscle biopsy in the diagnosis of undefined ataxia with cerebellar atrophy in children. Eur J Paediatr Neurol. (2012) 16-222:248–56. doi: 10.1016/j.ejpn.2011.07.016
19. Kellner U, Stöhr H, Weinitz S, Farmand G, Weber B. Mevalonate kinase deficiency associated with ataxia and retinitis pigmentosa in two brothers with mVK gene mutations. Ophthalmic Genet. (2017) 38:340–4. doi: 10.1080/13816810.2016.1227459
20. Bryant D, Lui Y, Datta S, Hariri H, Seda M, Anderson G, et al. SNX14 mutations affect endoplasmic reticulum associated neutral lipid metabolism in autosomal recessive spinocerebellar ataxia 20. Hum Mol Genet. (2018) 27:1927–40. doi: 10.1093/hmg/ddy101
21. Kim Y, Yum M, Jeong J, Choi S, Kim S, Yoon W, et al. A mild form of cOG5 defect showing early-Childhood-Onset friedreich's-Ataxia-Like phenotypes with isolated cerebellar atrophy. J Korean Med Sci. (2017) 32:1885–90. doi: 10.3346/jkms.2017.32.11.1885
22. Tranchant C, Anheim M. Movement disorders in mitochondrial diseases. Rev Neurol (Paris). (2016) 172:524–9. doi: 10.1016/j.neurol.2016.07.003
23. Mink J, Augustine E, Adams H, Marshall F, Kwon J. Classification and natural history of the neuronal ceroid lipofuscinoses. J Child Neurol. (2013) 28:1101–5. doi: 10.1177/0883073813494268
24. Darling A, Aguilera-Albesa S, Tello C, Serrano M, Tomás M, Camino-León R, et al. PLA2G6-associated neurodegeneration: new insights into brain abnormalities and disease progression. Parkinsonism Relat Disord. (2019) 61:179–86. doi: 10.1016/j.parkreldis.2018.10.013
25. Hoffjan S, Ibisler A, Tschentscher A, Dekomien G, Bidinost C, Rosa A. WDR45 mutations in rett (-Like) syndrome and developmental delay: case report and an appraisal of the literature. Mol Cell Probes. (2016) 30:44–9. doi: 10.1016/j.mcp.2016.01.003
26. Ferreira C, Gahl W. Lysosomal storage diseases. Transl Sci Rare Dis. (2017) 2:1–71. doi: 10.3233/TRD-160005
27. Steenweg M, Jakobs C, Errami A, van Dooren S, Bartolomé M, Aerssens P, et al. An overview of l-2-hydroxyglutarate dehydrogenase gene (L2HGDH) variants: a Genotype-Phenotype study. Hum Mutat. (2010) 31:380–90. doi: 10.1002/humu.21197
28. Gouider-Khouka N, Kraoua I, Benrhouma H, Fraj N, Rouissi A. Movement disorders in neuro-Metabolic diseases. Eur J Paediatr Neurol. (2010) 14:304–7. doi: 10.1016/j.ejpn.2009.11.005
29. van Egmond M, Lugtenberg C, Brouwer O, Contarino M, Fung V, Heiner-Fokkema R, et al. A post hoc study on gene panel analysis for the diagnosis of dystonia. Mov Disord. (2017) 32:569–75. doi: 10.1002/mds.26937
30. Neveling K, Feenstra I, Gilissen C, Hoefsloot L, Kamsteeg E, Mesenkamp A, et al. A post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat. (2013) 34:1721–6. doi: 10.1002/humu.22450
31. Montaut S, Tranchant C, Drouot N, Rudolf G, Guissart C, Tarabeux J, et al. Assessment of a targeted gene panel for identification of genes associated with movement disorders. JAMA Neurol. (2018) 75:1234–45. doi: 10.1001/jamaneurol.2018.1478
32. Reale C, Panteghini C, Carecchio M, Garavaglia B. The relevance of gene panels in movement disorders diagnosis: a lab perspective. Eur J Paediatr Neurol. (2018) 22:285–91. doi: 10.1016/j.ejpn.2018.01.013
33. Graziola F, Garone G, Stregapede F, Bosco L, Vigevano F, Curatolo P, et al. Diagnostic yield of a targeted next-Generation sequencing gene panel for pediatric-Onset movement disorders: a 3-Year cohort study. Front Genet. (2019) 29:10:1026. doi: 10.3389/fgene.2019.01026
34. Raucci U, Parisi P, Vanacore N, Garone G, Bondone C, Palmieri A, et al. Acute hyperkinetic movement disorders in italian paediatric emergency departments. Arch Dis Child. (2018) 103:790–94. doi: 10.1136/archdischild-2017-314464
35. Goraya J. Acute movement disorders in children: experience from a developing country. J Child Neurol. (2015) 30:406–11. doi: 10.1177/0883073814550828
36. Dale R, Singh H, Troedson C, Pillai S, Gaikiwari S, Kozlowaka K. A prospective study of acute movement disorders in children. Dev Med Child Neurol. (2010) 52:739–48. doi: 10.1111/j.1469-8749.2009.03598.x
37. Opladen T, López-Laso E, Cortès-Saladelafont E, Pearson T, Serap Sivri H, Yildiz Y, et al. Consensus guideline for the diagnosis and treatment of tetrahydrobiopterin (BH4) deficiencies. Orphanet J Rare Dis. (2020) 15:126. doi: 10.1186/s13023-020-01379-8
38. Huemer M, Diodato D, Schwahn B, Schiff M, Bandeira A, Benoist JF, et al. Guidelines for diagnosis and management of the cobalamin-related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ and mTHFR deficiency. J Inherit Metab Dis. (2017) 40:21–48. doi: 10.1007/s10545-016-9991-4
39. Maas R, Iwanicka-Pronicka K, Ucar S, Alhaddad B, AlSayed M, Al-Owain M, et al. Progressive deafness-Dystonia due to sERAC1 mutations: a Study of 67 cases. Ann Neurol. (2017) 82:1004–15. doi: 10.1002/ana.25110
40. Ebrahimi-Fakhari D, Hildebrandt C, Davis P, Rodan L, Anselm I, Bodamer O. The spectrum of movement disorders in childhood-onset lysosomal storage diseases. Mov Disord Clin Pract. (2017) 5:149–55. doi: 10.1002/mdc3.12573
41. Mostile G, Barone R, Nicoletti A, Rizzo R, Martinelli D, Sturiale L, et al. Hyperkinetic movement disorders in congenital disorders of glycosylation. Eur J Neurol. (2019) 26:1226–34. doi: 10.1111/ene.14007
42. Dhar SU, Scaglia F, Li FY, Smith L, Barshop BA, Eng CM, et al. Expanded clinical and molecular spectrum of guanidinoacetate methyltransferase (GAMT) deficiency. Mol Genet Metab. (2009) 96:38–43. doi: 10.1016/j.ymgme.2008.10.008
43. Anselm I, Alkuraya FS, Salomons GS, Jakobs C, Fulton AB, Mazumdar M, et al. X-linked creatine transporter defect: a Report on two unrelated boys with a severe clinical phenotype. J Inherit Metab Dis. (2006) 29:214–9. doi: 10.1007/s10545-006-0123-4
44. Furukawa Y, Kish S. Tyrosine hydroxylase deficiency. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2008). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1437/
45. Wassenberg T, Molero-Luis M, Jeltsch K, Hoffmann GF, Assmann B, Blau N, et al. Consensus guideline for the diagnosis and treatment of aromatic l-amino acid decarboxylase (AADC) deficiency. Orphanet J Rare Dis. (2017) 12:12. doi: 10.1186/s13023-016-0522-z
46. Kurian MA. SLC6A3-related dopamine transporter deficiency syndrome. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2006). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK442323/
47. Rilstone JJ, Alkhater RA, Minassian BA. Brain dopamine-Serotonin vesicular transport disease and its treatment. N Engl J Med. (2013) 368:543–50. doi: 10.1056/NEJMoa1207281
48. Furukawa Y. GTP Cyclohydrolase 1-deficient dopa-responsive dystonia. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2002). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1508/
49. Friedman J. Sepiapterin reductase deficiency. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2015). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK304122/
50. Anikster Y, Haack TB, Vilboux T, Pode-Shakked B, Thöny B, Shen N, et al. Biallelic mutations in dNAJC12 cause hyperphenylalaninemia, dystonia, intellectual disability. Am J Hum Genet. (2017) 100:257–66. doi: 10.1016/j.ajhg.2017.01.002
51. Olpin SE, Pollitt RJ, McMenamin J, Manning NJ, Besley G, Ruiter JP, et al. 2-methyl-3-hydroxybutyryl-CoA Dehydrogenase Deficiency in a 23year-old Man. J Inherit Metab Dis. (2002) 25:477–82. doi: 10.1023/a:1021251202287
52. Larson A, Goodman S. Glutaric Acidemia Type 1. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2019). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK546575/
53. Pope S, Artuch R, Heales S, Rahman S. Cerebral folate deficiency: analytical tests and differential diagnosis. J Inherit Metab Dis. (2019) 42:655–72. doi: 10.1002/jimd.12092
54. Ortigoza-Escobar JD, Alfadhel M, Molero-Luis M, Darin N, Spiegel R, de Coo IF, et al. Thiamine deficiency in childhood with attention to genetic causes: survival and outcome predictors. Ann Neurol. (2017) 82:317–30. doi: 10.1002/ana.24998
55. Schuelke M. Ataxia with vitamin E deficiency. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2005). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1241/
56. Tuschl K, Clayton PT, Gospe SM Jr, Gulab S, Ibrahim S, Singhi P, et al. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in sLC30A10, a manganese transporter in man. Am J Hum Genet. (2012) 90:457–66. doi: 10.1016/j.ajhg.2012.01.018
57. Wang D, De Vivo D. Pyruvate carboxylase deficiency. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2009). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK6852/
58. Castiglioni C, Verrigni D, Okuma C, Diaz A, Alvarez K, Rizza T, et al. Pyruvate dehydrogenase deficiency presenting as isolated paroxysmal exercise induced dystonia successfully reversed with thiamine supplementation. Case report and mini-review. Eur J Paediatr Neurol. (2015) 19:497–503. doi: 10.1016/j.ejpn.2015.04.008
59. Debray F, Lambert M, Gagne R, Maranda B, Laframboise R, MacKay N, et al. Pyruvate dehydrogenase deficiency presenting as intermittent isolated acute ataxia. Neuropediatrics. (2008) 39:20–3. doi: 10.1055/s-2008-1077084
60. Mellick G, Price L, Boyle R. Late-Onset presentation of pyruvate dehydrogenase deficiency. Mov Disord. (2004) 19:727–9. doi: 10.1002/mds.20063
61. Head RA, de Goede CG, Newton RW, Walter JH, McShane MA, Brown RM, et al. (2004) Pyruvate dehydrogenase deficiency presenting as dystonia in childhood. Dev Med Child Neurol. (2004) 46:710–2. doi: 10.1017/s0012162204001197
62. Carrozzo R, Verrigni D, Rasmussen M, de Coo R, Amartino H, Bianchi M, et al. Succinate-CoA ligase deficiency due to mutations in sUCLA2 and sUCLG1: phenotype and genotype correlations in 71 patients. J Inherit Metab Dis. (2016) 39:243–52. doi: 10.1007/s10545-015-9894-9
63. Balaji P, Viswanathan V, Chellathurai A, Panigrahi D. An interesting case of metabolic dystonia: l-2 hydroxyglutaric aciduria. Ann Indian Acad Neurol. (2014) 17:97. doi: 10.4103/0972-2327.128565
64. Melone MA, Tessa A, Petrini S, Lus G, Sampaolo S, di Fede G, et al. Revelation of a new mitochondrial DNA mutation (G12147A) in a MELAS/MERFF phenotype. Arch Neurol. (2004) 61:269–72. doi: 10.1001/archneur.61.2.269
65. Thorburn DR, Rahman J, Rahman S. Mitochondrial DNA-associated leigh syndrome and NARP. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2003). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1173/
66. Ucar SK, Mayr JA, Feichtinger RG, Canda E, Çoker M, Wortmann SB. Previously unreported biallelic mutation in dNAJC19: are sensorineural hearing loss and basal ganglia lesions additional features of dilated cardiomyopathy and ataxia (DCMA) syndrome? JIMD Rep. (2017) 35:39–45. doi: 10.1007/8904_2016_23
67. Wortmann SB, Wevers RA, de Brouwer APM. CLPB Deficiency. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2016). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK396257/
68. Vill K, Müller-Felber W, Gläser D, Kuhn M, Teusch V, Schreiber H, et al. SACS variants are a relevant cause of autosomal recessive hereditary motor and sensory neuropathy. Hum Genet. (2018) 137:911–9. doi: 10.1007/s00439-018-1952-6
69. Heimer G, Gregory A, Hogarth P, MECR-related neurologic disorder. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2006). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK540959/
70. Ozaki K, Doi H, Mitsui J, Sato N, Iikuni Y, Majima T, et al. A novel mutation in eLOVL4 leading to spinocerebellar ataxia (SCA) with the hot cross bun sign but lacking erythrokeratodermia: a Broadened spectrum of sCA34. JAMA Neurol. (2015) 72:797–805. doi: 10.1001/jamaneurol.2015.0610
71. Gowda VK, Srinivasan VM, Benakappa N, Benakappa A. Sialidosis type 1 with a novel mutation in the neuraminidase-1 (NEU1) gene. Indian J Pediatr. (2017) 84:403–4. doi: 10.1007/s12098-016-2286-9
72. Pattel MS, Callahan JW, Zhang S, Chan AK, Unger S, Levin AV, et al. Early-infantile galactosialidosis: prenatal presentation and postnatal follow-Up. Am J Med Genet. (1999) 85:38–47.
73. Rubboli G, Franceschetti S, Berkovic SF, Canafoglia L, Gambardella A, Dibbens LM, et al. Clinical and neurophysiologic features of progressive myoclonus epilepsy without renal failure caused by sCARB2 mutations. Epilepsia. (2011) 52:2356–63. doi: 10.1111/j.1528-1167.2011.03307.x
74. Ferdinandusse S, Kostopoulos P, Denis S, Rusch H, Overmars H, Dillmann U„, et al. Mutations in the gene encoding peroxisomal sterol carrier protein x (SCPx) cause leukencephalopathy with dystonia and motor neuropathy. Am J Hum Genet. (2006) 78:1046–52. doi: 10.1086/503921
75. Gordon-Lipkin E, Cohen JS, Srivastava S, Soares BP, Levey E, Fatemi A. ST3GAL5-Related disorders: a Deficiency in ganglioside metabolism and a genetic cause of intellectual disability and choreoathetosis. J Child Neurol. (2018) 33:825–31. doi: 10.1177/0883073818791099
76. Simpson MA, Cross H, Proukakis C, Priestman DA, Neville DC, Reinkensmeier G, et al. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-Of-Function mutation of gM3 synthase. Nat Genet. (2004) 36:1225–9. doi: 10.1038/ng1460
77. Kasapkara CS, Tümer L, Ezgü FS, Hasanoglu A, Race V, Matthijs G, et al. SRD5A3-CDG: a patient with a novel mutation. Eur J Paediatr Neurol. (2012) 16:554–6. doi: 10.1016/j.ejpn.2011.12.011
78. Dibbens L, Rubboli G. GOSR2: a Progressive myoclonus epilepsy gene. Epileptic Disord. (2017) 18:111–4. doi: 10.1684/epd.2016.0848
79. Lam C, Ferreira C, Krasnewich D, Toro C, Latham L, Zein WM, et al. Prospective phenotyping of nGLY1-CDDG, the first congenital disorder of deglycosylation. Genet Med. (2017) 19:160–8. doi: 10.1038/gim.2016.75
80. Leach E, Shevell M, Bowden K, Stockler-Ipsiroglu, van Karnebeek C. Treatable inborn errors of metabolism presenting as cerebral palsy mimics: systematic literature review. Orphanet J Rare Dis. (2014) 9:197. doi: 10.1186/s13023-014-0197-2
81. Ebrahimi-Fakhari D, van Karnebeek C, Münchau A. Movement disorders in treatable inborn errors of metabolism. Mov Disord. (2019) 34:598–613. doi: 10.1002/mds.27568
82. Brandsma R, Verschuuren-Bemelmans CC, Amrom D, Barisic N, Baxter O, Bertini E, et al. A clinical diagnostic algorithm for early onset cerebellar ataxia. Eur J Paediatr Neurol. (2019) 23:692–706. doi: 10.1016/j.ejpn.2019.08.004
83. Jinnah H, Lewis RF, Visser JE, Eddey GE, Barabas G, Harris JC. Ocular motor dysfunction in lesch-Nyhan disease. Pediatr Neurol. (2001) 24:200–4. doi: 10.1016/s0887-8994(00)00265-4
84. Ribeiro E, Pizarro M, Oliveira L, Caldas de Amorin R, Pinheiro T, Grieben U, et al. Juvenile neuronal ceroid-Lipofuscinosis: clinical and molecular investigation in a large family in brazil. Arq Neuropsiquiatr. (2011) 69:13–8. doi: 10.1590/s0004-282x2011000100004
85. Pellecchia M, Valente EM, Cif L, Salvi S, Albanese A, Scarano V, et al. The diverse phenotype and genotype of pantothenate kinase-associated neurodegeneration. Neurology. (2005) 64:1810–2. doi: 10.1212/01.WNL.0000161843.52641.EC
86. Goez H, Jakob F, Yager J. Lingual dyskinesia and tics: a Novel presentation of copper-Metabolism disorder. Pediatrics. (2011) 127:e505–8. doi: 10.1542/peds.2010-2391
87. Kurt B, Jaeken J, van Hove J, Lagae L, Löfgren A, Everman D, et al. A novel pOLG gene mutation in 4 children with alpers-like hepatocerebral syndromes. Arch Neurol. (2010) 67:239–44. doi: 10.1001/archneurol.2009.332
88. Sedel F, Friderici K, Nummy K, Caillaud C, Chabli A, Dürr A, et al. Atypical gilles de la tourette syndrome with β-Mannosidase deficiency. Arch Neurol. (2006) 63:129–31. doi: 10.1001/archneur.63.1.129
89. Dusi S, Valletta L, Haack TB, Tsuchiya Y, Venco P, Pasqualato S, et al. Exome sequence reveals mutations in coA synthase as a cause of neurodegeneration with brain iron accumulation. Am J Hum Genet. (2014) 94:11–22. doi: 10.1016/j.ajhg.2013.11.008
90. Carecchio M, Schneider S, Chan H, Lachmann R, Lee P, Murphy E, et al. Movement disorders in adult surviving patients with maple syrup urine disease. Mov Disord. (2011) 26:1324–8. doi: 10.1002/mds.23629
91. Ito D. BSCL2-related neurologic disorders/seipinopathy. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle (2005). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1307/
92. Kuchar L, Ledvinová J, Hrebícek M, Mysková H, Dvoráková L, Berná L, et al. Prosaposin deficiency and saposin b deficiency (activator-deficient metachromatic leukodystrophy): report on two patients detected by analysis of urinary sphingolipids and carrying novel pSAP gene mutations. Am J Med Genet A. (2009) 149ª:613–21. doi: 10.1002/ajmg.a.32712
93. Coughlin C, Swanson MA, Kronquist K, Acquaviva C, Hutchin T, Rodríguez-Pombo P, et al. The genetic basis of classic nonketotic hyperglycinemia due to mutations in GLDC and AMT. Genet Med. (2017) 19:104–11. doi: 10.1038/gim.2016.74
94. Kanekar S, Byler D. Characteristic mRI findings in neonatal nonketotic hyperglycinemia due to sequence changes in gLDC gene encoding the enzyme glycine decarboxylase. Metab Brain Dis. (2013) 28:717–20. doi: 10.1007/s11011-013-9415-1
95. Bindu PS, Nagappa M, Bharath RD. Isolated sulfite oxidase deficiency. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2017). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK453433/
96. Jurecka A, Zikanova M, Kmoch S, Tylki-Szymańska A. Adenylosuccinate lyase deficiency. J Inherit Metab Dis. (2015) 38:231–42. doi: 10.1007/s10545-014-9755-y
97. Ortez C, Jou C, Cortès-Saladelafont E, Moreno J, Pérez A, Ormazábal A, et al. Infantile parkinsonism and gABAergic hypotransmission in a patient with pyruvate carboxylase deficiency. Gene. (2013) 532:302–6. doi: 10.1016/j.gene.2013.08.036
98. Kovacs-Nagy R, Morin G, Nouri MA, Brandau O, Saadi NW, Nouri MA, et al. HTRA2 defect: a Recognizable inborn error of metabolism with 3-Methylglutaconic aciduria as discriminating feature characterized by neonatal movement disorder and epilepsy-Report of 11 patients. Neuropediatrics. (2018) 49:373–8. doi: 10.1055/s-0038-1667345
99. Jurecki E, Ueda K, Rohr F, Thompson A, Hussa C, Obernolte L, et al. Nutrition management guideline for propionic acidemia: an evidence- and consensus-Based approach. Mol Genet Metab. (2019) 126:341–54. doi: 10.1016/j.ymgme.2019.02.007
100. Bikker H, Bakker HD, Abeling NG, Poll-The BT, Kleijer WJ, Rosenblatt DS. A homozygous nonsense mutation in the methylmalonyl-CoA epimerase gene (MCEE) results in mild methylmalonic aciduria. Hum Mutat. (2006) 27:640–3. doi: 10.1002/humu.20373
101. Wolf B. Biotinidase Deficiency. 2000 Mar 24 [Updated 2016 Jun 9]. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2019). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1322/
102. Riley LG, Cowley MJ, Gayevskiy V, Roscioli T, Thorburn DR, Prelog K, et al. A sLC39A8 variant causes manganese deficiency, and glycosylation and mitochondrial disorders. J Inherit Metab Dis. (2017) 40:261–9. doi: 10.1007/s10545-016-0010-6
103. Wortmann SB, Kremer BH, Graham A, Willemsen MA, Loupatty FJ, Hogg SL, et al. 3-methylglutaconic aciduria type i redefined: a syndrome with late-onset leukoencephalopathy. Neurology. (2010) 75:1079–83. doi: 10.1212/WNL.0b013e3181f39a8a
104. Mérenet A, Wiame E, Marelli C, Lenglet T, Van Schaftingen E, Sedel F. A serine synthesis defect presenting with a charcot-Marie-Tooth-like polyneuropathy. Arch Neurol. (2012) 69:908–11. doi: 10.1001/archneurol.2011.1526
105. Almusafri F, Elamin HE, Khalaf TE, Ali A, Ben-Omran T, El-Hattab AW. Clinical, and molecular characterization of 6 children with glutamate-cysteine ligase deficiency causing hemolytic anemia. Blood Cells Mol Dis. (2017) 65:73–7. doi: 10.1016/j.bcmd.2017.05.011
106. Liskova P, Ulmanova O, Tesina P, Melsova H, Diblik P, Hansikova H, et al. Novel oPA1 missense mutation in a family with optic atrophy and severe widespread neurological disorder. Acta Ophthalmol. (2013) 91:e225–31. doi: 10.1111/aos.12038
107. Di Gregorio E, Borroni B, Giorgio E, Lacerenza D, Ferrero M, Lo Buono N, et al. ELOVL5 mutations cause spinocerebellar ataxia 38. Am J Hum Genet. (2014) 95:209–17. doi: 10.1016/j.ajhg.2014.07.001
108. Burnett JR, Hooper AJ, Hegele RA. Abetalipoproteinemia. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattl, WA: University of Washington, Seattle (2018). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK532447/
109. Jimenez-Jimenez F, Agúndez J, Martinez C, Navacerrada F, Plaza-Nieto JF, Pilo-de-la-Fuente B, et al. Hereditary coproporphyria associated with the q306X mutation in the coproporphyrin oxidase gene presenting with acute ataxia. Tremor Other Hyperkinet Mov. (2013). doi: 10.7916/D8DF6PXW
110. Bras J, Djaldetti R, Alves AM, Mead S, Darwent L, Lleo A, et al. Exome sequencing in a consanguineous family clinically diagnosed with early onset alzheimer's disease identifies an homozygous cTSF mutation. Neurobiol Aging. (2016) 46:236.e1. doi: 10.1016/j.neurobiolaging.2016.06.018
111. Yalçinkaya C, Apaydin H, Ozekmekçi S, Gibson KM. Delayed-onset dystonia associated with 3-oxothiolase deficiency. Mov Disord. (2001) 16:372–5. doi: 10.1002/mds.1060
112. Pode-Shakked N, Korman S, Pode-Shakked B, Landau Y, Kneller K, Abraham S, et al. Clues and challenges in the diagnosis of intermittent maple syrup urine disease. Eur J Med Genet. (2020) 63:103901. doi: 10.1016/j.ejmg.2020.103901
113. Iwama K, Iwata A, Shiina M, Mitsuhashi S, Miyatake S, Takata A, et al. A novel mutation in sLC1A3 causes episodic ataxia. J Hum Genet. (2018) 63:207–11. doi: 10.1038/s10038-017-0365-z
114. Ganetzky R, Stojinski C. Mitochondrial short-chain enoyl-CoA hydratase 1 deficiency. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2019). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK542806/
115. Saini A, Attri S, Sankhyan N, Singhi P. Hypomorphic citrullinaemia due to mutated aSS1 with episodic ataxia. BMJ Case Rep. (2018) 2018:bcr2017220193. doi: 10.1136/bcr-2017-220193
116. Paliwal D, Gupta P, Pradhen S. Gabapentin as a rescue drug in d-penicillamine-induced status dystonicus in patients with wilson disease. Neurol India. (2010) 57:761–3. doi: 10.4103/0028-3886.72184
117. Kurian M, McNeill A, Lin JP, Maher ER. Childhood disorders of neurodegeneration with brain iron accumulation (NBIA). Dev Med Child Neurol. (2011) 53:394–404. doi: 10.1111/j.1469-8749.2011.03955.x
118. Kyriagis M, Grattan-Smith P, Scheinberg A, Teo C, Nakaji N, Waugh M. Status dystonicus and hallervorden-Spatz disease: treatment with intrathecal baclofen and pallidotomy. J Paediatr Child Health. (2004) 40:322–5. doi: 10.1111/j.1440-1754.2004.00374.x
119. Balas I, Kovacs N, Hollody L. Staged bilateral stereotactic pallidothalamotomy for life-threatening dystonia in a child with hallervorden-spatz disease. Mov Disord. (2006) 21:82–5. doi: 10.1002/mds.20655
120. Mariotti P, Fasano A, Contarino MF, Della Marca G, Piastra M, Genovese O, et al. Management of status dystonicus: our experience and review of the literature. Mov Disord. (2007) 22:963–8. doi: 10.1002/mds.21471
121. Yildiz Y, Pektas E, Tokatli A, Haliloglu G. Hereditary dopamine transporter deficiency syndrome: challenges in diagnosis and treatment. Neuropediatrics. (2016) 48:49–52. doi: 10.1055/s-0036-1593372
122. Allen N, Lin JO, Lynch T, King M. Status dystonicus: a Practice guide. Dev Med Child Neurol. (2014) 56:105–12. doi: 10.1111/dmcn.12339
123. Plecko B, Paul K, Mills P, Clayton P, Paschke E, Maier O, et al. Pyridoxine responsiveness in novel mutations of the pNPO gene. Neurology. (2014) 82:1425–33. doi: 10.1212/WNL.0000000000000344
124. El-Hattab AW, Almannai M, Scaglia F. MELAS. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2001). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1233/
125. Evers C, Seitz A, Assmann B, Opladen T, Karch S, Hinderhofer K, et al. Diagnosis of coPAN by whole exome sequencing: waking up a sleeping tiger's eye. Am J Med Genet A. (2017) 173:1878–86. doi: 10.1002/ajmg.a.38252
126. Okamoto N, Wada S, Oga T, Kawabata Y, Baba Y, Habu D, et al. Hereditary ceruloplasmin deficiency with hemosiderosis. Hum Genet. (1996) 96:755–8. doi: 10.1007/BF02346185
127. Gautschi M, Merlini L, Calza AM, Hayflick S, Nouffer JM, Fluss J. Late diagnosis of fucosidosis in a child with progressive fixed dystonia, bilateral pallidal lesions and red spots on the skin. Eur J Paediatr Neurol. (2014) 18:516–9. doi: 10.1016/j.ejpn.2014.02.005
128. Lau NKC, Ching CK, Lee HHC, Chak WKM, Kwan Shing N, Hanchard NA, et al. First case of genetically confirmed cLN3 disease in chinese with cDNA sequencing revealing pathogenicity of a novel splice site variant. Clin Chim Acta. (2018) 486:151–5. doi: 10.1016/j.cca.2018.07.040
129. Pierson TM, Adams D, Bonn F, Martinelli P, Cherukuri PF, Teer JK, et al. Whole-exome sequencing identifies homozygous aFG3L2 mutations in a spastic ataxia-Neuropathy syndrome linked to mitochondrial m-AAA proteases. PLoS Genet. (2011) 7:e1002325. doi: 10.1371/journal.pgen.1002325
130. Hammer MB, Eleuch-Fayache G, Schottlaender LV, Nehdi H, Gibbs JR, Arepalli SK, et al. Mutations in gBA2 cause autosomal-Recessive cerebellar ataxia with spasticity. Am J Hum Genet. (2013) 92:245–51. doi: 10.1016/j.ajhg.2012.12.012
131. Yu HL, Shen Y, Sun YM, Zhang Y. Two novel mutations of pEX6 in one chinese zellweger spectrum disorder and their clinical characteristics. Ann Transl Med. (2019) 7:368. doi: 10.21037/atm.2019.06.42
132. Kumar KR, Wali G, Davis RL, Mallawaarachchi AC, Palmer EE, Gayevskiy, et al. Expanding the spectrum of pEX16 mutations and novel insights into disease mechanisms. Mol Genet Metab Rep. (2018) 20:16:46–51. doi: 10.1016/j.ymgmr.2018.07.003
133. Trinh J, Imhoff S, Dulovic-Mahlow M, Kandaswamy KK, Tadic V, Schäfer J, et al. Novel nAXE variants as a cause for neurometabolic disorder: implications for treatment. J Neurol. (2020) 267:770–82. doi: 10.1007/s00415-019-09640-2
134. Cheon CK, Lee BH, Ko JM, Kim HJ, Yoo HW. Novel mutation in sLC6A19 causing late-Onset seizures in hartnup disorder. Pediatr Neurol. (2010) 42:369–71. doi: 10.1016/j.pediatrneurol.2010.01.009
135. Cerone R, Holme E, Schiaffino MC, Caruso U, Maritano L, Romano C. Tyrosinemia type iII: diagnosis and ten-Year follow-Up. Acta Paediatr. (1997) 86:1013–5. doi: 10.1111/j.1651-2227.1997.tb15192.x
136. Schottmann G, Sarpong A, Lorenz C, Weinhold N, Gill E, Teschner L, et al. A movement disorder with dystonia and ataxia caused by a mutation in the hIBCH gene. Mov Disord. (2013) 31:1733–9. doi: 10.1002/mds.26704
137. Myers JH, Shook JE. Vomiting, ataxia, and altered mental status in an adolescent: late-Onset ornithine transcarbamylase deficiency. Am J Emerg Med. (1996) 14:553–7. doi: 10.1016/S0735-6757(96)90097-2
138. Keegan CE, Martin DM, Quint DJ, Gorski JL. Acute extrapyramidal syndrome in mild ornithine transcarbamylase deficiency: metabolic stroke involving the caudate and putamen without metabolic decompensation. Eur J Pediatr. (2003) 162:259–63. doi: 10.1007/s00431-002-1135-1
139. Morales-Briceño H, Chang FCF, Wong C, Mallawaarachchi A, Wolfe N, Pellegrino da Silva R, et al. Paroxysmal dyskinesias with drowsiness and thalamic lesions in gABA transaminase deficiency. Neurology. (2019) 92:94–7. doi: 10.1212/WNL.0000000000006744
140. Lyn CY, Weng WC, Lee WT. A novel mutation of aLDH5A1 gene associated with succinic semialdehyde dehydrogenase deficiency. J Child Neurol. (2015) 30:486–9. doi: 10.1177/0883073814544365
141. Schneider SA, Bhatia KP. Rare causes of dystonia parkinsonism. Curr Neurol Neurosci Rep. (2010) 10:431–9. doi: 10.1007/s11910-010-0136-0
142. Navarrete R, Leal F, Vega AI, Morais-López A, Garcia-Silva MT, Martín-Hernández E, et al. Value of genetic analysis for confirming inborn errors of metabolism detected through the spanish neonatal screening program. Eur J Hum Genet. (2019) 27:556–62. doi: 10.1038/s41431-018-0330-0
143. Hardies K, de Kovel CG, Weckhuysen S, Asselbergh B, Geuens T, Deconinck T, et al. Recessive mutations in sLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain. (2015) 138(Pt 11):3238–50. doi: 10.1093/brain/awv263
144. Yalaz K, Vanli L, Yilmaz E, Tokatli A, Anlar B. Phenylketonuria in pediatric neurology practice: a Series of 146 cases. Child Neurol. (2006) 21:987–90. doi: 10.1177/08830738060210111401
145. Yamanouchi H, Kasai H, Sakuragawa N, Kurokawa T. Palatal myoclonus in krabbe disease. Brain Dev. (1991) 13:355–8. doi: 10.1016/s0387-7604(12)80133-1
146. Crow YJ. Aicardi-goutières syndrome. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2005). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1475/
147. Christensen C, Walsh L. Movement disorders and neurometabolic diseases. Semin Pediatr Neurol. (2018) 25:82–91. doi: 10.1016/j.spen.2018.02.003
148. Koy A, Lin JP, Sanger TD, Marks WA, Mink JW, Timmermann L. Advances in management of movement disorders in children. Lancet Neurol. (2005) 15:719–35. doi: 10.1016/S1474-4422(16)00132-0
149. Mohammad SS, Paget SP, Dale RC. Current therapies and therapeutic decision making for childhood-Onset movement disorders. Mov Disord. (2019) 34:637–56. doi: 10.1002/mds.27661
150. Kojima K, Nakajima T, Taga N, Miyauchi A, Kato M, Matsumoto A, et al. Gene therapy improves motor and mental function of aromatic l-Amino acid decarboxylase deficiency. Brain. (2019) 142:322–33. doi: 10.1093/brain/awy331
151. Tseng CH, Chien YH, Lee NC, Hsu YC, Peng SF, Tseng WI, et al. Gene therapy improves brain white matter in aromatic l-Amino acid decarboxylase deficiency. Ann Neurol. (2019) 85:644–52. doi: 10.1002/ana.25467
152. Schulz A, Ajayi T, Specchio N, de Los Reyes E, Gissen P, Ballon D, et al. Study of intraventricular cerliponase alfa for cLN2 disease. N Engl J Med. (2018) 378:1898–907. doi: 10.1056/NEJMoa1712649
153. Ubeda Tikkanen A, Buxton K, Ullrich CK, Stone SS, Nimec DL. The palliative use of intrathecal baclofen in niemann-Pick disease type c. Pediatrics. (2019) 144:e20191438. doi: 10.1542/peds.2019-1438
154. Hidalgo ET, Orillac C, Hersh A, Harter DH, Rizzo WB, Weiner HL. Intrathecal baclofen therapy for the treatment of spasticity in sjögren-Larsson syndrome. J Child Neurol. (2017) 32:100–03. doi: 10.1177/0883073816671440
155. van der Veldt N, van Rappard DF, van de Pol LA, van der Knaap MS, van Ouwerkerk WJR, Becher JG, et al. Intrathecal baclofen in metachromatic leukodystrophy. Dev Med Child Neurol. (2019) 61:232–5. doi: 10.1111/dmcn.13919
156. Hori YS, Fukuhara T, Aoi M, Ochi M, Furujo M. Intrathecal baclofen pump implantation for type 2 gaucher disease. Pediatr Neurosurg. (2017) 52:331–5. doi: 10.1159/000479324
157. van Karnebeek C, Horvath G, Murphy T, Purtzki J, Bowden K, Sirrs S, et al. Deep brain stimulation and dantrolene for secondary dystonia in x-Linked adrenoleukodystrophy. JIMD Rep. (2014) 15:113–6. doi: 10.1007/8904_2014_305
158. Taira T, Kobayashi T, Hori T. Disappearance of self-mutilating behavior in a patient with lesch–Nyhan syndrome after bilateral chronic stimulation of the globus pallidus internus. J Neurosurg. (2003) 98:414–6. doi: 10.3171/jns.2003.98.2.0414
159. Pralong E, Pollo C, Coubes P, Bloch J, Roulet E, Tétreault MH, et al. Electrophysiological characteristics of limbic and motor globus pallidus internus (GPI) neurons in two cases of lesch–Nyhan syndrome. Neurophysiol Clin. (2005) 35:168–73. doi: 10.1016/j.neucli.2005.12.004
160. Martinez-Ramirez D, Hack N, Vasquez ML, Morita H, Giugni JC, Wolf JM, et al. Deep brain stimulation in a case of mitochondrial disease. Mov Disord Clin Pract. (2015) 3:139–45. doi: 10.1002/mdc3.12241
161. Madeo A, Di Rocco M, Brassier A, Bahi-Buisson N, De Lonlay P, Ceballos-Picot I. Clinical, biochemical and genetic characteristics of a cohort of 101 french and italian patients with hPRT deficiency. Mol Genet Metab. (2009) 127:147–57. doi: 10.1016/j.ymgme.2019.06.001
162. Deon LL, Kalichman MA, Booth CL, Slavin KV, Gaebler-Spira DJ. Pallidal deep-Brain stimulation associated with complete remission of self-injurious behaviors in a patient with lesch-Nyhan syndrome: a Case report. J Child Neurol. (2012) 27:117–20. doi: 10.1177/0883073811415853
163. Cif L, Biolsi B, Gavarini S, Saux A, Robles SG, Tancu C, et al. Antero-Ventral internal pallidum stimulation improves behavioral disorders in lesch–Nyhan disease. Mov Disord. (2007) 22:2126–9. doi: 10.1002/mds.21723
164. Chakraborti S, Hasegawa H, Lumsden DE, Ali W, Kaminska M, Lin JP, et al. Bilateral subthalamic nucleus deep brain stimulation for refractory total body dystonia secondary to metabolic autopallidotomy in a 4-year-old boy with infantile methylmalonic acidemia. J Neurosurg Pediatr. (2013) 12:374–9. doi: 10.3171/2013.7.PEDS1350
165. Beaulieu-Boire I, Aquino CC, Fasano A, Poon YY, Fallis M, Lang AE, et al. Deep brain stimulation in rare inherited dystonias. Brain Stimul. (2016) 9:905–10. doi: 10.1016/j.brs.2016.07.009
166. Air E, Ostrem JL, Sanger TD, Starr PA. Deep brain stimulation in children: experience and technical pearls. J Neurosurg Pediatr. (2011) 8:566–74. doi: 10.3171/2011.8.PEDS11153
167. Abel TJ, Dalm BD, Grossbach AJ, Jackson AW, Thomsen T, Greenlee JD. Lateralized effect of pallidal stimulation on self-mutilation in lesch-Nyhan disease. J Neurosurg Pediatr. (2014) 14:594–7. doi: 10.3171/2014.8.PEDS1451
168. Huebl J, Poshtiban A, Brücke C, Siegert S, Bock A, Koziara H, et al. Subthalamic and pallidal oscillatory activity in patients with neurodegeneration with brain iron accumulation type i (NBIA-I). Clin Neurophysiol. (2019) 130:469–73. doi: 10.1016/j.clinph.2018.12.012
169. Krause M, Fogel W, Tronnier V, Pohle S, Hörtnagel K, Thyen U, et al. Long-term benefit to pallidal deep brain stimulation in a case of dystonia secondary to pantothenate kinase-Associated neurodegeneration. Mov Disord. (2006) 21:2255–7. doi: 10.1002/mds.21166
170. Castelnau P, Cif L, Valente EM, Vayssiere N, Hemm S, Gannau A, et al. Pallidal stimulation improves pantothenate kinase-Associated neurodegeneration. Ann Neurol. (2005) 57:738–41. doi: 10.1002/ana.20457
171. Mikati MA, Yehya A, Darwish H, Karam P, Comair Y. Deep brain stimulation as a mode of treatment of early onset pantothenate kinase-Associated neurodegeneration. Eur J Paediatr Neurol. (2009) 13:61–4. doi: 10.1016/j.ejpn.2008.01.006
172. Mahoney R, Selway R, Lin JP. Cognitive functioning in children with pantothenate-Kinase-Associated neurodegeneration undergoing deep brain stimulation. Dev Med Child Neurol. (2011) 53:275–9. doi: 10.1111/j.1469-8749.2010.03815.x
173. Mahajan A, Sidiropoulos C. TPK1 mutation induced childhood onset idiopathic generalized dystonia: report of a rare mutation and effect of deep brain stimulation. J Neurol Sci. (2017) 376:42–3. doi: 10.1016/j.jns.2017.02.063
174. Liu Z, Liu Y, Yang Y, Wang L, Dou W, Guo J, et al. Subthalamic nuclei stimulation in patients with pantothenate kinase-Associated neurodegeneration (PKAN). Neuromodulation. (2017) 20:484–91. doi: 10.1111/ner.12549
175. Sharma MC, Aggarwal N, Bihari M, Goyal V, Gaikwed S, Vaishya, et al. Hallervorden spatz disease: mR and pathological findings of a rare case. Neurol India. (2005) 53:102–4. doi: 10.4103/0028-3886.15072
176. Shields DC, Sharma N, Gale JT, Eskandar EN. Pallidal stimulation for dystonia in pantothenate kinase-Associated neurodegeneration. Pediatr Neurol. (2007) 37:442–5. doi: 10.1016/j.pediatrneurol.2007.08.006
177. Isaac C, Wright I, Bhattacharyya D, Baxter P, Rowe J. Pallidal stimulation for pantothenate kinase-Associated neurodegeneration dystonia. Arch Dis Child. (2008) 93:239–40. doi: 10.1136/adc.2007.118968
178. Timmermann L, Pauls KA, Wieland K, Jech R, Kurlemann G, Sharma N, et al. Dystonia in neurodegeneration with brain iron accumulation: outcome of bilateral pallidal stimulation. Brain. (2010) 133(Pt 3):701–12. doi: 10.1093/brain/awq022
179. Ge M, Zhang K, Ma Y, Meng FG, Hu WH, Yang AC, et al. Bilateral subthalamic nucleus stimulation in the treatment of neurodegeneration with brain iron accumulation type 1. Stereotact Funct Neurosurg. (2011) 89:162–6. doi: 10.1159/000323374
180. Lim BC, Ki CS, Cho A, Hwang H, Kim KJ, Hwang YS, et al. Pantothenate kinase-Associated neurodegeneration in korea: recurrent r440P mutation in pANK2 and outcome of deep brain stimulation. Eur J Neurol. (2012) 19:556–61. doi: 10.1111/j.1468-1331.2011.03589.x
181. Adamovicová M, Jech R, Urgošík D, Spacková N, Krepelová A. Pallidal stimulation in siblings with pantothenate kinase-Associated neurodegeneration: four-Year follow-Up. Mov Disord. (2011) 26:184–7. doi: 10.1002/mds.23349
182. Elkaim LM, De Vloo P, Kalia SK, Lozano AM, Ibrahim GM. Deep brain stimulation for childhood dystonia: current evidence and emerging practice. Expert Rev Neurother. (2018) 18:773–84. doi: 10.1080/14737175.2018.1523721
183. Coene KLM, Kluijtmans LAJ, van der Heeft E, Engelke UFH, de Boer S, Hoegen B, et al. Next-generation metabolic screening: targeted and untargeted metabolomics for the diagnosis of inborn errors of metabolism in individual patients. J Inherit Metab Dis. (2018) 41:337–53. doi: 10.1007/s10545-017-0131-6
Keywords: dystonia, inborn error of metabolism, ataxia, myoclonus, chorea, athetosis, hypokinetic rigid syndrome
Citation: Ortigoza-Escobar JD (2020) A Proposed Diagnostic Algorithm for Inborn Errors of Metabolism Presenting With Movements Disorders. Front. Neurol. 11:582160. doi: 10.3389/fneur.2020.582160
Received: 10 July 2020; Accepted: 30 September 2020;
Published: 13 November 2020.
Edited by:
Renata Rizzo, University of Catania, ItalyReviewed by:
Wang-Tso Lee, National Taiwan University Hospital, TaiwanMario Mastrangelo, Policlinico Umberto I, Italy
Copyright © 2020 Ortigoza-Escobar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Darío Ortigoza-Escobar, am9ydGlnb3phQHNqZGhvc3BpdGFsYmFyY2Vsb25hLm9yZw==