 Christopher S. Ward1,2,3*
Christopher S. Ward1,2,3* Teng-Wei Huang2,4
Teng-Wei Huang2,4 Jose A. Herrera2,5†
Jose A. Herrera2,5† Rodney C. Samaco2,3
Rodney C. Samaco2,3 Christopher M. McGraw2,4†Diana E. Parra6E. Melissa Arvide6
Christopher M. McGraw2,4†Diana E. Parra6E. Melissa Arvide6 Aya Ito-Ishida2,3
Aya Ito-Ishida2,3 Xiangling Meng2,7Kerstin Ure2,3Huda Y. Zoghbi2,3,4,5,6,7,8
Xiangling Meng2,7Kerstin Ure2,3Huda Y. Zoghbi2,3,4,5,6,7,8 Jeffrey L. Neul2,3,4,5,6,7*
Jeffrey L. Neul2,3,4,5,6,7*- 1Department of Molecular Physiology and Biophysics, Baylor College of Medicine, Houston, TX, United States
- 2Jan and Dan Duncan Neurological Research Institute, Texas Children's Hospital, Houston, TX, United States
- 3Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, United States
- 4Program in Developmental Biology, Baylor College of Medicine, Houston, TX, United States
- 5Interdepartmental Program in Translational Biology and Molecular Medicine, Baylor College of Medicine, Houston, TX, United States
- 6Department of Pediatrics, Baylor College of Medicine, Houston, TX, United States
- 7Department of Neuroscience, Baylor College of Medicine, Houston, TX, United States
- 8Howard Hughes Medical Institute, Baylor College of Medicine, Houston, TX, United States
Rett Syndrome (RTT) is a neurodevelopmental disorder caused by loss of function of the transcriptional regulator Methyl-CpG-Binding Protein 2 (MeCP2). In addition to the characteristic loss of hand function and spoken language after the first year of life, people with RTT also have a variety of physiological and autonomic abnormalities including disrupted breathing rhythms characterized by bouts of hyperventilation and an increased frequency of apnea. These breathing abnormalities, that likely involve alterations in both the circuitry underlying respiratory pace making and those underlying breathing response to environmental stimuli, may underlie the sudden unexpected death seen in a significant fraction of people with RTT. In fact, mice lacking MeCP2 function exhibit abnormal breathing rate response to acute hypoxia and maintain a persistently elevated breathing rate rather than showing typical hypoxic ventilatory decline that can be observed among their wild-type littermates. Using genetic and pharmacological tools to better understand the course of this abnormal hypoxic breathing rate response and the neurons driving it, we learned that the abnormal hypoxic breathing response is acquired as the animals mature, and that MeCP2 function is required within excitatory, inhibitory, and modulatory populations for a normal hypoxic breathing rate response. Furthermore, mice lacking MeCP2 exhibit decreased hypoxia-induced neuronal activity within the nucleus tractus solitarius of the dorsal medulla. Overall, these data provide insight into the neurons driving the circuit dysfunction that leads to breathing abnormalities upon loss of MeCP2. The discovery that combined dysfunction across multiple neuronal populations contributes to breathing dysfunction may provide insight into sudden unexpected death in RTT.
Introduction
Rett syndrome (RTT, OMIM 312750) is a neurodevelopment disorder caused by mutations in the X-linked gene Methyl-CpG Binding Protein 2 (MECP2) that is characterized by initial normal development followed by regression manifesting as loss of acquired skills (1–3). Many affected individuals also develop autonomic and physiological deficits including disrupted breathing with bouts of hyperventilation and apnea (4, 5). Approximately one quarter of all deaths in RTT are sudden and unexpected, raising the possibility that breathing abnormalities contribute to some of these deaths (6). The abnormal breathing pattern may indicate deficits in the networks that maintain normal respiratory rhythmogenesis as well as those that modify its response to environmental stimuli (7).
Several mouse models have been generated that display features similar to the human disorder (8–11). While female mice heterozygous for Mecp2 mutations show many features reminiscent of the human disorder, the nature of the X-linked mutation also causes them to be mosaic for MeCP2 and less severely affected than hemizygous male animals. For these reasons, male mice are often used to help dissect anatomic and neuronal population effects of MeCP2 expression. Mice lacking MeCP2 function exhibit an abnormal response to hypoxia (12–14). Normally during acute hypoxia blood oxygen status is sensed by the carotid body located along the carotid bifurcation and relayed to brainstem respiratory centers through the nucleus tractus solitarius (NTS) (15–17). The expected response to an acute hypoxic challenge in adult mammals is an immediate increase in ventilation in an effort to increase gas exchange and blood oxygenation, within a timescale of minutes, ventilation then decreases as the failure to improve blood oxygen saturation persists and conservation of the remaining oxygen for essential body functions becomes critical (16–19). The increase in ventilation followed by ventilatory decline until ventilation approaches baseline levels is commonly referred to as the hypoxic ventilatory response, with the ventilatory decline stage often referred to as hypoxic ventilatory decline (HVD) (16, 20, 21).
However, in animals lacking Mecp2, the breathing rate during acute hypoxia remains elevated relative to that of wildtype littermates (12, 14, 22). In combination with disordered breathing characterized by bouts of hyperventilation interspersed with apnea, the persistent increase in ventilation in a hypoxic environment may be maladaptive and creates risk from increased metabolic demand without sufficient supply of oxygen (23). Furthermore, the maladaptive reflexive breathing response to external stimuli observed in these animals may reflect the underlying circuit-level disruption that might contribute ultimately to sudden unexpected death. A key unanswered question is the neuronal cell-type contribution to the abnormal hypoxia response in MeCP2 mutant animals. To investigate the underlying etiology of the abnormal HVD that occurs in mice lacking MeCP2, we used a combination of genetic and pharmacological methods to dissect the cellular contribution to the HVD. We identified that mice lacking MeCP2 acquire a deficit in HVD, presenting with a persistently increased breathing rate during exposure to hypoxia. Furthermore, we identified that MeCP2 function within excitatory, inhibitory, and modulatory neuronal populations is critical for a normal HVD.
Materials and Methods
Animals Used in Experiments
All research and animal care procedures were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee and housed in the Association for Assessment and Accreditation of Laboratory Animal Care-approved animal facility at Baylor College of Medicine. Mecp2Tm1Bird mice were obtained as a gift from Dr. Adrian Bird (University of Edinburgh, Edinburgh, UK, RRID:IMSR_JAX_007177) and backcrossed and maintained on a 129S6 background for >10 generations (8). Mecp2Tm1.1Bird mice were generated via Cre mediated recombination of Mecp2Tm1Bird mice and maintained on a 129S6 background. Mecp2Tm2Bird mice were obtained from The Jackson Laboratory and maintained on a C57BL/6J background (RRID:IMSR_JAX_006849) (9). Mecp2TM1.1Bird mice used for experiments were generated by crossing the congenic 129S6 Mecp2TM1.1Bird/+ female mice to wild-type male C57BL/6J mice to generate male 129B6F1 Mecp2+/Y (WT) and Mecp2Tm1.1Bird/Y (NULL) mice, and female Mecp2+/+ (WT) and Mecp2Tm1.1Bird/+ (HET) mice. Nestin-Cre (RRID:IMSR_JAX_003771) mice were maintained on a C57BL/6J background; Th-Cre (MGI:2677450), and VIAAT-Cre (MGI:4867735) and VGLUT2-Cre (RRID:IMSR_JAX_016963) mice were maintained on a FVB/NJ strain background. Cre lines were crossed to Mecp2Tm1Bird, or Mecp2Tm2Bird to generate male conditional knockout and conditional rescue mice and strain matched control littermates (24–27).
Unrestrained Whole Body Plethysmography
Unrestrained plethysmography in mice was performed similarly to methods previously described (14, 22). Mice were placed within unrestrained whole-body plethysmography chambers (Buxco), ~500 mL in volume with a continuous flow rate of 500 mL/min flushing the chambers with fresh air. Mice were allowed to acclimate for at least 20 min, and baseline breathing was then recorded for 30 min. Data were collected during each animals' session within the plethysmograph chambers without additional pre-data collection habituation sessions. To determine response to hypoxic gas (10% O2, balance N2), the chamber was then flushed with hypoxic gas for 20 min. Breathing rate was determined using a customized algorithm written in Matlab (RRID:SCR_001622) using the plethysmography signal files captured with Ponemah3 software (RRID:SCR_017107). To reduce the artifacts from excessive movement and sniffing behavior, breaths that exhibited an inspiratory time <0.03 s, an expiratory time >10 s, and a calculated exhaled tidal volume >150 or <50% of calculated inhaled tidal volume were excluded; breaths were then filtered with intervals excluded if they possessed >10% of their breaths above 500 breaths per minute (sliding 200 breath windows centered on the current breath). The peak breathing rate during hypoxia was calculated as the 90th percentile for breaths during the first 5 min of hypoxic challenge. Breathing parameters for each animal during baseline and as well as during the declined phase of hypoxic challenge (at 10 min from the onset of hypoxia until 15 min) were determined as the average instantaneous value over the recorded interval and then averaged across trials for animals subjected to repeated measurements. Additional parameters were collected during the baseline portion and include apnea index (apneas per 10,000 breaths, defined as a breath >0.5 s in duration and > 2x the overall baseline breath duration and >2x the duration of the 6 surrounding breaths), irregularity score calculated from breath duration changes (|[n]-[n−1]|/[n−1]), and uncompensated tidal volume calculated from the box flow of air into and out of the plethysmograph chamber. Respiratory parameters were then compared across genotypes, sex, or treatment as described in the text.
Pharmacological Manipulation of Hypoxic Breathing Response
Adult male 129B6F1 WT and NULL mice were treated with sterile phosphate buffered saline or drugs dissolved in sterile phosphate buffered saline by intraperitoneal injection immediately preceding plethysmography recordings of the hypoxic breathing response as described above. Following a minimum 48 h “wash-out” period, mice were tested with another drug and dosage combination. Drug and doses used include tiagabine at 3, 10, and 30 mg/kg; muscimol at 1, 2, and 3 mg/kg; baclofen at 1, 2, and 3 mg/kg; desipramine at 3, 10, and 30 mg/kg; ketamine at 10 and 30 mg/kg; and L-DOPA at 30, 60, or 100 mg/kg administered alone or with carbidopa at 10 mg/kg.
Histological Quantification of Neuronal Activity
Quantification of hypoxia induced neuronal activity and regions selected for analysis were adapted from methods used to look at hypoxia induced activity in the rat brain (28). To reduce artifacts of novelty and stress induced neuronal activation, male mice were habituated to the unrestrained whole body plethysmography chambers for at least 1 h a day for 4 days before being submitted to a gas challenge. On the day of the gas challenge 129B6F1 WT and NULL mice were placed in the plethysmography chambers and subjected to room air for 30 min to establish baseline breathing, mice were then treated for 3 h with hypoxic gas (10% O2, balance N2) or treated with room air to control for basal neuronal activity. Mice were then immediately anesthetized and processed for immuno-histology.
Male mice were deeply anesthetized by intraperitoneal injection with Avertin at a dose of 0.04 ml/g and then fixed by transcardiac perfusion with PBS followed by 4% paraformaldehyde in PBS. Tissues for histological analysis were harvested and fixed overnight in 4% paraformaldehyde in PBS and cryopreserved by overnight incubations in increasing concentrations of sucrose (up to 30% sucrose). Tissues were embedded in O.C.T. compound (Sakura) and sectioned. Sections were collected for at a thickness of 50 μm and stained as floating sections. Sections were blocked for 1 h in a PBS solution containing 10% serum (matched to the host used for the secondary antibodies) and 0.3% Triton X-100. Neuronal activation was assayed by immune-staining for c-Fos expression with a rabbit-anti-c-Fos antibody diluted 1:500 (RRID:AB_2106783), and a goat-anti-rabbit secondary antibody conjugated to dylight 549 (JaxIR). Primary and secondary antibody incubation was performed in the blocking solution for 12–48 h at 4°C or 6 h at room temperature. Sections were washed between incubations with PBS and 0.05% Triton X-100. If not included in the mounting medium, DAPI was included in the penultimate wash. Sections were mounted with Prolong mounting medium (Invitrogen) and imaged via epifluorescent microscopy (Zeiss M1 with Axiovision software, RRID:SCR_002677).
Quantification of neuronal activation bilaterally across regions in the hindbrain was performed via stereological estimation with an optical dissector excluding the upper surface of the tissue, a slide sampling fraction of 1/3 and an area fractionator of 1/4. Image processing and counting of neurons was performed using ImageJ software (29). NTS, locus coeruleus (LC), and Parabrachial Complex (PB) regions were identified based on their anatomic locations as defined in the Paxinos mouse brain atlas (30). Quantified neurons are expressed as the number of neurons bilaterally present in the region per mouse.
Statistics
All statistics were performed using SPSS (RRID:SCR_002865) on a PC. Parametric statistics were performed using ANOVA for the indicated factors. For conditions where factor had more than two levels, formalized post-hoc testing was performed using Student-Newman-Keuls or Bonferroni correction for multiple comparisons to detect pairwise differences.
Results
Loss of MeCP2 Function Impairs the Breathing Rate Response to Hypoxia
Mice lacking MeCP2 (male-NULL and female-HET) exhibit a higher breathing rate during exposure to hypoxia (10% O2, balance N2) than their wild-type littermates (14). We sought to better understand the nature of this abnormal breathing response using unrestrained whole body plethysmography. On a 129B6F1 isogenic strain background, we observed that similar to WT mice, NULL mice show a sharp increase in their breathing rate in response to acute hypoxia; however, NULL mice maintain a persistently elevated breathing rate as opposed to WT mice that exhibit HVD and decrease their breathing rate to near baseline levels as the exposure to hypoxia continues within the first 5 min after initiation of hypoxia (Figures 1A,B, Table 1).
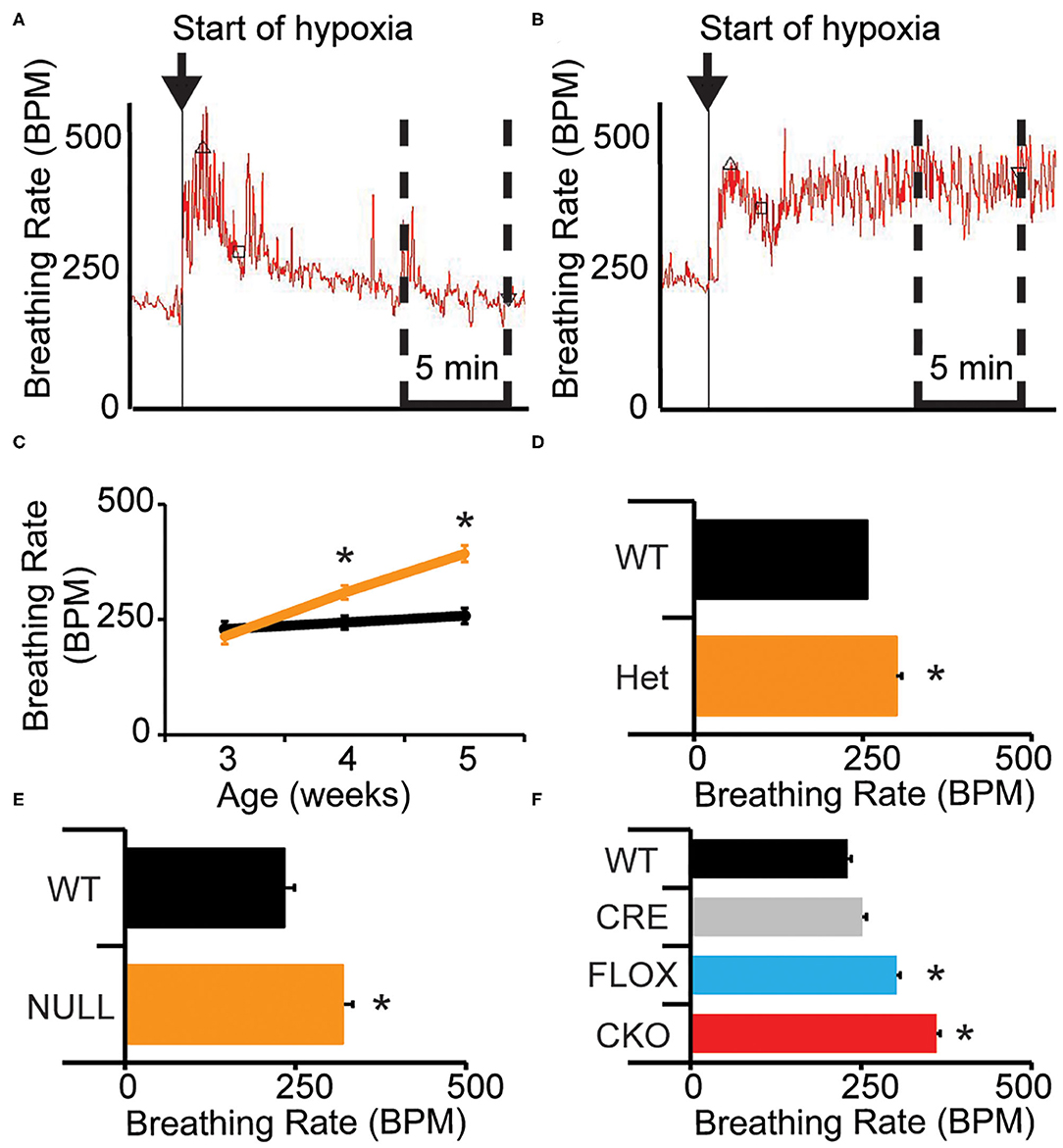
Figure 1. Loss of MeCP2 function disrupts the breathing rate response to acute hypoxia. (A) Wild-type mice exhibit a typical breathing rate response to exposure to hypoxia, with an immediate increase in breathing rate followed by a decline to near baseline breathing rates—representative image shown. Upward pointed triangle indicates timing of peak hypoxic breathing, defined as the 90th percentile for breathing rate during the first 5 min of hypoxic challenge, square indicates the third minute of the hypoxic challenge. (B) NULL mice exhibit an abnormal breathing rate response to hypoxia, with an immediate and persistent increase in breathing rate for the duration of exposure to hypoxia—representative image shown. The time segment used to determine the average “declined breathing rate” between 10 and 15 min after the onset of hypoxia is shown by the dashed box in (A,B). The “declined breathing rate” is presented in (C–F). (C) Male 129B6F1 NULL mice acquire the elevated “declined breathing rate” after 3 weeks of age (D). This elevated “declined breathing rate” is also present in aged female HET mice, (E) and in male NULL mice on a C57BL/6J background. (F) Removal of MeCP2 function from the nervous system with Nestin-Cre also causes the elevated breathing rate during hypoxia. (C–F) indicate mean +/– S.E.M. *p < 0.05 effect of genotype by ANOVA with Student-Newman-Keuls post-hoc correction for multiple comparisons for (F) (CKO vs. all other genotypes). Number of animals in each group are indicated in Tables 1, 2. Representative plethysmography traces are available (Supplementary Figures 1, 2).
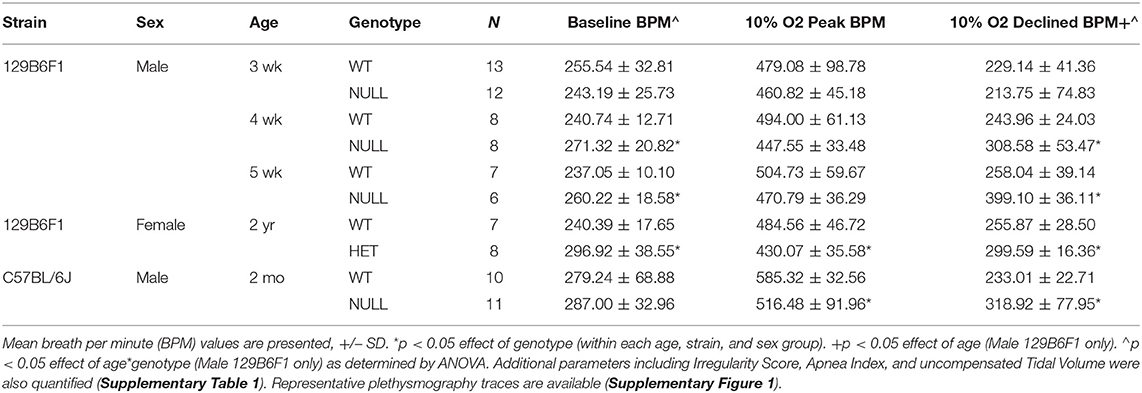
Table 1. Baseline and hypoxic breathing parameters in Mecp2 mutant mice grouped by age, strain, and sex.
Prior studies have demonstrated that the onset of apnea and disrupted breathing within this model are acquired during the first few weeks of life, progress across the lifespan of the animal, and can be exacerbated by hypoxic or hypercapnic challenge (31, 32). We sought to determine whether the deficit in HVD also is acquired as the animals age. We quantified HVD by determining the average “declined breathing rate” measured from 10 to 15 min after the onset of hypoxia. We observed normal patterns of HVD in NULL mice relative to control littermates at 3 weeks of age. By 4 weeks of age, NULL mice begin developing a significant HVD deficit that continues to worsen at 5 weeks of age. WT mice show similar patterns of HVD across the tested ages (Figure 1C, Table 1).
We also wanted to examine if this phenotype was robust enough to be observed across sexes and strain backgrounds. On a 129B6F1 strain background, 2 year old female HET mice reproduce the impaired HVD observed in the males (Figure 1D, Table 1). Similarly, male NULL mice raised on a C57BL/6J strain background also have an increased breathing rate during exposure to hypoxia (Figure 1E, Table 1). The trend toward a slight decrease in the peak breathing rate during hypoxic exposure observed in the 129B6F1 male NULL mice, is significant for the female HET mice and C57BL/6J male NULL mice (Table 1).
MeCP2 Function in Excitatory, Inhibitory, and Modulatory Neurons Critical for the Breathing Rate Response to Hypoxia
To better define in which neuronal cell-types MeCP2 expression is required for a normal HVD, we performed genetic removal and restoration of Mecp2. This produced wild-type (WT) mice, mice possessing just the Cre alleles (CRE), mice possessing just the Mecp2Tm1Bird allele (FLOX) or double mutant mice possessing both Cre and Mecp2Tm1Bird alleles (conditional knock out—CKO). The CKO mice are deficient for MeCP2 activity within the cell populations that expressed the cre-recombinase. For genetic rescue studies, we generated WT and CRE mice as well as mice possessing just the Mecp2Tm2Bird allele (STOP) or both Cre and Mecp2Tm2Bird alleles (RESC). The STOP mice do not express MeCP2 due to a Cre excisable STOP cassette within the endogenous Mecp2 locus, while RESC mice have MeCP2 activity solely restored to cells that have expressed the cre-recombinase.
CKO mice produced with Nestin-Cre, lack MeCP2 function within the nervous system and reproduce the impaired HVD observed in the NULL animals (Figure 1F, Table 2). Additionally, CKO mice with MeCP2 function removed from excitatory (VGLUT2-CKO), inhibitory (VIAAT-CKO) and modulatory neuronal populations (TH-CKO) all show significant impairment in HVD (Figures 2A,C,E, Table 2). Furthermore, RESC mice with MeCP2 function restored to excitatory (VGLUT2-RESC), inhibitory (VIAAT-RESC), and modulatory (TH-RESC) neuronal populations show significant improvement of their HVD relative to their STOP littermates (Figures 2B,D,F, Table 2).
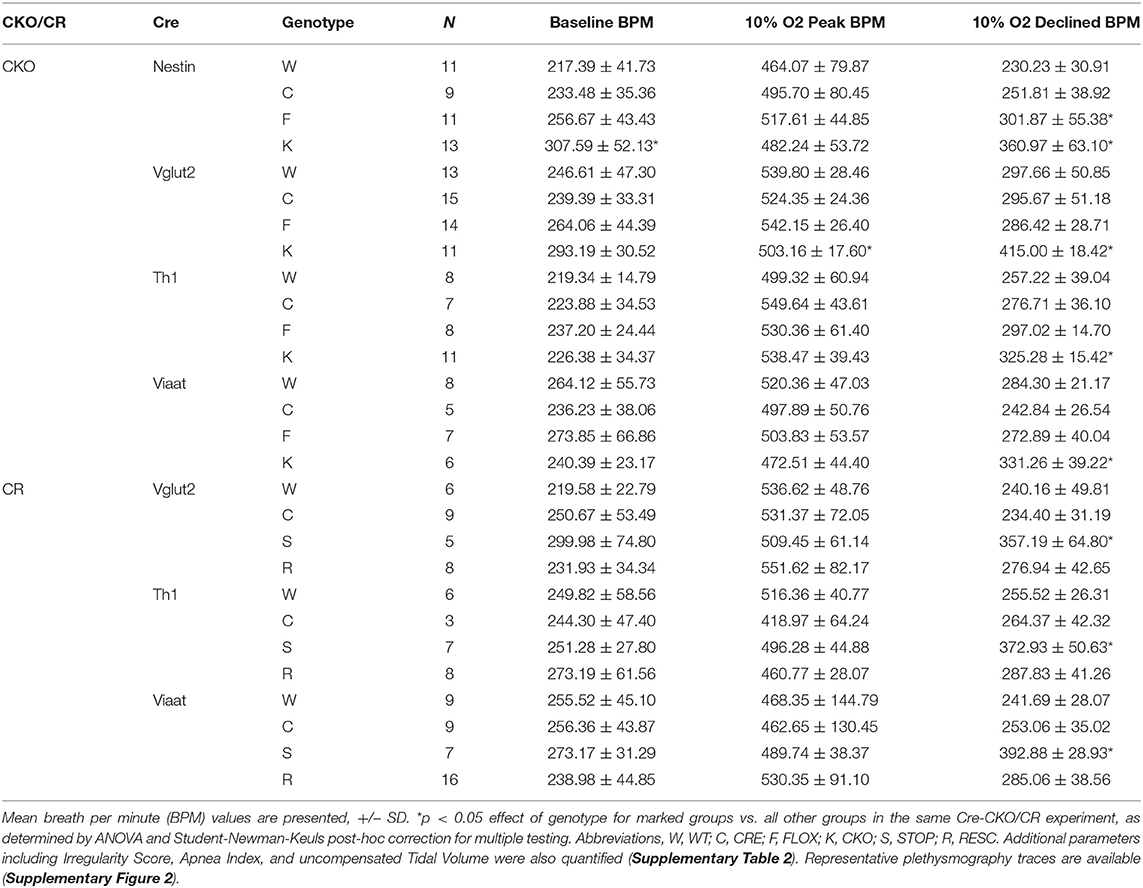
Table 2. Baseline and hypoxic breathing parameters from Mecp2 conditional knockout and conditional rescue mice.
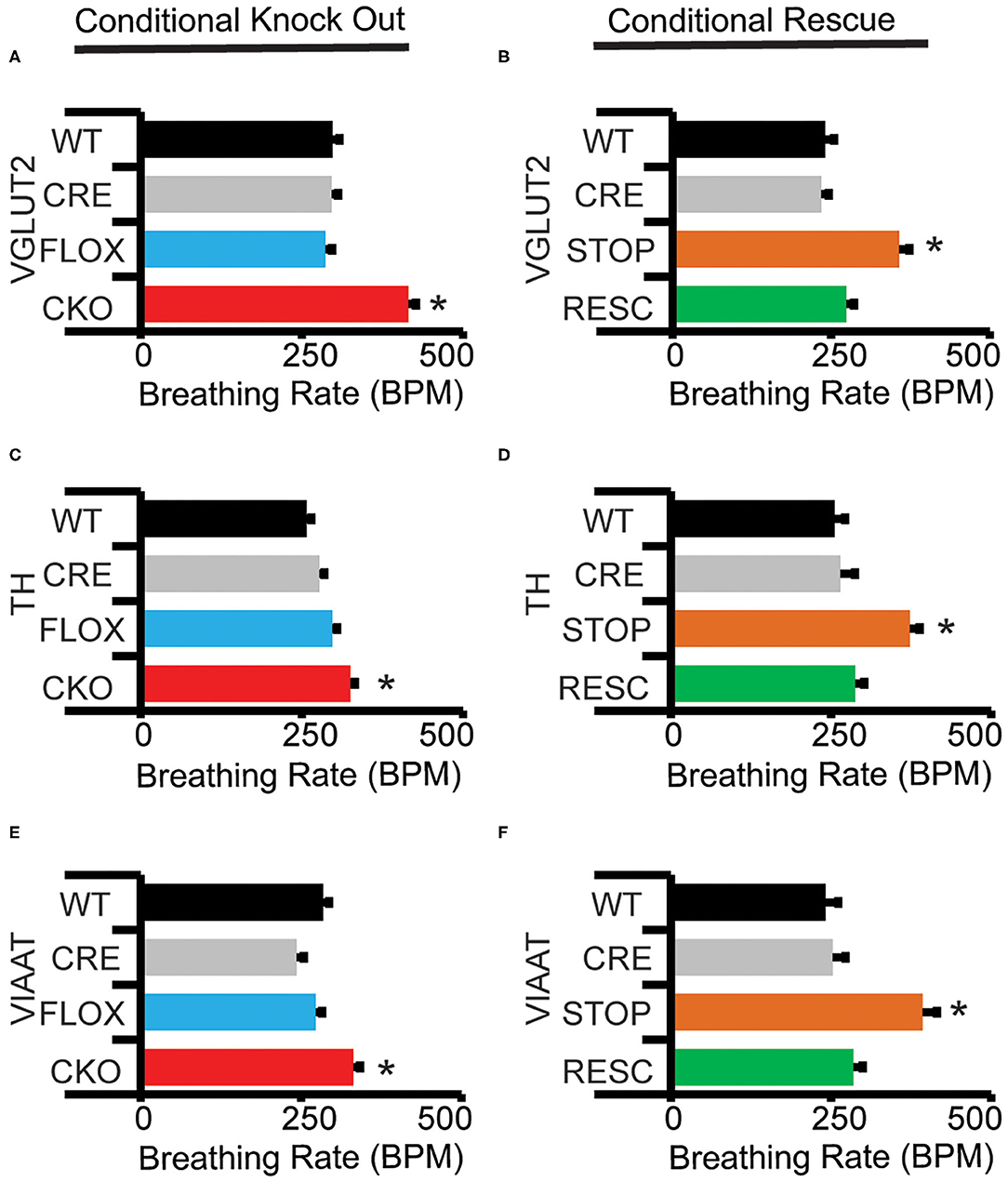
Figure 2. Conditional knock-out and conditional rescue of MeCP2 shows necessity and sufficiency of MeCP2 expression for hypoxic breathing decline. “Declined breathing rate” (between 10 and 15 min after the onset of hypoxia) data is presented for genetic conditional experiments generated with (A,B) VGLUT2-Cre, (C,D) TH1-Cre, and (E,F) VIAAT-Cre. Values indicate mean +/– S.E.M. *p < 0.05 effect of genotype by ANOVA with Student-Newman-Keuls post-hoc correction for multiple comparisons (indicated genotype vs. all other genotypes). ns not significant. Number of animals in each group is presented in Table 2. Representative plethysmography traces are available (Supplementary Figure 2).
Pharmacologic Manipulation of Excitatory, Inhibitory, and Modulatory Systems Affects the Breathing Rate Response to Hypoxia
Genetic manipulation of MeCP2 function within the excitatory, inhibitory, and modulatory populations demonstrated the critical requirement of MeCP2 function in these neuronal populations for normal HVD. To extend our understanding we pharmacologically manipulated these neurotransmitters to determine if the HVD could be improved within NULL mice. Previous work has shown that loss of MeCP2 function causes a cell-autonomous reduction of expression of the biosynthetic enzymes required for the synthesis of these specific neurotransmitters (25, 26, 33); therefore, we tested whether compounds that enhance signaling of these neurotransmitters (TH neurons: L-DOPA, carbidopa, desipramine; VIAAT neurons: tiagabine, baclofen, muscimol) would modulate the HVD. Additionally we tested ketamine, an NMDA receptor agonist, which has been reported to correct hyperexcitability within forebrain regions of mice lacking MeCP2 (34).
Treatment with L-DOPA bypasses the biosynthetic deficit in the production of dopamine and norepinephrine that is present in NULL mice due to decreased Tyrosine Hydroxylase expression (26). Efficacy of L-DOPA treatment can be improved by co-administration with carbidopa, a structural analog of L-DOPA that inhibits DOPA decarboxylase present in the gut, allowing more L-DOPA in peripheral circulation to cross the blood brain barrier where it can improve noradrenergic and dopaminergic neuron function. Low doses of L-DOPA (30 and 60 mg/kg) had little impact on HVD. However, increased levels of L-DOPA (100 mg/kg) decreased the breathing rate following several minutes of hypoxic exposure (Figure 3, Table 3). Carbidopa improved the response and reduced the dosage of L-DOPA required to modify the breathing rate during hypoxia; however, treatment with high doses of L-DOPA with carbidopa also diminished the peak breathing rate during hypoxia (Figure 3, Table 3).
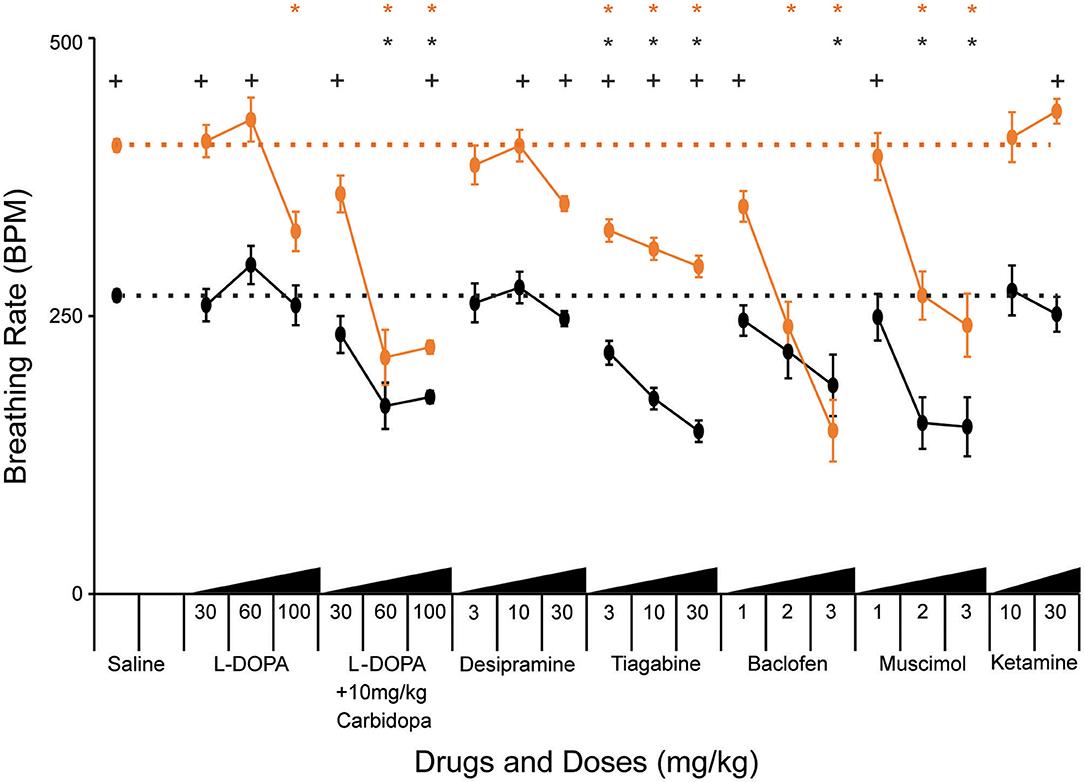
Figure 3. Pharmacological treatments modify breathing rate during acute hypoxia. “Declined breathing rate” (between 10 and 15 min after the onset of hypoxia) data is presented for 129B6F1 mice treated with saline and various concentrations of the indicated drugs 1 h before exposure to hypoxia. Values indicate mean +/– S.E.M. *p < 0.05 effect of drug vs. saline on breathing rate within genotype (black-WT, orange-NULL). +p < 0.05 effect of genotype within the indicated drug and dose. p-values determined by ANOVA with Bonferroni post-hoc correction for 61 multiple comparisons (40 for effect of drug vs. saline and 21 for effect of genotype at each drug and dose). Number of animals in each group is presented in Table 3.
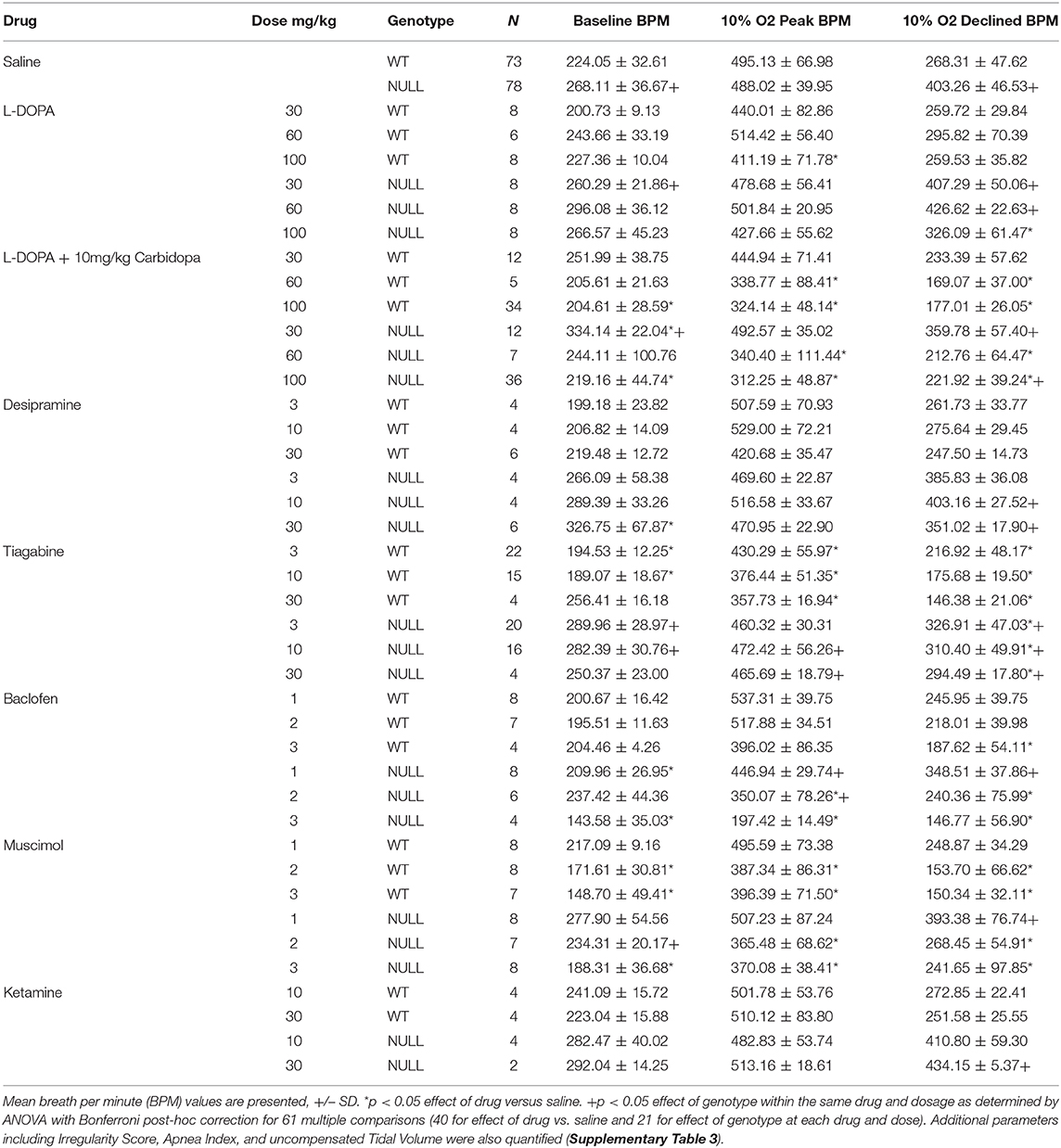
Table 3. Baseline and hypoxic breathing parameters following acute drug treatment.
Desipramine, a tricyclic antidepressant that inhibits reuptake of norepinephrine, has previously been tested in NULL mice and improves the incidence of apnea caused by loss of MeCP2 (13, 35). Treatment with desipramine (30 mg/kg) significantly improved HVD of NULL mice, with minimal impact on their peak breathing rate during hypoxia (Figure 3, Table 3).
Treatment of mice with tiagabine, a GABA reuptake inhibitor, improved HVD in NULL animals to levels closer to those of saline treated WT mice (Figure 3, Table 3). Similarly, the GABAA and GABAB receptor agonists muscimol and baclofen also improved the HVD of NULL animals. However, the dosages of muscimol and baclofen that showed significant reduction in breathing rate during exposure to hypoxia also caused significant decreases in the peak breathing rate during hypoxia. Dosages of tiagabine that improved HVD had less impact than muscimol or baclofen on the peak breathing rate during hypoxia (Figure 3, Table 3).
Both dosages of Ketamine that were tested showed no improvement in HVD, the higher 30 mg/kg dose trended toward a worsening of the impaired HVD (p = 0.01 ANOVA for effect of drug on NULL mice, not corrected for multiple comparisons) (Figure 3, Table 3).
While some treatments, such as L-DOPA without carbidopa, desipramine, baclofen, and ketamine, had little to no impact on the hypoxic breathing response in WT animals, several others such as L-DOPA with carbidopa, tiagabine, and muscimol also modified the WT hypoxic breathing response.
Mice Lacking MeCP2 Function Have Decreased Hypoxia Induced Neuronal Activity
Finally, we wanted to determine if loss of MeCP2 modified hypoxia induced neuronal activation as an explanation of the abnormal HVD. Hypoxia induced neuronal activation was assayed by immunostaining for c-Fos reactivity in mice that were treated with hypoxic gas compared to mice allowed by continue breathing room air. We performed stereological quantification of c-Fos immunoreactivity in regions that are activated by exposure to hypoxia including the LC, PB, and NTS. All three anatomic regions showed an increase in c-Fos positive nuclei in both WT and in NULL animals; however, NULL mice had a 20% deficit in hypoxia induced c-Fos expressing nuclei in the NTS (Figure 4).
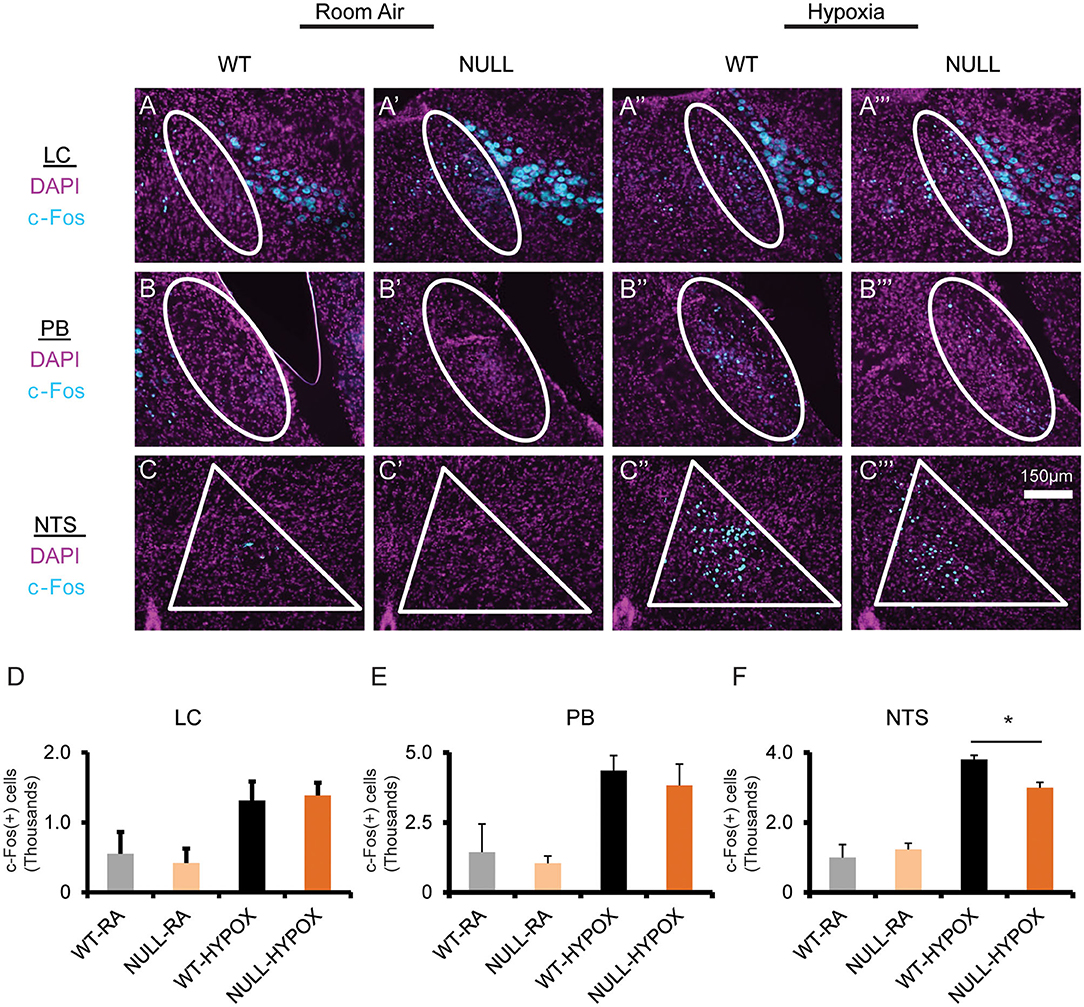
Figure 4. Mice lacking MeCP2 function exhibit decreased hypoxia induced neuronal activation. Treatment with hypoxia (10% O2, balance N2) for 3 h induces neuronal activation across several brainstem regions, as determined by immunofluorescent staining for c-Fos expression in coronal sections through the brainstem containing the locus coeruleus (LC) (A–A”'), Parabrachial Complex (PB) (B–B”'), and nucleus solitary tract (NTS) (C–C”'). Stereological quantification of c-Fos expressing nuclei identified an increase in neuronal activation in both WT and NULL mice during treatment with hypoxia in the LC (D), PB (E), and NTS (F). However, the quantity of neurons activated in the NTS of NULL mice was 20% lower than WT littermates (Room Air treated WT N = 2, Room Air treated NULL N = 2, Hypoxia treated WT N = 3, Hypoxia treated NULL N = 3). Indicated brain regions outlined in white. *p < 0.05 for indicated comparison by One-Way ANOVA with Student-Newman-Keuls post-hoc correction for multiple comparisons. RA, room air; HYPOX, hypoxia. Bar graphs indicate mean values, error bars indicate SEM.
Discussion
MeCP2 is required for an appropriate breathing rate response to acute hypoxic challenge. Genetic knockout and rescue of Mecp2 within glutamatergic, GABAergic, and dopaminergic/noradrenergic neurons indicates that MeCP2 function is required in a distributed set of neurons for a normal breathing response to hypoxia. Genetic knockout and rescue of Mecp2 within the caudal pons and medulla (using HoxB1-Cre) indicated that the critical neural circuitry for the normal HVD is within these brain regions (14, 22). In combination, this indicates that a neural circuit within the caudal pons and medulla comprised of excitatory, inhibitory, and dopaminergic/noradrenergic neurons underlie the HVD. Because within this region there are no dopaminergic neurons, it is likely that the MeCP2 requirement for normal HVD in TH-expressing neurons is solely within the noradrenergic neurons.
Other studies identified an increase in resting activity, as measured by c-Fos staining, within the NTS of NULL mice relative to WT controls; our current experiment is underpowered to confirm this in the room air condition (34). However, upon exposure to hypoxia, there is a clear increase in c-Fos expression in both genotypes, consistent with studies looking at brain regions activated by exposure to hypoxia (28). Interestingly, NULL mice have 20% fewer c-FOS expressing neurons within the NTS compared to WT controls. The c-FOS activation within the LC and PB did not show an effect size sufficient to be detected; however, the limited power of the present study prevents a definitive conclusion that these regions are not impacted in NULL mice. The impairment in hypoxia induced neuronal activation within the dorsal medulla's NTS, a key relay center in the processing of hypoxic stimuli, thus becomes an attractive candidate for contributions to the MeCP2 related impairment in HVD observed in NULL animals. The role for the NTS is also supported by previous anatomically focused conditional genetic studies which suggest key centers mediating the aberrant MeCP2 dependent hypoxic breathing phenotype do not extend beyond the rostral boundary of developmental expression of HoxB1 or HoxA4 (14, 22).
Excitatory/inhibitory balance within the NTS contributes to HVD (36). Inhibition of NMDA receptors within the NTS is thought to play a part in HVD, particularly via PDGF-beta signaling. This is due to observation of impaired HVD in mice heterozygous for a mutation in the PDGF-beta receptor as well as in mice lacking PDGF-beta expression in the brain (37, 38). Other studies have been able to demonstrate that treatment with the NMDA receptor agonist ketamine can ameliorate several phenotypes present in mice lacking MeCP2 including disrupted breathing and apnea (34, 39). We were unable to replicate the breathing improvement from ketamine treatment, potentially due to the limited number of animals tested, as well as to differences in dose and acute treatment vs. chronic dosing used in prior studies (Supplementary Table 3) (39). Similarly, we did not see any trend toward improvement of HVD from ketamine treatment.
Glutamatergic release in the NTS stimulated by hypoxia provides a pool of glutamate available for conversion to GABA and use for inhibitory signaling. The requirement of MeCP2 for GABA biosynthesis and GABAergic neuronal function suggest this as a likely contributor to the abnormal HVD observed in Mecp2 mutant mice (25). Consistent with this hypothesis, modulation of GABAergic function within the NTS of rats by microinjection with GABA agonists and antagonists modulates the ventilatory response to hypoxia (36).
The tyrosine hydroxylase neurons within the central nervous system also are involved in HVD, as blocking dopamine activity with haloperidol prevents HVD from occurring (40). Furthermore, A2/C2 adrenergic neurons within the NTS begin to lose expression of TH in the absence of MeCP2 by 1 month of age, correlating with the onset of the abnormal HVD observed in Mecp2 mutant mice (33). Optogenetic stimulation of C2 TH neurons genetically deficient for VGLUT2 suggest that the TH and VGLUT2 populations overlap and that TH dependent modulation of breathing is dependent on VGLUT2 expression (41).
The neuronal circuits that control the breathing response to hypoxia span multiple anatomic regions, and categories of neurons. The initially normal HVD observed in young NULL mice implies that MeCP2 function is not essential to the establishment of these circuits, but that it is critical across multiple components for their maintenance as the deficit manifests by 4 weeks of age. Pharmacological treatment of NULL mice targeting excitatory, inhibitory, and modulatory neurons suggests that the impairment in HVD is due to decreased activity across each of the populations.
While not the main objective of the current study, we were also able to monitor additional breathing parameters of baseline breathing including apnea and irregularity of breathing (Supplementary Tables 1–3). Overall, we did observe an expected increase in the incidence of apnea in NULL mice relative to WT animals as well as effects and trends indicating increased breathing irregularity scores in NULL mice. These non HVD outcome measures presented with large variation making definitive conclusions from the genetic and pharmacological studies underpowered with respect to apnea and breathing irregularity. However, we did note that some of the pharmacological treatments, such as L-DOPA with carbidopa, baclofen, and muscimol, modified breathing under sustained hypoxia and resulted in worsening of apnea as well as irregularity. Interestingly, these treatments also blunted the peak breathing response to hypoxia while tiagabine and desipramine reduced the breathing rate during sustained hypoxia while sparing the peak breathing response to hypoxia and did not appear to aggravate the incidence of apnea or breathing irregularity.
These experiments attempted to dissect the neuronal substrates modified by the absence of MeCP2 and result in impaired HVD. In this sense, the impaired HVD can be considered another biomarker used as an indicator for neurological impairment due to the loss of MeCP2. Our findings are consistent with the pleiotropic action of MeCP2, and with the distributed nature of the brainstem circuitry that regulates HVD (22), and provide insight into the neuronal cell and circuit basis of breathing abnormalities in RTT. As these breathing abnormalities may underlie the sudden and unexpected death seen in RTT, dissecting the pathophysiology and developing targeted therapeutic interventions may be able to help prevent these deaths.
It is important to note that while the experiments looking to dissect the HVD impairment were largely performed using male mice, we did observe the phenotype in female HET animals. Among the key differences between male and female animals with Mecp2 mutations is the mosaicism of MeCP2 expression within the neuronal populations under study due to Mecp2 being X-linked and subject to X-chromosome inactivation. Other studies have contrasted the effects of genetic studies between the male and female models. Typically, the phenotypes observed in the female models are less severe due to their mosaicism, but differential benefit may be observed depending on the system that is targeted for genetic restoration (24, 27). Specifically, this contrast is apparent within the genetic rescue of MeCP2 within the excitatory and inhibitory neuronal populations performed by Meng et al. and Ure et. al.; female mice showed greater benefit from rescue in the excitatory neurons while male mice showed greater benefit from rescue in the inhibitory neurons (24, 27). This effect is likely due to activity imbalances within their respective networks upon restoration of MeCP2 with network components expressing varying degrees of mosaicism. Overall, these and previous findings are encouraging regarding development of potential therapies for RTT because they provide additional evidence of neuronal circuits that remain present but are poorly tuned in the absence of MeCP2. Pharmacological or genetic therapies have the potential to re-adjust these circuits to a more normal state; however, full rescue will likely require broad acting interventions that target multiple neuronal populations to achieve balanced activity.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Baylor College of Medicine Institutional Animal Care and Use Committee.
Author Contributions
CW, T-WH, and JN contributed to conception and design of the study. CW, T-WH, JH, DP, and EA performed experiments and collected data. CW and CM developed analysis tools, CW analyzed the data. CM, RS, AI-I, XM, KU, and HZ, contributed to the genetic studies of Vglut2, Viaat, and Th conditional knock-out and rescue. CW and JN wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This project was supported by R01 HD062553 (JN), T32 NS043124 and F31 NS066601 (CW), U54 HD 083092 (BCM IDDRC Neurobehavioral Core), 5R01 NS057819 (HZ), F32 NS083137 (KU), UM1 HG006348, R01 DK114356, and R01 HL130249 (Mouse Metabolism and Phenotyping Core). HZ is an investigator with the Howard Hughes Medical Institute.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Components of the text and figures in this paper have appeared within a PhD thesis (CW). This project utilized resources from the Neurobehavioral Core, and the Mouse Metabolism and Phenotyping Core, which are subsidized by funding from NIH. The Mouse Metabolism and Phenotyping Core receives additional funding from Baylor College of Medicine as a member of its Advanced Technology Cores. The authors would like to thank Jonathan Merritt for thoughtful discussion over the data, David Corry and Paul Porter for technical assistance with plethysmography experiments, and Corey Reynolds for technical assistance with gas challenge experiments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2020.593554/full#supplementary-material
References
1. Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. (1999) 23:185–8. doi: 10.1038/13810
2. Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: rett's syndrome: report of 35 cases. Ann Neurol. (1983) 14:471–9. doi: 10.1002/ana.410140412
3. Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. (2010) 68:944–50. doi: 10.1002/ana.22124
4. Weese-Mayer DE, Lieske SP, Boothby CM, Kenny AS, Bennett HL, Silvestri JM, et al. Autonomic nervous system dysregulation: breathing and heart rate perturbation during wakefulness in young girls with rett syndrome. Pediatr Research. (2006) 60:443–9. doi: 10.1203/01.pdr.0000238302.84552.d0
5. Weese-Mayer DE, Lieske SP, Boothby CM, Kenny AS, Bennett HL, Ramirez JM. Autonomic dysregulation in young girls with rett syndrome during nighttime in-home recordings. Pediatr Pulmonol. (2008) 43:1045–60. doi: 10.1002/ppul.20866
6. Kerr AM, Armstrong DD, Prescott RJ, Doyle D, Kearney DL. Rett syndrome: analysis of deaths in the British survey. Eur Child Adolesc Psychiatry. (1997) 1:71–4.
7. Viemari JC, Roux JC, Tryba AK, Saywell V, Burnet H, Pena F, et al. Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J Neurosci. (2005) 25:11521–30. doi: 10.1523/JNEUROSCI.4373-05.2005
8. Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic rett syndrome. Nat Genet. (2001) 27:322–6. doi: 10.1038/85899
9. Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of rett syndrome. Science. (2007) 315:1143–7. doi: 10.1126/science.1138389
10. Pitcher MR, Herrera JA, Buffington SA, Kochukov MY, Merritt JK, Fisher AR, et al. Rett syndrome like phenotypes in the R255X Mecp2 mutant mouse are rescued by MECP2 transgene. Hum Mol Genet. (2015) 24:2662–72. doi: 10.1093/hmg/ddv030
11. Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. (2001) 27:327–31. doi: 10.1038/85906
12. Bissonnette JM, Knopp SJ. Separate respiratory phenotypes in methyl-CpG-binding protein 2 (Mecp2) deficient mice. Pediatr Res. (2006) 59:513–8. doi: 10.1203/01.pdr.0000203157.31924.4a
13. Roux JC, Dura E, Moncla A, Mancini J, Villard L. Treatment with desipramine improves breathing and survival in a mouse model for Rett syndrome. Eur J Neurosci. (2007) 25:1915–22. doi: 10.1111/j.1460-9568.2007.05466.x
14. Ward CS, Arvide EM, Huang TW, Yoo J, Noebels JL, Neul JL. MeCP2 is critical within HoxB1-derived tissues of mice for normal lifespan. J Neurosci. (2011) 31:10359–70. doi: 10.1523/JNEUROSCI.0057-11.2011
15. Kumar P, Prabhakar NR. Peripheral chemoreceptors: function and plasticity of the carotid body. Compr Physiol. (2012) 2:141–219. doi: 10.1002/cphy.c100069
16. Teppema LJ, Dahan A. The ventilatory response to hypoxia in mammals: mechanisms, measurement, and analysis. Physiol Rev. (2010) 90:675–754. doi: 10.1152/physrev.00012.2009
17. Guyenet PG, Regulation of breathing and autonomic outflows by chemoreceptors. Compr Physiol. (2014) 4:1511–62. doi: 10.1002/cphy.c140004
18. Steinback CD, Poulin MJ. Ventilatory responses to isocapnic and poikilocapnic hypoxia in humans. Respir Physiol Neurobiol. (2007) 155:104–13. doi: 10.1016/j.resp.2006.05.006
19. Easton PA, Slykerman LJ, Anthonisen NR. Ventilatory response to sustained hypoxia in normal adults. J Appl Physiol. (1985). (1986) 61:906–11. doi: 10.1152/jappl.1986.61.3.906
20. Pamenter ME, Powell FL. Time domains of the hypoxic ventilatory response and their molecular basis. Compr Physiol. (2016) 6:1345–85. doi: 10.1002/cphy.c150026
21. Powell FL, Milsom WK, Mitchell GS. Time domains of the hypoxic ventilatory response. Respir Physiol. (1998) 112:123–34. doi: 10.1016/S0034-5687(98)00026-7
22. Huang TW, Kochukov MY, Ward CS, Merritt J, Thomas K, Nguyen T, et al. Progressive changes in a distributed neural circuit underlie breathing abnormalities in mice lacking MeCP2. J Neurosci. (2016) 36:5572–86. doi: 10.1523/JNEUROSCI.2330-15.2016
23. Neubauer JA, Melton JE, Edelman NH. Modulation of respiration during brain hypoxia. J Appl Physiol. (1990) 68:441–51. doi: 10.1152/jappl.1990.68.2.441
24. Ure K, Lu H, Wang W, Ito-Ishida A, Wu Z, He LJ, et al. Restoration of Mecp2 expression in GABAergic neurons is sufficient to rescue multiple disease features in a mouse model of rett syndrome. Elife. (2016) 5:e15. doi: 10.7554/eLife.14198.015
25. Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and rett syndrome phenotypes. Nature. (2010) 468:263–9. doi: 10.1038/nature09582
26. Samaco RC, Mandel-Brehm C, Chao HT, Ward CS, Fyffe-Maricich SL, Ren J, et al. Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proc Natl Acad Sci USA. (2009) 106:21966–71. doi: 10.1073/pnas.0912257106
27. Meng X, Wang W, Lu H, He LJ, Chen W, Chao ES, et al. Manipulations of MeCP2 in glutamatergic neurons highlight their contributions to Rett and other neurological disorders. Elife. (2016) 5:e20. doi: 10.7554/eLife.14199.020
28. Teppema LJ, Veening JG, Kranenburg A, Dahan A, Berkenbosch A, Olievier C. Expression of c-fos in the rat brainstem after exposure to hypoxia and to normoxic and hyperoxic hypercapnia. J Comp Neurol. (1997) 388:169–90. doi: 10.1002/(SICI)1096-9861(19971117)388:2<169::AID-CNE1<>3.0.CO;2-#
29. Schneider CA, Rasband WS, Eliceiri KW. NIH image to imageJ: 25 years of image analysis. Nat Methods. (2012) 9:671–5. doi: 10.1038/nmeth.2089
30. Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. Amsterdam; Boston, MA: Elsevier Academic Press (2004)
31. Voituron N, Zanella S, Menuet C, Dutschmann M, Hilaire G. Early breathing defects after moderate hypoxia or hypercapnia in a mouse model of rett syndrome. Respir Physiol Neurobiol. (2009) 168:109–18. doi: 10.1016/j.resp.2009.05.013
32. Voituron N, Zanella S, Menuet C, Lajard AM, Dutschmann M, Hilaire G. Early abnormalities of post-sigh breathing in a mouse model of Rett syndrome. Respir Physiol Neurobiol. (2010) 170:173–82. doi: 10.1016/j.resp.2009.12.009
33. Roux JC, Dura E, Villard L. Tyrosine hydroxylase deficit in the chemoafferent and the sympathoadrenergic pathways of the Mecp2 deficient mouse. Neurosci Lett. (2008) 447:82–6. doi: 10.1016/j.neulet.2008.09.045
34. Kron M, Howell CJ, Adams IT, Ransbottom M, Christian D, Ogier M, et al. Brain activity mapping in Mecp2 mutant mice reveals functional deficits in forebrain circuits, including key nodes in the default mode network, that are reversed with ketamine treatment. J Neurosci. (2012) 32:13860–72. doi: 10.1523/JNEUROSCI.2159-12.2012
35. Zanella S, Mebarek S, Lajard AM, Picard N, Dutschmann M, Hilaire G. Oral treatment with desipramine improves breathing and life span in Rett syndrome mouse model. Respir Physiol Neurobiol. (2008) 160:116–21. doi: 10.1016/j.resp.2007.08.009
36. Tabata M, Kurosawa H, Kikuchi Y, Hida W, Ogawa H, Okabe S, et al. Role of GABA within the nucleus tractus solitarii in the hypoxic ventilatory decline of awake rats. Am J Physiol Regul Integr Comp Physiol. (2001) 281:R1411–9. doi: 10.1152/ajpregu.2001.281.5.R1411
37. Gozal D, Simakajornboon N, Czapla MA, Xue YD, Gozal E, Vlasic V, et al. Brainstem activation of platelet-derived growth factor-beta receptor modulates the late phase of the hypoxic ventilatory response. J Neurochem. (2000) 74:310–9. doi: 10.1046/j.1471-4159.2000.0740310.x
38. Tsunekawa S, Ohi Y, Ishii Y, Sasahara M, Haji A. Hypoxic ventilatory response in platelet-derived growth factor receptor-beta-knockout mice. J Pharmacol Sci. (2009) 110:270–5. doi: 10.1254/jphs.09102FP
39. Patrizi A, Picard N, Simon AJ, Gunner G, Centofante E, Andrews NA, et al. Chronic administration of the n-methyl-d-aspartate receptor antagonist ketamine improves rett syndrome phenotype. Biol Psychiatry. (2016) 79:755–64. doi: 10.1016/j.biopsych.2015.08.018
40. Tatsumi K, Pickett CK, Weil JV. Effects of haloperidol and domperidone on ventilatory roll off during sustained hypoxia in cats. J Appl Physiol. (1992) 72:1945–52. doi: 10.1152/jappl.1992.72.5.1945
41. Abbott SB, Holloway BB, Viar KE, Guyenet PG. Vesicular glutamate transporter 2 is required for the respiratory and parasympathetic activation produced by optogenetic stimulation of catecholaminergic neurons in the rostral ventrolateral medulla of mice in vivo. Eur J Neurosci. (2014) 39:98–106. doi: 10.1111/ejn.12421
Keywords: Rett, MeCP2, hypoxia, sudden death, biomarker, breathing abnormalities, genetic manipulation, pharmacological manipulation
Citation: Ward CS, Huang T-W, Herrera JA, Samaco RC, McGraw CM, Parra DE, Arvide EM, Ito-Ishida A, Meng X, Ure K, Zoghbi HY and Neul JL (2020) Loss of MeCP2 Function Across Several Neuronal Populations Impairs Breathing Response to Acute Hypoxia. Front. Neurol. 11:593554. doi: 10.3389/fneur.2020.593554
Received: 10 August 2020; Accepted: 06 October 2020;
Published: 30 October 2020.
Edited by:
Ana Paula Abdala, University of Bristol, United KingdomReviewed by:
Erica S. Levitt, University of Florida, United StatesMathias Dutschmann, University of Melbourne, Australia
Copyright © 2020 Ward, Huang, Herrera, Samaco, McGraw, Parra, Arvide, Ito-Ishida, Meng, Ure, Zoghbi and Neul. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher S. Ward, Y2hyaXN0b3BoZXIud2FyZEBiY20uZWR1; Jeffrey L. Neul, amVmZnJleS5sLm5ldWxAdnVtYy5vcmc=
†Present address: Jose A. Herrera, Department of Biology, South Texas College, Weslaco, TX, United States
Christopher M. McGraw, Department of Neurology, Massachusetts General Hospital and Harvard Medical School, Boston, MA, United States