 Yacov Balash
Yacov Balash Amos D. Korczyn
Amos D. Korczyn Nadejda Khmelev
Nadejda Khmelev Anda Eilam1
Anda Eilam1 Meital Adi
Meital Adi- 1Department of Neurology, Kaplan Medical Center, Rehovot, Israel
- 2Department of Neurology, Tel-Aviv University, Tel Aviv, Israel
Only a few case reports of stroke-like onset of Creutzfeldt-Jakob disease (CJD) have previously been published. We aimed to analyze the neurological, imaging, electroencephalographic (EEG), and laboratory features of patients with this very rare phenomenon. Here, we review the clinical characteristics, onset features, and clinical course variants of stroke-like CJD in 23 such patients. The median age of the patients was 71 years (range: 56–84 years); 12 were women. In 20 patients, CJD was sporadic. Thirteen patients developed apoplexy-like onset of symptoms, whereas the others had prodromal non-specific complaints. Most often the patients manifested with pyramidal signs (n = 13), ataxia (n = 9), and aphasia (n = 8). On MRI DWI sequence, all subjects had abnormal hyperintensities in various parts of the cerebral cortex, striatum, or thalamus, while EEG detected periodic triphasic waves only in 11. CSF 14-3-3 protein and total τ-protein were abnormal in 17 of 23 cases. All patients died, median lifespan being 2 months (range: 19 days−14 months). In conclusion, a complex of clinical, radiological, and laboratory manifestations of stroke-like onset of CJD is outlined. The clinical relationships between CJD and stroke are considered, in an attempt to highlight this rare presentation of an uncommon disease.
Introduction
Creutzfeldt-Jakob disease (CJD) is a rare neurodegenerative disease caused by accumulated misfolded proteins (prions, PrPSc), with their deposition in the cortex, striatum, and thalamus (1). The clinical hallmarks of the disease are progressive dementia, behavioral disorders, epileptic seizures, ataxia, myoclonus, and a variety of other movement disorders (2). Stroke-like onset of CJD is considered as very rare. Indeed, only a single case has been identified in a recent series of cases from China (3). But in an earlier important review 5.6% (30/532) of patients with “probable” or “definite” CJD collected in the UK from 1970 to 1993 had stroke-like onset (4).
In recent years, new observations of this neuropsychiatric syndrome have been reported using improved imaging, laboratory, and genetic methods. However, they were presented as scattered case reports.
To summarize and supplement the clinical picture of stroke-like appearance of CJD and to identify its relationships with actual cases of cerebrovascular accidents, we analyzed all published cases of the disease available in the literature and added a case seen by us.
Methods
Patients and Procedures
The clinical information was obtained from the published English and Japanese literature over the past 25 years and is supplemented by one original case. Cases were identified in EMBASE, PubMed, and Scopus biomedical databases, using keywords: Creutzfeldt-Jakob disease AND stroke, cerebro-vascular accident.
Inclusion criteria were as follows: (1) clinical situation when the onset of CJD was initially misdiagnosed as a stroke; (2) subsequent workup indicated probable or definite CJD according to the international criteria (5); or (3) detailed clinical case description including MRI, electroencephalogram (EEG), and cerebrospinal fluid (CSF) studies when available were consistent with the diagnosis of CJD. Data on brain biopsy and/or autopsy data, if performed, were included.
Statistical Analysis
Data were analyzed using a Microsoft Excel 2010 spreadsheet. Results were expressed as means with standard deviations (SD) or as median with interquartile range (IQR).
Results
Twenty-three clinical cases (cases 1–23) were analyzed. The mean age of the subjects was 70.1 years (SD = 7.3, median = 71, IQR: 66–75), of whom 12 (52.2%) were females. In 20 cases, sporadic CJD was diagnosed, in two cases, the disease was familial [cases 6 (6) and 7 (7)], and in another case, a mutation was identified characteristic of familial CJD, but no other cases were reported in his family [case 20 (8)] (Table 1).

Table 1. Demographic characteristics and clinical features in 23 patients with stroke-like onset of CJD.
Clinical Vignette (Case 23)
A 71-year-old right-handed woman was evaluated in the emergency department with a 2-day history of asthenia, dizziness, gait instability, speech disorders, and periodic involuntary jerky movements in her left arm. Past medical history included mild well-controlled hypertension. On admission, she was disorientated, her cranial nerves were normal, she had mild weakness and jerky dystonia with hyperactive tendon reflexes, and suspected hemi-ataxia were seen in the left arm. Plantar responses were flexor. A non-enhanced CT scan revealed a left parietal hypodense focus and cerebellar calcifications, which could not explain ipsilateral symptoms. At this stage the patient was clinically diagnosed with possible ischemic stroke in the right hemisphere. However, over a 3-week hospitalization period, her condition progressively deteriorated. She became negativistic and non-compliant. A psychiatric examination revealed inappropriate affect with euphoria and visual hallucinations. Her MoCA (9) score (day 5) was 19/30 and later declined to 16/30 (day 12). The myoclonic jerks (Supplementary Video 1) in the left arm were accompanied by rigidity, general weakness, incontinence, and eventually inability to stand and walk. No visual impairment or startle responses were seen. Repeated EEG (days 2, 5, and 10) disclosed diffuse delta activity with periodic triphasic sharp wave complexes mostly at the right fronto-parietal area in later records (days 5 and 10) (Figure 1). A brain MRI (day 6) showed multiple bilateral microvascular and macrovascular ischemic periventricular white matter lesions. DWI imaging (day 8) revealed restriction in diffusion with ribbon signs in the right parietal cortex (Figures 2A,B). This raised suspicion toward a stroke-like onset CJD.
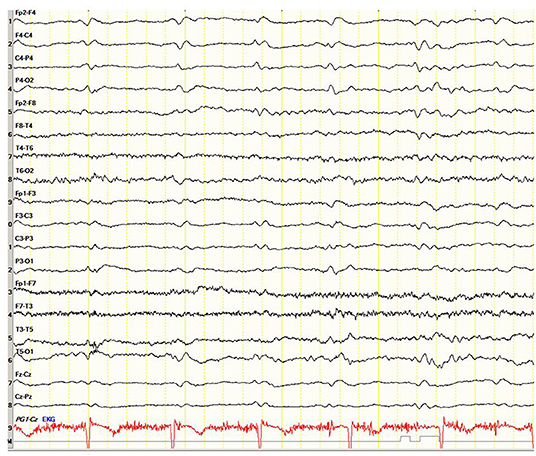
Figure 1. Case 23. Electroencephalogram showing periodic triphasic sharp wave complexes mostly at the right fronto-parietal area.
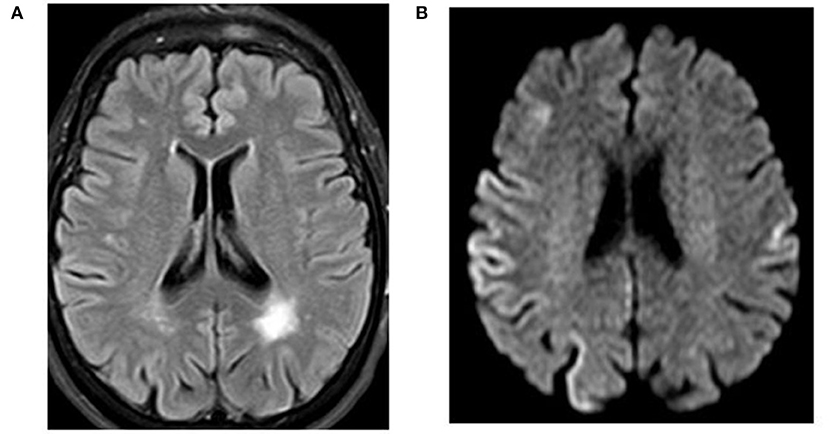
Figure 2. Case 23. Magnetic resonance imaging of the brain. (A) FLAIR indicating a left parietal lobe old infarction. (B) Diffusion-weighted magnetic resonance imaging with restricted diffusion at the right parietal cortex.
CSF exhibited no cells; glucose was 72 mg/dl, protein 34 mg/dl. However, total τ-protein was markedly elevated up to 2,524 pg/ml (normal level <240 pg/ml), suggesting a diagnosis of probable sporadic CJD according to the updated diagnostic criteria (5). Other laboratory data showed normal hematologic, biochemical, endocrine, and vitamin levels. Autoimmune workups, viral and bacterial screens including HIV and treponema pallidum, as well as a paraneoplastic antibodies panel, was negative. The patient was discharged with the diagnosis of probable sporadic CJD and died in a nursing home nearly 3 weeks later. Autopsy has not been performed.
Thirteen patients (56.5%) in this series presented with sudden apoplectic-like or wake up symptoms and (visual field defect or double vision, speech disorder, acute vertigo, and sudden hearing loss), motor disorders mono- or hemiparesis, dysmetria, dystonia, alien hand, or combined disorders (Figure 3). Ten other patients (43.5%) reported prodromal complaints (weakness, insomnia, personality changes, dysarthria, word finding difficulty, memory impairment or confusion, dizziness, and unsteadiness) lasting from several weeks to 6 months prior to an acute stroke-like deterioration, which in retrospect could be ascribed to incipient onset of CJD. Sixteen patients (69.6%) had one or more vascular risk factors most commonly hypertension (11 individuals, 47.8%), and two of them (8.7%) had a history of previous stroke or recurrent stroke-like symptoms [cases 4 (10), 9 (11)]. In several cases [7 (7), 13 (12), 16 (13), 18 (14), 20 (8)] the patients were discharged for outpatient or rehabilitation treatment after a temporary stabilization of their condition over 2–8 weeks.
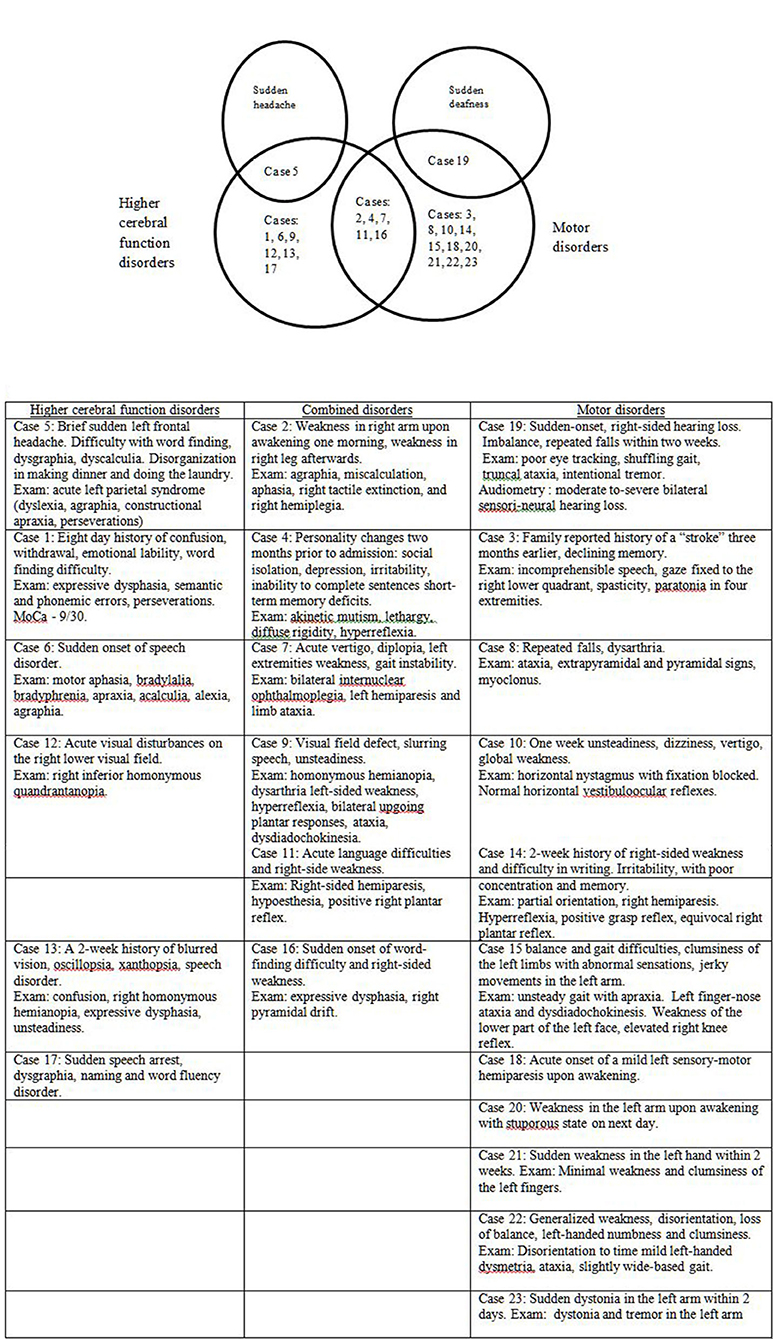
Figure 3. Initial neurological symptoms and signs in 23 patients with stroke-like onset of CJD.
Other differential diagnoses were antibody-mediated autoimmune encephalitis [cases 4 (10) and 11 (15)] treated with methylprednisolone pulse therapy and non-convulsive status epilepticus due to sharp delta activity [case 5 (16)] treated with levetiracetam and phenytoin.
The vast majority of patients, 20/23 (87%), showed a rapid progressive deterioration after the acute onset in both motor and cognitive state, followed by unresponsiveness, blindness, akinetic mutism, stupor, ataxia, generalized rigidity, spasticity, hyperreflexia, and myoclonus. In other 3 patients (13%) [cases 7 (7), 14 (17), 20 (8)] the deterioration to bedridden and mute condition was prolonged, lasting 9-14 months.
The mean survival of the 23 patients from the stroke-like onset was 4.2 months (SD = 4.1, median = 2, IQR: 1.1–5), range 19 days−14.2 months.
Brain MRI studies were performed in 22/23 patients (95.6%) and in 13 of them (59.1%) within 1–14 days from the onset of CJD symptoms. Four patients in this group had MRI not characteristic for CJD (18.2%). In four other patients, non-enhanced MRI images revealed unilateral or bilateral diffuse areas of abnormal high signal intensities with restriction in diffusion (ribbon sign) in various parts of the cerebral cortex (18.2%), and in the remaining five patients (22.7%), there was a combined lesion of the cortex and basal ganglia (hyperintensity of the head of caudate nucleus and putamen) on DWI sequence. In the remaining nine patients (40.9%), MRI images were performed later within two months from the onset of the apoplectic presentation. All of them were pathological, often with combined lesions of various parts of the cortex and basal ganglia (caudate nucleus, thalamus, and lentiform nucleus) (see Table 2).
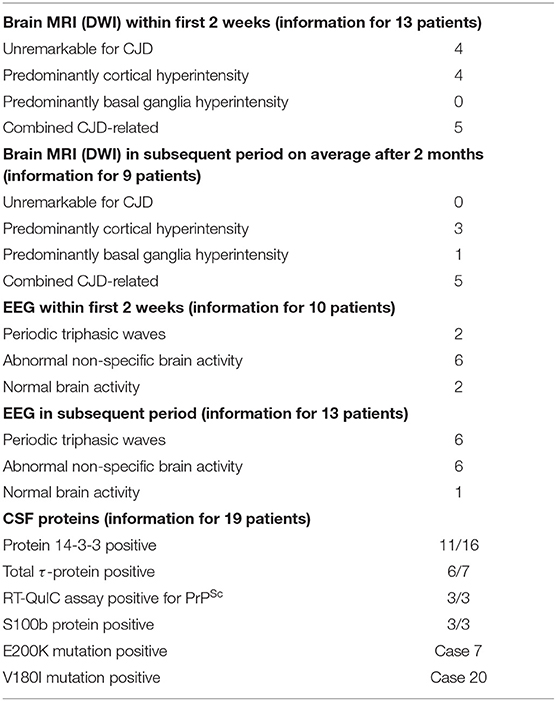
Table 2. Ancillary tests in 23 patients with stroke-like onset of CJD.
Along with the above mentioned MRI signs characteristic of CJD, in 4/22 cases (18.2%), T2 FLAIR in the first 3–4 days detected only scattered hyperintensities in the centrum semiovale [cases 8 (18), 9 (11), 15 (19), and 22 (20)] and in two other patients [cases 7 (7) and 19 (21)] DWI disclosed a combination of cortical ribboning hyperintensites with non-specific periventricular vascular changes. In the abovementioned studies [cases 7 (7) and 19 (21)], MRI performed during the first days revealed only an acute lesion at the left pontine-midbrain border compatible with an ischemic stroke and small vessel ischemic changes in periventricular areas of the brain but were otherwise normal. In case 7 (7), repeated MRI studies 3, 5, and 8 months after symptoms onset showed increasing DWI hyperintense lesions in the thalamus, basal ganglia, and anterior cingulate together with the gradual shrinkage and disappearance of the ischemic brainstem lesion in the last MRI (Table 2).
In one case, MRI changes characteristic of CJD were detected one month before the acute onset of the clinical picture, but the diagnosis was not made at that time due to the absence of clinical symptoms other than repeated falls [case 8 (18)].
Single or repeated EEG were recorded in all 23 studies. During the first 2 weeks after the CJD symptoms onset, the initial recordings were performed in 10/23 patients (43.5%). In two subjects, the EEG was within the normal range, in six, non-specific changes were registered (diffuse slowing with theta–delta activity, generalized dysrhythmia, focal abnormalities, and epileptiform discharges), while periodic sharp waves were observed in only two individuals. In a later period, on average 55 ± 4.5 days, in 6/23 patients (26.1%), the EEG changes remained non-specific, whereas in 6/23 cases, periodic triphasic waves were recorded (26.1%). In one patient, the EEG remained normal at 7 months after the onset of the disease shortly before death [case 8 (18)] (Table 2).
CSF 14-3-3 protein was studied in 16 patents and was positive in 11 (68.8%), τ-protein was measured in seven patients and was elevated on average to 1,924 ± 1,459 pg/ml in six cases (range: 789–4,219). RT-QuIC assay was positive in three cases: 1 (22), 2 (23), and 4 (10). S100B CSF protein was positive in three cases: 1 (22), 3 (24), and 9 (11).
Genetic analysis was abnormal in case 7 (7) (E200K). One other case revealed Val–>Ile at codon 180 [case 20 (8)].
Autopsy was performed in 9/23 cases (39.1%) and confirmed spongiform encephalopathy with diffuse synaptic pattern of prion protein immunoreactivity. None of them had changes consistent with an acute stroke. A brain biopsy, which verified the diagnosis, was performed in one patient. In general, these pathologically confirmed cases (Table 3) were not different from the other 13 patients.
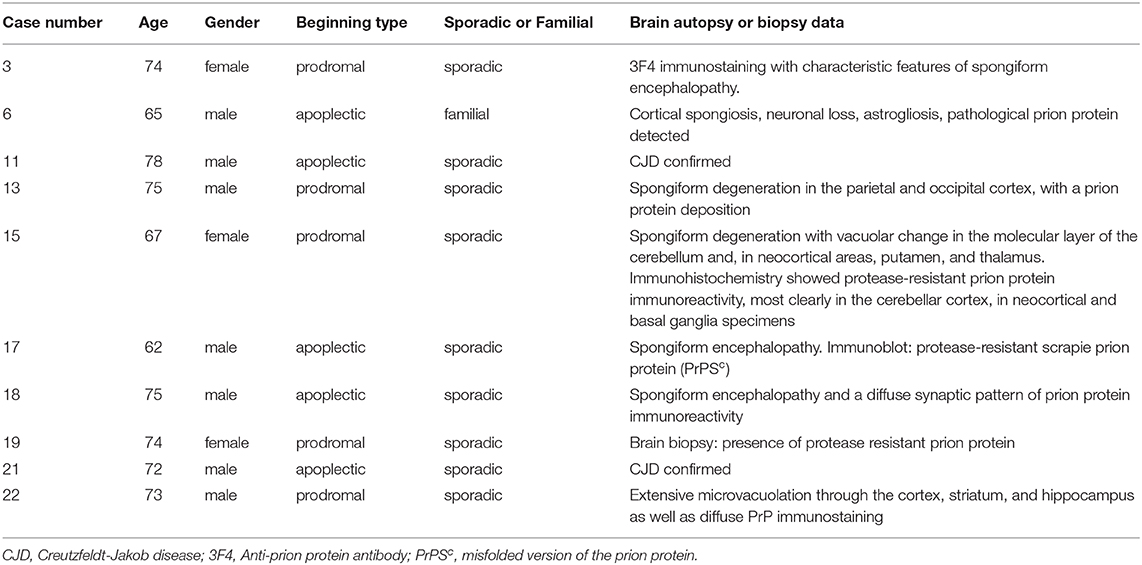
Table 3. Brain autopsy or biopsy data in 10 patients with apoplectic onset of CJD.
Discussion and Conclusions
In our case, as in the other cases cited, the sudden onset of neurological disorders suggested an acute stroke, but further developments and tests revealed the diagnosis of CJD.
The available data on the relationship between CJD and stroke, suggest several possibilities.
The apoplectic onset, presumably caused by local neuronal shutdown induced by rapid prion propagation, was initially diagnosed as stroke, and only later progressive clinical worsening supplanted by MRI, EEG, or CSF data indicated prion disease. No evidence for acute vascular brain lesion was observed on initial CT or MRI scans. This situation occurred in the majority of the cited cases: 1 (22), 4 (10), 6 (6), 8 (18), 10 (25), 12 (26), 13 (12), 14 (17), 17 (27), 20 (8), 21 (28), and 22 (20).
In our subject (case 23), as in some others [cases 2 (23), 9 (11), 15 (19), 16 (13), and 18 (14)], stroke-like CJD was initially diagnosed clinically as true vascular disease of the brain and CT or MRI imaging have suggested acute or chronic cerebrovascular disease. However, further clinical deterioration which occurred within 3–4 weeks forced the revision or repeat of MRI, which now revealed DWI cortical or subcortical hyperintensities and/or elevated CSF τ-protein or positive 14-3-3 protein. On initial MRI in some patients [cases 5 (16), 7 (7), 9 (11), and 19 (21)] with stroke-like onset CJD, there could be visible only vascular changes on FLAIR MRI studies without DWI features characteristic of CJD.
One patient was diagnosed with a stroke with progressive neurological deterioration. After her death, however, this diagnosis was not confirmed at autopsy, and instead, showed positive 3F4 immunostaining with characteristic features of spongiform encephalopathy along with PRNP gene sequence analysis with 129 polymorphism valine homozygosity (VV2) [case 3 (24)].
In another case, the clinical picture was apoplectic-like, the MRI data indicated CJD, showing abnormal high signal intensities in the contralateral frontal cerebral cortex, but both CSF protein 14-3-3 and PRNP gene analysis were negative. The diagnosis of CJD was established post-mortem with PRNP gene analysis where subtype MM/MV1 + 2C was uncovered [case 11 (15)].
Thus, the diagnosis of CJD should be considered in every stroke-like patient in whom the suspicion of stroke is not confirmed by MRI, particularly if a history of recent progressive cognitive, behavioral, or movement disorders can be elicited. Of course, the characteristic changes in DWI and FLAIR MRI should alert the clinicians to this possibility. In such situations, CSF τ-protein or RT-QuIC assay should be examined and repeat MRI should be performed.
It is remarkable that all the cases described in the literature had a stroke-like incidence of CJD at the disease onset. We have not seen a description of a stroke-like deterioration in a patient already diagnosed as CJD. However, this probably reflects reporting bias.
The causes of stroke-like onset CJD remain unknown. Possibly, they are related to the rate of prions accumulation, for which there may be a genetic reason (15). However, the PRNP gene sequence analysis in patients with stroke-like sporadic CJD was performed only in 2/23 studies [case 3 (24)—VV2 at the polymorphic codon 129; case 11 (15)—subtype MM/MV1 + 2C PrPSc-type accumulation, (29)], which did not allow us to draw any conclusions.
Currently, the gold standard method for definitive CJD diagnosis during life remains a brain biopsy. This is rarely done due to the risk of additional brain damage in the absence of curative treatment of the disease. Therefore, the developments of less invasive methods of intravital tissue diagnostics for prion diseases, such as olfactory mucosa washout or skin biopsies, remain relevant (30).
In general, our conclusions are similar to those of McNaughton and Will (4) from over 20 years ago, but the cases reported here show a wide spectrum of MRI results.
As expected in a retrospective case series, not all patients have been investigated in the same way. Moreover, this series cannot be regarded as representative. We presume that many cases are not diagnosed correctly and others are not reported.
This series of cases is reported in order to increase awareness of the possibility that CJD may present in an apoplectic form, which may lead to erroneous management. Since MRI is now more commonly performed in acute stroke, the characteristic ribbon-like cortical signal should indicate the correct diagnosis. While we have focused on the acute onset of symptoms, probably an important red flag should be the progressive clinical deterioration of the patient afterwards.
In conclusion, CJD may have an apoplectic onset and clinicians should be alert to this possibility to avoid the consequences of misdiagnosis.
Consent for Publication
We obtained written informed consent for publication from the family of our patient (case 23).
Author Contributions
YB contributed to the conception, execution, design, and preparation of the manuscript for publication. AK contributed to the conception, design, and review of the manuscript. NK contributed to the execution and preparation of the manuscript for publication. AE contributed to the execution and review of the manuscript. RG contributed to the conception, execution, and review of the manuscript. MA contributed to the design and review of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the patient's family for their cooperation and for permission to use this case for publication.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.613991/full#supplementary-material
Supplementary Video 1. Case 23. Myoclonic jerks in the left shoulder and arm in 71 year old woman with probable CJD.
Abbreviations
CJD, Creutzfeldt-Jakob's disease; CSF, cerebrospinal fluid; CT, computed tomography; CVA, cerebrovascular accident; DWI, diffusion-weighted magnetic resonance imaging; EEG, electroencephalogram; FLAIR, fluid-attenuated inversion recovery imaging; HIV, the human immunodeficiency viruses; MoCA, Montreal Cognitive Assessment; MRI, magnetic resonance imaging; PRNP, (prion protein) gene encoding for the major prion protein; PrPSc, scrapie isoform of the prion protein; RT-QuIC, real-time quaking-induced conversion assay.
References
1. Gambetti P, Puoti G, Cali I, Kong Q, Zou W. Creutzfeldt-Jakob disease. In: Verhagen Metman L, Kompoliti K, editors. Encyclopedia of Movement Disorders. Chicago, IL: Rush University Medical Center (2011). p. 263–9. doi: 10.1016/B978-0-12-374105-9.00166-0
2. Tee BL, Longoria Ibarrola EM, Geschwind MD. Prion disease. Neurol Clin. (2018) 36:865–97. doi: 10.1016/j.ncl.2018.07.005
3. Zhao W, Zhang JT, Xing XW, Huang DH, Tian CL, Jia WQ, et al. Chinese specific characteristics of sporadic Creutzfeldt-Jakob disease: a retrospective analysis of 57 cases. PLoS ONE. (2013) 8:e58442. doi: 10.1371/journal.pone.0058442
4. McNaughton HK, Will RG. Creutzfeldt–Jakob disease presenting acutely as stroke: an analysis of 30 cases. Neurol Infect Epidemiol. (1997) 2:19–24.
5. Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. (2009) 132:2659–68. doi: 10.1093/brain/awp191
6. Necpál J, Stelzer M, Koščová S, Patarák M. A corticobasal syndrome variant of familial Creutzfeldt-Jakob disease with stroke-like onset. Case Rep Neurol Med. (2016) 2016:4167391 Case6. doi: 10.1155/2016/4167391
7. Cohen OS, Kimiagar I, Korczyn AD, Nitsan Z, Appel S, Hoffmann C, et al. Unusual presentations in patients with E200K familial Creutzfeldt-Jakob disease. Eur J Neurol. (2016) 23:871–7 Case7. doi: 10.1111/ene.12955
8. Kamogawa K, Toi T, Okamoto K, Okuda B. A case of Creutzfeldt-Jakob disease with stroke-like episode as an initial symptom. Nihon Ronen Igakkai Zasshi. (2009) 46:458–61 Case20. doi: 10.3143/geriatrics.46.458
9. Nasreddine ZS, Phillips NA, Bédirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. (2005) 53:695–9. doi: 10.1111/j.1532-5415.2005.53221.x
10. Salazar R. Atypical presentation of probable Creutzfeldt-Jakob disease associated with anti-Zic4 antibody: literature review of neuronal antibodies in Creutzfeldt-Jakob disease. Clin Neurol Neurosurg. (2018) 168:72–6 Case4. doi: 10.1016/j.clineuro.2018.02.043
11. Ghadiri-Sani M, Sekhar A, Larner AJ. A stroke of ill-fortune: an unexpected diagnosis following a stroke-like event. Br J Hosp Med (Lond). (2015) 76:54–5 Case9. doi: 10.12968/hmed.2015.76.1.54
12. Hirst CL. Sporadic Creutzfeldt-Jakob disease presenting as a stroke mimic. Br J Hosp Med (Lond). (2011) 72:590–1 Case13. doi: 10.12968/hmed.2011.72.10.590
13. Winn S, Bhalla A. Sporadic Creutzfeldt-Jakob disease presenting as stroke mimic. Br J Hosp Med (Lond). (2008) 69:228–9 Case16. doi: 10.12968/hmed.2008.69.4.28981
14. Szabo K, Achtnichts L, Grips E, Binder J, Gerigk L, Hennerici M, et al. Stroke-like presentation in a case of Creutzfeldt-Jakob disease. Cerebrovasc Dis. (2004) 18:251–3 Case18. doi: 10.1159/000080109
15. Damato V, Cuccagna C, Costantini EM, Gaudino S, Colosimo C, Parchi P, et al. Creutzfeldt-Jakob disease manifesting as stroke mimic in a 78-year-old patient: pitfalls and tips in the diagnosis. J Neurol Sci. (2014) 346:343–4 Case11. doi: 10.1016/j.jns.2014.08.026
16. Sharma DK, Boggild M, van Heuven AW, White RP. Creutzfeldt-Jakob disease presenting as stroke: a case report and systematic literature review. Neurologist. (2017) 22:48–53 Case5. doi: 10.1097/NRL.0000000000000107
17. Ko KF, Lau WY, Cheng WK, Kwan MC, Yip LK. Creutzfeldt-Jakob disease with initial right hemiparesis masquerading as a stroke. Hong Kong Med J. (2010) 16:487–8 Case 14.
18. Magny E, Sagot C, Cohen-Bittan J, Makdessi S, Bertrand A, Verny M, et al. Probable Creutzfeldt-Jakob disease mimicking a perioperative stroke in an elderly adult. J Am Geriatr Soc. (2015) 63:1268–9 Case8. doi: 10.1111/jgs.13452
19. Lyytinen J, Sairanen T, Valanne L, Salmi T, Paetau A, Pekkonen E. Progressive stroke-like symptoms in a patient with sporadic Creutzfeldt-Jakob disease. Case Rep Neurol. (2010) 2:12–8 Case15. doi: 10.1159/000289177
20. Kleiner-Fisman G, Bergeron C, Lang AE. Presentation of Creutzfeldt-Jakob disease as acute corticobasal degeneration syndrome. Mov Disord. (2004) 19:948–9 Case22. doi: 10.1002/mds.20140
21. Krishna P, Bauer C. Hearing loss as the initial presentation of Creutzfeldt-Jakob disease. Ear Nose Throat J. (2004) 83:535–43 Case19. doi: 10.1177/014556130408300812
22. Podger H, Ipe A. An unusual presentation of sporadic Creutzfeldt-Jakob disease. Age Ageing. (2020) 49:490–2 Case1. doi: 10.1093/ageing/afaa013
23. Okamoto K, Abe T, Itoh Y. A case of Creutzfeldt-Jakob disease with stroke-like onset. J Stroke Cerebrovasc Dis. (2020) 29:104788 Case2. doi: 10.1016/j.jstrokecerebrovasdis.2020.104788
24. Oliver M, Dyke L, Rico A, Madruga M, Parellada J, Carlan SJ. Rapidly progressing sporadic Creutzfeldt-Jakob disease presenting as a stroke. Case Rep Neurol. (2018) 10:261–5 Case3. doi: 10.1159/000492613
25. Mantokoudis G, Saber Tehrani AS, Newman-Toker DE. An unusual stroke-like clinical presentation of Creutzfeldt-Jakob disease: acute vestibular syndrome. Neurologist. (2015) 19:96–8 Case10. doi: 10.1097/NRL.0000000000000019
26. Vachalová I, Gindl V, Heckmann JG. Acute inferior homonymous quandrantanopia in a 71-year-old woman. J Clin Neurosci. (2014) 21:683–5 Case12. doi: 10.1016/j.jocn.2013.05.015
27. Hohler AD, Flynn FG. Onset of Creutzfeldt-Jakob disease mimicking an acute cerebrovascular event. Neurology. (2006) 67:538–9 Case17. doi: 10.1212/01.wnl.0000228279.28912.75
28. Obi T, Takatsu M, Kitamoto T, Mizoguchi K, Nishimura Y. A case of Creutzfeldt-Jakob disease (CJD) started with monoparesis of the left arm. Rinsho Shinkeigaku. (1996) 36:1245–8 Case 21.
29. Parchi P, Strammiello R, Notari S, Giese A, Langeveld JP, Ladogana A, et al. Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol. (2009) 118:659–71. doi: 10.1007/s00401-009-0585-1
Keywords: Creutzfeldt-Jakob disease, stroke, cerebro-vascular accident, diagnosis, differential diagnosis
Citation: Balash Y, Korczyn AD, Khmelev N, Eilam A, Adi M and Gilad R (2021) Creutzfeldt-Jakob and Vascular Brain Diseases: Their Overlap and Relationships. Front. Neurol. 12:613991. doi: 10.3389/fneur.2021.613991
Received: 04 October 2020; Accepted: 04 January 2021;
Published: 25 February 2021.
Edited by:
Görsev Yener, Dokuz Eylul University, TurkeyReviewed by:
SangYun Kim, Seoul National University Bundang Hospital, South KoreaAntonio Giuliano Zippo, National Research Council (CNR), Italy
Copyright © 2021 Balash, Korczyn, Khmelev, Eilam, Adi and Gilad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yacov Balash, eWFjb3ZiYWxhc2hAZ21haWwuY29t