 Nadia Postupna1†
Nadia Postupna1† Shannon E. Rose1†Laura E. Gibbons2Natalie M. Coleman1Leanne L. Hellstern1Kayla Ritchie1Angela M. Wilson1Eiron Cudaback1Xianwu Li1Erica J. Melief1Allison E. Beller1
Shannon E. Rose1†Laura E. Gibbons2Natalie M. Coleman1Leanne L. Hellstern1Kayla Ritchie1Angela M. Wilson1Eiron Cudaback1Xianwu Li1Erica J. Melief1Allison E. Beller1 Jeremy A. Miller3Amber L. Nolan1Desiree A. Marshall1Rod Walker4
Jeremy A. Miller3Amber L. Nolan1Desiree A. Marshall1Rod Walker4 Thomas J. Montine5Eric B. Larson4
Thomas J. Montine5Eric B. Larson4 Paul K. Crane2Richard G. Ellenbogen6
Paul K. Crane2Richard G. Ellenbogen6 Edward S. Lein3,6
Edward S. Lein3,6 Kristen Dams-O'Connor7‡C. Dirk Keene1,6*‡
Kristen Dams-O'Connor7‡C. Dirk Keene1,6*‡- 1Department of Laboratory Medicine and Pathology, University of Washington School of Medicine, Seattle, WA, United States
- 2Department of Medicine, University of Washington School of Medicine, Seattle, WA, United States
- 3Allen Institute for Brain Science, Seattle, WA, United States
- 4Kaiser Permanente Washington Health Research Institute, Seattle, WA, United States
- 5Department of Pathology, Stanford University School of Medicine, Palo Alto, CA, United States
- 6Department of Neurological Surgery, University of Washington School of Medicine, Seattle, WA, United States
- 7Department of Neurology, Icahn School of Medicine at Mount Sinai, New York, NY, United States
The late neuropathological effects of traumatic brain injury have yet to be fully elucidated, particularly with respect to community-based cohorts. To contribute to this critical gap in knowledge, we designed a multimodal neuropathological study, integrating traditional and quantitative approaches to detect pathologic changes in 532 consecutive brain autopsies from participants in the Adult Changes in Thought (ACT) study. Diagnostic evaluation including assessment for chronic traumatic encephalopathy (CTE) and quantitative immunoassay-based methods were deployed to examine levels of pathological (hyperphosphorylated) tau (pTau) and amyloid (A) β in brains from ACT participants with (n = 107) and without (n = 425) history of remote TBI with loss of consciousness (w/LOC). Further neuropathological assessments included immunohistochemistry for α-synuclein and phospho-TDP-43 pathology and astro- (GFAP) and micro- (Iba1) gliosis, mass spectrometry analysis of free radical injury, and gene expression evaluation (RNA sequencing) in a smaller sub-cohort of matched samples (49 cases with TBI and 49 non-exposed matched controls). Out of 532 cases, only 3 (0.6%–none with TBI w/LOC history) showed evidence of the neuropathologic signature of chronic traumatic encephalopathy (CTE). Across the entire cohort, the levels of pTau and Aβ showed expected differences for brain region (higher levels in temporal cortex), neuropathological diagnosis (higher in participants with Alzheimer's disease), and APOE genotype (higher in participants with one or more APOE ε4 allele). However, no differences in PHF-tau or Aβ1−42 were identified by Histelide with respect to the history of TBI w/LOC. In a subset of TBI cases with more carefully matched control samples and more extensive analysis, those with TBI w/LOC history had higher levels of hippocampal pTau but no significant differences in Aβ, α-synuclein, pTDP-43, GFAP, Iba1, or free radical injury. RNA-sequencing also did not reveal significant gene expression associated with any measure of TBI exposure. Combined, these findings suggest long term neuropathological changes associated with TBI w/LOC may be subtle, involve non-traditional pathways of neurotoxicity and neurodegeneration, and/or differ from those in autopsy cohorts specifically selected for neurotrauma exposure.
Introduction
Traumatic brain injury (TBI), defined as a blow or jolt to the head or neck that results in altered brain function, is a major cause of morbidity and mortality and a public health crisis. It is estimated that TBI affects nearly three million people in the US per year, not including undiagnosed and untreated cases (1). Furthermore, at least 2% of the US population is living with chronic disability related to a prior head trauma (2, 3). Population-based studies suggest that this prevalence may be substantially higher (4).
The consequences of TBI are not necessarily limited to the injury sustained in the acute stage and frequently lead to chronic dysfunction, even in mild TBI (5). Progressive degeneration of the brain on imaging with time has also been identified in some cases (6). However, the late neuropathological features associated with TBI are not yet well-characterized. While many cases of significant exposure to repetitive head trauma (e.g., extended exposure to contact sports, military service, or domestic violence) are associated with chronic traumatic encephalopathy (CTE), with sulcal and perivascular accumulation of phosphorylated tau (pTau) in neurons and glia (7–10), there is less understanding of the effects of the most common type of TBI in the general population- single TBI or multiple isolated TBI(s) that are associated with altered mental status and/or loss of consciousness (LOC). These injuries are much more common than the prolonged exposure to repetitive concussive/subconcussive head trauma experienced in high level contact sport associated with the CTE neuropathologic change (7, 11, 12), and often result from common life activities, such as motor vehicle accidents or falls.
A growing interest in the late neuropathological/neurodegenerative impact of TBI is fueled by studies that link TBI with later development of dementia, the risk appearing to increase with the number and severity of TBIs (13–18). Lee et al. (19) reported that even a single mild TBI may increase the lifelong risk for dementia; a history of TBI also appears to lower the age of dementia onset (18, 20). Research supports a link between moderate and severe TBI and development of Alzheimer's disease (AD) dementia (13, 14, 21, 22). TBI exposure has also been associated with frontotemporal lobar degeneratin (FTLD) (23–25), amyotrophic lateral sclerosis (ALS) (26), and Parkinson's disease (PD), where early life TBI exposure was more strongly predictive of PD and Lewy body disease (LBD) (27). However, many well-controlled studies have not found any effect of prior TBI on risk of dementia or AD (25, 27–33).
To date, the pathology thought to characterize late structural impact of repetitive concussive/subconcussive neurotrauma is CTE (7, 34–36). The specific mechanism(s) linking this pattern of neurotrauma with the later sulcal and perivascular pTau deposition, the pathognomonic feature of CTE (9) has not been fully elucidated, but pathological forms of tau have been observed in brain tissue and CSF post-injury (34, 37, 38). Whether CTE neuropathology is unique to repetitive subconcussive/concussive head trama experienced by high level contact sport athletes, military service members, or those exposed to intimate partner violence, continues to be investigated (3). The pTau pathology of CTE and/or a history of TBI is also often associated with other pathologic protein depositions. Amyloid (A)β plaques have been detected in proximity to CTE-specific lesions (7, 8, 39). Accumulation of TAR DNA-binding protein 43 (TDP-43), a nuclear protein implicated in the development of FTLD-TDP and ALS, has also been described in cases of repetitive brain trauma, with or without CTE diagnosis in humans (8, 25, 40–42), as well as animal models (43). α-Synuclein, a synaptic protein characteristically present in Lewy bodies (LBs) and required for pathologic diagnosis of PD/Lewy body dementia, has also been found in the brains of patients with a history of remote TBI (27, 44), in CSF after acute TBI (45, 46), and in animal models of brain trauma (47). Persistent inflammation may also play an important role in the development of post-TBI pathology. Several studies have shown that the inflammatory process chronically persists in TBI patients following trauma (48–52).
Given the high prevalence of TBI with associated LOC in the general population, and the widespread participation in recreational activity and contact sports among children and young adults, it is essential to develop a better understanding of the long-term neuropathological effects of non-repetitive TBI. To address this important knowledge gap, we investigated the delayed neuropathological consequences of TBI in a community-based sample. We not only screened for the presence of CTE pathology in neocortical sections, but we also measured the levels of pathological proteins (including several forms of pathological tau, Aβ, α-synuclein, and TDP-43) with multiple modalities including immunohistochemistry in formalin-fixed paraffin-embedded (FFPE) and flash-frozen tissue, a multiplexed Luminex assay in flash frozen tissue, and Histelide in FFPE. The inflammatory response was also assessed by evaluating astrogliosis and microgliosis, and free radical injury/oxidative stress by measuring isoprostane species. Finally, bulk RNA-sequencing was evaluated to screen for a potential genetic signature of remote TBI.
Materials and Methods
Study Design and Experimental Cohorts
This study was approved by the University of Washington and Kaiser Permanente Research Institute Institutional Review Boards. All subjects were participants in the Adult Changes in Thought (ACT) study, described in detail elsewhere (53). Briefly, ACT study participants are community-dwelling members of the Kaiser Permanente health maintenance organization, aged 65 and older and without a dementia diagnosis at the time of enrollment. Information about TBI was obtained solely via self-report upon enrollment in the study using the single-item question, “Have you ever sustained a head injury so severe it resulted in loss of consciousness?” and at every study visit using the question “Since your last study visit, have you had a head injury so severe it resulted in loss of consciousness?” If a head injury w/LOC was reported, participants were then queried to determine the duration of LOC, age at injury, and whether medical attention was sought. Duration of LOC was classified as: a few seconds or less, a minute or less, 1–2 min, 3–5 min, 6–9 min, 10 minto 1 h, more than 1 h. Information about sport and military exposure was obtained from obituaries, which were available for 52% of cases. Cognitive status was assessed biannually by the Cognitive Abilities Screening Instrument (CASI) (54), which ranges from 0 to 100 and higher scores indicate better cognitive functioning. A CASI score of <86 indicated possible cognitive impairment and initiated a standardized dementia diagnostic evaluation, including a comprehensive neuropsychological battery and a neurological evaluation. All of these data were reviewed in a multidisciplinary consensus conference. Dementia diagnoses were assigned by consensus using DSM-IV criteria (55). All neuropathologic evaluations were performed according to current consensus protocols and methods at the time of autopsy. Apolipoprotein E (APOE; M12529) genotype was determined according to published methods (56).
To study the late neuropathological effects of TBI in ACT, we selected 532 consecutive ACT participant brain autopsies (TBIAll cohort), starting with the first ACT brain donor, from which subjects were divided into 2 groups by history of TBI with LOC: TBI group with 1-3 instances of TBI with LOC, duration ranging from a few seconds to more than an hour, n = 107, and an unexposed group with no history of TBI with LOC, n = 425. See Table 1 for cohort demographics.
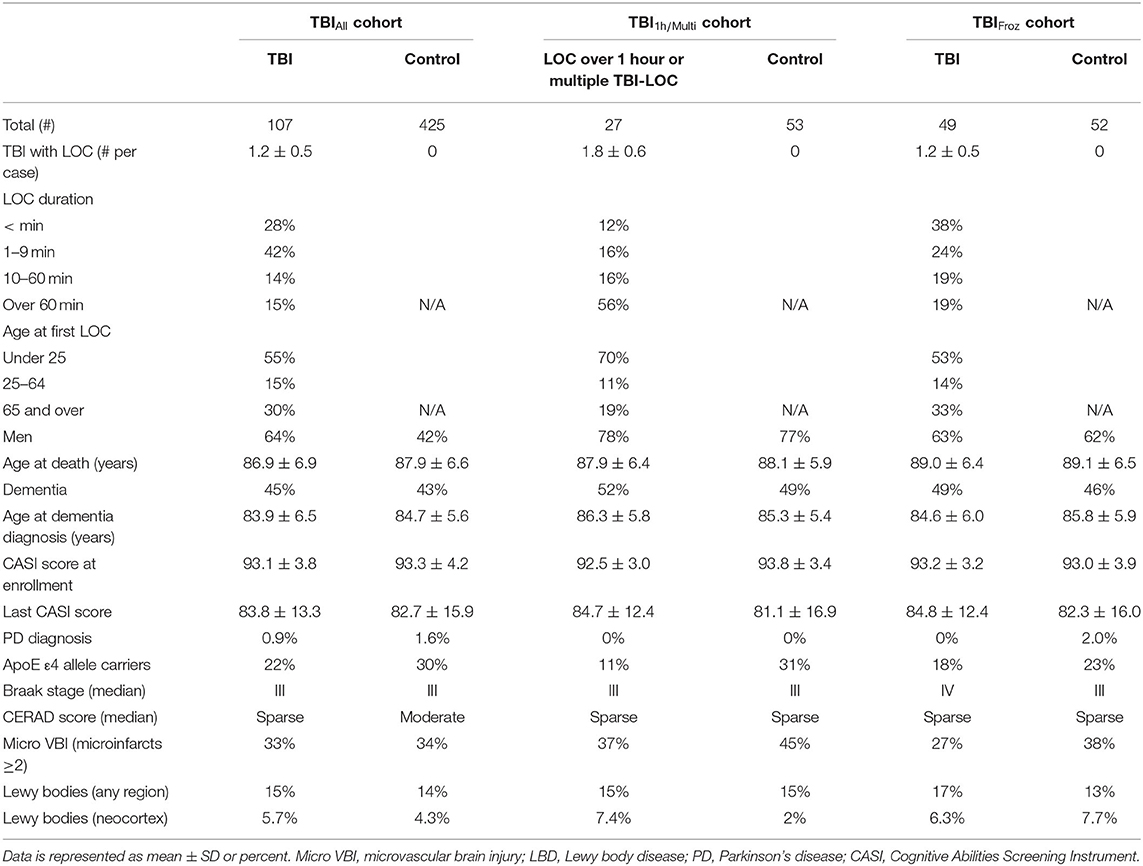
Table 1. Study cohorts.
Samples of FFPE brain tissue were obtained from middle frontal gyrus (MFG, Brodmann area nine), superior and middle temporal gyrus (SMTG), and inferior parietal lobule (IPL). Levels of pTau and Aβ1−42 were quantified in all three regions using the Histelide immunoassay (57). MFG, SMTG, and IPL sections immunolabeled for pTau were also screened for signs of CTE.
To focus in more depth on people with the highest TBI exposure, we used the original TBI cohort (TBIAll) to identify participants with the most significant TBI exposure, defined as a single TBI with LOC >1 h and/or >1 TBI with LOC (TBI1h/Multi, n = 27). Each of the 27 TBI1h/Multi participants was matched with two unexposed cases from the original control cohort, with matching by sex, age, year of death, and post-mortem interval (see Figure 1). We used the Histelide immunoassay to quantify pTau in an expanded set of brain regions in these individuals, including the contralateral MFG, SMTG and IPL (data from both sides were averaged), as well as occipital cortex (calcarine fissure), anterior cingulate cortex, and hippocampus with adjacent entorhinal cortex at the coronal level of lateral geniculate nucleus). See Table 2 for more information about the regions examined and analyses.
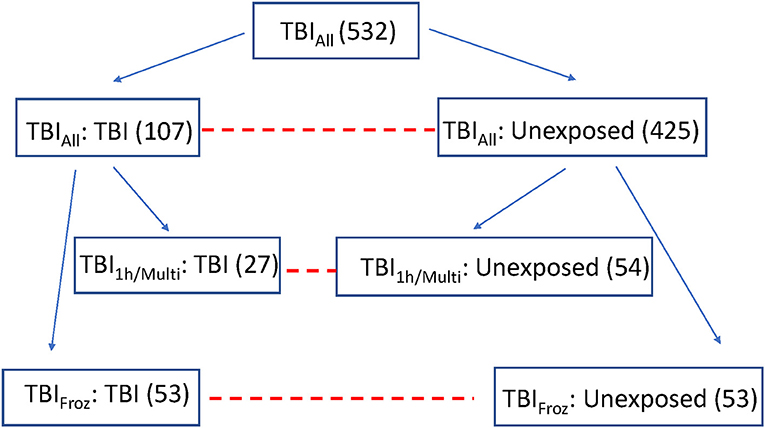
Figure 1. Overview of the experimental cohorts. Dashed lines connect comparison groups.

Table 2. Overview of experimental methods and analytes in respect to the experimental cohorts.
To study the effect of TBI on brain using methods not optimized for formalin fixation, such as RNA-sequencing, measurements of protein concentrations, and oxidative stress, we identified 53 ACT participant autopsies with TBI w/LOC history (as defined above) and fresh frozen samples derived at rapid autopsy (fresh dissection of the brain with samples flash frozen in liquid nitrogen occurs in donors with post-mortem interval < 8 h and is referred to as a rapid autopsy). Matched controls (53) with flash frozen tissue and no history of TBI w/LOC were selected using the same control matching algorithm as above (TBIFroz cohort). See Figure 1, Tables 1, 2 for more information. All experiments and analyses in the TBIFroz cohort were carried out in collaboration with the Allen Institute for Brain Science (www.alleninstitute.org) and all data are freely available at http://aging.brain-map.org.
We studied samples of FFPE brain tissue and tissue flash-frozen at the time of autopsy from SMTG, IPL, and hippocampus. Parietal white matter (PWM) samples were obtained from IPL blocks. Cortical and hippocampal frozen samples, and cortical FFPE tissue blocks were taken from the same side of the brain within cases; sidedness for frozen sampling was randomly chosen at autopsy. FFPE hippocampal samples were taken from the opposite side, as rapid protocols dictated complete sampling of rapid side hippocampus for freezing. To allow for measurement of protein concentrations and oxidative stress, and visualization of pathology by immunohistochemistry (IHC) to be carried out in the same tissue samples, two adjacent blocks of frozen SMTG, IPL, and hippocampal tissue were used, with one divided for isoprostane and Luminex studies, and the other for frozen section immunohistochemistry and RNA-sequencing. If only one block was available, the cortical blocks were sufficiently large (~1.5 g) to be divided and used for each assay, but hippocampal blocks were small and therefore used only for immunohistochemistry/RNA-sequencing and not available for protein/isoprostane measurements (11 cases). Punch biopsies (1 cm) were used to selectively collect PWM, SMTG and IPL gray matter. In all cases, punches of cerebral cortex were targeted to the depth of sulcus region and white matter punches were targeted to the deepest available layer in each tissue block. The hippocampus was sampled in its entirety; http://aging.brain-map.org for additional details.
Histelide in FFPE Tissue
Histelide immunoassays were carried out as previously published (57). Briefly, 5 μm sections of FFPE tissue were placed on charged microscope slides. Slides were deparaffinized, rehydrated and subjected to antigen retrieval. Slides were incubated in normal goat serum/bovine serum albumin (NGS/BSA) blocking solution, anti-pTau or anti-amyloid (A)β1−42 primary antibody (see Table 3), and alkaline phosphatase-conjugated goat secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Slides were then incubated in p-nitrophenyl phosphate (PNPP) solution, and the soluble chromogenic PNP product absorbance was read at 405 nm by a spectrophotometer (Molecular Devices SpectraMax). To allow for visualization of antibody deposits, the slides were then placed into insoluble chromogen nitro-blue tetrazolium/5-Bromo-4-chloro-3-indolyl phosphate (NBT/BCIP, Sigma-Aldrich, St. Louis, MO, USA) and coverslipped. Gray and white matter areas were traced using a Nikon 90 microscope and Stereo Investigator software (MBF Bioscience, Williston, VT). Background signal was calculated using negative control slides included in each run. PNPP signal minus background was normalized to gray matter area to give the signal intensity of gray matter (SIGM, Abs/cm2).
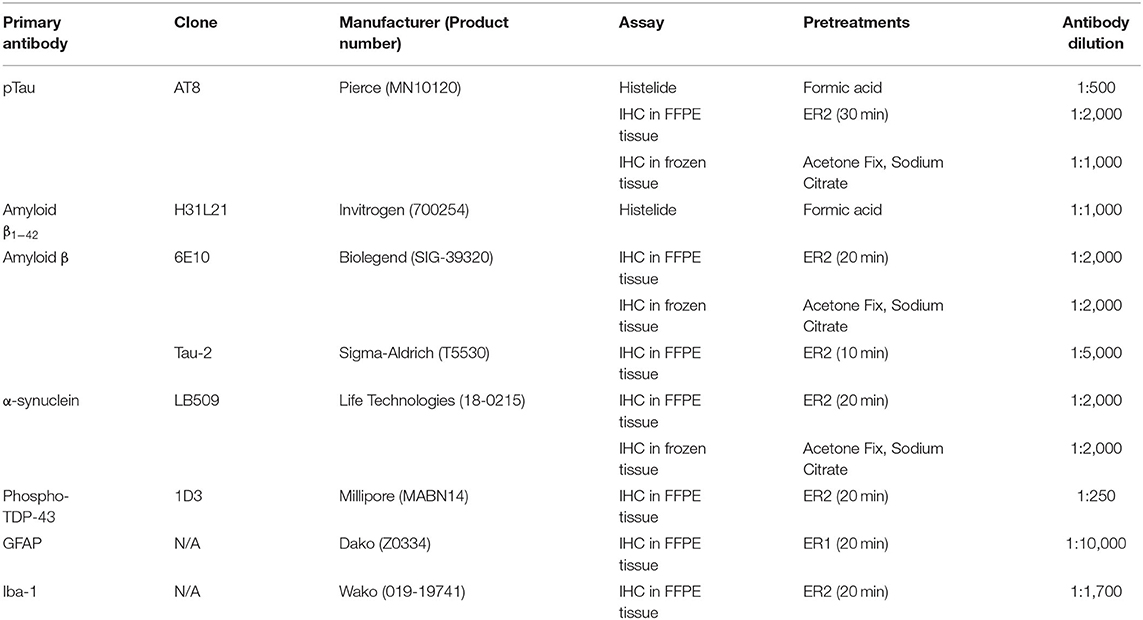
Table 3. Antibodies and pretreatments.
Histological Staining and IHC in FFPE Tissue
FFPE tissue blocks were cut at 5 μm thickness and processed using standard histological techniques. Sections of SMTG, IPL and hippocampus were stained with hematoxylin and eosin/Luxol Fast Blue (H&E/LFB) and immunolabeled for Aβ (clone 6E10), pTau (clone AT8), phosphorylated and non-phosphorylated pathologically modified tau (Tau-2), phospho-TDP-43 (1D3), GFAP, and Iba1 (see Table 3). More detailed descriptions of the methods are available at http://aging.brain-map.org.
Histological Staining and IHC in Flash-Frozen Tissue
Tissue blocks were cut at 25 μm thickness, processed using standard histological techniques and stained with cresyl violet for anatomical reference. The sections of SMTG, IPL and hippocampus were also immunolabeled using antibodies to Aβ (clone 6E10), pTau (AT8), and α-synuclein (LB509). For more details, see Table 3 and http://aging.brain-map.org.
Image Acquisition and Analysis
Histologically stained slides (frozen and FFPE) were scanned using the Leica ScanScope® automated slide scanner (Leica Biosystems). Images were acquired at 10x magnification. All images were first converted to JPEG 2,000 format, then oriented for consistency across slides and white balanced to achieve consistent white background intensities. Image analysis was carried out in a similar manner for frozen and FFPE tissue, except we applied a spectral filter to the FFPE tissue sections to attenuate the signal from the H&E/LFB stain. Expression density, defined as the percentage of the area within the region of interest (ROI) that was occupied by the IHC reaction product, was assessed algorithmically. To validate the metrics that were generated, data from antibodies used on both flash frozen and FFPE tissue was correlated. Good correlations were seen for both Aβ (R = 0.68, p = 6.5 × 10−51) and pTau (R = 0.78, p = 4.7 × 10−76). More detailed descriptions of all methods are available at http://aging.brain-map.org.
Luminex Assay
Flash-frozen tissue samples from 1 cm punch biopsies of gray matter from the SMTG, IPL, hippocampus, and of white matter from the IPL (PWM) were subjected to sequential extractions and centrifugation, and concentrations of pathological proteins pTau phosphorylated at threonine 181 (p181), amyloid β1−42, and α-synuclein, were measured by multiplexed Luminex assays (Luminex Corporation, Austin, TX). The data was analyzed using the LiquiChip Workstation (Qiagen, Hilden, Germany).
Measures of Oxidative Stress
Free radical injury was measured in the samples of flash-frozen SMTG and IPL using a stable isotope dilution assay with gas chromatography mass spectrometry (GC/MS) quantitation of isoprostanes according to the published protocol (58). GC/MS analysis was conducted using a 6890N Agilent gas chromatograph coupled to a 5,973-quadrupole mass spectrometer in the negative-ion mode. Areas under peaks for m/z 569.5 and 573.2 (internal standard) were manually integrated to quantify both analytes.
RNA-Sequencing and Analysis in Flash-Frozen Tissue
Samples of MFG, SMTG, IPL, and PWM and hippocampus were collected by manual microdissection in tissue adjacent to that processed for IHC. Specific areas for macrodissection were identified by neuroanatomists using images of tissue sections labeled with cresyl violet and were excised from the remaining frozen tissue block using a scalpel. For a detailed protocol please see Miller et al. (59). We used surrogate variable analysis (SVA) (60) to quantify significance of gene expression after normalizing by read depth only, and after controlling for RNA quality (59). Next, we repeated our analyses on RIN-normalized data using additional statistical tests including (1) two tailed student t-tests, (2) ANOVA, (3) and limma (61). Finally, we used SVA to assess whether any of the top principal components or module eigengenes described in Miller et al. (59) showed significant association with TBI. Statistical analyses for assessing significance of RNA-sequencing data are described in Miller et al. (59). Code for reproducing this part of the analysis has been added to https://github.com/AllenInstitute/agedbrain.
CTE Histology Screen
All sections of MFG, SMTG, and IPL immunolabeled with a pTau antibody as part of the Histelide assay were screened for an increase in tau at the depth of the sulcus. These cases were reviewed by a board-certified neuropathologist (A.L.N, C.D.K) and if an increase in tau accumulation in the deep gray matter at the sulcus was observed, additional evaluation of standard AT8 IHC stains was performed. These slides were evaluated for the pathognomonic lesion of CTE, according to consensus guidelines (9). The case was considered to have diagnostic neuropathologic evidence of CTE if any of the sections examined contained glial and neuronal pTau deposits at the depth of a sulcus in a clearly identified perivascular arrangement. Because of the limited number of regions analyzed, we did not assign a stage to any of the cases.
Data Analysis
Poisson regressions with robust standard errors were used because the outcome measures were all highly skewed. A relative risk of two means, for example, that the average measurement is estimated to be twice as high in the group of interest (TBI group) compared to the reference group (unexposed group). We adjusted for age at death, sex, education, and the presence of 1 or more APOE ε4 alleles. Alpha was set at 0.05, 2-sided. All statistical analyses were carried out in Stata 16.1.
Results
Cohort Characteristics
In the TBI group of the TBIAll cohort, 68 cases (64%) were male, and the average age at death was 86.9 ± 6.9 years. Thirty-six (34%) participants were diagnosed with AD, and 11 (10%) were diagnosed with LBD or PD. In the unexposed group, 179 (42%) were male, and the average age at death was 87.9 ± 6.6 years. One hundred forty-six participants (34%) were diagnosed with AD, 44 (10%) were diagnosed with LBD or PD, 2 (0.5%) were diagnosed with PSP and 1 with FTLD. The incidence of all-cause dementia appeared higher in the TBI group (45%, or 46/107), compared to the unexposed group (43% or 183/425), but the difference was not statistically significant. Likewise, average CASI score at study enrollment, last CASI score before death and average age at dementia diagnosis were not significantly different between the two groups (see Table 1). Over half of the participants in the TBI group (59/107, or 55%) sustained their first injury before the age of 25, and in 30% (32/107) the first TBI occurred after 65 years of age. For more details about the TBIAll cohort, as well as TBI1h/Multi and TBIFroz cohorts, see Table 1.
Histelide Measures of pTau and Aβ1−42 in TBIAll and TBI1h/Multi Cohorts
We used the Histelide method to measure pTau and Aβ1−42 levels in FFPE tissue samples from frontal, temporal and parietal cortex (MFG, SMTG, and IPL) in 532 ACT cases (TBIAll cohort): 107 cases with history of TBI exposure and 425 cases without history of TBI exposure. Data from a subset of these cases were reported earlier (57). There were no significant relationships between TBI exposure and any of the pTau or Aβ1−42 Histelide measures in MFG, SMTG or IPL, either for any TBI in the TBIAll sample (Figure 2, Table 4) or for those with LOC >1 h or multiple TBIs in the TBI1h/Multi sample (Figure 2, Table 5).
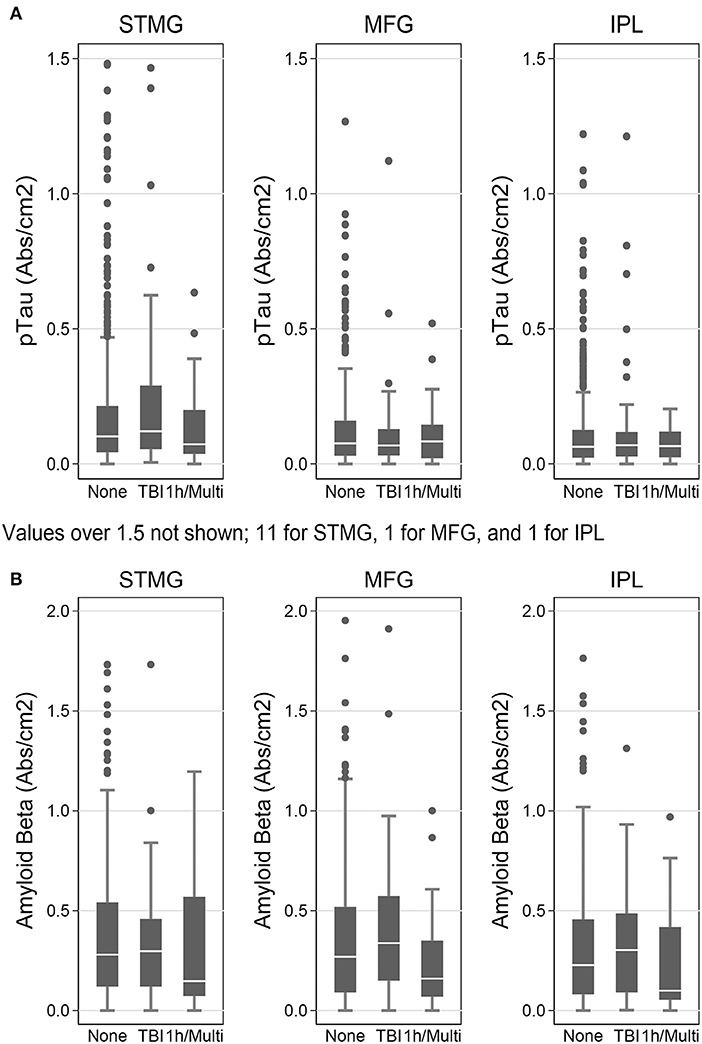
Figure 2. pTau and Aβ1−42 levels as measured by Histelide assay in the TBIAll cohort. pTau (A) and Aβ1−42 (B) were measured in the SMTG, MFG, and IPL. “TBI” indicates a history of a single TBI w/LOC of 1 h or less. “1h/Multi” indicates a history TBI w/LOC of more than 1 h or multiple TBIs. In the box-and-whiskers plots, the box spans the 25th to 75th percentiles, with the median indicated in white. The whiskers define 1½ times the inter-quartile range; individual observations more extreme than this are indicated with dots.
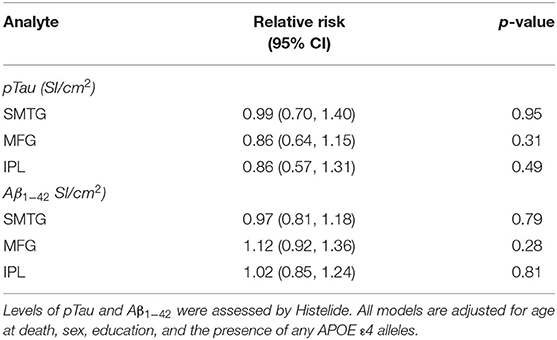
Table 4. Relative Risks with 95% confidence intervals for any TBI with LOC in the TBIAll cohort.
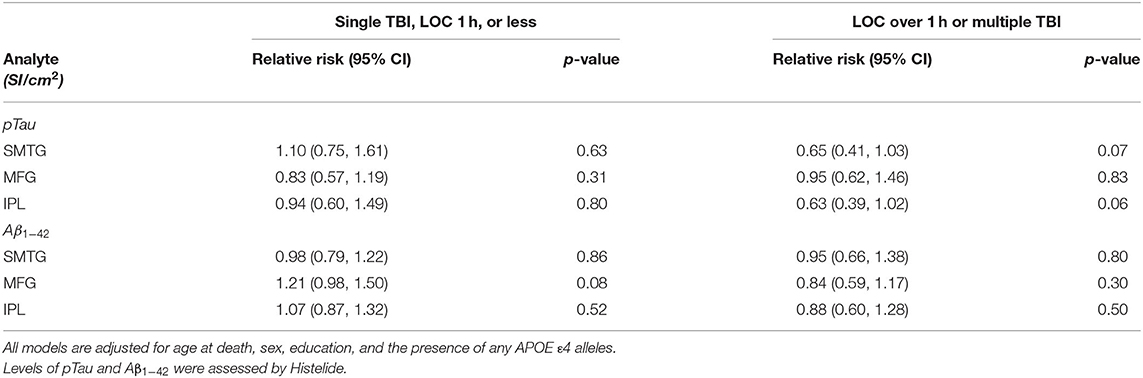
Table 5. Relative Risks with 95% confidence intervals for a single TBI-LOC lasting an hour or less and for LOC greater than an hour or multiple TBI-LOC in the TBIAll cohort.
In the 27 people with LOC >1 h or multiple TBI (TBI1h/Multi) and in 53 matched individuals with no history of TBI, pTau levels were measured in bilateral MFG, SMTG or IPL and 3 additional regions- occipital cortex, cingulate cortex, and hippocampus (with adjacent entorhinal cortex included). Again, no significant differences were identified (Supplementary Table 1).
PTau and Aβ1−42 Levels as Assessed by Immunohistochemistry and Luminex Assay in TBIFroz Cohort
Using immunohistochemistry, pTau signal (as a percentage of the tissue area occupied by the AT8 antibody immunostain) was measured in FFPE and frozen tissue samples and assessed with Tau-2 antibody in FFPE samples alone, from SMTG, IPL, hippocampus and PWM. As shown in Figures 3, 4A, Table 6, the only differences observed between people with a history of TBI w/LOC and people with no history of TBI were higher levels of pTau as assessed with AT8 and Tau-2 antibodies in FFPE tissue in hippocampus, as indicated by the higher risk ratios. PTau signal measured in frozen tissue samples by Luminex assay was not different between the two groups in any of the regions examined (Figure 4B).
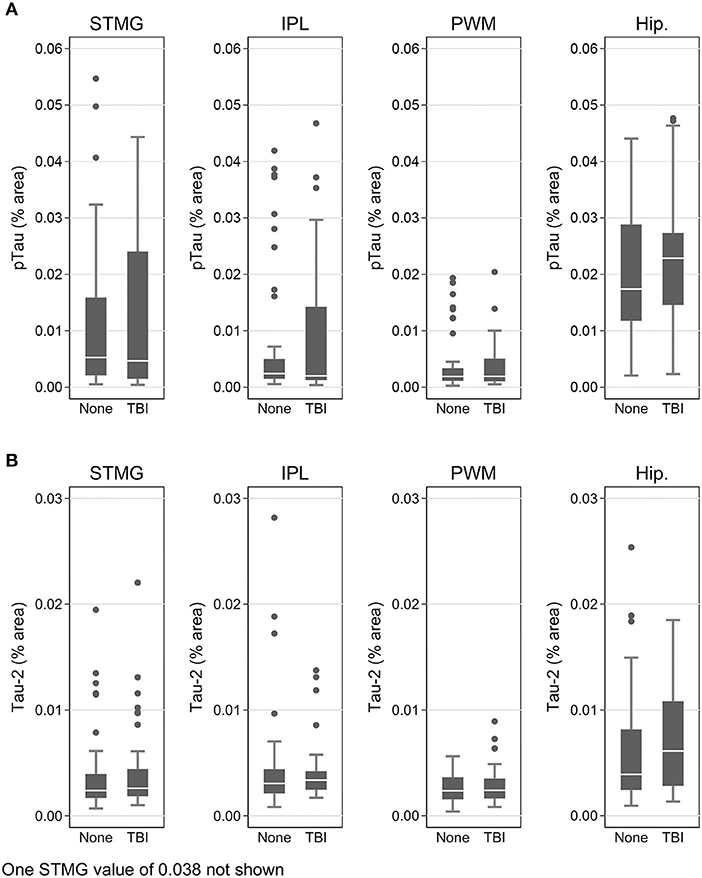
Figure 3. pTau levels in SMTG, IPL, hippocampus, and PWM in FFPE tissue samples in the TBIFroz cohort. Pathological tau (% area occupied by stain) in TBI cases and matched controls as measured by IHC with AT8 antibody (A), and Tau-2 antibody (B). In the box-and-whiskers plots, the box spans the 25th to 75th percentiles, with the median indicated in white. The whiskers define 1½ times the inter-quartile range; individual observations more extreme than this are indicated with dots.
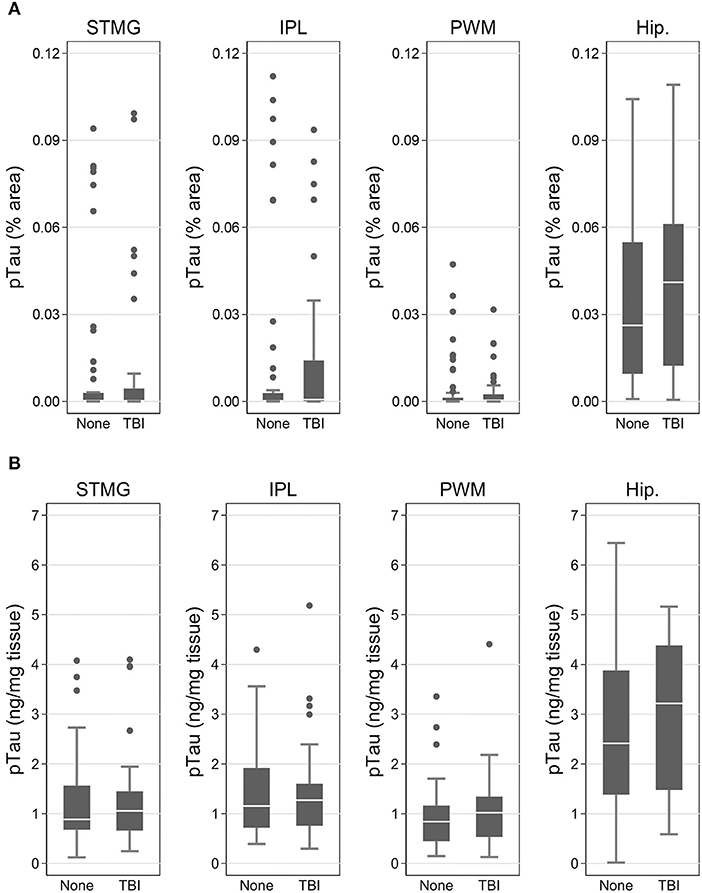
Figure 4. pTau levels in SMTG, IPL, hippocampus, and PWM in the in flash-frozen tissue samples TBIFroz cohort. pTau (% area occupied by stain) in TBI cases and matched controls as measured by IHC in frozen tissue sections using the AT8 antibody (A) and Luminex assay (B). In the box-and-whiskers plots, the box spans the 25th to 75th percentiles, with the median indicated in white. The whiskers define 1½ times the inter-quartile range; individual observations more extreme than this are indicated with dots.
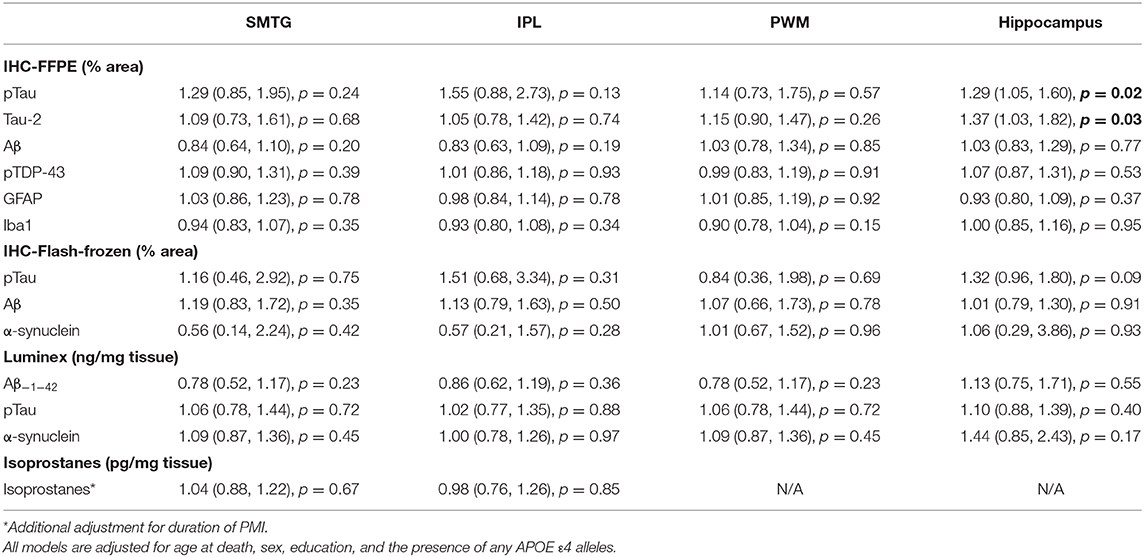
Table 6. Relative Risks with 95% confidence intervals TBI compared to no TBI, in the TBIFroz cohort.
We assessed Aβ signal (as a percentage of the tissue area occupied by the 6E10 antibody immunostain) in FFPE and frozen tissue samples from SMTG, IPL, hippocampus and PWM using immunohistochemistry, and measured levels of Aβ1−42 peptide in the samples of frozen tissue from the same four regions using a Luminex assay. TBI exposure was not associated with higher adjusted risk ratios for any of these measurements of Aβ compared to people without TBI (Table 6). The data is available at http://aging.brain-map.org/.
α-Synuclein, Phospho-TDP-43, Iba1 and GFAP as Assessed by Immunohistochemistry and Luminex Assay in TBIFroz Cohort
Levels of phospho-TDP-43, a pathological protein that has been implicated in CTE disease process and brain response to trauma (9) were measured in FFPE tissue in SMTG, IPL, hippocampus and PWM. We used immunohistochemistry and Luminex assay to measure levels of α-synuclein, a presynaptic protein present in Lewy bodies in frozen tissue samples in the same four regions. As shown in Table 6, TBI exposure was not associated with higher adjusted risk ratios for any of these measures compared to people without TBI.
To indirectly assess the extent of tissue reaction to injury and level of inflammation in the brains of people with a history of TBI and people without TBI history, we used immunohistochemistry to measure the levels of GFAP, a marker of astrocyte activation, and Iba1, a marker of activated microglia, in FFPE tissue samples from SMTG, IPL, hippocampus, and PWM. TBI exposure was not associated with higher adjusted risk ratios for any of these measures compared to people without TBI (Table 6). The data is available at http://aging.brain-map.org/
Measures of Free-Radical Damage in TBIFroz Cohort
Levels of free-radical damage in the cortical regions (SMTG and IPL) were assessed by measuring the concentrations of isoprostanes, products of free radical-catalyzed lipid peroxidation, in frozen tissue samples of SMTG and IPL. TBI exposure was not associated with higher adjusted risk ratios for isoprostane levels in any of these regions compared to people without TBI (see Table 6).
Validation of pTau and Amyloid β1−42 Measurements
To confirm the validity of our measurements of pTau and Aβ1−42, we performed several additional data analyses. Because most study participants showed at least some level of AD pathological change (see Table 1), and AD was the most common neuropathological diagnosis in this cohort, we wanted to see if the regional patterns of pTau and Aβ1−42 deposition that we observed were consistent with AD pathology. As expected, higher levels of pTau as measured by Histelide were found in the STMG [median 0.11, interquartile range (IQR) 0.48–0.24] than in the MFG (median 0.08, IQR 0.03–0.15; p < 0.0001) or the IPL (median 0.06, IQR 0.02–0.12; p < 0.0001), controlling for sex, education, age at death, and the presence of 1 or more APOE ε4 alleles. Lower levels of Aβ1−42 were found in the IPL (median 0.23, IQR 0.08–0.46) than in STMG (median 0.28, IQR 0.11–0.52; p < 0.0001) or the MFG (median 0.27, IQR 0.09–0.52; p = 0.0004), controlling for sex, education, age at death, and the presence of 1 or more APOE ε4 alleles.
To provide further validation of our results, we also stratified the data by the subjects' Braak stage, a staging system for AD based on the distribution of pTau-containing neurofibrillary tangles in the brain (62), and by CERAD score–another measure of AD pathology based on the density of Aβ-containing neuritic plaques in cerebral cortex (63). We hypothesized that in the temporal lobe samples (SMTG), final dementia status at the time of death and Braak stage should be associated with higher levels of both pTau and amyloid β1−42, and that the presence of 1 or more APOE ε4 alleles and CERAD score should be positively correlated with amyloid β1−42. As shown Figure 5, in the dataset obtained by Histelide, these relationships were all confirmed with our data (p-values 0.001–< 0.0001), and usually found in the MFG and IPL as well (Supplementary Table 2), adjusting for sex, education, age at death, and, except when it was the variable of interest, the presence of any APOE ε4 alleles.
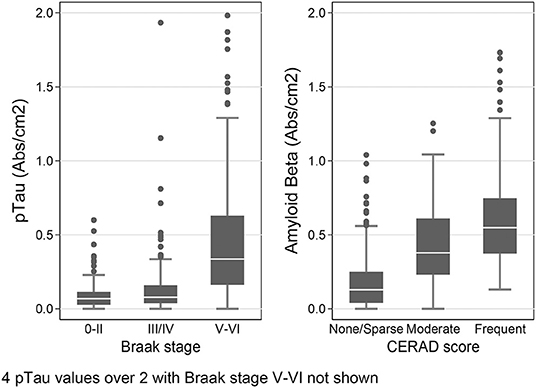
Figure 5. Histelide pTau levels stratified by Braak stage and Aβ1−42 levels (SI/cm2) by CERAD score in the SMTG in TBIAll cohort. In the box-and-whiskers plots, the box spans the 25th to 75th percentiles, with the median indicated in white. The whiskers define 1½ times the inter-quartile range; individual observations more extreme than this are indicated with dots.
Measures of pTau obtained by immunohistochemistry and Luminex assay were all associated with Braak stage V/VI, which is indicative of high level of AD neuropathological change, in the SMTG (p < 0.0001 compared to stages 0/II; Supplementary Table 3 and Figure 6). The contrast for pTau (IHC, FFPE) between stage III/IV and 0/II was also statistically significant (p = 0.02). Measures of Aβ1−42 were all associated with CERAD scores in the SMTG (p ≤ 0.0003 for Moderate and p < 0.0001 for Frequent, compared to None; Supplementary Table 3 and Figure 6). Aβ (IHC, FFPE) and Aβ1−42 (Luminex) also distinguished CERAD score Sparse from None (p = 0.03 and p = 0.008, respectively). All models were adjusted for age at death, sex, education, and the presence of any APOE ε4 alleles.
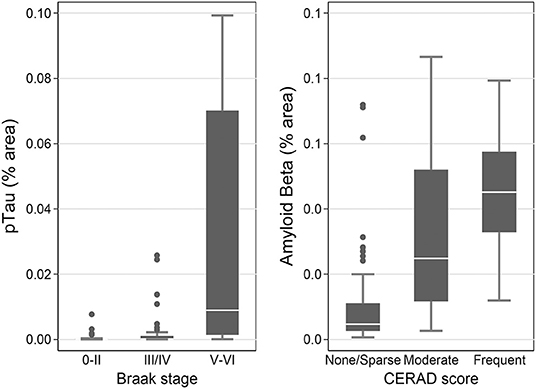
Figure 6. pTau levels stratified by Braak stage and Aβ levels by CERAD score as measured by IHC in the flash-frozen samples of STMG in TBIFroz cohort. In the box-and-whiskers plots, the box spans the 25th to 75th percentiles, with the median indicated in white. The whiskers define 1½ times the inter-quartile range; individual observations more extreme than this are indicated with dots.
RNA- Sequencing in TBIFroz Cohort
We did not find any genes with significant association to any measures of TBI in any of the regions based on several statistical tests. None of the statistical tests (see Methods) identified a single gene or component associated with TBI self-report, age at first TBI, longest loss of consciousness duration, or number of TBIs in any brain region tested, indicating that, using these methods, significant gene expression signatures of TBI are not identified in this data set (data not shown). The complete dataset is available at http://aging.brain-map.org/.
CTE Pathology in the TBIAll Cohort
To assess the prevalence of CTE pathology in our autopsy cohort, we used the Histelide slides immunolabeled with an antibody to pTau (AT8) to screen each section for increased density of sulcal pTau over other areas of the cortical section. Concerning regions with deposition of sulcal pTau in the deep gray matter were additionally evaluated with standard AT8 immunohistochemistry. Only three cases out of 532 (0.6%) demonstrated the neuropathologic features of CTE (Figures 7A–C). In all cases, CTE changes were only observed in a single region in temporal or parietal lobe (SMTG for one case and IPL for two cases).
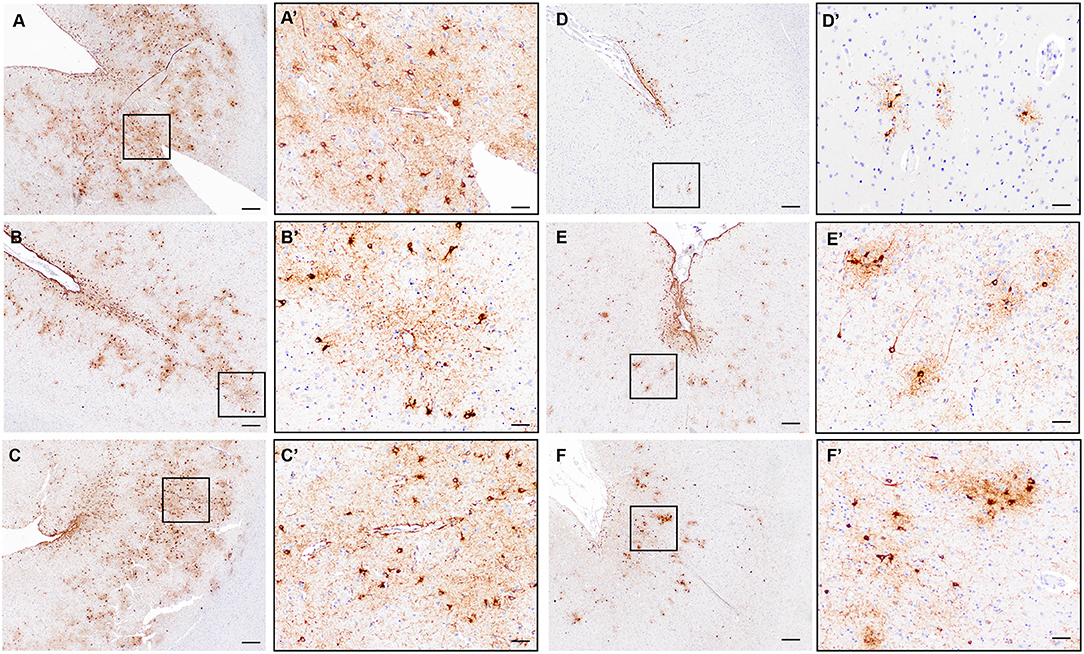
Figure 7. Evaluation of CTE neuropathologic change in the ACT cohort. (A–C) Representative images of the three cases with definite diagnostic features of CTE neuropathologic change characterized by clear perivascular pTau aggregates in neurons and glia at the depth of a sulcus and deep in the gray matter parenchyma. Black boxes indicate areas shown in (A'–C'). A/A'= Subject #2 in text, IPL; B/B'= Subject #1 in text, SMTG; C/C'=Subject #3 in text, IPL. (D–F) Representative images from cases without definite CTE but with irregular deep gray matter tau deposition. Distinct perivascular neuronal tau at the sulcal depth was not present to make a diagnosis of CTE. Black boxes indicate areas shown in (D'–F'). D = MFG; E = SMTG; F= IPL. Scale bars A-F = 200 μm; A'-F' = 50 μm.
Subject #1 was male, 82 years old and not demented at the time of death. Obituary was unavailable. Neuropathological diagnosis was AD. Subject #2 was male, 100 years old and demented at the time of death. Obituary was available, and there was no indication of either contact sport or military history in the obituary. The subject had depressive episodes later in life, was cognitively impaired in the last 7 years of life and sustained several falls during that time. Neuropathological diagnosis was vascular brain injury. Subject #3 was male, 94 years old and demented at the time of death. Obituary review indicated military service, but no contact sport exposure. The subject was treated for clinical late in life depression, was cognitively impaired in the last decade of life and sustained multiple falls during that time. Behavioral disturbances during the last 7 years of life were consistent with the clinical diagnosis of AD, which was later confirmed by neuropathological evaluation.
None of the three participants with CTE neuropathologic change had a history of previous TBI with LOC before developing dementia in the 8th or 9th decade of life, and none of the subjects were specifically evaluated for chronic traumatic encephalopathy syndrome (TES). Behavioral disturbances noted in subjects #2 and #3 were consistent with vascular/mixed dementia in case #2 and Alzheimer's type dementia in case #3. All three subjects were carriers of one copy of APOE ε4 allele.
Twenty-four additional participants showed pTau deposits at the depth of the sulcus, most often superficial subpial thorn-shaped astrocytes, in one or more cortical sections. Twenty of the 24 subjects were male, seven of them had a history of TBI. The average age at death was 86.9 years; nine were demented. Nine were diagnosed with AD, two with LBD and nine with vascular brain injury. Obituaries were available for 11 participants. According to the obituary data, four were military veterans and one played a contact sport. Three of these cases had additional deep gray matter tau deposition, but a convincing neuronal and perivascular distribution was not identified (Figures 7D–F), and therefore these cases were not considered to have definite CTE. Many of the remaining cases, as expected, had significant pTau pathology in a diffuse pattern in association with neuritic plaques consistent with AD neuropathologic change and lacked preferential accumulation of pTau in depth of sulcus.
Discussion
This study was designed to assess the delayed consequences of TBI with LOC in a community-based sample. We not only evaluated the cases for CTE neuropathologic change but performed a comprehensive multimodal assessment to examine for significant alterations in multiple pathologic proteins, inflammation and gene expression later in life.
We used the Histelide method to measure levels of pTau and Aβ1−42 in samples of frontal, temporal, and parietal cortex, and did not see a significant effect of history of prior TBI on levels of either protein. One caveat in assessing pTau and Aβ1−42 in the ACT autopsy cohort is that these are hallmark pathologic peptides of AD, which is highly prevalent due to the advanced age of most ACT decedents (64, 65). This confounding variable could have introduced variability to the data to obscure small differences related to TBI between groups. To address this potential impact, we designed and carried out additional experiments and analyses in a cohort of ACT participants with TBI w/LOC and non-exposed matched controls. Because there is a potential effect of previous neurotrauma on the development of AD (21, 66, 67) and other neurodegenerative diseases (26, 27, 68, 69), we chose not to match our subjects on markers of AD neuropathologic change or other neurodegenerative disease pathologies. Instead, we matched them on age and sex, factors that are known to affect the prevalence of AD (70–73) and can influence the rate of accumulation of pTau and Aβ in the brain (74, 75). However, we still did not detect any significant TBI w/LOC-related differences in pTau and Aβ1−42 levels, as assessed by Histelide, across the whole TBI-exposed ACT cohort and in a smaller subset of ACT participants with the most significant TBI exposures (TBI w/LOC>1 h and/or multiple TBI w/LOC). In another matched cohort of people with short PMI who had frozen tissue available for analyses, we also did not detect any differences in Aβ species between those with TBI w/LOC and the unexposed groups, but we did observe significantly higher levels of hippocampal, but not cortical, pTau in the TBI group in 2 out of 3 different types of assays. Given the total number of comparisons that were carried out, this result should be taken with caution. However, it may be in line with the increased tau pathology that has been reported in studies of more severe TBI at chronic time points after injury.
Our primary analyses relied on Histelide measures, a novel highly sensitive method of quantifying immunoreactivity in FFPE tissue. We conducted a series of sensitivity analyses to illustrate that Histelide is indeed quantifying the underlying distribution of pathology upon which the traditional gold-standard indices of neurodegenerative disease pathology are based. Each of our measurements of pTau and Aβ species were validated by analyzing subjects by AD neuropathologic change (63, 76), where we consistently observed a significant positive correlation between levels of pTau and Aβ and Braak stage and CERAD score. We detected elevated levels of both pathological proteins in subjects with dementia, as well as in carriers of APOE ε4 allele, trends that have been described previously (77, 78). Taken together, these observations confirm that the methods we used here were adequate to detect differences in the levels of pTau and Aβ in this cohort.
Chronic repetitive neurotrauma has been associated with increased pTau accumulation in the brain, at least in some cases (79–82), and pTau deposits are considered a major diagnostic feature of CTE (7, 8). However, much less is known about the effect of a single or a few mild concussive events. While a study by Uryu et al. reported increased pTau immunoreactivity in a small percentage of cases with recent TBI (83), Smith et al. did not find an increase in number of NFTs in acute fatal cases of a single TBI compared to age-matched controls (84). While post-concussive pTau accumulation has been previously demonstrated in some animal studies (82, 85, 86), other reports did not support this observation (87–89). In this study, history of remote TBI w/LOC does not appear to have a consistent, significant effect on cortical levels of pTau using quantitative approaches, but small effects of pTau related to TBI exposure could be masked by significant AD-neuropathological burden in this cohort.
We did not observe a relationship between the history of TBI with LOC and Aβ levels in the current study. APP, a precursor of amyloid β, is known to be involved in the acute response to brain injury (83), and several studies have reported increased accumulation of Aβ shortly after TBI in humans (38, 83, 90), although this phenomenon appears to occur only in a subset of cases with TBI exposure (81). A previous study by our group carried out in ACT, one of the largest studies on the subject, which included the majority of cases analyzed in the current report did not find an association between remote TBI with Aβ load as reflected by CERAD score (27). While Aβ deposits are common in severe CTE, they are often absent in milder cases (91, 92). Because none of the cases in this study had a known history of severe repetitive TBI or neuropathological changes suggestive of high stage CTE, it is not surprising that we did not observe a significant association between the history of TBI and cerebral Aβ.
We did not limit assessments to Aβ and pTau, but also measured levels of other pathologic peptides, such as phosphorylated α-synuclein and phospho-TDP-43, assessed markers of the inflammatory response (GFAP and Iba1), evaluated for oxidative stress (isoprostanes) and performed RNA-sequencing in an attempt to cast a wide net to detect TBI-related neuropathological changes in the ACT autopsy cohort. We did not detect any differences between groups defined by exposure to TBI w/LOC by any of the measurements in a smaller matched cohort with high quality frozen tissue samples (49 subject pairs), which might have provided insufficient statistical power to detect small differences.
All Histelide slides immunolabeled with pTau antibody were screened for neuropathological signs of CTE. For the whole cohort, MFG, SMTG, and IPL were screened, as standard samples taken for the entire cohort for recommended AD neuropathologic change workup. In a subset of cases with TBI w/LOC >1 h and/or multiple TBI w/LOC, additional regions, including some form the contralateral hemisphere, were also examined. However, superior frontal gyrus and temporal pole, sites recommended for CTE screening by NINDS/NIBIB consensus meeting criteria (9) are not routinely sampled and could not be assessed for this large number of cases. While we evaluated a minimum of three cortical regions in one hemisphere, recommended sampling protocols would have expanded that to five cortical regions bilaterally, or ~3 times more samples than our minimum per case. The three regions that we examined are known to be disproportionately affected by head trauma (8), and a very small subset of cases in this study (0.6%) displayed the neuropathologic changes that define CTE (each identified in a single brain region in all cases). The lack of a more extensive evaluation of additional regions of the brain may have contributed to the relatively low CTE prevalence in the current study, compared to earlier reports in other population-based studies: 11.9% in the study by Ling et al. (93), and 35% in the study by Noy et al. (94). However, even if we found three times more cases in the recommended 10 sampling areas, we would still be below 2% of the ACT cohort, and a more recent study, using diagnostic criteria similar to ours, reported no CTE cases in a community-based sample of 320 (95). Even if sensitivity to detect CTE was increased in a region not sampled here, it seems unlikely, based on the described distribution patterns of lesions of CTE, that the prevalence in the ACT cohort would approach levels seen in selected cohorts.
For the diagnosis of CTE neuropathology, cases of depth of sulcus pTau were not classified as CTE if the pTau was strictly astrocytic, was superficial/subpial only, or was not perivascular, all of which can be attributed to age-related tau astrogliopathy (ARTAG) (96, 97). Thus, of the 27 cases with depth of sulcus pTau lesions, 24 had features not diagnostic for CTE and more suggestive of ARTAG, a highly prevalent tauopathy of aging. None of the three CTE cases had a history of TBI w/LOC. Two of the three participants sustained multiple falls after developing dementia in in the 8th and 9th decade of life, but had no record of LOC. Observations from several large-scale neuropathology studies support the notion that diagnostic features of CTE can be present in cases without prior history of known head trauma with LOC. In the 2016 study by Noy and colleagues, more than one-third of cases displaying neuropathological signs of CTE with only focal sulcal deposition, similar to our cases, did not have an explicit history of head trauma (94). Similarly, in earlier reports by Ling et al. (93), Koga et al. (96), and Bieniek et al. (12) not every case displaying neuropathological signs of CTE had a documented history of head trauma. It is important to note that none of the cases in the current study had the level of pathologic changes suggestive of high stage CTE, but future consensus meetings are necessary to further refine diagnostic and staging criteria for CTE.
Several studies have described an association between neurotrauma and dementia in younger individuals. Nordstrom and colleagues (18) found a link between the history of TBI and the onset of dementia before the age of 65, while other authors reported increased risk of dementia and lower age of onset of cognitive impairment in subjects with moderate to severe TBI (13, 20). Because the enrollment in the ACT study is limited to individuals who do not have dementia at the age of 65, the ACT study is likely inadvertently selected against subjects who sustained a severe TBI or were exposed to repetitive head injury, and developed CTE and traumatic encephalopathy syndrome (TES) as a result. According to a previous report, CTE-associated cognitive symptoms could appear as early as in the 3rd decade of life, and over half of the subjects in the cognitive-predominant group in that study were demented at the time of death, which on average occurred at 69 years of age (98). The relatively short life span of subjects with symptomatic CTE [57 years in the study of Stern et al. (98)] creates significant difficulties in interpreting CTE findings in any cohort established to study brain aging. However, because of the high age and cognition cutoff of the ACT study, even people who sustained a single mild to moderated TBI, but had a higher than average susceptibility to the detrimental effect of neurotrauma on cognition, could have been excluded from the ACT. At present time, it is difficult to estimate the effect of this potential bias on our results. The issue is complicated by the fact that a number of studies reported no association between a history of TBI and development of AD, the most common type of dementia (33, 99, 100), and no association between TBI and dementia was reported earlier in the ACT cohort (33) and other smaller cohorts (101, 102). Another study in the ACT cohort, which included most of the cases analyzed here, did not find an association between the history of TBI and incidence of dementia or AD. The authors did report an association between TBI and incidence of PD, presence of LBD and microvascular injury (27), but these pathologies were less likely to affect the enrollment in the study unless accompanied by dementia. Another potential bias of the ACT study that warrants consideration is that all ACT study participants were members of an HMO and are therefore not a true representation of the general population. One can speculate that individuals in whom previous TBI significantly impacted general health and longevity were less likely to be recruited into ACT, creating a study bias toward participants, who were, on average, less susceptible to the pathological consequences of brain injury. This would be particularly true for individuals who sustained a severe and/or repeated TBI and developed TES, because as reported in an earlier study by Stern et al. participants with behavioral/mood variant of TES died at the average age of 51, and many of them developed symptoms decades earlier (98).
The assessment for TBI exposure history in ACT participants was designed at its inception three decades ago and does not perfectly conform to current concepts of TBI severity, which may affect direct comparisons to other studies. Although LOC duration of 30 min is commonly used to differentiate a mild from a moderate TBI, this time point was not available in the ACT study. Therefore, we chose LOC of >1 h to define the high TBI exposure group. Further, TBI ascertainment methods used in ACT prevented us from thoroughly characterizing subconcussive exposure and additional information (i.e., obituary) regarding military and contact sport participation was limited (although was not associated with any variables in this study in the participants for whom some exposure data was identified—data not shown). This problem is not unique to these types of assessments in cohorts not selected for TBI exposure and can lead to misclassification of participants with history of mild TBI without LOC and/or cases with repetitive subconcussive exposure being erroneously placed in the TBI unexposed group. Other studies before us faced the same issues. In their 2020 report, Bieniek et al. examined 750 autopsy cases from the Mayo Clinic Tissue Registry and relied on obituaries and high school yearbooks to assess sport participation (12). The authors found diagnostic CTE lesions in 1.3% of cases with no documented history of sports participation and in 5% of former athletes. We cannot directly compare our finding to the results of Bieniek et al. because of the uncertainty of sports exposure in the ACT study, where a little over half of the cases had an obituary available, while a presence of an obituary and/or yearbook was an inclusion criteria in the former study. Nevertheless, the prevalence of CTE in the ACT cohort as assessed by us appears lower than even in the unexposed group in the report of Bieniek and colleagues. A possible explanation for this discrepancy may be the relatively strict inclusion criteria of the ACT study, as described above. Average age at death in the study of Bieniek et al. was 68 and 64 y. o. for athletes and non-athletes, respectively, while the ACT study only accepted individuals that were at least 65 y. o. and non-demented at the time. Another potential factor could be the slight differences in the cortical regions examined (frontal and temporal, bilateral for at least one region in the study by Bieniek et al. vs. frontal, temporal and parietal in the current study).
Ongoing and future prospective cohort studies that include detailed methods to assess TBI exposure history will be essential to understand the degree to which false negative cases confound data analyses. Finally, the types of TBI exposures, particularly in sports and the military, have changed over the time period representing the lifespan of most of the ACT participants, and safety protocols and equipment have evolved as well. The relevant exposures studied in ACT need to be compared with those of subsequent generations to understand how changing exposure impacts future risk for neuropathological and functional outcomes.
Despite the aforementioned limitations, results from this community-based study allow us to draw several conclusions which should be applicable to the general population. We conclude that, on average, a remote single or rare TBI w/LOC is not associated with significant neurodegenerative disease-related changes in the brain in late life. Better characterization of TBI exposures, increasing numbers of cases and assay sensitivities, and extending sampling strategies to ensure inclusion of at risk brain regions, will be important to continue to explore relative risk for TBI-related late neuropathological changes. We also conclude that neuropathological features of CTE, which have been historically associated with the history of chronic repetitive brain trauma, are not highly prevalent in a community-based cohort of brain aging and neurodegeneration. Overall, while it appears that remote TBI w/LOC does not seem to leave a dramatic neuropathological signature on brain structure or its molecular composition, this finding raises more questions than it answers. The absence of clear general trends highlights the importance of uncovering differences in individual susceptibility to the effects of neurotrauma, with the goal of eventually developing targeted intervention and treatment strategies.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found at NIAGADS: https://www.niagads.org/datasets/ng00059.
Ethics Statement
The studies involving human participants were reviewed and approved by University of Washington Insitutional Review Board. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
CK, EL, and KD-O'C: study concept and design. SR, NC, LH, KR, AW, EC, XL, AB, JM, DM, KD-O'C, and CK: data acquisition. LG, SR, NP, JM, and EM: data analysis. CK, AN, and SR: neuropathological assessment. All authors: data interpretation and drafting of manuscript.
Funding
This work was funded by a Department of Defense Congressionally Directed Medical Research Program Grant #W81XWH-17-1-0330, NIH/NIA Grants R01AG061028, U01NS086625 (Late Effects of TBI study), U01AG006781 (Adult Changes in Thought Study), P50AG005136 (UW Alzheimer's Disease Research Center), a grant from the Paul Allen Family Foundation, and the Nancy and Buster Alvord Endowment.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Aimee Schantz, Tram Tran, Jim Pridgeon, Susan Sunkin, and Heather Hopkins for administrative support, Dr. Kathy Montine for helpful discussions, and the many dedicated individuals at the Allen Institute for Brain Science for outstanding support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.624696/full#supplementary-material
References
1. Centers for Disease Control and Prevention. Traumatic Brain Injury in the United States: Fact Sheet. Washington, DC: Centers for Disease Control and Prevention (2014).
2. Centers for Disease Control and Prevention. Report to Congress on Traumatic Brain Injury in the United States: Epidemiology and Rehabilitation. Atlanta, GA: Centers for Disease Control and Prevention (2015).
3. Centers for Disease Control and Prevention. TBI: Get the Facts. Washington, DC: Centers for Disease Control and Prevention (2019).
4. Whiteneck GG, Cuthbert JP, Corrigan JD, Bogner JA. Prevalence of self-reported lifetime history of traumatic brain injury and associated disability: a statewide population-based survey. J Head Trauma Rehabil. (2016) 31:E55–62. doi: 10.1097/HTR.0000000000000140
5. Nelson LD, Temkin NR, Dikmen S, Barber J, Giacino JT, Yuh E, et al. Recovery after mild traumatic brain injury in patients presenting to US Level I trauma centers: a transforming research and clinical knowledge in traumatic brain injury (TRACK-TBI) study. JAMA Neurol. (2019) 76:1049–59. doi: 10.1001/jamaneurol.2019.1313
6. Adnan A, Crawley A, Mikulis D, Moscovitch M, Colella B, Green R. Moderate-severe traumatic brain injury causes delayed loss of white matter integrity: evidence of fornix deterioration in the chronic stage of injury. Brain Inj. (2013) 27:1415–22. doi: 10.3109/02699052.2013.823659
7. McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. (2009) 68:709–35. doi: 10.1097/NEN.0b013e3181a9d503
8. McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. (2013) 136:43–64. doi: 10.1093/brain/aws307
9. McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. (2016) 131:75–86. doi: 10.1007/s00401-015-1515-z
10. Mez J, Daneshvar DH, Abdolmohammadi B, Chua AS, Alosco ML, Kiernan PT, et al. Duration of American Football play and chronic traumatic encephalopathy. Ann Neurol. (2020) 87:116–31. doi: 10.1002/ana.25611
11. Bieniek KF, Ross OA, Cormier KA, Walton RL, Soto-Ortolaza A, Johnston AE, et al. Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol. (2015) 130:877–89. doi: 10.1007/s00401-015-1502-4
12. Bieniek KF, Blessing MM, Heckman MG, Diehl NN, Serie AM, Paolini MA, et al. Association between contact sports participation and chronic traumatic encephalopathy: a retrospective cohort study. Brain Pathol. (2020) 30:63–74. doi: 10.1111/bpa.12757
13. Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, et al. Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology. (2000) 55:1158–66. doi: 10.1212/WNL.55.8.1158
14. Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer's disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry. (2003) 74:857–62. doi: 10.1136/jnnp.74.7.857
15. Suhanov AV, Pilipenko PI, Korczyn AD, Hofman A, Voevoda MI, Shishkin SV, et al. Risk factors for Alzheimer's disease in Russia: a case-control study. Eur J Neurol. (2006) 13:990–5. doi: 10.1111/j.1468-1331.2006.01391.x
16. Barnes DE, Kaup A, Kirby KA, Byers AL, Diaz-Arrastia R, Yaffe K. Traumatic brain injury and risk of dementia in older veterans. Neurology. (2014) 83:312–9. doi: 10.1212/WNL.0000000000000616
17. Gardner RC, Burke JF, Nettiksimmons J, Kaup A, Barnes DE, Yaffe K. Dementia risk after traumatic brain injury vs nonbrain trauma: the role of age and severity. JAMA Neurol. (2014) 71:1490–7. doi: 10.1001/jamaneurol.2014.2668
18. Nordstrom P, Michaelsson K, Gustafson Y, Nordstrom A. Traumatic brain injury and young onset dementia: a nationwide cohort study. Ann Neurol. (2014) 75:374–81. doi: 10.1002/ana.24101
19. Lee YK, Hou SW, Lee CC, Hsu CY, Huang YS, Su YC. Increased risk of dementia in patients with mild traumatic brain injury: a nationwide cohort study. PLoS ONE. (2013) 8:e62422. doi: 10.1371/journal.pone.0062422
20. Li W, Risacher SL, McAllister TW, Saykin AJ. Traumatic brain injury and age at onset of cognitive impairment in older adults. J Neurol. (2016) 263:1280–5. doi: 10.1007/s00415-016-8093-4
21. Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, et al. Head trauma as a risk factor for Alzheimer's disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol. (1991) 20(Suppl. 2):S28–35. doi: 10.1093/ije/20.Supplement_2.S28
22. Wang HK, Lin SH, Sung PS, Wu MH, Hung KW, Wang LC, et al. Population based study on patients with traumatic brain injury suggests increased risk of dementia. J Neurol Neurosurg Psychiatry. (2012) 83:1080–5. doi: 10.1136/jnnp-2012-302633
23. Jawaid A, Rademakers R, Kass JS, Kalkonde Y, Schulz PE. Traumatic brain injury may increase the risk for frontotemporal dementia through reduced progranulin. Neurodegener Dis. (2009) 6:219–20. doi: 10.1159/000258704
24. Kalkonde YV, Jawaid A, Qureshi SU, Shirani P, Wheaton M, Pinto-Patarroyo GP, et al. Medical and environmental risk factors associated with frontotemporal dementia: a case-control study in a veteran population. Alzheimers Dement. (2012) 8:204–10. doi: 10.1016/j.jalz.2011.03.011
25. Huang CH, Lin CW, Lee YC, Huang CY, Huang RY, Tai YC, et al. Is traumatic brain injury a risk factor for neurodegeneration? A meta-analysis of population-based studies. BMC Neurol. (2018) 18:184. doi: 10.1186/s12883-018-1187-0
26. Chen H, Richard M, Sandler DP, Umbach DM, Kamel F. Head injury and amyotrophic lateral sclerosis. Am J Epidemiol. (2007) 166:810–6. doi: 10.1093/aje/kwm153
27. Crane PK, Gibbons LE, Dams-O'Connor K, Trittschuh E, Leverenz JB, Keene CD, et al. Association of traumatic brain injury with late-life neurodegenerative conditions and neuropathologic findings. JAMA Neurol. (2016) 73:1062–9. doi: 10.1001/jamaneurol.2016.1948
28. Katzman R, Aronson M, Fuld P, Kawas C, Brown T, Morgenstern H, et al. Development of dementing illnesses in an 80-year-old volunteer cohort. Ann Neurol. (1989) 25:317–24. doi: 10.1002/ana.410250402
29. Williams DB, Annegers JF, Kokmen E, O'Brien PC, Kurland LT. Brain injury and neurologic sequelae: a cohort study of dementia, parkinsonism, and amyotrophic lateral sclerosis. Neurology. (1991) 41:1554–7. doi: 10.1212/WNL.41.10.1554
30. Launer LJ, Andersen K, Dewey ME, Letenneur L, Ott A, Amaducci LA, et al. Rates and risk factors for dementia and Alzheimer's disease: results from EURODEM pooled analyses. EURODEM Incidence Research Group and Work Groups European Studies of Dementia. Neurology. (1999) 52:78–84. doi: 10.1212/WNL.52.1.78
31. Mehta KM, Ott A, Kalmijn S, Slooter AJ, van Duijn CM, Hofman A, et al. Head trauma and risk of dementia and Alzheimer's disease: the rotterdam study. Neurology. (1999) 53:1959–62. doi: 10.1212/WNL.53.9.1959
32. Helmes E, Ostbye T, Steenhuis RE. Incremental contribution of reported previous head injury to the prediction of diagnosis and cognitive functioning in older adults. Brain Inj. (2011) 25:338–47. doi: 10.3109/02699052.2011.556104
33. Dams-O'Connor K, Gibbons LE, Bowen JD, McCurry SM, Larson EB, Crane PK. Risk for late-life re-injury, dementia and death among individuals with traumatic brain injury: a population-based study. J Neurol Neurosurg Psychiatry. (2013) 84:177–82. doi: 10.1136/jnnp-2012-303938
34. Asken BM, Sullan MJ, DeKosky ST, Jaffee MS, Bauer RM. Research gaps and controversies in chronic traumatic encephalopathy: a review. JAMA Neurol. (2017) 74:1255–62. doi: 10.1001/jamaneurol.2017.2396
35. Smith DH, Johnson VE, Trojanowski JQ, Stewart W. Chronic traumatic encephalopathy - confusion and controversies. Nat Rev Neurol. (2019) 15:179–83. doi: 10.1038/s41582-018-0114-8
36. McKee AC. The neuropathology of chronic traumatic encephalopathy: the status of the literature. Semin Neurol. (2020) 40:359–69. doi: 10.1055/s-0040-1713632
37. Zemlan FP, Rosenberg WS, Luebbe PA, Campbell TA, Dean GE, Weiner NE, et al. Quantification of axonal damage in traumatic brain injury: affinity purification and characterization of cerebrospinal fluid tau proteins. J Neurochem. (1999) 72:741–50. doi: 10.1046/j.1471-4159.1999.0720741.x
38. Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, et al. Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. (2004) 190:192–203. doi: 10.1016/j.expneurol.2004.06.011
39. Smith DH, Meaney DF, Shull WH. Diffuse axonal injury in head trauma. J Head Trauma Rehabil. (2003) 18:307–16. doi: 10.1097/00001199-200307000-00003
40. King A, Sweeney F, Bodi I, Troakes C, Maekawa S, Al-Sarraj S. Abnormal TDP-43 expression is identified in the neocortex in cases of dementia pugilistica, but is mainly confined to the limbic system when identified in high and moderate stages of Alzheimer's disease. Neuropathology. (2010) 30:408–19. doi: 10.1111/j.1440-1789.2009.01085.x
41. McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. (2010) 69:918–29. doi: 10.1097/NEN.0b013e3181ee7d85
42. McKee AC, Stein TD, Kiernan PT, Alvarez VE. The neuropathology of chronic traumatic encephalopathy. Brain Pathol. (2015) 25:350–64. doi: 10.1111/bpa.12248
43. Zhang J, Teng Z, Song Y, Hu M, Chen C. Inhibition of monoacylglycerol lipase prevents chronic traumatic encephalopathy-like neuropathology in a mouse model of repetitive mild closed head injury. J Cereb Blood Flow Metab. (2015) 35:706. doi: 10.1038/jcbfm.2014.247
44. Kenney K, Iacono D, Edlow BL, Katz DI, Diaz-Arrastia R, Dams-O'Connor K, et al. Dementia after moderate-severe traumatic brain injury: coexistence of multiple proteinopathies. J Neuropathol Exp Neurol. (2018) 77:50–63. doi: 10.1093/jnen/nlx101
45. Newell KL, Boyer P, Gomez-Tortosa E, Hobbs W, Hedley-Whyte ET, Vonsattel JP, et al. Alpha-synuclein immunoreactivity is present in axonal swellings in neuroaxonal dystrophy and acute traumatic brain injury. J Neuropathol Exp Neurol. (1999) 58:1263–8. doi: 10.1097/00005072-199912000-00007
46. Mondello S, Buki A, Italiano D, Jeromin A. alpha-Synuclein in CSF of patients with severe traumatic brain injury. Neurology. (2013) 80:1662–8. doi: 10.1212/WNL.0b013e3182904d43
47. Acosta SA, Tajiri N, de la Pena I, Bastawrous M, Sanberg PR, Kaneko Y, et al. Alpha-synuclein as a pathological link between chronic traumatic brain injury and Parkinson's disease. J Cell Physiol. (2015) 230:1024–32. doi: 10.1002/jcp.24830
48. Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. (2011) 70:374–83. doi: 10.1002/ana.22455
49. Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. (2013) 136:28–42. doi: 10.1093/brain/aws322
50. Smith C, Gentleman SM, Leclercq PD, Murray LS, Griffin WS, Graham DI, et al. The neuroinflammatory response in humans after traumatic brain injury. Neuropathol Appl Neurobiol. (2013) 39:654–66. doi: 10.1111/nan.12008
51. Kumar RG, Boles JA, Wagner AK. Chronic inflammation after severe traumatic brain injury: characterization and associations with outcome at 6 and 12 months postinjury. J Head Trauma Rehabil. (2015) 30:369–81. doi: 10.1097/HTR.0000000000000067
52. Bogoslovsky T, Wilson D, Chen Y, Hanlon D, Gill J, Jeromin A, et al. Increases of plasma levels of glial fibrillary acidic protein, tau, and amyloid beta up to 90 days after traumatic brain injury. J Neurotrauma. (2017) 34:66–73. doi: 10.1089/neu.2015.4333
53. Kukull WA, Higdon R, Bowen JD, McCormick WC, Teri L, Schellenberg GD, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol. (2002) 59:1737–46. doi: 10.1001/archneur.59.11.1737
54. Teng EL, Hasegawa K, Homma A, Imai Y, Larson E, Graves A, et al. The Cognitive Abilities Screening Instrument (CASI): a practical test for cross-cultural epidemiological studies of dementia. Int Psychogeriatr. (1994) 6:45–58; discussion 62. doi: 10.1017/S1041610294001602
55. American Psychiatric Association. Task Force on DSM-IDiagnostic V, and Statistical Manual of Mental Disorders: DSM-IV. 4. Washington, DC, American Psychiatric Association (1994).
56. Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. (1990) 31:545–8. doi: 10.1016/S0022-2275(20)43176-1
57. Postupna N, Keene CD, Crane PK, Gonzalez-Cuyar LF, Sonnen JA, Hewitt J, et al. Cerebral cortical Abeta42 and PHF-tau in 325 consecutive brain autopsies stratified by diagnosis, location, and APOE. J Neuropathol Exp Neurol. (2015) 74:100–9. doi: 10.1097/NEN.0000000000000153
58. Montine TJ, Montine KS, McMahan W, Markesbery WR, Quinn JF, Morrow JD. F2-isoprostanes in Alzheimer and other neurodegenerative diseases. Antioxid Redox Signal. (2005) 7:269–75. doi: 10.1089/ars.2005.7.269
59. Miller JA, Guillozet-Bongaarts A, Gibbons LE, Postupna N, Renz A, Beller AE, et al. Neuropathological and transcriptomic characteristics of the aged brain. Elife. (2017) 6. doi: 10.7554/eLife.31126.036
60. Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. (2007) 3:1724–35. doi: 10.1371/journal.pgen.0030161
61. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. (2015) 43:e47. doi: 10.1093/nar/gkv007
62. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. (1991) 82:239–59. doi: 10.1007/BF00308809
63. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. (1991) 41:479–86. doi: 10.1212/WNL.41.4.479
64. Sonnen JA, Larson EB, Haneuse S, Woltjer R, Li G, Crane PK, et al. Neuropathology in the adult changes in thought study: a review. J Alzheimers Dis. (2009) 18:703–11. doi: 10.3233/JAD-2009-1180
65. Cholerton B, Larson EB, Baker LD, Craft S, Crane PK, Millard SP, et al. Neuropathologic correlates of cognition in a population-based sample. J Alzheimers Dis. (2013) 36:699–709. doi: 10.3233/JAD-130281
66. Rasmusson DX, Brandt J, Martin DB, Folstein MF. Head injury as a risk factor in Alzheimer's disease. Brain Inj. (1995) 9:213–9. doi: 10.3109/02699059509008194
67. O'Meara ES, Kukull WA, Sheppard L, Bowen JD, McCormick WC, Teri L, et al. Head injury and risk of Alzheimer's disease by apolipoprotein E genotype. Am J Epidemiol. (1997) 146:373–84. doi: 10.1093/oxfordjournals.aje.a009290
68. Deutsch MB, Mendez MF, Teng E. Interactions between traumatic brain injury and frontotemporal degeneration. Dement Geriatr Cogn Disord. (2015) 39:143–53. doi: 10.1159/000369787
69. Gardner RC, Burke JF, Nettiksimmons J, Goldman S, Tanner CM, Yaffe K. Traumatic brain injury in later life increases risk for Parkinson disease. Ann Neurol. (2015) 77:987–95. doi: 10.1002/ana.24396
70. Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer's disease in the United States and the public health impact of delaying disease onset. Am J Public Health. (1998) 88:1337–42. doi: 10.2105/AJPH.88.9.1337
71. Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer's disease. Alzheimers Dement. (2007) 3:186–91. doi: 10.1016/j.jalz.2007.04.381
72. Riedel BC, Thompson PM, Brinton RD. Age, APOE and sex: triad of risk of Alzheimer's disease. J Steroid Biochem Mol Biol. (2016) 160:134–47. doi: 10.1016/j.jsbmb.2016.03.012
73. Mosconi L, Berti V, Quinn C, McHugh P, Petrongolo G, Varsavsky I, et al. Sex differences in Alzheimer risk: Brain imaging of endocrine vs chronologic aging. Neurology. (2017) 89:1382–90. doi: 10.1212/WNL.0000000000004425
74. Coughlin D, Irwin DJ. Emerging diagnostic and therapeutic strategies for tauopathies. Curr Neurol Neurosci Rep. (2017) 17:72. doi: 10.1007/s11910-017-0779-1
75. Lee SH, Cho H, Choi JY, Lee JH, Ryu YH, Lee MS, et al. Distinct patterns of amyloid-dependent tau accumulation in Lewy body diseases. Mov Disord. (2018) 33:262–72. doi: 10.1002/mds.27252
76. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. (2006) 112:389–404. doi: 10.1007/s00401-006-0127-z
77. Kok E, Haikonen S, Luoto T, Huhtala H, Goebeler S, Haapasalo H, et al. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann Neurol. (2009) 65:650–7. doi: 10.1002/ana.21696
78. Caselli RJ, Walker D, Sue L, Sabbagh M, Beach T. Amyloid load in nondemented brains correlates with APOE e4. Neurosci Lett. (2010) 473:168–71. doi: 10.1016/j.neulet.2010.02.016
79. Omalu B, Bailes J, Hamilton RL, Kamboh MI, Hammers J, Case M, et al. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in American athletes. Neurosurgery. (2011) 69:173–83. doi: 10.1227/NEU.0b013e318212bc7b
80. Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. (2012) 4:134ra160. doi: 10.1016/j.jalz.2012.05.592
81. Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. (2012) 22:142–9. doi: 10.1111/j.1750-3639.2011.00513.x
82. Tagge CA, Fisher AM, Minaeva OV, Gaudreau-Balderrama A, Moncaster JA, Zhang XL, et al. Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain. (2018) 141:422–58. doi: 10.1093/brain/awx350
83. Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, et al. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. (2007) 208:185–92. doi: 10.1016/j.expneurol.2007.06.018
84. Smith C, Graham DI, Murray LS, Nicoll JA. Tau immunohistochemistry in acute brain injury. Neuropathol Appl Neurobiol. (2003) 29:496–502. doi: 10.1046/j.1365-2990.2003.00488.x
85. Kane MJ, Angoa-Perez M, Briggs DI, Viano DC, Kreipke CW, Kuhn DM. A mouse model of human repetitive mild traumatic brain injury. J Neurosci Methods. (2012) 203:41–9. doi: 10.1016/j.jneumeth.2011.09.003
86. McAteer KM, Corrigan F, Thornton E, Turner RJ, Vink R. Short and long term behavioral and pathological changes in a novel rodent model of repetitive mild traumatic brain injury. PLoS ONE. (2016) 11:e0160220. doi: 10.1371/journal.pone.0160220
87. Mouzon B, Chaytow H, Crynen G, Bachmeier C, Stewart J, Mullan M, et al. Repetitive mild traumatic brain injury in a mouse model produces learning and memory deficits accompanied by histological changes. J Neurotrauma. (2012) 29:2761–73. doi: 10.1089/neu.2012.2498
88. Mannix R, Meehan WP, Mandeville J, Grant PE, Gray T, Berglass J, et al. Clinical correlates in an experimental model of repetitive mild brain injury. Ann Neurol. (2013) 74:65–75. doi: 10.1002/ana.23858
89. Winston CN, Noel A, Neustadtl A, Parsadanian M, Barton DJ, Chellappa D, et al. Dendritic spine loss and chronic white matter inflammation in a mouse model of highly repetitive head trauma. Am J Pathol. (2016) 186:552–67. doi: 10.1016/j.ajpath.2015.11.006
90. Roberts GW, Gentleman SM, Lynch A, Graham DI. beta A4 amyloid protein deposition in brain after head trauma. Lancet. (1991) 338:1422–3. doi: 10.1016/0140-6736(91)92724-G
91. Stein TD, Montenigro PH, Alvarez VE, Xia W, Crary JF, Tripodis Y, et al. Beta-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathol. (2015) 130:21–34. doi: 10.1007/s00401-015-1435-y
92. Rabinovici GD. Advances and gaps in understanding chronic traumatic encephalopathy: from pugilists to american football players. JAMA. (2017) 318:338–40. doi: 10.1001/jama.2017.9353
93. Ling H, Holton JL, Shaw K, Davey K, Lashley T, Revesz T. Histological evidence of chronic traumatic encephalopathy in a large series of neurodegenerative diseases. Acta Neuropathol. (2015) 130:891–3. doi: 10.1007/s00401-015-1496-y
94. Noy S, Krawitz S, Del Bigio RM. Chronic traumatic encephalopathy-like abnormalities in a routine neuropathology service. J Neuropathol Exp Neurol. (2016) 75:1145–54. doi: 10.1093/jnen/nlw092
95. Forrest SL, Kril JJ, Wagner S, Honigschnabl S, Reiner A, Fischer P, et al. Chronic traumatic encephalopathy (CTE) is absent from a European Community-based aging cohort while cortical aging-related tau astrogliopathy (ARTAG) is highly prevalent. J Neuropathol Exp Neurol. (2019) 78:398–405. doi: 10.1093/jnen/nlz017
96. Koga S, Dickson DW, Bieniek KF. Chronic traumatic encephalopathy pathology in multiple system atrophy. J Neuropathol Exp Neurol. (2016) 75:963–70. doi: 10.1093/jnen/nlw073
97. Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H, et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol. (2016) 131:87–102. doi: 10.1007/s00401-015-1509-x
98. Stern RA, Daneshvar DH, Baugh CM, Seichepine DR, Montenigro PH, Riley DO, et al. Clinical presentation of chronic traumatic encephalopathy. Neurology. (2013) 81:1122–9. doi: 10.1212/WNL.0b013e3182a55f7f
99. Xu W, Tan L, Wang HF, Jiang T, Tan MS, Tan L, et al. Meta-analysis of modifiable risk factors for Alzheimer's disease. J Neurol Neurosurg Psychiatry. (2015) 86:1299–306. doi: 10.1136/jnnp-2015-310548
100. Weiner MW, Crane PK, Montine TJ, Bennett DA, Veitch DP. Traumatic brain injury may not increase the risk of Alzheimer disease. Neurology. (2017) 89:1923–5. doi: 10.1212/WNL.0000000000004608
101. Ashman TA, Cantor JB, Gordon WA, Sacks A, Spielman L, Egan M, et al. A comparison of cognitive functioning in older adults with and without traumatic brain injury. J Head Trauma Rehabil. (2008) 23:139–48. doi: 10.1097/01.HTR.0000319930.69343.64
Keywords: chronic traumatic encephalopathy, histelide, Adult Changes in Thought study, amyloid–beta, traumatic brain injury, immunohistochemistry, hyperphosphorylated tau
Citation: Postupna N, Rose SE, Gibbons LE, Coleman NM, Hellstern LL, Ritchie K, Wilson AM, Cudaback E, Li X, Melief EJ, Beller AE, Miller JA, Nolan AL, Marshall DA, Walker R, Montine TJ, Larson EB, Crane PK, Ellenbogen RG, Lein ES, Dams-O'Connor K and Keene CD (2021) The Delayed Neuropathological Consequences of Traumatic Brain Injury in a Community-Based Sample. Front. Neurol. 12:624696. doi: 10.3389/fneur.2021.624696
Received: 01 November 2020; Accepted: 12 February 2021;
Published: 16 March 2021.
Edited by:
Thor Stein, Boston University, United StatesReviewed by:
Kimbra Kenney, Uniformed Services University of the Health Sciences, United StatesKevin Bieniek, The University of Texas Health Science Center at San Antonio, United States
Copyright © 2021 Postupna, Rose, Gibbons, Coleman, Hellstern, Ritchie, Wilson, Cudaback, Li, Melief, Beller, Miller, Nolan, Marshall, Walker, Montine, Larson, Crane, Ellenbogen, Lein, Dams-O'Connor and Keene. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: C. Dirk Keene, Y2RrZWVuZUB1dy5lZHU=
†These authors share first authorship
‡These authors share last authorship