 Aristide Merola1*
Aristide Merola1* Noelle Kobayashi2
Noelle Kobayashi2 Alberto Romagnolo3
Alberto Romagnolo3 Brenton A. Wright4Carlo Alberto Artusi3
Brenton A. Wright4Carlo Alberto Artusi3 Gabriele Imbalzano3
Gabriele Imbalzano3 Irene Litvan4
Irene Litvan4 Amber D. Van Laar5,6Krystof Bankiewicz7
Amber D. Van Laar5,6Krystof Bankiewicz7- 1Department of Neurology, Madden Center for Parkinson Disease and Other Movement Disorders, The Ohio State University Wexner Medical Center, Columbus, OH, United States
- 2The Ohio State University, Columbus, OH, United States
- 3Department of Neuroscience “Rita Levi Montalcini,” University of Turin, Turin, Italy
- 4Department of Neurosciences, Parkinson and Other Movement Disorders Center, University of California, San Diego, La Jolla, CA, United States
- 5Asklepios BioPharmaceutical Inc., Research Triangle, NC, United States
- 6Department of Neurology, University of Pittsburgh Medical Center, Pittsburgh, PA, United States
- 7Department of Neurological Surgery, College of Medicine, The Ohio State University, Columbus, OH, United States
Introduction: We sought to provide an overview of the published and currently ongoing movement disorders clinical trials employing gene therapy, defined as a technology aiming to modulate the expression of one or more genes to achieve a therapeutic benefit.
Methods: We systematically reviewed movement disorders gene therapy clinical trials from PubMed and ClinicalTrials.gov using a searching strategy that included Parkinson disease (PD), Huntington disease (HD), amino acid decarboxylase (AADC) deficiency, multiple system atrophy (MSA), progressive supranuclear palsy (PSP), dystonia, tremor, ataxia, and other movement disorders. Data extracted included study characteristics, investigational product, route of administration, safety/tolerability, motor endpoints, and secondary outcomes (i.e., neuroimaging, biomarkers).
Results: We identified a total of 46 studies focusing on PD (21 published and nine ongoing), HD (2 published and 5 ongoing), AADC deficiency (4 published and 2 ongoing), MSA (2 ongoing), and PSP (1 ongoing). In PD, intraparenchymal infusion of viral vector-mediated gene therapies demonstrated to be safe and showed promising preliminary data in trials aiming at restoring the synthesis of dopamine, enhancing the production of neurotrophic factors, or modifying the functional interaction between different nodes of the basal ganglia. In HD, monthly intrathecal delivery of an antisense oligonucleotide (ASO) targeting the huntingtin protein (HTT) mRNA proved to be safe and tolerable, and demonstrated a dose-dependent reduction of the cerebrospinal fluid levels of mutated HTT, while a small phase-I study testing implantable capsules of cells engineered to synthesize ciliary neurotrophic factor failed to show consistent drug delivery. In AADC deficiency, gene replacement studies demonstrated to be relatively safe in restoring catecholamine and serotonin synthesis, with promising outcomes. Ongoing movement disorders clinical trials are focusing on a variety of gene therapy approaches including alternative viral vector serotypes, novel recombinant genes, novel delivery techniques, and ASOs for the treatment of HD, MSA, and distinct subtypes of PD (LRRK2 mutation or GBA1 mutation carriers).
Conclusion: Initial phase-I and -II studies tested the safety and feasibility of gene therapy in PD, HD, and AADC deficiency. The ongoing generation of clinical trials aims to test the efficacy of these approaches and explore additional applications for gene therapy in movement disorders.
Introduction
Gene therapy is a rapidly evolving technology aiming to modulate the expression of one or more genes to achieve a clinical therapeutic benefit (1). Several gene therapy studies have now been conducted in movement disorders and other areas of neurology to target the pathogenesis of diseases, enhance the production of key enzymes involved in neurotransmitter metabolic pathways, or support the survival of specific cell populations.
Gene therapy uses vectors to convey complementary DNA (cDNA), guide-RNA, small interfering RNAs (siRNA), microRNAs, or docking sites for DNA binding proteins (2). The ability to vary the type of genetic modulation lends a gene therapy approach to potentially be applied to a broad range of clinical indications within the central nervous system (CNS) and beyond. Genes encoding therapeutic proteins can be delivered via cDNA to replace a missing or mutated protein, modulate the concentration of a particular enzyme, or enrich the cellular microenvironment by enhancing the production of neurotrophic factors. In contrast, gene silencing strategies may treat diseases with an associated toxic gain of function mutation by using siRNAs or microRNAs to interfere with and degrade mRNA, decreasing protein translation and the resultant protein production (3). More innovative techniques involve the addition or alteration of polyadenylation signals, splicing of introns, and open reading frames (ORF) to modulate levels of gene expression (4), or the use of an RNA-guided Cas9 nuclease to target a specific region of the genome for targeted editing (5).
In this systematic review, we sought to report the state-of-the-art advancements in gene therapy related to movement disorders by critically reviewing and discussing the results of published studies and ongoing clinical trials.
Methods
We conducted a systematic review in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) guideline (6), including both published data and available data from ongoing gene therapy clinical trials for the treatment of movement disorders. For the purpose of this review, gene therapy was defined as a therapeutic intervention aimed at modulating the expression of a gene though the use of DNA or RNA encoding a protein (i.e., trophic factor, enzyme), antisense oligonucleotide interfering with RNA translation, or DNA-/RNA-editing enzymes (1). In addition, we included studies based on the use of cells engineered to produce specific proteins, such as neurotrophic factors.
Search Methods
We searched for eligible studies in Pubmed and ClinicalTrials.gov, until October 2020 using the following search terms: “Parkinson Disease [MeSH Terms],” “Huntington Disease [MeSH Terms],” “Ataxia [MeSH Terms],” “Dystonia [MeSH Terms],” “Movement Disorders [MeSH Terms],” “genetic therapy [MeSH Terms],” “case report,” “AAV,” “adenovirus,” “adeno,” “lentivirus,” “ASO,” “antisense oligonucleotide,” “antisense” (Supplementary Table 1).
The “clinical trial” filter was applied for all searches except those including the term “case report.” Only studies referring to human subjects published in English were considered; no restrictions were applied to gender, age, ethnicity, follow-up duration, disease duration, or disease severity. The reference lists of retrieved articles were searched to obtain any other pertinent studies or search terms that were not captured by the original searching strategies.
Selection of Studies and Data Extraction
Abstracts and pertinent full-text articles were independently reviewed for eligibility criteria by one author (NK) and double-checked by two additional authors (CA; AR). Duplicated studies were identified and excluded. A data collection form was used to extract variables of interest from the selected studies. Particular attention was paid to studies that shared the same population or published data from the same cohort at different time-points: in these cases, data from all the available manuscripts were reported, specifying how many participants were included in more than one trial. Disagreements were anticipated to be settled by consensus.
Data collected included year of publication; study design; sample size; sex, age, and disease duration at the time of intervention; dose, target, and route of administration of the compound delivered; follow-up duration; and key adverse events (AEs). In addition, the following data were acquired for each individual population.
Parkinson Disease (PD)
Safety and tolerability, with particular attention to AE reporting; motor section of the Unified Parkinson's Disease Rating Scale (UPDRS-III); Levodopa Equivalent Daily Dose (LEDD) both before intervention and at follow-up. Depending on the endpoints evaluated and the outcome measures used in each trial, we reported changes in motor fluctuation scales (i.e., UPDRS-IV, or time of the waking day spent in Off-state, or in On-state without troublesome dyskinesia), in activities of daily living (ADL) scales [i.e., UPDRS-II, Schwab and England (S&E) scale], in quality of life (QoL) scales (i.e., Parkinson's disease Questionnaire 39—PDQ-39), in cognition scales (i.e., UPDRS-I, neuropsychological batteries), and changes in regional metabolic activity (i.e., PET ligand uptake). Finally, for post-mortem studies, we evaluated the enzymatic expression or immunoreactivity and the area of the targeted brain region covered by the compound delivered.
Huntington Disease (HD)
Safety and tolerability, with particular attention to AE reporting; changes in the Unified Huntington's Disease Rating Scale (UHDRS) and in the Total Functional Capacity Scale (TFC). Depending on the endpoints evaluated and the outcome measures used in each trial, we reported changes in functional, cognitive, and psychiatric outcomes, changes in regional metabolic activity (i.e., PET ligand uptake), and in pharmacokinetics variables.
Aromatic L-Amino Acid Decarboxylase (AADC) Deficiency
Safety and tolerability, with particular attention to AE reporting, and the following secondary endpoints: changes in motor, cognitive, and autonomic functions.
Data were expressed as a mean with standard deviation (SD) or proportion, as appropriate.
Assessment of Risk of Bias
Two investigators (NK; GI) independently assessed the quality appraisal of pertinent studies. A third investigator (C.A.A.) checked the quality assessment and any disagreement in the scoring was resolved by discussion between the three examiners. Given the heterogeneity of study designs, the risk of bias of individual studies was appraised utilizing the National Heart, Lung, and Blood Institute tools (NHLBI, Research Triangle Institute International. National Heart, Lung, and Blood Institute Quality Appraisal Tools), following the Cochrane handbook recommendations (7). Pooled studies were assessed to determine the overall quality of evidence (Tables 1, 2).
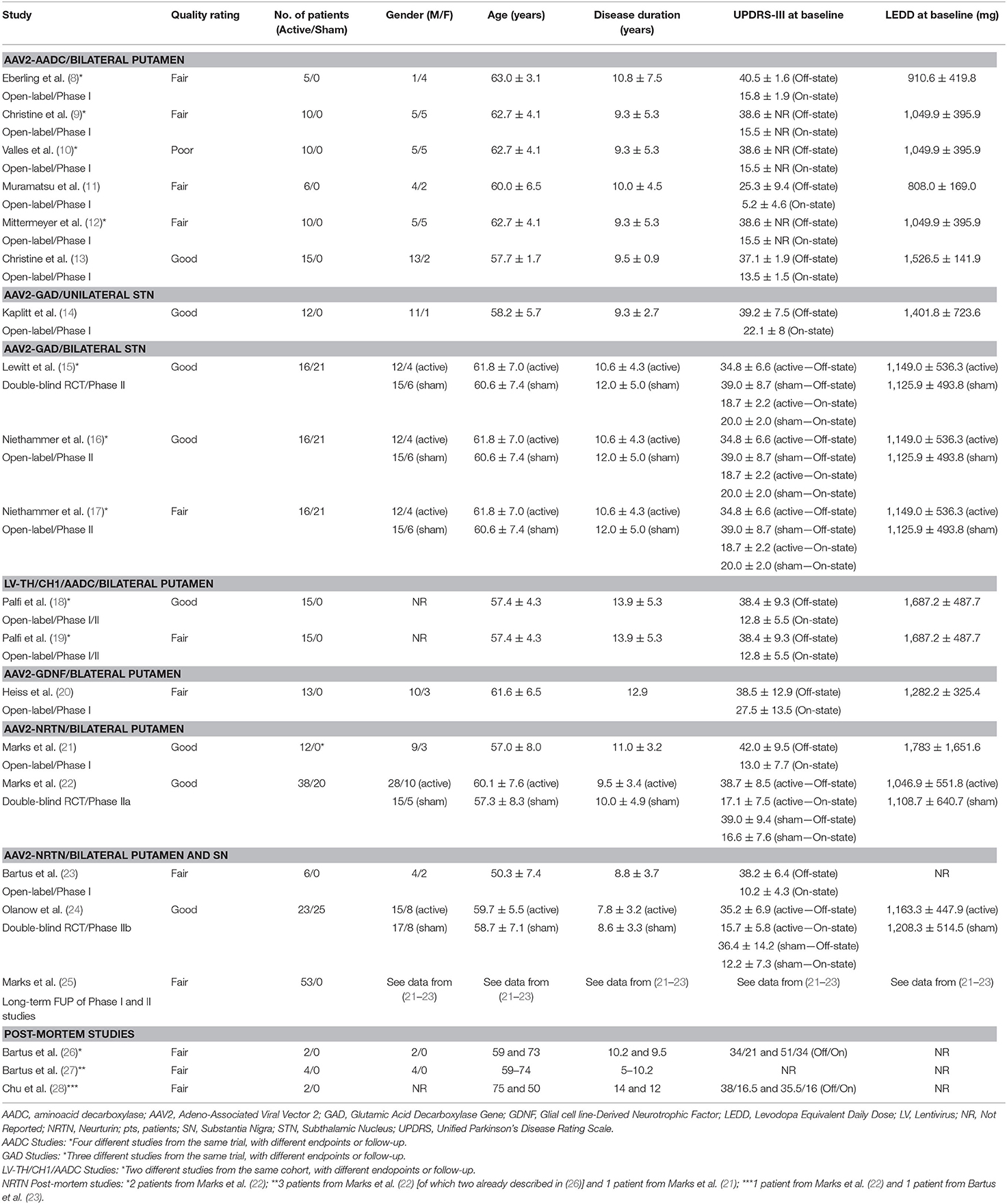
Table 1. Reviewed articles—baseline features—Parkinson disease.
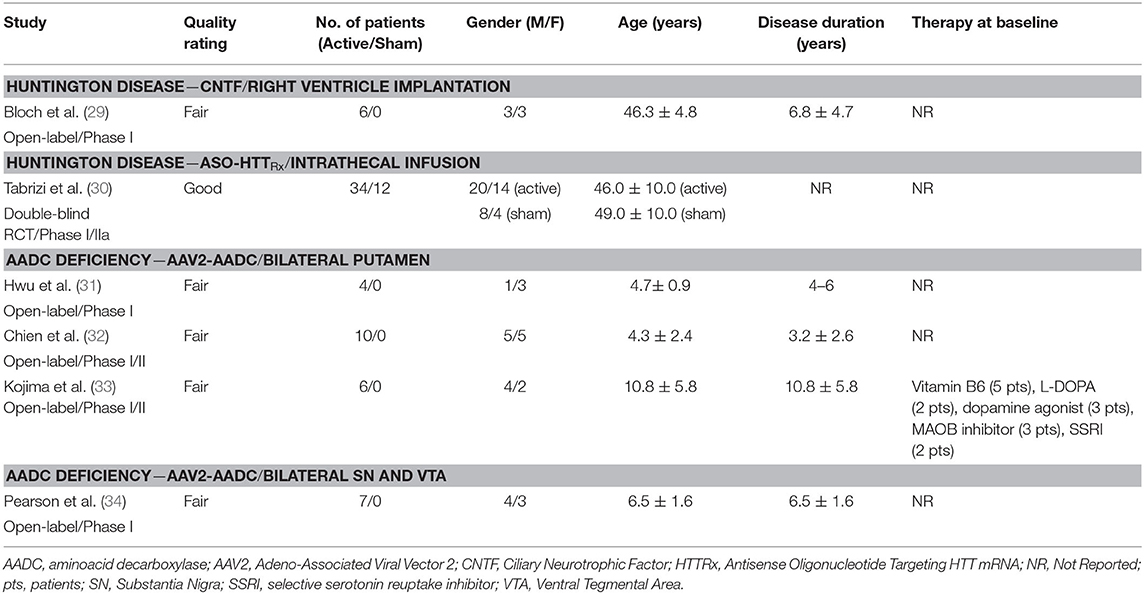
Table 2. Reviewed articles—baseline features—other diseases.
Results
Out of the 1,437 studies derived from the initial searching strategy (Figure 1), 27 published articles met full criteria (8–34) (Tables 1–6) and underwent data extraction and assessment for individual risk of bias. There were 21 studies focusing on PD (8–28), two on HD (29, 30), and four on AADC deficiency (31–34).
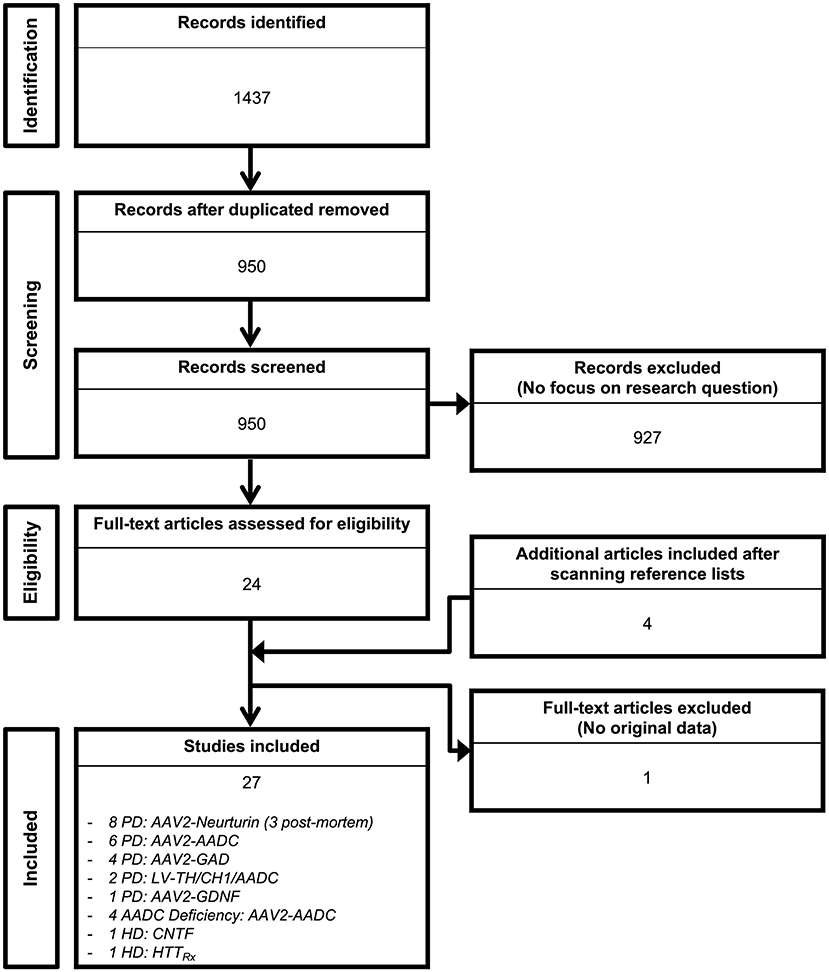
Figure 1. Search flow diagram. Graphical representation of the studies identified, screened for eligibility, and included in the review.
The searching strategy identified eight distinct gene therapy-based molecular approaches: AADC (n = 10 studies) (8–13, 31–34); Ciliary Neurotrophic Factor (CNTF; n = 1 study) (29); Glutamic acid decarboxylase (GAD; n = 4 studies) (14–17); Glial cell line-derived neurotrophic factor (GDNF; n = 1 study) (20); antisense oligonucleotide targeting HTT mRNA (HTTRx; n = 1 study) (30); Neurturin (NRTN; n = 8 studies) (21–28); and Prosavin (n = 2 studies), a multigene approach including tyrosine hydroxylase (TH), GTP-cyclohydrolase 1 (CH1), and AADC (18, 19).
There were 25 studies investigating intraparenchymal gene therapy delivery using Adeno-Associated Virus type 2 (AAV2; n = 19 PD studies, and n = 4 AADC deficiency study) (8–17, 20–28, 31–34) or lentivirus (LV; n = 2 PD studies) (18, 19), 1 study of direct ventricular implantation of polymer-encapsulated cells in HD (29), 1 study of ASO intrathecal infusion in HD (30).
Parkinson Disease
A total of 166 participants with PD received active treatment, and 66 underwent a sham procedure as part of the following studies: AAV2-AADC (n = 3 studies) (8–13), AAV2-GAD (n = 4 studies) (14–17), ProSavin (n = 2 studies) (18, 19), AAV2-GDNF (n = 1 study) (20), and AAV2-NRTN (n = 8 studies, three of which reporting post-mortem data) (21–28) (Tables 1, 3, 4). In all cases, the intraparenchymal delivery was performed via stereotactic neurosurgery.
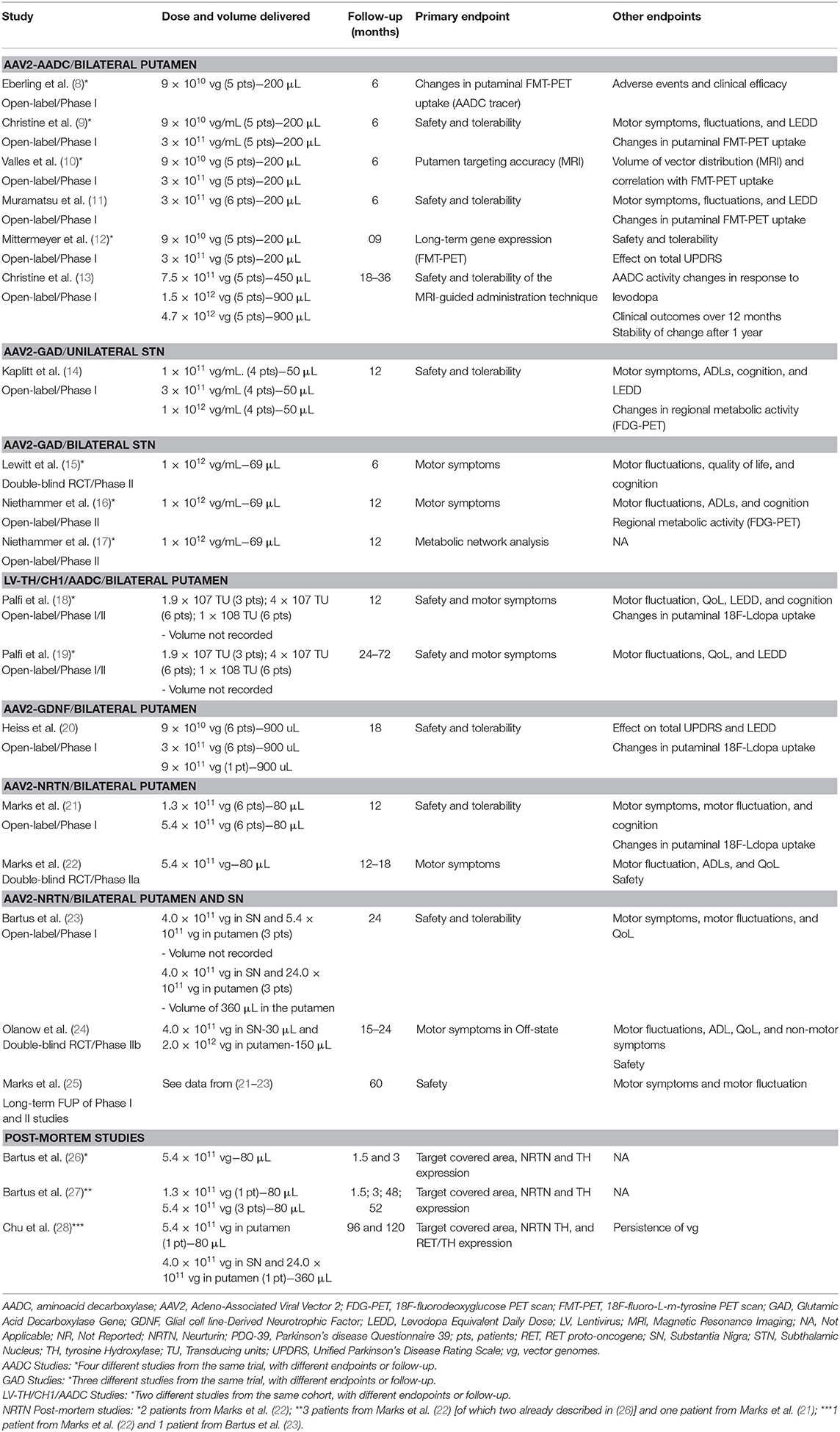
Table 3. Reviewed articles—sample characteristics and endpoints—Parkinson disease.
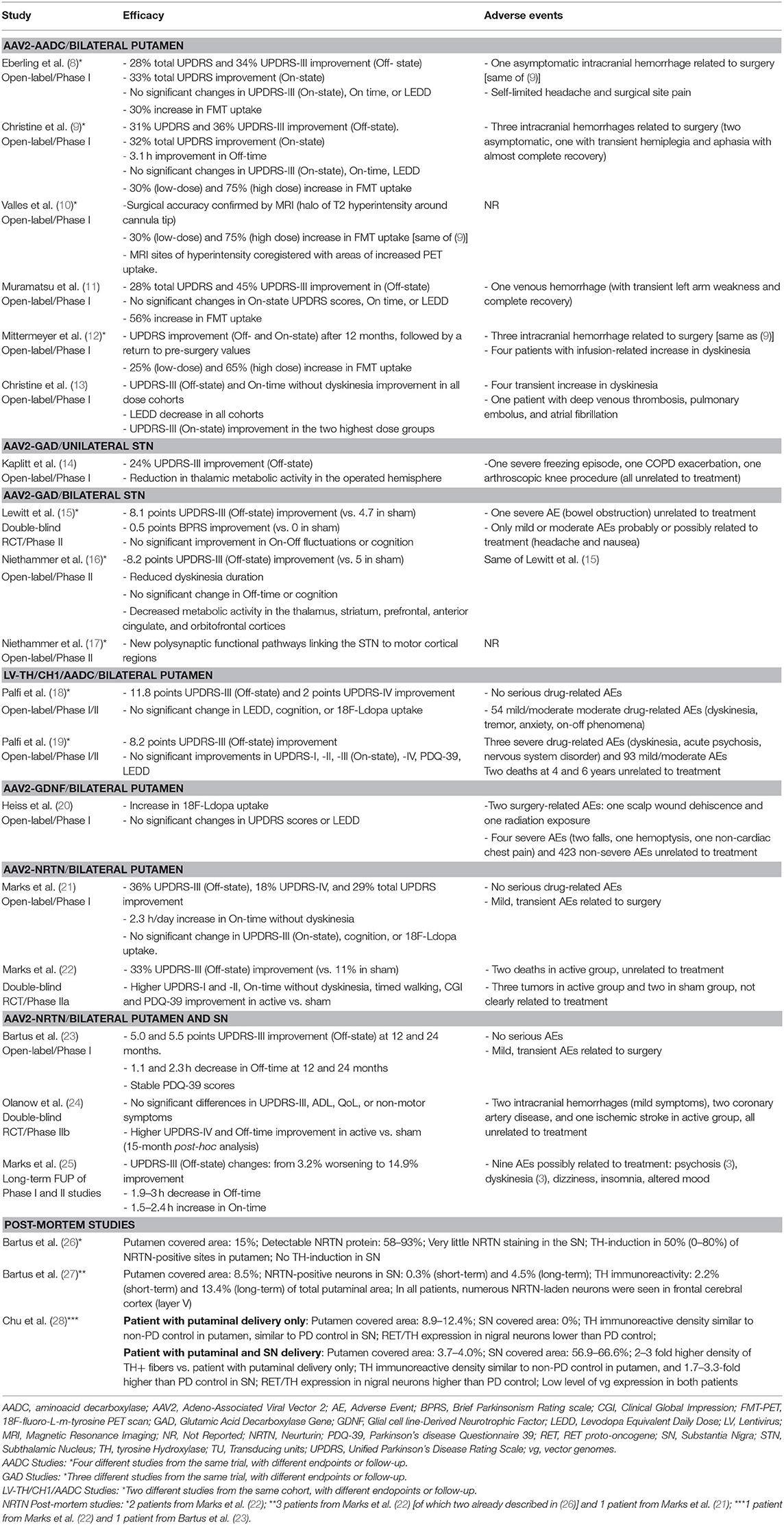
Table 4. Reviewed articles—efficacy and safety—Parkinson disease.
AAV2-AADC
A total of 31 participants (22 males and 9 females) received AAV2-AADC through intraputaminal bilateral infusions over 6 phase-I open-label studies (8–13) (Tables 1, 3, 4). Four studies (8–10, 12) reported data on the same cohort of participants.
Motor Outcomes
One study (9) found a 36% Off-state UPDRS-III improvement at 6 months, followed by a gradual return to the pre-surgical baseline values after 48 months of follow-up (12) (Table 4). A similar extent of Off-state UPDRS-III improvement (range of 21–46% improvement from baseline) was observed in two additional studies (11, 13), after 6 and 18–39 months, respectively, with a trend toward greater improvement in those treated with higher AAV2-AADC dosages (Table 4). No significant changes were observed in the On-state UPDRS-III scores. Three studies reported an improvement in Off-time of 0.6–3.1 h per day (9, 11, 13) and one study (13) showed an improvement in the On-time without dyskinesia of 1.5–3.3 h per day.
Other Efficacy Outcomes
Four studies reported data on Fluoro-m-tyrosine PET (FMT-PET) analysis (9–12) showing a 25–75% increase in the putaminal uptake, with a trend toward greater increases in those treated with higher dosages (Table 4). One study reported 18F-DOPA PET, which demonstrated a 13–79% increase in enzyme activity, with a trend toward greater increases in the higher dose cohorts (13) (Table 4).
Safety
There were five serious AEs related to surgery: three intracranial hemorrhages (two asymptomatic) (8, 9, 12), one venous hemorrhage (11), and one deep venous thrombosis and subsequent pulmonary embolus (13) (Table 4). A transient increase in dyskinesia was also reported in eight participants (12, 13).
AAV2-GAD
A total of 28 participants (23 males and 5 females) received AAV2-GAD through subthalamic bilateral (n = 16) or unilateral infusion (n = 12) within the context of 1 phase I open-label study (14), 1 phase II double-blinded, randomized controlled trial (RCT) (15), and 2 phase II open-label studies (16, 17) (Tables 1, 3, 4). Three studies (15–17) reported data on the same cohort of participants.
AAV2-GAD (Bilateral Subthalamic Infusion)
Motor outcomes. Two phase II trials reported greater improvement of Off-state UPDRS-III scores in the active group compared to the sham surgery group at 6 months (8.1 vs. 4.7 points) (15) and 12 months (8.2 vs. 5 points) (16), but no significant differences were observed in the On-state UPDRS-III scores between groups (Table 4). Neither of the studies were able to demonstrate significant improvement of motor fluctuations, based on participants' diary or total UPDRS-IV. However, one study (16) found a significantly greater improvement of the UPDRS-IV item 32 (duration of dyskinesia) in the active vs. sham group (Table 4).
Other efficacy outcomes. Two studies reported data on functional imaging changes following treatment, showing significant 18F-fluorodeoxyglucose (FDG) PET activity decline in several brain regions (16) and the formation of new pathways linking the STN to other cortical motor regions (17) (Table 4). There were no significant changes in the PDQ-39, UPDRS-II, or any of the neuropsychiatric measures evaluated in either phase-II study. However, one study (15) reported a significant improvement in the Brief Parkinsonism Rating scale (Table 4).
Safety data. One serious adverse event (bowel obstruction) was reported in the AAV2-GAD group (15) and classified as unrelated to treatment. All other AEs possibly related to AAV2-GAD or surgery (severity: mild-to-moderate; n = 28) resolved without residual deficit (Table 4).
AAV2-GAD (Unilateral Subthalamic Infusion)
Motor outcomes. The phase I study (14) showed a 24% improvement in Off-state UPDRS-III, and a 27% improvement in On-state UPDRS-III after 12 months (pooled data, without statistical comparisons amongst the three dosages) (Table 4). There was a non-significant trend toward UPDRS-IV improvement after the 12-month follow-up (Table 4).
Other efficacy outcomes. There were significant reductions in thalamic FDG uptake on PET scan in the treated hemispheres (Table 4). No significant differences were observed in the S&E scale, LEDD, or neuropsychological tests (Table 4).
Safety data. There were three serious AEs: a severe freezing episode, an exacerbation of chronic obstructive pulmonary disease, and an arthroscopic knee procedure. All of the reported AEs were transient and classified as unrelated to treatment (Table 4).
LV-TH/CH1/AADC (ProSavin)
A cohort of 15 male patients received LV-TH/CH1/AADC through stereotactic intraputaminal bilateral infusions, within the context of one phase I/II open-label, dose-escalation study (18), and subsequent long-term follow-up study of the same cohort (19) (Tables 1, 3, 4).
Motor Outcomes
There was an improvement in the Off-state UPDRS-III at 12 months (11.8 points) (18) and at long-term follow up (8.2 points) without a significant difference between the dose cohorts (19). No significant improvements in the On-state UPDRS-III were observed (Table 4). There was a two point improvement in the UPDRS-IV score at 12 months (18), which was not confirmed in the long-term follow-up study (19). No significant differences in mean dyskinesia rating scale scores were reported.
Other Efficacy Outcomes
There was an improvement in UPDRS-II (Off-state: 4 points; On-state: 2 points) and PDQ-39 (5.7 points) at 6 months (18). However, these findings were not confirmed during follow-up (19). No significant changes were observed in LEDD, neuropsychological tests, or 18F-DOPA PET uptake in either study.
Safety Data
There were three serious AEs: dyskinesia, acute psychosis, and an unspecified nervous system disorder. The safety profile was similar across all dose cohorts. Two deaths were reported after 4 and 6 years and were considered unrelated to the treatment (Table 4).
AAV2-GDNF
A total of 13 participants (10 males and 3 females) received AAV2-GDNF through MRI-guided intraputaminal bilateral infusions within the context of a phase I open-label, dose-escalation study (20) (Tables 1, 3, 4).
Motor Outcomes
No significant changes were reported in the On-state or Off-state UPDRS-III after 18 months (Table 4). No significant changes were reported in UPDRS-IV (Table 4).
Other Efficacy Outcomes
No significant changes were reported in UPDRS-I and -II scores, or LEDD, except for an improvement in Off-state UPDRS-II after 1 month of follow-up. Post-hoc analysis showed significant increase in 18F-DOPA putaminal uptake after 6 and 18 months of follow-up (median increase 36% and 54%, respectively; Table 4).
Safety Data
One serious AEs related to the surgical procedure was reported: scalp wound dehiscence that required debridement. No other AEs related to AAV2-GDNF or to the surgical procedure were reported (Table 4).
AAV2-NRTN
A total of 79 participants (56 males and 23 females) received AAV2-NRTN through stereotactic intraputaminal bilateral infusions (n = 50) or intraputaminal plus substantia nigra (SN) bilateral infusions (n = 29), within the context of 2 phase-I open-label studies (21, 23), 2 phase II double-blind RCTs (22, 24), and 1 observational long term follow-up study (25) (Tables 1, 3, 4). Three additional studies reported post-mortem data from participants treated with bilateral putamen (n = 5) or bilateral putamen plus SN (n = 1) AAV2-NRTN infusions (26–28).
AAV2-NRTN (Bilateral Putamen Infusion)
Motor outcomes. There was a 36% UPDRS-III (Off-state) improvement at 12 months in the phase I open-label trial (21), a greater improvement in active vs. sham group (33% vs. 11%) in the phase II RCT at 18 months (22). The 5-year long-term follow-up, however, demonstrated mixed results, ranging from a 3.2% worsening to a 14.9% improvement (25) of UPDRS III Off-state scores (Table 4). No significant differences in On-state UPDRS-III were reported (Table 4). Three studies (21, 22, 25) reported a significant increase in On-time without troublesome dyskinesia ranging from 1.5 to 2.7 h/day with a trend toward greater improvement with higher doses (Table 4), and a significantly higher improvement in the active vs. sham group (22). One study (21) showed a significant decrease in UPDRS-IV score (18%) (Table 4). Changes in Off-time were not significant in any of the studies (Table 4).
Other efficacy outcomes. The RCT (22) showed a greater improvement in the UPDRS-II (2.6 points) and PDQ-39 (6.9 points) in the active vs. sham groups at 12 months. No significant changes were reported on any of the cognitive tests or on 18F-DOPA PET (Table 4).
Safety data. Nine AEs were considered possibly related to AAV2-NRTN: psychosis, dizziness, insomnia, altered mood, and dyskinesia. Other transient AEs related to surgery were equally represented in both active and sham groups (Table 4). Two deaths and three cases of cancer were observed but considered unrelated to the treatment.
AAV2-NRTN (Bilateral Putamen and Substantia Nigra Infusion)
Motor outcomes. The phase I open-label, dose-escalation trial reported an improvement in the Off-state UPDRS-III at 12 months (5 points) and at 24 months (5.5 points) (23), while the phase-IIb RCT found no significant difference in UPDRS-III between active vs. sham group at 15–24 months (24) (Table 4). No data on On-state UPDRS-III were reported and no formal motor assessment data was provided in the 3-year follow-up study (25). There was a 1.1–2.3 h per day decrease in Off-time (23, 24). The phase-IIb RCT showed that this improvement, as well as the amelioration in the UPDRS-IV score, was significantly greater in the active vs. sham group (24) (Table 4). No significant changes in On-time were reported and no data were reported on motor fluctuations in the 3-year follow up (25) (Table 4).
Other efficacy outcomes. No significant differences were reported in the PDQ-39 (23, 24), UPDRS-II, or non-motor symptom questionnaire (24) (Table 4).
Safety data. There were seven serious AEs: two intracranial hemorrhages (mild symptoms), two coronary artery disease, an acute stroke, bradycardia, and orthostatic hypotension. All of the reported AEs were transient, classified as unrelated to the study drug, and resolved with no residual deficit (Table 4).
AAV2-NRTN (Post-Mortem Studies)
Three studies (26–28) reported post-mortem data from six participants treated with AAV2-NRTN (n = 5 putaminal infusion, n = 1 putaminal and SN infusion), evaluating target structure coverage, NRTN expression, and TH expression (Tables 1, 3, 4). Target coverage ranged from 8.5 to 15% of the putaminal volume, with NRTN expression limited to the transfected regions. TH expression, a surrogate measure for dopamine synthesis capacity, was limited to 2.2–13.4% of the total putaminal volume. The SN coverage in the case treated with putaminal and SN infusion was 56.9–66.6%, with a TH expression of 1.7–3.3 times higher than PD controls. The persistence of recombinant viral genome 8 and 10 years post-surgery, (via in situ hybridization) was lower than expected from murine models (28).
Huntington Disease
A total of 40 participants diagnosed with HD received active treatment and 12 received placebo as part of a study investigating CNTF (n = 1 study) (29) and another on ASO-HTTRx (n = 1 study) (30). In the CNTF study, capsules containing polymer-encapsulated BHK cell lines were implanted in the right lateral ventricle and in the ASO-HTTRx trial intrathecal infusions were used to deliver the study drug (Tables 2, 5, 6).
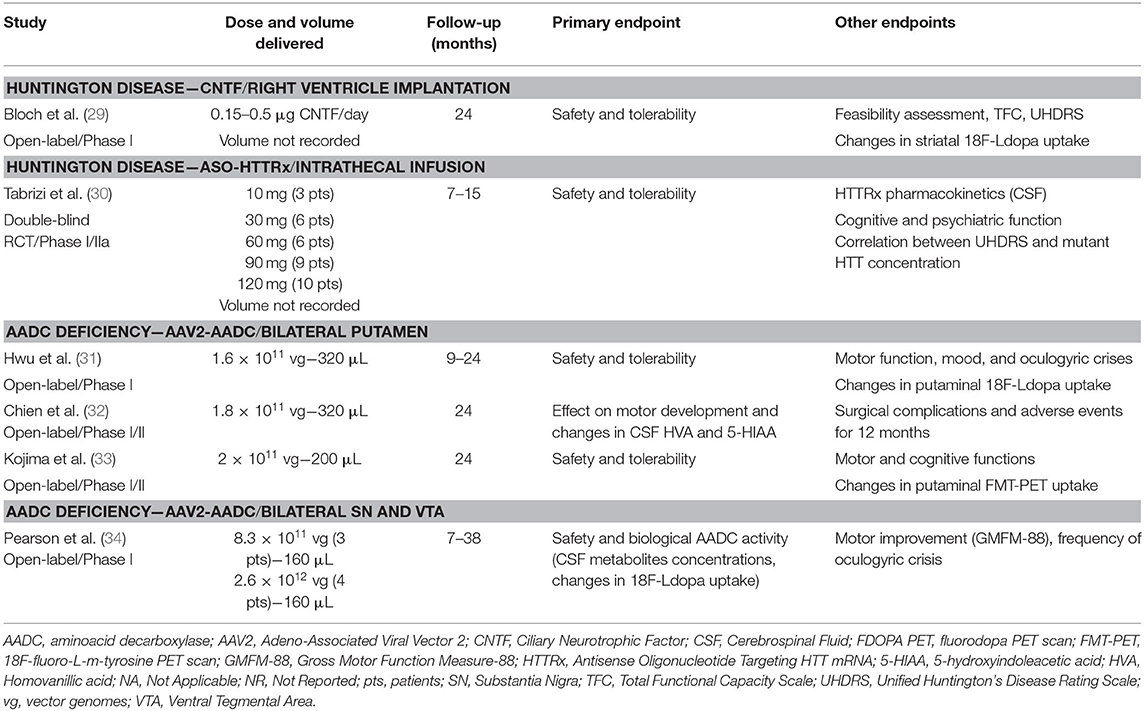
Table 5. Reviewed articles—sample characteristics and endpoints—other diseases.
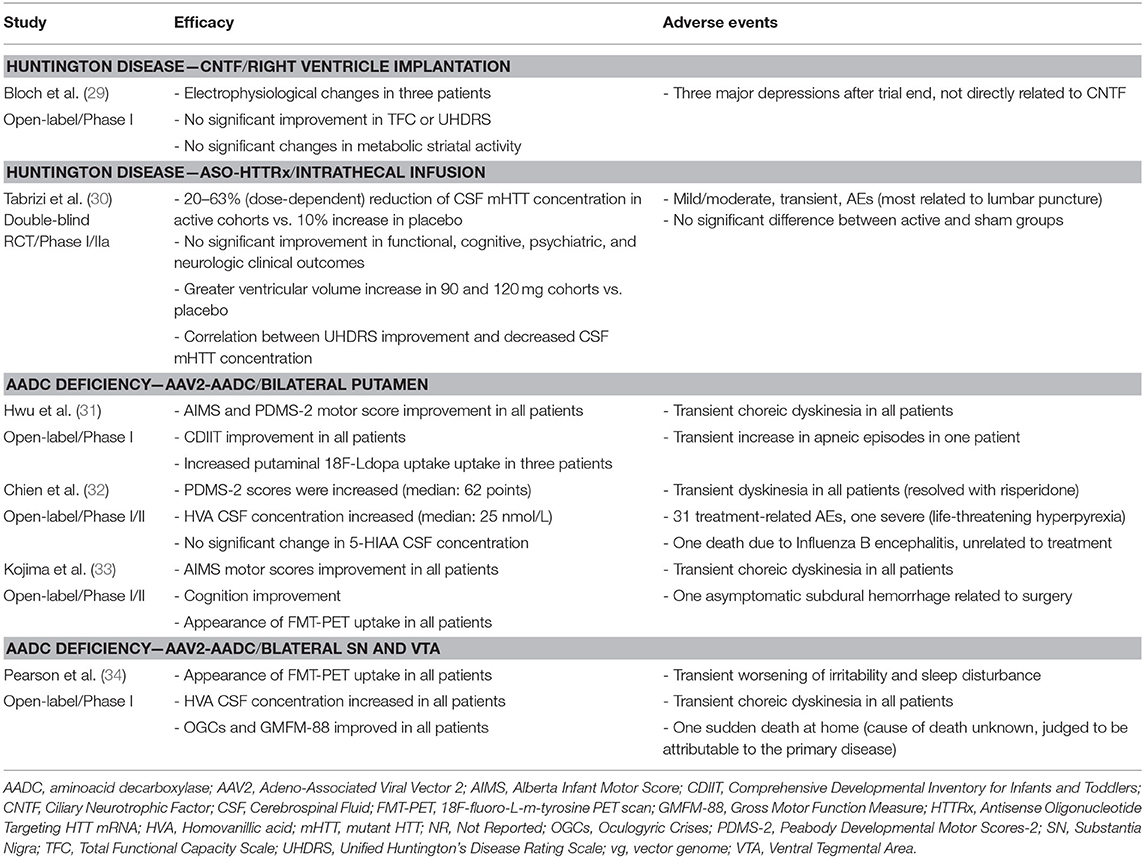
Table 6. Reviewed articles—efficacy and safety—other diseases.
CNTF
A total of six participants (three males and three females) were involved in a phase I open label study (29) and underwent implantation of capsules containing cells engineered to synthesize CNTF. Capsules were exchanged every 6 months for a total of four capsules (Tables 2, 5, 6).
Feasibility
The study demonstrated the feasibility of the procedure. Implantations were well tolerated, and capsule devices were able to remain in the CSF for months and be removed without significant difficulty. However, the capsules did not demonstrate reliable drug delivery: though all capsules released a detectable level of CNTF in the perioperative period, only 11 of 24 explanted capsules still secreted a detectable level after 6 months.
Motor Outcomes
No significant changes were reported in TFC or UHDRS. However, a trend toward a UHDRS amelioration was observed in those participants with capsules that were still actively secreting (Table 6).
Other Efficacy Outcomes
The study did not report any significant increase in striatal uptake by FDG-PET (Table 6). Three participants showed an improvement of electrophysiological parameters (recovery of normal range values for either somatosensory evoked potentials or for intracortical inhibitory response to motor cortex transcranial magnetic stimulation) (Table 6).
Safety Data
No AEs were considered related to CNTF or the surgical procedure. Following trial conclusion, three subjects displayed mood deterioration, in two cases requiring hospitalization; these AEs were most likely linked to the lack of any future therapeutic options and not directly related to CNTF (Table 6).
ASO-HTTRx
A total of 46 participants (n = 12 placebo, n =34 active with five therapeutic dosages) received 4 monthly intrathecal infusions of the ASO HTTRxvia lumbar spinal tap in a phase I/IIa, double-blind, dose-escalation RCT. Intrathecal infusions were performed every 4 weeks for four total doses and participants were followed-up for 7–15 months (30) (Tables 2, 5, 6).
ASO-HTTRx Pharmacokinetics
ASO-HTTRx was measurable in the CSF at therapeutic doses ≥30 mg, and CSF concentrations increased with escalating doses. Peak plasma concentrations were reached within 4 h and declined to <30% of the peak by 24 h. No accumulation of ASO-HTTRx was detected within the CSF or plasma was observed (Table 6).
Clinical Effficacy
No significant changes in UHDRS or HD Cognitive Assessment Battery were reported. However, a post-hoc analysis observed a correlation (no statistical data available) between CSF mutant HTT (mHTT) reduction and improvement in composite UHDRS (Table 6).
Other Outcome Measures
The study reported a dose-dependent decrease in mHTT concentration in the CSF ranging from −20/−42% in the active group as compared to a 10% increase in the placebo group (Table 6). Dose and time-dependent increases in ventricular volume on MRI in the highest dose groups were described (Table 6) as well as transient elevations of CSF neurofilament light chain (Table 6).
Safety Data
There was one serious AE: post-lumbar puncture headache. Mild-to-moderate AEs were reported in 98% of participants, most related to the lumbar puncture and equally represented in both the active and the placebo group (Table 6). Only 6% of AEs were considered treatment-related; all were transient and resolved with no residual deficit (Table 6).
AADC Deficiency
Twenty-seven participants (14 males and 13 females) received AAV2-AADC through intraputaminal bilateral infusions (n = 20) or SN and ventral tegmental area (VTA) (n = 7), in 2 phase I/II open-label studies, one phase I open-label study, and one compassionate use study, and were followed-up for 9–38 months (31–34) (Tables 2, 5, 6).
Motor Outcomes
All participants reported an amelioration in the Alberta Infant Motor Score, in the Peabody Developmental Motor Scores-2, or in the GMFM-88: Gross Motor Function Measure-88. There was a trend toward higher improvement in younger participants (no statistical data available) (Table 6). Swallowing and respiration were improved, dystonia attacks resolved, and oculogyric crises decreased in frequency and/or severity to varying degrees between individuals.
Mental Status
Cognitive scores (Kyoto Scale of Psychological Development-Cognitive-Adaptation and Language-Sociality, or Comprehensive Developmental Inventory for Infants and Toddlers) improved in all participants. However, across these studies, the greatest improvements were seen in the participants with a mild to moderate disease presentation (Table 6).
Other Efficacy Data
When available, imaging data showed more prominent increase in radiotracer uptake in regions that correlate with areas of infusions. FMT-PET putaminal uptake (33) or 18F-DOPA PET midbrain, putaminal, and caudate uptake (34) passed from absent to bilaterally present with high signal intensity, after up to 2-year follow-up, while in the study from Hwu and coll (31) three out of four participants showed an increased 18F-DOPA uptake, as compared to pre-treatment imaging (Table 6). CSF catechol analysis demonstrated levels that trended toward increased dopamine metabolites across these studies, but degree of change in serotonin metabolites and levodopa varied significantly across individuals both before and after treatment. Qualitative improvements in feeding, mood, autonomic functions, and sleep were also reported by majority of caregivers for these pediatric participants (31–34).
Safety Data
Three serious AEs were reported: one asymptomatic subdural hemorrhage related to the surgical procedure (33), one life-threatening hyperpyrexia (32), and one transient increase of apneic episodes (31). Two deaths due to Influenza B encephalitis (32) or to unknown cause (34) were considered unrelated to treatment. All participants experienced transient choreic movements weeks to months after surgery to varying degrees and most were managed with medication adjustments similar to what has been seen in the PD AAV2-AADC studies (Table 6).
Ongoing Studies
Our search on ClinicalTrials.gov retrieved nine ongoing studies on PD, five on HD, two on multiple system atrophy (MSA), two on AADC deficiency, and one on progressive supranuclear palsy (PSP) (Table 7).
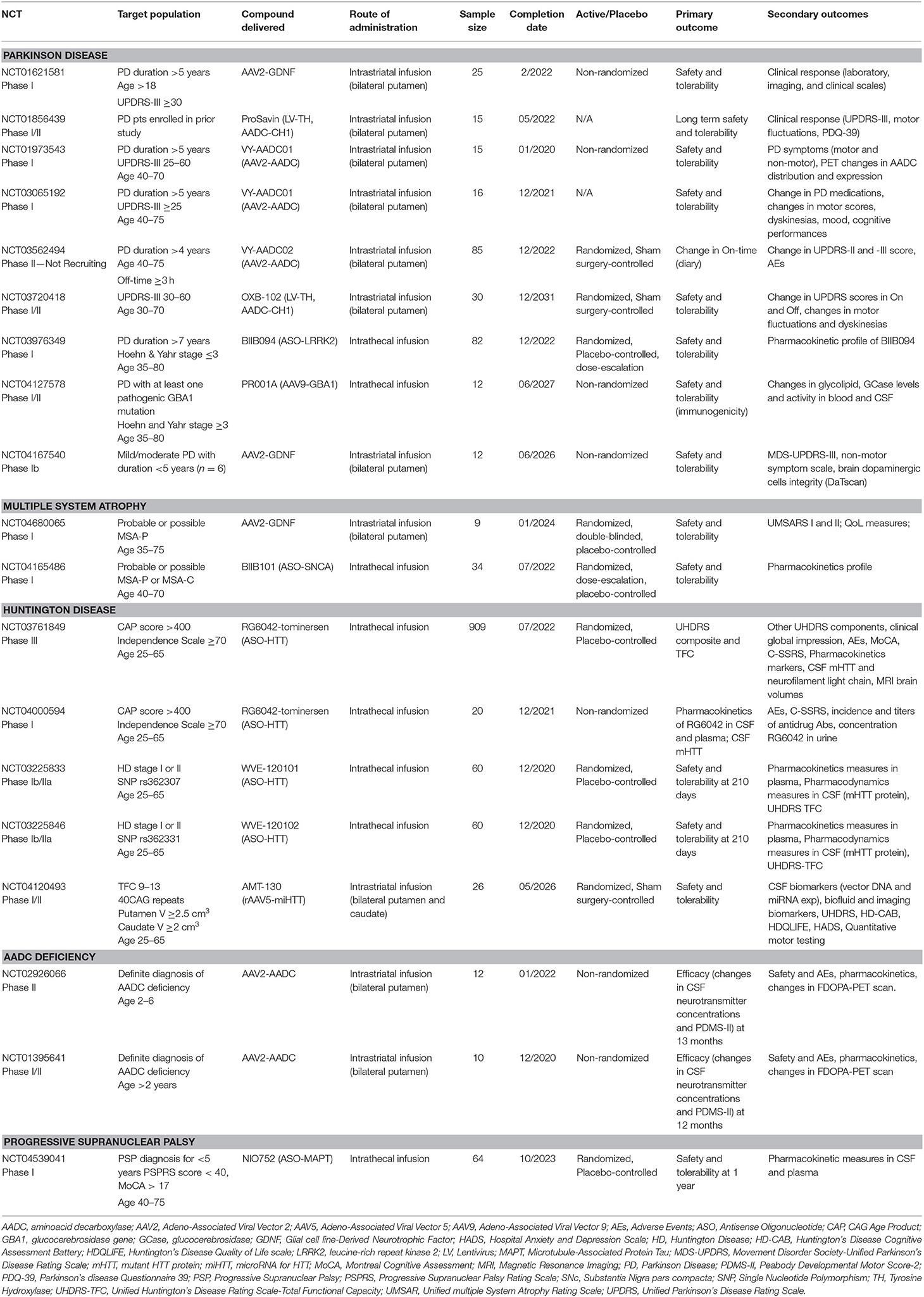
Table 7. Ongoing trials.
In PD, three studies will evaluate AAV2-AADC (one of which active but not recruiting due to asymptomatic MRI abnormalities seen at 12 months post-surgery), two AAV2-GDNF (one of them very close to completion), and one LV-TH/CH1/AADC (ProSavin) (Table 7). The remaining three studies will evaluate new compounds: OXB-102, an optimized version of ProSavin, expressing the same enzymes but with increased DA production (35); BIIB094, an ASO developed to reduce LRRK2 protein levels with repeated intrathecal infusions; and AAV9-GBA (PR001A), a vector-mediated delivery of a functional copy of GBA1 with the aim of constitutively increasing the GCase activity following a single infusion of the study drug. A total of 292 PD participants will take part in these studies.
In MSA, a randomized, double-blind, placebo-controlled phase I study is planned to enroll a total of nine participants (six receiving treatment and three receiving placebo) (Table 7). This first-in-human study will utilize MRI-guided bilateral intraputaminal infusions of AAV2-GDNF with the aim of restoring the putaminal GDNF deficiency found in post-mortem tissue of MSA patients. Safety and tolerability will be the primary endpoint for this phase I study. A phase I randomized, placebo-controlled, dose-escalation study will enroll 34 participants to receive repeated intrathecal infusions of ASO-SNCA (BIIB101) or placebo. The primary objective is safety and tolerability, and pharmacokinetic profile of BIIB101 is a secondary objective.
In PSP, a phase-I, multi-center, double-blind, placebo-controlled, multiple dose-escalation study will enroll 64 participants randomized to receive NIO752, an ASO developed to reduce the expression of Tau protein (Table 7). Patients will be divided into six cohorts and randomized to receive intrathecal NIO752 or placebo in a 3:1 ratio four times over 3 months. The primary outcome includes safety measures. Secondary outcomes will focus on pharmacokinetics and concentrations of NIO752 in the blood plasma and CSF.
In HD, four studies will evaluate the efficacy of intrathecally-infused ASOs targeting HTT mRNA (RG6042, WVE-120101, and WVE-120102), with the aim of reducing clinical meaures of HD progression and accumulation of the mutant HTT in CSF (Table 7). One study will evaluate rAAV5-miHTT (AMT-130) in a randomized, double-blind, dose-escalation, sham-surgery controlled study in early manifest HD participants. The primary outcome will be safety, but CSF levels of AMT-130, mutant huntingtin protein, and other biomarkers will be measured in addition to other disease-specific clinical scales. Safety and tolerability are the primary outcome. A total of 1,075 HD participants will take part in these studies.
In AADC deficiency, two studies will evaluate AAV2-AADC. The primary outcome measure will be biological and clinical efficacy (CSF neurotransmitter metabolite profile and motor scales). Secondary outcomes will include safety, pharmacokinetics profile, and changes in 18F-DOPA PET signal following gene transfer. A total of 22 participants are anticipated to be enrolled in these studies.
Discussion
Multiple clinical trials have examined the safety and preliminary efficacy of gene therapy for PD, HD, and AADC deficiency using complementary DNA sequences encapsulated into viral vectors (AAV2, LV), polymer-encapsulated cell lines engineered to synthesize neurotrophic factors (CNTF), or ASOs. Ongoing clinical trials are also exploring alternative viral vectors (AAV5, AAV9), recombinant genes (OXB-102, GBA1, and HTT), and novel antisense oligonucleotides (LRRK2, HTT, SNCA, and MAPT) for the treatment of several neurodegenerative disorders with a high unmet need to develop disease-modifying therapies.
Several PD gene therapy studies have demonstrated relative safety in multiple clinical trials aimed at restoring the synthesis of dopamine (8–13, 18, 19), enhancing the production of trophic factors (20–25), or modifying the functional interaction between different nodes of the basal ganglia (14–17). There was a variable improvement in the Off-state UPDRS-III with gene therapy approaches aiming at providing dopamine restoration (AAV2-AADC and LV-TH-CH1-AADC) or neuromodulating the basal ganglia network (AAV2-GAD). A variable extent of Off-state UPDRS-III reduction or stability of scores were also observed with gene therapy strategies based on enhancing the production of neurotrophic factors (AAV2-NRTN and AAV2-GDNF). Still, post-mortem studies (26–28) demonstrated limited target coverage in the putamina. These findings indicate the potential for greater clinical benefit by increasing infusion volumes to expand the amount of putaminal tissue exposed to the study drug with current volumes of administration now 4–5 times higher than earlier studies (36). The use of neuroimaging biomarkers, mostly FMT and 18F-DOPA PET, confirmed an increase in the AADC activity or dopamine storage terminal capacity in the AAV2-AADC and AAV2-GDNF trials (8–12, 20). Functional reorganization of the pathways linking the STN with cortical motor regions with AAV2-GAD was demonstrated through metabolic changes by FDG PET following treatment (14, 16, 17). The relative improvement demonstrated by neuroimaging suggests its employment as a key endpoint for gene therapy studies; however, the correlation between imaging and clinical change has been inconsistent and remains a challenge for the field. Ongoing trials aim to evaluate improved formulations of symptomatic and neurotrophic approaches and to test innovative recombinant genes or ASOs to treat specific genetic subtypes of PD, such as GBA1- and LRRK2-associated PD.
In HD, the ASO-HTTRx trial showed that monthly intrathecal delivery of the ASO was safe and tolerable, and demonstrated a dose-dependent reduction of CSF mHTT that persisted 2 months post dose (30). The study was not designed to assess clinical efficacy, but a post-hoc analysis suggested a correlation between improvement in composite UHDRS and CSF mHTT reduction. The ongoing multinational phase III study of the same RG6042 ASO (known as tominersen) will assess 909 participants, comparing clinical progression in those treated with RG6042 vs. placebo controls. Tominersen is a non-allele-specific ASO that reduces expression of both mutant and wild type HTT mRNA. Two phase Ib/IIa RCTs, designed to assess safety, tolerability, and pharmacokinetics, employ allele-specific ASOs, WVE-120101, and WVE-120102. These compounds target single nucleotide polymorphisms (SNPs) found in selected variants of the mHTT gene, thereby knocking down mHTT while preserving wtHTT expression. Though preclinical studies in non-human primates showed ASO target engagement in deep subcortical brain regions following lumbar intrathecal infusion (37), this has not yet been demonstrated in humans. Two studies, one ongoing and another planned (38) will assess the safety of direct subcortical administration of AAV-mediated siRNAs designed to persistently reduce mHTT expression after a single intracranial procedure.
Gene therapy studies have demonstrated to be relative safety in four AADC deficiency trials aimed at restoring catecholaminergic synthesis by constitutively enhancing AADC enzymatic capacity following a single infusion of AAV2-AADC. In the four reported studies, over a dozen serious AEs were reported, however, the majority of these events were expected following enhanced AADC activity (dyskinesia, orofacial dyskinesias) and other events have been reported with underlying disease progression (apneic episodes, feeding difficulties). Intriguingly, all studies reported promising results with amelioration of motor and cognitive performances, in particular in younger participants with milder baseline impairment (31–34). Notably, dysphagia and respiratory function improved, as well as dystonia and oculogyric crises; all of which are disease manifestations that have a high impact on quality of life as reported by caregivers of children with AADC deficiency. Further refinement of gene therapy delivery by directly targeting catecholaminergic midbrain nuclei, as compared to the downstream putaminal targets previously investigated, has recently demonstrated to be a promising approach to improve the magnitude of clinical effect in these neurodevelopmentally compromised children (34).
Multiple factors can influence the efficacy of gene therapy, with the route of administration being one of the most critical. The majority of trials for movement disorders have leveraged intraparenchymal administration to deliver the gene vector directly to the region of interest in the brain (8–25, 29–34). This technique substantially reduces the dose of vector required and reduces potential off-target spread into peripheral tissues, both of which limit the potential immunogenicity (39). In addition to achieving higher vector levels in target target brain regions as compared to intravenous or intrathecal administration routes, studies have also demonstrated intranetwork vector distribution through axonal transport to other brain regions via topographical connections (40, 41). Distribution volume within the target region of the brain can be further enhanced through the use of convection-enhanced delivery (CED), a positive pressure infusion system, to drive the regional spread of vector. This is a critical discovery as volume of coverage and resultant gene expression levels have demonstrated correlation in several humans and non-human studies (9, 13, 42, 43). When a broader delivery is required within the central nervous system, intrathecal, intracerebroventricular, subpial, and intracisternal infusions can also be utilized (Figure 2). Drawbacks to these techniques include higher required doses, lower target tissue levels of vector transduction, as well as more heterogeneous distribution. Finally, intravenous delivery of gene therapy for neurologic disorders are also being explored preclinically. This less invasive mode of delivery is an attractive option with the potential to transfer genes to the entire central nervous system (44). However, its use is limited by the blood brain barrier that requires substantially higher doses, carries the potential for antibody opsonization within the systemic circulation, and substantially increases the risk of immunogenicity and peripheral distribution of the vector (1, 39). Development of less immunogenic capsids and cell-specific enhancers will be critical to safely advance intravenous gene therapies, in particular for neurologic disorders.
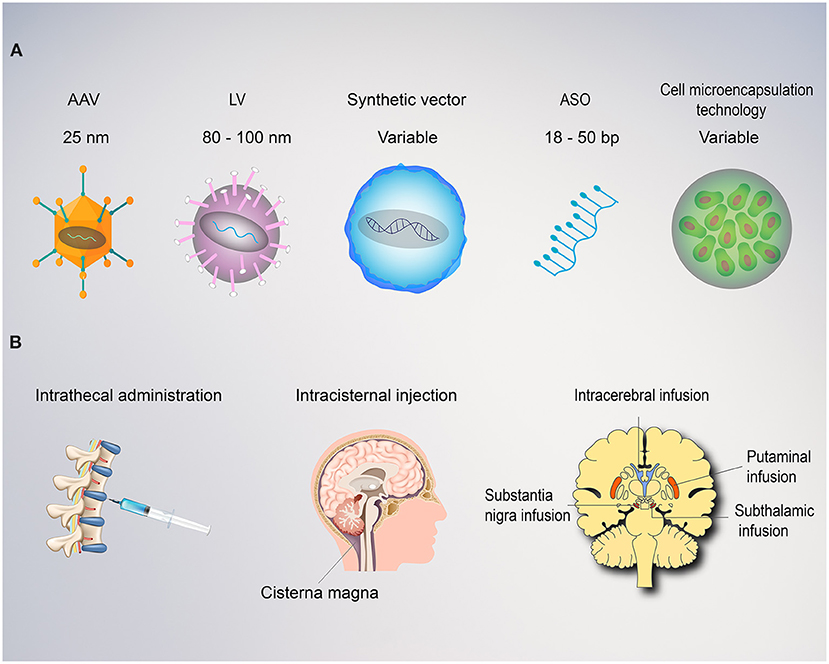
Figure 2. Vectors and route of administration for gene therapy in movement disorders. (A) Both viral and non-viral vectors can be employed for gene therapy. Adeno associated viruses (AAVs) are relatively small with ~4.0 kb packaging capacity and lower immunogenic potential when delivered directly to the brain. AAVs replicate primarily as episomes within the nucleus and rarely integrate into the genome with a low risk of insertional mutagenesis. Lentiviruses (LV) are larger with an 8.5 kb carrying capacity. LVs utilize reverse transcriptase, allowing the transgene to integrate into the host genome, providing stable, long-term transgene expression, but also carries a higher risk of insertional mutagenesis. Synthetic vectors are characterized by lower immunogenicity and larger carrying capacity, but poor delivery efficiency. Antisense oligonucleotides (ASO) are short, single-stranded synthetically derived oligonucleotides. In application for movement disorders, these are typically designed to bind mRNA to interfere with the mechanism of protein translation and thereby reduce levels of aberrant proteins. Cell microencapsulation technology employ cells genetically engineered to secrete proteins and encapsulated into an immunoisolating material that makes them suitable for transplantation. (B) The route of administration is a critical factor for the success of any movement disorders gene therapy approach. Intrathecal, intraventricular, or intracisternal magna injections provide wide distribution of the vector within the central nervous system, but poor diffusion into the deep regions of the target basal ganglia. This approach requires dilution into large volumes of cerebrospinal fluid and delivery of high titers with a consequent increase in cost and risk of immunogenicity. Intraparenchymal delivery ensure precise delivery and optimal vector distribution within the critical structures involved (i.e., putamina, substantia nigra, and subthalamic nucleus).
In conclusion, multiple phase-I and early phase-II studies have been critical in informing the development of the ongoing generation of gene therapy trials. Simultaneously, advancements in understanding viral and non-viral vector efficiency, specificity, and duration of transgene expression have helped select the most promising approaches to treat movement disorders symptoms or administer putative disease-modifying interventions. Still, several limitations should be considered in the interpretation of data. First, most of the studies are characterized by small sample size and many did not include a control group. Therefore, limited conclusions can be drawn on the impact of the placebo effect in these open-label studies, which is known to be especially strong in surgical/interventional trials. Additionally, although the length of follow-up is adequate to demonstrate the short-term safety profile for all evaluated compounds, there are few studies that examine the longterm safety profile, and no definitive conclusions can be drawn on the longterm clinical efficacy. This is even more significant when considering that neurotrophic factors are expected to manifest their neuroprotective effect over several years. Third, most of the trials involving intraputaminal administration for PD, HD, and AADC deficiency were affected by a substantial variability in target coverage, which have been deemed insufficient in a small number of post-hoc analyses and post-mortem immunohistochemical studies. This factor might have been critical in limiting the efficacy of the compounds delivered, and further refinement of delivery techniques are needed to improve reproducibility and consistency of intraparenchymal drug delivery. Fourth, the lack of cerebrospinal fluid biomarkers of PD progression and the preferential inclusion advanced PD patients prevented the ability to test the “disease-modifying” effect of gene therapy in PD. Although the lack of widely validated biomarkers of disease progression continues to represent a limitation in assessing the long-term projected effect of gene therapy in PD, currently ongoing studies are anticipated to provide additional data resulting from the inclusion of a population of early PD patients. Finally, the heterogeneity of study designs limits a direct comparison between the different therapeutic approaches and the possibility of running a meta-analysis.
Improvement in the molecular strategies used to modulate gene expression and target specificity, as well as increasing attention to a precision-medicine approach represent critical opportunities for improvement in upcoming clinical trials and are anticipated to represent the most significant innovations in the future of gene therapy.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
AM: conception, organization, and execution of research project: writing of the first draft and review, and critique of manuscript. NK: organization and execution of research project: writing of the first draft and review and critique of the manuscript organization and execution of research project, review, and critique of the manuscript. AR: organization and execution of research project, review, and critique of the manuscript. BW and AV: writing of sections of the first draft, review, and critique of the manuscript. CA, GI, and IL: execution of research draft, review, and critique of the manuscript. KB: conception, organization and execution of research project, review, and critique of manuscript. All the co-authors listed above gave their final approval of this manuscript version.
Conflict of Interest
AM is supported by NIH (KL2 TR001426) and has received speaker honoraria from CSL Behring, Abbvie, and Cynapsus Therapeutics. He has received grant support from Lundbeck. AR has received grant support and speaker honoraria from AbbVie, speaker honoraria from Chiesi Farmaceutici and travel grants from Lusofarmaco, Chiesi Farmaceutici, Medtronic, and UCB Pharma. BW has received grant support from Acadia and Tilray. CA has received travel grants from Zambon and Abbvie, and educational grants from Ralpharma and Neuraxpharm. IL research is supported by the National Institutes of Health grants: 2R01AG038791-06A, U01NS090259, U01NS100610, U01NS80818, R25NS098999, P20GM109025; U19 AG063911-1; 1R21NS114764-01A1; Parkinson Study Group, Michael J. Fox Foundation, Parkinson Foundation, Lewy Body Association, Roche, Abbvie, Biogen, EIP-Pharma, and Biohaven Pharmaceuticals. She was member of the Scientific Advisory Board of a Lundbeck and Corticobasal Degeneration Solutions. She receives her salary from the University of California San Diego and as Chief Editor of Frontiers in Neurology. AV has received grant support from Voyager Therapeutics and is employed by Asklepios BioPharmaceutical, Inc. KB is the founder of Brain Neurotherapy Bio and Voyager Therapeutics, gene companies. He has equity in Brain Neurotherapy Bio, Asklepios BioPharmaceutical, and Voyager Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.648532/full#supplementary-material
References
1. Chen W, Hu Y, Ju D. Gene therapy for neurodegenerative disorders: advances, insights and prospects. Acta Pharm Sin B. (2020) 10:1347–59. doi: 10.1016/j.apsb.2020.01.015
2. Liu C. Strategies for designing transgenic DNA constructs. Methods Mol Biol. (2013) 1027:183–201. doi: 10.1007/978-1-60327-369-5_8
3. Dana H, Chalbatani GM, Mahmoodzadeh H, Karimloo R, Rezaiean O, Moradzadeh A, et al. Molecular mechanisms and biological functions of siRNA. Int J Biomed Sci. (2017) 13:48–57.
4. Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M. Gene therapy comes of age. Science. (2018) 359:eaan4672. doi: 10.1126/science.aan4672
6. Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. (2009) 6:e1000097. doi: 10.1371/journal.pmed.1000097
7. Higgins JP, Altman DG, Gøtzsche PC, Jüni P, Moher D, Oxman AD, et al. The Cochrane Collaboration's tool for assessing risk of bias in randomised trials. BMJ. (2011) 343:d5928. doi: 10.1136/bmj.d5928
8. Eberling JL, Jagust WJ, Christine CW, Starr P, Larson P, Bankiewicz KS, et al. Results from a phase I safety trial of hAADC gene therapy for Parkinson disease. Neurology. (2008) 70:1980–3. doi: 10.1212/01.wnl.0000312381.29287.ff
9. Christine CW, Starr PA, Larson PS, Eberling JL, Jagust WJ, Hawkins RA, et al. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology. (2009) 73:1662–9. doi: 10.1212/WNL.0b013e3181c29356
10. Valles F, Fiandaca MS, Eberling JL, Starr PA, Larson PS, Christine CW, et al. Qualitative imaging of adeno-associated virus serotype 2-human aromatic L-amino acid decarboxylase gene therapy in a phase I study for the treatment of Parkinson disease. Neurosurgery. (2010) 67:1377–85. doi: 10.1227/NEU.0b013e3181f53a5c
11. Muramatsu S, Fujimoto K, Kato S, Mizukami H, Asari S, Ikeguchi K, et al. A phase I study of aromatic L-amino acid decarboxylase gene therapy for Parkinson's disease. Mol Ther. (2010) 18:1731–5. doi: 10.1038/mt.2010.135
12. Mittermeyer G, Christine CW, Rosenbluth KH, Baker SL, Starr P, Larson P, et al. Long-term evaluation of a phase 1 study of AADC gene therapy for Parkinson's disease. Hum Gene Ther. (2012) 23:377–81. doi: 10.1089/hum.2011.220
13. Christine CW, Bankiewicz KS, Van Laar AD, Richardson RM, Ravina B, Kells AP, et al. Magnetic resonance imaging-guided phase 1 trial of putaminal AADC gene therapy for Parkinson's disease. Ann Neurol. (2019) 85:704–14. doi: 10.1002/ana.25450
14. Kaplitt MG, Feigin A, Tang C, Fitzsimons HL, Mattis P, Lawlor PA, et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson's disease: an open label, phase I trial. Lancet. (2007) 369:2097–105. doi: 10.1016/S0140-6736(07)60982-9
15. LeWitt PA, Rezai AR, Leehey MA, Ojemann SG, Flaherty AW, Eskandar EN, et al. AAV2-GAD gene therapy for advanced Parkinson's disease: a double-blind, sham-surgery controlled, randomised trial. Lancet Neurol. (2011) 10:309–19. doi: 10.1016/S1474-4422(11)70039-4
16. Niethammer M, Tang CC, LeWitt PA, Rezai AR, Leehey MA, Ojemann SG, et al. Long-term follow-up of a randomized AAV2-GAD gene therapy trial for Parkinson's disease. JCI Insight. (2017) 2:e90133. doi: 10.1172/jci.insight.90133
17. Niethammer M, Tang CC, Vo A, Nguyen N, Spetsieris P, Dhawan V, et al. Gene therapy reduces Parkinson's disease symptoms by reorganizing functional brain connectivity. Sci Transl Med. (2018) 10:eaau0713. doi: 10.1126/scitranslmed.aau0713
18. Palfi S, Gurruchaga JM, Ralph GS, Lepetit H, Lavisse S, Buttery PC, et al. Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson's disease: a dose escalation, open-label, phase 1/2 trial. Lancet. (2014) 383:1138–46. doi: 10.1016/S0140-6736(13)61939-X
19. Palfi S, Gurruchaga JM, Lepetit H, Howard K, Ralph GS, Mason S, et al. Long-term follow-up of a phase I/II study of ProSavin, a lentiviral vector gene therapy for Parkinson's disease. Hum Gene Ther Clin Dev. (2018) 29:148–55. doi: 10.1089/humc.2018.081
20. Heiss JD, Lungu C, Hammoud DA, Herscovitch P, Ehrlich DJ, Argersinger DP, et al. Trial of magnetic resonance-guided putaminal gene therapy for advanced Parkinson's disease. Mov Disord. (2019) 34:1073–8. doi: 10.1002/mds.27724
21. Marks WJ Jr, Ostrem JL, Verhagen L, Starr PA, Larson PS, Bakay RA, et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson's disease: an open-label, phase I trial. Lancet Neurol. (2008) 7:400–8. doi: 10.1016/S1474-4422(08)70065-6
22. Marks WJ Jr, Bartus RT, Siffert J, Davis CS, Lozano A, Boulis N, et al. Gene delivery of AAV2-neurturin for Parkinson's disease: a double-blind, randomised, controlled trial. Lancet Neurol. (2010) 9:1164–72. doi: 10.1016/S1474-4422(10)70254-4
23. Bartus RT, Baumann TL, Siffert J, Herzog CD, Alterman R, Boulis N, et al. Safety/feasibility of targeting the substantia nigra with AAV2-neurturin in Parkinson patients. Neurology. (2013) 80:1698–701. doi: 10.1212/WNL.0b013e3182904faa
24. Olanow CW, Bartus RT, Baumann TL, Factor S, Boulis N, Stacy M, et al. Gene delivery of neurturin to putamen and substantia nigra in Parkinson disease: a double-blind, randomized, controlled trial. Ann Neurol. (2015) 78:248–57. doi: 10.1002/ana.24436
25. Marks WJ Jr, Baumann TL, Bartus RT. Long-term safety of patients with Parkinson's disease receiving rAAV2-neurturin (CERE-120) gene transfer. Hum Gene Ther. (2016) 27:522–7. doi: 10.1089/hum.2015.134
26. Bartus RT, Herzog CD, Chu Y, Wilson A, Brown L, Siffert J, et al. Bioactivity of AAV2-neurturin gene therapy (CERE-120): differences between Parkinson's disease and nonhuman primate brains. Mov Disord. (2011) 26:27–36. doi: 10.1002/mds.23442
27. Bartus RT, Kordower JH, Johnson EM, Brown L, Kruegel BR, Chu Y, et al. Post-mortem assessment of the short and long-term effects of the trophic factor neurturin in patients with α-synucleinopathies. Neurobiol Dis. (2015) 78:162–71. doi: 10.1016/j.nbd.2015.03.023
28. Chu Y, Bartus RT, Manfredsson FP, Olanow CW, Kordower JH. Long-term post-mortem studies following neurturin gene therapy in patients with advanced Parkinson's disease. Brain. (2020) 143:960–75. doi: 10.1093/brain/awaa020
29. Bloch J, Bachoud-Lévi AC, Déglon N, Lefaucheur JP, Winkel L, Palfi S, et al. Neuroprotective gene therapy for Huntington's disease, using polymer-encapsulated cells engineered to secrete human ciliary neurotrophic factor: results of a phase I study. Hum Gene Ther. (2004) 15:968–75. doi: 10.1089/hum.2004.15.968
30. Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, Wild EJ, Saft C, Barker RA, et al. Targeting Huntingtin expression in patients with Huntington's disease. N Engl J Med. (2019) 380:2307–16. doi: 10.1056/NEJMoa1900907
31. Hwu WL, Muramatsu S, Tseng SH, Tzen KY, Lee NC, Chien YH, et al. Gene therapy for aromatic L-amino acid decarboxylase deficiency. Sci Transl Med. (2012) 4:134ra61. doi: 10.1126/scitranslmed.3003640
32. Chien YH, Lee NC, Tseng SH, Tai CH, Muramatsu SI, Byrne BJ, et al. Efficacy and safety of AAV2 gene therapy in children with aromatic L-amino acid decarboxylase deficiency: an open-label, phase 1/2 trial. Lancet Child Adolesc Health. (2017) 1:265–73. doi: 10.1016/S2352-4642(17)30125-6
33. Kojima K, Nakajima T, Taga N, Miyauchi A, Kato M, Matsumoto A, et al. Gene therapy improves motor and mental function of aromatic l-amino acid decarboxylase deficiency. Brain. (2019) 142:322–33. doi: 10.1093/brain/awy331
34. Pearson TS, Gupta N, San Sebastian W, Imamura-Ching J, Viehoever A, Grijalvo-Perez A, et al. Gene therapy for aromatic L-amino acid decarboxylase deficiency by MR-guided direct delivery of AAV2-AADC to midbrain dopaminergic neurons. Nat Commun. [Preprint] (2020). doi: 10.21203/rs.3.rs-73870/v1
35. Badin RA, Binley K, Van Camp N, Jan C, Gourlay J, Robert C, et al. Gene Therapy for Parkinson's disease: preclinical evaluation of optimally configured TH:CH1 fusion for maximal dopamine synthesis. Mol Ther Methods Clin Dev. (2019) 14:206–16. doi: 10.1016/j.omtm.2019.07.002
36. Merola A, Van Laar A, Lonser R, Bankiewicz KS. Gene therapy for Parkinson's disease: contemporary practice and emerging concepts. Expert Rev Neurother. (2020) 20:577–90. doi: 10.1080/14737175.2020.1763794
37. McBride JL, Pitzer MR, Boudreau RL, Dufour B, Hobbs T, Ojeda SR, et al. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington's disease. Mol Ther. (2011) 19:2152–62. doi: 10.1038/mt.2011.219
38. Inc VT. Voyager Therapeutics Announces Preclinical Data for Huntington's Disease and Amyotrophic Lateral Sclerosis Programs at Congress of the European Society of Gene and Cell Therapy. GlobeNewswire News Room (2018). Available online at: https://www.globenewswire.com/news-release/2018/10/16/1621781/0/en/Voyager-Therapeutics-Announces-Preclinical-Data-for-Huntington-s-Disease-and-Amyotrophic-Lateral-Sclerosis-Programs-at-the-Congress-of-the-European-Society-of-Gene-and-Cell-Therapy.html (accessed December 21, 2020).
39. Deverman BE, Ravina BM, Bankiewicz KS, Paul SM, Sah DWY. Gene therapy for neurological disorders: progress and prospects. Nat Rev Drug Discov. (2018) 17:641–59. doi: 10.1038/nrd.2018.110
40. Cearley CN, Wolfe JH. A single injection of an adeno-associated virus vector into nuclei with divergent connections results in widespread vector distribution in the brain and global correction of a neyrodegenerative disease. J Neurosci. (2007) 27:9928–40. doi: 10.1523/JNEUROSCI.2185-07.2007
41. Kells AP, Forsayeth J, Bankiewicz KS. Glial-derived neurotrophic factor gene transfer for Parkinson's disease: anterograde distribution of AAV2 vectors in the primate brain. Neurobiol Dis. (2012) 48:228–35. doi: 10.1016/j.nbd.2011.10.004
42. Herzog CD, Dass B, Gasmi M, Bakay R, Stansell JE, Tuszynski M, et al. Transgene expression, bioactivity, and safety of CERE-120 (AAV2-neurturin) following delivery to the monkey striatum. Mol Ther. (2008) 16:1737–44. doi: 10.1038/mt.2008.170
43. Richardson RM, Bankiewicz KS, Christine CW, Van Laar AD, Gross RE, Lonser R, et al. Data-driven evolution of neurosurgical gene therapy delivery in Parkinson's disease. J Neurol Neurosurg Psychiatry. (2020) 91:1210–8. doi: 10.1136/jnnp-2020-322904
Keywords: movement disorder, multiple system atrophy, amino acid decarboxylase deficiency, ataxia, Huntington, Parkinson, gene therapy
Citation: Merola A, Kobayashi N, Romagnolo A, Wright BA, Artusi CA, Imbalzano G, Litvan I, Van Laar AD and Bankiewicz K (2021) Gene Therapy in Movement Disorders: A Systematic Review of Ongoing and Completed Clinical Trials. Front. Neurol. 12:648532. doi: 10.3389/fneur.2021.648532
Received: 11 January 2021; Accepted: 26 February 2021;
Published: 06 April 2021.
Edited by:
Steven Frucht, Mount Sinai Hospital, United StatesReviewed by:
Tommaso Schirinzi, University of Rome Tor Vergata, ItalyEmilia Mabel Gatto, Sanatorio de la Trinidad Miter, Argentina
Copyright © 2021 Merola, Kobayashi, Romagnolo, Wright, Artusi, Imbalzano, Litvan, Van Laar and Bankiewicz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aristide Merola, QXJpc3RpZGUuTWVyb2xhQG9zdW1jLmVkdQ==