 Yohei Mineharu
Yohei Mineharu Susumu Miyamoto
Susumu Miyamoto- Department of Neurosurgery, Kyoto University Graduate School of Medicine, Kyoto, Japan
Moyamoya disease is an idiopathic chronically progressive cerebrovascular disease, which causes both ischemic and hemorrhagic stroke. Genetic studies identified RNF213/Mysterin and GUCY1A3 as disease-causing genes. They were also known to be associated with non-moyamoya intracranial large artery disease, coronary artery disease and pulmonary artery hypertension. This review focused on these two molecules and their strong linker, calcineurin/NFAT signaling and caveolin to understand the pathophysiology of moyamoya disease and related vascular diseases. They are important regulators of lipid metabolism especially lipotoxicity, NF-κB mediated inflammation, and nitric oxide-mediated vascular protection. Although intimal thickening with fibrosis and damaged vascular smooth muscle cells are the distinguishing features of moyamoya disease, origin of the fibrous tissue and the mechanism of smooth muscle cell damages remains not fully elucidated. Endothelial cells and smooth muscle cells have long been a focus of interest, but other vascular components such as immune cells and extracellular matrix also need to be investigated in future studies. Molecular research on moyamoya disease would give us a clue to understand the mechanism preserving vascular stability.
Introduction
Moyamoya disease (MMD) is a progressive occlusive cerebrovascular disease of unknown etiology (1). Main treatment strategy is revascularization surgery to prevent cerebrovascular events, and antiplatelet therapy is another treatment option that has recently been reappraised (2, 3). However, there is no curative treatment that can prevent the progression of arterial stenosis. Understanding the molecular biology of MMD is needed to develop a new treatment strategy and proper biomarkers to evaluate therapeutic efficacy.
Genetics has contributed greatly to understanding the pathophysiology of MMD. In 2011, RNF213 (also called as Mysterin) was identified as the major susceptibility gene for MMD and p.R4810K mutation was found as a founder mutation that increases the risk of MMD by ~300 times (4). The mutation was detected in ~80% of patients with MMD in Japan, ~70% in Korea and ~30% in China. Although the same mutation was not detected in European patients, other mutations such as p.D4013N were found in ~10% (5). Thus, RNF213 is a major susceptibility gene for MMD and it has been recognized as a key molecule to understand the pathophysiology of MMD.
It has been reported that patients with MMD have not only cerebrovascular lesions but also extracranial lesions including coronary artery disease and pulmonary artery stenosis. Importantly, these lesions have common pathological features (6). In agreement with this finding, the p.R4810K mutation in RNF213 gene was also shown to be associated with coronary artery disease (7, 8), pulmonary artery hypertension (9), and renal artery stenosis (10). GUCY1A3 mutations were first detected in patients with quasi-MMD (syndromic MMD) with achalasia (11), but some cases show only moyamoya arteriopathy in the absence of achalasia (12). Of note, GUCY1A3 is also known to be associated with coronary artery disease (13) and pulmonary artery hypertension (14) as is the case with RNF213. Both RNF213 and GUCY1A3 are also associated with hypertension. These lines of evidence suggest that investigation of these two genes may give us some clues to elucidate the molecular pathogenesis of MMD and quasi-MMD.
In this narrative review, we will discuss characteristics of RNF213 and GUCY1A3 in terms of (1) genetics, (2) molecular structures and functions, (3) transcriptional regulation, (4) possible roles in vascular wall, (5) possible roles in vascular insults, and (6) other properties. Finally, we will propose a potential mechanism of MMD and future research directions.
Genetics of RNF213 and GUCY1A3
Susceptibility locus for MMD have been reported to reside on 3p, 8q23, and 17q25.3. Chromosome 17q locus was first identified by chromosome wide linkage analysis (15), and it was narrow down to the 3.5-Mb region at 17q25.3 (16) by linkage analysis of families with autosomal dominant inheritance with incomplete penetrance. In this locus, RNF213 was identified as the major susceptibility gene for MMD (4). In East Asian patients, the p.R4810K mutation is most common, and it is also found in 2% of the general population, which is in agreement with low penetrance (1/100–1/200) of the gene. Intrudingly, genotypes of RNF213 have impact on clinical features of MMD. The p.R4810K mutation was associated with severer form of the disease with higher frequency of bilateral involvement and posterior circulation involvement, and the homozygous p.R4810K mutation was associated with early-onset of the disease (17–19). The p.A4399T polymorphism, which is common in Chinese patients, is associated with hemorrhagic MMD (odds ratio = 2.8; 95% confidence interval, 1.2–6.5) (20). An intronic variant, rs9916351, was reported to be associated with early onset MMD in a Chinese population (21).
The p.R4810K mutation has been reported to be associated with various vascular phenotypes. First, it was shown to be associated with quasi-MMD (syndromic MMD) (22), moyamoya angiopathy with a known comorbidity such as Down syndrome or neurofibromatosis type I (NF1) (23). In addition, it was associated with other vascular diseases including non-moyamoya cerebral arteriopathy (24–26), coronary artery disease (7, 8) and pulmonary artery hypertension (9, 27). The p.R4810K mutation was also associated with hypertension in a dominant fashion (28), while rs9916351 was associated with hypertension in a recessive fashion (29).
Loci on 4q.32.1, 10q23.31, and Xp28 have been identified as loci for quasi-MMD, and GUCY1A3, ACTA2, and BRCC3 was found to be a disease associated gene for each locus (11, 30, 31). Genetic analysis of three families with autosomal recessive mode of inheritance identified GUCY1A3 as a cause of moyamoya angiopathy with achalasia, esophageal sphincter muscle contraction (32). Interestingly, some patients with moyamoya angiopathy associated with GUCY1A3 mutation do not have achalasia (12), meaning that the patient can be diagnosed as MMD but not quasi-MMD. In RNF213, the p.R4810K mutation was common to various vascular phenotypes including MMD, coronary artery disease and pulmonary artery hypertension, whereas different mutations were found in each phenotype for GUCY1A3. In detail, nonsense mutation p.R349X, p.E391KfsX, and c.1086 +1G>A splice donor site mutation were associated with moyamoya with achalasia (11). GUCY1A3 rs1842896 polymorphism was a risk factor for large artery arteriopathy stroke in a Southern Han Chinese population (33). Loss of function mutation p.L163FfsX was associated with an increased risk of myocardial infarction (13). Gain of function mutation p.A681T was associated with a reduced risk of pulmonary artery hypertension (14). A SNP, rs13139571, at the GUCY1A3-GUCY1B3 locus was associated with high blood pressure (34). Thus, different mutations or SNPs exhibit different phenotypes, although hypertension is a common phenotype for most mutations and polymorphisms in GUCY1A3.
Molecular Structure and Functions
Molecular Structure and Functions of RNF213/Mysterin
RNF213/Mysterin is a unique protein which has both functional AAA+ ATPase and E3 ligase. It is a large protein consisting of 5,207 amino acids with a size of 591-kDa (Figure 1). Isoform 3 which lacks exon 4 is the major isoform (NP_001243000.2) (4), and the p.R4810K mutation (rs112735431) is assigned as c.14429G>A. A recent cryo-EM analysis revealed the molecular structure of RNF213 (35, 36). It consists of three structural components including N-terminal structural motif (N-arm), AAA+ ATPase with Walker motif A and B, and E3 ligase with RING finger and NFX1-type zinc finger (Figure 1). Mutations associated with MMD are dominantly observed in E3 ligase.

Figure 1. Molecular structure of RNF213 and soluble guanylate cyclase (sGC) encoded by GUCY1A3. RNF213 consists of N-arm, AAA+ ATPase, and E3 ligase. E3 ligase contains two different types of zinc finger, RING and ZF2 (NFX1-type zinc finger). Not only mutations in the RING and ZF2, but also mutations in the AAA+ ATPase may disturb the function of E3 ligase. GUCY1A3 and GUCY1B3 encode the α1 and β1 subunits of soluble guanylate cyclase (sGC) to form a heterodimer.
E3 ligase plays a central role in post-translational modification by ubiquitination. It recruits an E2 ubiquitin-conjugating enzyme that has been loaded with ubiquitin, recognizes a protein substrate and assists or directly catalyzes the transfer of ubiquitin from the E2 to the protein substrate. Ubiquitin contains seven lysine (K) residues that, together with its amino terminus, provide eight attachment sites for further ubiquitin molecules (K0, K6, K11, K27, K29, K33 or K48, K63), thereby allowing the formation of polymeric chains (37). Variety of combination of branch sites and the length of polyubiquitin chain allow the system to be highly complex, and the system is called “ubiquitin codes.” Together with its various substrates, ubiquitin acts as a versatile cellular signal that controls a wide range of biological processes including protein degradation, DNA repair, endocytosis, autophagy, transcription, immunity, and inflammation. K63 linkages are known to regulate activation of the nuclear factor-kappa B (NF-κB) transcription factor, DNA repair, innate immune responses, clearance of damaged mitochondria, and protein sorting (38). RNF213 RING domain cooperates with Ubc13 E2 ubiquitin-conjugating enzyme to generate K63-linked polyubiquitin chains and induces NF-κB activation (Figure 2). MMD-associated mutations in the RING domain enhances NF-κB activation (39). Still, ubiquitination targets of RNF213 remain largely unknown, except for Nuclear factor of activated T-cells 1 (NFAT1) and filamin A (40).

Figure 2. Diverse biological functions of RNF213 and GUCY1A3. Biological functions and their related molecules were shown. Transcriptional regulators of RNF213 for each function were listed below. α-KGDDs represents α-Ketoglutarate-dependent dioxygenases; ECM, extracellular matrix; PDE, phosphodiesterase; RVF, Rift Valley fever.
The AAA+ superfamily of ATPases has diverse cellular functions including membrane fusion, proteolysis and DNA replication, and it works as molecular chaperon (41). RNF213 was considered to belong to the dynein ATPase family which has 6 AAA domains (35). It forms hexamer (42) and is similar to the resting states of dynein. However, it does not have motor activity along the microtubule but has trans-thiolation activity, which transfers ubiquitin from one substrate to another (36). This means that ATPase and E3 ligase work cooperatively. In fact, deletion or mutations of the ATPase domain reduced the function of E3 ligase (39, 43) (Figure 1). This may explain the reason why MMD is caused by both mutations in the ATPase and those in the E3 ligase.
Molecular Structure and Functions of Soluble Guanylate Cyclase Encoded by GUCY1A3
GUCY1A3 encodes the α1 subunit of soluble guanylate cyclase (sGC), which forms heterodimeric enzyme with ß1 subunit encoded by GUCY1B3 (Figure 1), and it is the major receptor for nitric oxide (NO) (Figures 2, 3). NO binds the heme iron of sGC to induce production of cyclic guanosine monophosphate (cGMP), which then activates the cGMP-dependent protein kinase (PKG) pathway (44). Murine retrovirus integration site 1 (MRVI1) encodes Inositol 1,4,5-Triphosphate Receptor Associated 1 (IRAG1) and plays a role as NO/PRKG1-dependent regulator of IP3-induced calcium release. Phosphorylation of MRVI1 by PRKG1 inhibits bradykinin and IP3-induced calcium release from intracellular stores, leading to inhibition of platelet activation and aggregation. It also mediates NO-dependent inhibition of calcium signaling, which contributes to NO-dependent relaxation of smooth muscle cells. Intrudingly, MRVI1 mutation was found to increase the risk of developing moyamoya angiopathy in patients with NF1. Specifically, p.P186S substitution (rs35857561) in MRVI1 was segregated with quasi-MMD in both the Italian and German NF1 families (45).

Figure 3. Regulation of nitric oxide signaling by RNF213 (hypothetical role) and GUCY1A3 (sGC) in endothelial cells and vascular smooth muscle cells. Endothelial cell stimulation induces calcium-calmodulin-calcineurin signaling, which produces eNOS via NFAT1. RNF213 degrades NFAT1, which may regulate eNOS production. NO is synthesized from L-Arginin by eNOS and diffuses to vascular smooth muscle cells. NO is catalyzed by soluble guanylate cyclase (sGC), and cIMP or cGMP transmit signaling to induce vasodilatation. sGC also contributes to prohibiting fibrinogenesis. Many molecules known to be associated with moyamoya disease such as caveolin-1 (CAV1), Down Syndrome Critical Region 1 (DSCR1), inositol 1,4,5-Triphosphate Receptor Associated 1 (IRAG1) is involved in this molecular network. ALDH2, aldehyde dehydrogenase 2; CaM, calmodulin; cIMP, Inosine 3', 5'-cyclic monophosphosphate; EC, endothelial cell; Hcy, homocysteine; ITP, inosine triphosphate; L-Arg, L-Arginin; MLCK, myosin light-chain kinase; PKD, protein kinase G; PGC-1α, PPARγ-coactivator-1α; PLC, phospholipase C; ROCK, Rho kinase; ROS, reactive oxygen species; VSMC, vascular smooth muscle cell.
The p.C517Y mutation in GUCY1A3 was found in patients with MMD without any evidence of achalasia (12). Sf9 cells expressing the GUCY1A3 p.C517Y mutation showed lower basal cGMP-forming activity and lower maximal NO-induced activity. Thus, the p.C517Y missense mutation led to a significantly blunted response in NO signaling and decreased cGMP production. This was in consistent with previously published observations on rat sGC-containing nitrosylated C516 (mouse homolog to human C517) to resulted in desensitization of sGC to NO activation (46).
Transcriptional Regulation
Transcriptional Regulation of RNF213: A Sensor for Mitochondrial Damage and Inflammation
Various factors have been reported to upregulate RNF213, and all of them were related to inflammation caused by mitochondrial dysfunction and infection. In patients with MMD, morphological abnormalities of mitochondria with higher reactive oxygen species (ROS) as well as elevated Ca2+ levels and reduced mitochondrial reductase activity was detected in circulating endothelial colony-forming cells (47). Recent evidence suggests that mitochondrial dysfunction upregulates RNF213 (Figure 2). Genetic ablation of several mitochondrial matrix factors such as the peptidase ClpP, the transcription factor Tfam increased Rnf213 expression in various organs in mice (48). RNF213 is also upregulated by poly(I:C), which triggers toll like receptor 3 (TLR3)-mediated responses to double-stranded RNA (dsRNA) toxicity (48) (Figure 2). Since dysfunctional mitochondria were recently reported to release immune-stimulatory dsRNA into the cytosol, RNA-dependent inflammation initiated by mitochondrial dysfunction or infections may increase the penetrance of patient mutations in RNF213. SAMHD1 loss of function mutations is known to cause inflammatory vasculopathy including moyamoya angiopathy. It is noteworthy that SAMHD1 works as a sensor of dsRNA and dsDNA at replication fork (49), and dysfunction of this gene is associated with excess interferon (IFN) production and senescence associated secretory phenotype (SASP). Tocilizumab, an interleukin (IL)-6 antagonist, was effective to reverse cerebral vasculopathy in a patient with homozygous SAMHD1 mutation (50). Therefore, regulation of inflammatory signals may be effective for RNF213-related disease as well.
In endothelial cells, RNF213 is also upregulated by type I IFN (51), and the combination of IFN-γ and tumor necrosis factor (TNF)-α synergistically activated transcription of RNF213 both in vitro and in vivo (52) (Figure 2). The transcription of RNF213 was regulated by phosphatidylinositol-3 kinase (PI3K)/AKT and dsRNA-dependent protein kinase R (PKR) pathways. Key et al. also showed that PKR inhibitor inhibited RNF213 expression; however, the effect was only seen in neuronal cell lines but not in fibroblast or HUVEC (48). These findings suggest that transcriptional regulation of RNF213 seems to be cell type- and context-dependent. In macrophage or adipocyte, another inflammatory component, lipopolysaccharide (LPS), was shown to upregulate RNF213, while peroxisome proliferator-activated receptor γ (PPARγ), an anti-inflammatory signal, downregulated it (53). In adipocyte, PTP1B mediated higher expression of RNF213 by TNF-α, while PTP1B suppresses E3 ligase activity of RNF213 in a breast cancer cell line under hypoxic condition (54) (Supplementary Figure 1). Again, regulation of RNF213 was cell-type and context-dependent.
Transcriptional Regulation of GUCY1A3
As compared with RNF213, transcriptional regulation of GUCY1A3 is not well-characterized. Investigation of the promoter activity of GUCY1A3 identified several consensus sequences including NFAT and NF-κB (p50) (55). Because both NFAT and NFκB are master regulators of inflammation, GUCY1A3 may also be regulated by inflammatory signals as is the case with RNF213. Another regulator of GUCY1A3 is eNOS. It has been reported that GUCY1A3 is upregulated by eNOS in the pulmonary vasculature (56). Upon chemical stimulation of sGC, Zinc finger E box-binding homeobox 1 transcription factor (ZEB1) binds to the promoter region of GUCY1A3 (57).
Functions of RNF213 and sGC in the Vascular Wall and Circulation
Histopathological Features of MMD
Histopathological features of MMD were intimal fibrous thickening without significant inflammatory cell infiltration or lipids, the tortuous internal elastic lamina, and disrupted ECs (58, 59). VSMC proliferation was seen in the internal carotid artery, whereas it was atrophic in the distal part of the lenticulostriate arteries (59). Moth-eaten change of the cells with increased extracellular matrix (ECM) was observed in the media, suggesting irregular atrophy and damage of the smooth muscle cells. We will discuss a possible link between these histopathological characteristics and the molecular functions of RNF213 and sGC.
Functions of RNF213 in ECs and VSMCs
Effect of the p.R4180K mutation in endothelial cells were first reported by Hitomi et al. who compared iPS-derived ECs from patients with MMD and unaffected carrier with the p.R4810K mutation and control individuals with wild type RNF213. Angiogenic ability (tube formation) of iPS-ECs from patients and mutation carriers were lower than those from wild-type subjects (60). Gene expression profiles showed that Securin was down-regulated, and knock-down of Securin also impaired the angiogenic activity. RNF213 p.D4013N, p.R4019C, and p.V4146A variants transfected into human umbilical vein endothelial cells had significantly decreased migratory abilities (61).
Effect of the p.R4810K in VSMCs were reported by Tokairin et al. By using VSMCs from iPS-derived neural crest stem cells, they showed that there was no significant difference in VSMC markers, cellular proliferation, migration, or contractile abilities between patients with the p.R4810K mutation and controls (62). Gene expression patterns of iPS-derived VSMCs were similar between patients and controls, whereas iPS-derived ECs displayed distinct patterns. Therefore, they speculated that pathological traits can be driven by naïve ECs predominantly and that VSMCs may require specific environmental factors. In agreement with this concept, RNF213 deficiency affected the cell growth of ECs but not VSMC or fibroblast (52). Likewise, peripheral blood-derived endothelial colony-forming cells but not smooth muscle progenitor cells were responsible for disturbed angiogenic activity when these cells from patients with MMD and healthy controls were co-cultured (63). Taken together, ECs is more likely to contributed to the pathogenesis of MMD, but this model cannot explain the damaged VSMCs as observed in patients with MMD. Therefore, there remains the possibility that even minimum changes in VSMCs caused by RNF213 mutation may affect the phenotype in long-term observation because MMD is a chronically progressive disease.
Although fibrous thickening is a distinguished pathological feature of affected arteries of patients with MMD, little is known about fibrosis in MMD. Hamauchi et al. showed that extracellular matrix (ECM) receptor-related genes, including integrin β3, were significantly downregulated in iPS-derived ECs from patients with the p.R4810K mutation (64). Masuo et al. investigated extracellular matrix secreted from ECs. They showed that iPS-derived ECs from patients with MMD produces less chondroitin sulfate as compared with those from controls (65). Because different types of chondroitin sulfate act differently on fibrillogenesis (e.g., versican enhances fibrillogenesis, while aggrecan, decorin, and lumican have the opposite effect), it remains unclear whether the reduced amount of chondroitin sulfate is associated with increased fibrosis in patients with MMD. Perivascular adipose tissue (66–68) is a well-known source of fibrous tissue in vascular disease, but it is scarce in the intracranial arteries. Thus, VSMCs and their dedifferentiated form are postulated to be responsible for fibrosis, and ECs may have some role in regulating fibrillogenesis of VSMCs via secretion of MMPs and ECM.
Functions of sGC in ECs, VSMCs, and Platelets
In contrast to the major role of RNF213 in ECs, sGC plays an important role in VSMC relaxation and inhibition of platelet aggregation (69). NO-driven cGMP production exerts an anti-atherogenic effects, including vasodilatation, inhibition of vascular smooth muscle proliferation, blockade of leukocyte recruitment, and anti-platelet activity. Although functions of GUCY1A3 mutation were not tested in VSMCs from patients with MMD, influence of sGC dysfunction in aortic or pulmonary smooth muscle cells have been well-studied. A single nucleotide polymorphism in GUCY1A3, rs7692387, was associated with coronary artery disease at genome-wide significance, and it interferes with binding of the transcription factor ZEB1 and impairs GUCY1A3 expression, leading to lower sGC levels and lower sGC activity after stimulation (57). Accordingly, inhibitory effects of sGC stimulator on VSMC migration were abolished in homozygous risk allele carriers. In agreement with these human data, lower Gucy1a3 expression correlated with more aortic atherosclerosis in a population of genetically diverse mice (70). Interestingly, sGC stimulation also has an anti-fibrotic effect (71). Therefore, dysfunctional mutations in GUCY1A3 may be a cause of fibrous thickening of intima in moyamoya angiopathy. In vitro and in vivo models of moyamoya angiopathy having the GUCY1A3 mutation should be developed and investigated.
NO and sGC are generally recognized as major regulators of platelet functions (72) (Figure 2). NO mediates inhibition of collagen-induced platelet aggregation and secretion via sGC. Hervé et al. compared platelet function between patients with moyamoya angiopathy with GUCY1A3 mutation and control individuals. Bleeding time and platelet count was not different, but inhibition of collagen-induced platelet aggregation and secretion by NO donor (PROLI NONOate or sodium nitroprusside) was significantly impaired in platelets derived from patients (11). The risk allele of coronary artery disease, rs7692387 in GUCY1A3, also impaired an inhibitory effect of platelet aggregation by NO donor. In agreement with human data, loss of the α1 subunit of sGC in mice leads to enhanced thrombus formation (13).
Functions of sGC are mostly studied in VSMCs and platelets, but sGC also plays a role in ECs. Pyriochou et al. demonstrated that overexpression of sGC promotes EC proliferation, migration, and tube-like network formation (73). Interestingly, pharmacological stimulation of PKG, a downstream signaling molecule of sGC-cGMP, actually have effects on ECs, VSMCs and platelets. It reduces neointimal hyperplasia, inhibits platelet aggregation, and facilitates re- endothelialization (74).
Functions of RNF213 and sGC in Vascular Insults
Vascular Insults Associated With MMD
As discussed above in the section of transcriptional regulation, RNF213 is upregulated by mitochondrial damage, LPS, poly I:C or type I IFNs, and GUCY1A3 is regulated by NF-κB and NFAT. This suggests that vascular insults such as infection or hypoxia should be associated with moyamoya angiopathy. In fact, recent evidences suggest that dyslipidemia, an elevated level of homocysteine and viral or bacterial infection have an impact on the progression of MMD. Therefore, it is important to clarify the functions of RNF213 and sGC in association with these vascular insults.
Dyslipidemia and Lipotoxicity
Association of lipid metabolism and MMD has been reported very recently. Ge et al. reported that the HDL cholesterol level was inversely associated with the risk of MMD (75). Church et al. reported that dyslipidemia was a risk factor for contralateral progression in patients with unilateral MMD (76). Hirano et al. also reported that dyslipidemia was associated with symptomization of asymptomatic patients with MMD (77). These findings suggest that lipid metabolism may be involved in the pathogenesis of MMD, although neither study showed direct association between RNF213 mutations and dyslipidemia.
In this context, functions of RNF213 in lipid metabolism are of particular interest (Supplementary Figure 1). Strikingly, it has been reported that RNF213 accelerate triglyceride accumulation in lipid droplet (LD) by eliminating adipose triglyceride lipase (ATGL) from LD, one of the major lipolytic molecule of triglyceride (42). The authors showed that the ubiquitin ligase and ATPase activities of RNF213 were both important for its proper LD targeting and its fat-stabilizing activity. Specifically, localization of RNF213 on lipid droplet and its fat-stabilizing activity were disturbed when Caucasian cysteine/histidine mutations (e.g., p.C3997Y) in the RING finger domain or mutations in the AAA+ ATPase domain were introduced. Of note, the p.D4013N mutation in the RING domain and p.R4810K in the E3 core at the C-terminal region, which are mutations found in familial MMD, did not alter the functions of RNF213. This may partly explain the mechanism of low penetrance of the p.D4013N and p.R4810K mutations and high penetrance of the cysteine/histidine mutations.
Lipotoxicity represents toxic effects by saturated fatty acid such as palmitate or stearate, and lipotoxic effect of palmitate on β cells in pancreas can be a cause of diabetes mellitus. It has been reported that ablation of RNF213 decreased lipotoxicity by palmitate (78). Lipid droplet itself does not exert lipotoxicity (79), while it reduces lipotoxicity through incorporating saturated fatty acid inside (80). In fact, RNF213 deficiency increases the activity of SCD1, a key enzyme that promotes palmitate detoxification and storage into triglycerides (78) (Supplementary Figure 1). Consistent with the protective effect against lipotoxicity, ablation of Rnf213 recovered insulin levels in Akita mouse, a diabetes model with impaired insulin production, and it improved glucose tolerance by protecting islet β cells (81). In patients with MMD, the p.R4810K mutation was inversely associated with diabetes mellitus (odds ratio = 0.35; 95% confidence interval, 0.18–0.68) (82). Inverse association of p.R4810K with diabetes was also found in patients with coronary artery disease (odds ratio = 0.34; 95% confidence interval, 0.12–0.79) (7). In respect of protection from diabetes, the p.R4810K is considered to have loss-of-function or dominant negative properties.
Vascular Insults by Homocysteine
Hypehomocysteinuria has been recognized as a cause of quasi-MMD, and recent study showed that homocysteine level is associated with an increased risk of MMD, especially for unilateral MMD (75). Duan et al. showed that rs9651118 in Methylenetetrahydrofolate reductase (MTHFR) gene and rs9651118 in Transcobalamin 2 (TCN2) gene was associated with a homocysteine level, and they were also associated with a risk of MMD (21). Alcohol intake or folate deficiency is associated with increased levels of homocysteine (Supplementary Figure 1), and it has recently been reported that daily alcohol drinking was an independent risk factor for contralateral progression of unilateral MMD and daily drinker with the p.R4810K mutation had significantly higher risk of progression (83). Homocysteine decreased the expression of CAV1 in coronary artery ECs, which induced translocation of NO from caveolae to non-caveolae fractions (Figure 3). As a result, homocysteine impairs NO release from ECs (84), leading to vascular dysfunction. Homocysteine has been shown to induce ER stress and apoptosis in a variety of cell types. Jeong et al. demonstrate that pharmacological inhibition of sGC almost completely abolished the protective effects of NO and nitrite, whereas pharmacological elevation of cellular cGMP, mimicked the protective action of NO donors in neurons (85). Thus, NO donors inhibit homocysteine-induced ER stress and apoptosis via NO-sGC-cGMP signaling pathway. In relation to the signaling of RNF213, Hcy upregulates PTP1B (86).
Viral and Bacterial Infection
Infection has been postulated as a risk factor for MMD, although there is no solid evidence to prove it. Quasi-MMD associated with meningitis was reviewed by Mikami et al. It was estimated to be 2.2% of all the quasi-MMD (87), and it was caused by various pathogens. Among them, varicella zoster virus infection draws attention due to the high frequency of reversible arteriopathy called transient cerebral arteriopathy (TCA) or focal cerebral arteriopathy (FCA), which sometimes progresses to moyamoya (88). Pathological examination of postinfectious vasculopathy with progression to moyamoya angiopathy following Streptococcus pneumoniae meningitis was reported by Czartoski et al., and they showed that inflammation or atherogenic features were absent in the lesion. Due to the progressive course, elevated anti-β2-glicoprotein 1 IgG titers, and transient response to immunomodulatory therapy, they speculated that the vasculopathy was likely to be mediated by an autoimmune process (89).
Involvement of RNF213 in viral infection has just recently been reported. Homozygous deletion of Rnf213 showed significantly shorter survival in C57BL/6 J mice lethally infected with Rift Valley fever virus (90), an enveloped negative single stranded RNA viruses. The mechanism of entry is dynamin-dependent, CAV1-mediated endocytosis (91). Because Rift Valley fever virus infection suppresses the response of IFN-β or other IFN-related molecules (92, 93), type-I IFN mediated upregulation of Rnf213 is not likely to be the mechanism of action against the virus. More recently, it has been shown that RNF213 restricts the proliferation of cytosolic Salmonella and is essential for the generation of the bacterial ubiquitin coat, which initiates antibacterial autophagy (43). The ubiquitylation of LPS on Salmonella requires the AAA+ ATPase domain and newly identified NFX1-type zinc finger domain (ZF2 in Figure 1). Together with the fact that RNF213 is upregulated by LPS, RNF213 would contribute to antibacterial immune reactions. However, no patient mutation tested in the study including p.R4810K mutation affected the ubiquitination of LPS.
Other Properties
Inflammation via NF-κB Signaling
RNF213 protects cells from ER stress and inflammation by lipotoxicity. Depletion of RNF213 stabilizes ER stress gene expression, normalizes the cellular lipidome, and blocks NF-κB pathway during palmitate exposure (78). Recent studies showed that RNF213 selectively cooperates with Ubc13 (E2 enzyme) to generate K63-linked polyubiquitin chains, but not K48-linked ones (39, 94). K63 linkages are known to regulate activation of the NF-κB transcription factor, DNA repair, innate immune responses, clearance of damaged mitochondria, and protein sorting (38). Interestingly, BRCC3, whose deletion was associated with X-linked syndromic moyamoya, is a E3 ligase that specifically cleaves K63-linked polyubiquitin chains (31). This molecule regulates the abundance of these polyubiquitin chains in chromatin and plays a role in the DNA damage response.
Importantly, most mutations in the RING domain found in patients with MMD reduced E3 ligase activity and many of them induced NF-κB activation (39). These mutations that induce NF-κB activation included not only Caucasian cysteine/histidine mutations but also proline mutations (p.P4007R in a Chinese patient and P4033L in a Caucasian patient). These mutations also induced apoptosis in a NF-κB dependent manner. However, p.D4013N mutation did not affect either E3 ligase activity or NF-κB activation. Importantly, critical point mutations in both the Walker A and B of the AAA domains, completely abrogated NF-κB activation by RNF213 mutation in the RING domain. Thus, inflammation via NF-κB pathway was enhanced by patient mutations in RNF213, while it was suppressed in the absence of RNF213. In respect of NF-κB activation, RNF213 mutations should have gain of function properties.
RNF213 also controls mitochondrial functions, cell cycle, or differentiation and maturation of immune cells (Figure 2, details are described in Supplementary Material). These properties will also play a role in the development of MMD.
Potential Mechanism in MMD
Possible Interaction of RNF213 and sGC
Molecular networks that potentially connects RNF213 and sGC are shown in Figure 4. The key molecule is NFAT1, which is a ubiquitin target of RNF213 downstream of non-canonical WNT/Ca2+ signaling (40). Activation of calcineurin/NFAT signaling by VEGF in human endothelial progenitor cells leads to increased eNOS protein expression and NO production (95). NFAT may also regulate the expression of sGC through binding of NFAT1 to the consensus sequence in GUCY1A3 (55). Activation of PI3K/AKT leads to inactivation of glycogen synthase kinase (GSK)-3β, which induces degradation of NFAT1 by the proteasome (96). Because PI3K/AKT was reported to be an upstream regulator of RNF213 expression in ECs (52), the effect of PI3K/AKT on NFAT1 may be mediated by RNF213. Upregulation of NFAT1 is also mediated by S-nitrosylation of RNF213 (97). S-nitrosylation is post-translational modification adding a nitrosyl group to the reactive thiol group of a cysteine to form S-nitrosothiol, which is a key mechanism in transferring NO-mediated signals. S-nitrosylation of ubiquitin ligase leads to its auto-ubiquitination and, as a consequence, increases its substrate levels. NFAT is, in turn, regulated by sGC (98) via activation of PKG, which phosphorylates GSK-3β.
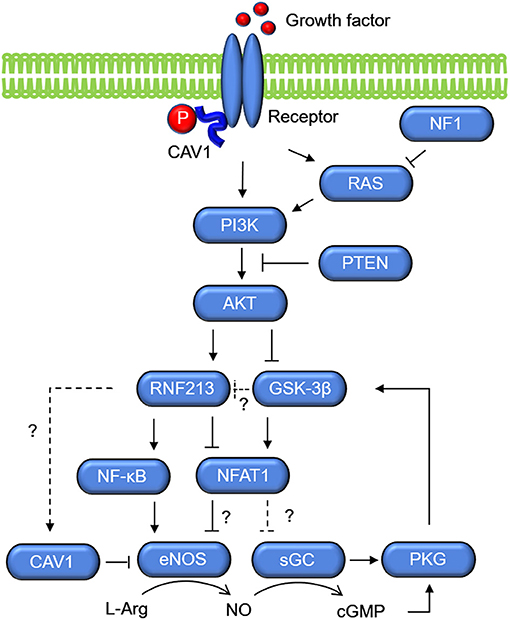
Figure 4. Molecular networks of RNF213 and GUCY1A3 (sGC). Growth factors activate PI3K/AKT, which upregulate the expression of RNF213 and eNOS. Activation of PI3K/AKT leads to inactivation of GSK-3β, which increases degradation of NFAT1 by the proteasome possibly via ubiquitination by RNF213. As opposed to the Ca+/Calmodulin/NFAT1 signaling shown in Figure 3, NFAT1 is assumed to suppress eNOS expression in the context of PI3K/AKT. GSK-3β, glycogen synthase kinase-3β; PKG, protein kinase G; sGC, soluble guanylate cyclase.
Another key molecule that regulates NO signaling is caveolin. The integral membrane protein caveolin-1 (CAV1) is ~21–24 kDa and is found primarily in 50–100-nm flask-shaped invaginations called plasma membrane caveolae, where it acts as a scaffold to organize multiple molecular complexes that regulate a variety of cellular events. CAV1 is regulated by a signal mediated through Ca2+/calcineurin/NFAT (99). Importantly, CAV1 was reported to be associated not only with MMD, but also with coronary artery disease and pulmonary artery hypertension. The function of CAV1 is well-characterized in pulmonary artery hypertension (100–102). CAV1 level was decreased in patients with MMD as compared with either patients with intracranial atherosclerotic stroke or healthy subjects, and it was markedly decreased in RNF213 R4810K variant carriers (103). However, it remains unclear whether RNF213 has direct, e.g., CAV1 is a target of ubiquitination by RNF213, or indirect association with CAV1. CAV1 is associated with eNOS release (Figure 4). NO is generated by eNOS release and it is metabolized by sGC. Direct binding of eNOS to the scaffolding domain of CAV1 is a well-accepted mechanism for inactivating eNOS (104). The absence of CAV1 is thought to promote eNOS dysfunction associated with cerebrovascular disease.
Potential Mechanism of MMD
Based on the molecular functions and networks of RNF213 and sGC encoded by GUCY1A3 as discussed above, we propose a potential mechanism in MMD as shown in Figure 3. Under the physiological condition, EC stimulation by growth factors or shear stress induces Ca2+-calmodulin signaling. Calmodulin accelerates eNOS dissociation from CAV1 and upregulates eNOS production via Calcineurin/NAFT1. RNF213, which degrades NFAT1 through ubiquitin proteasome system, is not activated without pathological stimulations. eNOS then produces NO from L-Arginine, and NO diffuses into VSMCs. In VSMCs, NO activates sGC to produce cGMP. cGMP then activates PKG, which induces relaxation of VSMCs. Under the pathological condition where viral infection causes mitochondria destruction, RNF213 is upregulated and prohibits eNOS production by degrading NFAT1. Patient mutations in RNF213 may sustain the inflammation even after resolution of the infection, and it causes sustained cGMP signaling impairment. The same situation can be caused by GUCY1A3 mutations. Impaired cGMP signaling leads to VSMC proliferation, impaired vasodilation, and fibrosis as well as endothelial dysfunction. These events will account for intimal hyperplasia with fibrous thickening, a typical pathological feature of MMD. Because impaired cGMP signaling can cause VSMC dedifferentiation and endothelial-to-mesenchymal transition, these cells can be potential sources of fibrosis.
Impaired protection from vascular insults is also a potential mechanism of arterial stenosis in MMD. As mentioned in the previous section, sGC acts protective against homocysteine, and RNF213 regulates lipotoxicity and has antiviral and antibacterial properties. Viral or bacterial infection causes type I IFN production and mitochondrial dysfunction, and they increase the RNF213 expression. When patient mutations induce dysfunction of RNF213 or sGC, vascular damage caused by homocysteine, dyslipidemia or infection may be amplified, leading to chronic inflammation. RNF213 mutations also induce inflammation via NF-κB. This may cause damaged VSMCs, which is another typical pathological feature of MMD.
In terms of inflammation, autoimmune conditions with Th17+ T cell polarization seem to be associated with MMD as discussed in the Supplementary Material. However, the effect of RNF213 and GUCY1A3 mutations have been studied mostly in vascular cells (ECs and VSMCs) but not in immune cells. To understand the precise mechanism of MMD, investigation of functions of RNF213 and GUCY1A3 in immune cells especially T cell, B cell, neutrophil and dendritic cell will be needed.
Potential Therapeutic Strategy for MMD
Based on these potential mechanisms, we propose several therapeutic strategies that includes lipid and homocysteine regulation (Mediterranean or Japanese traditional diet, restriction of alcohol intake, and lipid/homocysteine lowering drugs), control of inflammation (avoidance of hypoxic condition and anti-inflammatory drugs), and pharmacological stimulation of eNOS-sGC-cGMP pathway. Candidates for pharmacological treatment of MMD would be anti-inflammatory drugs (e.g., COX-2 inhibitor or anti-IL-6 antibody), lipid lowering (e.g., statins or PCSK9 inhibitor) and homocysteine lowering drugs (e.g., folate or vitamin B12). Riociguat, a stimulator for sGC that is used to treat pulmonary artery hypertension, may be another option. However, there is not enough evidence that proves functionality and interaction of the candidate molecules, which account for the phenotypes of MMD. More precise molecular networks should be identified to develop a new treatment strategy.
Conclusions
In 10 years after identification of RNF213 as a susceptibility gene for MMD, function of RNF213 has been gradually unraveled. It plays a key role in lipid metabolism, oxygen consumption, cell cycle control and inflammation, and it contributes to the maintenance of vascular cells. GUCY1A3 is a regulator of platelet function and VSMC contraction via NO-sGC-cGMP pathway. Both mutations in RNF213 and GUCY1A3 cause not only MMD, but also non-moyamoya intracranial arterial diseases, coronary artery disease, and pulmonary artery hypertension. They have significant interaction with CAV1 and NFAT1, both of which have diverse molecular functions including immune regulation and cell cycle control. Functions of RNF213 and GUCY1A3 in ECs and VSMCs have been well-studied, but a precise mechanism of intimal thickening, fibrosis and its origin remain unresolved. Involvement of other vascular components such as inflammatory cells, platelet and extracellular matrix need to be further investigated.
Author Contributions
YM conceived and designed the study, performed the literature search, and drafted the paper. SM reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by Grants in Aid for Scientific Research (B) to SM (19H03770).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.687088/full#supplementary-material
References
1. Kuroda S, Houkin K. Moyamoya disease: current concepts and future perspectives. Lancet Neurol. (2008) 7:1056–66. doi: 10.1016/S1474-4422(08)70240-0
2. Ye F, Li J, Wang T, Lan K, Li H, Yin H, et al. Efficacy and safety of antiplatelet agents for adult patients with ischemic moyamoya disease. Front Neurol. (2021) 11:e608000. doi: 10.3389/fneur.2020.608000
3. Miyoshi K, Chida K, Kobayashi M, Kubo Y, Yoshida K, Terasaki K, et al. Two-year clinical, cerebral hemodynamic, and cognitive outcomes of adult patients undergoing medication alone for symptomatically ischemic moyamoya disease without cerebral misery perfusion: a prospective cohort study. Neurosurgery. (2019) 84:1233–1241. doi: 10.1093/neuros/nyy234
4. Liu W, Morito D, Takashima S, Mineharu Y, Kobayashi H, Hitomi T, et al. Identification of RNF213 as a Susceptibility Gene for Moyamoya Disease and Its Possible Role in Vascular Development. PLoS ONE. (2011) 6:e22542. doi: 10.1371/journal.pone.0022542
5. Guey S, Kraemer M, Hervé D, Ludwig T, Kossorotoff M, Bergametti F, et al. Rare RNF213 variants in the C-terminal region encompassing the RING-finger domain are associated with moyamoya angiopathy in Caucasians. Eur J Hum Genet. (2017) 25:995–1003. doi: 10.1038/ejhg.2017.92
6. Ikeda E. Systemic vascular changes in spontaneous occlusion of the circle of Willis. Stroke. (1991) 22:1358–62. doi: 10.1161/01.STR.22.11.1358
7. Morimoto T, Mineharu Y, Ono K, Nakatochi M, Ichihara S, Kabata R, et al. Significant association of RNF213 p.R4810K, a moyamoya susceptibility variant, with coronary artery disease. PLoS ONE. (2017) 12:e0175649. doi: 10.1371/journal.pone.0175649
8. Koyama S, Ito K, Terao C, Akiyama M, Horikoshi M, Momozawa Y, et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat Genet. (2020) 52:1169–77. doi: 10.1038/s41588-020-0705-3
9. Kobayashi H, Kabata R, Kinoshita H, Morimoto T, Ono K, Takeda M, et al. Rare variants in RNF213, a susceptibility gene for moyamoya disease, are found in patients with pulmonary hypertension and aggravate hypoxia-induced pulmonary hypertension in mice. Pulm Circ. (2018) 8:204589401877815. doi: 10.1177/2045894018778155
10. Pinard A, Fiander MDJ, Cecchi AC, Rideout AL, Azouz M, Fraser SM, et al. Association of de novo RNF213 variants with childhood onset moyamoya disease and diffuse occlusive vasculopathy. Neurology. (2021) 96:e1783–91. doi: 10.1212/WNL.0000000000011653
11. Hervé D, Philippi A, Belbouab R, Zerah M, Chabrier S, Collardeau-Frachon S, et al. Loss of α1β1 soluble guanylate cyclase, the major nitric oxide receptor, leads to moyamoya and achalasia. Am J Hum Genet. (2014) 94:385–94. doi: 10.1016/j.ajhg.2014.01.018
12. Wallace S, Guo DC, Regalado E, Mellor-Crummey L, Bamshad M, Nickerson DA, et al. Disrupted nitric oxide signaling due to GUCY1A3 mutations increases risk for moyamoya disease, achalasia and hypertension. Clin Genet. (2016) 90:351–60. doi: 10.1111/cge.12739
13. Erdmann J, Stark K, Esslinger UB, Rumpf PM, Koesling D, DeWit C, et al. Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature. (2013) 504:432–6. doi: 10.1038/nature12722
14. Wilkins MR, Aldashev AA, Wharton J, Rhodes CJ, Vandrovcova J, Kasperaviciute D, et al. α1-A680T variant in GUCY1A3 as a candidate conferring protection from pulmonary hypertension among kyrgyz highlanders. Circ Cardiovasc Genet. (2014) 7:920–9. doi: 10.1161/CIRCGENETICS.114.000763
15. Yamauchi T, Tada M, Houkin K, Tanaka T, Nakamura Y, Kuroda S, et al. Linkage of familial moyamoya disease (spontaneous occlusion of the circle of Willis) to chromosome 17q25. Stroke. (2000) 31:930–5. doi: 10.1161/01.str.31.4.930
16. Mineharu Y, Liu W, Inoue K, Matsuura N, Inoue S, Takenaka K, et al. Autosomal dominant moyamoya disease maps to chromosome 17q25.3. Neurology. (2008) 70:2357–63. doi: 10.1212/01.wnl.0000291012.49986.f9
17. Miyatake S, Miyake N, Touho H. Homozygous c 14576G> A variant of RNF213 predicts early-onset and severe form of moyamoya disease. Neurology. (2012) 78:803–10. doi: 10.1212/WNL.0b013e318249f71f
18. Moteki Y, Onda H, Kasuya H, Yoneyama T, Okada Y, Hirota K, et al. Systematic validation of RNF213 coding variants in Japanese patients with moyamoya disease. J Am Hear Assoc. (2015) 4:e001862. doi: 10.1161/JAHA.115.001862
19. Wang Y, Zhang Z, Wei L, Zhang Q, Zou Z, Yang L, et al. Predictive role of heterozygous p.R4810K of RNF213 in the phenotype of Chinese moyamoya disease. Neurology. (2020) 94:e678–86. doi: 10.1212/wnl.0000000000008901
20. Wu Z, Jiang H, Zhang L, Xu X, Zhang X, Kang Z, et al. Molecular analysis of RNF213 gene for moyamoya disease in the Chinese Han population. PLoS ONE. (2012) 7:e48179. doi: 10.1371/journal.pone.0048179
21. Duan L, Wei L, Tian Y, Zhang Z, Hu P, Wei Q, et al. Novel susceptibility loci for moyamoya disease revealed by a genome-wide association study. Stroke. (2018) 49:11–8. doi: 10.1161/STROKEAHA.117.017430
22. Morimoto T, Mineharu Y, Kobayashi H, Harada KH, Funaki T, Takagi Y, et al. Significant association of the RNF213 p.R4810K polymorphism with quasi-moyamoya disease. J Stroke Cerebrovasc Dis. (2016) 25:2632–6. doi: 10.1016/j.jstrokecerebrovasdis.2016.07.004
23. Koizumi A, Kobayashi H, Hitomi T, Harada KH, Habu T, Youssefian S. A new horizon of moyamoya disease and associated health risks explored through RNF213. Environ Health Prev Med. (2016) 21:55–70. doi: 10.1007/s12199-015-0498-7
24. Miyawaki S, Imai H, Takayanagi S, Mukasa A, Nakatomi H, Saito N. Identification of a genetic variant common to moyamoya disease and intracranial major artery stenosis/occlusion. Stroke. (2012) 43:3371–4. doi: 10.1161/STROKEAHA.112.663864
25. Kamimura T, Okazaki S, Morimoto T, Kobayashi H, Harada K, Tomita T, et al. Prevalence of RNF213 p.R4810K variant in early-onset stroke with intracranial arterial stenosis. Stroke. (2019) 50:1561–3. doi: 10.1161/STROKEAHA.118.024712
26. Okazaki S, Morimoto T, Kamatani Y, Kamimura T, Kobayashi H, Harada K, et al. Moyamoya disease susceptibility variant RNF213 p.R4810K increases the risk of ischemic stroke attributable to large-artery atherosclerosis. Circulation. (2019) 139:295–8. doi: 10.1161/circulationaha.118.038439
27. Fukushima H, Takenouchi T, Kosaki K. Homozygosity for moyamoya disease risk allele leads to moyamoya disease with extracranial systemic and pulmonary vasculopathy. Am J Med Genet A. (2016) 170:2453–6. doi: 10.1002/ajmg.a.37829
28. Koizumi A, Kobayashi H, Liu W, Fujii Y, Senevirathna STMLD, Nanayakkara S, et al. P.R4810K, a polymorphism of RNF213, the susceptibility gene for moyamoya disease, is associated with blood pressure. Env Heal Prev Med. (2013) 18:121–9. doi: 10.1007/s12199-012-0299-1
29. Liu S, Liu M, Li Q, Liu X, Wang Y, Mambiya M, et al. Association of single nucleotide polymorphisms of MTHFR, TCN2, RNF213 with susceptibility to hypertension and blood pressure. Biosci Rep. (2019) 39:1–11. doi: 10.1042/BSR20191454
30. Guo D-C, Papke CL, Tran-Fadulu V, Regalado ES, Avidan N, Johnson RJ, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. (2009) 84:617–27. doi: 10.1016/j.ajhg.2009.04.007
31. Miskinyte S, Butler MG, Hervé D, Sarret C, Nicolino M, Petralia JD, et al. Loss of BRCC3 deubiquitinating enzyme leads to abnormal angiogenesis and is associated with syndromic moyamoya. Am J Hum Genet. (2011) 88:718–28. doi: 10.1016/j.ajhg.2011.04.017
32. Guey S, Tournier-Lasserve E, Hervé D, Kossorotoff M. Moyamoya disease and syndromes: from genetics to clinical management. Appl Clin Genet. (2015) 8:49–68. doi: 10.2147/TACG.S42772
33. Li J li, Liu L yu, Jiang D dong, Jiang Y ying, Zhou G qiu, Mo D can, et al. Associations between GUCY1A3 genetic polymorphisms and large artery atherosclerotic stroke risk in Chinese Han population: A case-control study. Lipids Heal Dis. (2019) 18:233. doi: 10.1186/s12944-019-1177-2
34. Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. (2011) 478:103–9. doi: 10.1038/nature10405
35. Ahel J, Lehner A, Vogel A, Schleiffer A, Meinhart A, Haselbach D, et al. Moyamoya disease factor RNF213 is a giant E3 ligase with a dynein-like core and a distinct ubiquitin-transfer mechanism. Elife. (2020) 9:1–23. doi: 10.7554/eLife.56185
36. Ahel J, Fletcher A, Grabarczyk DB, Roitinger E, Deszcz L. E3 ubiquitin ligase RNF213 employs a non- canonical zinc finger active site and is allosterically regulated by ATP. bioRxiv. (2021). https://doi.org/10.1101/2021.05.10.443411
37. Husnjak K, Dikic I. Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Ann Rev Biochem. (2012) 81:291–322. doi: 10.1146/annurev-biochem-051810-094654
38. Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol. (2016) 18:579–86. doi: 10.1038/ncb3358
39. Takeda M, Tezuka T, Kim M, Choi J, Oichi Y, Kobayashi H, et al. Moyamoya disease patient mutations in the RING domain of RNF213 reduce its ubiquitin ligase activity and enhance NFκB activation and apoptosis in an AAA+ domain-dependent manner. Biochem Biophys Res Commun. (2020) 525:668–74. doi: 10.1016/j.bbrc.2020.02.024
40. Scholz B, Korn C, Wojtarowicz J, Mogler C, Augustin I, Boutros M, et al. Endothelial RSPO3 controls vascular stability and pruning through non-canonical WNT/Ca2+/NFAT signaling. Dev Cell. (2016) 36:79–93. doi: 10.1016/j.devcel.2015.12.015
41. Ogura T, Wilkinson AJ. AAA+ superfamily ATPases: common structure-diverse function. Genes Cells. (2001) 6:575–97. doi: 10.1046/j.1365-2443.2001.00447.x
42. Sugihara M, Morito D, Ainuki S, Hirano Y, Ogino K, Kitamura A, et al. The AAA+ ATPase/ubiquitin ligase mysterin stabilizes cytoplasmic lipid droplets. J Cell Biol. (2019) 218:949–60. doi: 10.1083/jcb.201712120
43. Otten EG, Werner E, Crespillo-casado A, Boyle KB, Dharamdasani V, Pathe C, et al. Ubiquitylation of lipopolysaccharide by RNF213 during bacterial infection. Nature. (2021) 594:111–6. doi: 10.1038/s41586-021-03566-4
44. Zhang Y, Janssens SP, Wingler K, Schmidt HHHW, Moens AL. Modulating endothelial nitric oxide synthase: a new cardiovascular therapeutic strategy. Am J Psysiol Hear Circ Physiol. (2011) 301:H634–46. doi: 10.1152/ajpheart.01315.2010
45. Santoro C, Giugliano T, Kraemer M, Torella A, Schwitalla C, Cirillo M, et al. Whole exome sequencing identifies MRVI1 as a susceptibility gene for moyamoya syndrome in neurofibromatosis type 1. PLoS ONE. (2018) 13:e0200446. doi: 10.1371/journal.pone.0200446
46. Crassous PA, Couloubaly S, Huang C, Zhou Z, Baskaran P, Kim DD, et al. Soluble guanylyl cyclase is a target of angiotensin II-induced nitrosative stress in a hypertensive rat model. Am J Physiol. (2012) 303:2012. doi: 10.1152/ajpheart.00138.2012
47. Choi JW, Son SM, Mook-Jung I, Moon YJ, Lee JY, Wang KC, et al. Mitochondrial abnormalities related to the dysfunction of circulating endothelial colony-forming cells in moyamoya disease. J Neurosurg. (2018) 129:1151–9. doi: 10.3171/2017.5.JNS17147
48. Key J, Maletzko A, Kohli A, Gispert S, Torres-Odio S, Wittig I, et al. Loss of mitochondrial ClpP, Lonp1, and Tfam triggers transcriptional induction of Rnf213, a susceptibility factor for moyamoya disease. Neurogenetics. (2020) 21:187–203. doi: 10.1007/s10048-020-00609-2
49. Coquel F, Silva MJ, Técher H, Zadorozhny K, Sharma S, Nieminuszczy J, et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature. (2018) 557:57–61. doi: 10.1038/s41586-018-0050-1
50. Henrickson M, Wang H. Tocilizumab reverses cerebral vasculopathy in a patient with homozygous SAMHD1 mutation. Cli Rheumatol. (2017) 36:1445–51. doi: 10.1007/s10067-017-3600-2
51. Kobayashi H, Matsuda Y, Hitomi T, Okuda H, Shioi H, Matsuda T, et al. Biochemical and functional characterization of RNF213 (Mysterin) R4810K, a susceptibility mutation of moyamoya disease, in angiogenesis in vitro and in vivo. J Am Hear Assoc. (2015) 4:e002146. doi: 10.1161/JAHA.115.002146
52. Ohkubo K, Sakai Y, Inoue H, Akamine S, Ishizaki Y, Matsushita Y, et al. Moyamoya disease susceptibility gene RNF213 links inflammatory and angiogenic signals in endothelial cells. Sci Rep. (2015) 5:13191. doi: 10.1038/srep13191
53. Sarkar P, Thirumurugan K. New insights into TNFα/PTP1B and PPARγ pathway through RNF213- a link between inflammation, obesity, insulin resistance and Moyamoya disease. Gene. (2021) 771:145340. doi: 10.1101/2020.07.07.192153
54. Banh RS, Iorio C, Marcotte R, Xu Y, Cojocari D, Rahman AA, et al. PTP1B controls non-mitochondrial oxygen consumption by regulating RNF213 to promote tumour survival during hypoxia. Nat Cell Biol. (2016) 18:803–13. doi: 10.1038/ncb3376
55. Marro ML, Peiró C, Panayiotou CM, Baliga RS, Meurer S, Schmidt HHHW, et al. Characterization of the human α1β1 soluble guanylyl cyclase promoter: Key role for NF-κb(p50) and CCAAT-binding factors in regulating expression of the nitric oxide receptor. J Biol Chem. (2008) 283:20027–36. doi: 10.1074/jbc.M801223200
56. Li D, Laubach VE, Johns RA. Upregulation of lung soluble guanylate cyclase during chronic hypoxia is prevented by deletion of eNOS. Am J Physiol Lung Cell Mol Physiol. (2001) 281:369–76. doi: 10.1152/ajplung.2001.281.2.l369
57. Kessler T, Wobst J, Wolf B, Eckhold J, Vilne B, Hollstein R, et al. Functional characterization of the GUCY1A3 coronary artery disease risk locus. Circulation. (2017) 136:476–89. doi: 10.1161/CIRCULATIONAHA.116.024152
58. Lin R, Xie Z, Zhang J, Xu H, Su H, Tan X, et al. Clinical and immunopathological features of Moyamoya disease. PLoS ONE. (2012) 7:386. doi: 10.1371/journal.pone.0036386
59. Takebayashi S, Matsuo K, Kaneko M. Ultrastructural studies of cerebral arteries and collateral vessels in moyamoya disease. Stroke. (1984) 15:728–32. doi: 10.1161/01.STR.15.4.728
60. Hitomi T, Habu T, Kobayashi H, Okuda H, Harada KH, Osafune K, et al. Downregulation of Securin by the variant RNF213 R4810K reduces angiogenic activity of induced pluripotent stem cell-derived vascular endothelial cells from moyamoya patients. Biochem Biophys Res Commun. (2013) 438:13–9. doi: 10.1016/j.bbrc.2013.07.004
61. Kobayashi H, Brozman M, Kyselová K, Viszlayová D, Morimoto T, Roubec M, et al. RNF213 rare variants in Slovakian and Czech moyamoya disease patients. PLoS ONE. (2016) 11:759. doi: 10.1371/journal.pone.0164759
62. Tokairin K, Hamauchi S, Ito M, Kazumata K, Sugiyama T, Nakayama N, et al. Vascular smooth muscle cell derived from IPS cell of moyamoya disease - comparative characterization with endothelial cell transcriptome. J Stroke Cerebrovasc Dis. (2020) 29:105305. doi: 10.1016/j.jstrokecerebrovasdis.2020.105305
63. Phi JH, Suzuki N, Moon YJ, Park AK, Wang KC, Lee JY, et al. Chemokine ligand 5 (CCL5) derived from endothelial colony-forming cells (ECFCs) mediates recruitment of smooth muscle progenitor cells (SPCs) toward critical vascular locations in moyamoya disease. PLoS ONE. (2017) 12:9714. doi: 10.1371/journal.pone.0169714
64. Hamauchi S, Shichinohe H, Uchino H, Yamaguchi S, Nakayama N, Kazumata K, et al. Cellular functions and gene and protein expression profiles in endothelial cells derived from Moyamoya disease-specific iPS cells. PLoS One. (2016) 11:e163561. doi: 10.1371/journal.pone.0163561
65. Matsuo M, Nadanaka S, Soga M, Sugiyama T, Serigano S, Shimano K, et al. Vulnerability to shear stress caused by altered peri - endothelial matrix is a key feature of Moyamoya disease. Sci Rep. (2021) 11:1–12. doi: 10.1038/s41598-021-81282-9
66. Chang L, Garcia-Barrio MT, Chen YE. Perivascular adipose tissue regulates vascular function by targeting vascular smooth muscle cells. Arter Thromb Vasc Biol. (2020) 40:1094–109. doi: 10.1161/ATVBAHA.120.312464
67. Brown NK, Zhou Z, Zhang J, Zeng R, Wu J, Eitzman DT, et al. Perivascular adipose tissue in vascular function and disease: A review of current research and animal models. Arter Thromb Vasc Biol. (2014) 34:1621–30. doi: 10.1161/ATVBAHA.114.303029
68. Cheng CK, Bakar HA, Gollasch M, Huang Y. Perivascular adipose tissue: the sixth man of the cardiovascular system. Cardiovasc Drugs Ther. (2018) 32:481–502. doi: 10.1007/s10557-018-6820-z
69. Wobst J, Schunkert H, Kessler T. Genetic alterations in the NO-cGMP pathway and cardiovascular risk. Nitric Oxide. (2018) 76:105–12. doi: 10.1016/j.niox.2018.03.019
70. Bennett BJ, Davis RC, Civelek M, Orozco L, Wu J, Qi H, et al. Genetic architecture of atherosclerosis in mice: a systems genetics analysis of common inbred strains. PLoS Genet. (2015) 11:5711. doi: 10.1371/journal.pgen.1005711
71. Sandner P, Stasch JP. Anti-fibrotic effects of soluble guanylate cyclase stimulators and activators: A review of the preclinical evidence. Respir Med. (2017) 122:S1–9. doi: 10.1016/j.rmed.2016.08.022
72. Zhang G, Xiang B, Dong A, Skoda RC, Daugherty A, Smyth SS, et al. Biphasic roles for soluble guanylyl cyclase (sGC) in platelet activation. Blood. (2011) 118:3670–9. doi: 10.1182/blood-2011-03-341107
73. Pyriochou A, Beis D, Koika V, Potytarchou C, Papadimitriou E, Zhou Z, et al. Soluble guanylyl cyclase activation promotes angiogenesis. J Pharmacol Exp Ther. (2006) 319:663–71. doi: 10.1124/jpet.106.108878
74. Baburski AZ, Sokanovic SJ, Andric SA, Kostic TS. Aging has the opposite effect on cAMP and cGMP circadian variations in rat Leydig cells. J Comp Physiol B. (2017) 187:613–23. doi: 10.1007/s00360-016-1052-7
75. Ge P, Zhang Q, Ye X, Liu X, Deng X, Wang J, et al. Modifiable risk factors associated with moyamoya disease: a case-control study. Stroke. (2020) 51:2472–9. doi: 10.1161/STROKEAHA.120.030027
76. Church EW, Bell-Stephens TE, Bigder MG, Gummidipundi S, Han SS, Steinberg GK. Clinical course of unilateral moyamoya disease. Neurosurgery. (2020) 0:1–7. doi: 10.1093/neuros/nyaa284
77. Hirano Y, Miyawaki S, Imai H, Hongo H, Ohara K, Dofuku S, et al. Association between the onset pattern of adult moyamoya disease and risk factors for stroke. Stroke. (2020) 51:3124–8. doi: 10.1161/STROKEAHA.120.030653
78. Piccolis M, Bond LM, Kampmann M, Pulimeno P, Chitraju C, Jayson CBK, et al. Probing the global cellular responses to lipotoxicity caused by saturated fatty acids. Mol Cell. (2019) 74:32–44.e8. doi: 10.1016/j.molcel.2019.01.036
79. Plötz T, Hartmann M, Lenzen S, Elsner M. The role of lipid droplet formation in the protection of unsaturated fatty acids against palmitic acid induced lipotoxicity to rat insulin-producing cells. Nutr Metab. (2016) 13:1–11. doi: 10.1186/s12986-016-0076-z
80. Listenberger LL, Han X, Lewis SE, Cases S, Farese R V., Ory DS, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. PNAS. (2003) 100:3077–82. doi: 10.1073/pnas.0630588100
81. Kobayashi H, Yamazaki S, Takashima S, Liu W, Okuda H, Yan J, et al. Ablation of Rnf213 retards progression of diabetes in the Akita mouse. Biochem Biophys Res Commun. (2013) 432:519–25. doi: 10.1016/j.bbrc.2013.02.015
82. Bang OY, Ryoo S, Kim SJ, Yoon CH, Cha J, Yeon JY, et al. Adult moyamoya disease: a burden of intracranial stenosis in East Asians? PLoS ONE. (2015) 10:e0130663. doi: 10.1371/journal.pone.0130663
83. Mineharu Y, Takagi Y, Koizumi A, Morimoto T, Funaki T, Hishikawa T, et al. Genetic and non-genetic factors for contralateral progression of unilateral moyamoya disease:the first report from the SUPRA Japan Study Group. J Neurosurg. (2021).
84. Meye C, Schumann J, Wagner A, Gross P. Effects of homocysteine on the levels of caveolin-1 and eNOS in caveolae of human coronary artery endothelial cells. Atherosclerosis. (2007) 190:256–63. doi: 10.1016/j.atherosclerosis.2006.03.009
85. Jeong SO, Son Y, Lee JH, Choi SW, Kim SH, Cheong YK, et al. Both nitric oxide and nitrite prevent homocysteine-induced endoplasmic reticulum stress and subsequent apoptosis via cGMP-dependent pathway in neuronal cells. Biochem Biophys Res Commun. (2017) 493:164–9. doi: 10.1016/j.bbrc.2017.09.054
86. Liu P, Huang J, Zhong L. Role and mechanism of homocysteine in affecting hepatic protein-tyrosine phosphatase 1B. Biochim Biophys Acta Gen Subj. (2019) 1863:941–9. doi: 10.1016/j.bbagen.2019.03.007
87. Mikami T, Suzuki H, Komatsu K, Mikuni N. Influence of inflammatory disease on the pathophysiology of moyamoya disease and quasi-moyamoya disease. Neurol Med Chir. (2019) 59:1–10. doi: 10.2176/nmc.ra.2019-0059
88. Braun KPJ, Bulder MMM, Chabrier S, Kirkham FJ, Uiterwaal CSP, Tardieu M, et al. The course and outcome of unilateral intracranial arteriopathy in 79 children with ischaemic stroke. Brain. (2009) 132:544–57. doi: 10.1093/brain/awn313
89. Czartoski T, Hallam D, Lacy JM, Chun MR, Becker K. Postinfectious vasculopathy with evolution to moyamoya syndrome. J Neurol Neurosurg Psychiatry. (2005) 76:256–9. doi: 10.1136/jnnp.2004.041046
90. Houzelstein D, Simon-Chazottes D, Batista L, Tokuda S, Langa Vives F, Flamand M, et al. The ring finger protein 213 gene (Rnf213) contributes to Rift Valley fever resistance in mice. Mamm Genome. (2021) 1:30–7. doi: 10.1007/s00335-020-09856-y
91. Harmon B, Schudel BR, Maar D, Kozina C, Ikegami T, Tseng C-TK, et al. Rift valley fever virus strain MP-12 enters mammalian host cells via caveola-mediated endocytosis. J Virol. (2012) 86:12954–70. doi: 10.1128/jvi.02242-12
92. Ikegami T. Molecular biology and genetic diversity of Rift Valley fever virus. Antivir Res. (2012) 95:293–310. doi: 10.1016/j.antiviral.2012.06.001
93. McElroy AK, Nichol ST. Rift Valley fever virus inhibits a pro-inflammatory response in experimentally infected human monocyte derived macrophages and a pro-inflammatory cytokine response may be associated with patient survival during natural infection. Virology. (2012) 422:6–12. doi: 10.1016/j.virol.2011.09.023
94. Habu T, Harada KH. UBC13 is an RNF213-associated E2 ubiquitin-conjugating enzyme, and Lysine 63-linked ubiquitination by the RNF213-UBC13 axis is responsible for angiogenic activity. FASEB BioAdv. (2021) 49:347–55. doi: 10.1096/fba.2019-00092
95. Yang L, Guan H, He J, Zeng L, Yuan Z, Xu M, et al. VEGF increases the proliferative capacity and eNOS/NO levels of endothelial progenitor cells through the calcineurin/NFAT signalling pathway. Cell Biol Intern. (2012) 36:21–7. doi: 10.1042/cbi20100670
96. Yoeli-lerner M, Chin YR, Hansen CK, Toker A. Akt / protein kinase B and glycogen synthase kinase-3 B signaling pathway regulates cell migration through the NFAT1 transcription factor. Mol Cancer Res. (2009) 7:425–33. doi: 10.1158/1541-7786.MCR-08-0342
97. Amal H, Gong G, Gjoneska E, Lewis SM, Wishnok JS, Tsai L, et al. S-nitrosylation of E3 ubiquitin-protein ligase RNF213 alters non-canonical Wnt / Ca + 2 signaling in the P301S mouse model of tauopathy. Transl Psychiatr. (2019) 9:44. doi: 10.1038/s41398-019-0388-7
98. Pilz RB, Casteel DE. Regulation of gene expression by cyclic GMP. Circ Res. (2003) 93:1034–46. doi: 10.1161/01.RES.0000103311.52853.48
99. Yang XY, Huang CC, Kan QM, Li Y, Liu D, Zhang XC, et al. Calcium regulates caveolin-1 expression at the transcriptional level. Biochem Biophys Res Commun. (2012) 426:334–41. doi: 10.1016/j.bbrc.2012.08.079
100. Patel HH, Zhang S, Murray F, Suda RYS, Head BP, Yokoyama U, et al. Increased smooth muscle cell expression of caveolin-1 and caveolae contribute to the pathophysiology of idiopathic pulmonary arterial hypertension. FASEB J. (2007) 21:2970–9. doi: 10.1096/fj.07-8424com
101. Bakhshi FR, Mao M, Shajahan AN, Piegeler T, Chen Z, Chernaya O, et al. Nitrosation-dependent caveolin 1 phosphorylation, ubiquitination, and degradation and its association with idiopathic pulmonary arterial hypertension. Pulm Circ. (2014) 3:816–30. doi: 10.1086/674753.I
102. Mathew R. Pulmonary hypertension and metabolic syndrome: Possible connection, PPARγ and Caveolin-1. World J Cardiol. (2014) 6:692. doi: 10.4330/wjc.v6.i8.692
103. Bang OY, Chung J-W, Kim SJ, Oh MJ, Kim SY, Cho YH, et al. Caveolin-1, ring finger protein 213, and endothelial function in Moyamoya disease. Int J Stroke. (2016) 11:999–1008. doi: 10.1177/1747493016662039
Keywords: moyamoya disease, RNF213 mutation, GUCY1A3, CAV1 (caveolin-1), calcineurein/NFAT pathway, nitric oxide, inflammation, vascular
Citation: Mineharu Y and Miyamoto S (2021) RNF213 and GUCY1A3 in Moyamoya Disease: Key Regulators of Metabolism, Inflammation, and Vascular Stability. Front. Neurol. 12:687088. doi: 10.3389/fneur.2021.687088
Received: 28 March 2021; Accepted: 24 June 2021;
Published: 26 July 2021.
Edited by:
Yuxiang Gu, Fudan University, ChinaReviewed by:
Gerrit M. Grosse, Hannover Medical School, GermanyHugues Chabriat, Université Paris Diderot, France
Copyright © 2021 Mineharu and Miyamoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yohei Mineharu, bWluZWhhcnVAa3VocC5reW90by11LmFjLmpw