 Jorge Diogo Da Silva1,2,3*
Jorge Diogo Da Silva1,2,3* Marta Daniela Costa1,2
Marta Daniela Costa1,2 Bruno Almeida1,2
Bruno Almeida1,2 Fátima Lopes1,2
Fátima Lopes1,2 Patrícia Maciel1,2†
Patrícia Maciel1,2† Andreia Teixeira-Castro1,2†
Andreia Teixeira-Castro1,2†- 1Life and Health Sciences Research Institute (ICVS), School of Medicine, University of Minho, Braga, Portugal
- 2ICVS/3B's—Portuguese Government Associate Laboratory, Braga, Guimarães, Portugal
- 3Pediatrics Department, Hospital of Santa Maria Maior, Barcelos, Portugal
Diseases of neurodevelopment mostly exhibit neurological and psychiatric symptoms that go from very mild to extremely severe. While the etiology of most cases of neurodevelopmental disease is still unknown, the discovery of underlying genetic causes is rapidly increasing, with hundreds of genes being currently implicated as disease-causing. Here, we report a clinical case of a patient with a previously undiagnosed syndrome comprising severe global developmental delay, intellectual disability, and behavioral disorders (such as attention-deficit/hyperactivity disorder, autism spectrum disorder and recurrent bouts of aggressive behavior). After genetic testing, a pathogenic variant was detected in the GNB1 gene, which codes for the G-protein subunit β1. The detected variant (c.217G>A, p.A73T) has not been previously reported in any of the 58 published cases of GNB1 encephalopathy. However, it localizes to the mutational hotspot in exons 6 and 7 in which 88% of all missense mutations occur. An in silico model predicts that this mutation is likely to disrupt the WD40 domain of the GNB1 protein, which is required for its interaction with other G-proteins and, consequently, for downstream signal transduction. In conclusion, we reported an additional GNB1 encephalopathy patient, bearing a novel mutation, taking another step toward a better understanding of its clinical presentation and prospective development of treatments for the disease.
Introduction
Knowledge on the genetic causes of neurodevelopmental diseases has been increasingly growing. Among the hundreds of genes that have been established as neurodevelopmental disease-causing, genes that encode for guanine nucleotide-binding proteins (G proteins) have recently been included (1, 2). Specifically, mutations in GNB1, which encodes for the G protein subunit β1, have been recently described as causal of a neurodevelopmental syndrome (1, 3). The Gβ subunit forms a heterotrimer with Gα and Gγ in the steady-state which is bound to a membrane G protein-coupled receptor (GPCR) (4). Upon binding of a specific extracellular ligand, there is separation of the Gα from the Gβγ dimer, and consequently activation of an intracellular signaling cascade (4). This is a ubiquitous signaling pathway, which is in line with the fact that its components, including GNB1, are ubiquitously expressed (5). In neurons, GNB1 is also widely expressed in all brain regions, but enriched in rod photoreceptor cells of the retina (5).
Pathogenic variants in GNB1 cause a heterogeneous neurodevelopmental syndrome named GNB1 encephalopathy (OMIM: 616973) whose unifying characteristic is a global development delay (GDD), present in 100% of patients (3, 6). Manifestations that are present in at least 50% of patients include moderate-to-severe intellectual disability (ID), abnormal muscle tone, abnormal vision, epilepsy, and gastrointestinal abnormalities (3, 6). Several other neuropsychiatric and non-neuropsychiatric symptoms have also been reported, albeit less frequently (3, 6). Nevertheless, clinical data is very limited since only 58 patients with clinical-impacting GNB1 variants have been reported (1, 3, 7–19). Moreover, this syndrome can easily overlap with many conditions, being clinically indistinguishable from other neurodevelopmental disorders.
An interesting finding is that, out of the 51 patients with pathogenic missense variants, 25 (49%) are located in exon 6, and 20 (39%) are in exon 7, indicating the presence of a mutational hotspot (1, 3, 7–19). Of note, 13 patients (25%) bear a p.(Ile80Thr) variant, which further supports such hypothesis (Table 1). As previously suggested, these mutations are likely to affect residues from the interacting interface of the GNB1 protein, compromising its binding to the other G protein subunits (9). Here, we describe a Portuguese patient with GDD bearing the first pathogenic GNB1 missense mutation affecting residue alanine 73 (A73), provide an in silico prediction of how it affects protein function and integrate the clinical findings with the previously described cases.
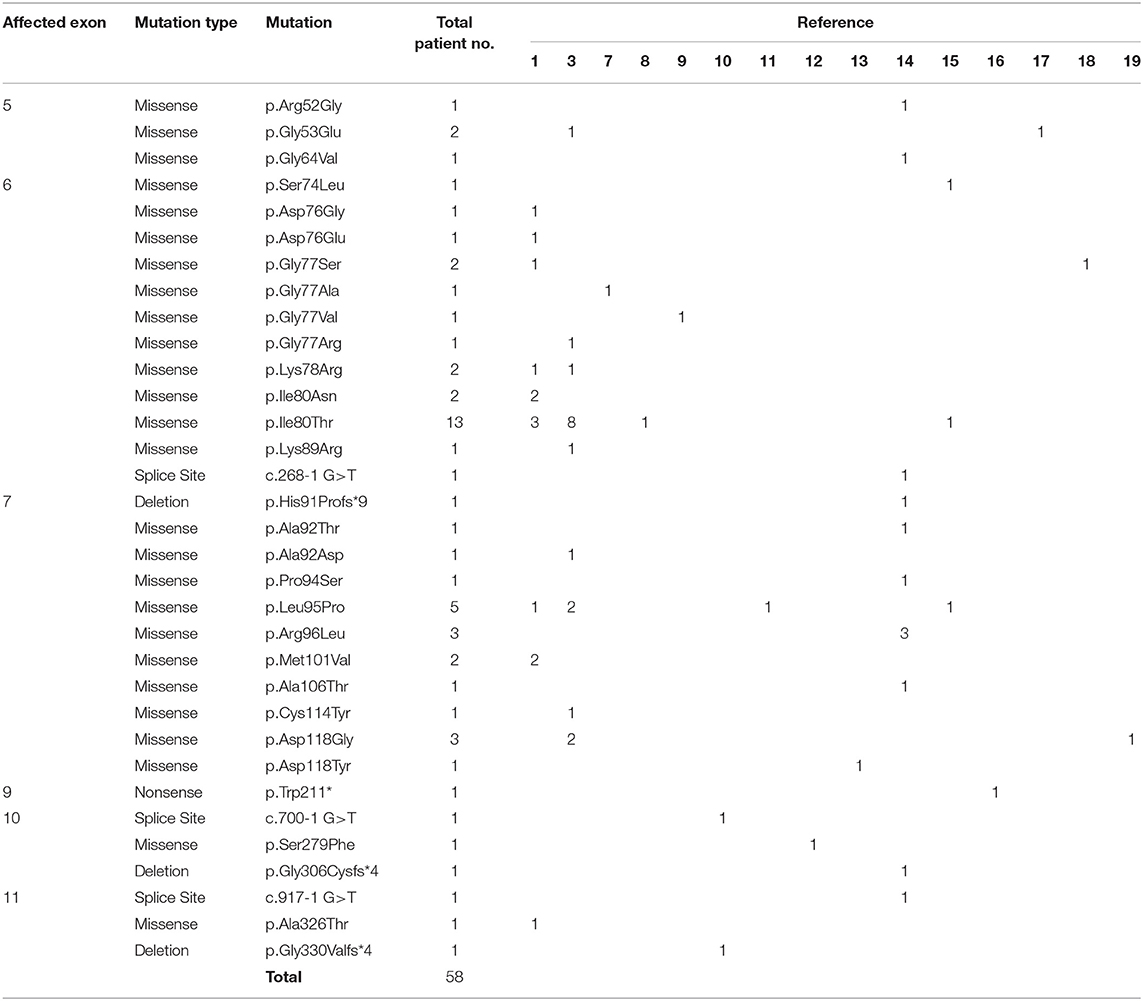
Table 1. List of all patients with clinically relevant GNB1 mutations currently published.
Case Report
A 15 year 8-month old female patient was born at 36 weeks gestation with a weight of 2880 g (P21), length of 47 cm (P12), and head circumference of 33 cm (P23). Pregnancy was uncomplicated, and birth was by cesarean section due to prolonged labor, but otherwise unremarkable, with normal Apgar scores. Metabolic screening and otoacoustic emissions were normal. Parents (mother with 30 and father with 33 years of age) were non-consanguineous and healthy, and the patient has a healthy younger sibling. Regarding developmental milestones, the patient showed a social smile at 2 months, sat unsupported at 9 months, walked autonomously at 17 months, spoke the first intelligible words at 4 years and built sentences at 5 years of age, indicating a global developmental delay (GDD), which was further supported by the Griffiths III Mental Development Scale assessment (general quotient of 52 at 9 years of age). Pubertal development is adequate for the current age.
In addition to GDD, the patient fulfilled clinical criteria for autism spectrum disorder (ASD) at 18 months of age, due to impaired social interaction, restricted interests, and repetitive behavior. Throughout development, the patient has additionally met the criteria for other behavioral disorders, namely attention-deficit/hyperactivity disorder (ADHD) and oppositional defiant disorder. Moreover, the patient was diagnosed with mild-to-moderate intellectual disability (IQ of 52 as measured by the WISC-III). Finally, the patient met the criteria for a mixed language disorder, as well as developmental coordination disorder. No other specific neurological symptoms were observed, namely dystonia (or other movement disorders), sensorineural hearing loss, visual symptoms or epilepsy. Furthermore, no structural abnormalities were observed in two brain MRIs (one at 3 years and another at 15 years of age), nor electroencephalographic changes (studied at 18 months of age). No dysmorphic features were observed.
At the current age of 15, the most impacting symptoms are related with behavior, with the patient showing repeated bouts of auto and hetero-aggressive behavior, attention deficit and a frequently obsessive conduct. Multiple treatment regimens have been tried, including several drug types, namely antipsychotics, antidepressants, mood stabilizers and stimulants, albeit with mixed results in the management of the psychiatric disorder. Multiple episodes of extrapyramidal adverse effects of antipsychotics have also been observed in the patient.
Considering the clinical presentation of the patient, genetic testing was undertaken. A karyotype was normal, and a comparative genomic hybridization (CGH) array was carried out using the Agilent 180K oligo-array (Amadid 023363, Agilent, Santa Clara, CA). The array-CGH detected 2 CNVs that were deemed not pathogenic, as both were inherited from the patient's healthy parents [arr [hg18] 9q32 (114,200,278-114,248,375) X3 mat, 17q23.3 (59,293,888-59,356,442) X1 pat].
Following this initial approach, a pre-defined panel of genes whose mutations are known to cause GDD/ID (Supplementary Table 1) was sequenced by massive parallel sequencing, using the SureSelectXT Target Enrichment System (Agilent, Santa Clara, CA) for preparation of the DNA library, followed by sequencing using a MiSeq system (Illumina, San Diego, CA). Two heterozygote intronic variants were detected, one in the SETD5 (NM_001282539.1; rs924035385) and one in the TNIK gene (NM_015028.3; rs192028546), both previously reported in the general population. Two heterozygote missense mutations were also detected, namely in the HIVEP2 (NM_006734.3; c.5866G>A; p.Gly1956Arg) and GNB1 (NM_001282539.1; c.217G>A; p.Ala73Thr) genes, with both genes having been implicated in autosomal dominant GDD. However, the HIVEP2 variant is categorized as likely benign, with in silico predictions strongly supporting a neutral effect of the mutation in protein function (Condel, 0.317; SIFT, 0.630; Polyphen-2, 0.013; Massessor, −0.205). In contrast, the GNB1 variant is predicted to be deleterious by several in silico tools (Condel, 0.558; SIFT, 0.000; Polyphen-2, 0.976; Massessor, 1.000). Moreover, and as previously described, GNB1 is highly intolerant to genetic variation, with disruptions to protein structure leading to profound changes in its function (1). Finally, the GNB1 variant was confirmed by Sanger sequencing in the patient and shown to occur de novo, since it was not detected in either parent (Figure 1A). Therefore, this novel GNB1 variant can be categorized as pathogenic (as it fulfills 2 strong criteria for pathogenicity, PS2 and PS3) (21). Interestingly, this variant maps to exon 6, one of the two GNB1 exons with mutational hotspots (Figure 1B).
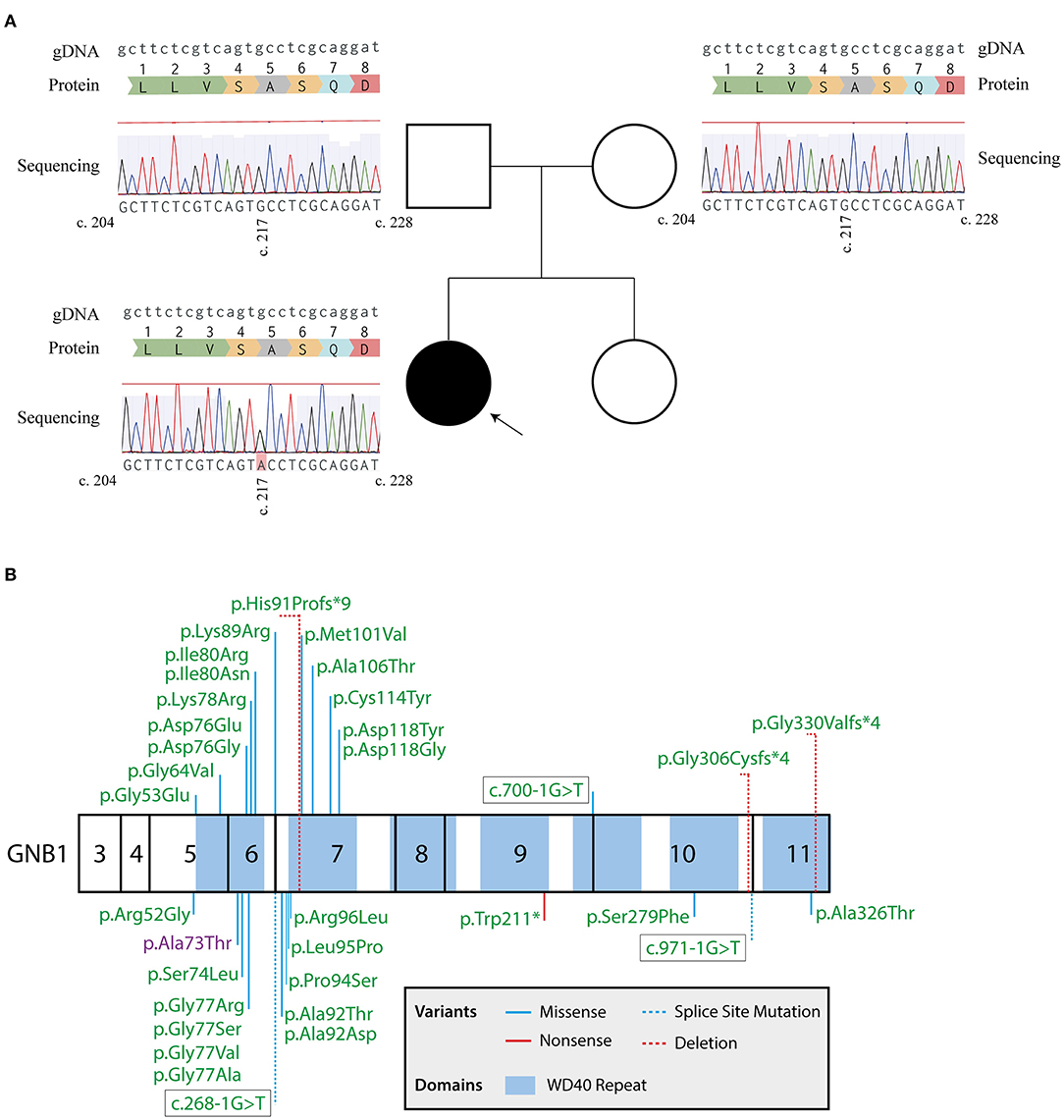
Figure 1. Sanger sequencing confirms a de novo GNB1 variant. (A) Genogram of the proband with the respective results for GNB1 sequencing of the patient and each parent. The c.217G>A heterozygous mutation was detected in the proband but not in the parents, indicating a de novo occurring variant. Results from Sanger sequencing were aligned with the genomic GNB1 sequence (NG_047052.1) using the MAFFT 7 algorithm (20). The gDNA lane represents the genomic GNB1 sequence; the protein lane represents the translation of amino acids from GNB1 exon 6; the sequencing lane represents the results from Sanger sequencing of each patient, with a c.217G>A variant in the proband. (B) Mutation map of all GNB1 variants implicated in GNB1 encephalopathy to the date of the study. Numbers inside each box represent the translated exons of GNB1 (exons 1, 2, and 12 are not translated). Whenever possible, the mutation is represented by the change in protein structure. The variant detected in the patient is highlighted in purple.
GNB1 (Uniprot: P62873) is an extremely well-conserved protein. In fact, the amino acid sequence is identical between humans and other mammals, such as mouse, rat and bovine, and differs only by 4 residues from zebrafish (Figure 2A). This demonstrates that almost no mutations were selected during evolution, indicating that any mutation could possibly have a dramatic structural impact with consequent loss of function. A73 is located in a core beta sheet from a WD40 repeat motif that is part of the circularized β-propeller WD40 domain of the GNB1 protein (Figure 2B). WD40 domains are mainly involved in the assembly of large molecular complexes and the WD motifs act as protein interaction scaffolds. A73 lies within 4A from 9 residues: I58, Y59, A60, S72, S74, K78, C79, V100 and C103 (Figure 2C, left). Substitution of the non-polar A73 for the polar hydroxyl residue threonine (A73T, Figure 2C, right) might locally impact the structure. It is particularly important for lysine 78, a residue whose mutation (K78R) was already identified as causal of GNB1 encephalopathy (1). Moreover, simulation of the A73T substitution, using the highest probable side chain orientations for threonine, revealed an additional residue (T102, Figure 2C, right) in the proximity (within 4A) that belongs to an adjacent WD40 motif. This indicates that any destabilization caused by the A73T substitution might have a broader impact on the WD40 domains of GNB1. Interestingly, while A73 is not involved in the formation of the Gβγ dimer (Figure 2D), K78 establishes a polar contact with D26 from Gα (Figure 2E), helping in the stabilization of the Gα N-terminal helix near the β-propeller domain of Gβ. A destabilization of this region might therefore impact the Gα-Gβγ interaction, with important functional consequences. Given the position of A73 within the WD40 motif and the functionality of the WD40 domain, a mutation in this residue could also impact G protein-effector interactions.
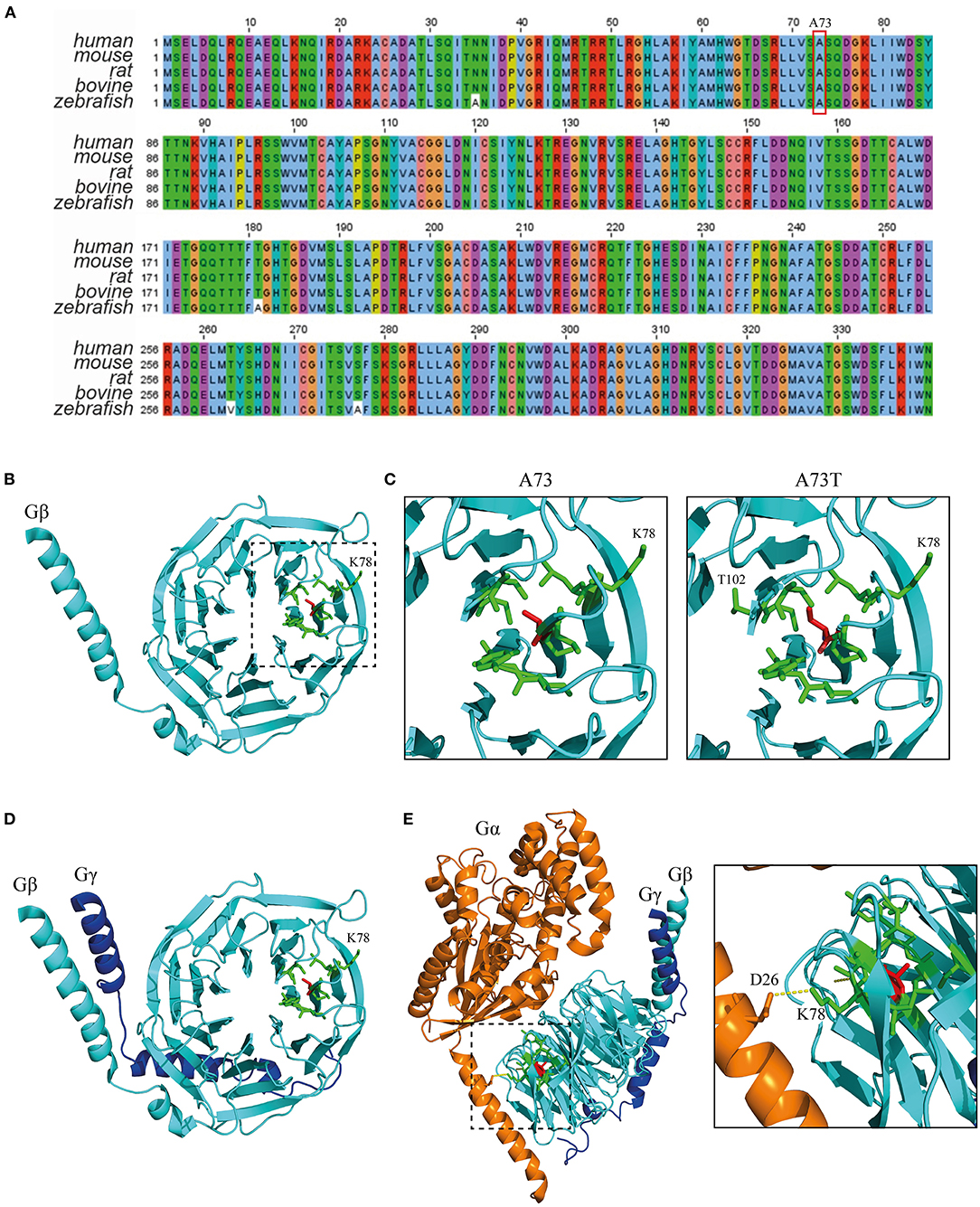
Figure 2. The A73T mutation might compromise a WD40 repeat from the WD40 domain of GNB1. (A) Sequence alignment of GNB1 protein from different species demonstrating that the amino acid sequence is almost unchanged across evolution. The alignment was performed using Jalview (Version 2) software (22). (B) Three-dimensional structure of GNB1 (pdb −1gp2, chain B). A73 (red sticks) is located in a core β-sheet from an WD repeat (boxed) that composes the circularized β-propeller WD40 domain of GNB1. Represented in green sticks are the residues that distance less than 4A of A73. (C) Three-dimensional structure detail of the WD repeat encompassing A73 residue (left) and simulated A73T mutagenesis (right). Residues that are within 4A distance that might be stabilized by A73 are represented as green sticks. Upon A73T mutagenesis, using the highest probable side chain orientations for threonine, an additional residue (T102) from an adjacent WD40 repeat appears within 4A distance, indicating that A73T might have an impact on this region. (D) Three-dimensional structure detail of the Gβγ dimer, showing that A73 is unlikely to be required for dimer formation. (E) Three-dimensional structure detail of the Gαβγ trimer. The inset represents the interaction between Gα D26 and Gβ K78 that is required for trimer assembly, which is potentially affected by the A73T mutation. Structural modeling was performed using PyMOL (23).
Discussion
We have described a new case of GNB1 encephalopathy, namely a patient with profound GDD, ID and a complex behavioral disorder. Genetic testing uncovered a novel pathogenic de novo GNB1 mutation which affects a different residue from all other described patients, with in silico predictions indicating that the abnormal protein is likely to be functionally compromised. Similarly to more than 50% of the described series (6), the presented patient displays developmental delay and ID. Less frequent manifestations that were also observed in the patient are behavioral issues (usually ADHD and ASD). Frequent GNB1 encephalopathy symptoms that were not observed are abnormal muscle tone, abnormal vision and epilepsy. Nonetheless, the sole finding that is common to 100% of patients is GDD.
This is the first described patient with a substitution on residue A73, which is encoded in exon 6 of the GNB1 gene. This supports the hypothesis of the presence of a mutational hotspot spanning exons 6 and 7, as 88% of all missense mutations map to his region. Functional studies have shown that GPCR-dependent signal transduction that requires Gβ1 activity is decreased by all exon 6–7 mutations described to the date of the study, suggesting that this is a critical region for protein function (9). Nevertheless, mutations in other regions also disrupt protein function and cause a similar disorder. In fact, GNB1 has several WD40 domains that assemble to form a tridimensional β-propeller interface to which Gγ proteins bind, and an opposite β-barrel surface for Gα binding (24). Therefore, key residues that span most of the length of the protein have been implicated in these interactions, suggesting that mutations outside of the predicted hotspot can be equally damaging.
One of the effector interactions that could be destabilized by the A73T substitution is the interaction of GNB1 with the β-adrenergic receptor kinase 1, GRK2. GNB1 tyrosine 59 is important for GNB1:GRK2 interaction, since it stabilizes K663 and M664 from GRK2 (25). Any structural impact on Y59 caused by the A73T mutation could impact GNB1:GRK2 interaction. Interestingly, Y59 is also part of the interaction surface between GNB1 and phosducin, a protein involved in the regulation of visual phototransduction (26).
In conclusion, we have described a case of GNB1 encephalopathy with a de novo mutation which affects a residue that has not been previously implicated in disease. While other variants might contribute to the observed phenotype, it is extremely likely that the GNB1 variant is the key alteration that caused disease. In fact, the patient presents with key aspects of the disorder and has a mutation that is very likely to disrupt protein function. The addition of new cases to the literature of this condition are important (27), not only to expand the understanding of this rare syndrome, but also in the hope of working toward potential treatments.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author/s.
Ethics Statement
Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
JDS: drafting/revision of the manuscript for content, including medical writing for content, major role in the acquisition of data, study concept or design, and analysis or interpretation of data. MDC and FL: drafting/revision of the manuscript for content, including medical writing for content, major role in the acquisition of data and analysis or interpretation of data. BA: drafting/revision of the manuscript for content, including medical writing for content and analysis or interpretation of data. PM and AT-C: drafting/revision of the manuscript for content, including medical writing for content, study concept or design, and analysis or interpretation of data. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by National funds, through the Foundation for Science and Technology (FCT) - project UIDB/50026/2020 and UIDP/50026/2020 and by the project NORTE-01-0145-FEDER-000039, supported by Norte Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are thankful to the patient and her family for participating in the study. We would also like to thank Dr. Vânia Miranda and Dr. Carla Carvalho for helping with the evaluation of clinical information.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.735549/full#supplementary-material
References
1. Petrovski S, Küry S, Myers CT, Anyane-Yeboa K, Cogné B, Bialer M, et al. Germline de novo mutations in GNB1 cause severe neurodevelopmental disability, hypotonia, and seizures. Am J Hum Genet. (2016) 98:1001–10. doi: 10.1016/j.ajhg.2016.03.011
2. Marcé-Grau A, Dalton J, López-Pisón J, García-Jiménez MC, Monge-Galindo L, Cuenca-León E, et al. GNAO1 encephalopathy: further delineation of a severe neurodevelopmental syndrome affecting females. Orphanet J Rare Dis. (2016) 11:1–9. doi: 10.1186/s13023-016-0416-0
3. Hemati P, Revah-Politi A, Bassan H, Petrovski S, Bilancia CG, Ramsey K, et al. Refining the phenotype associated with GNB1 mutations: Clinical data on 18 newly identified patients and review of the literature. Am J Med Genet A. (2018) 176:2259–75. doi: 10.1002/ajmg.a.40472
4. Freissmuth M, Casey PJ, Gilman AG. G proteins control diverse pathways of transmembrane signaling 1. FASEB J. (1989) 3:2125–31. doi: 10.1096/fasebj.3.10.2546847
5. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. (2015) 347:1260419. doi: 10.1126/science.1260419
6. Revah-Politi A, Sands TT, Colombo S, Goldstein DB, Anyane-Yeboa K. GNB1 Encephalopathy. Seattle, WA: University of Washington (2020).
7. Peng J, Wang Y, He F, Chen C, Wu L, Yang L, et al. Novel West syndrome candidate genes in a Chinese cohort. CNS Neurosci Ther. (2018) 24:1196–206. doi: 10.1111/cns.12860
8. Jones HF, Morales-Briceño H, Barwick K, Lewis J, Sanchis-Juan A, Raymond FL, et al. Myoclonus-dystonia caused by GNB1 mutation responsive to deep brain stimulation. Mov Disord. (2019) 34:1079–80. doi: 10.1002/mds.27708
9. Lohmann K, Masuho I, Patil DN, Baumann H, Hebert E, Steinrücke S, et al. Novel GNB1 mutations disrupt assembly and function of G protein heterotrimers and cause global developmental delay in humans. Hum Mol Genet. (2017) 26:1078–86. doi: 10.1093/hmg/ddx018
10. Endo W, Ikemoto S, Togashi N, Miyabayashi T, Nakajima E, Hamano S, et al. Phenotype-genotype correlations in patients with GNB1 gene variants, including the first three reported Japanese patients to exhibit spastic diplegia, dyskinetic quadriplegia, and infantile spasms. Brain Dev. (2020) 42:199–204. doi: 10.1016/j.braindev.2019.10.006
11. Hildebrand MS, Jackson VE, Scerri TS, Van Reyk O, Coleman M, Braden RO, et al. Severe childhood speech disorder: gene discovery highlights transcriptional dysregulation. Neurology. (2020) 94:e2148–67. doi: 10.1212/WNL.0000000000009441
12. Basel-Salmon L, Orenstein N, Markus-Bustani K, Ruhrman-Shahar N, Kilim Y, Magal N, et al. Improved diagnostics by exome sequencing following raw data reevaluation by clinical geneticists involved in the medical care of the individuals tested. Genet Med. (2019) 21:1443–51. doi: 10.1038/s41436-018-0343-7
13. Guo H, Duyzend MH, Coe BP, Baker C, Hoekzema K, Gerdts J, et al. Genome sequencing identifies multiple deleterious variants in autism patients with more severe phenotypes. Genet Med. (2019) 21:1611–20. doi: 10.1038/s41436-018-0380-2
14. Steinrücke S, Lohmann K, Domingo A, Rolfs A, Bäumer T, Spiegler J, et al. Novel GNB1 missense mutation in a patient with generalized dystonia, hypotonia, and intellectual disability. Neurol Genet. (2016) 2:e106. doi: 10.1212/NXG.0000000000000106
15. Brett M, Lai AH, Ting T-W, Tan A-M, Foo R, Jamuar S, et al. Acute lymphoblastic leukemia in a child with a de novo germline gnb1 mutation. Am J Med Genet A. (2016) 173:550–2. doi: 10.1002/ajmg.a.38026
16. Lecoquierre F, Duffourd Y, Vitobello A, Bruel A-L, Urteaga B, Coubes C, et al. Variant recurrence in neurodevelopmental disorders: the use of publicly available genomic data identifies clinically relevant pathogenic missense variants. Genet Med. (2019) 21:2504–11. doi: 10.1038/s41436-019-0518-x
17. Szczałuba K, Biernacka A, Szymańska K, Gasperowicz P, Kosińska J, Rydzanicz M, et al. Novel GNB1 de novo mutation in a patient with neurodevelopmental disorder and cutaneous mastocytosis: clinical report and literature review. Eur J Med Genet. (2018) 61:157–60. doi: 10.1016/j.ejmg.2017.11.010
18. Schultz-Rogers L, Masuho I, Pinto e Vairo F, Schmitz CT, Schwab TL, Clark KJ, et al. Haploinsufficiency as a disease mechanism in GNB1-associated neurodevelopmental disorder. Mol Genet Genomic Med. (2020) 8:e1477. doi: 10.1002/mgg3.1477
19. Lansdon LA, Saunders CJ. Genotype-phenotype correlation in GNB1-related neurodevelopmental disorder: potential association of p.Leu95Pro with cleft palate. Am J Med Genet A. (2021) 185:1341–3. doi: 10.1002/ajmg.a.62080
20. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. (2013) 30:772–80. doi: 10.1093/molbev/mst010
21. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–23. doi: 10.1038/gim.2015.30
22. Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. (2009) 25:1189–91. doi: 10.1093/bioinformatics/btp033
24. Waldschmidt H V, Homan KT, Cato MC, Cruz-Rodríguez O, Cannavo A, Wilson MW, et al. Structure-based design of highly selective and potent G protein-coupled receptor kinase 2 inhibitors based on paroxetine. J Med Chem. (2017) 60:3052–69. doi: 10.1021/acs.jmedchem.7b00112
25. Tesmer JJG, Tesmer VM, Lodowski DT, Steinhagen H, Huber J. Structure of human G protein-coupled receptor kinase 2 in complex with the kinase inhibitor balanol. J Med Chem. (2010) 53:1867–70. doi: 10.1021/jm9017515
26. Gaudet R, Bohm A, Sigler PB. Crystal structure at 2.4 angstroms resolution of the complex of transducin betagamma and its regulator, phosducin. Cell. (1996) 87:577–88. doi: 10.1016/S0092-8674(00)81376-8
Keywords: neurodevelopment, G proteins, intellectual disability, GNB1, neurogenetics
Citation: Da Silva JD, Costa MD, Almeida B, Lopes F, Maciel P and Teixeira-Castro A (2021) Case Report: A Novel GNB1 Mutation Causes Global Developmental Delay With Intellectual Disability and Behavioral Disorders. Front. Neurol. 12:735549. doi: 10.3389/fneur.2021.735549
Received: 02 July 2021; Accepted: 27 August 2021;
Published: 27 September 2021.
Edited by:
Jie Shen, Brigham and Women's Hospital and Harvard Medical School, United StatesReviewed by:
Tu-Hsueh Yeh, Taipei Medical University, TaiwanArtur Francisco Schumacher-Schuh, Federal University of Rio Grande do Sul, Brazil
Copyright © 2021 Da Silva, Costa, Almeida, Lopes, Maciel and Teixeira-Castro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jorge Diogo Da Silva, am9yZ2UuZGNyLnNpbHZhQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work and share senior authorship