 Yongzhi Xie1†
Yongzhi Xie1† Zhiqiang Lin1†
Zhiqiang Lin1† Xiaobo Li1
Xiaobo Li1 Lei Liu1Shunxiang Huang1Huadong Zhao1
Lei Liu1Shunxiang Huang1Huadong Zhao1 Binghao Wang1
Binghao Wang1 Wanqian Cao1
Wanqian Cao1 Zhengmao Hu2
Zhengmao Hu2 Jifeng Guo3
Jifeng Guo3 Lu Shen3
Lu Shen3 Beisha Tang3
Beisha Tang3 Ruxu Zhang1*
Ruxu Zhang1*- 1Department of Neurology, The Third Xiangya Hospital, Central South University, Changsha, China
- 2Hunan Key Laboratory of Medical Genetics, Center for Medical Genetics, School of Life Sciences, Central South University, Changsha, China
- 3Department of Neurology, Xiangya Hospital, Central South University, Changsha, China
Background and Aims: Charcot-Marie-Tooth (CMT) disease is a clinically and genetically heterogeneous group of inherited peripheral neuropathies. The wide phenotypic variability may not be completely explained by a single mutation.
Aims and Methods: To explore the existence of concomitant variants in CMT, we enrolled 189 patients and performed molecular diagnosis by application of next-generation sequencing combined with multiplex ligation-dependent probe amplification. We conducted a retrospective analysis of patients harboring coinherited variants in different genes.
Results: Four families were confirmed to possess variants in two genes, accounting for 2.1% (4/189) of the total in our cohort. One CMT1 patient with PMP22 duplication and MPZ variant (c.286A>C, p.K96Q) exhibited moderate neuropathy with infantile onset, while her father possessing MPZ variant was mildly affected with adolescence onset. A CMT2 patient with heterozygous variants in MFN2 (c.613_622delGTCACCACAG, p.V205Sfs*26) and GDAP1 (c.713G>T, p.W238L) exhibited childhood onset mild phenotype, while his mother with MFN2 variant developed bilateral pes cavus only. A CMT2 patient with heterozygous variants in MFN2 (c.839G>A, p.R280H) and GDAP1 (c.3G>T, p.M1?) presented infantile onset and rapid progression, while her father with MFN2 variant presented with absence of deep tendon reflexes. One sporadic CMT2 patient with early onset was confirmed harboring de novo MFN2 variant (c.1835C>T, p.S612F) and heterozygous GDAP1 variant (c.767A>G, p.H256R).
Conclusion: Our results suggest that the possibility of concomitant variants was not uncommon and should be considered when significant intrafamilial clinical heterogeneity is observed.
Introduction
Charcot-Marie-Tooth (CMT) disease is the most common inherited neuropathy with an estimated prevalence of 1 in 2,500 (1). It is a clinically and genetically heterogeneous group of disorders that is characterized by progressive weakness and atrophy of the extremities and loss of sensory function (2). Neurophysiological findings in median nerve differentiate CMT into two groups: demyelinating CMT (CMT1) with slow motor nerve conduction velocity (MNCV) (<38 m/s) and axonal CMT (CMT2) with normal or a slight reduction of MNCV (≥ 38 m/s) (2). It is generally accepted that CMT is a monogenic condition. Up till now, more than 100 causative genes have been reported as causal for CMT (http://neuromuscular.wustl.edu/time/hmsn.html), among which PMP22 and MFN2 are the most common causative genes for CMT1 and CMT2, respectively (3). Of note, patients with PMP22 duplication or MFN2 mutation, such as p.I126S and p.R94W, have a heterogeneous clinical presentation in terms of age at onset, disease severity, and clinical progression (4, 5). Thus, a single mutation may not completely explain the intrafamilial heterogeneity of CMT. Next-generation sequencing (NGS) affords opportunities to detect the co-occurrence of variants in dual or multiple genes, providing insights into the wide phenotypic variability of the disease.
In this study, we screened 189 Chinese CMT families by using multiplex ligation probe amplification (MLPA) combined with NGS technologies. We are further reporting families with significant intrafamilial phenotypic heterogeneity in the presence of concomitant heterozygous MFN2 and GDAP1 variants and a PMP22 duplication combined with an MPZ variant.
Materials and Methods
Patients and Clinical Analysis
We recruited 189 unrelated Chinese CMT families from the outpatient neurology clinic of the Third Xiangya Hospital from 2016 to 2020. All the patients were diagnosed by two experienced neurologists according to the CMT diagnostic criteria formulated by the European CMT Consortium (6). Electrophysiological examinations were performed on probands and available family members. Patients were classified into CMT1 (median MNCV <38 m/s) and CMT2 (median MNCV≥38 m/s) subtype accordingly (2). Disease severity was evaluated with the application of the CMT neuropathy score (CMTNS) (7). This study was approved by the Ethics Committee of the Third Xiangya Hospital of Central South University. Written informed consent was obtained from all the participants.
Genetic Analysis
Genomic DNA was isolated from peripheral blood obtained from all participants using standard Phenol-Chloroform procedures. We first utilized the application of MLPA (P033 kit, MRC Holland, the Netherlands) for the detection of PMP22 duplication in patients with CMT1. The target NGS was a well-suited and cost-effective strategy for efficient molecular diagnosis of CMT (8–11). Thus, inherited peripheral neuropathy multigene panel (Supplementary Table 1) sequencing was further applied in patients with CMT1 who failed to achieve molecular diagnosis and in patients with CMT2. For patients with concomitant variants, whole-exome sequencing (WES) was carried out to exclude other potential genetic variants. The sample was captured by SureSelect Human All ExonV5 Kit (Agilent). Genomic DNA sequencing was performed on the IlluminaHiseq 2500 platform (San Diego, CA, USA).
Data Analysis for the Determination of Pathogenic Mutations
All the variants were filtered against the following population database: Genome Aggregation Database (gnomAD), 1,000 Genome project (1,000 genomes), and dbSNP129. In silico analyses were performed by Mutation Taster, PolyPhen-2, SIFT, and CADD (Combined Annotation Dependent Depletion) to predict the biological relevance of the amino acid changes and phylogenetic conservation of the mutation sites. Cosegregation analysis was performed utilizing Sanger sequencing to verify the variants. All variants were interpreted according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines (12).
Results
Genetic Findings and Analysis
Four families were confirmed to possess variants in two distinct CMT genes. One CMT1 family (F1) harbored a heterozygous PMP22 duplication and an MPZ variant (c.286A>C, p.K96Q) (Figure 1A). Three families with CMT2 carried simultaneous heterozygous variants in MFN2 and GDAP1, among which F2 possessed variants c.613_622delGTCACCACAG (p.V205Sfs*26) in MFN2 and c.713G>T (p.W238L) in GDAP1, F3 harbored variants c.839G>A (p.R280H) in MFN2 and c.3G>T (p.M1?) in GDAP1, and F4 with de novo c.1835 C>T (p.S612F) variant in MFN2 and c.767 A>G (p.H256R) variant in GDAP1 (Figures 1B–D). All the variants were absent or very rare in gnomAD, 1000 Genomes and dbSNP129 (Table 1), were predicted to be damaging by utilizing the application of bioinformatics tool (Table 1), and were well conserved among different species (Figure 1E). We classified them according to the ACMG standards and guidelines and illustrated the results of in silico analysis and predicted the pathogenicity of these variants in Table 1. WES was further performed in two families with CMT2 (F2 and F3), and no other potential disease-causing variants were identified.
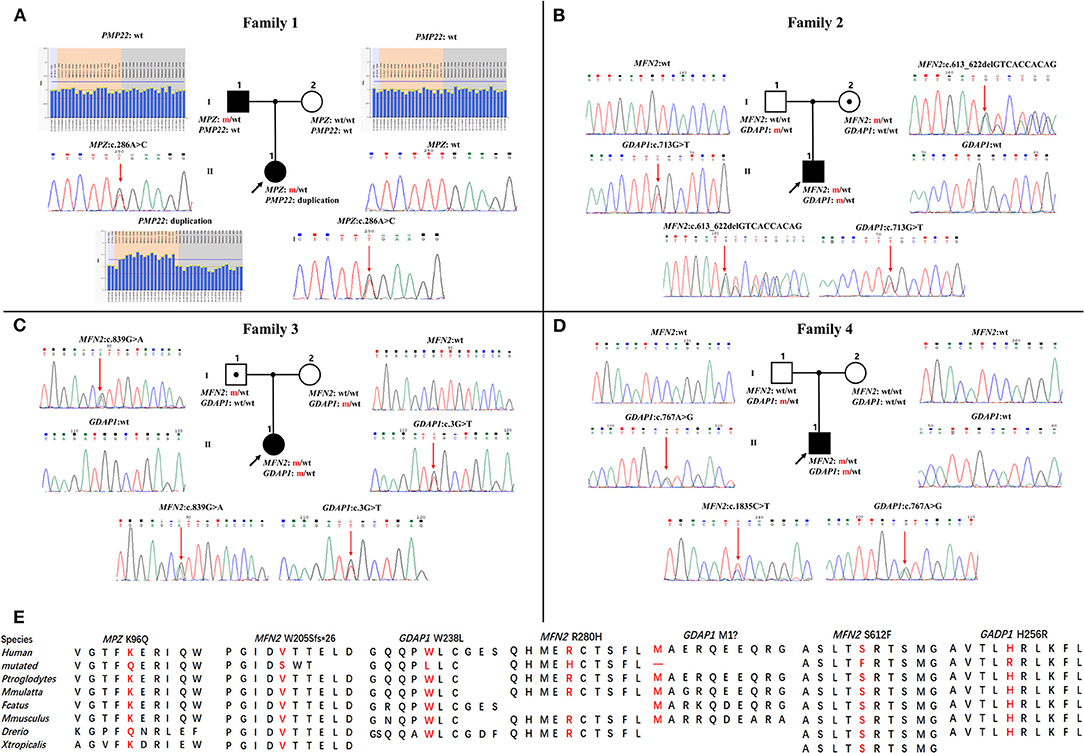
Figure 1. Pedigrees and genomic sequencing results are shown for 4 families with concomitant variants. (A) The proband in family 1 has a PMP22 duplication and an MPZ c.286A>C (p.K96Q) variant. (B) The proband in family 2 has a combination of heterozygous variants in MFN2 (c.613_622delGTCACCACAG, p.V205Sfs*26) and GDAP1 (c.713G>T, p.W238L). (C) The proband in family 3 possessing heterozygous variants in MFN2 (c.839G>A, p.R280H) and GDAP1 (c.3G>T, p.M1?). (D) The proband in family 4 with de novo heterozygous MFN2 c.1835 C>T (p.S612F) variant and heterozygous GDAP1 c.767A>G (p.H256R) variant. Clinical status: open symbols, unaffected family members; filled symbol, patients; black dot in open symbols, subclinical subjects with electromyographic or clinical examination perturbation; the black arrow, proband; wt, wild-type alleles; m, mutated alleles; the red arrow, mutated sites. (E) Conservation of amino acids at mutation sites in the different species.
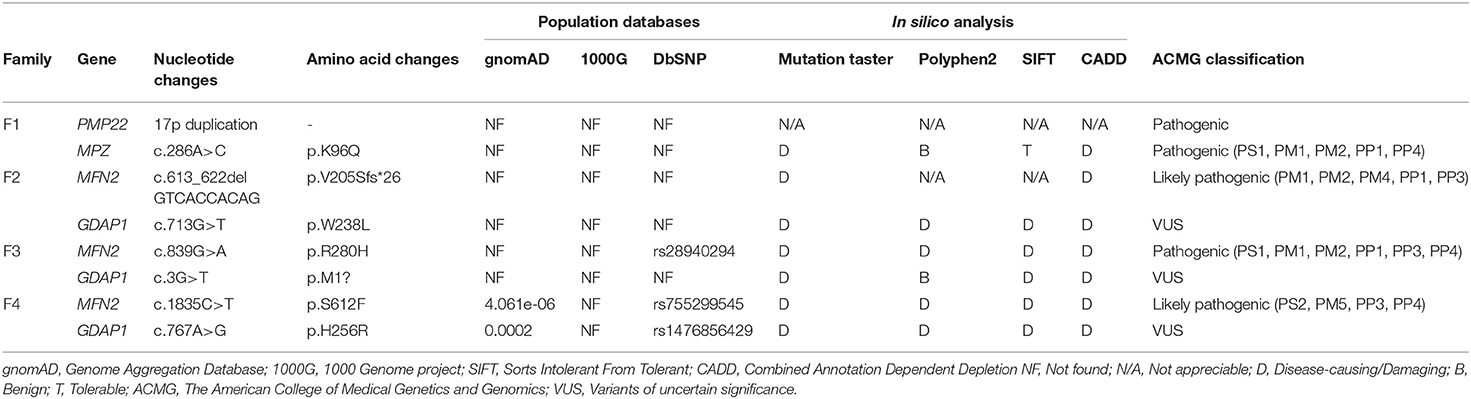
Table 1. Bioinformatics analysis and ACMG classification of the variants detected in 4 families.
We further made frequency comparisons of GDAP1 variants between patients with CMT and controls (gnomAD) using Pearson chi-squared test. In our cohort, the c.713G>T variant was identified in 1/189 patients (T = 1/378), the c.3G>T variant was identified in 2/189 patients (T = 2/378), and the c.767A>G variant was identified in 4/189 patients (G = 4/378). These three GDAP1 variants are more frequent in patients with CMT than in controls (p < 0.0001).
Clinical Features of Four Families With Concomitant Variants
F1. The proband, a 6-year-old girl, harboring PMP22 duplication and MPZ variant c.286A>C (p.K96Q) presented with delayed motor milestones and walked independently until 3 years of age. Subsequently, she developed frequent falls and had difficulty walking upstairs or downstairs. The symptoms progressed into her upper limbs with difficulty in buttoning at age 6. Neurological examination revealed atrophy and weakness of the distal upper (scored 3/5) and lower extremities (scored 3/5) and decreased superficial sensations under the ankle. Tendon reflexes were absent in the upper and lower limbs. She presented with bilateral pes cavus deformity and walked with a steppage gait. Electrophysiological studies revealed that the amplitudes of compound muscle action potentials (CMAPs) and sensory nerve action potentials (SNAPs) were absent in both the upper and lower limbs. The CMTNS was 15. Her father, a 28-year-old male, with a single MPZ variant, noticed his high-arched feet at age 15. He was presented with the reduced athletic ability (running and jumping) compared with his peers and was unable to maintain balance during squatting at age 16. The weakness was gradually progressive without the involvement of the upper extremities. Neurological examination revealed the distal weakness of his lower limbs (scored 4/5), normal sensations, bilateral pes cavus, and steppage gait. Tendon reflexes were absent in the lower limbs and reduced in the bilateral upper limbs. Electrophysiological studies revealed that the CMAPs amplitudes were decreased in the upper limbs and absent in the lower limbs and the SNAPs amplitudes were absent in the upper and lower limbs. His CMTNS was 6.
F2. The proband, a 22-year-old male, harboring heterozygous variants in MFN2 (c.613_622delGTCACCACAG, p.V205Sfs*26) and GDAP1 (c.713G>T, p.W238L) presented with an inability to stand on his heels since age 5. The patient noticed atrophy of the distal lower limbs at 14 years of age and had difficulty in climbing stairs at age 20. Neurological examination showed symmetrical muscle weakness and atrophy in the distal upper (scored 4/5) and lower limbs (scored 0/5), normal sensations, absent knee jerks, and ankle reflexes, pes cavus, and steppage gait. Electrophysiological studies revealed decreased amplitudes of CMAPs and SNAPs in the upper and lower limbs. The CMTNS was 9. His mother harboring the same MFN2 variants was subclinical with pes cavus as the only clinical presentation.
F3. The proband with heterozygous variants in MFN2 (c.839G>A, p.R280H) and GDAP1 (c.3G>T, p.M1?) was firstly evaluated at the age of 3. She had delayed motor milestones, and frequently sprained her ankle while walking at age 2. On examination, she had symmetrical muscle weakness and atrophy in the distal upper (scored 3/5) and lower limbs (scored 0/5), normal sensations of all modalities, absent ankle reflexes, normal knee jerks, bilateral pes cavus, and steppage gait. Electrophysiological studies revealed that the amplitudes of CMAPs were normal in upper limbs and reduced in lower limbs, while the SNAPs amplitudes were decreased in both the upper and lower limbs. The CMTNS was 16. His father carrying a heterozygous variant in MFN2 (c.839G>A, p.R280H) had subclinical neuropathy with diminished tendon reflexes in the upper and lower limbs as the only clinical sign. Electrophysiological examination revealed slightly decreased CMAP amplitudes in the tibial never and decreased SNAP amplitudes in the upper and lower limbs.
F4. The sporadic patient, a 29-year-old male, harboring heterozygous de novo MFN2 (c.1835C>T, p.S612F) variant and heterozygous GDAP1 (c.767A>G, p.H256R) variant noticed foot drop at age 4 and distal atrophy of the calves muscle at age 5. He developed bilateral weakness and atrophy of intrinsic hand muscle at age 7. Neurological examination at age 29 revealed severe atrophy of the distal upper and lower extremities, with reduced strength in foot dorsiflexion (scored 2/5), foot plantarflexion (scored 2/5), and finger abduction (scored 4/5). The sensory examinations were normal. Tendon reflexes were absent in the lower limbs and reduced in the upper limbs. He presented with bilateral pes cavus deformity and steppage gait. Electrophysiological studies showed that the amplitudes of CMAPs and SNAPs were reduced in the upper limbs and absent in the lower limbs. The CMTNS was 12. His father and mother were healthy.
Detailed clinical features of the 4 families are summarized in Table 2. Detailed nerve conduction studies are summarized in Supplementary Table 2.
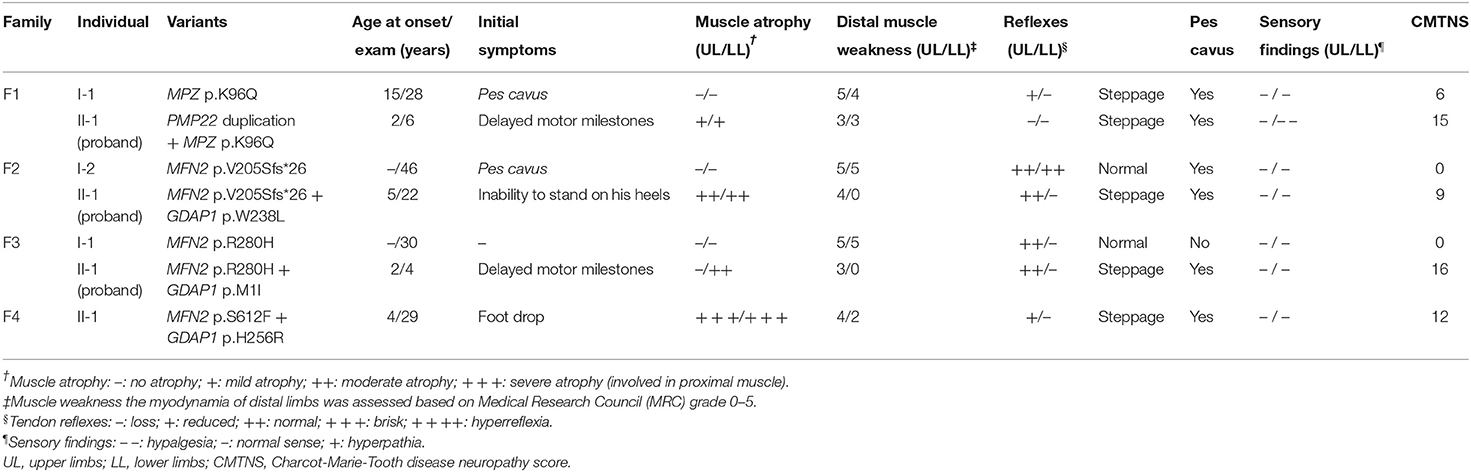
Table 2. Clinical features of four families with concomitant variants in this study.
Discussion
In this study, the patient with CMT1 harboring a de novo PMP22 duplication and an inherited MPZ variant presented with an earlier disease onset and a severer phenotype than her father harboring only the MPZ variant (Figure 1A). Each variant was sufficient to cause disease, suggesting a cumulative “double trouble” effect of these two concomitant variants (Figure 2A). MPZ is the major structural protein of peripheral myelin expressed by Schwann cells (more than 50%) (13). Mutation in MPZ was associated with unfolded protein response activation and protein aggregates, leading to the apoptosis and demyelination of the Schwann cell and altered axonal interaction (13, 14). PMP22 is also highly expressed in myelinating Schwann cell (2–5%) and directly contribute to myelin organization (15). PMP22 duplication disrupts the development and maintenance of normal myelin in the Schwann cell (16). Thus, simultaneous variants in these two myelin-related genes might exert double deleterious effects in Schwann cells, which finally leads to more severe defects in the myelination of axons. Of interest, the co-occurrence of PMP22 duplication and variant in another CMT gene (GJB1, LITAF) had also been reported (17, 18). Considering PMP22 duplication as the most common cause of CMT1 patients (approximately 70%) (3, 19, 20), one of the changes observed in cases with CMT1 with variants in two genes is usually the PMP22 duplication.
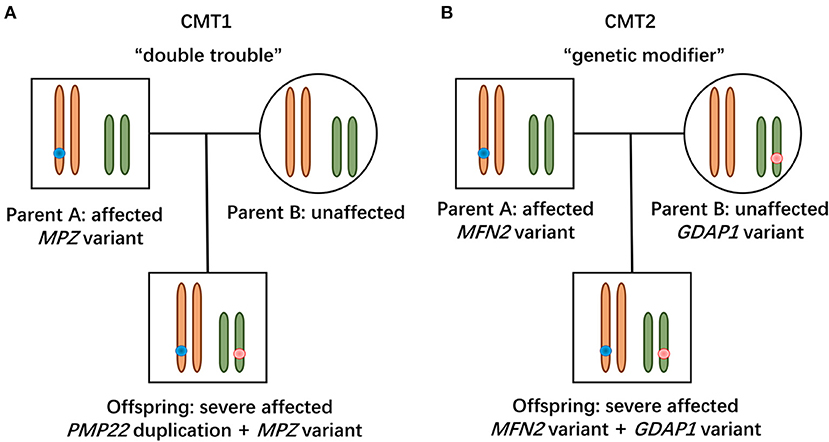
Figure 2. Modes of concomitant variants in CMT. (A) Patient with CMT1 harboring de novo PMP22 duplication and inherited MPZ variant presented with a severe phenotype, suggesting a “double trouble” effect. (B) CMT2 patients possessing concomitant variants in MFN2 and GDAP1 were severely affected, indicating that the heterozygous state of GADP1 variants serves as a “genetic modifier.”
Two patients with concomitant variants in MFN2 and GDAP1 were severer affected than their parents harboring a single MFN2 variant (Figures 1B,C). Heterozygous MFN2 variants were the primary mutations that could establish the diagnosis. The variant c.3G>T in GDAP1 changed the initiation codon, which has been reported in previous literature (21–23). The GDAP1 c.713G>T missense change occurred at the same position (p.W238) as another pathogenic variant observed (21, 24). The bioinformatic analysis supported the deleterious effect of both variants. Of note, WES ruled out other candidate genetic variants that could be involved in phenotypic expression in these families. Taken together, the heterozygous state of GADP1 variants could increase the disease severity in association with the inherited MFN2 variant, indicating their role of “genetic modifier” (Figure 2B). Another patient with CMT2 with de novo MFN2 variant and inherited heterozygous GDAP1 variant (Figure 1D) presented with early disease onset and moderate phenotype. The variant c.767A>G in GDAP1 is widely recognized as causative of autosomal recessive (AR) CMT (25–27) and might also modulate phenotypic expression in this case. MFN2 and GDAP1 serve as a protein partners in regulating mitochondrial dynamics (MFN2 for fusion, GDAP1 for fission) and are involved in the same mitochondrial function such as energy coupling (28, 29). Mutant GDAP1 exerts a loss of function mechanism and aggravate mitochondrial damage (dynamics dysfunction and energy deficit) caused by MFN2 mutation, which could explain the exacerbation of the CMT2A phenotype. The deficits in mitochondrial function might be their underlying mechanism which still needs to be further studied. MFN2 and GDAP1 are among the more frequent causative genes of CMT2, and this might be the reason why concomitant variants in these two genes have been repeatedly encountered in patients with variants in two CMT2 genes. Of interest, the frameshift p.V205Sfs*26 variant in MFN2 associated with subclinical neuropathy in our study suggested that MFN2 is not sensitive to haploinsufficiency. Previous reports have described patients with heterozygous state of MFN2 variants (p.E308X, p.V160fs*26 and del ex7-8) developing a subclinical phenotype or asymptomatic presentation (30, 31), indicating that null variants in MFN2 might be associated with a minimal phenotype.
This study identified a 2.1% (4/189) prevalence of concomitant variants in our cohort. The concomitant variants in families with CMT have been documented in several studies. Recently, a Japanese cohort study identified 5 out of 1,005 families with CMT harboring coinherited variants, accounting for 0.5% (5/1,005) of the total, in which the “double trouble” effect of concomitant variants were the underlying causes (32). Compared with the “genetic modifier” effect of GDAP1 variants observed in this study, the coexistence of homozygous AR-CMT2K (p.Q163X) or heterozygous autosomal dominant-CMT2K (p.H123R, p.E222K, and p.R120W) variant in GDAP1 and heterozygous variant in MFN2 had been reported associating with a more severe phenotype, suggesting the “double trouble” effect of concomitant MFN2 and GDAP1 variants (28, 30, 33, 34). Moreover, concomitant variants were also related to true digenic inheritance (DI) that has been defined as the coinheritance of two nonallelic mutations, both are indispensable to establish the diagnosis (35). The existence of true DI was reported in a family with CMT with heterozygous MFN2 p.L741V and GDAP1 p.Q163* variants (36). These results suggest that the occurrence of concomitant variants was not uncommon in CMT.
There was an important limitation to this study. Because we conducted a retrospective analysis of target NGS data, genetic variants in non-CMT-causing genes could not be detected. WES (or whole-genome sequencing [WGS]) would detect all possible genetic modifiers. The application of WES (WGS) in all the families with phenotypic variabilities could provide further insights into the clinical and genetic heterogeneity of CMT.
In summary, the coexistence of variants in PMP22/MPZ and MFN2/GDAP1 related to more severe phenotypes accounted for 2.1% of patients with CMT in our cohort. Cumulative deficits on myelination or mitochondrial function might be their underlying mechanism. The possibility of the coexistence of variants in distinct causative genes should be taken into consideration when significant clinical heterogeneity is observed.
Data Availability Statement
The data presented in this study were deposited in Figshare open access repository doi: 10.6084/m9.figshare.17192927. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Third Xiangya Hospital of Central South University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
RZ designed and conceptualized study. YX, ZL, XL, LL, BW, WC, JG, LS, and RZ contributed patient material and clinical data. SH and HZ contributed acquisition of neurophysiological data. ZL and ZH interpreted the genetic data. YX provide the first draft of the manuscript. RZ and BT revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study is funded by the National Natural Science Foundation of China (81771366 and 82001338) and the China International Medical Foundation (CIMF-Z-2016-20-1801).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would thank the patients and their family members for their participation in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.736704/full#supplementary-material
References
1. Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin Genet. (1974) 6:98–118. doi: 10.1111/j.1399-0004.1974.tb00638.x
2. Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. (1980) 103:259–80. doi: 10.1093/brain/103.2.259
3. Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol. (2011) 69:22–33. doi: 10.1002/ana.22166
4. Kim YH, Chung HK, Park KD, Choi KG, Kim SM, Sunwoo IN, et al. Comparison between clinical disabilities and electrophysiological values in Charcot-Marie-Tooth 1A patients with PMP22 duplication. J Clin Neurol. (2012) 8:139–45. doi: 10.3988/jcn.2012.8.2.139
5. Bombelli F, Stojkovic T, Dubourg O, Echaniz-Laguna A, Tardieu S, Larcher K, et al. Charcot-Marie-Tooth disease type 2A: from typical to rare phenotypic and genotypic features. JAMA Neurol. (2014) 71:1036–42. doi: 10.1001/jamaneurol.2014.629
6. De Jonghe P, Timmerman V, Van Broeckhoven C. 2nd Workshop of the European CMT Consortium: 53rd ENMC International Workshop on Classification and Diagnostic Guidelines for Charcot-Marie-Tooth Type 2 (CMT2-HMSN II) and Distal Hereditary Motor Neuropathy (distal HMN-Spinal CMT) 26-28 September 1997, Naarden, The Netherlands. Neuromuscul Disord. (1998) 8:426–31. doi: 10.1016/S0960-8966(98)00025-X
7. Murphy SM, Herrmann DN, McDermott MP, Scherer SS, Shy ME, Reilly MM, et al. Reliability of the CMT neuropathy score (second version) in Charcot-Marie-Tooth disease. J Peripher Nerv Syst. (2011) 16:191–8. doi: 10.1111/j.1529-8027.2011.00350.x
8. Nam SH, Hong YB, Hyun YS, Nam da E, Kwak G, Hwang SH, et al. Identification of genetic causes of inherited peripheral neuropathies by targeted gene panel sequencing. Mol Cells. (2016) 39:382–8. doi: 10.14348/molcells.2016.2288
9. Wang W, Wang C, Dawson DB, Thorland EC, Lundquist PA, Eckloff BW, et al. Target-enrichment sequencing and copy number evaluation in inherited polyneuropathy. Neurology. (2016) 86:1762–71. doi: 10.1212/WNL.0000000000002659
10. Cortese A, Wilcox JE, Polke JM, Poh R, Skorupinska M, Rossor AM, et al. Targeted next-generation sequencing panels in the diagnosis of Charcot-Marie-Tooth disease. Neurology. (2020) 94:e51–e61. doi: 10.1212/WNL.0000000000008672
11. Padilha JPD, Brasil CS, Hoefel AML, Winckler PB, Donis KC, Brusius-Facchin AC, et al. Diagnostic yield of targeted sequential and massive panel approaches for inherited neuropathies. Clin Genet. (2020) 98:185–90. doi: 10.1111/cge.13793
12. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
13. Shy ME. Peripheral neuropathies caused by mutations in the myelin protein zero. J Neurol Sci. (2006) 242:55–66. doi: 10.1016/j.jns.2005.11.015
14. Bai Y, Wu X, Brennan KM, Wang DS, D'Antonio M, Moran J, et al. Myelin protein zero mutations and the unfolded protein response in Charcot Marie Tooth disease type 1B. Ann Clin Transl Neurol. (2018) 5:445–55. doi: 10.1002/acn3.543
15. Mittendorf KF, Marinko JT, Hampton CM, Ke Z, Hadziselimovic A, Schlebach JP, et al. Peripheral myelin protein 22 alters membrane architecture. Sci Adv. (2017) 3:e1700220. doi: 10.1126/sciadv.1700220
16. Li J, Parker B, Martyn C, Natarajan C, Guo J. The PMP22 gene and its related diseases. Mol Neurobiol. (2013) 47:673–98. doi: 10.1007/s12035-012-8370-x
17. Hodapp JA, Carter GT, Lipe HP, Michelson SJ, Kraft GH, Bird TD. Double trouble in hereditary neuropathy: concomitant mutations in the PMP-22 gene and another gene produce novel phenotypes. Arch Neurol. (2006) 63:112–7. doi: 10.1001/archneur.63.1.112
18. Sinkiewicz-Darol E, Lacerda AF, Kostera-Pruszczyk A, Potulska-Chromik A, Sokołowska B, Kabzińska D, et al. The LITAF/SIMPLE I92V sequence variant results in an earlier age of onset of CMT1A/HNPP diseases. Neurogenetics. (2015) 16:27–32. doi: 10.1007/s10048-014-0426-9
19. Murphy SM, Laura M, Fawcett K, Pandraud A, Liu YT, Davidson GL, et al. Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry. (2012) 83:706–10. doi: 10.1136/jnnp-2012-302451
20. Fridman V, Bundy B, Reilly MM, Pareyson D, Bacon C, Burns J, et al. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J Neurol Neurosurg Psychiatry. (2015) 86:873–8. doi: 10.1136/jnnp-2014-308826
21. DiVincenzo C, Elzinga CD, Medeiros AC, Karbassi I, Jones JR, Evans MC, et al. The allelic spectrum of Charcot-Marie-Tooth disease in over 17,000 individuals with neuropathy. Mol Genet Genomic Med. (2014) 2:522–9. doi: 10.1002/mgg3.106
22. Liu C, Yan Y, Zhao J, Ha L, Xu X. [Identification of a novel c.1A>G variant of GDAP1 gene in a pedigree affected with autosomal recessive fibula atrophy]. Zhonghua Yi Xue Yi Chuan Xue Za. (2020) 37:1244–6. doi: 10.3760/cma.j.cn511374-20191210-00630
23. Xie Y, Lin Z, Liu L, Li X, Huang S, Zhao H, et al. Genotype and phenotype distribution of 435 patients with Charcot-Marie-Tooth disease from central south China. Eur J Neurol. (2021) 28:3774–83. doi: 10.1111/ene.15024
24. Antoniadi T, Buxton C, Dennis G, Forrester N, Smith D, Lunt P, et al. Application of targeted multi-gene panel testing for the diagnosis of inherited peripheral neuropathy provides a high diagnostic yield with unexpected phenotype-genotype variability. BMC Med Genet. (2015) 16:84. doi: 10.1186/s12881-015-0224-8
25. Zhang RX, Tang BS, Zi XH, Luo W, Xia K, Pan Q, et al. [Mutation analysis of ganglioside-induced differentiation associated protein-1 gene in Chinese Charcot-Marie-Tooth disease]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2004) 21:207–10. doi: 10.3760/j.issn:1003-9406.2004.03.004
26. Chung KW, Hyun YS, Lee HJ, Jung HK, Koo H, Yoo JH, et al. Two recessive intermediate Charcot-Marie-Tooth patients with GDAP1 mutations. J Peripher Nerv Syst. (2011) 16:143–6. doi: 10.1111/j.1529-8027.2011.00329.x
27. Fu J, Dai S, Lu Y, Wu R, Wang Z, Yuan Y, et al. Similar clinical, pathological, and genetic features in Chinese patients with autosomal recessive and dominant Charcot-Marie-Tooth disease type 2K. Neuromuscul Disord. (2017) 27:760–5. doi: 10.1016/j.nmd.2017.04.001
28. Cassereau J, Casasnovas C, Gueguen N, Malinge MC, Guillet V, Reynier P, et al. Simultaneous MFN2 and GDAP1 mutations cause major mitochondrial defects in a patient with CMT. Neurology. (2011) 76:1524–6. doi: 10.1212/WNL.0b013e318217e77d
29. Eijkenboom I, Vanoevelen JM, Hoeijmakers JGJ, Wijnen I, Gerards M, Faber CG, et al. A zebrafish model to study small-fiber neuropathy reveals a potential role for GDAP1. Mitochondrion. (2019) 47:273–81. doi: 10.1016/j.mito.2019.01.002
30. Vital A, Latour P, Sole G, Ferrer X, Rouanet M, Tison F, et al. A French family with Charcot-Marie-Tooth disease related to simultaneous heterozygous MFN2 and GDAP1 mutations. Neuromuscul Disord. (2012) 22:735–41. doi: 10.1016/j.nmd.2012.04.001
31. Polke JM, Laurá M, Pareyson D, Taroni F, Milani M, Bergamin G, et al. Recessive axonal Charcot-Marie-Tooth disease due to compound heterozygous mitofusin 2 mutations. Neurology. (2011) 77:168–73. doi: 10.1212/WNL.0b013e3182242d4d
32. Yoshimura A, Yuan JH, Hashiguchi A, Ando M, Higuchi Y, Nakamura T, et al. Genetic profile and onset features of 1005 patients with Charcot-Marie-Tooth disease in Japan. J Neurol Neurosurg Psychiatry. (2019) 90:195–202. doi: 10.1136/jnnp-2018-318839
33. Kostera-Pruszczyk A, Kosinska J, Pollak A, Stawinski P, Walczak A, Wasilewska K, et al. Exome sequencing reveals mutations in MFN2 and GDAP1 in severe Charcot-Marie-Tooth disease. J Peripher Nerv Syst. (2014) 19:242–5. doi: 10.1111/jns.12088
34. Anghelescu C, Francou B, Cardas R, Guiochon-Mantel A, Aubourg P, Servais L, et al. Targeted exomes reveal simultaneous MFN2 and GDAP1 mutations in a severe Charcot-Marie-Tooth disease type 2 phenotype. Eur J Neurol. (2017) 24:e15–e6. doi: 10.1111/ene.13250
35. Deltas C. Digenic inheritance and genetic modifiers. Clin Genet. (2018) 93:429–38. doi: 10.1111/cge.13150
Keywords: Charcot-Marie-Tooth diseases, concomitant variants, intrafamilial clinical heterogeneity, double trouble, genetic modifier
Citation: Xie Y, Lin Z, Li X, Liu L, Huang S, Zhao H, Wang B, Cao W, Hu Z, Guo J, Shen L, Tang B and Zhang R (2022) One PMP22/MPZ and Three MFN2/GDAP1 Concomitant Variants Occurred in a Cohort of 189 Chinese Charcot-Marie-Tooth Families. Front. Neurol. 12:736704. doi: 10.3389/fneur.2021.736704
Received: 05 July 2021; Accepted: 16 December 2021;
Published: 28 January 2022.
Edited by:
Albena Jordanova, University of Antwerp, BelgiumReviewed by:
Liena Elbaghir Omer Elsayed, University of Khartoum, SudanMeng-Han Tsai, Kaohsiung Chang Gung Memorial Hospital, Taiwan
Copyright © 2022 Xie, Lin, Li, Liu, Huang, Zhao, Wang, Cao, Hu, Guo, Shen, Tang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruxu Zhang, emhhbmdydXh1QHZpcC4xNjMuY29t
†These authors have contributed equally to this work