 Lorenzo Nanetti1
Lorenzo Nanetti1 Daniela Di Bella1
Daniela Di Bella1 Stefania Magri1
Stefania Magri1 Mario Fichera1
Mario Fichera1 Elisa Sarto1
Elisa Sarto1 Anna Castaldo1
Anna Castaldo1 Alessia Mongelli1Silvia Baratta1Silvia Fenu2
Alessia Mongelli1Silvia Baratta1Silvia Fenu2 Marco Moscatelli3Maria Teresa Bonati4
Marco Moscatelli3Maria Teresa Bonati4 Andrea Martinuzzi5
Andrea Martinuzzi5 Caterina Mariotti1*
Caterina Mariotti1* Franco Taroni1
Franco Taroni1- 1Unit of Medical Genetics and Neurogenetics, Fondazione Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Neurologico Carlo Besta, Milan, Italy
- 2Unit of Rare Neurodegenerative and Neurometabolic Diseases, Fondazione Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Neurologico Carlo Besta, Milan, Italy
- 3Unit of Neuroradiology, Fondazione Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Neurologico Carlo Besta, Milan, Italy
- 4Unit of Medical Genetics, Institute for Maternal and Child Health Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Burlo Garofalo, Trieste, Italy
- 5Conegliano Research Center, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Eugenio Medea, Conegliano, Italy
A wide spectrum of neurodegenerative diseases has been associated with pathogenic variants in the PNPLA6 (patatin-like phospholipase domain-containing protein 6) gene, including spastic paraplegia type 39, Gordon—Holmes, Boucher—Neuhauser, Oliver—Mc Farlane, and Laurence—Moon syndromes. These syndromes present variable and overlapping clinical symptoms, encompassing cerebellar ataxia, hypogonadotropic hypogonadism, chorioretinal dystrophy, spastic paraplegia, muscle wasting, peripheral neuropathy, and cognitive impairment. In the present study, we performed a wide genetic screening in 292 patients presenting with ataxia or spastic paraplegia using a probe-based customized gene panel, covering >200 genes associated with spinocerebellar diseases. We identified six novel and four recurrent PNPLA6 gene variants in eight patients (2.7%). Six patients presented an infantile or juvenile onset (age < 18), and two patients had an adult onset. Cerebellar ataxia was observed in seven patients and spastic paraplegia in one patient. Progression of cerebellar symptoms was slow in all patients, who retained ambulation even after a mean disease duration of 15 years. Brain MRI showed cerebellar atrophy in 6/8 patients, more pronounced in superior and dorsal vermis lobules (I to VII). Additional clinical features included hypogonadotropic hypogonadism (5/8), growth hormone deficiency (2/8), peripheral axonal neuropathy (4/8), cognitive impairment (3/8), chorioretinal dystrophy (2/8), and bilateral vestibular areflexia with a reduced visual vestibule-ocular reflex (1/8). In accordance with previous studies, chorioretinal dystrophy was the most frequent presenting symptom in early onset patients, hypogonadotropic hypogonadism in juvenile onset cases, and cerebellar ataxia in adult patients. One patient had an initial clinical presentation compatible with Cerebellar Ataxia with Neuropathy and Vestibular Areflexia Syndrome (CANVAS), but no pathological expansions in the RFC1 gene. In conclusion, patients with PNPLA6 variants present a variable age of onset spanning from infancy to adulthood, and each clinical symptom has an age-dependent manifestation thus requiring a multi-systemic diagnostic approach. The description of patients presenting very late-onset cerebellar ataxia suggests that PNPLA6 genetic screening should also be considered in the diagnostic workout of adult cerebellar ataxia.
Introduction
Pathogenic variants in the PNPLA6 gene(-encoding patatin-like phospholipase domain containing protein 6) have been demonstrated to cause a number of variable neurodegenerative diseases. This gene codes for the enzyme neuropathy target esterase (NTE), an endoplasmic reticulum-localized lysophospholipase that deacylates phosphatidylcholine and lysophosphatidylcholine, and has been studied for its involvement in the pathogenesis of organophosphorus compound-induced delayed neuropathy (1).
Pathogenic variants in the PNPLA6 gene have been originally described in patients presenting a neurological phenotype characterized by early-onset spastic paraplegia, motor neuropathy, and distal muscle wasting. The disease was classified among the groups of the Hereditary Spastic Paraplegias as SPG type 39 (SPG39, MIM612020) (1–3).
Recently, bi-allelic PNPLA6 gene variants have been detected in patients affected by two clinical syndromes described more than 50 years ago and named Gordon—Holmes (GH, MIM212840) and Boucher—Neuhauser (BN, MIM215470) (4). The typical features of the two clinical syndromes are cerebellar ataxia and hypogonadotropic hypogonadism (HH), and BN is distinguished from GH for the presence of chorioretinal dystrophy (5).
Genetic screening in large series of cases with neurodegenerative diseases further widened the clinical phenotype associated with PNPLA6 gene variants, identifying patients with trichomegaly, chorioretinal atrophy, and multiple pituitary hormone deficiencies, including growth hormone (G-H), gonadotrophins, and thyroid-stimulating hormone (TSH). This particular phenotype was named Oliver—Mc Farlane syndrome (OMcF; MIM 275400) (6–9). Pathogenic variants in the PNPLA6 gene were also demonstrated to cause the Laurence—Moon syndrome (LM, MIM 245800) that includes all the symptoms of the Oliver—Mc Farlane syndrome plus neurological symptoms, such as ataxia, spastic paraplegia, and neuropathy (6). Pure cerebellar ataxia and isolated congenital Leber amaurosis or retinitis were also described in single families (7, 10, 11).
It is not yet fully understood why pathogenic variants in the PNPLA6 gene are associated with a wide spectrum of phenotypes. The NTE plays a critical role in phosphatidylcholine metabolism, membrane phospholipid trafficking, and maintenance of axonal integrity (12).
NTE enzymatic activity was demonstrated to be reduced in patient fibroblasts and mouse models (6, 13); however, a clear relationship between the variability in the NTE activity and the phenotypical variability in patients with PNPLA6 gene variants has not been entirely established (6, 13).
In the present study, we describe the clinical phenotype of 8 Italian patients carrying novel and recurrent pathogenic PNPLA6 gene variants and manifesting variable neurological and extra-neurological features.
Methods
From 2015 to 2020, we investigated 292 index Italian patients recruited in our referral center for spinocerebellar diseases. The subjects were selected on the basis of the following criteria: (1) progressive ataxic syndrome or spastic paraplegia; (2) exclusion of infectious, autoimmune, or cerebrovascular diseases; (3) absence of pathogenic variants associated with spinocerebellar ataxia (SCA) types 1, 2, 3, 6, 7, and 17, Friedreich ataxia, and hereditary spastic paraplegia types 4 and 7 (SPG4; SPG7).
Genomic DNA was extracted from venous peripheral blood lymphocytes by standard procedures. NGS-targeted resequencing analysis was performed using a probe-based customized gene panel (Illumina Nextera Rapid Capture Custom Kit - Illumina Inc., San Diego, CA, USA) or an Agilent SureSelect QXT custom kit (Agilent Technologies, Santa Clara, CA, USA), including >200 genes associated with ataxia and spastic paraplegia (Supplementary Table 1).
Written informed consent was obtained from all the participants (or guardians of participants) in the study in accordance with the local Ethic Committee Board.
Results
Eight out of the 292 index patients (2.7%) were demonstrated to carry homozygous or compound heterozygous PNPLA6 pathogenic or likely pathogenic gene variants. Clinical and genetic characteristics of patients carrying PNPLA6 gene variants are summarized in Table 1.
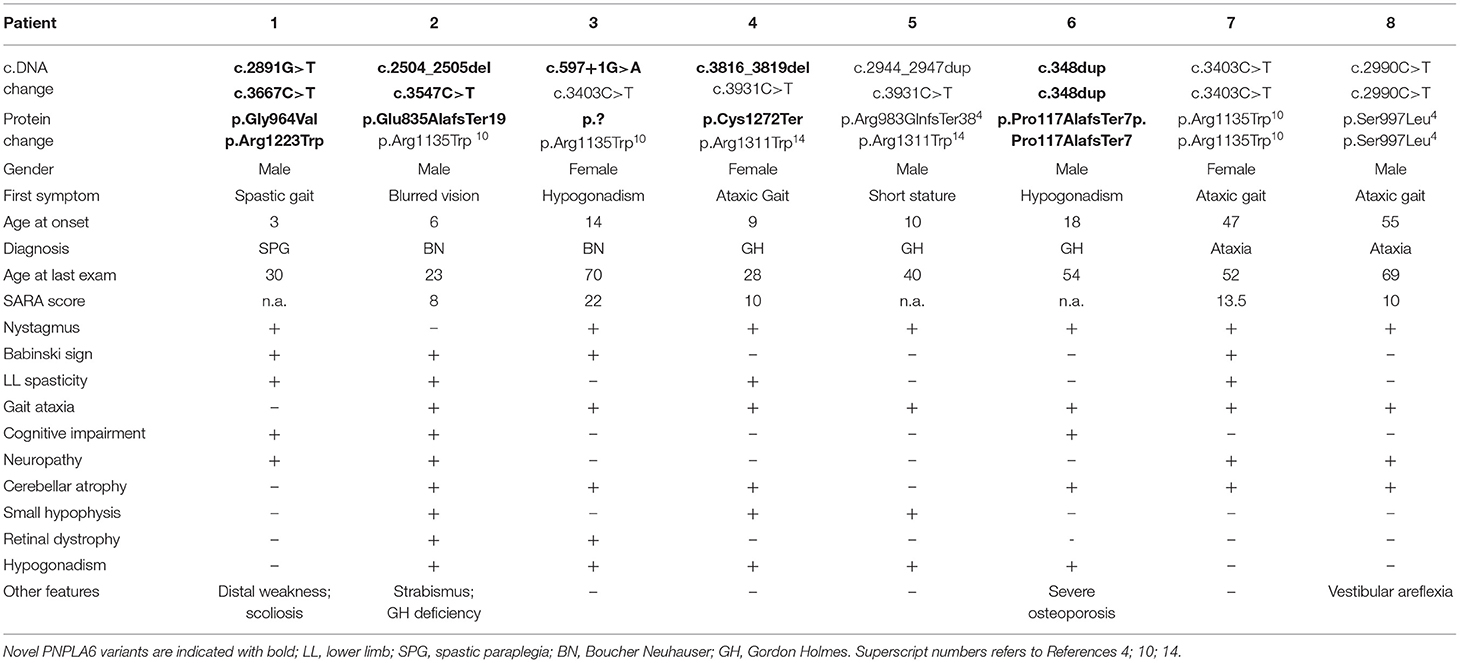
Table 1. Clinical and genetic characterization of eight patients with PNPLA6 gene variants.
The patients carried four previously described pathogenic PNPLA6 variants (4, 10, 14) and six novel variants. Three novel variants were truncating, two were missense, and one variant was predicted to affect a splice site (Table 1).
The novel missense variants c.2891G > T and c.3667C > T were in trans in Patient 1, and were absent (GnomAD-genome) or very rare (GnomAD-exome: T = 0.000008) in control population databases. According to ACMG criteria, both variants can be classified as Likely Pathogenic (Class 4) variants (15).
The splice site variant c.597 + 1G > A was in trans with the previously described c.3403C > T missense variant in Patient 3 (10), was absent in control population databases (GnomAD-exome and genome), predicted to “break/abolish the donor site” (Human Splicing Finder), and classified as “pathogenic” (Class 5 variant) (15).
Clinical presentations of our subjects spanned through the clinical spectrum of neurodegenerative diseases associated with PNPLA6 gene variants: one patient manifested spastic paraplegia at age 3; one patient had chorioretinal dystrophy at age 6 and was subsequently diagnosed as having BN syndrome; one subject had chorioretinal dystrophy and HH at age 14 and was also diagnosed as BN; three patients had gait ataxia associated with HH or G-H deficiency (ages 9, 10, and 18) and were diagnosed as having GH syndrome; two subjects presented a late-onset cerebellar ataxia at ages 47 and 55 (Table 1).
Clinical diagnosis was highly dependent on the initial symptom of the patient and on the age at the onset. In fact, in infantile cases, the onset was mainly characterized by visual defects; in juvenile cases, the first clinical signs were associated with hormone deficiencies; and, in late-onset cases, the predominant presentation was characterized by cerebellar symptoms.
However, during the course of the disease, all the subjects developed common additional clinical symptoms: 7/8 patients manifested a progressive cerebellar syndrome, 5/8 hypogonadotropic hypogonadism, 4/8 peripheral axonal neuropathy and/or lower limb spasticity, 2/8 chorioretinal dystrophy, and 1 subject had bilateral vestibular areflexia with poor visual vestibule-ocular reflex (VVOR).
Cognitive impairment was reported in 3/8 patients: two subjects (Patients 1 and 2, Table 1) showed intellectual deficit at ages 5 and 6, in concomitance with other clinical manifestations of the disease. Both subjects necessitated dedicated learning support at school. In the third subject (Patient 8), mild cognitive impairment was disclosed in adulthood and ascertained by neurocognitive tests (ENB2, brief neuropsychological examination-2).
Cerebellar symptoms were the most recurrent clinical features, having a mean age of onset 31 (range 9–55), and very slow progression. All the patients were ambulatory at the last examination and had a mean SARA score of 12.7 (range 8–22) after mean disease duration of 15 years (range 2–30). Two index patients had a very late-onset cerebellar ataxia at age 47 (pt 7) and 55 (pt 8) and no additional PNPLA6-associated clinical features, in particular chorioretinal dystrophy or hypogonadotropic hypogonadism. Patient 7 had spastic ataxia complicated by peripheral motor neuropathy. The patient had a sibling aged 59, affected by cerebellar ataxia since age 51. No other clinical or genetic data are available as he refused any clinical evaluation.
Patient 8 had an initial clinical presentation compatible with Cerebellar Ataxia with Neuropathy and Vestibular Areflexia Syndrome (CANVAS), manifesting ataxia with cerebellar atrophy, axonal neuropathy, and vestibular areflexia. For this subject, the test for pathogenic repeat expansions in RFC1 was performed in parallel with NGS analysis, and the result excluded the diagnosis of CANVAS.
Brain MRI scans demonstrated cerebellar atrophy in 6/7 ataxic patients with more pronounced atrophy in superior and dorsal vermal lobules (I to VII), compared to caudal lobules (VIII-X) (Figure 1).
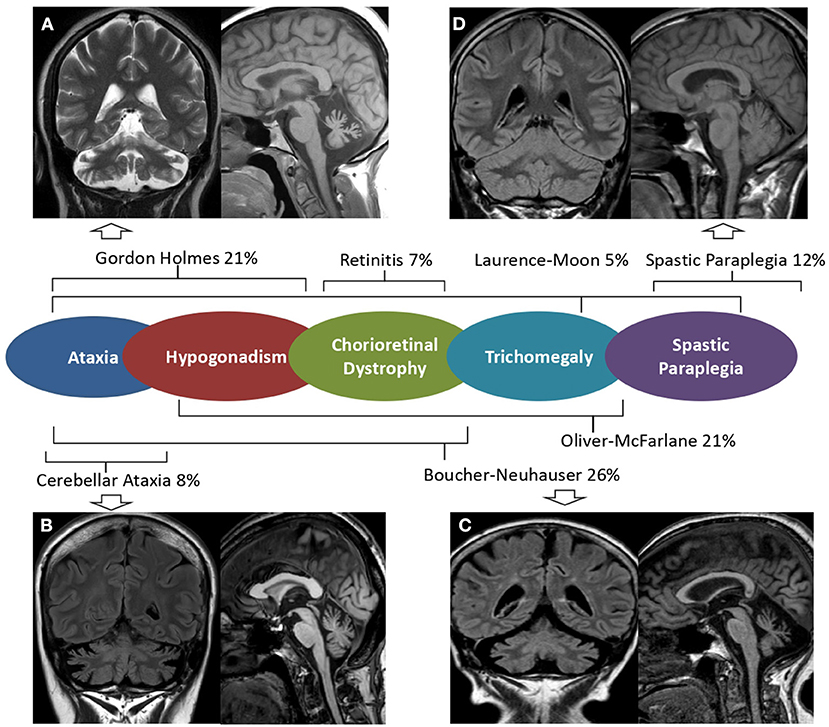
Figure 1. Brain MRI in PNLA6-associated phenotypes. Mid-sagittal T1 weighted and coronal FLAIR or T2 w brain MRI images of patients with PNPLA6-associated clinical phenotypes. Severe cerebellar atrophy and slightly hyperintense dentate nuclei are shown in patients with Gordon Holmes syndrome [(A), Patient 4], late-onset cerebellar ataxia [(B), Patient 7], and Boucher Neuhauser syndrome [(C), Patient 3]. No MRI abnormalities are detectable in patients presenting Spastic Paraplegia [(D), Patient 1]. A central scheme summarizes the frequency of the syndromic diagnoses reported in the literature.
Pituitary hormone deficiencies were also observed in patients with ataxia: hypogonadotropic hypogonadism in 5 out of 7 patients, and G-H deficiency in 2 patients. Brain MRI detected a reduced volume hypophysis in 5 out of 7 patients.
Patient 1 had a “not-ataxic” phenotype characterized by early-onset (age 3) progressive spastic paraplegia with cognitive impairment (QI = 49), and scoliosis (Table 1). At age 15, hand muscle weakness and amyotrophy were observed, and, in the subsequent years, lower limb distal muscles were affected, and progressive muscle wasting was detected. Nerve conduction studies revealed an axonal motor neuropathy more pronounced at median nerves and lower limb motor nerves. The disease progression was slow, and the patient was able to walk without support at the last examination (age 30). No cerebellar symptoms, hypogonadotropic hypogonadism, or retinal dystrophy were observed, and cerebellar atrophy was not detected at brain MRI (Figure 1).
Discussion
Up to date, PNPLA6 pathogenic gene variants were identified worldwide in 76 patients from 49 families. We revised the literature and summarized clinical (Table 2) and genetic (Supplementary Table 2) findings described in all reported PNPLA6 patients.
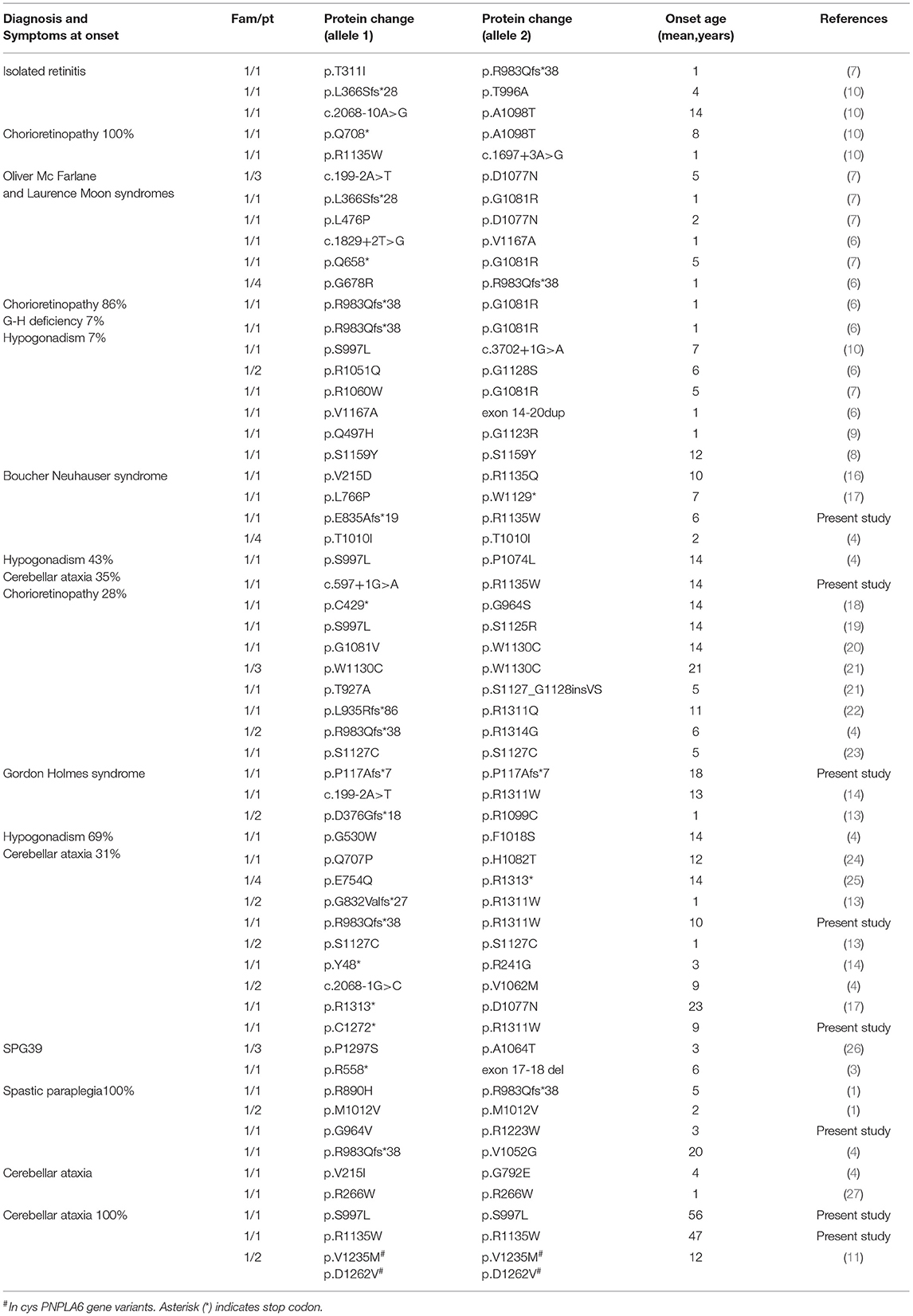
Table 2. Clinical/genetic characterization of patients reported in the literature grouped for initial syndromic diagnosis.
The age at the onset was highly variable, with the majority of patients (70%) presenting an infantile onset, ranging from 0 to 8 years (53/76 patients). Juvenile-onset was reported in 27% of cases (range, 9–18 years, 19/76 patients), and only 6% of cases manifested the disease in adulthood (4/76 patients) (Figure 2).
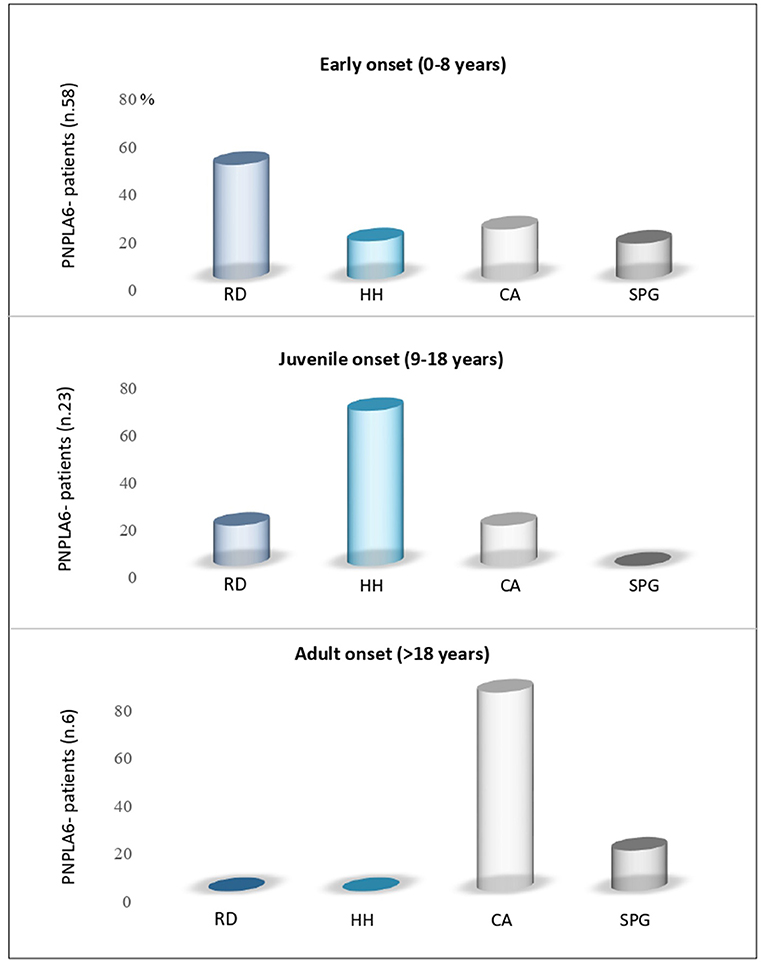
Figure 2. Age-dependent PNPLA6-associated clinical symptoms. Review of the literature for manifesting symptoms reported in patients with PNPLA6 variants: Subjects with an early onset had predominantly retinal dystrophy (RD); juvenile cases had hypogonadotropic hypogonadism (HH) as presenting symptom, and adult cases had cerebellar ataxia (CA) or spastic paraplegia (SPG).
The presenting phenotype seems to be age-dependent. In fact, chorioretinal dystrophy was the most frequent clinical presentation in infantile cases (51%), hypogonadotropic hypogonadism in juvenile-onset cases (68%), and cerebellar ataxia in late-onset cases (75%) (Figure 2).
Initial clinical diagnosis reported in the literature was very heterogeneous and probably influenced by the large number of syndromes described in the last decades before the identification of the PNPLA6-associated phenotypes.
In fact, the syndromic diagnosis in patients reported in the literature was BN in 26% of patients, GH in 21%, Oliver—Mc Farlane Syndrome in 21%, Hereditary Spastic Paraplegia in 12%, cerebellar ataxia in 8%, and Laurence—Moon Syndrome 5%. In addition, four patients had a diagnosis of isolated retinitis (5%), and a single case was diagnosed as having Leber congenital amaurosis (Table 2).
Irrespectively of the clinical presentation at diagnosis, the clinical features observed during the course of the diseases largely overlap in most of the patients with PNPLA6 variants. Hypogonadotropic hypogonadism was found in 74% of cases and has been detected in all PNPLA6-associated phenotypes except in SPG39. Cerebellar ataxia was described in 68% of the patients encompassing all the syndromes associated with PNPLA6. Chorioretinal dystrophy was present in 65% of cases, being always observed in BN, LM, and OMF syndromes, and nearly absent in GH, spastic paraplegia, and late-onset cerebellar ataxia phenotypes (Figure 3). Late-onset cases have a pure spinocerebellar phenotype and do not manifest visual or endocrinological deficits.
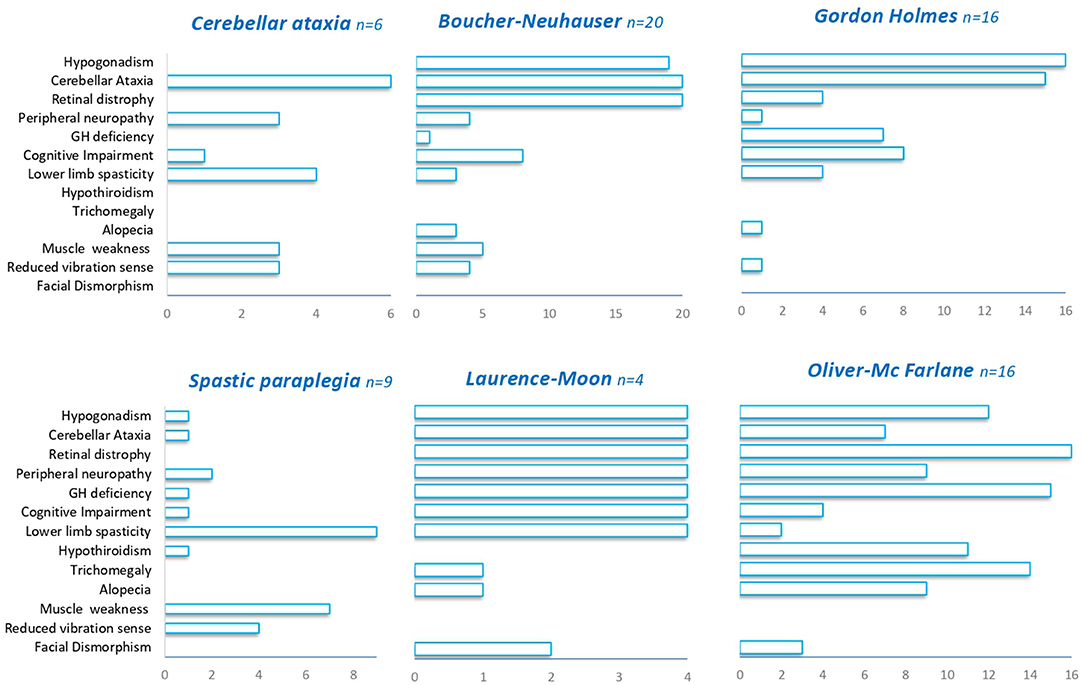
Figure 3. Clinical features in PNPLA6-associated phenotypes. A summary of the different clinical features (Y-axis) observed in PNPLA6-associated phenotypes.
In our series, we screened patients with either juvenile or an adult onset; we, therefore, observed hypogonadotropic hypogonadism and cerebellar ataxia as predominant presenting symptoms (88 and 63% of patients), while chorioretinal dystrophy was observed only in two patients (25%) (Table 1). Brain MRI confirmed that cerebellar atrophy is more pronounced in the superior and dorsal lobules than in caudal lobules (Figure 1). We also describe two unrelated patients with very late-onset cerebellar ataxia at age 47 (pt 7) and 55 (pt 8), and no chorioretinal dystrophy or hypogonadotropic hypogonadism (Table 1). Patient 8 had an initial clinical presentation compatible with CANVAS that was never described in patients with PNPLA6 gene variants. Notably, poor VVOR suppression was previously described in two patients diagnosed as having BN. Deik et al. described a Spanish-Italian woman presenting amenorrhea during adolescence, followed by gait ataxia, retinal dystrophy, axonal neuropathy, and poor VVOR suppression manifested in the sixth decade (19). In addition, Tarnutzer et al. reported the case of a man presenting cerebellar ataxia, hypogonadotropic hypogonadism, retinal dystrophy, and poor VVOR suppression in the third decade (14).
Regarding the genetic variants identified in our series and in previous PNPLA6 cases, we confirmed that pathogenic variants are identified across all the gene regions, although the majority are identified in the Phospholipid Esterase Domain (EST) (Supplementary Table 2).
Not only different variants were associated with different presenting phenotypes, but even the same recurrent PNPLA6 gene variant can be found in subjects with different clinical manifestations.
For example, the most frequent variant worldwide, the c.2944_2947dupAGCC, has been previously identified in several cases, including our Patient 5. The patients presented different syndromes, including BN (4), GH (pt 5), Spastic Paraplegia (4), Oliver—Mc Farlane syndrome (6), Laurence—Moon syndrome (6), and Leber congenital amaurosis (7). The variability of clinical PNPLA6-phenotypes was further confirmed for the c.3403C > T (p.Arg1135Trp) variant. This PNPLA6 variant is very frequent in our patients (4/16 alleles, 25%), and it was previously identified in a Chinese patient affected by BN (10).
It has to be noted, however, that recurrent variants were variably associated in trans with different pathogenic PNPLA6 gene variants, thus suggesting a different and probably interrelating contribution of both PNPLA6 alleles to the final disease phenotype.
It has been demonstrated that PNPLA6 knock-out mice are embryonic lethal, and a pathogenic variant creating an early stop codon in human PNPLA6 has only been identified in compound heterozygous patients (28).
Truncating variants represent 18% (13/72) of all PNPLA6 gene variants, and homozygous patients always carried missense variants, localized both in the EST domain and in Cyclic Nucleotide Binding-Homology (CNB) regions (28).
Previous studies suggested that the localization of PNPLA6 variants could impact the development of different clinical phenotypes, with certain variants toward N terminal, causing preferentially ataxia and spastic paraplegia, while variants toward C terminal cause neurological symptoms with hypogonadism (26). Analyzing the different distributions and types of PNPLA6 variants within the protein domains, we observed that missense variants in the EST domain are more frequent in phenotypes associated with chorioretinal dystrophy (63–65%) (Figure 4). Truncating variants have the same frequency in all phenotypes (25–40%), except for cerebellar ataxia in which truncating variants were not described. However, it remains extremely challenging to provide more accurate genotype-phenotype information and a precise correlation with the NTE residual enzymatic activity.
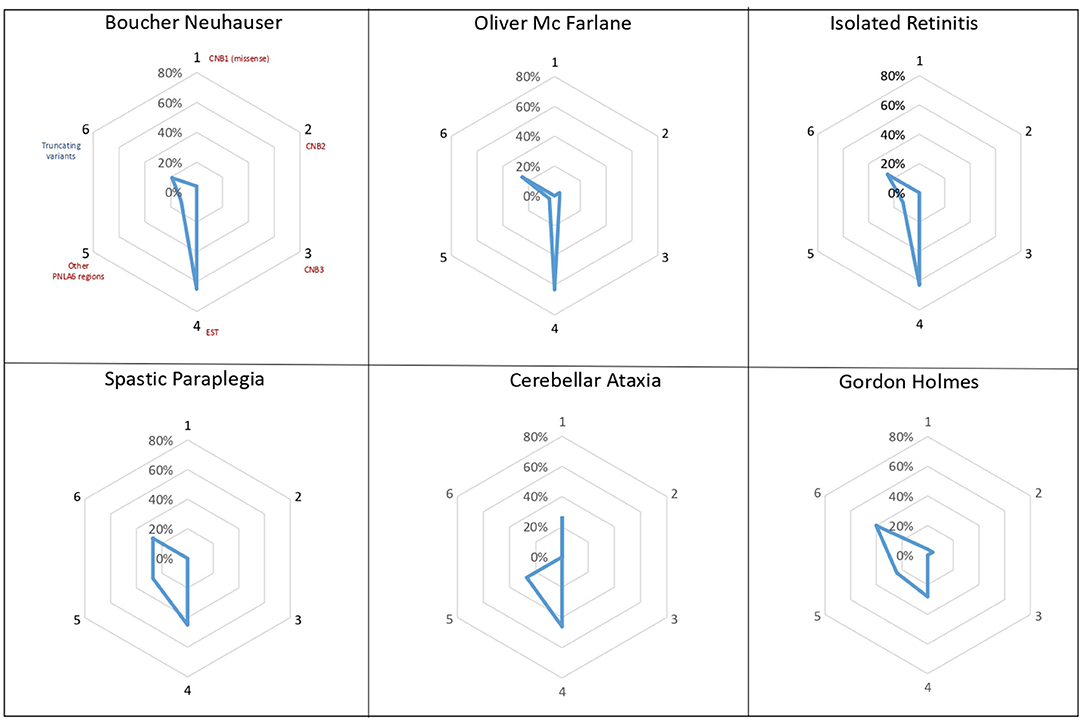
Figure 4. PNPLA6 gene variants. Diagrams illustrate the frequency of PNPLA6 gene variant types in patients described in the literature. Missense variants (in red) have been grouped according to their domain localization: the Cyclic Nucleotide Binding-Homology (CNB) domains (1-2-3), the Phospholipid Esterase (EST) domain (4), or other PNPLA6 regions (5). Truncating variants (in blue) have been reported as a unique group, irrespectively to their localization within the PNPLA6 gene.
In conclusion, PNPLA6 gene variants cause a wide spectrum of clinical syndromes and a great variability in the age of onset spanning from early infancy to adulthood. The complexity of the diagnosis and clinical management of the patients requires the collaboration of several specialists depending on the age of the patient and the specific clinical features (pediatrician, ophthalmologist, endocrinologist, geneticist, and adult neurologist).
The clinical presentation greatly influences the diagnostic workout. For example, early-onset cases are often evaluated by ophthalmologists and initially screened for genes associated with visual impairment. Juvenile- and adult-onset cases are likely to be evaluated by different physicians depending on the specific presenting symptom (29–31). We suggest that PNPLA6 gene screening should also be considered in the diagnostic workout of patients with late-onset cerebellar signs, including patients presenting a CANVAS-like phenotype, when pathological expansions in the RFC1 gene are not detected.
Data Availability Statement
Anonymized data which is not published within this article will be made available by request from any qualified investigator. The data presented in this study are deposited in Zenodo repository (https://zenodo.org), accession number 10.5281/zenodo.5793958. Requests to access these datasets should be directed to dWZmaWNpb3JpY2VyY2FAaXN0aXR1dG8tYmVzdGEuaXQ=.
Ethics Statement
The studies involving human participants were reviewed and approved by Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
LN, DD, SM, CM, and FT: study design, conception, execution, writing of the draft, and review manuscript. MF, ES, AC, AMo, SF, MM, SB, MB, and AMa: data collection and writing of the draft. All authors contributed to the article and approved the submitted version.
Funding
This work was partly funded by grants CP 20/2018 (Care4NeuroRare) from the Fondazione Regionale per la Ricerca Biomedica (FRRB) and RF-2018-12367768 from the Italian Ministry of Health to FT.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
LN, DD, SM, MF, CM, and FT are members of the European Reference Network for Rare Neurological Disorders (ERN-RND), Project ID No. 739510. SF is a member of the European Reference Network for Rare Neuromuscular Diseases (ERN EURO-NMD).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2021.793547/full#supplementary-material
References
1. Rainier S, Bui M, Mark E, Thomas D, Tokarz D, Ming L, et al. Neuropathy target esterase gene mutations cause motor neuron disease. Am J Hum Genet. (2008) 82:780–5. doi: 10.1016/j.ajhg.2007.12.018
2. Rainier S, Albers JW, Dyck PJ, Eldevik OP, Wilcock S, Richardson RJ, et al. Motor neuron disease due to neuropathy target esterase gene mutation: clinical features of the index families. Muscle Nerve. (2011) 43:19–25. doi: 10.1002/mus.21777
3. Yoon G, Baskin B, Tarnopolsky M, Boycott KM, Geraghty MT, Sell E, et al. Autosomal recessive hereditary spastic paraplegia-clinical and genetic characteristics of a well-defined cohort. Neurogenetics. (2013) 14:181–8. doi: 10.1007/s10048-013-0366-9
4. Synofzik M, Gonzalez MA, Lourenco CM, Coutelier M, Haack TB, Rebelo A, et al. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain. (2014) 137:69–77. doi: 10.1093/brain/awt326
5. Boucher BJ, Gibberd FB. Familial ataxia, hypogonadism and retinal degeneration. Acta Neurol Scand. (1969) 45:507–10. doi: 10.1111/j.1600-0404.1969.tb01261.x
6. Hufnagel RB, Arno G, Hein ND, Hersheson J, Prasad M, Anderson Y, et al. Neuropathy target esterase impairments cause Oliver-McFarlane and Laurence-Moon syndromes. J Med Genet. (2015) 52:85–94. doi: 10.1136/jmedgenet-2014-102856
7. Kmoch S, Majewski J, Ramamurthy V, Cao S, Fahiminiya S, Ren H, et al. Mutations in PNPLA6 are linked to photoreceptor degeneration and various forms of childhood blindness. Nat Commun. (2015) 6:5614. doi: 10.1038/ncomms6614
8. Patsi O, De Beaufort C, Kerschen P, Cardillo S, Soehn A, Rautenberg M, et al. new PNPLA6 mutation presenting as Oliver McFarlane syndrome. J Neurol Sci. (2018) 392:1–2. doi: 10.1016/j.jns.2018.06.016
9. Liu F, Ji Y, Li G, Xu C, Sun Y. Identification of Oliver-McFarlane syndrome caused by novel compound heterozygous variants of PNPLA6. Gene. (2020) 761:145027. doi: 10.1016/j.gene.2020.145027
10. Wu S, Sun Z, Zhu T, Weleber RG, Yang P, Wei X, et al. Novel variants in PNPLA6 causing syndromic retinal dystrophy. Exp Eye Res. (2021) 202:108327. doi: 10.1016/j.exer.2020.108327
11. Wiethoff S, Bettencourt C, Paudel R, Madon P, Liu YT, Hersheson J, et al. Pure cerebellar ataxia with homozygous mutations in the PNPLA6 gene. Cerebellum. (2017) 16:262–7. doi: 10.1007/s12311-016-0769-x
12. Chang P, He L, Wang Y, Heier C, Wu Y, Huang F. Characterization of the interaction of neuropathy target esterase with the endoplasmic reticulum and lipid droplets. Biomolecules. (2019) 9:848. doi: 10.3390/biom9120848
13. Topaloglu AK, Lomniczi A, Kretzschmar D, Dissen GA, Kotan LD, McArdle CA, et al. Loss-of-function mutations in PNPLA6 encoding neuropathy target esterase underlie pubertal failure and neurological deficits in Gordon Holmes syndrome. J Clin Endocrinol Metab. (2014) 99:2067–75. doi: 10.1210/jc.2014-1836
14. Tarnutzer AA, Gerth-Kahlert C, Timmann D, Chang DI, Harmuth F, Bauer P, et al. Boucher-Neuhäuser syndrome: cerebellar degeneration, chorioretinal dystrophy and hypogonadotropic hypogonadism: two novel cases and a review of 40 cases from the literature. J Neurol. (2015) 262:194–202. doi: 10.1007/s00415-014-7555-9
15. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
16. Donaldson L, Tarnopolsky MA, Martin JA, Rodriguez AR. Severe chorioretinal atrophy in Boucher-Neuhauser syndrome. Can J Ophthalmol. (2020) 55:e26–8. doi: 10.1016/j.jcjo.2019.07.001
17. Teive HAG, Camargo CHF, Sato MT, Shiokawa N, Boguszewski CL, Raskin S, et al. Different cerebellar ataxia phenotypes associated with mutations of the PNPLA6 gene in Brazilian patients with recessive ataxias. Cerebellum. (2018) 17:380–5. doi: 10.1007/s12311-017-0909-y
18. DeNaro BB, Dhrami-Gavazi E, Rubaltelli DM, Freund KB, Lee W, Yannuzzi LA, et al. Chorioretinal changes in a genetically confirmed case of boucher-neuhäuser syndrome. Retin Cases Brief Rep. (2021) 15:179–84. doi: 10.1097/ICB.0000000000000769
19. Deik A, Johannes B, Rucker JC, Sánchez E, Brodie SE, Deegan E, et al. Compound heterozygous PNPLA6 mutations cause Boucher-Neuhauser syndrome with late-onset ataxia. J Neurol. (2014) 261:2411–23. doi: 10.1007/s00415-014-7516-3
20. Zheng R, Zhao Y, Wu J, Wang Y, Liu JL, Zhou ZL, et al. A novel PNPLA6 compound heterozygous mutation identified in a Chinese patient with Boucher-Neuhauser syndrome. Mol Med Rep. (2018) 18:261–7. doi: 10.3892/mmr.2018.8955
21. Koh K, Kobayashi F, Miwa M, Shindo K, Isozaki E, Ishiura H, et al. Novel mutations in the PNPLA6 gene in Boucher-Neuhauser syndrome. J Hum Genet. (2015) 60:217–20. doi: 10.1038/jhg.2015.3
22. Langdahl JH, Frederiksen AL, Nguyen N, Brusgaard K, Juhl CB. Boucher Neuhauser Syndrome - A rare cause of inherited hypogonadotropic hypogonadism. A case of two adult siblings with two novel mutations in PNPLA6. Eur J Med Genet. (2017) 60:105–9. doi: 10.1016/j.ejmg.2016.11.003
23. Dogan M, Eröz R, Öztürk E. Chorioretinal dystrophy, hypogonadotropic hypogonadism, and cerebellar ataxia: Boucher-Neuhauser syndrome due to a homozygous (c.3524C>G (p.Ser1175Cys)) variant in PNPLA6 gene. Ophthalmic Genet. (2021) 42:276–82. doi: 10.1080/13816810.2021.1894461
24. Locci S, Bianchi S, Tessa A, Santorelli FM, Mignarri A. Gordon Holmes syndrome caused by two novel mutations in the PNPLA6 gene. Clin Neurol Neurosurg. (2021) 207:106763. doi: 10.1016/j.clineuro.2021.106763
25. Salgado P, Carvalho R, Brandão AF, Jorge P, Ramos C, Dias D, et al. Gordon Holmes syndrome due to compound heterozygosity of two new PNPLA6 variants - A diagnostic challenge. eNeurological Sci. (2018) 14:9–12. doi: 10.1016/j.ensci.2018.11.022
26. Sen K, Finau M, Ghosh P. Bi-allelic variants in PNPLA6 possibly associated with Parkinsonian features in addition to spastic paraplegia phenotype. J Neurol. (2020) 267:2749–53. doi: 10.1007/s00415-020-10028-w
27. Emekli AS, Samanci B, Simşir G, Hanagasi HA, Gürvit H, Bilgiç B, et al. novel PNPLA6 mutation in a Turkish family with intractable Holmes tremor and spastic ataxia. Neurol Sci. (2021) 42:1535–9. doi: 10.1007/s10072-020-04869-6
28. Sunderhaus ER, Law AD, Kretzschmar D. Disease-associated PNPLA6 mutations maintain partial functions when analyzed in drosophila. Front Neurosci. (2019) 13:1207. doi: 10.3389/fnins.2019.01207
29. Burdon KP. The utility of genomic testing in the ophthalmology clinic: a review. Clin Exp Ophthalmol. (2021) 49:615–25. doi: 10.1111/ceo.13970
30. Millar AC, Faghfoury H, Bieniek JM. Genetics of hypogonadotropic hypogonadism. TranslAndrol Urol. (2021) 10:1401–9. doi: 10.21037/tau.2020.03.33
Keywords: cerebellar ataxia, spastic paraplegia, hypogonadotropic hypogonadism, chorioretinal dystrophy, Gordon Holmes syndrome, Boucher Neuhauser syndrome, Oliver Mc Farlane syndrome
Citation: Nanetti L, Di Bella D, Magri S, Fichera M, Sarto E, Castaldo A, Mongelli A, Baratta S, Fenu S, Moscatelli M, Bonati MT, Martinuzzi A, Mariotti C and Taroni F (2022) Multifaceted and Age-Dependent Phenotypes Associated With Biallelic PNPLA6 Gene Variants: Eight Novel Cases and Review of the Literature. Front. Neurol. 12:793547. doi: 10.3389/fneur.2021.793547
Received: 12 October 2021; Accepted: 02 December 2021;
Published: 06 January 2022.
Edited by:
Mathias Toft, University of Oslo, NorwayReviewed by:
Kuntal Sen, Children's National Hospital, United StatesThiago Cardoso Vale, Juiz de Fora Federal University, Brazil
Copyright © 2022 Nanetti, Di Bella, Magri, Fichera, Sarto, Castaldo, Mongelli, Baratta, Fenu, Moscatelli, Bonati, Martinuzzi, Mariotti and Taroni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caterina Mariotti, Y2F0ZXJpbmEubWFyaW90dGlAaXN0aXR1dG8tYmVzdGEuaXQ=