 Udo Bartsch1
Udo Bartsch1 Stephan Storch2*
Stephan Storch2*- 1Department of Ophthalmology, Experimental Ophthalmology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 2University Children's Research@Kinder-UKE, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
The neuronal ceroid lipofuscinoses (NCLs) are a group of childhood-onset neurodegenerative lysosomal storage disorders mainly affecting the brain and the retina. In the NCLs, disease-causing mutations in 13 different ceroid lipofuscinoses genes (CLN) have been identified. The clinical symptoms include seizures, progressive neurological decline, deterioration of motor and language skills, and dementia resulting in premature death. In addition, the deterioration and loss of vision caused by progressive retinal degeneration is another major hallmark of NCLs. To date, there is no curative therapy for the treatment of retinal degeneration and vision loss in patients with NCL. In this review, the key findings of different experimental approaches in NCL animal models aimed at attenuating progressive retinal degeneration and the decline in retinal function are discussed. Different approaches, including experimental enzyme replacement therapy, gene therapy, cell-based therapy, and immunomodulation therapy were evaluated and showed encouraging therapeutic benefits. Recent experimental ocular gene therapies in NCL animal models with soluble lysosomal enzyme deficiencies and transmembrane protein deficiencies have shown the strong potential of gene-based approaches to treat retinal dystrophies in NCLs. In CLN3 and CLN6 mouse models, an adeno-associated virus (AAV) vector-mediated delivery of CLN3 and CLN6 to bipolar cells has been shown to attenuate the retinal dysfunction. Therapeutic benefits of ocular enzyme replacement therapies were evaluated in CLN2 and CLN10 animal models. Since brain-targeted gene or enzyme replacement therapies will most likely not attenuate retinal neurodegeneration, there is an unmet need for treatment options additionally targeting the retina in patients with NCL. The long-term benefits of these therapeutic interventions aimed at attenuating retinal degeneration and vision loss in patients with NCL remain to be investigated in future clinical studies.
Introduction
This review focuses on experimental approaches aimed at attenuating the progression of retinal degeneration in the different animal models of neuronal ceroid lipofuscinoses (NCLs). The NCLs are caused by defects in 13 different genes (CLN1–CLN8, CLN10–CLN14) encoding soluble lysosomal proteins (CLN1, CLN2, CLN5, CLN10, CLN11, and CLN13), membrane proteins located in the ER (CLN6 and CLN8), ER-Golgi intermediate compartment (CLN8) or lysosomes (CLN3, CLN7, and CLN12), and cytosolic proteins associated with synaptic vesicles (CLN4) or the plasma membrane (CLN14, Table 1) (1). The majority of affected gene products play important roles for lysosomal biogenesis and function: soluble lysosomal enzymes involved in lysosomal protein degradation (palmitoyl-protein thioesterase 1/PPT1, tripeptidyl-peptidase 1/TPP1, cathepsin D, and cathepsin F), soluble lysosomal proteins with unknown function (CLN5), and polytopic lysosomal membrane proteins (CLN3, CLN7, and CLN12). The primary function of the lysosomal membrane protein CLN3 is unclear (2). CLN7 was recently shown to function as an endolysosomal chloride channel (3). CLN6 and CLN8 form a complex in the endoplasmic reticulum (ER) which is required for the biosynthetic transport of a subset of newly synthesized soluble lysosomal proteins from the ER to the Golgi apparatus (4, 5). Biochemically, defects in NCL genes lead to lysosomal and autophagic dysfunction and subsequent accumulation of autofluorescent ceroid lipopigments. Based on the age of patients at the onset of first symptoms, the NCLs have been classified into congenital, infantile, late infantile, juvenile, and adult NCL phenotypes (6, 7). The clinical symptoms include seizures, progressive neurological decline, deterioration of motor and language skills, and dementia resulting in premature death (8). All disease-causing mutations and sequence variations in the CLN genes are summarized in the NCL mutation database (https://www.ucl.ac.uk/ncl-disease), and genotype-phenotype correlations are discussed in a recent review (9). With the exception of CLN4, CLN12, and CLN13 disease, deterioration and loss of vision is another major hallmark of NCLs (Table 1) (10). However, the deterioration of vision does not always appear as the first symptom in different NCLs (11). In rare cases, patients with CLN3 and CLN7 disease present with non-syndromic retina degeneration (12–14). To date, there are no curative therapies for the treatment of neurodegeneration in the brain and the retina, and patients rely on palliative treatment (15). An enzyme replacement therapy (ERT) using the intracerebroventricular infusions of recombinant TPP1 (cerliponase alfa) every 2 weeks has been shown to decelerate the disease progression and was recently approved for the treatment of patients with CLN2 disease (16).
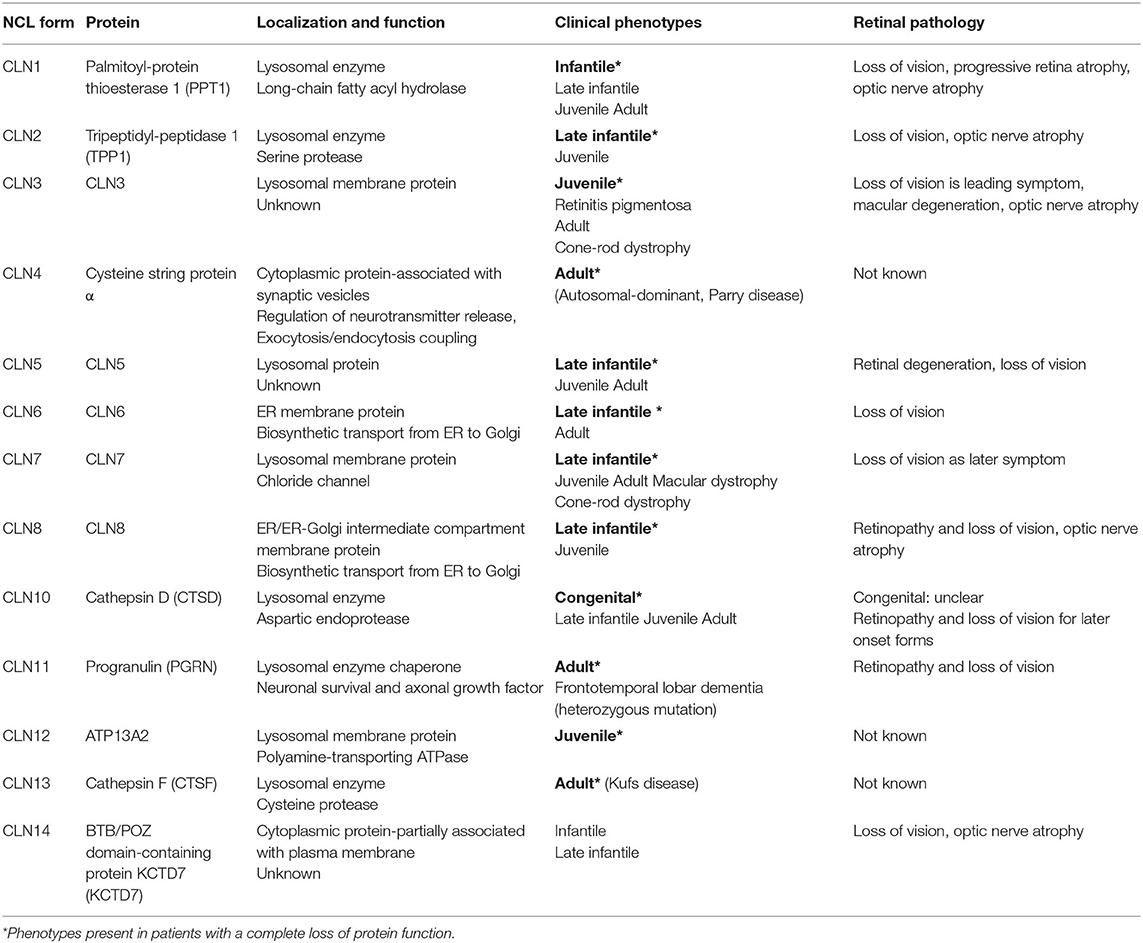
Table 1. Summary of NCL forms, localization and function of gene products and retinal pathology in human patients.
Naturally occurring and gene-targeted mouse models and large animal models, such as dogs, sheep, and macaques, allowed the age-dependent morphological, biochemical, and functional analyses of retinal pathologies in NCL (17, 18). The pathomechanisms leading to the degeneration of neuronal cells in different retinal cell layers and the loss of retinal function are not well understood. CLN3 has been suggested to be required for the phagocytosis of photoreceptor outer segments by retinal pigment epithelial (RPE) cells (19). In line with this notion, lysosomal storage and increased numbers of mature autophagosomes and basal phagolysosomes were found in the retinal pigment epithelium of Cln3Δex1−6 mice, a CLN3 mouse model (20).
Preclinical studies targeting the brain of NCL animal models using ERT and gene therapy demonstrated a delayed onset and an attenuated progression of neuroinflammation and neurodegeneration (21). However, intracerebroventricular ERT and brain-targeted gene therapies are unlikely to prevent or attenuate neurodegeneration in the retina (15, 22). Therefore a combination of the brain- and eye-directed therapy might be required to prevent neurodegeneration in both the brain and the retina (23).
Gene Therapy
Gene therapies for lysosomal storage disorders and NCLs are designed to correct the primary genetic defect (21). Experimental adeno-associated virus (AAV) vector-based brain-targeted gene therapies have been evaluated in animal models for CLN1, CLN2, CLN3, CLN5, CLN6, CLN7, CLN8, CLN10, and CLN11 disease (21, 24, 25). Based on the promising results of the preclinical studies, some of these brain-targeted gene therapies are currently being tested in clinical trials in CLN2, CLN3, and CLN6 patients (21). Ocular gene therapy is an emerging field. In general, gene therapies for soluble lysosomal enzyme deficiencies involve the cross-correction of non-transduced cells, and therapeutic benefits might thus be achieved with a relatively low number of successfully transduced cells. Gene therapies for membrane protein deficiencies, in comparison, will most likely require higher numbers of transduced cells to achieve therapeutic benefits (23). The most widely used viral vectors for ocular gene therapy are based on AAVs since they mediate stable, long-term transgene expression, and produce only minor immune responses (26). Preclinical ocular gene therapy studies in NCL animal models have mainly used intravitreally administered AAV vectors with different serotypes prior to the onset of the retinal pathology (27). Ocular gene therapy studies have been performed in animal models targeting soluble lysosomal protein deficiencies (CLN1, CLN5, CLN10, and CLN11) but also transmembrane protein defects (CLN3 and CLN6) (Table 2) and showed encouraging results. To date, the long-term therapeutic benefits of ocular gene therapy in human patients with NCL are unknown since no clinical trials are ongoing or have been completed (21).

Table 2. Preclinical ocular-targeted gene therapies for NCLs.
CLN1 Disease
The CLN1 mouse model is characterized by the progressive loss of photoreceptors starting at 3 months of age and decreased retinal functions compared with wild type mice as measured by electroretinogram (ERG) recordings (28). Intravitreal administration of an AAV2 vector carrying human PPT1 cDNA led to a five-fold increase in PPT1 enzymatic activities compared with age-matched wild type mice (28). Although a better organization of the photoreceptor layer and improved retinal function as measured by ERG recordings were detected in AAV2-PPT1 treated mutant mice, the progression of the retinal dystrophy was only retarded but not completely prevented.
CLN2 Disease
A naturally occurring TPP1-deficient Dachshund model recapitulates the key features of human CLN2 disease, including ataxia, tremor, progressive brain atrophy, loss of vision, and a reduced life span (34). For CLN2 disease, there are no reports on the efficacy of experimental ocular gene therapies in animal models. A single pre-symptomatic intraventricular injection of an AAV2 vector harboring canine TPP1 (cTPP1) cDNA into the CLN2 Dachshund model led to the reduced storage of autofluorescent material and decreased astrocytosis in the brain, and delayed onset of cognitive deficits and extended lifespan of the mutant dogs (35). However, TPP1 was not detected in photoreceptors and retinal pigment epithelial cells. In the treated CLN2 mutant dogs, the retinal degeneration and reduction of ERG b-wave amplitudes were not prevented by the brain-directed administration of AAV2-cTPP1 compared with untreated dogs (36). These data suggest that AAV-mediated brain-targeted gene therapy is not sufficient to treat the retinal degeneration and loss of vision in CLN2 dog models.
CLN3 Disease
The Cln3 knock-in mouse model (Cln3Δex7/8) genetically recapitulates the 1 Kb deletion mutation of exons 7 and 8 found in 85% of human patients with CLN3 disease (37). Retinal degeneration in Cln3Δex7/8 mice is relatively mild leading to the loss of bipolar cells and a progressive reduction of the b-wave amplitudes beginning from 12 months of age (29, 38). These data demonstrate a progressive reduction of inner retinal function in the retinas of mutant mice. An ocular gene therapy using the intravitreal injections of an AAV7m8 vector harboring human CLN3 in postnatal Cln3Δex7/8 mice led to the improved survival of bipolar cells and retinal function (29). In contrast, a subretinal injection using an AAV2/8-mCln3 vector targeting photoreceptors and retinal pigment epithelial cells did not attenuate the loss of bipolar cells and the decline in inner retinal function suggesting that the expression of CLN3 in photoreceptors was not therapeutic in the mutant mice. Of note, the Cln3Δex7/8 mice do not fully recapitulate the retinal phenotype observed in patients with CLN3 disease. The number of photoreceptors was unchanged in the 15-month-old Cln3Δex7/8 mice whereas photoreceptors in the retinal postmortem tissues of patients with CLN3 disease are almost completely lost (7).
CLN5 Disease
Murray and colleagues reported on the first successful intravitreal gene therapy in a large NCL animal model (30). They used the naturally occurring CLN5-deficient Borderdale sheep which recapitulates the key features of human CLN5 disease, such as motor and cognitive decline, progressive neurodegeneration in the brain and the retina, and loss of vision (39). The sheep eye represents a good model for the human eye because of its similar morphology and size. CLN5-deficient Borderdale sheep received a single intravitreal injection of an AAV9 vector harboring ovine CLN5 cDNA at 3 months of age and were analyzed at 18 months of age (30). AAV9-CLN5-treated eyes showed minor lysosomal storage and neuroinflammation and intact retinal layers with a thickness comparable to that of the control sheep (30). In addition, the measurements of retinal functions in treated eyes showed ERG amplitudes nearly comparable with amplitudes in wild type control sheep (30).
CLN6 Disease
The Cln6 nclf mouse is a naturally occurring mouse model of CLN6 disease (40). The main pathological features in the retina of Cln6 nclf mice include the loss of photoreceptors, early-onset reactive gliosis, accumulation of lysosomal storage material in multiple retinal cell layers, and the increased expression of soluble lysosomal enzymes (41). Surprisingly, an AAV2/8 vector-mediated gene transfer of human or mouse CLN6 did not prevent the loss of photoreceptors and did not preserve photoreceptor functions in Cln6 nclf mice. In contrast, an AAV2.7m8 vector-mediated bipolar cell-specific expression of CLN6 prevented the loss of photoreceptors and preserved their function (31). Data indicated that Cln6 deficiency in bipolar cells is the cause of photoreceptor degeneration in the Cln6 nclf mouse. White and colleagues reported for the first time that brain-targeted gene therapy in Cln6 nclf mice attenuated retinal pathology. Intracerebroventricular gene therapy in Cln6 nclf mice reduced the pathology in visual centers of the brain and in the retina (42). The intracerebroventricular injection of an AAV9-CBV-CLN6 vector into postnatal day 1 (P1) Cln6 nclf mice reduced the degeneration of photoreceptors in 3-, 6-, and 9-month-old mice compared with untreated controls. In striking contrast, a brain-directed AAV-mediated expression of cathepsin D in a CLN10 mouse model prevented the accumulation of ceroid lipofuscin, the activation of microglia, and neurodegeneration in brain tissues, but not the rapidly progressing retinal degeneration (43). Intravitreal injection of an AAV9 vector encoding CLN6 into a naturally occurring CLN6-deficient South Hampshire sheep model led to a minor reduction of lysosomal storage and retinal atrophy and had no beneficial effects on retinal function as indicated by unaltered ERG amplitudes compared with untreated contralateral eyes (30).
CLN10 Disease
The CLN10 mouse deficient in the lysosomal protease cathepsin D (CTSD) is a model for the most severe NCL form, congenital NCL. Cathepsin D knockout (Ctsd ko) mice are characterized by an early-onset loss of photoreceptor cells and a subsequent loss of all other retinal nerve cell types, the accumulation of storage material, lysosomal dysfunction, reduced autophagic flux, reactive astrogliosis and microgliosis, and a shortened lifespan with premature death at P26 (44, 45). The Ctsd ko mouse is a valuable model to study the efficacy of experimental ocular therapies due to the early-onset and most rapid progression of retinal degeneration in all NCL forms. Intravitreal administration of an AAVsh10 vector harboring mouse Ctsd transduced retinal glial cells and RPE cells (32). Biochemical and morphological analyses of the AAV-treated retinas revealed a restoration of CTSD enzymatic activities close to wild type levels, a complete reduction of lysosomal storage material, the absence of lysosomal hypertrophy, and the preservation of photoreceptor and rod bipolar cells. However, this gene therapy study was unable to clarify whether the retinal function was preserved due to ethical issues related to experiments on severely affected animals at the end stage of the disease.
CLN11 Disease
In a mouse model for CLN11 disease, the progranulin knockout (Pgrn ko) mouse, autofluorescent storage material accumulation, and the degeneration of photoreceptors and retinal ganglion cells become apparent in 12-month-old mutant mice (46). In a recent study, the therapeutic benefits of intravenous administration of an AAV9.2YF-Pgrn vector were compared with the intravitreal delivery of an AAV2.7m8-Pgrn vector into Pgrn ko mice (33). Systemically administered AAV9 vectors cross the blood-retina- and blood-brain-barriers until 7 days of age. Intravenous delivery of an AAV9.2YF vector encoding murine PGRN into P3 or P4 Pgrn ko mice led to a reduction of autofluorescent ceroid lipopigments and attenuated the thinning of the outer nuclear layer and the total retina in 12-month-old mutant mice (33). Interestingly, intravitreal injection of an AAV2.7m8-Pgrn vector into 1- or 6-month-old Pgrn ko mice reduced lipofuscin lipopigments, decreased microglial infiltration, but did not attenuate retinal neurodegeneration. These data suggest that both the route and time of AAV administration are crucial to achieving therapeutic benefits in the retina of Pgrn ko mice.
Enzyme Replacement Therapy
Ocular enzyme replacement therapies (ERTs) rely on the intravitreal administration of a recombinant soluble lysosomal enzyme and its uptake via mannose 6-phosphate receptors, delivery to lysosomes, and cross-correction (47). Therapeutic benefits of ocular ERTs have been tested in animal models for CLN2 and CLN10 diseases (15).
Periodic intravitreal injections of recombinant TPP1 starting at 12 weeks of age into the CLN2 Dachshund dog model led to decreased neurodegeneration in the inner nuclear layer and inhibited declines in ERG amplitudes (48). Intravitreal TPP1 administration also prevented focal retinal detachments in the mutant dogs. A single intravitreal injection of recombinant CTSD into P7 and P14 CLN10 mutant mice partially attenuated lysosomal dysfunction and reduced reactive microgliosis but failed to prevent the photoreceptor loss and retinal degeneration (49). The data suggest that the regular intravitreal administration of a recombinant lysosomal enzyme may be a therapeutic option to treat retinal degeneration and vision loss at least in some NCL forms. A new clinical trial (Clinical Trial gov Identifier: NCT05152914) is currently enrolling patients to test the therapeutic efficacy of intravitreal ERT (Cerliponase alfa) in CLN2 disease.
Cell-Based Therapy
Therapeutic benefits of cell-based ERTs were evaluated in NCL animal models (15). Cell transplantation into the retina of NCL models deficient in soluble lysosomal proteins is based on the rationale that the grafted cells secrete the missing lysosomal proteins (donor cells) followed by their internalization via mannose 6-phosphate receptors by surrounding defective acceptor cells (47). The lysosomal enzyme-mannose 6-phosphate receptor complexes are internalized and lysosomal enzymes are finally targeted to lysosomes where they are proteolytically activated. Preclinical experiments evaluated the benefits of transplanted stem cells overexpressing the missing lysosomal enzyme. Stem cells transduced with an AAV2-vector carrying the human PPT1 cDNA were intravitreally implanted at early disease stages into the CLN2 Dachshund model (50). A single injection of these modified stem cells inhibited the pathological changes in retinal morphology and retinal function suggesting that genetically modified stem cells might serve as useful vehicles for a long-term intraocular administration of the soluble lysosomal protein in NCLs. In another approach, neural stem cells that were transduced ex vivo with a lentiviral vector harboring murine Ctsd cDNA were intravitreally implanted into the CLN10 mouse model of the CLN10 mouse model (32). In treated retinas, the restoration of CTSD enzymatic activities to 44% of wild type levels, a partial decrease of lysosomal storage material, and reduced microgliosis and astrocytosis compared with untreated Ctsd knockout retinas were detected. However, the degeneration of different retinal cell types was not prevented by the implanted stem cells. In summary, the data suggest that intravitreal injection of genetically modified stem cells may be an encouraging approach to attenuate retinal degeneration for some NCL forms with soluble lysosomal enzyme deficiencies.
Immunomodulation
Previous studies showed that the genetic inhibition of the adaptive or innate immune system led to disease-ameliorating effects in the CNS of Ppt1 ko/Cln3Δex1−6 ko and Ppt1 ko mice, respectively (51, 52). In line with these findings, the treatment of Ppt1 ko or Cln3Δex1−6 ko mice with immunosuppressive drugs showed therapeutic benefits (53, 54). Oral administration of the immunomodulators fingolimod and teriflunomide prevented retinal thinning in Ppt1 ko mice and attenuated retinal thinning in Cln3Δex1−6 ko mice (55). In a genetically modified Cln3Δex7/8 knock-in mouse susceptible to light damage, light exposure resulted in pathological changes, including retinal neurodegeneration, activation of microglia, and accumulation of autofluorescent storage material (56). Treatment of the mutant mice with the antibiotic and anti-inflammatory drug minocycline prior to light stress led to reduced photoreceptor loss and decreased amounts of autofluorescent storage material (56). Based on the strong reactive gliosis present in Cln6 nclf retinas, mutant mice were treated with the natural immunomodulators curcumin and docosahexanoic acid [DHA, (57)]. In the curcumin- and DHA-treated Cln6 nclf mice, reactive gliosis was attenuated and the decline in visual acuity and ERG amplitudes was delayed when compared with untreated mutant mice.
Discussion
Retinal degeneration and loss of vision are among the major hallmarks of NCLs of NCLs. With the exception of one study on Cln6 nclf mice, brain-targeted therapies in NCL animal models had no therapeutic impact on retinal degeneration and loss of retinal function. Therefore, there is an unmet need to design novel eye-targeted therapies. The therapeutic efficacy of eye-targeted experimental therapies in NCL animal models, including gene therapy, enzyme replacement therapy, cell-based therapy, and immunomodulation, were evaluated in the past. Recent experimental ocular gene therapies on animal models with soluble lysosomal enzyme deficiencies (CLN1, CLN5, CLN10, and CLN11) and transmembrane protein deficiencies (CLN3 and CLN6) have shown the strong potential of gene therapeutic approaches to effectively treat NCL-related retinopathies. A major breakthrough in the experimental gene therapy approaches was the identification of the specific cell types that have to be targeted to achieve therapeutic benefit. In the CLN3 and CLN6 mouse models, the AAV-mediated bipolar cell-specific delivery of CLN3 and CLN6 was successful in preventing the loss of photoreceptors and bipolar cells, respectively, and to partly preserve retinal function. Furthermore, the intravitreal injection of an AAV9-CLN5 vector into a CLN5 sheep model largely prevented retinal degeneration and loss of retinal function. Finally, results from a recent study suggest that an AAV-mediated CTSD expression in the retina of a CLN10 mouse model is more potent in preventing retinal degeneration than intravitreal ERT mediated by the injections of recombinant CTSD or by transplantation of neural stem cells overexpressing CTSD. While the results of the eye-targeted therapies are encouraging, most studies have started the treatment prior to the onset of the retinal pathology. Future work thus needs to evaluate whether the treatment strategies are still effective when they are started at the initial or advanced stages of the retinal dystrophy. For clinical applications, gene therapies have the advantage of single dosing compared with enzyme replacement therapies which require repeated administration. Combined therapies targeting the brain and the retina separately may attenuate neurological symptoms and additionally vision loss in patients with NCL. The long-term benefits of these experimental ocular treatment options have to be evaluated in patients with NCL in future clinical studies.
Author Contributions
UB and SS interpreted the data and wrote the review. Both authors contributed to the article and approved the submitted version.
Funding
The laboratory works of the authors were supported by funding from the Mila's Miracle Foundation (SS), the Stiftung zur Förderung der Universitätsmedizin Hamburg, and the Ernst and Berta Grimmke Stiftung (UB).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mole SE, Cotman SL. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim Biophys Acta. (2015) 1852:2237–41. doi: 10.1016/j.bbadis.2015.05.011
2. Cotman SL, Lefrancois S. CLN3, at the crossroads of endocytic trafficking. Neurosci Lett. (2021) 762:136117. doi: 10.1016/j.neulet.2021.136117
3. Wang Y, Zeng W, Lin B, Yao Y, Li C, Hu W, et al. CLN7 is an organellar chloride channel regulating lysosomal function. Sci Adv. (2021) 7:eabj9608. doi: 10.1126/sciadv.abj9608
4. Bajaj L, Sharma J, di Ronza A, Zhang P, Eblimit A, Pal R, et al. A CLN6-CLN8 complex recruits lysosomal enzymes at the ER for Golgi transfer. J Clin Invest. (2020) 130:4118–32. doi: 10.1172/JCI130955
5. di Ronza A, Bajaj L, Sharma J, Sanagasetti D, Lotfi P, Adamski CJ, et al. CLN8 is an endoplasmic reticulum cargo receptor that regulates lysosome biogenesis. Nat Cell Biol. (2018) 20:1370–7. doi: 10.1038/s41556-018-0228-7
6. Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology. (2012) 79:183–91. doi: 10.1212/WNL.0b013e31825f0547
7. Mole SE, Williams RE, Goebel HH. The Neuronal Ceroid Lipofuscinoses (Batten disease). 2nd ed Oxford: Oxford University Press. (2011).
8. Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases - clinical perspectives. Biochim Biophys Acta. (2013) 1832:1801–6. doi: 10.1016/j.bbadis.2013.04.008
9. Gardner E, Mole SE. The genetic basis of phenotypic heterogeneity in the neuronal ceroid lipofuscinoses. Front Neurol. (2021) 12:754045. doi: 10.3389/fneur.2021.754045
10. Specchio N, Ferretti A, Trivisano M, Pietrafusa N, Pepi C, Calabrese C, et al. Neuronal ceroid lipofuscinosis: potential for targeted therapy. Drugs. (2021) 81:101–23. doi: 10.1007/s40265-020-01440-7
11. Radke J, Stenzel W, Goebel HH. Human NCL neuropathology. Biochim Biophys Acta. (2015) 1852:2262–6. doi: 10.1016/j.bbadis.2015.05.007
12. Ku CA, Hull S, Arno G, Vincent A, Carss K, Kayton R, et al. Detailed clinical phenotype and molecular genetic findings in CLN3-associated isolated retinal degeneration. JAMA Ophthalmol. (2017) 135:749–60. doi: 10.1001/jamaophthalmol.2017.1401
13. Khan KN, El-Asrag ME, Ku CA, Holder GE, McKibbin M, Arno G, et al. Specific alleles of CLN7/MFSD8, a protein that localizes to photoreceptor synaptic terminals, cause a spectrum of nonsyndromic retinal dystrophy. Invest Ophthalmol Vis Sci. (2017) 58:2906–14. doi: 10.1167/iovs.16-20608
14. Roosing S, van den Born LI, Sangermano R, Banfi S, Koenekoop RK, Zonneveld-Vrieling MN, et al. Mutations in MFSD8, encoding a lysosomal membrane protein, are associated with nonsyndromic autosomal recessive macular dystrophy. Ophthalmology. (2015) 122:170–9. doi: 10.1016/j.ophtha.2014.07.040
15. Kohlschütter A, Schulz A, Bartsch U, Storch S. Current and emerging treatment strategies for neuronal ceroid lipofuscinoses. CNS Drugs. (2019) 33:315–25. doi: 10.1007/s40263-019-00620-8
16. Schulz A, Ajayi T, Specchio N, de Los Reyes E, Gissen P, Ballon D, et al. Study of intraventricular Cerliponase alfa for CLN2 Disease. N Engl J Med. (2018) 378:1898–907. doi: 10.1056/NEJMoa1712649
17. Bond M, Holthaus SM, Tammen I, Tear G, Russell C. Use of model organisms for the study of neuronal ceroid lipofuscinosis. Biochim Biophys Acta. (2013) 1832:1842–65. doi: 10.1016/j.bbadis.2013.01.009
18. Shacka JJ. Mouse models of neuronal ceroid lipofuscinoses: useful pre-clinical tools to delineate disease pathophysiology and validate therapeutics. Brain Res Bull. (2012) 88:43–57. doi: 10.1016/j.brainresbull.2012.03.003
19. Tang C, Han J, Dalvi S, Manian K, Winschel L, Volland S, et al. A human model of Batten disease shows role of CLN3 in phagocytosis at the photoreceptor-RPE interface. Commun Biol. (2021) 4:161. doi: 10.1038/s42003-021-01682-5
20. Wavre-Shapton ST, Calvi AA, Turmaine M, Seabra MC, Cutler DF, Futter CE, et al. Photoreceptor phagosome processing defects and disturbed autophagy in retinal pigment epithelium of Cln3Δex1-6 mice modelling juvenile neuronal ceroid lipofuscinosis (Batten disease). Hum Mol Genet. (2015) 24:7060–74. doi: 10.1093/hmg/ddv406
21. Liu W, Kleine-Holthaus SM, Herranz-Martin S, Aristorena M, Mole SE, Smith AJ, et al. Experimental gene therapies for the NCLs. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165772. doi: 10.1016/j.bbadis.2020.165772
22. Mole SE, Anderson G, Band HA, Berkovic SF, Cooper JD, Kleine Holthaus SM, et al. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. Lancet Neurol. (2019) 18:107–16. doi: 10.1016/S1474-4422(18)30368-5
23. Murray SJ, Mitchell NL. Ocular therapies for neuronal ceroid lipofuscinoses: more than meets the eye. Neural Regen Res. (2022) 17:1755–6. doi: 10.4103/1673-5374.332148
24. Johnson TB, White KA, Brudvig JJ, Cain JT, Langin L, Pratt MA, et al. AAV9 gene therapy increases lifespan and treats pathological and behavioral abnormalities in a mouse model of CLN8-Batten Disease. Mol Ther. (2021) 29:162–75. doi: 10.1016/j.ymthe.2020.09.033
25. Chen X, Dong T, Hu Y, Shaffo FC, Belur NR, Mazzulli JR, et al. AAV9/MFSD8 gene therapy is effective in preclinical models of neuronal ceroid lipofuscinosis type 7 disease. J Clin Invest. (2022) 22:46228. doi: 10.1172/JCI146286
26. Askou AL, Jakobsen TS, Corydon TJ. Retinal gene therapy: an eye-opener of the 21st century. Gene Ther. (2021) 28:209–16. doi: 10.1038/s41434-020-0168-2
27. Schön C, Biel M, Michalakis S. Retinal gene delivery by adeno-associated virus (AAV) vectors: strategies and applications. Eur J Pharm Biopharm. (2015) 95:343–52. doi: 10.1016/j.ejpb.2015.01.009
28. Griffey M, Macauley SL, Ogilvie JM, Sands MS. AAV2-mediated ocular gene therapy for infantile neuronal ceroid lipofuscinosis. Mol Ther. (2005) 12:413–21. doi: 10.1016/j.ymthe.2005.04.018
29. Kleine Holthaus SM, Aristorena M, Maswood R, Semenyuk O, Hoke J, Hare A, et al. Gene therapy targeting the inner retina rescues the retinal phenotype in a mouse model of CLN3 Batten disease. Hum Gene Ther. (2020) 31:709–18. doi: 10.1089/hum.2020.038
30. Murray SJ, Russell KN, Melzer TR, Gray SJ, Heap SJ, Palmer DN, et al. Intravitreal gene therapy protects against retinal dysfunction and degeneration in sheep with CLN5 Batten disease. Exp Eye Res. (2021) 207:108600. doi: 10.1016/j.exer.2021.108600
31. Kleine Holthaus SM, Ribeiro J, Abelleira-Hervas L, Pearson RA, Duran Y, Georgiadis A, et al. Prevention of photoreceptor cell loss in a Cln6(nclf) mouse model of Batten disease requires CLN6 gene transfer to bipolar cells. Mol Ther. (2018) 26:1343–53. doi: 10.1016/j.ymthe.2018.02.027
32. Liu J, Bassal M, Schlichting S, Braren I, Di Spiezio A, Saftig P, et al. Intravitreal gene therapy restores the autophagy-lysosomal pathway and attenuates retinal degeneration in cathepsin D-deficient mice. Neurobiol Dis. (2022) 164:105628. doi: 10.1016/j.nbd.2022.105628
33. Zin EA, Han D, Tran J, Morisson-Welch N, Visel M, Kuronen M, et al. Outcomes of progranulin gene therapy in the retina are dependent on time and route of delivery. Mol Ther Methods Clin Dev. (2021) 22:40–51. doi: 10.1016/j.omtm.2021.05.009
34. Katz ML, Coates JR, Sibigtroth CM, Taylor JD, Carpentier M, Young WM, et al. Enzyme replacement therapy attenuates disease progression in a canine model of late-infantile neuronal ceroid lipofuscinosis (CLN2 disease). J Neurosci Res. (2014) 92:1591–8. doi: 10.1002/jnr.23423
35. Katz ML, Tecedor L, Chen Y, Williamson BG, Lysenko E, Wininger FA, et al. AAV gene transfer delays disease onset in a TPP1-deficient canine model of the late infantile form of Batten disease. Sci transl med. (2015) 7:313ra180. doi: 10.1126/scitranslmed.aac6191
36. Whiting REH, Jensen CA, Pearce JW, Gillespie LE, Bristow DE, Katz ML. Intracerebroventricular gene therapy that delays neurological disease progression is associated with selective preservation of retinal ganglion cells in a canine model of CLN2 disease. Exp Eye Res. (2016) 146:276–82. doi: 10.1016/j.exer.2016.03.023
37. Cotman SL, Vrbanac V, Lebel LA, Lee RL, Johnson KA, Donahue LR, et al. Cln3(Deltaex7/8) knock-in mice with the common JNCL mutation exhibit progressive neurologic disease that begins before birth. Hum Mol Genet. (2002) 11:2709–21. doi: 10.1093/hmg/11.22.2709
38. Staropoli JF, Haliw L, Biswas S, Garrett L, Holter SM, Becker L, et al. Large-scale phenotyping of an accurate genetic mouse model of JNCL identifies novel early pathology outside the central nervous system. PLoS ONE. (2012) 7:e38310. doi: 10.1371/journal.pone.0038310
39. Frugier T, Mitchell NL, Tammen I, Houweling PJ, Arthur DG, Kay GW, et al. A new large animal model of CLN5 neuronal ceroid lipofuscinosis in Borderdale sheep is caused by a nucleotide substitution at a consensus splice site (c571 + 1G>A) leading to excision of exon 3. Neurobiol Dis. (2008) 29:306–15. doi: 10.1016/j.nbd.2007.09.006
40. Bronson RT, Donahue LR, Johnson KR, Tanner A, Lane PW, Faust JR. Neuronal ceroid lipofuscinosis (nclf), a new disorder of the mouse linked to chromosome 9. Am J Med Genet. (1998) 77:289–97.
41. Bartsch U, Galliciotti G, Jofre GF, Jankowiak W, Hagel C, Braulke T. Apoptotic photoreceptor loss and altered expression of lysosomal proteins in the nclf mouse model of neuronal ceroid lipofuscinosis. Invest Ophthalmol Vis Sci. (2013) 54:6952–9. doi: 10.1167/iovs.13-12945
42. White KA, Nelvagal HR, Poole TA, Lu B, Johnson TB, Davis S, et al. Intracranial delivery of AAV9 gene therapy partially prevents retinal degeneration and visual deficits in CLN6-Batten disease mice. Mol Ther Methods Clin Dev. (2021) 20:497–507. doi: 10.1016/j.omtm.2020.12.014
43. Shevtsova Z, Garrido M, Weishaupt J, Saftig P, Bahr M, Luhder F, et al. CNS-expressed cathepsin D prevents lymphopenia in a murine model of congenital neuronal ceroid lipofuscinosis. Am J Pathol. (2010) 177:271–9. doi: 10.2353/ajpath.2010.091267
44. Koike M, Shibata M, Ohsawa Y, Nakanishi H, Koga T, Kametaka S, et al. Involvement of two different cell death pathways in retinal atrophy of cathepsin D-deficient mice. Mol Cell Neurosci. (2003) 22:146–61. doi: 10.1016/S1044-7431(03)00035-6
45. Bassal M, Liu J, Jankowiak W, Saftig P, Bartsch U. Rapid and progressive loss of multiple retinal cell types in cathepsin D-deficient mice-an animal model of CLN10 disease. Cells. (2021) 10:696. doi: 10.3390/cells10030696
46. Hafler BP, Klein ZA, Jimmy Zhou Z, Strittmatter SM. Progressive retinal degeneration and accumulation of autofluorescent lipopigments in Progranulin deficient mice. Brain Res. (2014) 1588:168–74. doi: 10.1016/j.brainres.2014.09.023
47. Platt FM. d'Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat Rev Dis Primers. (2018) 4:27. doi: 10.1038/s41572-018-0025-4
48. Whiting REH, Pearce JW, Vansteenkiste DP, Bibi K, Lim S, Robinson Kick G, et al. Intravitreal enzyme replacement preserves retinal structure and function in canine CLN2 neuronal ceroid lipofuscinosis. Exp Eye Res. (2020) 197:108130. doi: 10.1016/j.exer.2020.108130
49. Marques ARA, Di Spiezio A, Thiessen N, Schmidt L, Grotzinger J, Lüllmann-Rauch R, et al. Enzyme replacement therapy with recombinant pro-CTSD (cathepsin D) corrects defective proteolysis and autophagy in neuronal ceroid lipofuscinosis. Autophagy. (2020) 16:811–25. doi: 10.1080/15548627.2019.1637200
50. Tracy CJ, Whiting RE, Pearce JW, Williamson BG, Vansteenkiste DP, Gillespie LE, et al. Intravitreal implantation of TPP1-transduced stem cells delays retinal degeneration in canine CLN2 neuronal ceroid lipofuscinosis. Exp Eye Res. (2016) 152:77–87. doi: 10.1016/j.exer.2016.09.003
51. Groh J, Ribechini E, Stadler D, Schilling T, Lutz MB, Martini R. Sialoadhesin promotes neuroinflammation-related disease progression in two mouse models of CLN disease. Glia. (2016) 64:792–809. doi: 10.1002/glia.22962
52. Groh J, Kühl TG, Ip CW, Nelvagal HR, Sri S, Duckett S, et al. Immune cells perturb axons and impair neuronal survival in a mouse model of infantile neuronal ceroid lipofuscinosis. Brain. (2013) 136:1083–101. doi: 10.1093/brain/awt020
53. Macauley SL, Wong AM, Shyng C, Augner DP, Dearborn JT, Pearse Y, et al. An anti-neuroinflammatory that targets dysregulated glia enhances the efficacy of CNS-directed gene therapy in murine infantile neuronal ceroid lipofuscinosis. J Neurosci. (2014) 34:13077–82. doi: 10.1523/JNEUROSCI.2518-14.2014
54. Seehafer SS, Ramirez-Montealegre D, Wong AM, Chan CH, Castaneda J, Horak M, et al. Immunosuppression alters disease severity in juvenile Batten disease mice. J Neuroimmunol. (2011) 230:169–72. doi: 10.1016/j.jneuroim.2010.08.024
55. Groh J, Berve K, Martini R. Fingolimod and teriflunomide attenuate neurodegeneration in mouse models of neuronal ceroid lipofuscinosis. Mol Ther. (2017) 25:1889–99. doi: 10.1016/j.ymthe.2017.04.021
56. Dannhausen K, Mohle C, Langmann T. Immunomodulation with minocycline rescues retinal degeneration in juvenile neuronal ceroid lipofuscinosis mice highly susceptible to light damage. Dis Model Mech. (2018) 11:dmm033597. doi: 10.1242/dmm.033597
57. Mirza M, Volz C, Karlstetter M, Langiu M, Somogyi A, Ruonala MO, et al. Progressive retinal degeneration and glial activation in the CLN6 (nclf) mouse model of neuronal ceroid lipofuscinosis: a beneficial effect of DHA and curcumin supplementation. PLoS ONE. (2013) 8:e75963. doi: 10.1371/annotation/ae907882-62e0-4803-8c00-35b30a649fe9
Keywords: neuronal ceroid lipofuscinoses, NCL, Batten disease, retinal degeneration, enzyme replacement therapy, gene therapy, lysosomal storage disorder
Citation: Bartsch U and Storch S (2022) Experimental Therapeutic Approaches for the Treatment of Retinal Pathology in Neuronal Ceroid Lipofuscinoses. Front. Neurol. 13:866983. doi: 10.3389/fneur.2022.866983
Received: 31 January 2022; Accepted: 17 March 2022;
Published: 18 April 2022.
Edited by:
Alessandro Simonati, University of Verona, ItalyReviewed by:
Nadia Lesley Mitchell, Lincoln University, New ZealandSara Mole, University College London, United Kingdom
Copyright © 2022 Bartsch and Storch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephan Storch, c3RvcmNoQHVrZS5kZQ==