 Jun-Hui Yuan
Jun-Hui Yuan Yujiro HiguchiAkihiro Hashiguchi
Yujiro HiguchiAkihiro Hashiguchi Masahiro AndoAkiko Yoshimura
Masahiro AndoAkiko Yoshimura Tomonori NakamuraYu HiramatsuYusuke Sakiyama
Tomonori NakamuraYu HiramatsuYusuke Sakiyama Hiroshi Takashima*
Hiroshi Takashima*- Department of Neurology and Geriatrics, Graduate School of Medical and Dental Sciences, Kagoshima University, Kagoshima, Japan
Introduction: Genetic factors are recognized as the major reason for patients with periodic paralysis. The goal of this study was to determine the genetic causes of periodic paralysis in Japan.
Methods: We obtained a Japanese nationwide case series of 119 index patients (108 men and 11 women) clinically suspected of periodic paralysis, and a gene panel analysis, targeting CACNA1S, SCN4A, and KCNJ2 genes, was conducted.
Results: From 34 cases, 25 pathogenic/likely pathogenic/unknown significance variants were detected in CACNA1S (nine cases), SCN4A (19 cases), or KCNJ2 (six cases), generating a molecular diagnostic rate of 28.6%. In total, seven variants have yet been found linked to periodic paralysis previously. The diagnostic yield of patients with hypokalemic and hyperkalemic periodic paralyzes was 26.2 (17/65) and 32.7% (17/52), respectively. A considerably higher yield was procured from patients with than without positive family history (18/25 vs. 16/94), onset age ≤20 years (24/57 vs. 9/59), or recurrent paralytic attacks (31/94 vs. 3/25).
Discussion: The low molecular diagnostic rate and specific genetic proportion of the present study highlight the etiological complexity of patients with periodic paralysis in Japan.
1. Introduction
Periodic paralysis (PP) is a rare skeletal muscle channelopathy induced by abnormal excitability of the sarcolemma, leading to episodes of flaccid paralysis in the extremities of patients. Symptoms commonly appear in the first or second decade, usually upon awakening in the middle of the night or early morning, and last for hours (occasionally days) before gradually disappearing. The calculated minimum point prevalence rates of PP have been reported at 0.38–0.69/100,000 in the UK and the Netherlands (1, 2).
Clinically, patients with PP with decreased serum potassium level (<3.5 mmol/L) during the paralytic attacks are subtyped as hypokalemic periodic paralysis (hypoPP), genetically linked to the mutations in CACNA1S (encoding α1-subunit of the skeletal muscle L-type calcium channel Cav1.1; hypoPP1) or SCN4A (encoding α1-subunit of voltage-gated sodium channel Nav1.4; hypoPP2) (3, 4). However, naming of the subtype of patients with PP with normal-range or high serum potassium levels is controversial, and in the present study, we refer to multiple recent publications and group these patients with serum potassium level ≥3.5 mmol/L as hyperkalemic periodic paralysis (hyperPP) (1, 2, 5, 6). SCN4A is the causative gene of hyperPP as well (7), and it is also responsible for SCN4A-related non-dystrophic myotonia, characterized by a heterogeneous phenotypic spectrum of myotonia (8, 9). Furthermore, mutations in the KCNJ2 gene (encoding inward-rectifier potassium channel Kir2.1), which have been linked to Andersen-Tawil syndrome (ATS), could also result in a PP phenotype, although typically accompanied by ventricular arrhythmias and dysmorphism (10).
To date, large-group genetic studies concerning both hypoPP and hyperPP are inadequate, and only a few studies have covered all three abovementioned genes, CACNA1S, SCN4A, and KCNJ2. The genetic diagnostic rate of overall patients with PP remains unclear, which is estimated to be 64.1% in the USA or 56.6% in China (11, 12). In this study, among 119 index patients with PP, referring to broad diagnostic criteria, we present a low molecular diagnostic rate in Japan and reassess multiple clinical features associated with the diagnostic yield.
2. Materials and methods
2.1. Sample collection
This is a monocentric retrospective study that included a nationwide case series of 148 patients clinically suspected of PP in Japan (ranging from January 1999 to January 2022). All patients were examined by their attending doctors from the departments of neurology/pediatrics of local hospitals and then referred to our laboratory for genetic testing. The included criteria are acute-onset flaccid paralysis that resolves spontaneously or with potassium treatment within hours or days, without disturbance of consciousness and respiratory muscle involvement. Patients with hyperthyroidism, renal diseases (primary aldosteronism or IgA nephropathy), or gastrointestinal disorders were exempted. Ultimately, we collected 119 consecutive unrelated index patients in this project. Therein, 25 cases with more than one affected individual in their pedigrees were grouped as familial PP (FPP); 94 cases without any positive family history were grouped as sporadic PP (SPP). Within the FPP, 21 pedigrees were considered as autosomal dominant inheritance, encompassing more than one affected individual from ≥2 generations, while the inheritance pattern was not clear in the other four pedigrees. The inclusion and exclusion flowchart are illustrated in Figure 1A.
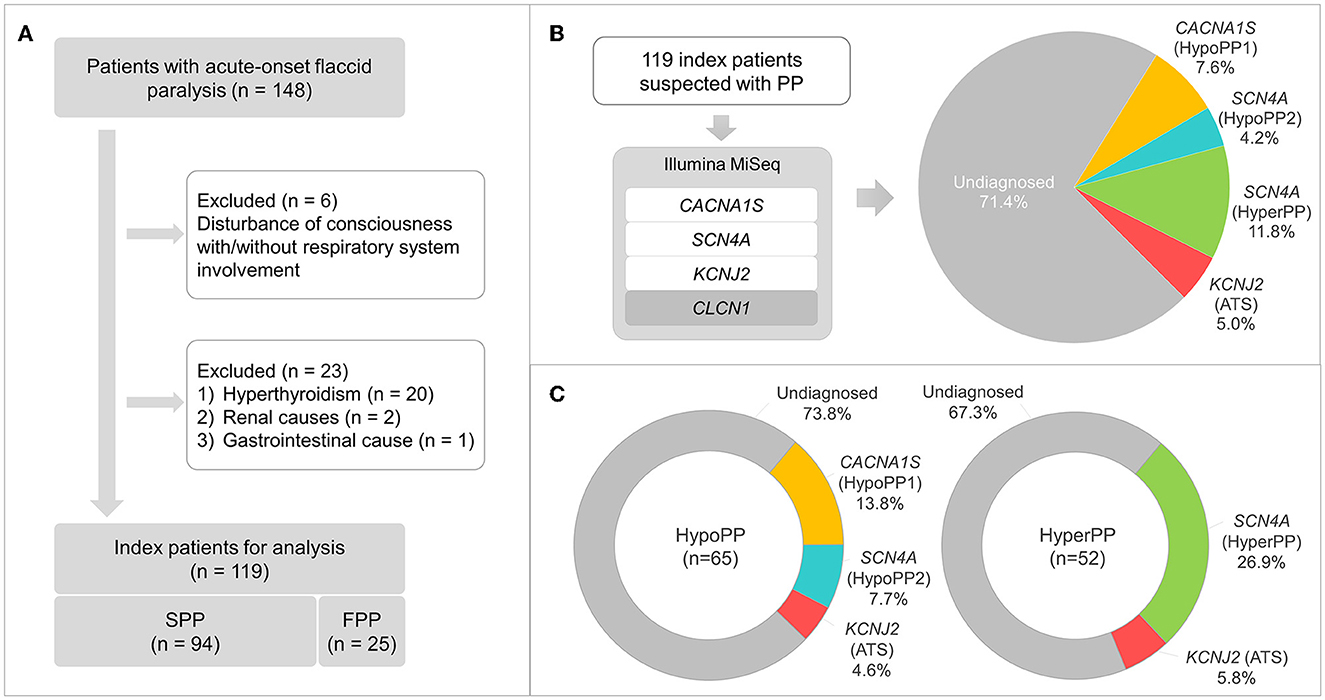
Figure 1. Patient selection flowchart, gene panel sequencing workflow, and findings. (A) Inclusion and exclusion criteria of patients clinically suspected with periodic paralysis (PP), and 119 index patients, consisting of 94 and 25 cases with sporadic (SPP) and familial PP (FPP), respectively, are selected for the following analysis. (B) Gene panel sequencing with Illumina MiSeq demonstrates a molecular diagnostic rate of 28.6%, and the genetic proportions are presented. (C) Genetic proportions of hypoPP (n = 65) and hyperPP (n = 52) patients.
This research was authorized by the institutional review board of Kagoshima University (Application ID: 490). All patients/parents and their available family members provided informed consent for their participation in this study.
2.2. Genomic DNA isolation and Sanger sequencing
Blood samples were collected from patients and any of their available family members. Genomic DNA was extracted from peripheral blood lymphocytes using DNA extraction kits following the corresponding manufacturer's protocols. For DNA samples collected before 2013, Sanger sequencing was conducted on the voltage–sensor coding exons of CACNA1S and SCN4A, as well as the coding region of the KCNJ2 gene, according to the initially published procedures (13).
2.3. Gene panel sequencing on Illumina MiSeq
All samples, whether previously sequenced by Sanger sequencing or obtained after 2013, were subjected to NGS-based gene panel sequencing. Primers were designed using the Primer 3 program, covering all coding exons and exon–intron junctions of CACNA1S (NM_000069.3), SCN4A (NM_000334.4), KCNJ2 (NM_000891.3), and CLCN1 (NM_000083.3). After multiplex polymerase chain reaction (PCR) (Qiagen Multiplex PCR Kit; Qiagen GmbH, Hilden, Germany), amplicons were pooled together and sequenced on the Illumina MiSeq platform (Figure 1A). Low coverage amplicons (reading depth <10) and suspected variants were subsequently substantiated by Sanger sequencing.
2.4. Variant annotation and interpretation
Sequencing data alignment to human reference genome GRCh37, variant processing/annotation, and analysis were conducted via CLC Genomics Workbench (Qiagen, Hilden, Germany), Ensembl-VEP, and in-house R scripts. All variants were contrasted against two population databases, including the East Asian population in Genome Aggregation Database (gnomAD_EAS v2.1.1; https://gnomad.broadinstitute.org) and the Japanese Multi Omics Reference Panel (jMorp; https://jmorp.megabank.tohoku.ac.jp/202102/), as well as the Human Gene Mutation Database (HGMD 2022.2, Qiagen). In total, five in silico prediction scores were enrolled using dbNSFP (v4.0), consisting of SIFT, PolyPhen2, PROVEAN, FATHMM, and Condel (14). All suspected variants were interpreted using a modified American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) classification and ClinGen Expert Panel consensus approaches (15, 16) (Supplementary Table 1). Only pathogenic (P), likely pathogenic (LP), and variant of unknown significance (VUS) variants are described here.
2.5. Statistical analysis
To contrast the frequencies for categorical variables, a two-tailed Fisher's exact test was conducted using a GraphPad online tool (https://www.graphpad.com/quickcalcs/contingency1/). A p-value of <0.05 was deemed substantial. The odds ratio and significance values of the variants in jMorp (ToMMo 38KJPN) were calculated using MedCalc (https://www.medcalc.org/calc/odds_ratio.php).
3. Results
3.1. Clinical analyses
Among 119 index cases with suspected PP, male and female patients accounted for 108 and 11, respectively. Based on the serum potassium levels during attacks, these patients were classified as hypoPP (65 cases) and hyperPP (52 cases), and two cases lacked serum potassium records. Approximately half of these patients had their first paralytic attack at the age ≤20 years (57 cases), while the other half had their onset age >20 years (59 cases). No onset record was available from three patients. There were 94 cases with two or more paralytic attacks and 25 cases with only one attack before the genetic screening. Clinical data of all patients are summarized in Supplementary Table 2.
3.2. Genetic findings
P/LP/VUS variants in the CACNA1S (nine cases), SCN4A (19 cases), or KCNJ2 (six cases) genes were discovered in 34 cases, generating a detection rate of 28.6%. All these variants are listed in Table 1 along with their classification basis. Within 19 cases carrying SCN4A variants, respectively, five and 14 cases had hypoPP and hyperPP phenotypes. The diagnosed patients' detailed genetic proportions were 7.6 (hypoPP1), 4.2 (hypoPP2), 11.8 (hyperPP), and 5.0% (ATS) (Figure 1B). No P/LP/VUS variants were found in the CLCN1 gene.
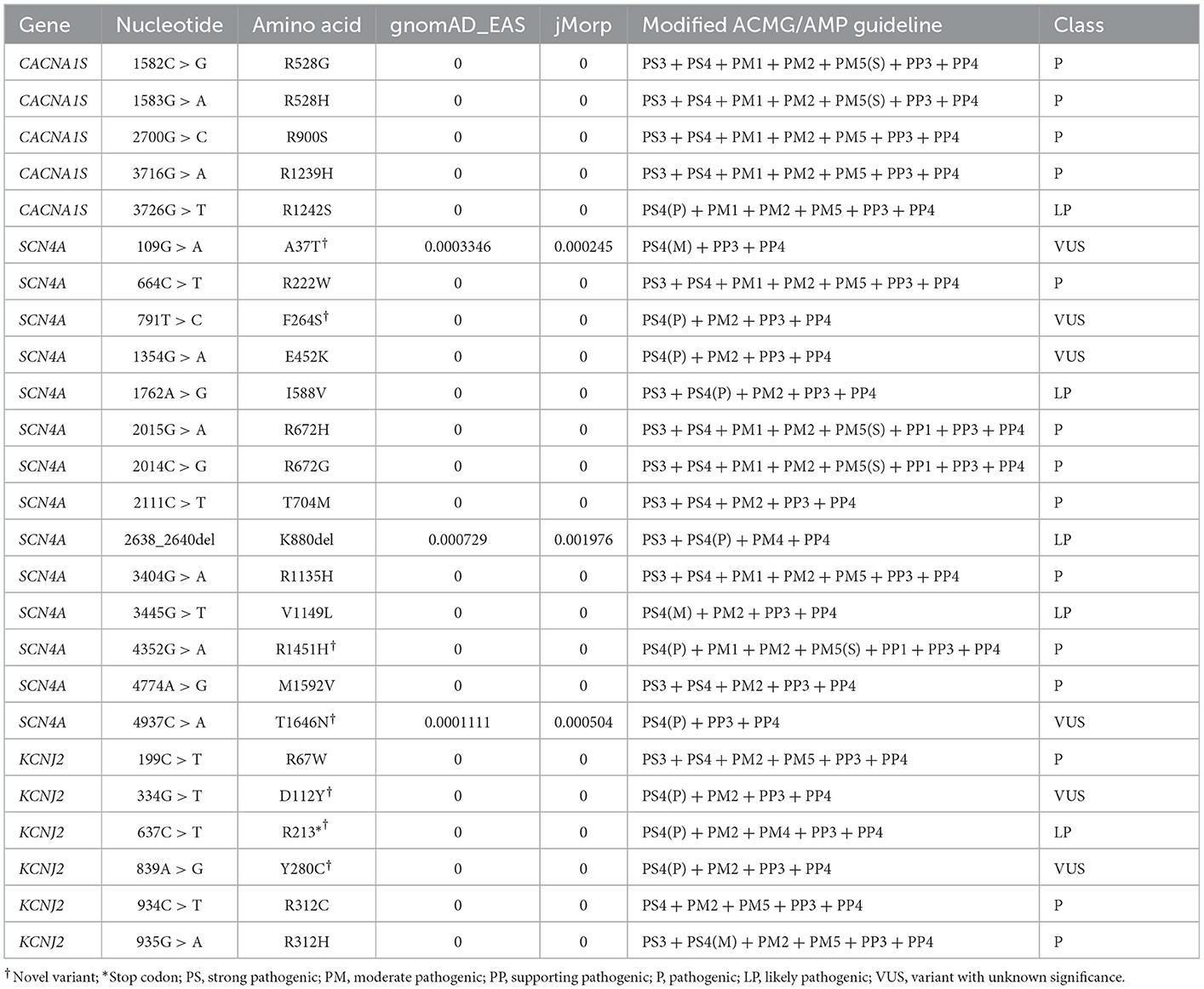
Table 1. All variants detected from 34 Japanese patients with PP.
The molecular diagnostic percentage of patients with hypoPP and hyperPP was 26.2 (17/65) and 32.7% (17/52), respectively (Figure 1C). The percentage between male and female patients was 29/108 and 5/11 (p > 0.05) (Figure 2A). Patients with FPP (18/25) were found easier to receive a genetic diagnosis than patients with SPP (16/94) (p < 0.0001) (Figure 2B). Pedigree sequencing of other affected/unaffected family members was available from seven pedigrees with FPP, and co-segregation of variants was verified from all but the pedigree carrying p.V1149L variant (LP) in SCN4A. Within this pedigree, the same variant was detected from the asymptomatic mother of the proband as well, suggesting a lower penetrance in female (Supplementary Table 2).
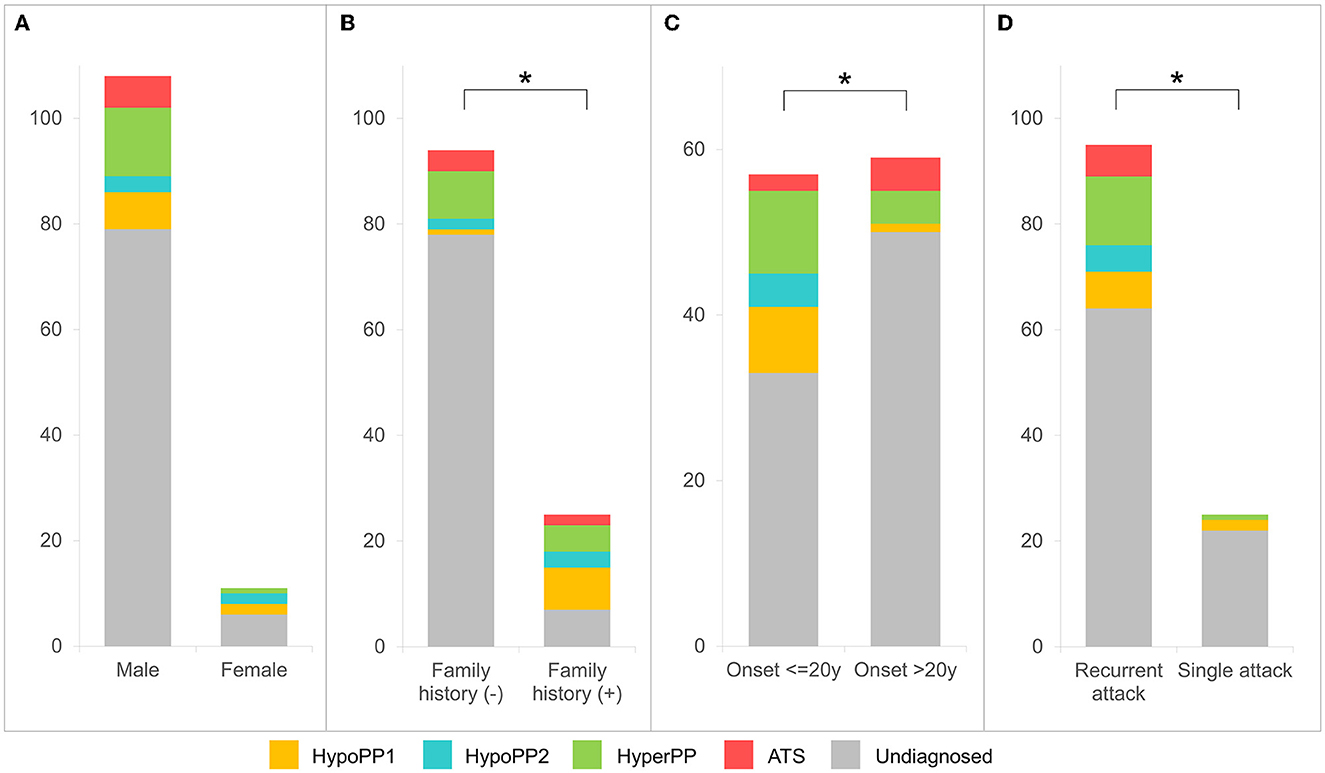
Figure 2. Clinical and molecular diagnostic rate analyses for patients with PP. Molecular diagnostic rates are observed higher in female patients [(A) p > 0.05], patients with positive family history [(B) p < 0.0001], with onset age ≤20 years [(C) p < 0.01], or patients who experienced recurrent paralytic attacks [(D) p < 0.05].
A higher diagnostic rate was observed in patients with onset age ≤20 years than that of later onset (>20 years), at 24/57 vs. 9/59 (p < 0.01) (Figure 2C). Otherwise, the positive rate of patients with recurrent paralytic attacks (31/94) was detected as higher than those who experienced only a single attack (3/25) (p < 0.05) (Figure 2D).
3.3. CACNA1S variants
Within nine patients with hypoPP, we found five initially reported variants within the CACNA1S gene, comprising p.R528H (five cases; P), p.R528G (one case; P), p.R900S (one case; P), p.R1239H (one case; P), and p.R1242S (one case; LP). All of these variants are located in voltage–sensor domains of the CaV1.1 protein (Figure 3A).
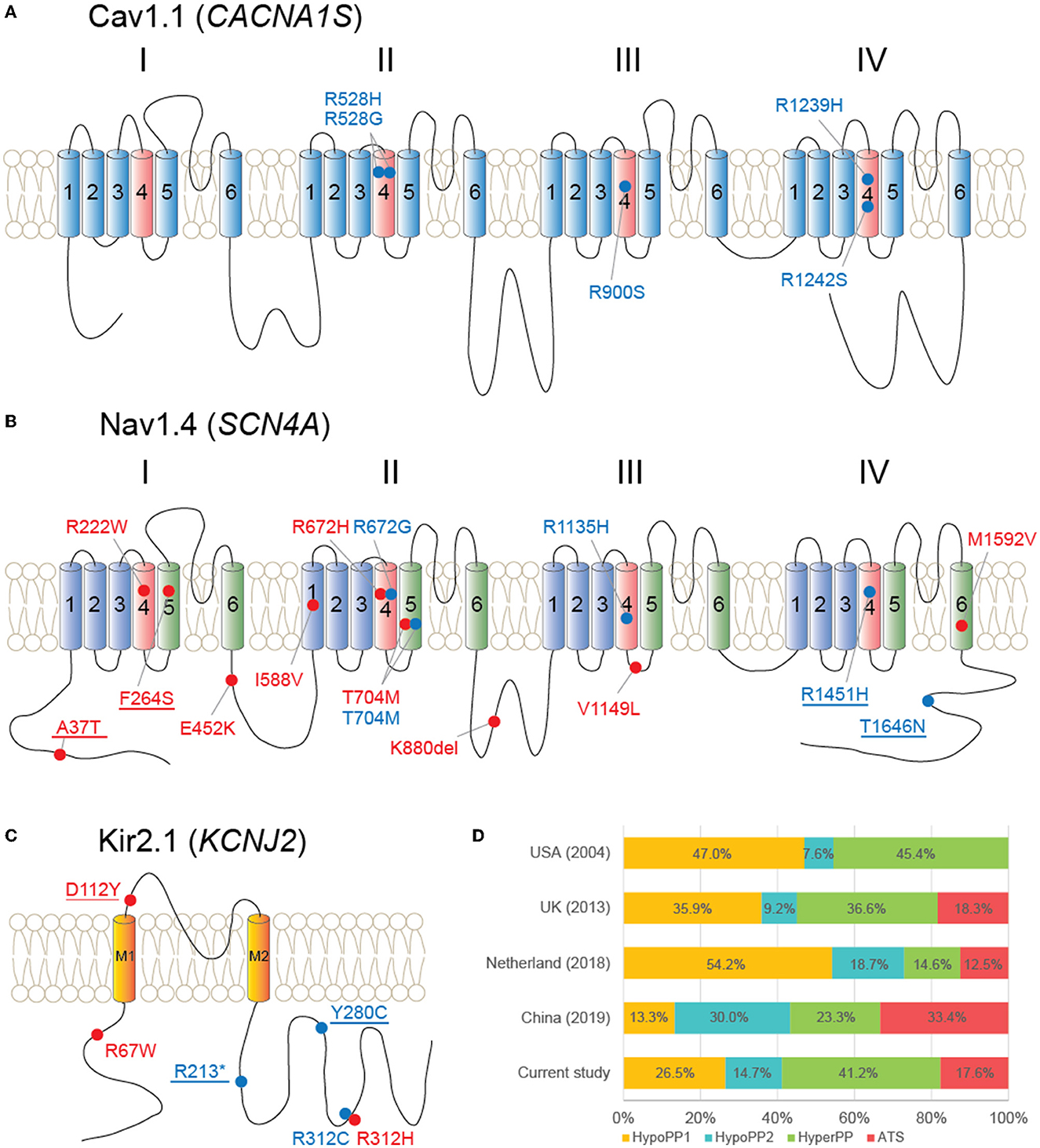
Figure 3. Schematic diagrams of proteins and variants detected in this study and literature review. (A) Cav1.1 protein and five variants locate at its voltage–sensor domains (light red color). (B) Nav1.4 and 14 variants scatter throughout the protein. (C) Kir2.1 and location of six variants. Red dot and label: hyperPP phenotype; blue dot and label: hypoPP phenotype; underline: novel variants. (D) Literature review of genetic proportions from large-group studies of multiple countries, covering all CACNA1S, SCN4A, and KCNJ2 genes.
3.4. SCN4A variants
From five patients with hypoPP, five distinct SCN4A variants were detected, including p.R672G (P), p.T704M (P), p.R1135H (P), p.R1451H (novel; P), and p.T1646N (VUS). Therein, p.T1646N could also be discovered from gnomAD_EAS (allele frequency = 0.0001) and jMorp databases (allele frequency = 0.0005; odds ratio = 8.37, p = 0.0362).
SCN4A variants were found in 14 patients clinically suspected with hyperPP as well, including p.A37T (three cases; novel; VUS), p.R222W (two cases; P), p.F264S (one case; novel; VUS), p.E452K (one case; VUS), p.R672H (one case; P), p.T704M (two cases; P), p.K880del (one case; LP), p.V1149L (one case; LP), and p.M1592V (one case; P) (Figure 3B). Within the four VUS variants, p.A37T and p.K880del were found on the population databases, with frequencies of 0.0003 and 0.0007 on gnomAD_EAS, and 0.0002 (odds ratio = 52.02, p < 0.0001) and 0.002 (odds ratio = 2.13, p = 0.45) on jMorp, respectively. Therein, a recent functional analysis of p.K880del revealed a weak functional effect on Nav1.4, increasing the excitability of the sarcolemma, which could represent a potential pathogenic factor (17).
3.5. KCNJ2 variants
In total, six different KCNJ2 variants were found in six patients with either hypoPP (three cases) or hyperPP (three cases) phenotype. The variants were p.R67W (P), p.D112Y (novel; VUS), p.R213* (novel; LP), p.R280C (novel; VUS), p.R312C (P), and p.R312H (P) (Figure 3C). There was no discernible skeletal deformity in any of these patients. In four cases, electrocardiogram (ECG) data were available, and none of them revealed ventricular arrhythmias.
4. Discussion
In this study, based on our relaxed enrollment criteria, we collected 119 unrelated index patients with clinically suspected PP, and the genetic diagnosis was only procured from 28.6% of them. However, even using the more stringent criteria involving recurrent paralytic attacks, the diagnostic rate was 33.0% (31/94). Both rates were significantly lower than previous studies conducted in the USA and China (11, 12). In terms of hypoPP, the diagnostic rate was 26.2%, which was also much lower than in several western countries (64.3–89.2%) (12, 18–20), but higher than in a Taiwan study (12.5%) (21) (Table 2). In contrast, 18 out of 25 (72.0%) patients with FPP received a molecular diagnosis, showing a much higher yield than patients with SPP (17.0%; p < 0.0001). Otherwise, patients with early onset (≤20 years) were found to be more amenable to molecular diagnosis than that of late-onset cases (24/57 vs. 9/59).

Table 2. Literature review of large-group studies for patients with PP.
Taken together, the aforementioned differences between present and previous studies may be contributed by but not limited to a high proportion of late-onset PP (59/116) and SPP (94/119) in our case series. This is comparable to the Taiwan study with by far the lowest diagnostic rate, where 93.8% (60/64) of their cases were SPP (21). Furthermore, we also noted that the diagnostic rates of PP were concurrently lower in Asia than that of studies in Europe or the USA, and thus, a racial difference should be taken into account as well.
When compared to multiple large-group studies that covered all CACNA1S, SCN4A, and KCNJ2 genes, the genetic proportion of our diagnosed patients had the following characteristics: (1) hypoPP1 was more common than hypoPP2 (9:5), which was comparable to a recent Japanese study (4:3) (5), but not as noticeable as the difference observed in western countries; (2) hyperPP (41.2%) was more common than either hypoPP phenotype alone (Figure 3D). Our case series included 108 men and 11 women, for a gender ratio of about 10:1. This gender disparity could be explained by females' lower penetrance, which is consistent with previous findings (18, 22, 23). Though without substantial variation, female patients (45.5%) were more likely than male patients (26.9%) to receive a molecular diagnosis.
All five CACNA1S gene variants were found at arginines of S4 voltage sensors in domains II (p.R528H/G), III (p.R900S), and IV (p.R1239H, p.R1242S) of Cav1.1. Functional assessments have been conducted for all these variants except p.R1242S. Reduced amplitude of inward Ca2+ currents was observed from all of the four variants, and an abnormal gating pore leak current was detected from p.R528H/G and p.R1239H (24–26). These changes would result in susceptibility to recurrent episodes of depolarization-induced loss of excitability and weakness in HypoPP (27).
Unlike CACNA1S, SCN4A variants associated with both hyperPP and hypoPP2 were found throughout the protein Nav1.4. It is of note that multiple SCN4A variants, previously reported from patients with hypoPP (p.R222W and p.R672H) or non-dystrophic myotonia (p.E452K) (19, 28, 29), developed hyperPP phenotype in our patients. These findings, together with the p.T704M variant, which is associated with both hyperPP and hypoPP2 phenotypes in the present study, highlight the phenotypic heterogeneity of sodium channelopathies. Mutations associated with hyperPP produce the gain-of-function changes for Nav1.4, commonly exhibiting defects of fast and/or slow inactivation, and occasionally showing an enhancement of activation (27). In contrast, multiple mechanisms have been elucidated from hypoPP2 mutations, consisting of loss-of-function changes of Nav1.4, such as enhanced inactivation and decoupling of voltage-sensor displacement to channel opening (29, 30), as well as the gating pore “leakage” current (31). Reduced Nav1.4 currents may also contribute to the reduced excitability of the muscle membrane, leading to paralysis. Among novel SCN4A variants (p.A37T, p.F264S, p.R1451H, and p.T1646N), p.A37T and p.T1646N locate at cytoplasmic N or C terminus of Nav1.4, the domains where multiple variants have been reported, and a p.F1705I variant was found causing fast inactivation defects (32).
Despite PP, ventricular arrhythmias, and dysmorphism being identified as the triad of ATS, patients frequently lack one or more features of the classic triad. As demonstrated in our patients, among all six patients carrying KCNJ2 variants, none of them showed any noticeable dysmorphic features or electrocardiographic abnormalities. This PP-only phenotype complicates the clinical diagnosis of ATS and emphasizes the importance of genetic screening for the KCNJ2 gene in patients with isolated PP. Our outcomes also indicate that the frequency of ATS with PP-only phenotype may be underestimated, referring to a previous report in Japan (2/57) (33). Mutations of KCNJ2 locate throughout the Kir2.1 protein, and cellular analyses revealed the loss-of-function and mostly with a dominant-negative effect on lowering the inward rectifier current, which subsequently depolarizes resting membrane potential and leads to paralysis (10, 34).
On the other hand, SPP is predominant in our case series (94:25), and the etiology requires further research. Recently, within a molecularly undiagnosed Japanese SPP cohort, disease susceptibility was confirmed for nine single-nucleotide variants (SNVs), discovered in genome-wide association studies from SPP and/or thyrotoxic PP in Asian populations (35–40). All of these SNVs are found on chromosome 17 downstream of the KCNJ2 gene, with strong linkage disequilibrium, implying a genetic basis for the undiagnosed SPP.
In this study, we adopted a relatively broad inclusion criterion and obtained a low diagnostic rate (28.6%) from a case series of patients with PP. As indicated in our subsequent statistical analyses, the diagnostic yield could be improved using a more stringent enrollment criterion, such as positive family history, early-onset, and recurrent paralysis. However, since disease-associated variants were also identified from sporadic, atypical, or first-onset cases, we decided to involve all of them in this study. We could not exclude the possibility that the part of our patients was actually not PP, particularly those patients with hyperPP. Another limitation of this study is that the pathogenicity of the VUS variants has not been functionally verified, whereas the possible existence of benign variants would make the diagnostic rate even lower.
In summary, we evaluate the low molecular diagnostic rate and specific genetic proportion of a large Japanese case series of patients suspected of PP. Our outcomes outline the racial diversity and etiological complexity of patients with PP in Japan. Future research should attempt to explore other possible causes of undiagnosed PP, the pathogenicity of detected variants in known PP disease-causing genes, particularly VUS variants, and the pathogenesis of SPP-associated SNVs.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Institutional Review Board of Kagoshima University (Application ID: 490). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author contributions
HT conceptualized the study. J-HY and AY conducted the genetic experiments and analyzed the data. J-HY, YujH, AH, MA, TN, YuH, and YS participated in the clinical data acquisition and analysis. J-HY drafted the original manuscript. All authors revised the manuscript and approved the final version.
Funding
This work was supported by the grants-in-aid from the Research Committee of Ataxia, Health Labor Sciences Research Grant, the Ministry of Health, Labor and Welfare, Japan (20317603 and 201610002B). This research was also supported by the research program for conquering intractable disease from the Japan Agency for Medical Research and Development (AMED) (201442014A and 201442071A) and JSPS KAKENHI Grants (JP18H02742, JP20K16604, JP21K15702, JP21H02842, JP22K07495, and JP22K15713).
Acknowledgments
The authors thank all the patients and their families for participating in this study. We are grateful to Shunichi Sakoda and Kimiyoshi Arimura for their contribution and to all the neurologists and pediatricians for their clinical evaluation of patients and for providing support. We appreciate Tomoko Ohnishi at Kagoshima University, for her excellent technical assistance. We also thank the Division of Gene Research, Research Support Center, Kagoshima University, for the use of their facilities.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1078195/full#supplementary-material
References
1. Horga A, Raja Rayan DL, Matthews E, Sud R, Fialho D, Durran SC, et al. Prevalence study of genetically defined skeletal muscle channelopathies in England. Neurology. (2013) 80:1472–5. doi: 10.1212/WNL.0b013e31828cf8d0
2. Stunnenberg BC, Raaphorst J, Deenen JCW, Links TP, Wilde AA, Verbove DJ, et al. Prevalence and mutation spectrum of skeletal muscle channelopathies in the Netherlands. Neuromuscul Disord. (2018) 28:402–7. doi: 10.1016/j.nmd.2018.03.006
3. Bulman DE, Scoggan KA, van Oene MD, Nicolle MW, Hahn AF, Tollar LL, et al. A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology. (1999) 53:1932–6. doi: 10.1212/WNL.53.9.1932
4. Ptacek LJ, Tawil R, Griggs RC, Engel AG, Layzer RB, Kwiecinski H, et al. Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell. (1994) 77:863–8. doi: 10.1016/0092-8674(94)90135-X
5. Sasaki R, Nakaza M, Furuta M, Fujino H, Kubota T, Takahashi MP. Mutation spectrum and health status in skeletal muscle channelopathies in Japan. Neuromuscul Disord. (2020) 30:546–53. doi: 10.1016/j.nmd.2020.06.001
7. Ptacek LJ, George AL, Griggs RC, Tawil R, Kallen RG, Barchi RL, et al. Identification of a mutation in the gene causing hyperkalemic periodic paralysis. Cell. (1991) 67:1021–7. doi: 10.1016/0092-8674(91)90374-8
8. McClatchey AI, McKenna-Yasek D, Cros D, Worthen HG, Kuncl RW, DeSilva SM, et al. Novel mutations in families with unusual and variable disorders of the skeletal muscle sodium channel. Nat Genet. (1992) 2:148–52. doi: 10.1038/ng1092-148
9. Ptacek LJ, George AL, Barchi RL, Griggs RC, Riggs JE, Robertson M, et al. Mutations in an S4 segment of the adult skeletal muscle sodium channel cause paramyotonia congenita. Neuron. (1992) 8:891–7. doi: 10.1016/0896-6273(92)90203-P
10. Plaster NM, Tawil R, Tristani-Firouzi M, Canun S, Bendahhou S, Tsunoda A, et al. Mutations in Kir21 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell. (2001) 105:511–9. doi: 10.1016/S0092-8674(01)00342-7
11. Luo S, Xu M, Sun J, Qiao K, Song J, Cai S, et al. Identification of gene mutations in patients with primary periodic paralysis using targeted next-generation sequencing. BMC Neurol. (2019) 19:92. doi: 10.1186/s12883-019-1322-6
12. Miller TM, Dias da Silva MR, Miller HA, Kwiecinski H, Mendell JR, Tawil R, et al. Correlating phenotype and genotype in the periodic paralyses. Neurology. (2004) 63:1647–55. doi: 10.1212/01.WNL.0000143383.91137.00
13. Arimura K, Arimura Y, Ng AR, Sakoda S, Higuchi I. Muscle membrane excitability after exercise in thyrotoxic periodic paralysis and thyrotoxicosis without periodic paralysis. Muscle Nerve. (2007) 36:784–8. doi: 10.1002/mus.20865
14. Liu X, Li C, Mou C, Dong Y, Tu Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. (2020) 12:103. doi: 10.1186/s13073-020-00803-9
15. Gelb BD, Cave H, Dillon MW, Gripp KW, Lee JA, Mason-Suares H, et al. ClinGen's RASopathy expert panel consensus methods for variant interpretation. Genet Med. (2018) 20:1334–45. doi: 10.1038/gim.2018.3
16. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
17. Shibano M, Kubota T, Kokubun N, Miyaji Y, Kuriki H, Ito Y, et al. Periodic paralysis due to cumulative effects of rare variants in SCN4A with small functional alterations. Muscle Nerve. (2022) 66:757–61. doi: 10.1002/mus.27725
18. Brugnoni R, Canioni E, Filosto M, Pini A, Tonin P, Rossi T, et al. Mutations associated with hypokalemic periodic paralysis: from hotspot regions to complete analysis of CACNA1S and SCN4A genes. Neurogenetics. (2022) 23:19–25. doi: 10.1007/s10048-021-00673-2
19. Matthews E, Labrum R, Sweeney MG, Sud R, Haworth A, Chinnery PF, et al. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology. (2009) 72:1544–7. doi: 10.1212/01.wnl.0000342387.65477.46
20. Sternberg D, Maisonobe T, Jurkat-Rott K, Nicole S, Launay E, Chauveau D, et al. Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. Brain. (2001) 124:1091–9. doi: 10.1093/brain/124.6.1091
21. Sung CC, Cheng CJ, Lo YF, Lin MS, Yang SS, Hsu YC, et al. Genotype and phenotype analysis of patients with sporadic periodic paralysis. Am J Med Sci. (2012) 343:281–5. doi: 10.1097/MAJ.0b013e31822b430c
22. Ke Q, Luo B, Qi M, Du Y, Wu W. Gender differences in penetrance and phenotype in hypokalemic periodic paralysis. Muscle Nerve. (2013) 47:41–5. doi: 10.1002/mus.23460
23. Li FF, Li QQ, Tan ZX, Zhang SY, Liu J, Zhao EY, et al. A novel mutation in CACNA1S gene associated with hypokalemic periodic paralysis which has a gender difference in the penetrance. J Mol Neurosci. (2012) 46:378–83. doi: 10.1007/s12031-011-9596-1
24. Fuster C, Perrot J, Berthier C, Jacquemond V, Allard B. Elevated resting H(+) current in the R1239H type 1 hypokalaemic periodic paralysis mutated Ca(2+) channel. J Physiol. (2017) 595:6417–28. doi: 10.1113/JP274638
25. Wu F, Quinonez M, Cannon SC. Gating pore currents occur in CaV1.1 domain III mutants associated with HypoPP. J Gen Physiol. (2021) 153:2946. doi: 10.1085/jgp.202112946
26. Wu F, Quinonez M, DiFranco M, Cannon SC. Stac3 enhances expression of human Ca(V)11 in Xenopus oocytes and reveals gating pore currents in HypoPP mutant channels. J Gen Physiol. (2018) 150:475–89. doi: 10.1085/jgp.201711962
27. Cannon SC. Channelopathies of skeletal muscle excitability. Compr Physiol. (2015) 5:761–90. doi: 10.1002/cphy.c140062
28. Dupre N, Chrestian N, Bouchard JP, Rossignol E, Brunet D, Sternberg D, et al. Clinical, electrophysiologic, and genetic study of non-dystrophic myotonia in French-Canadians. Neuromuscul Disord. (2009) 19:330–4. doi: 10.1016/j.nmd.2008.01.007
29. Jurkat-Rott K, Mitrovic N, Hang C, Kouzmekine A, Iaizzo P, Herzog J, et al. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc Natl Acad Sci U S A. (2000) 97:9549–54. doi: 10.1073/pnas.97.17.9549
30. Mi W, Rybalchenko V, Cannon SC. Disrupted coupling of gating charge displacement to Na+ current activation for DIIS4 mutations in hypokalemic periodic paralysis. J Gen Physiol. (2014) 144:137–45. doi: 10.1085/jgp.201411199
31. Sokolov S, Scheuer T, Catterall WA. Gating pore current in an inherited ion channelopathy. Nature. (2007) 446:76–8. doi: 10.1038/nature05598
32. Wu FF, Gordon E, Hoffman EP, Cannon SC. A C-terminal skeletal muscle sodium channel mutation associated with myotonia disrupts fast inactivation. J Physiol. (2005) 565:371–80. doi: 10.1113/jphysiol.2005.082909
33. Kimura H, Zhou J, Kawamura M, Itoh H, Mizusawa Y, Ding WG, et al. Phenotype variability in patients carrying KCNJ2 mutations. Circ Cardiovasc Genet. (2012) 5:344–53. doi: 10.1161/CIRCGENETICS.111.962316
34. Andelfinger G, Tapper AR, Welch RC, Vanoye CG, George AL, Benson DW. KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. Am J Hum Genet. (2002) 71:663–8. doi: 10.1086/342360
35. Cheung CL, Lau KS, Ho AY, Lee KK, Tiu SC, Lau EY, et al. Genome-wide association study identifies a susceptibility locus for thyrotoxic periodic paralysis at 17q243. Nat Genet. (2012) 44:1026–9. doi: 10.1038/ng.2367
36. Chu PY, Cheng CJ, Tseng MH, Yang SS, Chen HC, Lin SH. Genetic variant rs623011 (17q24.3) associates with non-familial thyrotoxic and sporadic hypokalemic paralysis. Clin Chim Acta. (2012) 414:105–8. doi: 10.1016/j.cca.2012.08.004
37. Jongjaroenprasert W, Phusantisampan T, Mahasirimongkol S, Mushiroda T, Hirankarn N, Snabboon T, et al. A genome-wide association study identifies novel susceptibility genetic variation for thyrotoxic hypokalemic periodic paralysis. J Hum Genet. (2012) 57:301–4. doi: 10.1038/jhg.2012.20
38. Li X, Yao S, Xiang Y, Zhang X, Wu X, Luo L, et al. The clinical and genetic features in a cohort of mainland Chinese patients with thyrotoxic periodic paralysis. BMC Neurol. (2015) 15:38. doi: 10.1186/s12883-015-0290-8
39. Nakaza M, Kitamura Y, Furuta M, Kubota T, Sasaki R, Takahashi MP. Analysis of the genetic background associated with sporadic periodic paralysis in Japanese patients. J Neurol Sci. (2020) 412:116795. doi: 10.1016/j.jns.2020.116795
Keywords: periodic paralysis, CACNA1S, SCN4A, KCNJ2, gene panel sequencing
Citation: Yuan J-H, Higuchi Y, Hashiguchi A, Ando M, Yoshimura A, Nakamura T, Hiramatsu Y, Sakiyama Y and Takashima H (2023) Gene panel analysis of 119 index patients with suspected periodic paralysis in Japan. Front. Neurol. 14:1078195. doi: 10.3389/fneur.2023.1078195
Received: 24 October 2022; Accepted: 09 January 2023;
Published: 26 January 2023.
Edited by:
Rossella Tupler, University of Modena and Reggio Emilia, ItalyReviewed by:
Roope Mannikko, University College London, United KingdomXiaoyan Hao, First Affiliated Hospital of Zhengzhou University, China
Copyright © 2023 Yuan, Higuchi, Hashiguchi, Ando, Yoshimura, Nakamura, Hiramatsu, Sakiyama and Takashima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroshi Takashima,  dGhpcm9zaGlAbTMua3VmbS5rYWdvc2hpbWEtdS5hYy5qcA==
dGhpcm9zaGlAbTMua3VmbS5rYWdvc2hpbWEtdS5hYy5qcA==