Abstract
Pathogenic variants in the ATP7A gene, which encodes a transmembrane copper-transporting P-type ATPase, underlie Menkes disease, a rare X-linked recessive disorder of copper metabolism. We report a 3-year-old boy presenting with progressive neurodegeneration, refractory epilepsy, connective tissue abnormalities, and characteristic kinky hair. Whole-exome sequencing and confirmatory analysis identified a retropseudogene insertion (~500 bp) in exon 3 of ATP7A, displaying the hallmarks of target-primed reverse transcription. PCR and RNA-seq revealed a marked reduction in ATP7A transcript levels in the patient. This case underscores the diagnostic relevance of retropseudogene insertions in disease genes and highlights their role in human pathology.
Introduction
Menkes disease (MD, OMIM #309400) is a fatal multisystem disorder of copper metabolism, primarily caused by intragenic variants or partial deletions in the ATP7A gene (OMIM #300011). Affected individuals typically present with early-onset neurological deficits, intractable epilepsy, connective tissue dysfunction, skeletal abnormalities, distinctive kinky hair, and urological complications due to loss of ATP7A gene function (1). Reported prevalence varies from 1 in 354,507 to 1 in 40,000 based on clinically confirmed cases, while predictive estimates suggest a frequency as high as 1 in 8,664 live male births. Most patients with the classic MD phenotype succumb in early childhood, often from vascular complications or recurrent respiratory infections.
The ATP7A gene, located on chromosome Xq13.3, comprises 23 exons and encodes a 1,500-amino-acid transmembrane copper-transporting ATPase (transcript NM_000052.7). Inactivation of ATP7A impairs intestinal copper absorption, systemic copper distribution, and the activity of copper-dependent enzymes, including cytochrome C oxidase and dopamine β-hydroxylase (2). Approximately 80% of pathogenic ATP7A variants are point mutations, while copy number alterations involving the entire gene account for the remaining 20% (3). Nevertheless, more than half of the identified variants have been classified as variants of uncertain significance (4), highlighting the need to explore additional molecular mechanisms underlying MD.
Transposable elements (TEs), a diverse class of mobile DNA elements, constitute nearly two-thirds of the human genome and play crucial roles in genome evolution and gene regulation (5). Through embedded regulatory sequences such as promoters, enhancers, and open reading frames, TEs can modulate protein-coding gene expression (6). However, TE activity may also be deleterious. Insertions into exonic or noncoding regions can disrupt coding sequences, alter RNA splicing, or cause deletions, thereby producing frameshifts and loss of function (LoF) (7). To date, more than 120 TE-mediated insertions have been implicated in human diseases, including hemophilia, Dent’s disease, neurofibromatosis, and various cancers (8). Retrotransposons, in particular, propagate via a “copy-and-paste” mechanism, creating additional insertions across the genome.
In this report, we describe a retropseudogene insertion generated through retrotransposition in exon 3 of ATP7A, identified in a proband presenting with the classic MD phenotype. Sequence analysis revealed that the insertion introduced multiple premature stop codons, while PCR and RNA-seq confirmed markedly reduced ATP7A transcript expression. This case expands the mutational spectrum of ATP7A and emphasizes the role of retropseudogene insertions in human genetic disease.
Materials and methods
Patient
The proband was a 3-year-old male, born to non-consanguineous parents after an uncomplicated pregnancy and delivery, with no history of perinatal asphyxia. Developmental regression was first noted at 3 months of age, followed by global developmental delay and medically refractory seizures (10–20 per day) that were unresponsive to antiepileptic drugs. As the disease progressed, both seizure frequency and seizure types increased. Electroencephalography (EEG) revealed epileptic spasms, tonic spasms, and focal seizures of mixed types (Supplementary Figure 1).
In addition to neurological symptoms, the patient developed urological complications, including multiple bladder diverticula and recurrent urinary tract infections. Physical examination demonstrated hypopigmented skin, fragile kinky hair (Supplementary Figure 2), skin and joint laxity, a high and narrow palatal vault, and generalized hypotonia. Laboratory testing showed reduced serum copper and ceruloplasmin levels (2.0 μmol/L and 50 mg/L, respectively; normal ranges:11.0–23.6 μmol/L and 200–600 mg/L).
Brain magnetic resonance imaging (MRI) demonstrated delayed white matter myelination and diffuse symmetrical cerebral atrophy, while magnetic resonance angiography (MRA) revealed tortuous cerebral vessels (Figure 1). Copper-histidine therapy (0.5 mL daily) was initiated at 11 months of age. Following treatment, serum copper (20.7 μmol/L) and ceruloplasmin (320 mg/L) normalized. Despite biochemical correction, neurological deterioration and seizures persisted, with only partial improvement in hair pigmentation and texture. At the age of 3 years, the patient remained unable to raise his head and exhibited profound deficits in gaze fixation, rolling over, and chewing.
Figure 1

Magnetic resonance angiography results of the proband. The proband’s MRA revealed that the intracranial blood vessels were highly coiled, resembling a screw cone.
Molecular genetic testings
After obtaining written informed consent from the patient’s guardian, peripheral blood samples were collected from the proband, who was clinically diagnosed with Menkes disease, as well as from his first- and second-degree relatives. A comprehensive panel of molecular genetic analyses was performed, including whole-exome sequencing (WES), Sanger sequencing, copy number variation (CNV) analysis, multiplex ligation-dependent probe amplification (MLPA), Manta structural variant analysis, chromosome karyotyping, targeted sequence analysis, polymerase chain reaction (PCR), and RNA sequencing (RNA-seq). These stepwise investigations allowed us to elucidate the molecular genetic etiology of the disease and to provide accurate genetic counseling for the family (9).
Results
WES identified a missense variant in ATP7A (c.4390A > G, p. Ile1464Val) in the proband. According to ACMG-AMP guidelines, this variant was classified as benign/likely benign. It has been reported as a polymorphism in the Japanese population (10), and the proband’s family segregation analysis excluded its pathogenicity (Supplementary Figure 3). Structural variant analysis using Manta revealed two chromosomal breakpoints on chromosomes 22 and X, the latter where ATP7A is located. However, no deletions, duplications, or balanced chromosomal translocations were detected by CNV analysis, MLPA, or karyotyping. Given the X-linked recessive inheritance of MD and the limitations of detecting noncoding variants, we performed deep targeted WES and transposable element (TE) analysis to further investigate potential pathogenic variants.
TE analysis revealed an insertion of approximately 500 bp within exon 3 of ATP7A in the proband (Supplementary Figure 4). Further in silico review with Integrative Genomics Viewer (IGV) revealed multiple hallmarks of target-primed reverse transcription (TPRT)-mediated retrotransposition, including soft-clipped reads, poly(A) tails, and a flanking target site duplication (TSD) (Figure 2). Targeted sequencing of junction amplicons resolved the insertion at single-nucleotide resolution, demonstrating a full-length retrotransposon with an uninterrupted poly(A) tail of at least 100 bp at the 3′ end and a 16 bp TSD (GACAATAATCCCTTCT) flanking the site (Figure 3). These findings confirmed that the insertion occurred via TPRT in the reverse orientation. In silico reading-frame analysis further indicated that the insertion introduced multiple premature stop codons, truncating the ATP7A transcript and abolishing its functional capacity.
Figure 2

Identification of the insertion generated through retrotransposition in ATP7A exon 3. Integrative Genomics Viewer (IGV) analysis revealed an insertion generated through retrotransposition within exon 3 of the ATP7A gene in the proband, which was absent in his father; the mother exhibited mosaicism. The non-reference insertion was supported by soft-clipped reads (colored bases) spanning the insertion breakpoints (red dashed lines). Hallmarks of target-primed reverse transcription (TPRT)–mediated retrotransposition were evident, including a target site duplication (TSD) and a poly(A) tail. A sharp decrease in coverage depth was observed at the junction of the insertion.
Figure 3
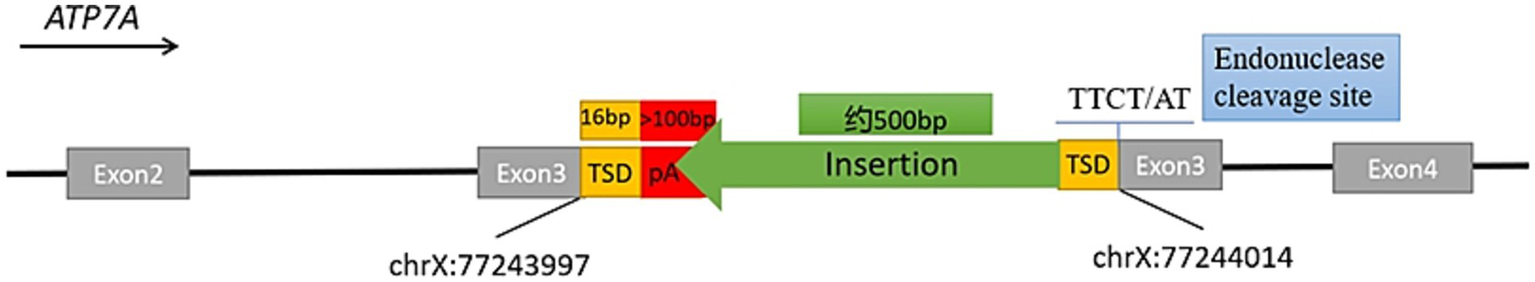
A schematic diagram of the insertion in exon 3 of the ATP7A gene. The schematic diagram illustrates the fully resolved exonic insertion at single-base resolution. The ~500 bp insertion generated through retrotransposition contained a 16-bp target site duplication (TSD), a poly(A) tail exceeding 100 bp, and a LINE-1 endonuclease cleavage motif (5′–TTCT/AT–3′).
To confirm the insertion, full-length PCR was performed using primers flanking the affected region. An additional ~500 bp product was observed in the proband compared with DNA from unaffected donors. PCR with primers spanning upstream and downstream regions of the insertion further validated the event (Figure 4).
Figure 4
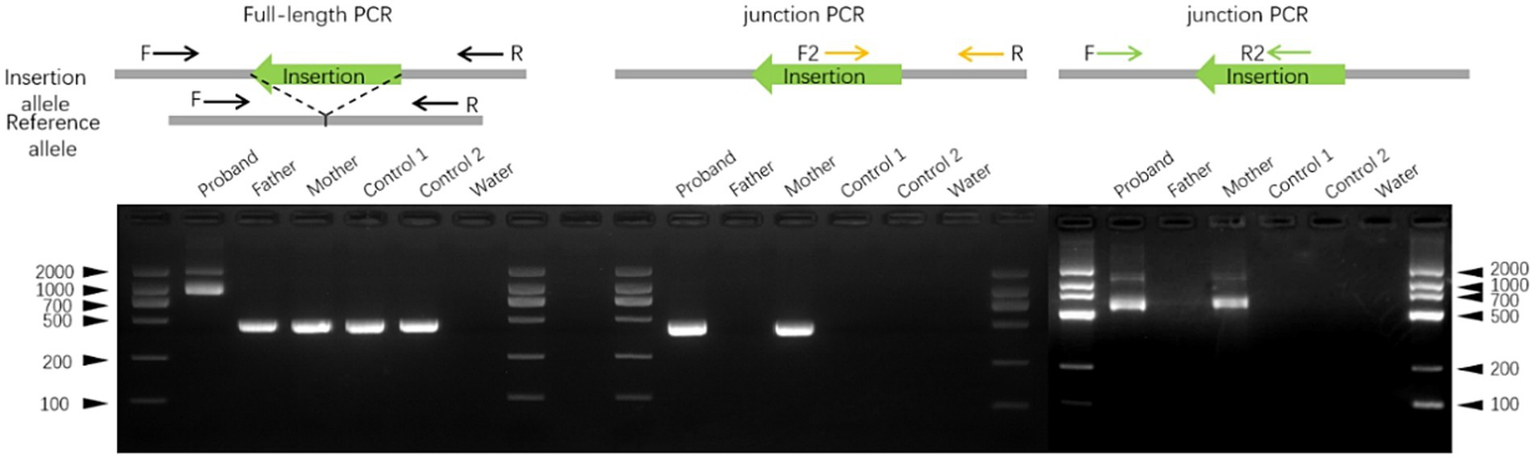
Validation of the insertion in ATP7A exon three by PCR. Representative gel electrophoresis images from full-length PCR and PCR using primers flanking both ends of the transposable element confirmed the exonic insertion. Genomic DNA (gDNA) was extracted from blood samples of the proband, his parents, and two unrelated controls.
Finally, we examined the effect of the insertion on ATP7A expression using RNA sequencing of peripheral blood. After alignment with STAR and normalization using RPKM, we observed comparable expression levels of ATP7A in both parents, whereas the proband exhibited markedly reduced transcript abundance. This reduction in expression is consistent with the predicted pathogenic mechanism and correlates with the clinical phenotype of Menkes disease.
Discussion
Retrotransposons can profoundly influence chromosome integrity and gene expression, thereby contributing to hereditary disorders (11, 12). Their transpositions can disrupt genomic structure and function, and more than 150 de novo retrotransposition events have been reported to date (13). However, these mobile element insertions (MEIs) are often larger than the read lengths of short-read sequencing platforms and are therefore frequently missed by routine genetic testing methods (14). For example, a recent long-read sequencing study identified a 2.8 kb SVA retrotransposon insertion deep within an intron of ATP7A in a 16-year-old boy with occipital horn syndrome (OHS) (15). Similarly, exome and Sanger sequencing have been used to characterize an SVA-F retrotransposon in SMN1 intron 7 as a novel mutational cause of spinal muscular atrophy (16), and PCR combined with Sanger sequencing identified an SVA retrotransposon insertion in ITGB3 in a family with Glanzmann thrombasthenia (17).
In the present study, we identified a novel insertion in exon 3 of ATP7A in a patient with Menkes disease. Sequence analysis demonstrated that the insertion carried hallmarks of LINE-1–mediated retrotransposition, including a 16-bp TSD, a poly(A) tail exceeding 100 bp, and a LINE-1 endonuclease cleavage motif (5′–TTCT/AT–3′). PCR confirmed the insertion, and RNA-seq revealed significant downregulation of ATP7A expression in the proband. We hypothesize that the insertion introduced a strong cryptic donor splice site, leading to aberrant splicing and degradation of the transcript via nonsense-mediated decay (NMD). The generation of premature termination codons (PTCs) is consistent with the observed depletion of ATP7A expression and provides a plausible molecular mechanism (18).
LINE-1 is the only autonomous transposable element currently active in humans, capable of mobilizing not only itself but also non-autonomous elements such as Alu, SVA, and occasionally cellular RNAs to form retropseudogenes (19). Some processed mRNAs are reverse transcribed and integrated into a staggered chromosome break by the enzymatic machinery of LINE-1 non-LTR retrotransposon, called retropseudogenes (20, 21). Although most retropseudogenes likely represent dead-on-arrival gene copies, they can still influence the evolution and function of genes. These retrotranspositions utilize LINE–1–encoded proteins to relocate to new genomic locations via a coupled reverse-transcription integration mechanism, referred to as TPRT (22, 23). Hallmarks of such events include remnants of poly(A) tails and the presence of TSDs (19). Although many retropseudogenes are transcriptionally inactive, LINE-1-mediated retrotransposition events have long been recognized as drivers of genomic instability, particularly when they occur within coding or regulatory regions. In this case, BLAST and RepeatMasker analyses revealed that this retrotransposon is neither an SVA nor an Alu, but rather a retropseudogene. Sequence alignment demonstrated that the retropseudogene shares 97.7% sequence similarity with a region on chromosome 22, allowing the source of the retrocopy to be traced. The proband’s mother was identified as a mosaic carrier, with amplicon sequencing confirming ~2% chimerism, whereas the father harbored no variants. Notably, the mother’s mosaic status suggests the retrotransposition event occurred during gametogenesis or early embryogenesis, after which it was transmitted in a mosaic state. Functionally, this retropseudogene localizes to exon 3 of ATP7A, which encodes the metal-binding domains (MBDs) harboring the copper-binding GMXCXXC motifs essential for protein function (24). Disruption of this domain likely results in a truncated, non-functional protein.
In conclusion, our findings expand the mutational spectrum of ATP7A by identifying a novel retrotransposition-derived, exon-disrupting retropseudogene insertion as a pathogenic cause of Menkes disease. This case highlights the diagnostic challenges of detecting mobile element insertions and underscores the importance of considering such events in unresolved genetic etiologies. Advances in bioinformatics pipelines and re-analysis strategies, particularly those incorporating long-read sequencing, will be essential for improving the detection of MEIs in hereditary disorders.
Statements
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1330472.
Ethics statement
The studies involving humans were approved by ethics committee of the First Affiliated Hospital of Harbin Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
MK: Data curation, Investigation, Software, Writing – original draft. BR: Conceptualization, Project administration, Writing – review & editing. MX: Investigation, Software, Writing – review & editing. LC: Supervision, Validation, Writing – review & editing. XX: Conceptualization, Data curation, Project administration, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We are deeply grateful to the patient and his family for their participation and support. Technical assistance was provided by Shanghai Nyuen Biotechnology Co., Ltd.
Conflict of interest
BR and LC were employed by Shanghai Nyuen Biotechnology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2025.1680208/full#supplementary-material
References
1.
Fujisawa C Kodama H Sato Y Mimaki M Yagi M Awano H et al . Early clinical signs and treatment of Menkes disease. Mol Genet Metab Rep. (2022) 31:100849. doi: 10.1016/j.ymgmr.2022.100849
2.
Maung MT Carlson A Olea-Flores M Elkhadragy L Schachtschneider KM Navarro-Tito N et al . The molecular and cellular basis of copper dysregulation and its relationship with human pathologies. FASEB J. (2021) 35:e21810. doi: 10.1096/fj.202100273RR
3.
Ramani PK Parayil Sankaran B . Menkes kinky hair disease StatPearls. Treasure Island (FL): StatPearls Publishing LLC (2022).
4.
Mhaske A Dileep KV Kumar M Poojary M Pandhare K Zhang KYJ et al . ATP7A clinical genetics resource - a comprehensive clinically annotated database and resource for genetic variants in ATP7A gene. Comput Struct Biotechnol J. (2020) 18:2347–56. doi: 10.1016/j.csbj.2020.08.021
5.
Luqman-Fatah A Nishimori K Amano S Fumoto Y Miyoshi T . Retrotransposon life cycle and its impacts on cellular responses. RNA Biol. (2024) 21:11–27. doi: 10.1080/15476286.2024.2409607
6.
Enriquez-Gasca R Gould PA Rowe HM . Host gene regulation by transposable elements: the new, the old and the ugly. Viruses. (2020) 12:1089. doi: 10.3390/v12101089
7.
Ciomborowska-Basheer J Staszak K Kubiak MR Makałowska I . Not so dead genes-retrocopies as regulators of their disease-related progenitors and hosts. Cells. (2021) 10:912. doi: 10.3390/cells10040912
8.
Niu Y Teng X Zhou H Shi Y Li Y Tang Y et al . Characterizing mobile element insertions in 5675 genomes. Nucleic Acids Res. (2022) 50:2493–508. doi: 10.1093/nar/gkac128
9.
Devine SE . Emerging opportunities to study Mobile element insertions and their source elements in an expanding universe of sequenced human genomes. Genes (Basel). (2023) 14:1923. doi: 10.3390/genes14101923
10.
Ogawa A Yamamoto S Takayanagi M Kogo T Kanazawa M Kohno Y . An Ile/Val polymorphism at codon 1464 of the ATP7A gene. J Hum Genet. (1999) 44:423–4. doi: 10.1007/s100380050194
11.
Takahashi Ueda M . Retrotransposon-derived transcripts and their functions in immunity and disease. Genes Genet Syst. (2024) 98:305–19. doi: 10.1266/ggs.23-00187
12.
Zhang W Huang C Yao H Yang S Jiapaer Z Song J et al . Retrotransposon: an insight into neurological disorders from perspectives of neurodevelopment and aging. Transl Neurodegener. (2025) 14:14. doi: 10.1186/s40035-025-00471-y
13.
Borges-Monroy R Chu C Dias C Choi J Lee S Gao Y et al . Whole-genome analysis reveals the contribution of non-coding de novo transposon insertions to autism spectrum disorder. Mob DNA. (2021) 12:28. doi: 10.1186/s13100-021-00256-w
14.
Won D Hwang JY Shim Y Byeon SH Lee J Lee CS et al . In silico identification of a common mobile element insertion in exon 4 of RP1. Sci Rep. (2021) 11:13381. doi: 10.1038/s41598-021-92834-4
15.
Yano N Chong PF Kojima KK Miyoshi T Luqman-Fatah A Kimura Y et al . Long-read sequencing identifies an SVA_D retrotransposon insertion deep within the intron of ATP7A as a novel cause of occipital horn syndrome. J Med Genet. (2024) 61:950–8. doi: 10.1136/jmg-2024-110056
16.
Vezain M Thauvin-Robinet C Vial Y Coutant S Drunat S Urtizberea JA et al . Retrotransposon insertion as a novel mutational cause of spinal muscular atrophy. Hum Genet. (2023) 142:125–38. doi: 10.1007/s00439-022-02473-6
17.
Zhang J Tang J Li G Li N Wang J Yao R et al . SINE-VNTR-alu retrotransposon insertion as a novel mutational event underlying Glanzmann thrombasthenia. J Thromb Haemost. (2023) 21:3597–607. doi: 10.1016/j.jtha.2023.08.012
18.
Cheng MM Cao YY . The NMD escape mechanism and its application in disease therapy. Yi Chuan. (2020) 42:354–62. doi: 10.16288/j.yczz.19-335
19.
Ghanim GE Hu H Boulanger J Nguyen THD . Structural mechanism of LINE-1 target-primed reverse transcription. Science. (2025) 388. doi: 10.1126/science.ads8412
20.
Wan L Su S Liu J Zou B Jiang Y Jiao B et al . The Spatio-temporal expression profiles of silkworm pseudogenes provide valuable insights into their biological roles. Evol Bioinformatics Online. (2024) 20:261814. doi: 10.1177/11769343241261814
21.
Ma Y Liu S Gao J Chen C Zhang X Yuan H et al . Genome-wide analysis of pseudogenes reveals HBBP1's human-specific essentiality in erythropoiesis and implication in β-thalassemia. Dev Cell. (2021) 56:478–493.e11. doi: 10.1016/j.devcel.2020.12.019
22.
Lagisquet J Zuber K Gramberg T . Recognize yourself-innate sensing of non-LTR retrotransposons. Viruses. (2021) 13:94. doi: 10.3390/v13010094
23.
Wilkinson ME Frangieh CJ Macrae RK Zhang F . Structure of the R2 non-LTR retrotransposon initiating target-primed reverse transcription. Science. (2023) 380:301–8. doi: 10.1126/science.adg7883
24.
Horn N Wittung-Stafshede P . ATP7A-regulated enzyme metalation and trafficking in the Menkes disease puzzle. Biomedicine. (2021) 9:33917579. doi: 10.3390/biomedicines9040391
Summary
Keywords
Menkes disease, ATP7A , retropseudogene, retrotransposon, premature termination codon
Citation
Kou M, Ren B, Xing M, Chen L and Xu X (2025) Retropseudogene insertion generated through retrotransposition in the ATP7A gene results in premature stop codons and a case of Menkes disease. Front. Neurol. 16:1680208. doi: 10.3389/fneur.2025.1680208
Received
07 August 2025
Revised
01 October 2025
Accepted
06 November 2025
Published
27 November 2025
Volume
16 - 2025
Edited by
Huifang Shang, Sichuan University, China
Reviewed by
Jose Laffita Mesa, Karolinska Institutet (KI), Sweden
Takeshi Yoshida, Kyoto University, Japan
Updates
Copyright
© 2025 Kou, Ren, Xing, Chen and Xu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lijuan Chen, chenlijuan@nhwa-group.com; Xiangping Xu, xxp562005@163.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.