 Yu-Jie Gu
Yu-Jie Gu Peng-Yu Wang
Peng-Yu Wang Qing-Qing Fu
Qing-Qing Fu Jia-He Lai1
Jia-He Lai1 Xiang-Hong Liu
Xiang-Hong Liu Bao-Zhu Guan
Bao-Zhu Guan- 1Department of Neurology, Ganzhou People's Hospital, Ganzhou, China
- 2Department of Neurology, Institute of Neuroscience, Key Laboratory of Neurogenetics and Channelopathies of Guangdong Province and the Ministry of Education of China, The Second Affiliated Hospital, Guangzhou Medical University, Guangzhou, China
Background: The CHD2 gene is one of the most common causative genes of developmental and epileptic encephalopathy (DEE). With the advent of high-throughput sequencing, identifying CHD2 variants has increased, necessitating evaluation of the gene-specific performance of widely used tools, as genome-wide benchmarks may mask such heterogeneity.
Methods: The dataset of pathogenic and control CHD2 missense variants was curated from ClinVar, HGMD, and PubMed databases. Tools included SIFT, SIFT4G, Polyphen2_HDIV, Polyphen2_HVAR, MutationAssessor, PROVEAN, MetaSVM, MetaLR, MetaRNN, M-CAP, MutPred2, PrimateAI, DEOGEN2, BayesDel_addAF, BayesDel_noAF, ClinPred, LIST-S2, ESM1b, AlphaMissense, and fathmm-XF_coding. The in silico tools were evaluated based on accuracy, sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), Matthews correlation coefficient (MCC), F-score, and area under the ROC curve (AUC).
Result: A total of 27 missense variants which were classified as pathogenic or likely pathogenic were used as a positive set, and 57 missense variants were used as a negative set. The top tools in accuracy are MutPred2, ESM1b, AlphaMissense, and PROVEAN. In terms of the MCC and F score, the higher degree was observed in MutPred2 and AlphaMissense (MCC score >0.8). ClinPred, AlphaMissense, and BayesDel_addAF had a higher AUC score (AUC > 0.99). SIFT, SIFT4G, Polyphen2_HDIV, Polyphen2_HVAR, ClinPred, and AlphaMissense scores exhibited a distinct bimodal distribution. While scores from other predictors showed a wider distribution range.
Conclusion: Our study highlights the significant variation in the performance of different in silico tools for predicting CHD2 missense variant pathogenicity. Given its overall performance, MutPred2 and AlphaMissense may be the preferred choice for clinical application in CHD2-associated DEE, providing possible reference in optimizing genetic diagnosis and classification of CHD2 missense variants.
1 Introduction
Developmental and epileptic encephalopathy (DEE) represents a heterogeneous group of early-onset epilepsy disorders characterized by refractory seizures, accompanied by cognitive decline or regression associated with ongoing seizure activity (1). This debilitating condition significantly impacts the quality of life of patients and their families, necessitating robust diagnostic and therapeutic strategies. In recent years, significant advancements have been made in understanding the genetic underpinnings of DEE, with the identification of various causative genes, including CHD2 (2).
The CHD2 gene, located on chromosome 15q26, encodes the chromodomain helicase DNA binding protein 2, which is abundantly expressed in the brain, particularly in the neocortex, hippocampus, cerebellum, and olfactory bulb (3, 4). As chromatin remodelers, CHD2 proteins play crucial roles in gene regulation and neuronal development (2). Mutations in the CHD2 gene have been implicated in a range of neurodevelopmental disorders, including DEE, autism spectrum disorder (ASD), intellectual disability (ID), and attention deficit hyperactivity disorder (ADHD) (2, 4–10).
With the advent of high-throughput sequencing technologies, variant screening in patients with DEE has led to the discovery of numerous CHD2 variants, many of which are missense variants (2, 11–26). However, determining the pathogenicity of these variants remains a significant challenge due to the time-consuming and costly nature of functional evaluations. In this context, bioinformatics tools have emerged as indispensable resources for predicting the functional significance of variants (27, 28). These tools leverage various methodologies, such as sequence conservation, amino acid property analysis, and structural modeling, to classify variants into categories including “pathogenic/deleterious” or “benign/tolerant” (27, 29–41).
Despite the proliferation of in silico tools, their performance in predicting the pathogenicity of CHD2 missense variants varies widely. Several studies have evaluated the accuracy, sensitivity, and specificity of these tools, with inconsistent results (42). Therefore, there is a pressing need for a comprehensive evaluation of the performance of these tools specifically for CHD2 missense variants to guide clinical decision-making and improve patient outcomes.
In this study, we aimed to evaluate the prediction performance of 20 in silico tools for CHD2 missense variants. By collecting a dataset of pathogenic and control variants, we assessed accuracy, sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), Matthews correlation coefficient (MCC), F-score, and area under the receiver operating characteristic curve (AUC) of each tool. Our findings reveal significant differences in the performance of these tools in several aspects, with MutPred2 and AlphaMissense maybe the first line tool overall. This study provides valuable insights into the strengths and limitations of various in silico tools, facilitating more informed and accurate predictions of CHD2 missense variant pathogenicity. Furthermore, our findings provide clues for clinical gene interpretation, highlighting the potential of in silico tools to improve the diagnosis and management of DEE and related neurodevelopmental disorders.
2 Materials and methods
2.1 Variants collection and analysis
To systematically evaluate the performance of in silico tools in predicting the pathogenicity of CHD2 missense variants, variants were initially divided into two groups: pathogenic and control. Pathogenic variants were meticulously curated from the Human Gene Mutation Database (HGMD) and PubMed using the query: CHD2 AND (variant OR mutation OR polymorphism). Subsequently, a rigorous selection criterion was applied, included only those variants accompanied by clinical information, explainable origins for genetic diseases, and the American College of Medical Genetics and Genomics (ACMG) criteria. The control variants were retrieved from the ClinVar database, which were classified as “benign” or “likely benign” by ACMG criteria and reviewed by our panels. The data review cutoff date was August 31, 2025.
2.2 In silico prediction
A panel of 20 bioinformatics tools was selected for this study, chosen based on their widespread use and established effectiveness in predicting the functional impact of missense variants. The selection of these tools was based on three primary criteria: (1) inclusion in the ACMG variant interpretation guidelines or recommendation by clinical genetics consortia, (2) the availability of well-defined and recommended score thresholds for pathogenicity classification; (3) the ability to generate a valid score for at least 80% of the variants included in this study; (4) or demonstrated performance in published comparative studies of variant pathogenicity prediction. All prediction scores were obtained from the pre-computed databases (62) to ensure reproducibility. These tools included:
1) SIFT (Sorting Intolerant from Tolerant): a tool utilizes sequence conservation and amino acid properties to predict the impact of amino acid substitutions on protein function (31).
2) SIFT 4G: a faster version of SIFT (Sorting Intolerant from Tolerant), capable of providing predictions for a large number of organisms (41).
3) PolyPhen-2_HDIV: the HumDiv-trained version (HDIV) is optimized for assessing rare alleles potentially involved in complex traits, fine-mapping regions identified through genome-wide association studies, and evolutionary analyses where even mildly deleterious variants are considered damaging (43).
4) PolyPhen-2_HVAR: the HumVar-trained version (HVAR) is specifically designed for the diagnosis of Mendelian diseases, aiming to distinguish strongly deleterious mutations from the remaining human variation, including the presence of mildly damaging alleles (43).
5) Mutation Assessor: a tool predicts functional impact based on the evolutionary conservation of affected amino acids in protein homologs (34).
6) PROVEAN (Protein Variation Effect Analyzer): a tool combines evolutionary conservation, neural network models, and the BLOSUM62 scoring matrix to assess variant impact (28).
7) MetaSVM: an ensemble-based predictor for missense variant deleteriousness. It integrates nine individual deleterious prediction scores along with the maximum minor allele frequency (44).
8) MetaLR: an ensemble scoring method for deleterious missense mutations, which demonstrated the value of combining information from multiple orthologous approaches (44).
9) MetaRNN: a deep recurrent neural network–based ensemble models that integrate 28 high-level annotation features, including multiple functional prediction scores, evolutionary conservation metrics, and allele frequency information, to predict the pathogenic likelihood of human nonsynonymous SNVs and non-frameshift indels (45).
10) M-CAP: a pathogenicity classifier for rare missense variants that integrates existing prediction scores and additional genomic features within a high-sensitivity model, optimized for clinical use (46).
11) MutPred2: a machine learning–based predictor that employs an ensemble of neural networks trained on large sets of pathogenic and putatively neutral variants (47).
12) PrimateAI: a tool uses deep neural networks to predict the clinical impact of human mutations, incorporating primary sequence and protein structure information (40).
13) DEOGEN2: a tool integrates heterogeneous information about molecular effects, domains, gene relevance, and protein interactions to predict variant deleteriousness (33).
14) BayesDel_addAF/BayesDel_noAF: a Bayesian ensemble-based meta-predictor that estimates the deleteriousness of both coding and non-coding variants, including single-nucleotide variants and small insertions or deletions (48).
15) ClinPred: a machine learning–based predictor for disease-associated missense variants that integrates existing pathogenicity scores with population allele frequency from gnomAD database and is trained on ClinVar data to achieve highly accurate and robust pathogenicity classification across diverse disease contexts (49).
16) LIST-S2: the successor to LIST, quantifies conservation across species and predicts variant deleteriousness, not limited to human sequences (32).
17) ESM1b: a 650-million-parameter protein language model trained on 250 million protein sequences from diverse organisms using a masked language modeling objective, in which randomly masked residues are predicted based on their surrounding sequence context (50).
18) AlphaMissense: an adaptation of AlphaFold, which trained on databases of population frequencies of human and primate variants, incorporating structural context and evolutionary conservation as parameters for predicting missense variant pathogenicity (27).
19) FATHMM-XF: the enhanced version of functional analysis through hidden Markov models with additional features, improving predictions for single-nucleotide variants across the genome (35).
2.3 Evaluation of predictive performance
To accurately assess the predictive performance of these in silico tools, we calculated a series of critical metrics, including true positives (TP), true negatives (TN), false positives (FP), and false negatives (FN). From these metrics, we derived additional indices such as accuracy, sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV). Furthermore, we computed the F-score, which balances precision and recall, and the Matthews correlation coefficient (MCC), which measures the quality of binary classifications. The F score was defined as , where precision (P)= and recall (R)= .
The Matthews correlation coefficient (MCC) score was calculated using the following equation , with the score ranging from −1 to 1; −1 indicates a completely wrong binary classifier, while 1 indicates a completely correct binary classifier.
Additionally, ROC curves were plotted for each tool, with the pathogenic missense variants serving as the gold standard for positive samples and the control missense variants with no relevant phenotypic reports serving as the gold standard for negative samples. The area under the ROC curve (AUC) provides a quantitative measure of the overall predictive performance of each tool.
2.4 Statistical analysis
Statistical analyses were conducted using R (4.5.1). For comparisons between two independent samples, the choice of statistical test was contingent upon the normality of the data. Normality was assessed using the Shapiro–Wilk test. If the data followed a normal distribution, Student’s t-test was employed. Conversely, for non-normally distributed data, the Mann–Whitney U test (also known as the Wilcoxon rank-sum test) was utilized. A p-value < 0.05 was considered statistically significant for all comparisons.
3 Results
3.1 Performance evaluation of in silico tools
In this study, we assessed the predictive capabilities of 20 different in silico tools specifically tailored for analyzing CHD2 missense variants. Variants were meticulously curated from reliable sources: pathogenic variants were sourced from HGMD and PubMed, supported by robust clinical data, while control variants were retrieved from the ClinVar database (2, 11–26). This meticulous variant collection yielded a dataset comprising 27 pathogenic and 57 control variants, totaling 84 variants for comprehensive analysis (Supplementary Table S1).
Our analysis revealed substantial variability in the performance of these tools across various metrics (Figure 1 and Table 1). Accuracy, a crucial benchmark, ranged from a low of 58.3% (M-CAP) to a remarkable high of 95.4% (MutPred2). This striking difference underscores the importance of selecting the appropriate tool for variant interpretation. Sensitivity and specificity were evaluated, which are equally pivotal in assessing tool performance. Sensitivity, reflecting the ability to correctly identify pathogenic variants, varied from 70.4% (PrimateAI) to 100% (PROVEAN, MetaSVM, MetaLR, M-CAP, DEOGEN2, ClinPred, LIST-S2, and ESM1b). Conversely, specifically, indicating the capacity to accurately discern control variants, ranged from 16.7% (M-CAP) to 98.3% (MutPred2).
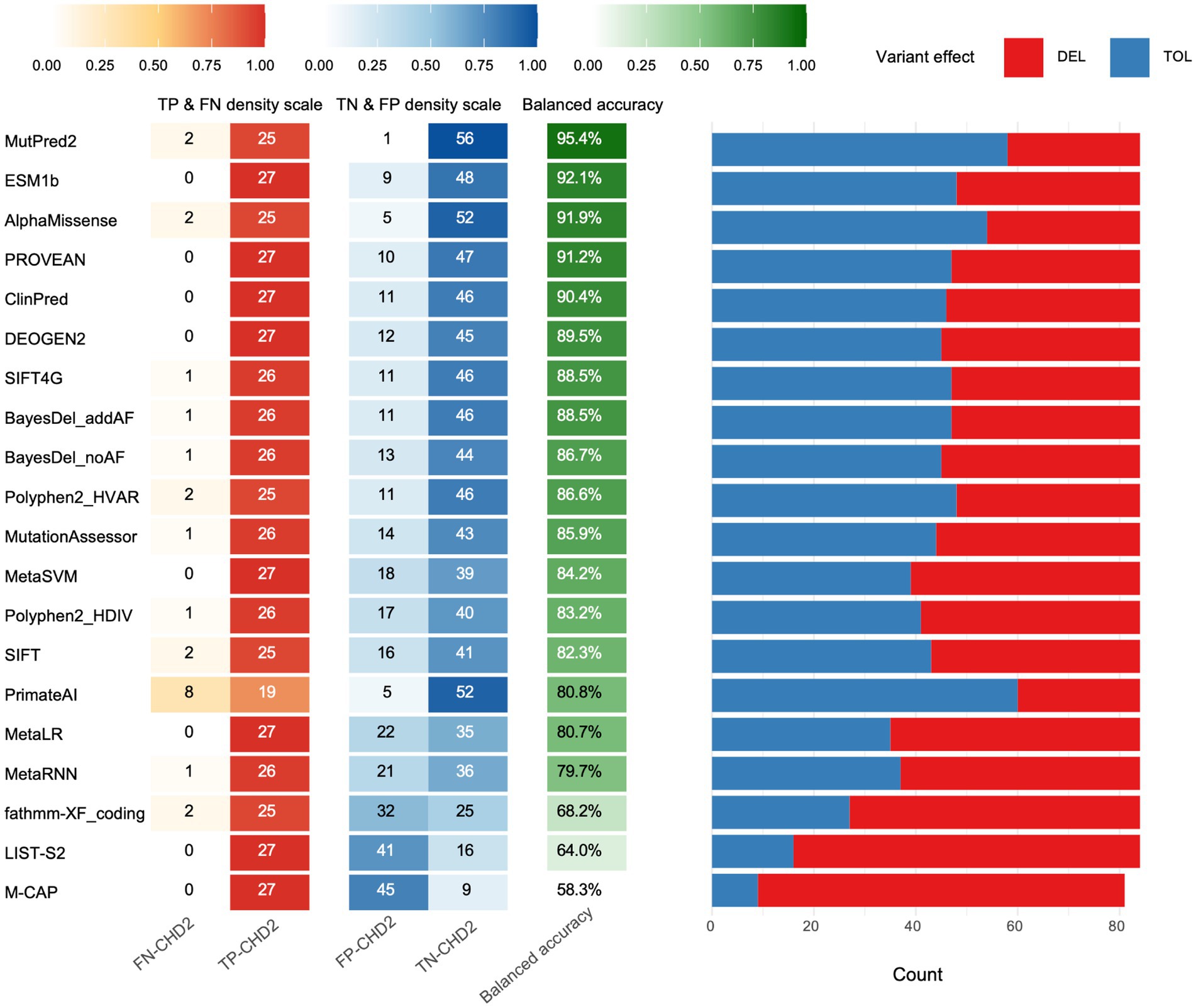
Figure 1. Performance evaluation of algorithms. For each tool, the left heat-map panel reports the rates of true positives (TP) and false negatives (FN) for pathogenic variants and true negatives (TN) and false positives (FP) for control variants. The middle column gives the resulting balanced accuracy (balanced acc.), which was used to rank the tools from top to bottom. The right stacked bar shows the proportion of variants each algorithm called deleterious (DEL, red) or tolerated (TOL, blue) using recommended thresholds.
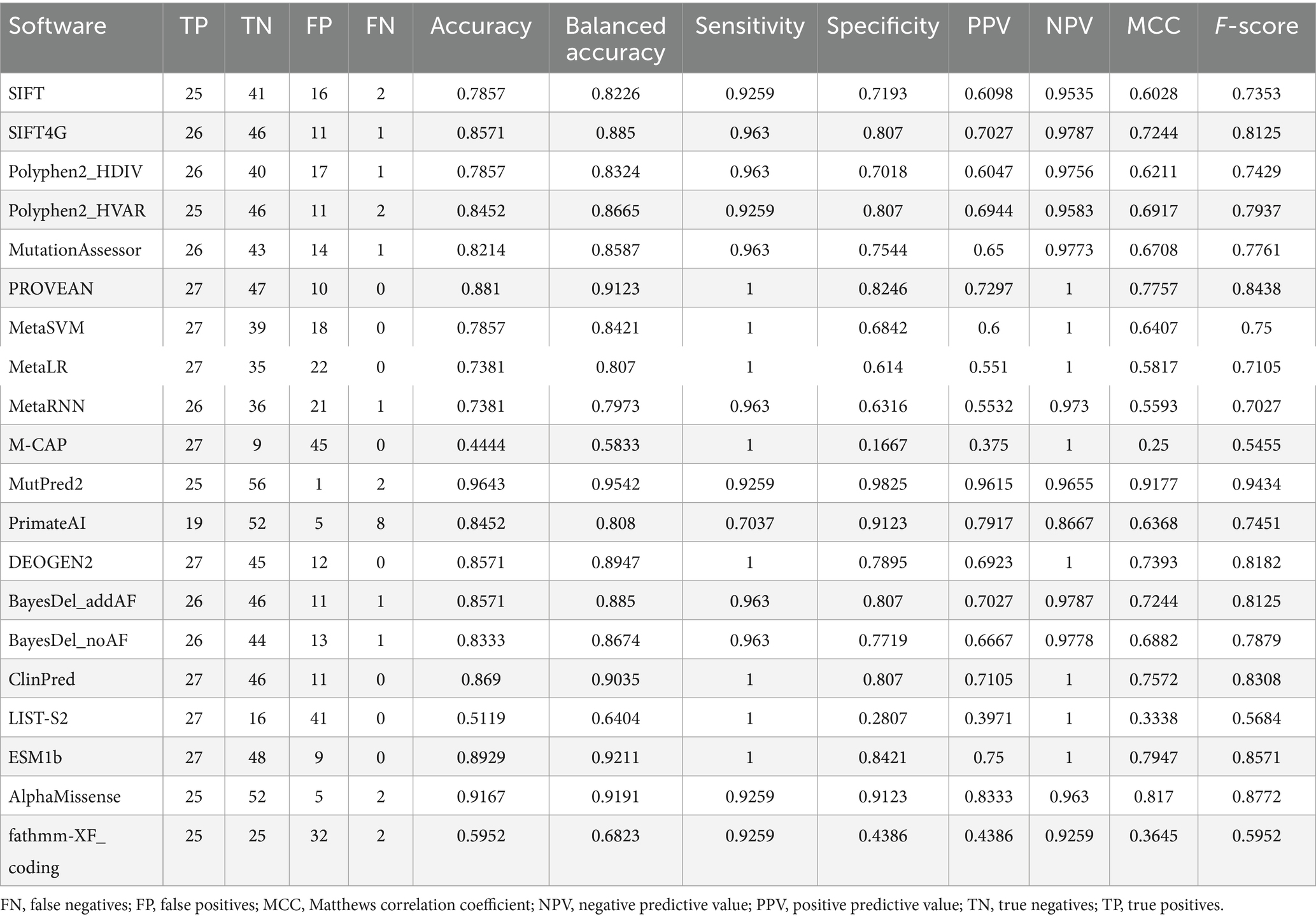
Table 1. Performance of in silico tools for the prediction of CHD2 missense variants.
To delve deeper into the predictive prowess of these tools, we examined additional metrics such as PPV and NPV (Table 1). PPV, signifying the proportion of true positives among all predicted positives, ranged from 37.5% (M-CAP) to 96.2% (MutPred2). Similarly, NPV, representing the proportion of true negatives among all predicted negatives, spanned from 86.7% (PrimateAI) to 100% (ESM1b, PROVEAN, ClinPred, DEOGEN2, MetaSVM, MetaLR, LIST-S2, M-CAP, and SIFT4G).
To further gauge the quality of binary classifications produced by these tools, we calculated the Matthews correlation coefficient (MCC) and F-score. Both metrics provided consistent rankings, with MutPred2 topping the charts, exhibiting an MCC of 0.918 and an F-score of 0.943.
3.2 ROC curves for the in silico tools
To visually represent and compare the predictive performance of the tools, we generated ROC curves. The area under the curve (AUC), a quantitative measure derived from the ROC curve, provides an overall assessment of a tool’s diagnostic accuracy. Consistent with our previous findings, the ROC curve indicated that ClinPred (0.9948) emerged as the preeminent tool with an AUC value of 0.9948, followed by AlphaMissense (0.9929), BayesDel_addAF (0.9929), M-CAP (0.9856), MutPred2 (0.9851), BayesDel_noAF (0.9851), SIFT4G (0.9838), MetaRNN (0.9825), DEOGEN2 (0.9825), PROVEAN (0.9779), ESM1b (0.9773), MetaSVM (0.9734), MetaLR (0.9623), Polyphen2_HVAR (0.9561), SIFT (0.9539), PrimateAI (0.9526), LIST-S2 (0.9418), MutationAssessor (0.9379), Polyphen2_HDIV (0.9230) and fathmm-XF_coding (0.7745) (Figure 2). These AUC values reflect the exceptional ability of ClinPred, AlphaMissense, BayesDel_addAF to discriminate between pathogenic and control variants.
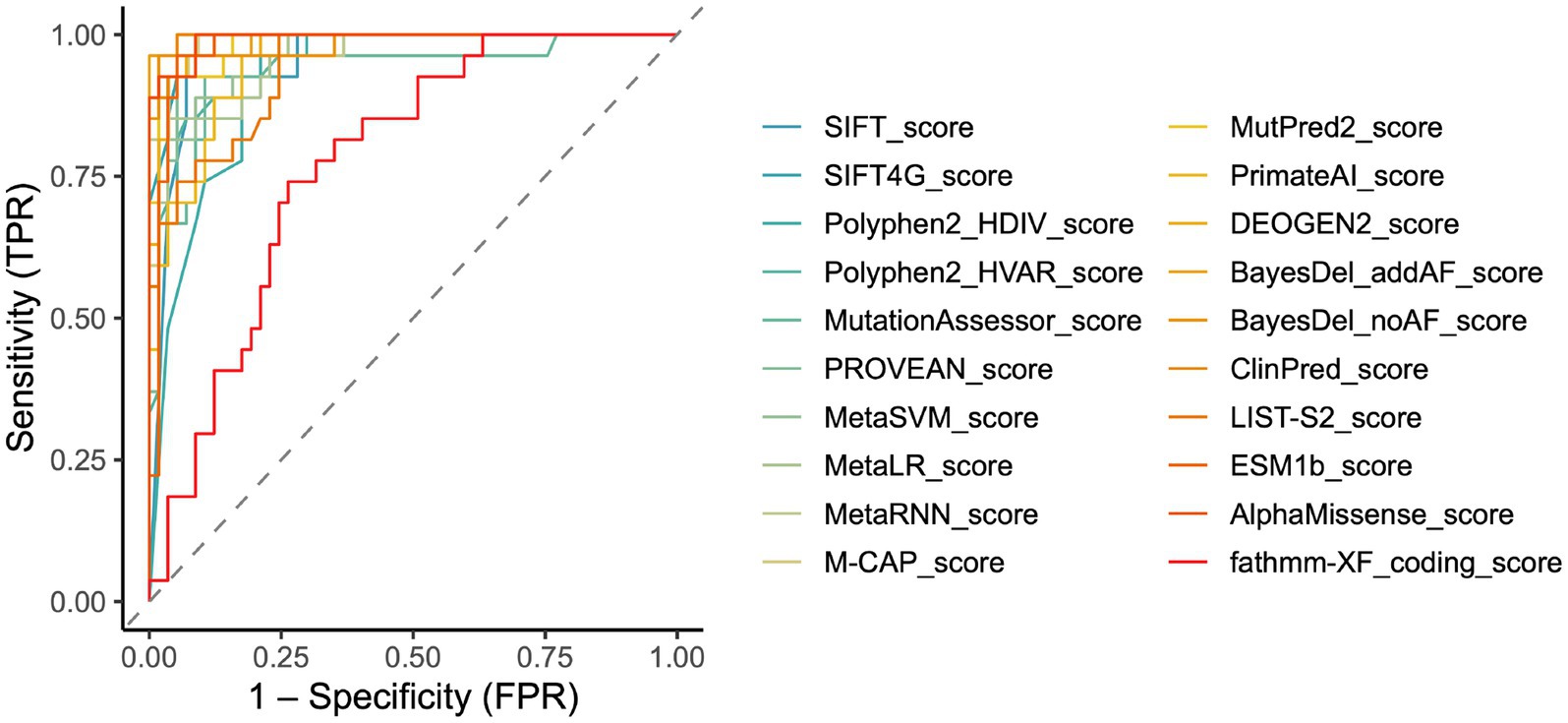
Figure 2. Receiver operating characteristic curve (ROC) performance and optimal thresholds of in silico tools for CHD2 variants. Combined ROC curves showed each color is one tool, and the dashed line meant no discrimination. All curves are provided in Supplementary Figure S1.
3.3 Distributions of the prediction scores for the in silico tools
The prediction scores of 20 in silico tools on a continuous scale, applying a binary “cutoff point” and classifying variants as “pathogenic” or “control.” We, therefore, visualized the scores generated by these in silico tools in scatter plots. Most variant pathogenicity scores significantly differed between control and pathogenic groups (all p-value < 0.0001) (Figure 3). Interestingly, the SIFT, SIFT4G, PolyPhen2_HDIV, PolyPhen2_HVAR, ClinPred, and AlphaMissense scores displayed a distinct bimodal distribution, highlighting their ability to clearly distinguish pathogenic from control variants. In contrast, scores derived from other predictors exhibited broader and more continuous distributions, reflecting greater overlap between variant categories and suggesting varying levels of discriminative performance among prediction algorithms.
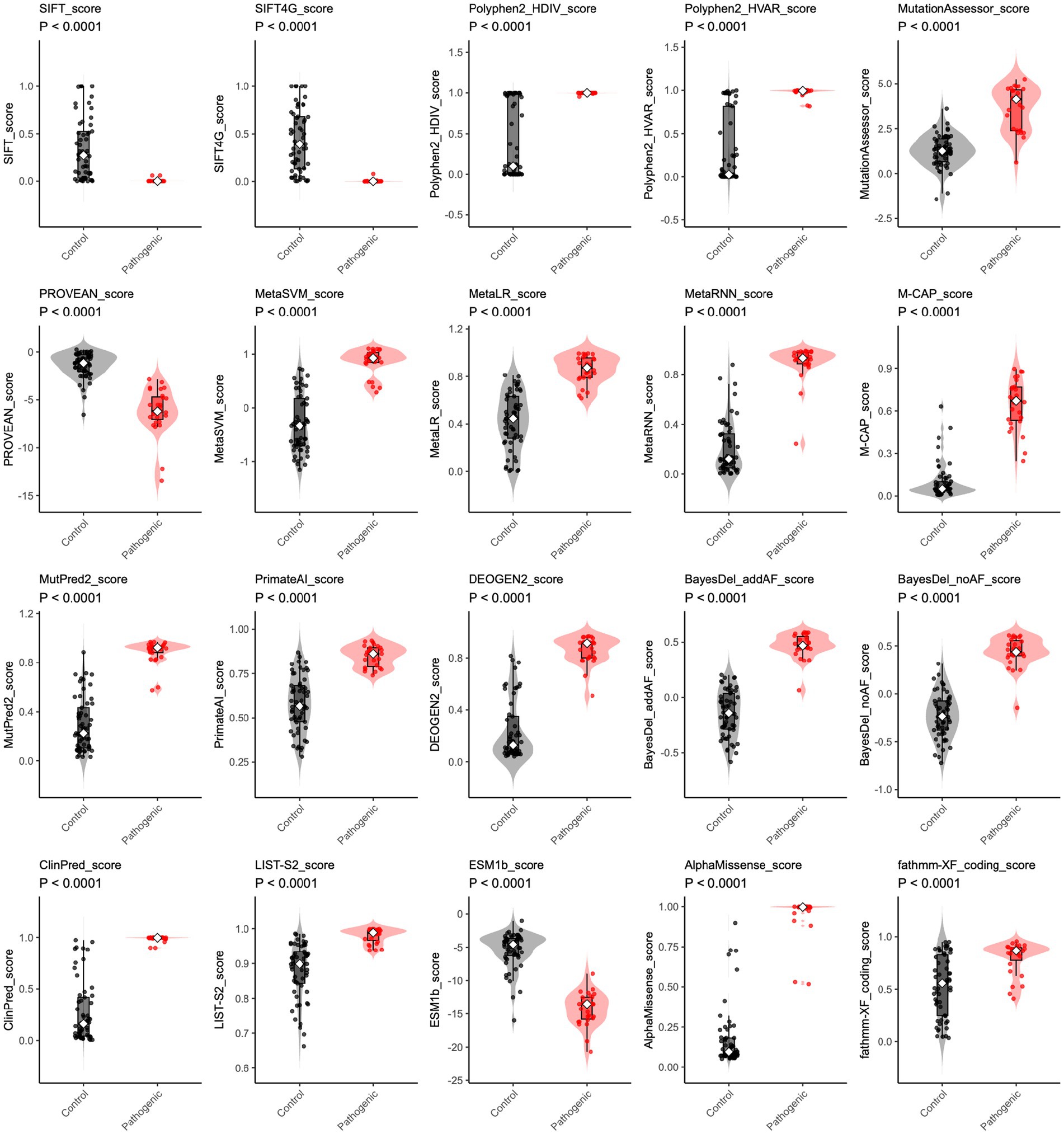
Figure 3. The distribution of in silico prediction raw scores for CHD2 variants. For each algorithm (including SIFT, SIFT4G, Polyphen2_HDIV, Polyphen2_HVAR, MutationAssessor, PROVEAN, MetaSVM, MetaLR, MetaRNN, M-CAP, MutPred2, PrimateAI, DEOGEN2, BayesDel_addAF, BayesDel_noAF, ClinPred, LIST-S2, ESM1b, AlphaMissense, and fathmm-XF_coding), raw scores are shown for control (gray) and pathogenic (red) variants. Violin plots depict the score distribution. Higher rank scores indicate more deleterious predictions. Group differences were tested using a two-sided Wilcoxon rank-sum test; all comparisons were significant (p < 0.0001).
4 Discussion
In this study, we benchmarked 20 in silico predictors on 27 pathogenic and 57 control CHD2 missense variants show substantial performance heterogeneity across tools. While MutPred2 achieved the highest balanced accuracy (≈95.4%). AlphaMissense, together with ClinPred and BayesDel_addAF, delivered near-perfect discrimination by ROC analysis. Given its consistently strong performance across multiple metrics, MutPred2 and AlphaMissense may be possible as the practical first line predictor for clinical triage of CHD2 variants in DEE. This finding is particularly significant given the increasing number of CHD2 variants identified in patients with DEE, as accurate prediction of variant pathogenicity is crucial for guiding clinical decision-making and treatment strategies.
The ranking of predictive tools differed significantly depending on the metric used to quantify performance and shown that no single predictive tool is best at all times. This is illustrated, for example, by the fact that MutPred2 had the best-balanced accuracy (0.954) and Matthews correlation coefficient (MCC = 0.918) while other tools, including ESM1b, PROVEAN, and ClinPred were superior in terms of sensitivity (1.000). Their perfect sensitivity was however limited by lower specificity and higher false positive rate. MutPred2 and AlphaMissense, on the other hand, performed well and generated a generally balanced result with regard to a number of key metrics, resulting in a top ranking in accuracy, correlation and discrimination. This elevates AlphaMissense to the status of a superb overall performer, which is to be trusted for overall balance between the identification of true pathogenic variants and minimum false positives.
We also assessed predictor performance from a ranking ability perspective and assessed ROC/AUC. This approach illuminates the principle that excellent discrimination is not tantamount to the best possible performance at a fixed or default threshold. A salient example in this case is M-CAP, which had perfect sensitivity, but poor specificity (0.167), and balanced accuracy (0.583) at its default cutoff, misclassifying 45 of 57 control variants. This shows that having the ability to rank pathogenic variants highly is not automatically correlated with good classification accuracy at a fixed, predetermined cutoff. Other predictors such as ClinPred (AUC = 0.995), AlphaMissense (AUC = 0.993), and BayesDel_addAF (AUC = 0.993), while also ranking well, also had excellent performance scores in terms of threshold classifications and therefore are the more useful predictors for direct clinical application.
An important consideration in the choice of the predictors is the trade-off between sensitivity and specificity, since our results show that it is often possible to obtain a high sensitivity at the expense of specificity. For example, ESM1b, PROVEAN and ClinPred identified all 27 pathogenic variants correctly (sensitivity = 1.000) but classified, respectively, 9, 10 and 11 of the benign/likely benign variants as pathogenic (specificity = 0.842, 0.825 and 0.807). This trade-off is particularly important for M-CAP and LIST-S2 in which case perfect sensitivity was also obtained, but specificities were very low (0.167 and 0.281, respectively). This very large false positive rate has great clinical significance as it could lead to unnecessary follow-up investigations or increased anxiety on the part of the patient. It would seem therefore that a balanced predictor or a combination of those that are complementary may be preferred for clinical use.
The performance patterns of these tools in our analysis offer important insights. The high accuracy of AlphaMissense and ESM-1b, for instance, strongly emphasizes that the structural integrity and sequence conservation of CHD2 proteins are critical determinants of their function, and that disruptions to these features are a major driver of pathogenicity. Their top-perform validated that methods focusing on these fundamental biochemical properties are highly effective. Additionally, ClinPred’s consistently high ranking in ROC evaluations underscores the importance and power of the “meta-predictor” strategy, which combines multiple, diverse algorithms to forge a more robust and accurate consensus. This suggests that future advancements may lie not only in developing novel individual predictors but also in intelligently integrating the strengths of existing ones.
This study indicates that the new predictors in use now, in particular those generated by deep learning and protein language models such as AlphaMissense (27) and ESM1b (50), tend to outperform many of the older generation predictors. This indicates the quick advancement in this area and goes some way to proving the need for frequent re-evaluation of those tools used in clinical practice. Guidelines such as those issued by the ACMG/AMP need to be continually evaluated in order to better reflect the capabilities of these predictors so that the best and most trustworthy evidence is always being used to classify variants. At the same time, it is well known that the ability of any particular predictor to accurately assess variants differs from gene to gene. This study gives a necessary gene specific assessment of CHD2 and also draws attention to the vital importance of carrying out similar assessments on other clinically significant disease genes before any single predictor tool is used in a diagnostic context on a widespread basis.
The limitations of in silico tools must be appreciated and their predictions treated with caution. Predictive ability can be influenced by a number of factors, not least the quality and heterogeneity of the training datasets, methodology of prediction and the features of the variants studied. Validation of the predictions of these tools by experimental means, e.g., functional assays or clinical data is therefore essential in order to ascertain their reliability and accuracy. Furthermore, the classification is imbalanced between pathogenic and control variants. The imbalance reflected the real-world distribution of variants but may lead to inflated specificity value and potentially bias accuracy measurements. The study highlighted a focus on metrics insensitive to data imbalance, such as balanced accuracy, MCC rankings, and ROC/AUC results. However, sensitivity and specificity rankings serve only as suggestive indicators that necessitate prospective larger-scale clinical validation.
Missense variants are the most common variants in the real world, which may account for a major part of disease-causative variants. Recent studies showed missense variants is frequently associated with epilepsy in established genes of neurodevelopmental disorders, such as several genes reported in recent studies such as ACTB (51), APC2 (52), BCOR (53), CCDC22 (54), DLG3 (55), EP400 (56) FRMPD4 (57), GABRA1 (58), SZT2 (59), TANC2 (60), and KCNK4 (61). This study highlights the need for continued development of in silico pathogenicity predictors; as DEE genetics advances, more accurate and reliable tools are critical for clinical decision-making. Integrating these gene-specific functional datasets with curated clinical evidence provides the most direct way to assess and improve gene-specific predictor performance and reduce bias.
In conclusion, the study provides valuable insights into the performance of various in silico tools for predicting the pathogenicity of CHD2 missense variants. By identifying MutPred2 and AlphaMissense as the first-line predictor, the study has demonstrated the potential of advanced bioinformatics methods to improve the accuracy and reliability of variant pathogenicity assessments. However, it is important to recognize the limitations of these tools and to validate their predictions using experimental methods. In the future, we look forward to the continued development and refinement of in silico tools to better serve the needs of the clinical community and improve patient care and outcomes.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Author contributions
Y-JG: Data curation, Formal analysis, Investigation, Resources, Writing – original draft, Writing – review & editing. P-YW: Methodology, Software, Writing – review & editing. Q-QF: Writing – review & editing. J-HL: Writing – review & editing. XC: Writing – review & editing. X-HL: Conceptualization, Funding acquisition, Writing – review & editing. B-ZG: Data curation, Funding acquisition, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the CAAE Epilepsy Research Fund -Shenji Fund, grant number “CS-2025-038.”
Acknowledgments
We would like to express our gratitude to the generative AI technology (ChatGPT 5.0 and Gemini 2.5 Pro) for its assistance in refining the language and enhancing the clarity of this paper.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that Gen AI was used in the creation of this manuscript. Generative AI was used to assist with language editing and to improve the clarity and readability of the manuscript. It was not used for data generation, analysis, or interpretation of the scientific results. The authors reviewed and edited all AI-generated text and take full responsibility for the final content of the manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2025.1729387/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | All ROC curves. ROC curves were presented with AUC and 95% CI, including SIFT, SIFT4G, Polyphen2_HDIV, Polyphen2_HVAR, MutationAssessor, PROVEAN, MetaSVM, MetaLR, MetaRNN, M-CAP, MutPred2, PrimateAI, DEOGEN2, BayesDel_addAF, BayesDel_noAF, ClinPred, LIST-S2, ESM1b, AlphaMissense, and fathmm-XF_coding_rawscore.
References
1. Berg, AT, Berkovic, SF, Brodie, MJ, Buchhalter, J, Cross, JH, van Emde Boas, W, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE commission on classification and terminology, 2005-2009. Epilepsia. (2010) 51:676–85. doi: 10.1111/j.1528-1167.2010.02522.x
2. Carvill, GL, Heavin, SB, Yendle, SC, McMahon, JM, O'Roak, BJ, Cook, J, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. (2013) 45:825–30. doi: 10.1038/ng.2646
3. Woodage, T, Basrai, MA, Baxevanis, AD, Hieter, P, and Collins, FS. Characterization of the CHD family of proteins. Proc Natl Acad Sci USA. (1997) 94:11472–7. doi: 10.1073/pnas.94.21.11472
4. Kulkarni, S, Nagarajan, P, Wall, J, Donovan, DJ, Donell, RL, Ligon, AH, et al. Disruption of chromodomain helicase DNA binding protein 2 (CHD2) causes scoliosis. Am J Med Genet A. (2008) 146A:1117–27. doi: 10.1002/ajmg.a.32178
5. Rauch, A, Wieczorek, D, Graf, E, Wieland, T, Endele, S, Schwarzmayr, T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. (2012) 380:1674–82. doi: 10.1016/S0140-6736(12)61480-9
6. Suls, A, Jaehn, JA, Kecskés, A, Weber, Y, Weckhuysen, S, Craiu, DC, et al. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet. (2013) 93:967–75. doi: 10.1016/j.ajhg.2013.09.017
7. Carvill, GL, and Mefford, HC. CHD2-related neurodevelopmental disorders. In: MP Adam, J Feldman, GM Mirzaa, RA Pagon, SE Wallace, and A Amemiya, editors. Gene reviews (R). Seattle (WA): University of Washington. (1993). Available at: https://www.ncbi.nlm.nih.gov/books/NBK333201/
8. Lamar, KJ, and Carvill, GL. Chromatin Remodeling proteins in epilepsy: lessons from CHD2-associated epilepsy. Front Mol Neurosci. (2018) 11:208. doi: 10.3389/fnmol.2018.00208
9. Neale, BM, Kou, Y, Liu, L, Ma’ayan, A, Samocha, KE, Sabo, A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. (2012) 485:242–5. doi: 10.1038/nature11011
10. O'Roak, BJ, Stessman, HA, Boyle, EA, Witherspoon, KT, Martin, B, Lee, C, et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun. (2014) 5:5595. doi: 10.1038/ncomms6595
11. van der Ven, AT, Johannsen, J, Kortüm, F, Wagner, M, Tsiakas, K, Bierhals, T, et al. Prevalence and clinical prediction of mitochondrial disorders in a large neuropediatric cohort. Clin Genet. (2021) 100:766–70. doi: 10.1111/cge.14061
12. Routier, L, Verny, F, Barcia, G, Chemaly, N, Desguerre, I, Colleaux, L, et al. Exome sequencing findings in 27 patients with myoclonic-atonic epilepsy: is there a major genetic factor? Clin Genet. (2019) 96:254–60. doi: 10.1111/cge.13581
13. Lebrun, N, Parent, P, Gendras, J, Billuart, P, Poirier, K, and Bienvenu, T. Autism spectrum disorder recurrence, resulting of germline mosaicism for a CHD2 gene missense variant. Clin Genet. (2017) 92:669–70. doi: 10.1111/cge.13073
14. De Maria, B, Balestrini, S, Mei, D, Melani, F, Pellacani, S, Pisano, T, et al. Expanding the genetic and phenotypic spectrum of CHD2-related disease: from early neurodevelopmental disorders to adult-onset epilepsy. Am J Med Genet A. (2022) 188:522–33. doi: 10.1002/ajmg.a.62548
15. Piccolo, B, Gennaro, E, and Pisani, F. A new CHD2 variant: not only severe epilepsy-a case report. Acta Neurol Belg. (2022) 122:1653–6. doi: 10.1007/s13760-021-01820-0
16. Chen, J, Zhang, J, Liu, A, Zhang, L, Li, H, Zeng, Q, et al. CHD2-related epilepsy: novel mutations and new phenotypes. Dev Med Child Neurol. (2020) 62:647–53. doi: 10.1111/dmcn.14367
17. Stranneheim, H, Lagerstedt-Robinson, K, Magnusson, M, Kvarnung, M, Nilsson, D, Lesko, N, et al. Integration of whole genome sequencing into a healthcare setting: high diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Med. (2021) 13:40. doi: 10.1186/s13073-021-00855-5
18. Kaplanis, J, Samocha, KE, Wiel, L, Zhang, Z, Arvai, KJ, Eberhardt, RY, et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. (2020) 586:757–62. doi: 10.1038/s41586-020-2832-5
19. Peng, J, Pang, N, Wang, Y, Wang, XL, Chen, J, Xiong, J, et al. Next-generation sequencing improves treatment efficacy and reduces hospitalization in children with drug-resistant epilepsy. CNS Neurosci Ther. (2019) 25:14–20. doi: 10.1111/cns.12869
20. Alkelai, A, Greenbaum, L, Shohat, S, Povysil, G, Malakar, A, Ren, Z, et al. Genetic insights into childhood-onset schizophrenia: the yield of clinical exome sequencing. Schizophr Res. (2023) 252:138–45. doi: 10.1016/j.schres.2022.12.033
21. Brunet, T, Jech, R, Brugger, M, Kovacs, R, Alhaddad, B, Leszinski, G, et al. De novo variants in neurodevelopmental disorders-experiences from a tertiary care center. Clin Genet. (2021) 100:14–28. doi: 10.1111/cge.13946
22. Benson, KA, White, M, Allen, NM, Byrne, S, Carton, R, Comerford, E, et al. A comparison of genomic diagnostics in adults and children with epilepsy and comorbid intellectual disability. Eur J Hum Genet. (2020) 28:1066–77. doi: 10.1038/s41431-020-0610-3
23. French, CE, Dolling, H, Mégy, K, Sanchis-Juan, A, Kumar, A, Delon, I, et al. Refinements and considerations for trio whole-genome sequence analysis when investigating mendelian diseases presenting in early childhood. HGG Adv. (2022) 3:100113. doi: 10.1016/j.xhgg.2022.100113
24. Galizia, EC, Myers, CT, Leu, C, de Kovel, CGF, Afrikanova, T, Cordero-Maldonado, ML, et al. CHD2 variants are a risk factor for photosensitivity in epilepsy. Brain. (2015) 138:1198–208. doi: 10.1093/brain/awv052
25. Shin, S, Lee, J, Kim, YG, Ha, C, Park, JH, Kim, JW, et al. Genetic diagnosis of children with neurodevelopmental disorders using whole genome sequencing. Pediatr Neurol. (2023) 149:44–52. doi: 10.1016/j.pediatrneurol.2023.09.003
26. Jiao, Q, Sun, H, Zhang, H, Wang, R, Li, S, Sun, D, et al. The combination of whole-exome sequencing and copy number variation sequencing enables the diagnosis of rare neurological disorders. Clin Genet. (2019) 96:140–50. doi: 10.1111/cge.13548
27. Cheng, J, Novati, G, Pan, J, Bycroft, C, Žemgulytė, A, Applebaum, T, et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science. (2023) 381:eadg7492. doi: 10.1126/science.adg7492
28. Choi, Y, Sims, GE, Murphy, S, Miller, JR, and Chan, AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. (2012) 7:e46688. doi: 10.1371/journal.pone.0046688
29. Ioannidis, NM, Rothstein, JH, Pejaver, V, Middha, S, McDonnell, SK, Baheti, S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. (2016) 99:877–85. doi: 10.1016/j.ajhg.2016.08.016
30. Ionita-Laza, I, McCallum, K, Xu, B, and Buxbaum, JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet. (2016) 48:214–20. doi: 10.1038/ng.3477
31. Kumar, P, Henikoff, S, and Ng, PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. (2009) 4:1073–81. doi: 10.1038/nprot.2009.86
32. Malhis, N, Jacobson, M, Jones, SJM, and Gsponer, J. LIST-S2: taxonomy based sorting of deleterious missense mutations across species. Nucleic Acids Res. (2020) 48:W154–61. doi: 10.1093/nar/gkaa288
33. Raimondi, D, Tanyalcin, I, Ferté, J, Gazzo, A, Orlando, G, Lenaerts, T, et al. DEOGEN2: prediction and interactive visualization of single amino acid variant deleteriousness in human proteins. Nucleic Acids Res. (2017) 45:W201–6. doi: 10.1093/nar/gkx390
34. Reva, B, Antipin, Y, and Sander, C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. (2011) 39:e118. doi: 10.1093/nar/gkr407
35. Rogers, MF, Shihab, HA, Mort, M, Cooper, DN, Gaunt, TR, and Campbell, C. FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Bioinformatics. (2018) 34:511–3. doi: 10.1093/bioinformatics/btx536
36. Schwarz, JM, Rödelsperger, C, Schuelke, M, and Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. (2010) 7:575–6. doi: 10.1038/nmeth0810-575
37. Shihab, HA, Gough, J, Cooper, DN, Stenson, PD, Barker, GLA, Edwards, KJ, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. (2013) 34:57–65. doi: 10.1002/humu.22225
38. Shihab, HA, Rogers, MF, Gough, J, Mort, M, Cooper, DN, Day, INM, et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. (2015) 31:1536–43. doi: 10.1093/bioinformatics/btv009
39. Steinhaus, R, Proft, S, Schuelke, M, Cooper, DN, Schwarz, JM, and Seelow, D. MutationTaster2021. Nucleic Acids Res. (2021) 49:W446–51. doi: 10.1093/nar/gkab266
40. Sundaram, L, Gao, H, Padigepati, SR, McRae, JF, Li, Y, Kosmicki, JA, et al. Predicting the clinical impact of human mutation with deep neural networks. Nat Genet. (2018) 50:1161–70. doi: 10.1038/s41588-018-0167-z
41. Vaser, R, Adusumalli, S, Leng, SN, Sikic, M, and Ng, PC. SIFT missense predictions for genomes. Nat Protoc. (2016) 11:1–9. doi: 10.1038/nprot.2015.123
42. Li, J, Zhao, T, Zhang, Y, Zhang, K, Shi, L, Chen, Y, et al. Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. (2018) 46:7793–804. doi: 10.1093/nar/gky678
43. Adzhubei, IA, Schmidt, S, Peshkin, L, Ramensky, VE, Gerasimova, A, Bork, P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
44. Dong, C, Wei, P, Jian, X, Gibbs, R, Boerwinkle, E, Wang, K, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. (2015) 24:2125–37. doi: 10.1093/hmg/ddu733
45. Li, C., Zhi, D, Wang, K, and Liu, X. MetaRNN: differentiating rare pathogenic and rare benign missense SNVs and In Dels using deep learning. Genome medicine, (2022) 14:115. doi: 10.1186/s13073-022-01120-z
46. Jagadeesh, KA, Wenger, AM, Berger, MJ, Guturu, H, Stenson, PD, Cooper, DN, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. (2016) 48:1581–6. doi: 10.1038/ng.3703
47. Pejaver, V, Urresti, J, Lugo-Martinez, J, Pagel, KA, Lin, GN, Nam, HJ, et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat Commun. (2020) 11:5918. doi: 10.1038/s41467-020-19669-x
48. Pejaver, V, Byrne, AB, Feng, BJ, Pagel, KA, Mooney, SD, Karchin, R, et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am J Hum Genet. (2022) 109:2163–77. doi: 10.1016/j.ajhg.2022.10.013
49. Alirezaie, N, Kernohan, KD, Hartley, T, Majewski, J, and Hocking, TD. ClinPred: prediction tool to identify disease-relevant nonsynonymous single-nucleotide variants. Am J Hum Genet. (2018) 103:474–83. doi: 10.1016/j.ajhg.2018.08.005
50. Brandes, N, Goldman, G, Wang, CH, Ye, CJ, and Ntranos, V. Genome-wide prediction of disease variant effects with a deep protein language model. Nat Genet. (2023) 55:1512–22. doi: 10.1038/s41588-023-01465-0
51. Yan, HJ, Wang, PY, Liu, WH, Gu, YJ, Pan, JC, Li, H, et al. De novo ACTB variant associated with juvenile-onset temporal lobe epilepsy with Favorable outcomes. Hum Mutat. (2025) 2025:9951922. doi: 10.1155/humu/9951922
52. Jin, L, Li, Y, Luo, S, Peng, Q, Zhai, QX, Zhai, JX, et al. Reprint of: recessive APC2 missense variants associated with epilepsies without neurodevelopmental disorders. Seizure. (2024) 116:87–92. doi: 10.1016/j.seizure.2024.03.006
53. Li, X, Bian, WJ, Liu, XR, Wang, J, Luo, S, Li, BM, et al. BCOR variants are associated with X-linked recessive partial epilepsy. Epilepsy Res. (2022) 187:107036. doi: 10.1016/j.eplepsyres.2022.107036
54. He, YL, Ye, YC, Wang, PY, Liang, XY, Gu, YJ, Zhang, SQ, et al. CCDC22 variants caused X-linked focal epilepsy and focal cortical dysplasia. Seizure. (2024) 123:1–8. doi: 10.1016/j.seizure.2024.10.007
55. He, YY, Luo, S, Jin, L, Wang, PY, Xu, J, Jiao, HL, et al. DLG3 variants caused X-linked epilepsy with/without neurodevelopmental disorders and the genotype-phenotype correlation. Front Mol Neurosci. (2024) 16:1290919. doi: 10.3389/fnmol.2023.1290919
56. Luo, SW, and Peng-Yu,. Variants in Ep400, encoding a chromatin remodeler, cause epilepsy with neurodevelopmental disorders. Am J Hum Genet. (2025) 112:1–19. doi: 10.1016/j.ajhg.2024.11.010
57. Li, RK, Li, H, Tian, MQ, Li, Y, Luo, S, Liang, XY, et al. Investigation of FRMPD4 variants associated with X-linked epilepsy. Seizure. (2024) 116:45–50. doi: 10.1016/j.seizure.2023.05.014
58. Liu, WH, Luo, S, Zhang, DM, Lin, ZS, Lan, S, Li, X, et al. De novo GABRA1 variants in childhood epilepsies and the molecular subregional effects. Front Mol Neurosci. (2024) 16:1321090. doi: 10.3389/fnmol.2023.1321090
59. Luo, S, Ye, XG, Jin, L, Li, H, He, YY, Guan, BZ, et al. SZT2 variants associated with partial epilepsy or epileptic encephalopathy and the genotype-phenotype correlation. Front Mol Neurosci. (2023) 16:1162408. doi: 10.3389/fnmol.2023.1162408
60. Luo, S, Zhang, WJ, Jiang, M, Ren, RN, Liu, L, Li, YL, et al. De novo TANC2 variants caused developmental and epileptic encephalopathy and epilepsy. Epilepsia. (2025) 66:2365–78. doi: 10.1111/epi.18358
61. Yan, HJ, Liu, WH, Xu, MX, Wang, PY, Gu, YJ, Li, H, et al. De novo KCNK4 variant caused epilepsy with febrile seizures plus, neurodevelopmental abnormalities, and hypertrichosis. Front Genet. (2025) 16:1499716. doi: 10.3389/fgene.2025.1499716
Keywords: missense variant, in silico tools, CHD2, MutPred2, AlphaMissense, developmental and epileptic encephalopathy, optimizing genetic diagnosis
Citation: Gu Y-J, Wang P-Y, Fu Q-Q, Lai J-H, Chen X, Liu X-H and Guan B-Z (2025) Epilepsy-associated CHD2 missense variants and optimization strategies for genetic diagnosis: a comparative analysis of algorithms. Front. Neurol. 16:1729387. doi: 10.3389/fneur.2025.1729387
Edited by:
Miriam Kessi, Central South University, ChinaReviewed by:
Mingqiang Li, University of South China, ChinaTieshi Zhu, Zhanjiang Central Hospital, China
Sifen Xie, Guangdong 999 Brain Hospital, China
Copyright © 2025 Gu, Wang, Fu, Lai, Chen, Liu and Guan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bao-Zhu Guan, YnpndWFuQHFxLmNvbQ==; Xiang-Hong Liu, bHhoNzE3NkAxMjYuY29t
†These authors have contributed equally to this work
‡ORICD: Peng-Yu Wang, orcid.org/0009-0001-6848-2021