





- Howard Hughes Medical Institute, Department of Biological Sciences, Columbia University, New York, NY, USA
Two-photon microscopy is often performed at slow frame rates due to the need to serially scan all points in a field of view with a single laser beam. To overcome this problem, we have developed two optical methods that split and multiplex a laser beam across the sample. In the first method a diffractive optical element (DOE) generates a fixed number of beamlets that are scanned in parallel resulting in a corresponding increase in speed or in signal-to-noise ratio in time-lapse measurements. The second method uses a computer-controlled spatial light modulator (SLM) to generate any arbitrary spatio-temporal light pattern. With an SLM one can image or photostimulate any predefined region of the image such as neurons or dendritic spines. In addition, SLMs can be used to mimic a large number of optical transfer functions including light path corrections as adaptive optics.
We review here our recent efforts (Nikolenko et al., 2008; Watson et al., 2009) to improve two-photon microscopic imaging. These efforts focus on the use of multiple beamlets and seek to better utilize the power available from lasers to improve imaging speed, signal-to-noise ratio and flexibility. While our motivation was to utilize two-photon microscopy to better understand how neurons and neuronal circuits contribute to brain function, the methods here are applicable beyond that specific question. In this focused review we concentrate on these approaches and their context, but we do not intend to review comprehensively other fast imaging techniques. For that purpose, we refer readers to recent general reviews of the field (Saggau, 2006; Grewe and Helmchen, 2009).
Early Approaches to Imaging of Neuronal Activity
Calcium indicator dyes have led to the advent of time lapse microscopic techniques for the study of neuronal and neural circuit function (Tsien, 1981; Grynkiewicz et al., 1985). After loading these dyes into populations of neurons, experimenters are able to measure action potential generation in neuronal ensembles via their fluorescence (Yuste and Katz, 1991; Smetters et al., 1999; Goldberg and Yuste, 2009). Such imaging was initially performed using wide-field excitation, that is, simply bathing the whole field of view in large amounts of excitation light and imaging fluorescent emission with a camera as a detector (Yuste and Katz, 1991; Smetters et al., 1999). This is straightforward and easy to instantiate but provides limited spatial resolution of imaging, especially in the direction parallel to the optical axis (axial or Z-resolution as referred later).
To overcome this problem, one can use confocal microscopy – a method wherein a pinhole in front of the detector improves the axial resolution of detection (White et al., 1987). Confocal microscopy traditionally relied on raster scanning a single excitation beam progressively across all points in the sample. This scanning process limits imaging to ∼0.2–1 s per frame for the majority of commercial systems. To increase speed, the confocal spinning disk with camera-based detection was developed (Nipkow, 1885; Egger and Petran, 1967). In this system, the imaging process is parallelized: multiple beamlets (each with an individual imaging pinhole) scan the sample at the same time, thus reducing the time necessary to scan whole field of view. The modern instantiations of this system allow for imaging at relatively high speeds (30 Hz or more) and are well suited to many basic imaging experiments in vitro (Ikegaya et al., 2004).
All of these methods use linear (one-photon) absorption of excitation light and suffer from significant photobleaching rendering the fluorophore less fluorescent over time. This limits the total amount of time one can image a particular sample, therefore limiting experimental flexibility as well as data yields. Beyond photobleaching, the excitation light in one-photon systems is typically in UV or visible part of spectrum – i.e. at a wavelength that is easily absorbed and scattered by tissue decreasing the resolution and power of incident light.
Two-Photon Microscopy – Single Beam Methods
Two-photon imaging was developed partly as a response to these challenges (Denk et al., 1990). In this system, fluorophores are excited by absorption of a pair of photons each with wavelength double that of a photon capable of individually exciting a given fluorophore alone. Only at a very small (roughly 1 cubic micron) region surrounding the exact point of focus is light effectively absorbed to fully excite fluorescence. Thus, the non-linear absorption of light naturally provides axial resolution. This therefore limits photobleaching as well as any possible photodamage to points where the signal is acquired from. Two-photon imaging allows high spatial resolution imaging deep into tissues, including in vivo imaging in the mammalian brain (Denk et al., 1994), and also provides a means by which to carry out photochemical manipulations of cells and circuits (Denk, 1994).
Time lapse imaging using two-photon systems has traditionally relied on the same serial scanning as confocal imaging has and therefore suffers from the same time limitations on signal acquisition. While faster scanning regimes (as discussed below) represent a simple and common approach to this problem, such approaches reduce dwell time per pixel and can compromise signal-to-noise ratio. Furthermore, simply increasing excitation power is frequently not an option because above a certain intensity chromophore saturation and biological tissue photodamage occur. Consequently many experimenters use a laser power just below the photodamage and/or chromophore saturation threshold to maximize signal without inducing damage. In some cases this means much of the available laser power is not used, though in deep tissue imaging this is often not the case since more power must be used to overcome scattering.
Pulse Splitting
A few methods aimed at improving two-photon imaging utilize on alteration of the excitation beam itself. Of these, the technique of splitting ultrafast pulses in time by increasing repetition frequency (Ji et al., 2008) stands out and allows application of greater total laser power to the sample without oversaturation or photodamage. Pulse splitting utilizes a beam splitter with delaying mirrors to split each laser pulse into two smaller pulses. Each new pulse excites one quarter the fluorescence photons excited by the original pulse (the two-photon fluorescence process is proportional to square of excitation power), but there are twice as many of pulses, so without a change in the laser output power, this system creates half as much total excitation. On the other hand, by increasing the laser power, one can induce more total excitation and greater signal without crossing damage thresholds or saturating chromophores. This is true since one may double laser output to render each new pulse as powerful as the original pulse but there remain twice as many pulses (as long as the interpulse interval is sufficient to allow molecules of fluorophore to physically relax, this has not been a problem in practice, given that fluorescence lifetime of typical dyes is in 0.5–20 ns range). This approach of changing the properties of the laser beam itself may be combined with other methods discussed below.
Enhanced Scanning Methods
We and others have concentrated on optimizing the scanning aspects of two-photon microscopy. Traditionally “frames” in a time lapse movie were created by scanning the focused laser across the microscopic field in a series of parallel lines. The scanning occurred by control of the angle of the laser path using galvanometer mirrors which pivot about fixed axes. Resonant mirror systems have gained some favor since they allow for very high speed (up to several KHz) scanning in one axis for faster scanning of each line in the image (Tsien, 1995). This technology is based on galvanometer mirrors that resonantly sweep back-and-forth at a fixed frequency when activated and can produce very nice full frame scanning results (Rochefort et al., 2009), though it does not allow for flexibility in how the scanners are used, restricting at least one axis to linear back-and-forth scanning. Another fast scanning method utilizes acousto-optical devices which control the beam path not with mirrors but with crystals whose refractive properties are very quickly altered by varying sound waves passing through them (Goldstein et al., 1990). These devices allow for flexible fast scanning and can be optimized to produce very useful results (Otsu et al., 2008), but have the limitation that only a narrow range of angles can be scanned without degradation of image quality.
Random Access Scanning
A different strategy is to only scan the points of interest rather than all of the pixels in a frame (Nikolenko et al., 2007). In this method the galvanometer mirror is commanded to jump from one point of interest (i.e., cell body) to another in series until all points of interest are sampled and then repeat the sampling cycle – each cycle representing a “frame”. With relatively small numbers of points of interest this can allow for very fast scans, though the method slows down significantly with larger numbers of points. This method has been optimized in recent work wherein the momentum of the scan mirrors was taken into account such that scanning either occurs with maximal optimized acceleration and deceleration between points (Lillis et al., 2008) or as a single smooth movement at constant speed passing through all of the necessary points of interest without stopping (Gobel and Helmchen, 2007), thereby improving the efficiency of the method yet more. These authors later expanded this method into the third dimension using a Z-scanning objective adaptor (Gobel et al., 2007).
Two-Photon Microscopy – Multibeam Methods
A fundamentally different approach is described below: multibeam scanning. While some applications, such as deep tissue imaging require use of large amounts of power to compensate for scattering, many other applications use as little as 1/20th of the available laser power in order to keep single beam power below threshold for saturation and photodamage. To more efficiently utilize the available laser power in such circumstances we developed a multibeam approach in which the source laser is split into multiple beamlets using a diffractive element (Watson et al., 2009). These many beamlets are then simultaneously directed to different parts of the tissue (Sacconi et al., 2003, Watson et al., 2009). This allows use of the full power of the laser to either speed up imaging (similar to multibeam confocal imaging described above) or to increase sampling per frame and therefore signal-to-noise ratio.
A particularly interesting though not yet practical approach takes this notion to its fullest conceptual extent (Oron et al., 2005). In this method, the laser simultaneously illuminates the entire field of view continuously. In a sense this is like “bathing” the sample in epifluorescent excitation but relies on two-photon excitation and on focusing the excitation pulse in time, therefore providing excellent Z-resolution. As might be imagined, the limitation of this method is average laser power – despite the high power of currently available lasers, this method demands so many photons per unit time that fluorescence intensities are very low and imaging is necessarily slow to allow sufficient integration. However in the future, with more powerful lasers this may be a promising approach.
Diffractive Optical Elements and Spatial Light Modulators
Our initial approach used diffractive optical elements (DOEs) to spatially multiplex beamlets to improve imaging. Each of these beamlets has the same power as the single beam in a traditional optimized system. There exists a well-made and highly functional commercial variation on this technology (Nielsen et al., 2001) which uses multiple beam splitters rather than a DOE to create beamlets. This technology has been used to produce useful scientific observations (Kurtz et al., 2006) however it is over 100-fold more expensive than the method we describe (Watson et al., 2009) and does not clearly outperform our design.
A DOE is an optical device – essentially a diffractive grating that allows splitting of a single beam from a coherent source (laser) into multiple beamlets using the effects of diffraction. In practice the DOE is usually a piece of transparent medium (glass, plastic, fused silica, etc.) with embossed pattern on its surface. A single laser beam is directed to the thin element and small variations in thickness of at different points in the element perturb the phase of electromagnetic waves to allow interference to create a specific final output pattern of beamlets, for instance a linear array as in Figure 1 (O’Shea, 2004).
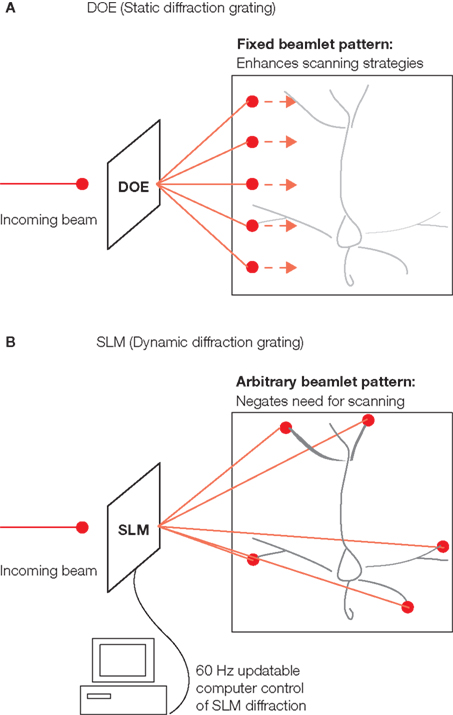
Figure 1 Multibeam two-photon excitation with diffractive optical elements (DOE) or spatial light modulators (SLM). Both the DOE and the SLM use diffraction to create an output pattern of multiple beamlets of light from a single input beam. (A) DOE is a static diffraction grating which creates a single particular output pattern, in this case a linear array of evenly spaced beamlets. These beamlets can be used to increase speed and/or increase sampling of imaging given that they allow for simultaneous exposure of multiple full power excitation points either in parallel or in series over portions of the field. (B) The SLM is dynamically controllable via computer and can create arbitrary patterns of output light to fit the spatial aspects of particular imaging fields (i.e., targeting particular neurites or cells). Furthermore, the SLM can have a new diffraction grating pattern thereby creating a new output pattern of beamlets every 16.7 ms (or even faster depending on type of the used SLM). SLMs allow scanless microscopy for both imaging as well as photostimulation experiments all at multiple spatial points simultaneously.
Besides DOEs, which have an imprinted fixed pattern in them, we have used spatial light modulators (SLMs) (Nikolenko et al., 2008). SLMs are essentially DOEs made not of an etched medium, but instead a dynamic medium which can be controlled by a computer to create arbitrary diffraction patterns. This allows for a great deal of flexibility and customizability and we view it as particularly powerful for neuro-scientific experiments (Figures 1A vs 1B).
We utilize beam multiplexing approaches in three different ways: (1) Signal boost: increasing signal by oversampling, (2) Speed boost: increasing speed with parallel scanning, and (3) to create scanless dynamic SLM-based system; all of which will be described in detail in the following sections.
Signal Boost
In the first method, we use a diffractive optical element (DOE) combined with a standard galvanometer mirror scanners to essentially oversample the field of view to improve signal reliability. In the case of a DOE creating n beamlets, this has the effect of exposing each point in the scanned field to n-fold as much excitation per image frame, without exposing those points to n-fold as much excitation light at once. This therefore avoids non-linear photodamage effects. Thus, the signal each point in the created image is a more reliable indicator of the true brightness of the underlying sample. Given that we use a photomultiplier tube as a detector the brightness of each pixel in the resultant image is actually the total brightness produced at an instant in time, which is in this case the result of emission from multiple points in the sample excited by the DOE. The improvement in signal-to-noise ratio is therefore achieved with the tradeoff of a decrease lateral spatial resolution: blurring occurs in accordance with the spatial spread of the beamlets swept. This can be mitigated by choosing a DOE or a modifying telescope after the DOE such that the beamlets are spread over a smaller spatial range. For our application of imaging neuronal cell bodies, the beamlets are easily spaced to a distance less than 1 cell body diameter (in our case 7.5 μm with 20× objective though this will change with different objectives) and the loss of spatial resolution is minimal compared with the increase in signal-to-noise ratio from the cell as a whole. This trade-off may be more limiting in situations where more scattering is present either due to insufficient power of excitation or decreased resolution of signal sources. Signal boost mode has been demonstrated to be effective in improving signal-to-noise ratios during frame scans (Watson et al., 2009), line scans (Watson et al., 2009) and random access scans (Nikolenko et al., 2007). This simple approach requires minimal modification of the imaging apparatus. It is also very flexible and capable of acting synergistically with almost any imaging approach or modification.
Speed Boost
A second use of the DOE is one we have called “speed boost” (Watson et al., 2009). This is a method for making full frame scans by using beamlets spread equidistantly over the entire frame. These beamlets each scan a fraction or stripe of the field of interest, and by doing so simultaneously will quickly cover the full field. n equally spaced beamlets can thereby scan a field in roughly 1/n the time needed to scan with a single beam. This method requires only small modification of existing scanning setups: (1) setting up the DOE and (2) setting up an imaging device which spatially resolves light coming from many sources at once such as a camera or multi-anode PMT (without modification of scanning software). Using this method we were able to create high quality images at video rate with good signal-to-noise ratios. We were able to very effectively detect action potential-related calcium fluxes in neurons at high speed as shown in Figure 2. While this strategy is inexpensive and easily combined with other techniques it is reliant on large amounts of laser power and is only useful in situations where extra power is available.
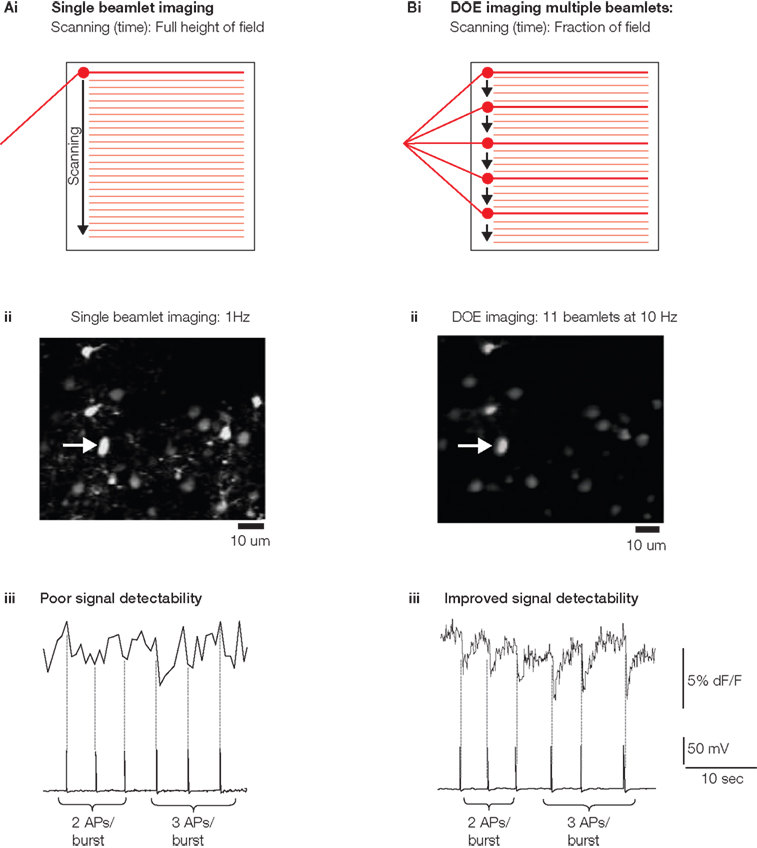
Figure 2. Diffractive optical element (DOE)-based high-speed imaging. Adapted from Watson et al. (2009). (Ai) schematic of traditional single beam imaging requiring that a single beam scan of all lines to be imaged necessitating a relatively long scan time per frame. (Bi) Multiple beamlets created by the DOE (each as powerful as the single beamlet used for single beam imaging) simultaneously sweep the field with each beamlet covering a small portion of the territory to quickly excite the entire field of view. (Aii) Image created by scanning across a population of calcium indicator-loaded cells (Fura 2-AM) using single beam scanning and photomultiplier tube detection. Scanning at this resolution and mirror sweep speed was 1 Hz at the fastest. (Bii) DOE 10 Hz scanning of same field of with 11 beamlets and with light gathering using a CCD camera at the same mirror sweep speed. (Aiii) Calcium indicator fluorescence recorded with single beam imaging from a patch clamped neuron indicated by arrow in (Aii). Above: calcium indicator fluorescence versus time of the patch clamped neuron. Below: electrophysiological tracing recorded simultaneously from the same cell as was being imaged, demonstrating periods of induced action potential generation. The first three action potential bursts have two action potentials per burst and are undetectable in the calcium indicator brightness trace, the later bursts have three action potentials each give somewhat clearer signals in the calcium indicator tracing. (Biii) Imaging of calcium transients at 10 Hz using DOE imaging. Bursts of either two or three action potentials are easily detectible in this higher speed scheme, likely due increased sampling rate. All imaging at 800-nm light with detection at 510 nm.
Spatial Light Modulator-Based Scanless Two-Photon Microscopy
We and others have developed another multibeam approach which uses SLMs in one- photon (Lutz et al., 2008) and two-photon regimes (Nikolenko et al., 2008). SLMs not only allow for truly simultaneous monitoring of fluorescence at multiple targets, but also allow for simultaneous photomanipulation of targets in a customizable and temporally dynamic manner. The core of this method is the SLM itself which acts as a programmable DOE to construct a custom light or beamlet pattern (Figures 3A,B), and can in fact dynamically change that pattern over time (at 60 Hz in our case). SLMs can create any pattern of light within the output resolution of the SLM itself (256 × 256 in our case) since they are made of dynamic controllable materials such as liquid crystal which allow the diffractive properties of each “pixel” of the SLM to be modified by electrical signals from a computer. It is important that as opposed to micromirror technology (Wang et al., 2007) in which patterns can be created by blocking out light at individual pixels to varying degrees (a purely subtractive process), the phase-only SLM (and DOE) preserves incident light by creating both destructive and constructive interference. For example, a beamlet pattern on a 256 × 256 output image in which only three pixels are allowed to be bright (i.e., three beamlets) keeps 50% of the input power in the output pattern, versus <1% with micromirror devices. An SLM approach was developed for one-photon microscopy by (Lutz et al., 2008), in parallel to the two-photon approach we developed (Nikolenko et al., 2008).
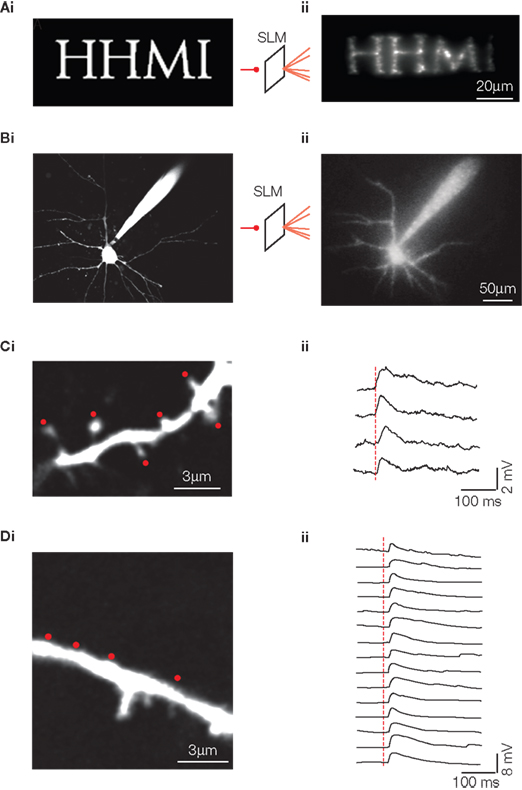
Figure 3. Spatial light modulator (SLM) two-photon imaging and photostimulation. (Ai) Arbitrary pattern of desired output from SLM in the shape of letters “HHMI”. (Aii) Pattern by SLM from a single circular input beam, projected through 60× 0.9NA objective into agar block filled with Alexa 488 fluorescent dye. (Bi) Similar to (Ai), however more complex desired output pattern of output from SLM in the shape of a patch clamped neuron. This image was thresholded and binarized to create a final output template. (Bii) SLM-created pattern projected into Alexa 488-filled agar block under 40× 0.8NA objective. (Ci) Basal dendrite from a layer 5 pyramidal neuron in an acute slice of mouse neocortex. The neuron is loaded with Alexa-488 and the slice bathed in MNI-glutamate. The SLM was used to create a diffraction pattern placing beamlets adjacent to individual spines of the dendrite (red dots). Pulses of excitation light delivered simultaneously at these points using the SLM triggered uncaging of MNI-glutmate. (Cii) Whole cell patch clamp recording at cellular soma of integrated potentials created by simultaneous uncaging at the spots indicated. Each horizontal trace represents a repetition of the stimulation and reveals the uncaging potentials recorded at the soma. (Di) Similar to (C), however now in a new neuron with uncaging targeted to locations near a dendritic shaft rather than near spines. (Dii) Patch clamp recordings of multiple trials of multi-point simultaneous uncaging revealing the uncaging potentials recorded at the soma. See Nikolenko et al. (2008) for more detailed description of the methods used for these recordings.
Imaging-based uses of the SLM include simultaneous time lapse imaging of a fixed set of multiple targets. This is accomplished by delivering excitation light to all targets continuously while monitoring emission at all those sites at once using a camera. The number of excitation points which may be specified and still receive sufficient excitation power to give good signals is currently limited by the amount of power produced by modern lasers (roughly 20 neurons in brain slice experiments). Liquid crystal SLMs can update their output pattern every 16.7 ms (or more often, depending on the model of SLM) for faster than video rate updating of projected patterns. This temporal flexibility means the SLM may also be useful as a multiplexed random access scanner. In this mode it would update to a new set of multiple targets (rather than a single target) every 16.7 ms, thus allowing allow for random access scanning across large numbers of targets. This may be of particular interest in neuronal integration or circuit integration studies.
Perhaps the most unique application of the SLM, however, is for induction of photochemical effects such as uncaging of neuro-active compounds at multiple points simultaneously (Figures 3C,D). Caged neurotransmitters such as glutamate (Matsuzaki et al., 2001; Fino et al., 2009) and GABA (Rial Verde et al., 2008) are coming into increased use for manipulation of neurons (Araya et al., 2006; Nikolenko et al., 2008). The SLM allows for stimulation or inhibition of multiple neurites or neurons at once to achieve large-scale yet highly specific neuro-excitatory/-inhibitory effects One may be able to perform studies of whole neuron or whole circuit integrative properties via stimulation of specific targets chosen based on known properties of those targets (i.e., connectivity at spines, or firing properties of neurons) (Nikolenko et al., 2008).
Summary
In conclusion, we have explored methods that exploit the idea of using diffraction to optimize two-photon microscopy. We have used static DOE as well as active, computer-controlled SLM to dynamically control the distribution of excitation light on the sample. While these multibeam methods are currently limited in applications with large power requirements, we have proven their practicality and functionality. Not only are they currently useful in non-power-limited settings but stand to be equally useful in power-limited applications when more powerful lasers become available. We have devised several novel experimental schemes for improving either speed and/or sensitivity of imaging, specifically applicable to non-linear (two-photon) modalities. The use of computer-controlled phase-only SLM also allows for photostimulation of multiple regions of the sample while largely preserving excitation power. In addition, a newly developed imaging technique relies on SLM to continuously illuminate preselected regions of the sample acquire time lapse signals from those regions faster than video rate. The proposed and demonstrated techniques entail insertion of simple optical elements into a two-photon imaging rig and thus can be easily and inexpensively implemented and added to existing experimental equipment, including commercial and custom two-photon microscopes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the laboratory for and comments. Supported by the Kavli Institute for Brain Science, the NINDS and the NEI.
Key Concepts
Fluorescent molecule which binds to calcium and whose fluorescence spectrum is altered by whether or not it is bound to calcium. By monitoring fluorescence of a population of these molecules it is possible to assess changes in calcium concentration in a compartment, such as a cell. “Calcium imaging” is imaging utilizing a calcium indicator dye and can be used to detect action potential firing since each spike increases the intracellular free calcium concentration.
Process wherein fluorescent molecules lose their ability to fluoresce after being exposed to excitation light, due to chemical changes induced in those molecules by the excitation light itself.
High resolution imaging technique wherein rather than a single photon exciting molecules to fluoresce, the energies of two long-wavelength low-energy photons are combined. Spatial resolution improved by this non-linear excitation since fluorescence only occurs at the regions of very highest photon concentration thereby, for most practical applications, restricting that excitation to within 1 micron of the focal point. In addition, near-infrared light is typically used for two-photon excitation, thus allowing for deeper tissue penetration and imaging of thick biological specimens (including in vivo imaging) due to reduced unspecific scattering and absorption at longer wavelengths.
Emission of photons by molecules after those molecules absorb and are therefore “excited” by photons. This process occurs via excitation of an orbital electron of the molecule to a higher quantum state (upon absorption of excitation photon). With subsequent relaxation of that same electron back to its ground state, a low energy photon is emitted.
Small mirrors mounted on electrical devices which rotate based on the current passing through them (galvanometers) – used in these applications to direct a laser beam. Mirrors are mounted in very close proximity and orthogonally and by rotating independently these mirrors fully control the two dimensional angle of the outgoing laser beam.
Many lasers produce pulses or groups of photons in bursts at some frequency rather than a continuous stream of light. Included in this group are the “ultrafast” lasers (usually based on titanium–sapphire) used in two-photon microscopy; these typically produce pulses of approximately 140 fs duration and at 80 MHz repetition rate. Ultrafast pulsed lasers effectively “pack” the energy of continuous radiation into very short pulses, thus producing extremely high instantaneous intensity of light necessary for efficient non-linear excitation.
Change in direction of propagation of waves such as light (electromagnetic wave) upon encountering an inhomogeneity in its propagation medium. By selectively diffracting different cross-sectional regions of an incoming beam wavefront, those waves will “interfere” or superimpose with each other such that they either cancel each other or reinforce each other to create a desired 2D output pattern.
References
Araya, R., Eisenthal, K. B., and Yuste, R. (2006). Dendritic spines linearize the summation of excitatory potentials. Proc. Natl. Acad. Sci. USA 103, 18779–18804.
Denk, W. (1994). Two-photon scanning photochemical microscopy: mapping ligand-gated ion channel distributions. Proc. Natl. Acad. Sci. USA 91, 6629–6633.
Denk, W., Delaney, K. R., Gelperin, A., Kleinfeld, D., Strowbridge, B. W., Tank, D. W., and Yuste, R. (1994). Anatomical and functional imaging of neurons using 2-photon laser scanning microscopy. J. Neurosci. Meth. 54, 151–162.
Denk, W., Strickler, J. H., and Webb, W. W. (1990). Two-photon laser scanning fluorescence microscopy. Science 248, 73–76.
Egger, M., and Petran, M. (1967). New reflected-light microscope for viewing unstained brain and ganglion cells Science 157, 305–307.
Fino, E., Araya, R., Peterka, D. S., Salierno, M., Etchenique, R., and Yuste, R. (2009). RuBi-glutamate: two-photon and visible-light photoactivation of neurons and dendritic spines. Front. Neural Circuits 3, 2. doi:10.3389/neuro.04.002.2009.
Gobel, W., and Helmchen, F. (2007). New angles on neuronal dendrites in vivo. J. Neurophysiol. 98, 3770–3779.
Gobel, W., Kampa, B. M., and Helmchen, F. (2007). Imaging cellular network dynamics in three dimensions using fast 3D laser scanning. Nat. Methods 4, 73–79.
Goldberg, J. H., and Yuste, R. (2009). Two-photon calcium imaging of spines and dendrites. CSH Protoc. 6, pdb.prot5231.
Goldstein, S. R., Hubin, T., Rosenthal, S., and Washburn, C. (1990). A confocal video-rate laser-beam scanning reflected-light microscope with no moving parts. J. Microsc. 157, 29–38.
Grewe, B. F., and Helmchen, F. (2009). Optical probing of neuronal ensemble activity. Curr. Opin. Neurobiol. 19, 520–529.
Grynkiewicz, G., Poenie, M., and Tsien, R. Y. (1985). A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450.
Ikegaya, Y., Aaron, G., Cossart, R., Aronov, D., Lampl, I., Ferster, D., and Yuste, R. (2004). Synfire chains and cortical songs: temporal modules of cortical activity. Science (New York, NY) 304, 559–564.
Ji, N., Magee, J. C., and Betzig, E. (2008). High-speed, low-photodamage nonlinear imaging using passive pulse splitters. Nat. Methods 5, 197–202.
Kurtz, R., Fricke, M., Kalb, J., Tinnefeld, P., and Sauer, M. (2006). Application of multiline two-photon microscopy to functional in vivo imaging. J. Neurosci. Methods 151, 276–286.
Lillis, K. P., Eng, A., White, J. A., and Mertz, J. (2008). Two-photon imaging of spatially extended neuronal network dynamics with high temporal resolution. J. Neurosci. Methods 172, 178–184.
Lutz, C., Otis, T., DeSars, V., Charpak, S., DiGregorio, D., and Emiliani. V. (2008). Holographic photolysis of caged neurotransmitters. Nat. Methods 5, 821–827.
Matsuzaki, M., Ellis-Davies, G. C., Nemoto, T., Miyashita, Y., Iino, M., and Kasai, H. (2001). Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 4, 1086–1092.
Nielsen, T., Fricke, M., Hellweg, D., and Andresen, P. (2001). High efficiency beam splitter for multifocal multiphoton microscopy. J. Microsc. 201, 368–376.
Nikolenko, V., Poskanzer, K. E., and Yuste, R. (2007). Two-photon photostimulation and imaging of neural circuits. Nat. Methods 4, 943–950.
Nikolenko, V., Watson, B. O., Araya, R., Woodruff, A., Peterka, D. S., and Yuste, R. (2008). SLM Microscopy: scanless two-photon imaging and photostimulation using spatial light modulators. Front. Neural Circuits 2–5, 1–14. doi:10.3389/neuro.04.005.2008.
Nipkow, P. (1885). Elektrisches Teleskop. vol. Patentschrift 30105 Germany: Kaiserliches Patentamt, B.
O’Shea, D. C., and Society of Photo-optical Instrumentation Engineers. (2004). Diffractive Optics: Design, Fabrication, and Test. Bellingham, Washington: SPIE Press.
Oron, D., Tal, E., and Silberberg, Y. (2005). Scanningless depth-resolved microscopy. Opt. Express 13, 1468–1476.
Otsu, Y., Bormuth, V., Wong, J., Mathieu, B., Dugue, G. P., Feltz, A., and Dieudonne, S. (2008). Optical monitoring of neuronal activity at high frame rate with a digital random-access multiphoton (RAMP) microscope. J. Neurosci. Methods 173, 259–270.
Rial Verde, E. M., Zayat, L., Etchenique, R., and Yuste, R. (2008). Photorelease of GABA with visible light using an inorganic caging group. Front. Neural Circuits 2:2. doi:10.3389/neuro.3304.3002.3200.
Rochefort, N. L., Garaschuk, O., Milos, R. I., Narushima, M., Marandi, N., Pichler, B., Kovalchuk, Y., and Konnerth, A. (2009). Sparsification of neuronal activity in the visual cortex at eye-opening. Proc. Natl. Acad. Sci. USA 106, 15049–15054.
Sacconi L, F. E., Antolini, R., Taghizadeh, M. R., Choudhury, A., and Pavone, F. S. (2003). Multiphoton multifocal microscopy exploiting a diffractive optical element. Opt. Lett. 28, 1918–1920.
Saggau, P. (2006). New methods and uses for fast optical scanning. Curr. Opin. Neurobiol. 16, 543–550.
Smetters, D., Majewska, A., and Yuste, R. (1999). Detecting action potentials in neuronal populations with calcium imaging. Methods (San Diego, CA) 18, 215–221.
Tsien, R. Y. (1981). A non-disruptive technique for loading calcium buffers and indicators into cells. Nature 290, 527–528.
Tsien, R. Y., and Bacskai, B.J. (1995). “Video-rate confocal microscopy,” in Handbook of Biological Confocal Microscopy, ed. J. B. Pawley (New York: Plenum Press), 459–478.
Wang, S., Szobota, S., Wang, Y., Volgraf, M., Liu, Z., Sun, C., Trauner, D., Isacoff, E. Y., and Zhang, X. (2007). All optical interface for parallel, remote, and spatiotemporal control of neuronal activity. Nano Lett. 7, 3859–3863.
Watson, B. O., Nikolenko, V., and Yuste, R. (2009). Two-photon imaging with diffractive optical elements. Front Neural Circuits 3, 6. doi:10.3389/neuro.04.006.2009.
White, J. G., Amos, W. B., and Fordham, M. (1987). An evaluation of confocal versus conventional imaging of biological structures by fluorescence light microscopy. J. Cell Biol. 105, 41–48.
Keywords: diffractive optical element, spatial light modulator, two-photon microscopy, imaging, calcium imaging
Citation: Watson BO, Nikolenko V, Araya R, Peterka DS, Woodruff A and Yuste R (2010) Two-photon microscopy with diffractive optical elements and spatial light modulators. Front. Neurosci. 4:29. doi: 10.3389/fnins.2010.00029
Received: 23 February 2010;
Paper pending published: 23 March 2010;
Accepted: 28 April 2010;
Published online: 15 September 2010
Edited by:
Gero Miesenboeck, Oxford University, UKReviewed by:
Alan Fine Dalhousie University, CanadaFritjof Helmchen University of Zurich, Switzerland
Copyright: © 2010 Watson, Nikolenko, Araya, Peterka, Woodruff and Yuste. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Brendon Watson, Department of Biological Sciences, Columbia University, 1212 Amsterdam Avenue, Box 2435, New York, NY 10027.Ym93NEBjb2x1bWJpYS5lZHU=