


- 1 Department of Molecular Neurology, SYGNIS Bioscience, Heidelberg, Germany
- 2 Faculty of Medicine, University of Strasbourg, Strasbourg, France
- 3 Institut National de la Santé et de la Recherche Médicale U692, Strasbourg, France
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease that results in progressive loss of motoneurons, motor weakness and death within 3–5 years after disease onset. Therapeutic options remain limited despite substantial number of approaches that have been tested clinically. Many neurotrophic growth factors are known to promote the survival of neurons and foster regeneration in the central nervous system. Various neurotrophic factors have been investigated pre-clinically and clinically for the treatment of ALS. Although pre-clinical data appeared promising, no neurotrophic factors succeeded yet in a clinical phase III trial. In this review we discuss the rationale behind those factors, possible reasons for clinical failures, and argue for a renewal of hope in this powerful class of drugs for the treatment of ALS.
ALS: A Multifaceted Disease
Amyotrophic lateral sclerosis (ALS) is an incurable fatal motoneuron disease with a lifetime risk of 1:800. It is characterized by progressive weakness, muscle wasting and death ensuing 3–5 years after diagnosis (Mitchell and Borasio, 2007 ). Since the first description of ALS by Charcot, around 150 years ago, and despite tremendous work, the cause(s) of ALS remain(s) unclear. A number of genes have been identified that, when mutated, cause familial ALS (Valdmanis et al., 2009 ). In addition, several polymorphisms have been identified that pose a genetic risk of developing sporadic ALS (Dunckley et al., 2007 ; Schymick et al., 2007 ). From these data, and a wealth of pre-clinical and clinical investigations it is clear that the etiopathogenesis and pathophysiology of ALS is complex with many players involved that lead to the functional decline of the motor pathway (Pasinelli and Brown, 2006 ; Rothstein, 2009 ). On the tissue level, it is clear that cells beyond motoneurons are involved in ALS pathophysiology. This involves astrocytes, which are reactive and apparently toxic in ALS (Schiffer et al., 1996 ; Nagai et al., 2007 ; Blackburn et al., 2009 ). Also, reactive microglia and protective T-cell infiltration have been noted as players in non-cell-autonomous toxicity (Engelhardt and Appel, 1990 ; Xiao et al., 2007 ; Beers et al., 2008 ). On the motoneuron level, it is a matter of ongoing research where the disease process is initiated, candidates being the neuromuscular junction (NMJ) (Fischer et al., 2004 ), the axon (Collard et al., 1995 ; Strom et al., 2008 ), mitochondria (Dupuis et al., 2004 ; Bacman et al., 2006 ; Martin, 2006 ), or the cell nucleus (Kinoshita et al., 2009 ). On the molecular level, a lot of possible culprits have been followed including glutamate excitotoxicity (Corona et al., 2007 ), reactive oxygen species (Carter et al., 2009 ), lipid metabolism (Dupuis et al., 2008a ), and others.
Currently, the only available treatment option is riluzole that prolongs life by 2–3 months, with questionable functional improvement (Miller et al., 2007 ). The broad spectrum of molecular and cellular players in ALS obviously makes drug development difficult. We argue that in the absence of one specific causative mechanism identified, protection of the motoneuron with multipotent growth factors remains the most rational approach available.
Discovery and Activities of Growth Factors
The story of growth factors in the nervous system was started by Rita Levi-Montalcini, Viktor Hamburger, and Stanley Cohen with the discovery of the nerve growth factor (NGF) in the 1950’s (Levi-Montalcini and Hamburger, 1951 ; Levi-Montalcini et al., 1954 ; Cohen and Levi-Montalcini, 1957 ; Levi-Montalcini and Cohen, 1960 ), for which Levi-Montalcini and Cohen received the Nobel prize in 1986. Interestingly, the Nobel prize in Medicine and Physiology was awarded for the “discovery of growth factors”, which at that time already acknowledged the scope of their work for opening up a large area of research and potential therapeutic approaches. Presently, the group of proteins regarded as neurotrophic growth factors has immensely grown, and a lot has been learned about common and specialized activities of these factors. Some of them, the neurotrophins, were first described as acting on neuronal cells, whereas others, more recently described neurotrophic factors, were initially discovered in a non-neuronal context.
Neurotrophic growth factors have an array of activities in the nervous system that consist of functions in development, plasticity, neurogenesis, disease and injury. Today it is clear that these activities are reaching far beyond the original “neurotrophic theory”, which postulated that only neurons that encounter these tissue target-derived factors in a critical phase of their development will survive, and that the availability of limited amounts of survival factors is a key factor in the development of the nervous system and other organs.
One essential activity of these factors is an intrinsic autocrine survival effect that is modulated during development (Lamballe et al., 1994 ; Valdes-Sanchez et al., 2010 ). Other effects are stimulation of neurite outgrowth and axon guidance (Tuttle and O’Leary, 1998 ; Huang and Reichardt, 2003 ), securing correct connectivity in the brain and spinal cord. Moreover, growth factors in the nervous system influence dendrite and synapse formation, thus directly impacting on plasticity mechanisms (Lo, 1998 ; Vicario-Abejon et al., 2002 ). An additional basic activity of many growth factors in the nervous system are influences on the proliferation, migration and differentiation of stem cells in the developing and adult nervous system (Cattaneo and McKay, 1990 ; Kuipers and Bramham, 2006 ).
These basic properties are also employed by the nervous system in the case of disease and injury, and thus constitute a powerful endogenous repair and maintenance system. The potential for a therapeutic application of these factors has been realized early on, and the last two decades have seen a number of approaches to exploit this potential in several neurological disorders, one of the most prominent being ALS. In the following we review growth factors that have been studied pre-clinically and clinically in ALS.
Brain-Derived Neurotrophic Factor
Brain-derived neurotrophic factor (BDNF) together with the related factors NGF, Neurotrophin-3 (NT-3), and Neurotrophin-4/5 (NT-4/5) belongs to the group of “Neurotrophic factors” or “Neurotrophins”, which constitute the most narrowly defined and oldest group of nervous growth factors. This narrow definition is justified as these factors are all related to the first member of that family (NGF), and share related receptors (trkA,B,C; p75NTR). BDNF is historically the second neurotrophic factor directly described as such (Barde et al., 1982 ). Several groups published promising results on rescuing axotomized motoneurons (Sendtner et al., 1992a ,b ; Yan et al., 1992 ; Gimenez y Ribotta et al., 1997 ) and on improving the phenotype of wobbler mutant mice (Ikeda et al., 1995 ), a model for motoneuron disorders (Table 1 ). In addition, BDNF protects neurons from in vivo excitotoxicity (Bemelmans et al., 2006 ), a mode of action of relevance to ALS.
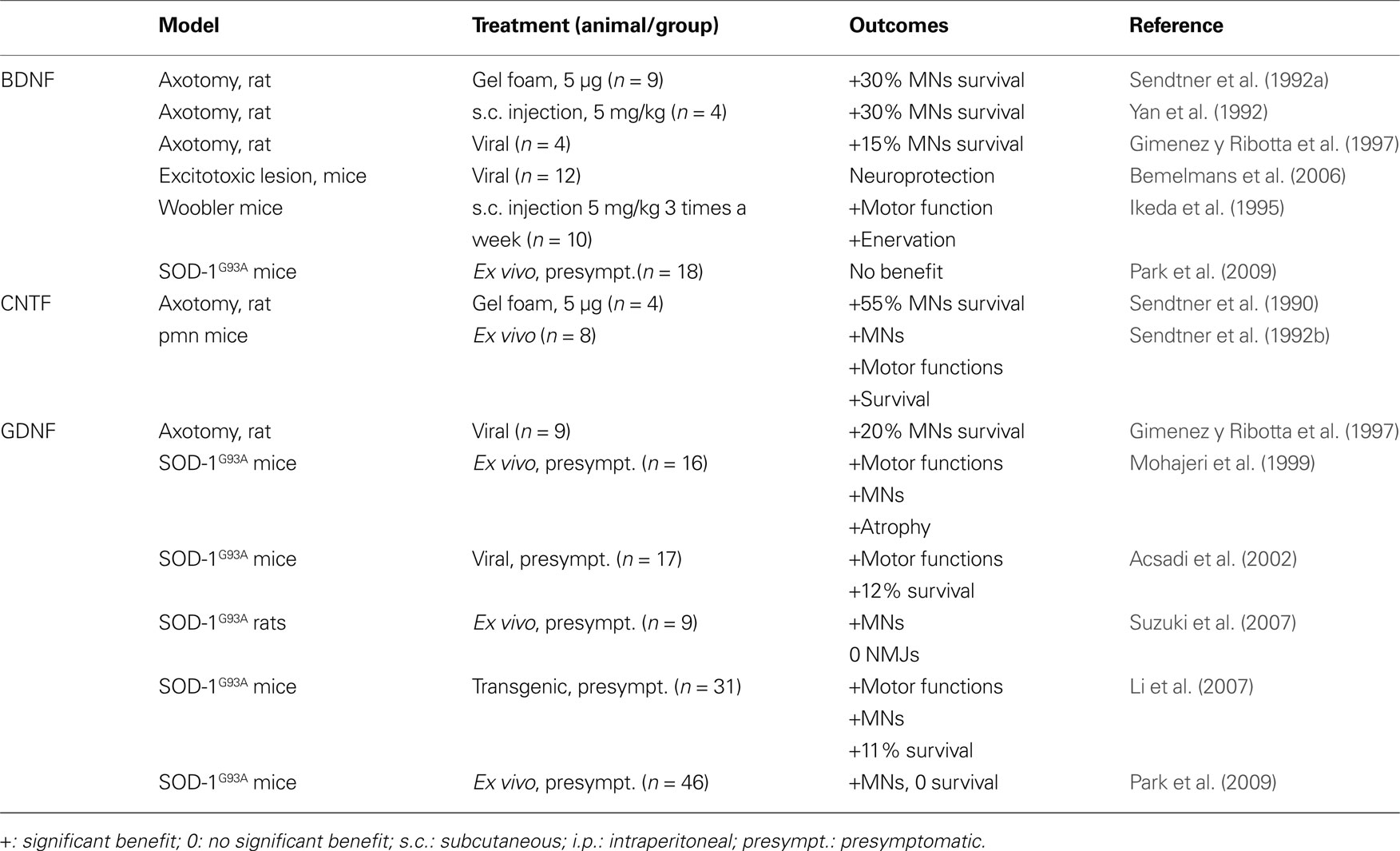
Table 1. Listed are classic neurotrophins that have been evaluated in animal models related to ALS. Given are the respective animal model, the application mode of the factor, and the outcome of the experiment.
Several clinical studies were conducted after those encouraging data (Table 3 ). More than 1000 patients were enrolled in a trial published in 1999 (The BDNF Study Group (Phase III) 1999 ). In this trial, the tolerability of BDNF was shown to be better than of Ciliary neurotrophic factor (CNTF), despite some adverse effects such as diarrhea. While the results failed to show an amelioration for patients’ condition or survival after treatment with BDNF, it appeared that a subgroup of patients responded positively to the treatment and demonstrated a higher survival rate. This subgroup consisted of patients having the lowest vital capacity and receiving the highest dose of BDNF, suggesting that BDNF could be beneficial for patients in advanced stages of the disease. The investigators suggested to use higher doses or intrathecal delivery to deliver BDNF to the diseased spinal cord. One year later, a phase 1 trial was published, showing the feasibility of intrathecal delivery of BDNF. Indeed, delivering BDNF with this method leads to a detectable level of BDNF in the cerebrospinal fluid, especially at the lumbar level (Ochs et al., 2000 ). The method being feasible, two clinical trial were conducted in 2003 and 2005. In 2003, eleven ALS patients were enrolled in a trial to receive an intrathecal delivery of BDNF. Another trial with intrathecal delivery was conducted in 2005 with 13 patients (Kalra et al., 2003 ; Beck et al., 2005 ). Both trials were too small to detect any meaningful efficacy signal.
The failure of BDNF in clinical trials could be explained by several reasons. First of all, it remains controversial if and to what degree BDNF passes the BBB (Pardridge et al., 1994 ; Poduslo and Curran, 1996 ; Pan et al., 1998 ), rendering it problematic to treat the spinal cord by subcutaneous delivery. The lack of any efficacy signals in the relatively small number of patients treated by direct delivery of BDNF to the central nervous system (CNS) might however point to additional problems. The second concern with BDNF is that it might not have been the most ideal growth factor to test clinically. It appears that BDNF could have the potential to exacerbate the disease to some extent, as p75, a receptor for BDNF, appears involved in ALS pathophysiology (Lowry et al., 2001 ; Dupuis et al., 2008b ). Indeed, when p75 is not bound to trk receptors, it might induce apoptosis in both animal models of ALS and patients. A report in 2004 actually showed that an antagonist for p75 slows progression of ALS in SOD-1 animals (Turner et al., 2004 ). Thus, the potential beneficial effect of BDNF could be concealed by the concomitant activation of pro-apoptotic pathways through the p75 receptor. Doubts in the suitability of BDNF for ALS treatment are also supported by animal experiments conducted after the clinical trials. A study performed on SOD-1 transgenic mice using a gene therapy approach with BDNF among others neurotrophic factors (Park et al., 2009 ) could not observe any positive effects of BDNF neither on disease progression nor on survival, questioning the rationale for clinical trials with BDNF in ALS.
Ciliary Neurotrophic Factor
After the discovery of NGF, a number of other, unrelated growth factors in the nervous system were discovered, that have a similar scope of activities, but do not belong to the group of “neurotrophins” in its restricted sense. The most well-known of these are the Ciliary neurotrophic factor (CNTF) (Lin et al., 1989 ) and the glial cell-line derived neurotrophic factor (GDNF) (Lin et al., 1993 ). The history of CNTF in ALS is similar to the one of BDNF. CNTF was one of the earliest neurotrophic factor shown to have a direct neuroprotective effect on degenerating motoneurons in stress-induced conditions (Table 1 ). Two studies in 1990 demonstrated that CNTF protects motoneurons in cell culture (Arakawa et al., 1990 ), and in vivo after axotomy-induced apoptosis in the rodent (Sendtner et al., 1990 ). Those results were strengthened by the beneficial effect of CNTF in a mouse model of neuronopathy with motoneuron degeneration (Sendtner et al., 1992b ), and by increased vulnerability of spinal motoneurons in a knock-out mouse line for CNTF (Masu et al., 1993 ). These results were published in the early 90s, prior to the development of mutated super oxide dismutase 1 (SOD-1) transgenic mice in 1994 (Gurney et al., 1994 ), today the most frequently employed mouse model for ALS. Because of these promising effects on motoneurons, CNTF went directly to the clinic, and became the first growth factor investigated clinically for ALS. Two clinical trials were conducted in 1996 and enrolled 570 and 730 patients, respectively (Table 3 ). The investigators focused mainly on daily living activities outcomes and survival (ALS CNTF Treatment Study Group, 1996 ; Miller et al., 1996 ). Unfortunately, both of those trials reported no observable benefit of treatment. Moreover, adverse effects, especially for the doses over 5 μg/kg, were severe, leading to the cessation of treatment in many patients.
What could be the reasons to explain the lack of efficiency of CNTF after such promising pre-clinical outcomes? Apart from possible general aspects of failure discussed further below (e.g. lacking translatability of rodent findings) we see two main specific reasons. First of all, it has to be pointed out that despite CNTF’s ability to cross the blood-brain barrier (Pan et al., 1999 ), a pharmacokinetic study in the rodent showed that CNTF has a half-life of only 2.9 min after intravenous injection. Due to this low half-life, subcutaneous application of CNTF appeared not appropriate to target the central nervous system. Secondly, the doses used in animals were about 1 mg/kg, whereas the highest safe dose in the human is 200 times lower than that (5 μg/kg). This limited dose and short half-life of CNTF could easily account for the inefficiency of CNTF in patients. No further clinical projects were pursued with CNTF in ALS after these two trials.
Glial Cell-Line Derived Neurotrophic Factor
Glial cell-line derived neurotrophic factor (GDNF) was studied a few years later than CNTF and BDNF. As it was the case for the other neurotrophic factors, the pre-clinical results were encouraging, with rescue of motoneurons after axotomy-induced degeneration and numerous positive in vitro data, (Buj-Bello , 1995 ; Gimenez y Ribotta et al., 1997 ; Bilak et al., 2001 ; Kosuge et al., 2009 ). including excitotoxity-induced stress (Kosuge et al., 2009 ). Unlike BDNF and CNTF, GDNF effects were tested in SOD-1 animals in a number of studies prior to initiation of clinical studies (Table 1 ). The outcomes of those studies argued for a benefit of GDNF treatment despite several controversial data. GDNF treated motoneurons of SOD-1 animals are clearly protected (Mohajeri et al., 1999 ; Acsadi et al., 2002 ; Li et al., 2007 ; Suzuki et al., 2007 ; Park et al., 2009 ). The effects of GDNF on survival and functional outcomes appear more problematic, as well as the rescue of neuromuscular junctions. While some studies report a benefit on disease progression and/or survival (Mohajeri et al., 1999 ; Acsadi et al., 2002 ; Li et al., 2007 ), others find no effect on disease progression and integrity of neuromuscular junctions (Li et al., 2007 ; Suzuki et al., 2007 ; Park et al., 2009 ). With respect to extrapolation to the clinic, it has to be mentioned that Park and associates were the only ones who initiated treatment at the onset of the disease, and they observed a protection of motoneurons but not an increase in survival.
Glial cell-line derived neurotrophic factor has not yet been tested clinically for the treatment of ALS patients. Clinical observations demonstrated that the level of GDNF is increased in the cerebrospinal fluid of ALS patients (Grundstrom et al., 2000 ), and that its receptor is expressed in the spinal cord, even at late stages of the disease (Mitsuma et al., 1999 ). Due to its in vivo neuroprotective effects on motoneurons GDNF remains a potential candidate for a clinical trial in ALS, despite controversial efficacy data on survival. However, as there is evidence that GDNF does not cross the BBB (Kastin et al., 2003 ), a future challenge remains in developing a protocol to resolve this issue, such as using an intrathecal delivery device for GDNF. Although there were signs of biological efficacy of GDNF in Parkinson’s disease (PD) (Gill et al., 2003 ; Patel et al., 2005 ), the further development of direct intracerebral delivery in PD was halted by the originator of the drug due to possible safety issues.
Insulin-Like Growth Factor-1
Beside the classic neurotrophins, other growth factors (“Non-classical’’ neurotrophic factors) show prominent neuronal activity but were first described as acting in other compartments of the body. Insulin-like growth factor-1 (IGF-1) has a multitude of actions and targets in the body, and the discovery of its effect on neurons dates from the end of the 80’s (Shemer et al., 1987 ). Neurons in the central nervous systems express functional receptors of insulin-like growth factor 1 (IGF-1) (Shemer et al., 1987 ). In vitro and in vivo, IGF-1 was able to rescue motoneurons (Caroni, 1993 ; Li et al., 1994 ; Bilak et al., 2001 ) and to reduce glutamate excitotoxicity (Nakao et al., 1996 ; Vincent et al., 2004 ). Therefore IGF-1 became an interesting drug candidate for ALS.
Several clinical studies with IGF-1 in ALS were conducted early, before testing IGF-1 in SOD-1 animals (Table 3 ). In the late 90’s, two investigations were published with different conclusions (Lai et al., 1997 ; Borasio et al., 1998 ). Lai et al. enrolled 266 patients and administrated IGF-1 subcutaneously. The conclusion of the investigators were that IGF-1 is safe, slows progression of motor-impairment, and increases quality of life. In a second trial, Borasio et al. enrolled 183 patients with the same mode of administration. The investigators focused on disease progression but found no improvement after treatment. Indeed, IGF-1 treatment appeared to be safe but did not improve quality of life of the patients nor motor functions. Additionally, a pilot study was conducted with intrathecal delivery of IGF-1 (Nagano et al., 2005b ). Investigators claimed that the treatment was beneficial for the patients. Two groups of patients were compared, receiving high or low doses of IGF-1 and it was observed that the motor functions decline was slowed down with the high dose of IGF-1 compared to the low dose. However, as this study was not placebo-controlled, the results should be considered with caution.
Some years later, pre-clinical reports investigating the effects of IGF-1 in SOD-1 animals were published (Table 2 ). The majority of the reports argue in favor of a potential protective effect in ALS, with an increase in survival and motor function parameters (Kaspar et al., 2003 ; Dobrowolny et al., 2005 ; Nagano et al., 2005a ; Lepore et al., 2007 ; Dodge et al., 2008 ; Franz et al., 2009 ). Also, IGF-1 mediates NMJ regeneration in a non-ALS related animal model (Apel et al., 2009 ), although some contradictory results are published (Grumbles et al., 2009 ). Nevertheless, two studies did not observe an improvement of survival in the SOD-1 transgenic model after IGF-1 treatment (Messi et al., 2007 ; Franz et al., 2009 ), and the work of Lepore et al. (2007) suggested that IGF-1 effect were only observed in male mice. At the same time, a new clinical trial was initiated, with subcutaneous administration of IGF-1. This clinical trial enrolled 330 patients and studied the effects of IGF-1 on muscular strength and survival (Sorenson et al., 2008 ). Unfortunately, IGF-1 treated patients did not improve relative to the control patients.
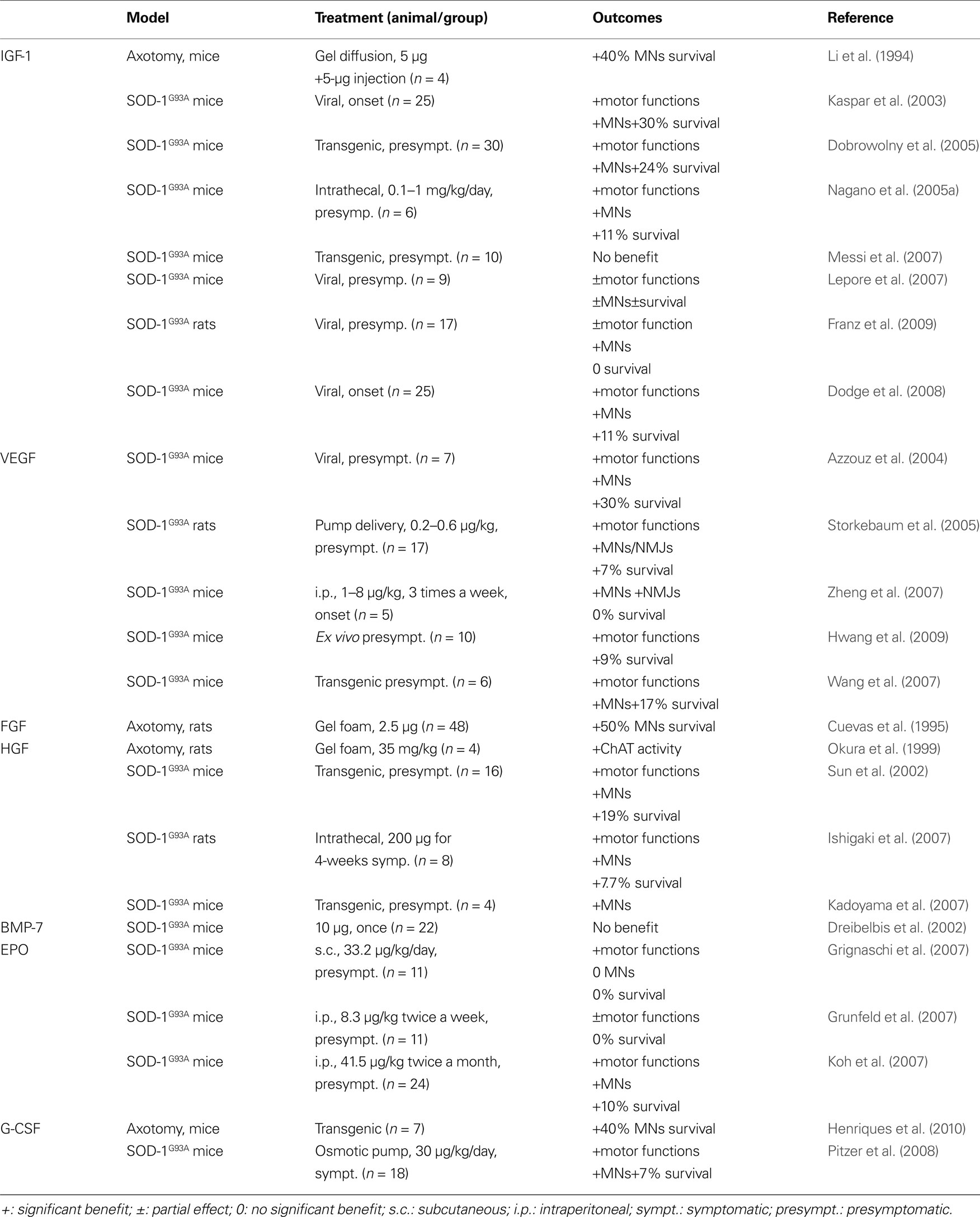
Table 2. Listed are all non-classical neurotrophic factors that have been evaluated in animal models related to ALS. Given are the respective animal model, the application mode of the factor, and the outcome of the experiment.
There may be explanations for the lack of success of this trial apart from the conclusion that IGF-1 is ineffective in ALS, especially since clinical observations show that the distribution of IGF-1 is increased at the spinal level of ALS patients (Wilczak et al., 2003 ; Corbo et al., 2010 ). One point of critique against the pre-clinical work is that no subcutaneous administration of IGF-1 was performed, indeed, only genetic therapies (Kaspar et al., 2003 ; Lepore et al., 2007 ; Dodge et al., 2008 ; Franz et al., 2009 ) or intrathecal administration (Nagano et al., 2005a ) were used to treat SOD-1 animals. A genetic approach can not be translated into subcutaneous delivery in patients, due to the high and constant expression of IGF-1 over time and the intrathecal administration which directly delivers IGF-1 to the CNS in contrast to the subcutaneous injections studied in the large trials. Also, treatment in the SOD-1 model ideally should be started at later time points during disease progression, and not before the disease onset.
Indeed, as Howe et al. (2009) observed, it is highly unlikely that meaningful levels of IGF-1 were reached in the CSF and CNS tissue with the dose of IGF-1 used in the latest clinical trial (Sorenson et al., 2008 ).
Vascular Endothelial Growth Factor
Discovered in the late 80’s as a growth factor for vascular endothelial cells, Vascular endothelial growth factor (VEGF) induces endothelial cell growth and angiogenesis (Connolly et al., 1989 ). The first evidence for neurotrophic proprieties of VEGF arose in the early 2000’s, with studies on neuronal regeneration (Sondell et al., 1999 , 2000 ; Hobson et al., 2000 ). VEGF was implicated as a growth factor possibly involved in ALS by the discovery that mice with deletion of the HIF-response element in the VEGF promoter developed an ALS-like phenotype (Oosthuyse et al., 2001 ). Subsequent genetic analyses however failed to produce clear evidence for an effect of VEGF polymorphisms on the risk of developing ALS in the human (Gros-Louis et al., 2003 ; Lambrechts et al., 2003 ; Van Vught et al., 2005 ; Chen et al., 2006 ; Fernandez-Santiago et al., 2006 ; Zhang et al., 2006 ). However, apart from the genetic discussion, VEGF promotes survival of cells over expressing a mutated SOD1 G93A protein (Li et al., 2003 ; Lunn et al., 2009 ) and ameliorates effects of glutamate excitotoxicity (Tolosa et al., 2008 ; Bogaert et al., 2009 ). In rodents (Table 2 ), VEGF treatment leads to a delay of disease onset, an improvement of motor functions, protection of motoneurons and neuromuscular junctions, and increase in survival (Azzouz et al., 2004 ; Storkebaum et al., 2005 ; Wang et al., 2007 ; Zheng et al., 2007 ; Hwang et al., 2009 ).
These data are promising for a clinical investigation of VEGF in ALS. Beside the pre-clinical evidence for a potential benefit of VEGF in the treatment of ALS, clinical observations did show that VEGF is down regulated in the late stage of ALS (Devos et al., 2004 ), and restoring its level therefore could bring benefit for the patients. The potential of VEGF as a therapeutic agent is further strengthened by its ability to cross the BBB (Wang et al., 1996 ; Dobrogowska et al., 1998 ; Yang et al., 2009 ). There are no published clinical trials so far, but a clinical trial is ongoing in Belgium and is currently recruiting patients. Clearly, angiogenic side effects of VEGF treatment will have to be monitored closely.
The Fibroblast Growth Factors
The Fibroblast Growth Factors (FGF) proteins are growth factors with homologous structure that promote angiogenesis and the proliferation of fibroblasts. Members of this family were tested for their effects on motoneurons. It was demonstrated that FGF-1 and FGF-2 can protect motoneurons after axotomy-induced apoptosis, and that they support neuronal regeneration (Cuevas et al., 1995 ; Piehl et al., 1995 ; Klimaschewski et al., 1999 ) (Table 2 ). However, one recent report points out that, in ALS, FGF-1 might activate motoneuron apoptosis by astrocytes, thereby possibly contributing to ALS pathophysiology (Cassina et al., 2005 ). More work is needed to establish if the FGFs can be drug candidates for ALS.
Hepatocyte Growth Factor
Hepatocyte Growth Factor (HGF) was discovered in 1984 as a growth factor acting on the liver (Nakamura et al., 1984 ). This growth factor exerts anti-apoptotic activity in the liver after endotoxin-induced hepatic failure (Kosai et al., 1999 ). Neuronal effects consist of activity in the guidance and survival of neurons in the developing nervous system (Ebens et al., 1996 ). Moreover, HGF is protective on motoneurons in culture and in vivo after axotomy (Wong et al., 1997 ; Okura et al., 1999 ). In rodent models of ALS, HGF reduces motoneuron degeneration and increases survival (Sun et al., 2002 ; Ishigaki et al., 2007 ; Kadoyama et al., 2007 ) (Table 2 ).
Hepatocyte Growth Factor appears to be a good candidate for the treatment of ALS, especially since HGF levels in ALS patients appear to be dysregulated (Tsuboi et al., 2002 ). However, additional pre-clinical work is required. Indeed, more insight at the level of the neuromuscular junctions is welcome since it seems that HGF can have an influence on the formation of the NMJs (Madhavan and Peng, 2006 ; Tatsumi et al., 2009 ) but appears to be inefficient on their regeneration unless used in conjunction with other growth factors (Grumbles et al., 2009 ). It can be also of interest for the design of a clinical trial to investigate whether a systemic delivery of HGF can reproduce the promising results observed after intrathecal administration in rodents.
Bone Morphogenetic Proteins
The Bone Morphogenetic Proteins (BMPs) are a family of growth factors influencing bone formation. BMPs have potent actions in the nervous system (Ebendal et al., 1998 ). A recent report on a drosophila model of ALS suggests an implication of the BMP signaling pathway in the pathophysiology of the disease (Ratnaparkhi et al., 2008 ). Only one report on transgenic ALS models was published with one of the BMPs, BMP-7 (Dreibelbis et al., 2002 ). The protocol used to treat the mice was certainly ambitious with a single injection of BMP-7 which likely resulted in the negative outcome of this experiment (Table 2 ). BMPs appear as an interesting growth factor family for the treatment of ALS, however, much more work is needed to come to a judgement whether BMPS are clinical candidates for ALS.
Erythropoietin
The first hematopoietic growth factor identified as a possible neuronal growth factor was Erythropoietin (EPO). Research on its ability to counteract effects of hypoxia in general led to the discovery of the expression of EPO and its receptor in the central nervous system (Lewczuk et al., 2000 ; Siren et al., 2001a ,b ; Ehrenreich et al., 2004 ). EPO is able to counteract apoptosis of neurons after hypoxia and ischemia. It is also able to enhance regeneration of the NMJs in rats after crush injury (Toth et al., 2008 ).
Recently, EPO was tested in SOD-1 transgenic animals (Table 2 ). Three studies were published in 2007 by three different groups. All report a beneficial effect of EPO (Koh et al., 2007 ; Grignaschi et al., 2007 ; Grunfeld et al., 2007 ). Koh et al. describe protection of motoneurons against degeneration and an increase in survival of the SOD-1 animals after treatment. In contrast, despite a delay on symptom progression, Grignaschi et al. and Grunfeld et al. did not observe an extension of survival. Previous work on EPO in brain injury demonstrated that EPO is able to cross the BBB (Statler et al., 2007 ), making it a good candidate to target the CNS. Especially carbamylated EPO (CEPO) might be a good candidate for ALS treatment, as it lacks hematopoietic effects of EPO while preserving neuroprotective properties (Leist et al., 2004 ; Mennini et al., 2006 ; Kirkeby et al., 2008 ). However, more pre-clinical work is needed before concluding on the relevance of EPO for ALS.
Thrombopoietin
Thrombopoietin (TPO) is a glycoprotein involved in the regulation of the thrombocytosis. Recently the receptor for TPO has been detected in the central nervous system (Yang et al., 2004 ). In contrast to other hematopoietic factors with neuroprotective activity, TPO induces apoptosis in neurons and inhibits neuronal differentiation (Ehrenreich et al., 2005 ; Samoylenko et al., 2008 ). Despite those negative results, the effects of TPO were investigated in the SOD-1 transgenic mouse model. TPO did not show any benefit in this model (Caraganis et al., 2008 ).
Granulocyte-Colony Stimulating Factor
Granulocyte-colony stimulating factor (G-CSF) is a glycoprotein responsible for the generation of neutrophilic granulocytes. G-CSF is currently used clinically in the treatment of chemotherapy-induced neutropenia. Recently, neuroprotective and regenerative properties in the brain and in the spinal cord have been described (Schneider et al., 2005 ). The G-CSF receptor is expressed in diverse areas of the CNS with a predominantly neuronal localization, including spinal motoneurons. In rodent models for stroke, G-CSF displays anti-apoptotic properties. In addition, G-CSF induces neuronal differentiation of neural progenitor cells, and is able to cross the brain blood barrier (Schneider et al., 2005 ). G-CSF enhances neurite outgrowth in vitro, and preserves tract integrity in spinal cord injury (Pitzer et al., 2010 ). It also enhances axonal regeneration and muscle reinnervation after axotomy in rodents (Pan et al., 2009 ).
We investigated the effects of G-CSF on animal models of motoneuron diseases (Table 2 ), including axotomized neonatal mice (Henriques et al., 2010 ) and SOD-1 transgenic animals (Pitzer et al., 2008 ). Subcutaneous delivery of G-CSF, as well as transgenic over expression, is able to slow down disease progression and increase survival of SOD1 mice by rescuing motoneurons.
Curiously, the potential of G-CSF in ALS was investigated in patients before our pre-clinical study was published (Table 3 ). Up to now, three clinical pilot studies were conducted with G-CSF in ALS patients showing that subcutaneous G-CSF treatment is reasonably tolerated in the doses given (Cashman et al., 2008 ; Zhang et al., 2008 ; Nefussy et al., 2009 ). Effects of G-CSF on disease progression can not be deduced from these studies since the size of patient cohorts was too small. Nevertheless, in one trial, an interesting trend for slowing down disease progression was reported (Nefussy et al., 2009 ). Similarly, Zhang et al. (2008) report on a delay in motor decline. Another clinical trial used G-CSF indirectly by treating ALS patients with an autologous transplantation of hematopoietic stem cells into the cerebrospinal fluid. The conclusion of the investigators was that the treatment results in an improvement of the patients’ quality of life and a delay of disease progression (Martinez et al., 2009 ). It is not clear whether the reported benefit might be caused by treatment with G-CSF or by the transplantation of stem cells, or both.
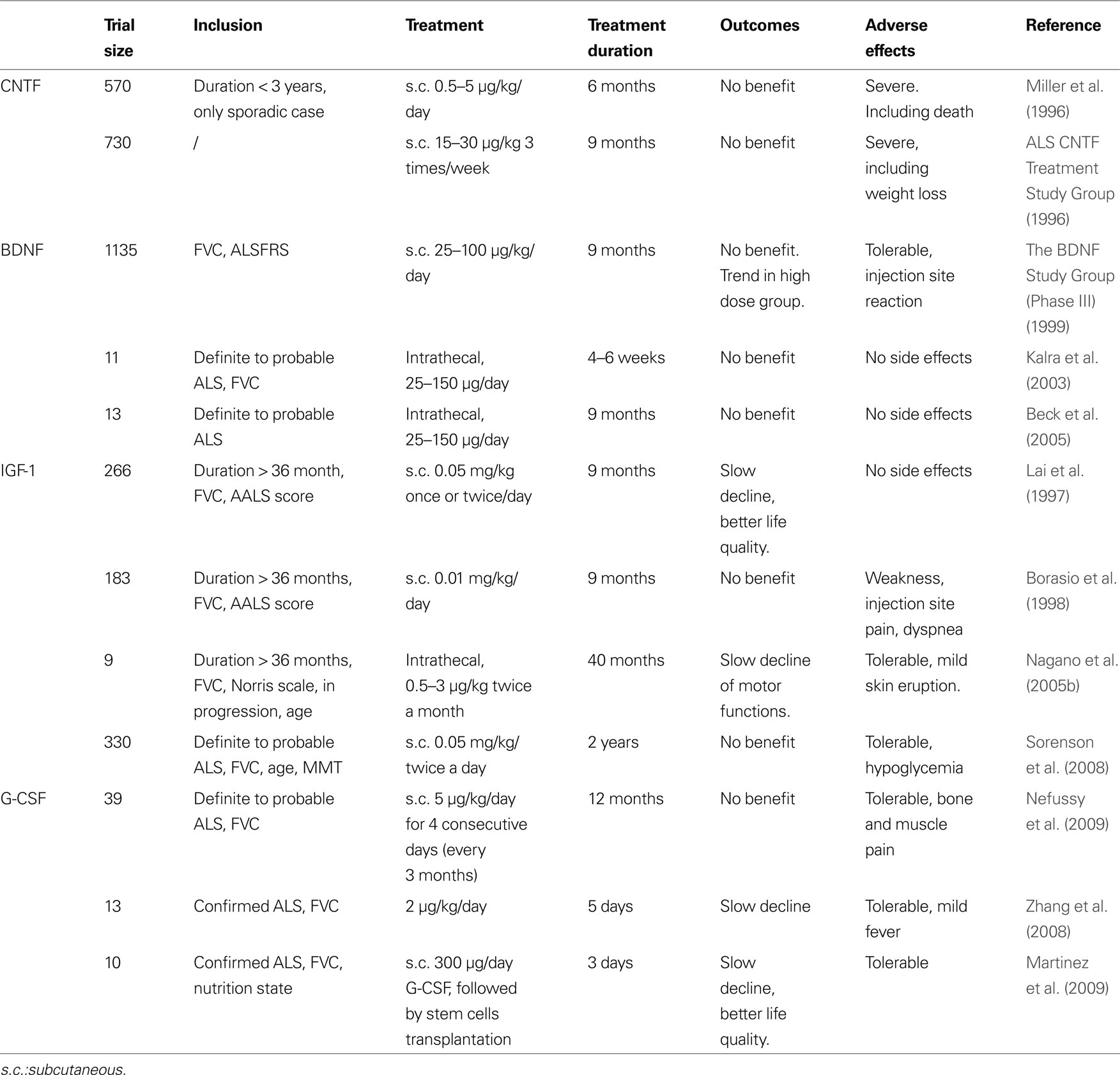
Table 3. Given are all growth factors that have been in any clinical trial in ALS patients, inclusion criteria, application modes and doses, and outcome.
Due to its good tolerability (Morstyn et al., 1988 ; Nefussy et al., 2009 ), its capability to cross the BBB (Schneider et al., 2005 ; Zhao et al., 2007 ), a clinically translatable dose range, and the initial promising hints from the published clinical studies, G-CSF is a good candidate for a larger clinical trial in ALS.
General Reasons for Clinical Failures
The demonstration of therapeutic effects of neurotrophic factors for various CNS disease conditions did raise great enthusiasm about their use as therapeutic agents in the 80’s and early 90’s. Therefore, a number of clinical trials in neurodegenerative conditions in the peripheral and central nervous system have been conducted. The fact that these early studies did not immediately bear striking success was likewise received with great disappointment. Today we can better weigh the opportunities and pitfalls of growth factor treatment in the nervous system, and come to a balanced understanding why individual factors have failed, and why others may be successful. The clinical failure of growth factors has to be viewed with great diligence, and is in no way ascribable to one single reason especially in ALS. We have grouped the likely main causative factors below:
Selection of Clinical Candidates was Based on Insufficient Pre-Clinical Efficacy Data
Because of the failure to find clinical efficacy so far, the value of pre-clinical data has been questioned. There are two aspects to this issue. First, the models used may not be predictive for the working of a drug in the human system. This is of course an irrefutable argument, which can’t be proven or disproven in the absence of any clinical positive outcome seen so far. This argument has in particular been raised with the recent clinical data from the minocycline trial where actually the treatment worsened patients’ outcome (Couzin, 2007 ; Gordon et al., 2007b ; Carri, 2008 ; Leigh et al., 2008 ), in contrast to successful treatment of SOD1-transgenic mouse models (Kriz et al., 2002 ; Van Den Bosch et al., 2002 ; Zhu et al., 2002 ; Zhang et al., 2003 ). However, before drawing that conclusion it has to be considered that effects of minocycline in conjunction with the glutamatergic system and interactions with riluzole have simply not been studied closely enough before starting the trial (Manev and Manev, 2009 ; Milane et al., 2009 ).
Second, animal models may be predictive, but have not been conducted with sufficient rigorousness and diligence to really tell us the truth about efficacy of a drug candidate in the rodent system. Reasons for this include insufficient blinding procedures, neglect of copy number issues in transgenic models, insufficient number of animals used, inadequate statistical methods, and inadequate independent replication of study results (Scott et al., 2008 ).
An example may have been BDNF, where pre-clinical studies were done in motoneuron disease models (axotomy, pmn and wobbler mice) that were available at the time. After the clinical trial, investigations in ALS animal models showed that a BDNF-based treatment does not work in the SOD1-transgenic model.
Doses and/or Application Mode were Inadequate
For a chronic CNS disease like ALS, the application and dose chosen should guarantee a therapeutic level of the drug in the brain and spinal cord. For a motoneuron-supportive principle like growth factors, the drug level should likely be held constant over time. In all major clinical trials done so far, this was not the case, or has not been investigated. Examples for that include BDNF, CNTF and IGF-1. For CNTF, the dose showing benefit for motoneurons in vivo was three orders of magnitude lower in the human case. In addition, s.c. injected CNTF has a plasma half-life time of only a few minutes, and does not reach the target tissues. A similar scenario can be drawn for BDNF, with questionable delivery of the growth factors to the CNS. For IGF-1, CSF levels reached with the chosen subcutaneous dose of 0.05 mg/kg bid likely are more than an order of magnitude below endogenous levels in the CSF, and thus therapeutically meaningless (Howe et al., 2009 ).
Problems with Clinical Studies in ALS
A number of issues complicate clinical studies in ALS, particularly with growth factors. In placebo-controlled studies, patients want to receive the verum drug, and will often undertake attempts to find out whether they receive verum, and then try to obtain the drug, or simply discontinue the trial. For growth factors this is often easier than for small-molecule drugs (e.g. EPO, IGF-1). This might obscure evaluation of the trial in the end. Otherwise, trials must be designed in a way that ensures that all patients will receive drug at some point, which might limit the rigorousness or power of the study (e.g. cross-over design). Moreover, patients might try to take part in several studies at once to obtain the maximum of possibly disease-modifying new treatments. This might provoke interactions between experimental treatments, and obscure treatment effects as well.
Finally, the number of patients available for trials is limited, and there are no good surrogate parameters with increased power over measures like the ALSFRS that would allow to conduct thorough dose-finding studies, implying that the doses tested in large trials have to be chosen by extrapolation from animal efficacy data, pharmacokinetics, and safety considerations.
Lessons Learned
The need for a therapy is urgent. However, urgency should not lead to rushed decisions for initiating clinical trials that will be negative because necessary key data were lacking. Every negative trial in the field will lead to further frustration of physicians, patients, and funding agencies, and undermine the most powerful weapon we have, hope for finding a real therapeutic option for the patients.
On the pre-clinical side, we need a consensus of what are minimal requirements or an assessment strategy for a pre-clinical efficacy package. In the field of stroke, this has been done by the so-called STAIR (Stroke Therapy Academic Industry Roundtable) criteria (Stroke Therapy Academic Industry Roundtable II (STAIR-II) 2001 ; Fisher, 2003 ; Fisher et al., 2005 , 2007 , 2009 ; Saver et al., 2009 ). Attempts to do so have been initiated by the ALS TDI with the idea of a unified test battery (Scott et al., 2008 ). However, it is also clear that without a precedence of successful translation of pre-clinical data to the patient (with the possible exception of Riluzole) it is very difficult to select strict cut-off criteria for pre-clinical efficacy, as these might pose a danger for unwillfully discarding exactly those drugs that would have worked in the human. In the absence of proof for the predictiveness of all models used, we should use a combination of different animal models that monitor different aspects of ALS disease pathophysiology, e.g. two different SOD1-mutant models, possibly in mice and rats, and axotomy models. Ideally, a dose-effect relationship should be established. All tests have to be conducted in a rigorously blinded and randomized manner, and ideally be performed in at least two independent labs. If novel animal models become available, they should be immediately tested for effects.
In addition, we have to have a very good understanding of how the drug in question works, and if it addresses several of the mechanisms likely relevant to ALS. For example, growth factors should show some efficacy against cell death, both apoptotic and excitotoxic, demonstrated on motoneurons, other primary neurons and/or motoneuron lines. Also, effects on the integrity of the NMJ should be ascertained. Only VEGF is known to preserve the innervations of NMJs in ALS (Zheng et al., 2007 ). For other growth factors, even if there is no direct evidence in an ALS model so far, some of them may have a beneficial effect on the NMJs and their regeneration, such as G-CSF (Pan et al., 2009 ), IGF-1 (Apel et al., 2009 ) and EPO (Toth et al., 2008 ). The available mode of action and animal model data should then be reviewed by an expert panel with the aim to decide whether the evidence package as a whole is sufficient to initiate clinical development. This appears advantageous to us in contrast to relying on single strict cut-off criteria, as each growth factor has a specific spectrum of activities that in each case will generate a blend of mode of actions and animal data that have to be appreciated on a case by case basis.
On the pharmacokinetic side, we have to be certain that the protein in question passes the BBB, and reaches motoneurons in sufficient concentration, and over a reasonable time period. The delivery route and formulation need to be fitted to that purpose. Measurements of the protein levels in the CSF should be performed within the trial. Indeed, drug delivery to the CNS appears to be the most crucial problem to address, because pharmacokinetic problems otherwise preclude to reach a concise conclusion of whether a specific growth factor is indeed without effect in ALS trials. This appears to be the case with all growth factors that have been tested in larger clinical trials (BDNF, CNTF, IGF-1).
Novel delivery tools and application forms should also be explored, such as gene therapy with a viral injection or cellular transplants (for review, see Hedlund et al., 2007 ; Bjorklund and Kordower, 2010 ). Those approaches have made progress in terms of safety and efficacy at pre-clinical level (Vercelli et al., 2008 ; Hester et al., 2009 ; Kim et al., 2010 ), and indeed clinical trials with cellular therapies have been recently conducted in ALS patients with no major safety concerns (Deda et al., 2009 ; Mazzini et al., 2009 ). Likewise, viral delivery has been explored in patients with Parkinon’s disease (Kaplitt et al., 2007 ). These approaches are promising, but remain much more complicated in terms of safety than using pure proteins. The side effects of the delivered growth factors have to be well characterized prior to gene therapy, with special regard to long term CNS delivery. One principal disadvantage of many of those delivery systems is that it is difficult to stop delivery should any serious safety problems emerge. One potentially attractive but also difficult delivery method, intrathecal delivery, should be further improved as a application route that would largely eliminate peripheral adverse effects.
Finally, in clinical trials we should continue to try and identify meaningful surrogate parameters that would allow a rational dose-finding strategy with relatively small patient numbers (e.g. EMG, MR tensor imaging, biomarkers). Genome wide association studies (GWAS) in sporadic ALS should be intensified to allow genome-based subgroup analyses in trials (Dunckley et al., 2007 ; Schymick et al., 2007 ; Valdmanis et al., 2009 ). Finally, innovative trial designs that may allow to judge early on whether a drug likely has no efficacy (futility designs), or smaller trials against cleverly controlled database controls need to be further explored (Bryan et al., 2003 ; Moore and Miller, 2004 ; Palesch and Tilley, 2004 ; Shefner et al., 2004 ; Cheung et al., 2006 ; Czaplinski et al., 2006 ; Gordon et al., 2007a , 2008 ; Schoenfeld and Cudkowicz, 2008 ; Gordon, 2009 ; Simmons, 2009 ; Aggarwal et al., 2010 ).
Why are Growth Factors Still the Most Rational Approach to ALS?
The largest consensus on ALS is that its origin(s) remain(s) unknown despite tremendous work since decades. As long as the precise etiopathology(-ies) of the disease is not be known, no specific and fully rational therapy can be designed to treat ALS. However, in ALS, disease progression is due to the disruption of the motor unit, whatever the triggering factor(s) may be. As long as we do not have more insights into the disease pathway(s), the most rational strategy remains to rescue and to strengthen the motor units as a whole, from the neuromuscular junctions up to the motoneuron cell body. In that respect, neurotrophic growth factors are the best tool we have. As detailed before, almost all growth factors are able to rescue the motoneuron cell bodies in the spinal cord once they reach the CNS.
Despite the considerable number of clinical trials undertaken with growth factors in ALS, it appears in retrospect that the hypothesis of growth factors for the treatment of ALS has never really been put to test in the human system, as all trials were inadequate in dosing and/or application of the protein in question.
There are growth factors with excellent rationale on the shelf, that should be evaluated further with stringent criteria. One particularly interesting group are the hematopoietic growth factors EPO and G-CSF: These appear to have many of the above discussed properties of nervous growth factors, but may be advantageous with regard to the above identified problematic issues. In particular, they have an excellent history of safety, a well-known pharmacokinetic profile, pass the blood-brain barrier, and the doses efficacious in animal models can be easily translated into the clinic.
Conflict of Interest Statement
Armin Schneider is inventor on patent applications claiming the use of G-CSF for the treatment of neurodegenerative conditions.
References
Acsadi, G., Anguelov, R. A., Yang, H., Toth, G., Thomas, R., Jani, A., Wang, Y., Ianakova, E., Mohammad, S., Lewis, R. A., and Shy, M. E. (2002). Increased survival and function of SOD1 mice after glial cell-derived neurotrophic factor gene therapy. Hum. Gene Ther. 13, 1047–1059.
Aggarwal, S. P., Zinman, L., Simpson, E., McKinley, J., Jackson, K. E., Pinto, H., Kaufman, P., Conwit, R. A., Schoenfeld, D., Shefner, J., and Cudkowicz, M. (2010). Safety and efficacy of lithium in combination with riluzole for treatment of amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 9, 481–488.
ALS CNTF Treatment Study Group (1996). A double-blind placebo-controlled clinical trial of subcutaneous recombinant human ciliary neurotrophic factor (rHCNTF) in amyotrophic lateral sclerosis. Neurology 46, 1244–1249.
Apel, P. J., Ma, J., Callahan, M., Northam, C. N., Alton, T. B., Sonntag, W. E., and Li, Z. (2009). Effect of locally delivered IGF-1 on nerve regeneration during aging: an experimental study in rats. Muscle Nerve. 41, 335–341.
Arakawa, Y., Sendtner, M., and Thoenen, H. (1990). Survival effect of ciliary neurotrophic factor (CNTF) on chick embryonic motoneurons in culture: comparison with other neurotrophic factors and cytokines. J. Neurosci. 10, 3507–3515.
Azzouz, M., Ralph, G. S., Storkebaum, E., Walmsley, L. E., Mitrophanous, K. A., Kingsman, S. M., Carmeliet, P., and Mazarakis, N. D. (2004). VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature 429, 413–417.
Bacman, S. R., Bradley, W. G., and Moraes, C. T. (2006). Mitochondrial involvement in amyotrophic lateral sclerosis: trigger or target? Mol. Neurobiol. 33, 113–131.
Barde, Y. A., Edgar, D., and Thoenen, H. (1982). Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1, 549–553.
Beck, M., Flachenecker, P., Magnus, T., Giess, R., Reiners, K., Toyka, K. V., and Naumann, M. (2005). Autonomic dysfunction in ALS: a preliminary study on the effects of intrathecal BDNF. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 6, 100–103.
Beers, D. R., Henkel, J. S., Zhao, W., Wang, J., and Appel, S. H. (2008). CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc. Natl. Acad. Sci. U.S.A. 105, 15558–15563.
Bemelmans, A. P., Husson, I., Jaquet, M., Mallet, J., Kosofsky, B. E., and Gressens, P. (2006). Lentiviral-mediated gene transfer of brain-derived neurotrophic factor is neuroprotective in a mouse model of neonatal excitotoxic challenge. J. Neurosci. Res. 83, 50–60.
Bilak, M. M., Corse, A. M., and Kuncl, R. W. (2001). Additivity and potentiation of IGF-I and GDNF in the complete rescue of postnatal motor neurons. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2, 83–91.
Bjorklund, T., and Kordower, J. H. (2010). Gene therapy for Parkinson’s disease. Mov. Disord. 25(Suppl. 1), S161–S173.
Blackburn, D., Sargsyan, S., Monk, P. N., and Shaw, P. J. (2009). Astrocyte function and role in motor neuron disease: a future therapeutic target? Glia 57, 1251–1264.
Bogaert, E., Van Damme, P., Poesen, K., Dhondt, J., Hersmus, N., Kiraly, D., Scheveneels, W., Robberecht, W., and Van Den Bosch, L. (2009). VEGF protects motor neurons against excitotoxicity by upregulation of GluR2. Neurobiol. Aging. (in press).
Borasio, G. D., Robberecht, W., Leigh, P. N., Emile, J., Guiloff, R. J., Jerusalem, F., Silani, V., Vos, P. E., Wokke, J. H., and Dobbins, T. (1998). A placebo-controlled trial of insulin-like growth factor-I in amyotrophic lateral sclerosis. European ALS/IGF-I Study Group. Neurology 51, 583–586.
Bryan, W. W., Hoagland, R. J., Murphy, J., Armon, C., Barohn, R. J., Goodpasture, J. C., Miller, R. G., Parry, G. J., Petajan, J. H., Ross, M. A., and Stromatt, S. C. (2003). Can we eliminate placebo in ALS clinical trials? Amyotroph Lateral Scler. Other Motor Neuron Disord. 4, 11–15.
Buj-Bello, A., Buchman, V. L., Horton, A., Rosenthal, A., and Davies, A. M. (1995). GDNF is an age-specific survival factor for sensory and autonomic neurons. Neuron 15, 821–828.
Caraganis, A., Benn, S., Cudkowicz, M., and Brown, R. H. Jr. (2008). Thrombopoietin is ineffective in a mouse model of motor neuron disease. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 9, 354–358.
Caroni, P. (1993). Activity-sensitive signaling by muscle-derived insulin-like growth factors in the developing and regenerating neuromuscular system. Ann. N. Y. Acad. Sci. 692, 209–222.
Carri, M. T. (2008). Minocycline for patients with ALS. Lancet Neurol. 7, 118–119; author reply 120–111.
Carter, B. J., Anklesaria, P., Choi, S., and Engelhardt, J. F. (2009). Redox modifier genes and pathways in amyotrophic lateral sclerosis. Antioxid. Redox Signal. 11, 1569–1586.
Cashman, N., Tan, L. Y., Krieger, C., Madler, B., Mackay, A., Mackenzie, I., Benny, B., Nantel, S., Fabros, M., Shinobu, L., Yousefi, M., and Eisen, A. (2008). Pilot study of granulocyte colony stimulating factor (G-CSF)-mobilized peripheral blood stem cells in amyotrophic lateral sclerosis (ALS). Muscle Nerve 37, 620–625.
Cassina, P., Pehar, M., Vargas, M. R., Castellanos, R., Barbeito, A. G., Estevez, A. G., Thompson, J. A., Beckman, J. S., and Barbeito, L. (2005). Astrocyte activation by fibroblast growth factor-1 and motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J. Neurochem. 93, 38–46.
Cattaneo, E., and McKay, R. (1990). Proliferation and differentiation of neuronal stem cells regulated by nerve growth factor. Nature 347, 762–765.
Chen, W., Saeed, M., Mao, H., Siddique, N., Dellefave, L., Hung, W. Y., Deng, H. X., Sufit, R. L., Heller, S. L., Haines, J. L., Pericak-Vance, M., and Siddique, T. (2006). Lack of association of VEGF promoter polymorphisms with sporadic ALS. Neurology 67, 508–510.
Cheung, Y. K., Gordon, P. H., and Levin, B. (2006). Selecting promising ALS therapies in clinical trials. Neurology 67, 1748–1751.
Cohen, S., and Levi-Montalcini, R. (1957). Purification and properties of a nerve growth-promoting factor isolated from mouse sarcoma 180. Cancer Res. 17, 15–20.
Collard, J. F., Cote, F., and Julien, J. P. (1995). Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature 375, 61–64.
Connolly, D. T., Heuvelman, D. M., Nelson, R., Olander, J. V., Eppley, B. L., Delfino, J. J., Siegel, N. R., Leimgruber, R. M., and Feder, J. (1989). Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J. Clin. Invest. 84, 1470–1478.
Corbo, M., Lunetta, C., Magni, P., Dozio, E., Ruscica, M., Adobbati, L., and Silani, V. (2010). Free insulin-like growth factor (IGF)-1 and IGF-binding proteins-2 and -3 in serum and cerebrospinal fluid of amyotrophic lateral sclerosis patients. Eur. J. Neurol. 17, 398–404.
Corona, J. C., Tovar-y-Romo, L. B., and Tapia, R. (2007). Glutamate excitotoxicity and therapeutic targets for amyotrophic lateral sclerosis. Expert Opin. Ther. Targets 11, 1415–1428.
Couzin, J. (2007). Clinical research. ALS trial raises questions about promising drug. Science 318, 1227.
Cuevas, P., Carceller, F., and Gimenez-Gallego, G. (1995). Acidic fibroblast growth factor prevents post-axotomy neuronal death of the newborn rat facial nerve. Neurosci. Lett. 197, 183–186.
Czaplinski, A., Haverkamp, L. J., Yen, A. A., Simpson, E. P., Lai, E. C., and Appel, S. H. (2006). The value of database controls in pilot or futility studies in ALS. Neurology 67, 1827–1832.
Deda, H., Inci, M. C., Kurekci, A. E., Sav, A., Kayihan, K., Ozgun, E., Ustunsoy, G. E., and Kocabay, S. (2009). Treatment of amyotrophic lateral sclerosis patients by autologous bone marrow-derived hematopoietic stem cell transplantation: a 1-year follow-up. Cytotherapy 11, 18–25.
Devos, D., Moreau, C., Lassalle, P., Perez, T., De Seze, J., Brunaud-Danel, V., Destee, A., Tonnel, A. B., and Just, N. (2004). Low levels of the vascular endothelial growth factor in CSF from early ALS patients. Neurology 62, 2127–2129.
Dobrogowska, D. H., Lossinsky, A. S., Tarnawski, M., and Vorbrodt, A. W. (1998). Increased blood-brain barrier permeability and endothelial abnormalities induced by vascular endothelial growth factor. J. Neurocytol. 27, 163–173.
Dobrowolny, G., Giacinti, C., Pelosi, L., Nicoletti, C., Winn, N., Barberi, L., Molinaro, M., Rosenthal, N., and Musaro, A. (2005). Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J. Cell Biol. 168, 193–199.
Dodge, J. C., Haidet, A. M., Yang, W., Passini, M. A., Hester, M., Clarke, J., Roskelley, E. M., Treleaven, C. M., Rizo, L., Martin, H., Kim, S. H., Kaspar, R., Taksir, T. V., Griffiths, D. A., Cheng, S. H., Shihabuddin, L. S., and Kaspar, B. K. (2008). Delivery of AAV-IGF-1 to the CNS extends survival in ALS mice through modification of aberrant glial cell activity. Mol. Ther. 16, 1056–1064.
Dreibelbis, J. E., Brown, R. H. Jr., Pastuszak, K. A., Smith, E. R., Kaplan, P. L., and Cudkowicz, M. E. (2002). Disease course unaltered by a single intracisternal injection of BMP-7 in ALS mice. Muscle Nerve 25, 122–123.
Dunckley, T., Huentelman, M. J., Craig, D. W., Pearson, J. V., Szelinger, S., Joshipura, K., Halperin, R. F., Stamper, C., Jensen, K. R., Letizia, D., Hesterlee, S. E., Pestronk, A., Levine, T., Bertorini, T., Graves, M. C., Mozaffar, T., Jackson, C. E., Bosch, P., McVey, A., Dick, A., Barohn, R., Lomen-Hoerth, C., Rosenfeld, J., O’Connor D, T., Zhang, K., Crook, R., Ryberg, H., Hutton, M., Katz, J., Simpson, E. P., Mitsumoto, H., Bowser, R., Miller, R. G., Appel, S. H., and Stephan, D. A. (2007). Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N. Engl. J. Med. 357, 775–788.
Dupuis, L., Corcia, P., Fergani, A., Gonzalez De Aguilar, J. L., Bonnefont-Rousselot, D., Bittar, R., Seilhean, D., Hauw, J. J., Lacomblez, L., Loeffler, J. P., and Meininger, V. (2008a). Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 70, 1004–1009.
Dupuis, L., Pehar, M., Cassina, P., Rene, F., Castellanos, R., Rouaux, C., Gandelman, M., Dimou, L., Schwab, M. E., Loeffler, J. P., Barbeito, L., and Gonzalez de Aguilar, J. L. (2008b). Nogo receptor antagonizes p75NTR-dependent motor neuron death. Proc. Natl. Acad. Sci. U.S.A. 105, 740–745.
Dupuis, L., Gonzalez de Aguilar, J. L., Oudart, H., de Tapia, M., Barbeito, L., and Loeffler, J. P. (2004). Mitochondria in amyotrophic lateral sclerosis: a trigger and a target. Neurodegener. Dis. 1, 245–254.
Ebendal, T., Bengtsson, H., and Soderstrom, S. (1998). Bone morphogenetic proteins and their receptors: potential functions in the brain. J. Neurosci. Res. 51, 139–146.
Ebens, A., Brose, K., Leonardo, E. D., Hanson, M. G. Jr., Bladt, F., Birchmeier, C., Barres, B. A., and Tessier-Lavigne, M. (1996). Hepatocyte growth factor/scatter factor is an axonal chemoattractant and a neurotrophic factor for spinal motor neurons. Neuron 17, 1157–1172.
Ehrenreich, H., Hasselblatt, M., Knerlich, F., von Ahsen, N., Jacob, S., Sperling, S., Woldt, H., Vehmeyer, K., Nave, K. A., and Siren, A. L. (2005). A hematopoietic growth factor, thrombopoietin, has a proapoptotic role in the brain. Proc. Natl. Acad. Sci. U.S.A. 102, 862–867.
Ehrenreich, H., Timner, W., and Siren, A. L. (2004). A novel role for an established player: anemia drug erythropoietin for the treatment of cerebral hypoxia/ischemia. Transfus. Apher. Sci. 31, 39–44.
Engelhardt, J. I., and Appel, S. H. (1990). IgG reactivity in the spinal cord and motor cortex in amyotrophic lateral sclerosis. Arch. Neurol. 47, 1210–1216.
Fernandez-Santiago, R., Sharma, M., Mueller, J. C., Gohlke, H., Illig, T., Anneser, J., Munch, C., Ludolph, A., Kamm, C., and Gasser, T. (2006). Possible gender-dependent association of vascular endothelial growth factor (VEGF) gene and ALS. Neurology 66, 1929–1931.
Fischer, L. R., Culver, D. G., Tennant, P., Davis, A. A., Wang, M., Castellano-Sanchez, A., Khan, J., Polak, M. A., and Glass, J. D. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240.
Fisher, M. (2003). Recommendations for advancing development of acute stroke therapies: Stroke Therapy Academic Industry Roundtable 3. Stroke 34, 1539–1546.
Fisher, M., Albers, G. W., Donnan, G. A., Furlan, A. J., Grotta, J. C., Kidwell, C. S., Sacco, R. L., and Wechsler, L. R. (2005). Enhancing the development and approval of acute stroke therapies: Stroke Therapy Academic Industry roundtable. Stroke 36, 1808–1813.
Fisher, M., Feuerstein, G., Howells, D. W., Hurn, P. D., Kent, T. A., Savitz, S. I., and Lo, E. H. (2009). Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke 40, 2244–2250.
Fisher, M., Hanley, D. F., Howard, G., Jauch, E. C., and Warach, S. (2007). Recommendations from the STAIR V meeting on acute stroke trials, technology and outcomes. Stroke 38, 245–248.
Franz, C. K., Federici, T., Yang, J., Backus, C., Oh, S. S., Teng, Q., Carlton, E., Bishop, K. M., Gasmi, M., Bartus, R. T., Feldman, E. L., and Boulis, N. M. (2009). Intraspinal cord delivery of IGF-I mediated by adeno-associated virus 2 is neuroprotective in a rat model of familial ALS. Neurobiol. Dis. 33, 473–481.
Gill, S. S., Patel, N. K., Hotton, G. R., O’Sullivan, K., McCarter, R., Bunnage, M., Brooks, D. J., Svendsen, C. N., and Heywood, P. (2003). Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat. Med. 9, 589–595.
Gimenez y Ribotta, M., Revah, F., Pradier, L., Loquet, I., Mallet, J., and Privat, A. (1997). Prevention of motoneuron death by adenovirus-mediated neurotrophic factors. J. Neurosci. Res. 48, 281–285.
Gordon, P. H. (2009). A placebo arm is not always necessary in clinical trials of amyotrophic lateral sclerosis. Muscle Nerve 39, 858–860.
Gordon, P. H., Cheng, B., Montes, J., Doorish, C., Albert, S. M., and Mitsumoto, H. (2007a). Outcome measures for early phase clinical trials. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 8, 270–273.
Gordon, P. H., Moore, D. H., Miller, R. G., Florence, J. M., Verheijde, J. L., Doorish, C., Hilton, J. F., Spitalny, G. M., MacArthur, R. B., Mitsumoto, H., Neville, H. E., Boylan, K., Mozaffar, T., Belsh, J. M., Ravits, J., Bedlack, R. S., Graves, M. C., McCluskey, L. F., Barohn, R. J., and Tandan, R. (2007b). Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 6, 1045–1053.
Gordon, P. H., Cheung, Y. K., Levin, B., Andrews, H., Doorish, C., Macarthur, R. B., Montes, J., Bednarz, K., Florence, J., Rowin, J., Boylan, K., Mozaffar, T., Tandan, R., Mitsumoto, H., Kelvin, E. A., Chapin, J., Bedlack, R., Rivner, M., McCluskey, L. F., Pestronk, A., Graves, M., Sorenson, E. J., Barohn, R. J., Belsh, J. M., Lou, J. S., Levine, T., Saperstein, D., Miller, R. G., and Scelsa, S. N. (2008). A novel, efficient, randomized selection trial comparing combinations of drug therapy for ALS. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 9, 212–222.
Grignaschi, G., Zennaro, E., Tortarolo, M., Calvaresi, N., and Bendotti, C. (2007). Erythropoietin does not preserve motor neurons in a mouse model of familial ALS. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 8, 31–35.
Gros-Louis, F., Laurent, S., Lopes, A. A., Khoris, J., Meininger, V., Camu, W., and Rouleau, G. A. (2003). Absence of mutations in the hypoxia response element of VEGF in ALS. Muscle Nerve 28, 774–775.
Grumbles, R. M., Sesodia, S., Wood, P. M., and Thomas, C. K. (2009). Neurotrophic factors improve motoneuron survival and function of muscle reinnervated by embryonic neurons. J. Neuropathol. Exp. Neurol. 68, 736–746.
Grundstrom, E., Lindholm, D., Johansson, A., Blennow, K., and Askmark, H. (2000). GDNF but not BDNF is increased in cerebrospinal fluid in amyotrophic lateral sclerosis. Neuroreport 11, 1781–1783.
Grunfeld, J. F., Barhum, Y., Blondheim, N., Rabey, J. M., Melamed, E., and Offen, D. (2007). Erythropoietin delays disease onset in an amyotrophic lateral sclerosis model. Exp. Neurol. 204, 260–263.
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., Caliendo, J., Hentati, A., Kwon, Y. W., Deng, H. X., Chen, W., Zhai, P., Sufit, R. L., and Siddique, T. (1994). Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264, 1772–1775.
Hedlund, E., Hefferan, M. P., Marsala, M., and Isacson, O. (2007). Cell therapy and stem cells in animal models of motor neuron disorders. Eur. J. Neurosci. 26, 1721–1737.
Henriques, A., Pitzer, C., Dupuis, L., and Schneider, A. (2010). G-CSF protects motoneurons against axotomy-induced apoptotic death in neonatal mice. BMC Neurosci. 11, 25.
Hester, M. E., Foust, K. D., Kaspar, R. W., and Kaspar, B. K. (2009). AAV as a gene transfer vector for the treatment of neurological disorders: novel treatment thoughts for ALS. Curr. Gene Ther. 9, 428–433.
Hobson, M. I., Green, C. J., and Terenghi, G. (2000). VEGF enhances intraneural angiogenesis and improves nerve regeneration after axotomy. J. Anat. 197, 591–605.
Howe, C. L., Bergstrom, R. A., and Horazdovsky, B. F. (2009). Subcutaneous IGF-1 is not beneficial in 2-year ALS trial. Neurology 73, 1247; author reply 1247–1248.
Huang, E. J., and Reichardt, L. F. (2003). Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642.
Hwang, D. H., Lee, H. J., Park, I. H., Seok, J. I., Kim, B. G., Joo, I. S., and Kim, S. U. (2009). Intrathecal transplantation of human neural stem cells overexpressing VEGF provide behavioral improvement, disease onset delay and survival extension in transgenic ALS mice. Gene Ther. 16, 1234–1244.
Ikeda, K., Klinkosz, B., Greene, T., Cedarbaum, J. M., Wong, V., Lindsay, R. M., and Mitsumoto, H. (1995). Effects of brain-derived neurotrophic factor on motor dysfunction in wobbler mouse motor neuron disease. Ann. Neurol. 37, 505–511.
Ishigaki, A., Aoki, M., Nagai, M., Warita, H., Kato, S., Kato, M., Nakamura, T., Funakoshi, H., and Itoyama, Y. (2007). Intrathecal delivery of hepatocyte growth factor from amyotrophic lateral sclerosis onset suppresses disease progression in rat amyotrophic lateral sclerosis model. J. Neuropathol. Exp. Neurol. 66, 1037–1044.
Kadoyama, K., Funakoshi, H., Ohya, W., and Nakamura, T. (2007). Hepatocyte growth factor (HGF) attenuates gliosis and motoneuronal degeneration in the brainstem motor nuclei of a transgenic mouse model of ALS. Neurosci. Res. 59, 446–456.
Kalra, S., Genge, A., and Arnold, D. L. (2003). A prospective, randomized, placebo-controlled evaluation of corticoneuronal response to intrathecal BDNF therapy in ALS using magnetic resonance spectroscopy: feasibility and results. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 4, 22–26.
Kaplitt, M. G., Feigin, A., Tang, C., Fitzsimons, H. L., Mattis, P., Lawlor, P. A., Bland, R. J., Young, D., Strybing, K., Eidelberg, D., and During, M. J. (2007). Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: an open label, phase I trial. Lancet 369, 2097–2105.
Kaspar, B. K., Llado, J., Sherkat, N., Rothstein, J. D., and Gage, F. H. (2003). Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science 301, 839–842.
Kastin, A. J., Akerstrom, V., and Pan, W. (2003). Glial cell line-derived neurotrophic factor does not enter normal mouse brain. Neurosci. Lett. 340, 239–241.
Kim, H., Kim, H. Y., Choi, M. R., Hwang, S., Nam, K. H., Kim, H. C., Han, J. S., Kim, K. S., Yoon, H. S., and Kim, S. H. (2010). Dose-dependent efficacy of ALS-human mesenchymal stem cells transplantation into cisterna magna in SOD1-G93A ALS mice. Neurosci. Lett. 468, 190–194.
Kinoshita, Y., Ito, H., Hirano, A., Fujita, K., Wate, R., Nakamura, M., Kaneko, S., Nakano, S., and Kusaka, H. (2009). Nuclear contour irregularity and abnormal transporter protein distribution in anterior horn cells in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 68, 1184–1192.
Kirkeby, A., Torup, L., Bochsen, L., Kjalke, M., Abel, K., Theilgaard-Monch, K., Johansson, P. I., Bjorn, S. E., Gerwien, J., and Leist, M. (2008). High-dose erythropoietin alters platelet reactivity and bleeding time in rodents in contrast to the neuroprotective variant carbamyl-erythropoietin (CEPO). Thromb. Haemost. 99, 720–728.
Klimaschewski, L., Meisinger, C., and Grothe, C. (1999). Localization and regulation of basic fibroblast growth factor (FGF-2) and FGF receptor-1 in rat superior cervical ganglion after axotomy. J. Neurobiol. 38, 499–506.
Koh, S. H., Kim, Y., Kim, H. Y., Cho, G. W., Kim, K. S., and Kim, S. H. (2007). Recombinant human erythropoietin suppresses symptom onset and progression of G93A-SOD1 mouse model of ALS by preventing motor neuron death and inflammation. Eur. J. Neurosci. 25, 1923–1930.
Kosai, K., Matsumoto, K., Funakoshi, H., and Nakamura, T. (1999). Hepatocyte growth factor prevents endotoxin-induced lethal hepatic failure in mice. Hepatology 30, 151–159.
Kosuge, Y., Sekikawa-Nishida, K., Negi, H., Ishige, K., and Ito, Y. (2009). Characterization of chronic glutamate-mediated motor neuron toxicity in organotypic spinal cord culture prepared from ALS model mice. Neurosci. Lett. 454, 165–169.
Kriz, J., Nguyen, M. D., and Julien, J. P. (2002). Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 10, 268–278.
Kuipers, S. D., and Bramham, C. R. (2006). Brain-derived neurotrophic factor mechanisms and function in adult synaptic plasticity: new insights and implications for therapy. Curr. Opin. Drug Discov. Dev. 9, 580–586.
Lai, E. C., Felice, K. J., Festoff, B. W., Gawel, M. J., Gelinas, D. F., Kratz, R., Murphy, M. F., Natter, H. M., Norris, F. H., and Rudnicki, S. A. (1997). Effect of recombinant human insulin-like growth factor-I on progression of ALS. A placebo-controlled study. The North America ALS/IGF-I Study Group. Neurology 49, 1621–1630.
Lamballe, F., Smeyne, R. J., and Barbacid, M. (1994). Developmental expression of trkC, the neurotrophin-3 receptor, in the mammalian nervous system. J. Neurosci. 14, 14–28.
Lambrechts, D., Storkebaum, E., Morimoto, M., Del-Favero, J., Desmet, F., Marklund, S. L., Wyns, S., Thijs, V., Andersson, J., van Marion, I., Al-Chalabi, A., Bornes, S., Musson, R., Hansen, V., Beckman, L., Adolfsson, R., Pall, H. S., Prats, H., Vermeire, S., Rutgeerts, P., Katayama, S., Awata, T., Leigh, N., Lang-Lazdunski, L., Dewerchin, M., Shaw, C., Moons, L., Vlietinck, R., Morrison, K. E., Robberecht, W., Van Broeckhoven, C., Collen, D., Andersen, P. M., and Carmeliet, P. (2003). VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat. Genet. 34, 383–394.
Leigh, P. N., Meininger, V., Bensimon, G., Cudkowicz, M., and Robberecht, W. (2008). Minocycline for patients with ALS. Lancet Neurol. 7, 119–120; author reply 120–111.
Leist, M., Ghezzi, P., Grasso, G., Bianchi, R., Villa, P., Fratelli, M., Savino, C., Bianchi, M., Nielsen, J., Gerwien, J., Kallunki, P., Larsen, A. K., Helboe, L., Christensen, S., Pedersen, L. O., Nielsen, M., Torup, L., Sager, T., Sfacteria, A., Erbayraktar, S., Erbayraktar, Z., Gokmen, N., Yilmaz, O., Cerami-Hand, C., Xie, Q. W., Coleman, T., Cerami, A., and Brines, M. (2004). Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science 305, 239–242.
Lepore, A. C., Haenggeli, C., Gasmi, M., Bishop, K. M., Bartus, R. T., Maragakis, N. J., and Rothstein, J. D. (2007). Intraparenchymal spinal cord delivery of adeno-associated virus IGF-1 is protective in the SOD1G93A model of ALS. Brain Res. 1185, 256–265.
Levi-Montalcini, R., and Cohen, S. (1960). Effects of the extract of the mouse submaxillary salivary glands on the sympathetic system of mammals. Ann. N. Y. Acad. Sci. 85, 324–341.
Levi-Montalcini, R., and Hamburger, V. (1951). Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J. Exp. Zool. 116, 321–361.
Levi-Montalcini, R., Meyer, H., and Hamburger, V. (1954). In vitro experiments on the effects of mouse sarcomas 180 and 37 on the spinal and sympathetic ganglia of the chick embryo. Cancer Res. 14, 49–57.
Lewczuk, P., Hasselblatt, M., Kamrowski-Kruck, H., Heyer, A., Unzicker, C., Siren, A. L., and Ehrenreich, H. (2000). Survival of hippocampal neurons in culture upon hypoxia: effect of erythropoietin. Neuroreport 11, 3485–3488.
Li, B., Xu, W., Luo, C., Gozal, D., and Liu, R. (2003). VEGF-induced activation of the PI3-K/Akt pathway reduces mutant SOD1-mediated motor neuron cell death. Brain Res. 111, 155–164.
Li, L., Oppenheim, R. W., Lei, M., and Houenou, L. J. (1994). Neurotrophic agents prevent motoneuron death following sciatic nerve section in the neonatal mouse. J. Neurobiol. 25, 759–766.
Li, W., Brakefield, D., Pan, Y., Hunter, D., Myckatyn, T. M., and Parsadanian, A. (2007). Muscle-derived but not centrally derived transgene GDNF is neuroprotective in G93A-SOD1 mouse model of ALS. Exp. Neurol. 203, 457–471.
Lin, L. F., Doherty, D. H., Lile, J. D., Bektesh, S., and Collins, F. (1993). GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 260, 1130–1132.
Lin, L. F., Mismer, D., Lile, J. D., Armes, L. G., Butler, E. T. III, Vannice, J. L., and Collins, F. (1989). Purification, cloning, and expression of ciliary neurotrophic factor (CNTF). Science 246, 1023–1025.
Lo, D. C. (1998). Instructive roles of neurotrophins in synaptic plasticity. Prog. Brain Res. 117, 65–70.
Lowry, K. S., Murray, S. S., McLean, C. A., Talman, P., Mathers, S., Lopes, E. C., and Cheema, S. S. (2001). A potential role for the p75 low-affinity neurotrophin receptor in spinal motor neuron degeneration in murine and human amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2, 127–134.
Lunn, J. S., Sakowski, S. A., Kim, B., Rosenberg, A. A., and Feldman, E. L. (2009). Vascular endothelial growth factor prevents G93A-SOD1-induced motor neuron degeneration. Dev. Neurobiol. 69, 871–884.
Madhavan, R., and Peng, H. B. (2006). HGF induction of postsynaptic specializations at the neuromuscular junction. J. Neurobiol. 66, 134–147.
Manev, H., and Manev, R. (2009). Interactions with GluR1 AMPA receptors could influence the therapeutic usefulness of minocycline in ALS. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1–2.
Martin, L. J. (2006). Mitochondriopathy in Parkinson disease and amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 65, 1103–1110.
Martinez, H. R., Gonzalez-Garza, M. T., Moreno-Cuevas, J. E., Caro, E., Gutierrez-Jimenez, E., and Segura, J. J. (2009). Stem-cell transplantation into the frontal motor cortex in amyotrophic lateral sclerosis patients. Cytotherapy 11, 26–34.
Masu, Y., Wolf, E., Holtmann, B., Sendtner, M., Brem, G., and Thoenen, H. (1993). Disruption of the CNTF gene results in motor neuron degeneration. Nature 365, 27–32.
Mazzini, L., Ferrero, I., Luparello, V., Rustichelli, D., Gunetti, M., Mareschi, K., Testa, L., Stecco, A., Tarletti, R., Miglioretti, M., Fava, E., Nasuelli, N., Cisari, C., Massara, M., Vercelli, R., Oggioni, G. D., Carriero, A., Cantello, R., Monaco, F., and Fagioli, F. (2009). Mesenchymal stem cell transplantation in amyotrophic lateral sclerosis: a Phase I clinical trial. Exp. Neurol. 223, 229–237.
Mennini, T., De Paola, M., Bigini, P., Mastrotto, C., Fumagalli, E., Barbera, S., Mengozzi, M., Viviani, B., Corsini, E., Marinovich, M., Torup, L., Van Beek, J., Leist, M., Brines, M., Cerami, A., and Ghezzi, P. (2006). Nonhematopoietic erythropoietin derivatives prevent motoneuron degeneration in vitro and in vivo. Mol. Med. 12, 153–160.
Messi, M. L., Clark, H. M., Prevette, D. M., Oppenheim, R. W., and Delbono, O. (2007). The lack of effect of specific overexpression of IGF-1 in the central nervous system or skeletal muscle on pathophysiology in the G93A SOD-1 mouse model of ALS. Exp. Neurol. 207, 52–63.
Milane, A., Tortolano, L., Fernandez, C., Bensimon, G., Meininger, V., and Farinotti, R. (2009). Brain and plasma riluzole pharmacokinetics: effect of minocycline combination. J. Pharm. Pharm. Sci. 12, 209–217.
Miller, R. G., Mitchell, J. D., Lyon, M., and Moore, D. H. (2007). Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. CD001447.
Miller, R. G., Petajan, J. H., Bryan, W. W., Armon, C., Barohn, R. J., Goodpasture, J. C., Hoagland, R. J., Parry, G. J., Ross, M. A., and Stromatt, S. C. (1996). A placebo-controlled trial of recombinant human ciliary neurotrophic (rhCNTF) factor in amyotrophic lateral sclerosis. rhCNTF ALS Study Group. Ann. Neurol. 39, 256–260.
Mitsuma, N., Yamamoto, M., Li, M., Ito, Y., Mitsuma, T., Mutoh, T., Takahashi, M., and Sobue, G. (1999). Expression of GDNF receptor (RET and GDNFR-alpha) mRNAs in the spinal cord of patients with amyotrophic lateral sclerosis. Brain Res. 820, 77–85.
Mohajeri, M. H., Figlewicz, D. A., and Bohn, M. C. (1999). Intramuscular grafts of myoblasts genetically modified to secrete glial cell line-derived neurotrophic factor prevent motoneuron loss and disease progression in a mouse model of familial amyotrophic lateral sclerosis. Hum. Gene Ther. 10, 1853–1866.
Moore, D. H., II, and Miller, R. G. (2004). Improving efficiency of ALS clinical trials using lead-in designs. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 5(Suppl. 1), 57–60.
Morstyn, G., Campbell, L., Souza, L. M., Alton, N. K., Keech, J., Green, M., Sheridan, W., Metcalf, D., and Fox, R. (1988). Effect of granulocyte colony stimulating factor on neutropenia induced by cytotoxic chemotherapy. Lancet 1, 667–672.
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., and Przedborski, S. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622.
Nagano, I., Ilieva, H., Shiote, M., Murakami, T., Yokoyama, M., Shoji, M., and Abe, K. (2005a). Therapeutic benefit of intrathecal injection of insulin-like growth factor-1 in a mouse model of Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 235, 61–68.
Nagano, I., Shiote, M., Murakami, T., Kamada, H., Hamakawa, Y., Matsubara, E., Yokoyama, M., Moritaz, K., Shoji, M., and Abe, K. (2005b). Beneficial effects of intrathecal IGF-1 administration in patients with amyotrophic lateral sclerosis. Neurol. Res. 27, 768–772.
Nakamura, T., Nawa, K., and Ichihara, A. (1984). Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem. Biophys. Res. Commun. 122, 1450–1459.
Nakao, N., Odin, P., Lindvall, O., and Brundin, P. (1996). Differential trophic effects of basic fibroblast growth factor, insulin-like growth factor-1, and neurotrophin-3 on striatal neurons in culture. Exp. Neurol. 138, 144–157.
Nefussy, B., Artamonov, I., Deutsch, V., Naparstek, E., Nagler, A., and Drory, V. E. (2009). Recombinant human granulocyte-colony stimulating factor administration for treating amyotrophic lateral sclerosis: A pilot study. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1–7.
Ochs, G., Penn, R. D., York, M., Giess, R., Beck, M., Tonn, J., Haigh, J., Malta, E., Traub, M., Sendtner, M., and Toyka, K. V. (2000). A phase I/II trial of recombinant methionyl human brain derived neurotrophic factor administered by intrathecal infusion to patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1, 201–206.
Okura, Y., Arimoto, H., Tanuma, N., Matsumoto, K., Nakamura, T., Yamashima, T., Miyazawa, T., and Matsumoto, Y. (1999). Analysis of neurotrophic effects of hepatocyte growth factor in the adult hypoglossal nerve axotomy model. Eur. J. Neurosci. 11, 4139–4144.
Oosthuyse, B., Moons, L., Storkebaum, E., Beck, H., Nuyens, D., Brusselmans, K., Van Dorpe, J., Hellings, P., Gorselink, M., Heymans, S., Theilmeier, G., Dewerchin, M., Laudenbach, V., Vermylen, P., Raat, H., Acker, T., Vleminckx, V., Van Den Bosch, L., Cashman, N., Fujisawa, H., Drost, M. R., Sciot, R., Bruyninckx, F., Hicklin, D. J., Ince, C., Gressens, P., Lupu, F., Plate, K. H., Robberecht, W., Herbert, J. M., Collen, D., and Carmeliet, P. (2001). Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat. Genet. 28, 131–138.
Palesch, Y. Y., and Tilley, B. C. (2004). An efficient multi-stage, single-arm Phase II futility design for ALS. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 5(Suppl. 1), 55–56.
Pan, H. C., Wu, H. T., Cheng, F. C., Chen, C. H., Sheu, M. L., and Chen, C. J. (2009). Potentiation of angiogenesis and regeneration by G-CSF after sciatic nerve crush injury. Biochem. Biophys. Res. Commun. 382, 177–182.
Pan, W., Banks, W. A., Fasold, M. B., Bluth, J., and Kastin, A. J. (1998). Transport of brain-derived neurotrophic factor across the blood-brain barrier. Neuropharmacology 37, 1553–1561.
Pan, W., Kastin, A. J., Maness, L. M., and Brennan, J. M. (1999). Saturable entry of ciliary neurotrophic factor into brain. Neurosci. Lett. 263, 69–71.
Pardridge, W. M., Kang, Y. S., and Buciak, J. L. (1994). Transport of human recombinant brain-derived neurotrophic factor (BDNF) through the rat blood-brain barrier in vivo using vector-mediated peptide drug delivery. Pharm. Res. 11, 738–746.
Park, S., Kim, H. T., Yun, S., Kim, I. S., Lee, J., Lee, I. S., and Park, K. I. (2009). Growth factor-expressing human neural progenitor cell grafts protect motor neurons but do not ameliorate motor performance and survival in ALS mice. Exp. Mol. Med. 41, 487–500.
Pasinelli, P., and Brown, R. H. (2006). Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 7, 710–723.
Patel, N. K., Bunnage, M., Plaha, P., Svendsen, C. N., Heywood, P., and Gill, S. S. (2005). Intraputamenal infusion of glial cell line-derived neurotrophic factor in PD: a two-year outcome study. Ann. Neurol. 57, 298–302.
Piehl, F., Ji, R. R., Cullheim, S., Hokfelt, T., Lindholm, D., and Hughes, R. A. (1995). Fibroblast growth factors regulate calcitonin gene-related peptide mRNA expression in rat motoneurons after lesion and in culture. Eur. J. Neurosci. 7, 1739–1750.
Pitzer, C., Klussmann, S., Kruger, C., Letellier, E., Plaas, C., Dittgen, T., Kirsch, F., Stieltjes, B., Weber, D., Laage, R., Martin-Villalba, A., and Schneider, A. (2010). The hematopoietic factor granulocyte-colony stimulating factor improves outcome in experimental spinal cord injury. J. Neurochem. 113, 930–942.
Pitzer, C., Kruger, C., Plaas, C., Kirsch, F., Dittgen, T., Muller, R., Laage, R., Kastner, S., Suess, S., Spoelgen, R., Henriques, A., Ehrenreich, H., Schabitz, W. R., Bach, A., and Schneider, A. (2008). Granulocyte-colony stimulating factor improves outcome in a mouse model of amyotrophic lateral sclerosis. Brain 131, 3335–3347.
Poduslo, J. F., and Curran, G. L. (1996). Permeability at the blood-brain and blood-nerve barriers of the neurotrophic factors: NGF, CNTF, NT-3, BDNF. Brain Res. 36, 280–286.
Ratnaparkhi, A., Lawless, G. M., Schweizer, F. E., Golshani, P., and Jackson, G. R. (2008). A Drosophila model of ALS: human ALS-associated mutation in VAP33A suggests a dominant negative mechanism. PLoS ONE 3, e2334. doi:10.1371/journal.pone.0002334.
Rothstein, J. D. (2009). Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann. Neurol. 65(Suppl 1), S3–S9.
Samoylenko, A., Byts, N., Rajalingam, K., von Ahsen, N., Rapp, U. R., Ehrenreich, H., and Siren, A. L. (2008). Thrombopoietin inhibits nerve growth factor-induced neuronal differentiation and ERK signalling. Cell. Signal. 20, 154–162.
Saver, J. L., Albers, G. W., Dunn, B., Johnston, K. C., and Fisher, M. (2009). Stroke Therapy Academic Industry Roundtable (STAIR) recommendations for extended window acute stroke therapy trials. Stroke 40, 2594–2600.
Schiffer, D., Cordera, S., Cavalla, P., and Migheli, A. (1996). Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J. Neurol. Sci. 139(Suppl.), 27–33.
Schneider, A., Kruger, C., Steigleder, T., Weber, D., Pitzer, C., Laage, R., Aronowski, J., Maurer, M. H., Gassler, N., Mier, W., Hasselblatt, M., Kollmar, R., Schwab, S., Sommer, C., Bach, A., Kuhn, H. G., and Schabitz, W. R. (2005). The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J. Clin. Invest. 115, 2083–2098.
Schoenfeld, D. A., and Cudkowicz, M. (2008). Design of phase II ALS clinical trials. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 9, 16–23.
Schymick, J. C., Talbot, K., and Traynor, B. J. (2007). Genetics of sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 16, R233–R242.
Scott, S., Kranz, J. E., Cole, J., Lincecum, J. M., Thompson, K., Kelly, N., Bostrom, A., Theodoss, J., Al-Nakhala, B. M., Vieira, F. G., Ramasubbu, J., and Heywood, J. A. (2008). Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 9, 4–15.
Sendtner, M., Holtmann, B., Kolbeck, R., Thoenen, H., Barde, Y. A. (1992a). Brain-derived neurotrophic factor prevents the death of motoneurons in newborn rats after nerve section. Nature 360, 757–759.
Sendtner, M., Schmalbruch, H., Stockli, K. A., Carroll, P., Kreutzberg, G. W., and Thoenen, H. (1992b). Ciliary neurotrophic factor prevents degeneration of motor neurons in mouse mutant progressive motor neuronopathy. Nature 358, 502–504.
Sendtner, M., Kreutzberg, G. W., and Thoenen, H. (1990). Ciliary neurotrophic factor prevents the degeneration of motor neurons after axotomy. Nature 345, 440–441.
Shefner, J. M., Cudkowicz, M. E., Zhang, H., Schoenfeld, D., and Jillapalli, D. (2004). The use of statistical MUNE in a multicenter clinical trial. Muscle Nerve 30, 463–469.
Shemer, J., Raizada, M. K., Masters, B. A., Ota, A., and LeRoith, D. (1987). Insulin-like growth factor I receptors in neuronal and glial cells. Characterization and biological effects in primary culture. J. Biol. Chem. 262, 7693–7699.
Siren, A. L., Fratelli, M., Brines, M., Goemans, C., Casagrande, S., Lewczuk, P., Keenan, S., Gleiter, C., Pasquali, C., Capobianco, A., Mennini, T., Heumann, R., Cerami, A., Ehrenreich, H., and Ghezzi, P. (2001a). Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc. Natl. Acad. Sci. U.S.A. 98, 4044–4049.
Siren, A. L., Knerlich, F., Poser, W., Gleiter, C. H., Bruck, W., and Ehrenreich, H. (2001b). Erythropoietin and erythropoietin receptor in human ischemic/hypoxic brain. Acta Neuropathol. 101, 271–276.
Sondell, M., Lundborg, G., and Kanje, M. (1999). Vascular endothelial growth factor has neurotrophic activity and stimulates axonal outgrowth, enhancing cell survival and Schwann cell proliferation in the peripheral nervous system. J. Neurosci. 19, 5731–5740.
Sondell, M., Sundler, F., and Kanje, M. (2000). Vascular endothelial growth factor is a neurotrophic factor which stimulates axonal outgrowth through the flk-1 receptor. Eur. J. Neurosci. 12, 4243–4254.
Sorenson, E. J., Windbank, A. J., Mandrekar, J. N., Bamlet, W. R., Appel, S. H., Armon, C., Barkhaus, P. E., Bosch, P., Boylan, K., David, W. S., Feldman, E., Glass, J., Gutmann, L., Katz, J., King, W., Luciano, C. A., McCluskey, L. F., Nash, S., Newman, D. S., Pascuzzi, R. M., Pioro, E., Sams, L. J., Scelsa, S., Simpson, E. P., Subramony, S. H., Tiryaki, E., and Thornton, C. A. (2008). Subcutaneous IGF-1 is not beneficial in 2-year ALS trial. Neurology 71, 1770–1775.
Statler, P. A., McPherson, R. J., Bauer, L. A., Kellert, B. A., and Juul, S. E. (2007). Pharmacokinetics of high-dose recombinant erythropoietin in plasma and brain of neonatal rats. Pediatr. Res. 61, 671–675.
Storkebaum, E., Lambrechts, D., Dewerchin, M., Moreno-Murciano, M. P., Appelmans, S., Oh, H., Van Damme, P., Rutten, B., Man, W. Y., De Mol, M., Wyns, S., Manka, D., Vermeulen, K., Van Den Bosch, L., Mertens, N., Schmitz, C., Robberecht, W., Conway, E. M., Collen, D., Moons, L., and Carmeliet, P. (2005). Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat. Neurosci. 8, 85–92.
Stroke Therapy Academic Industry Roundtable II (STAIR-II). (2001). Recommendations (for) clinical trial evaluation of acute stroke therapies. Stroke 32, 1598–1606.
Strom, A. L., Gal, J., Shi, P., Kasarskis, E. J., Hayward, L. J., and Zhu, H. (2008). Retrograde axonal transport and motor neuron disease. J. Neurochem. 106, 495–505.
Sun, W., Funakoshi, H., and Nakamura, T. (2002). Overexpression of HGF retards disease progression and prolongs life span in a transgenic mouse model of ALS. J. Neurosci. 22, 6537–6548.
Suzuki, M., McHugh, J., Tork, C., Shelley, B., Klein, S. M., Aebischer, P., and Svendsen, C. N. (2007). GDNF secreting human neural progenitor cells protect dying motor neurons, but not their projection to muscle, in a rat model of familial ALS. PLoS ONE 2, e689. doi:10.1371/journal.pone.0000689.
Tatsumi, R., Sankoda, Y., Anderson, J. E., Sato, Y., Mizunoya, W., Shimizu, N., Suzuki, T., Yamada, M., Rhoads, R. P., Jr., Ikeuchi, Y., and Allen, R. E. (2009). Possible implication of satellite cells in regenerative motoneuritogenesis: HGF upregulates neural chemorepellent Sema3A during myogenic differentiation. Am. J. Physiol. Cell Physiol. 297, C238–C252.
The BDNF Study Group (Phase III) (1999). A controlled trial of recombinant methionyl human BDNF in ALS. Neurology 52, 1427–1433.
Tolosa, L., Mir, M., Asensio, V. J., Olmos, G., and Llado, J. (2008). Vascular endothelial growth factor protects spinal cord motoneurons against glutamate-induced excitotoxicity via phosphatidylinositol 3-kinase. J. Neurochem. 105, 1080–1090.
Toth, C., Martinez, J. A., Liu, W. Q., Diggle, J., Guo, G. F., Ramji, N., Mi, R., Hoke, A., and Zochodne, D. W. (2008). Local erythropoietin signaling enhances regeneration in peripheral axons. Neuroscience 154, 767–783.
Tsuboi, Y., Kakimoto, K., Akatsu, H., Daikuhara, Y., and Yamada, T. (2002). Hepatocyte growth factor in cerebrospinal fluid in neurologic disease. Acta Neurol. Scand. 106, 99–103.
Turner, B. J., Murray, S. S., Piccenna, L. G., Lopes, E. C., Kilpatrick, T. J., and Cheema, S. S. (2004). Effect of p75 neurotrophin receptor antagonist on disease progression in transgenic amyotrophic lateral sclerosis mice. J. Neurosci. Res. 78, 193–199.
Tuttle, R., and O’Leary, D. D. (1998). Neurotrophins rapidly modulate growth cone response to the axon guidance molecule, collapsin-1. Mol. Cell. Neurosci. 11, 1–8.
Valdes-Sanchez, T., Kirstein, M., Perez-Villalba, A., Vega, J. A., and Farinas, I. (2010). BDNF is essentially required for the early postnatal survival of nociceptors. Dev. Biol. 339, 465–476.
Valdmanis, P. N., Daoud, H., Dion, P. A., and Rouleau, G. A. (2009). Recent advances in the genetics of amyotrophic lateral sclerosis. Curr. Neurol. Neurosci. Rep. 9, 198–205.
Van Den Bosch, L., Tilkin, P., Lemmens, G., and Robberecht, W. (2002). Minocycline delays disease onset and mortality in a transgenic model of ALS. Neuroreport 13, 1067–1070.
Van Vught, P. W., Sutedja, N. A., Veldink, J. H., Koeleman, B. P., Groeneveld, G. J., Wijmenga, C., Uitdehaag, B. M., de Jong, J. M., Baas, F., Wokke, J. H., and Van den Berg, L. H. (2005). Lack of association between VEGF polymorphisms and ALS in a Dutch population. Neurology 65, 1643–1645.
Vercelli, A., Mereuta, O. M., Garbossa, D., Muraca, G., Mareschi, K., Rustichelli, D., Ferrero, I., Mazzini, L., Madon, E., and Fagioli, F. (2008). Human mesenchymal stem cell transplantation extends survival, improves motor performance and decreases neuroinflammation in mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 31, 395–405.
Vicario-Abejon, C., Owens, D., McKay, R., and Segal, M. (2002). Role of neurotrophins in central synapse formation and stabilization. Nat. Rev. Neurosci. 3, 965–974.
Vincent, A. M., Mobley, B. C., Hiller, A., and Feldman, E. L. (2004). IGF-I prevents glutamate-induced motor neuron programmed cell death. Neurobiol. Dis. 16, 407–416.
Wang, W., Merrill, M. J., and Borchardt, R. T. (1996). Vascular endothelial growth factor affects permeability of brain microvessel endothelial cells in vitro. Am. J. Physiol. 271, C1973–C1980.
Wang, Y., Mao, X. O., Xie, L., Banwait, S., Marti, H. H., Greenberg, D. A., and Jin, K. (2007). Vascular endothelial growth factor overexpression delays neurodegeneration and prolongs survival in amyotrophic lateral sclerosis mice. J. Neurosci. 27, 304–307.
Wilczak, N., de Vos, R. A., and De Keyser, J. (2003). Free insulin-like growth factor (IGF)-I and IGF binding proteins 2, 5, and 6 in spinal motor neurons in amyotrophic lateral sclerosis. Lancet 361, 1007–1011.
Wong, V., Glass, D. J., Arriaga, R., Yancopoulos, G. D., Lindsay, R. M., and Conn, G. (1997). Hepatocyte growth factor promotes motor neuron survival and synergizes with ciliary neurotrophic factor. J. Biol. Chem. 272, 5187–5191.
Xiao, Q., Zhao, W., Beers, D. R., Yen, A. A., Xie, W., Henkel, J. S., and Appel, S. H. (2007). Mutant SOD1(G93A) microglia are more neurotoxic relative to wild-type microglia. J. Neurochem. 102, 2008–2019.
Yan, Q., Elliott, J., and Snider, W. D. (1992). Brain-derived neurotrophic factor rescues spinal motor neurons from axotomy-induced cell death. Nature 360, 753–755.
Yang, J. P., Liu, H. J., Cheng, S. M., Wang, Z. L., Cheng, X., Yu, H. X., and Liu, X. F. (2009). Direct transport of VEGF from the nasal cavity to brain. Neurosci. Lett. 449, 108–111.
Yang, M., Xia, W. J., Li, K., Pong, N. H., Chik, K. W., Li, C. K., Ng, M. H., Ng, H. K., Fung, K. P., and Fok, T. F. (2004). Identification of TPO receptors on central nervous system-a preliminary report. Zhongguo Shi Yan Xue Ye Xue Za Zhi 12, 494–497.
Zhang, W., Narayanan, M., and Friedlander, R. M. (2003). Additive neuroprotective effects of minocycline with creatine in a mouse model of ALS. Ann. Neurol. 53, 267–270.
Zhang, Y., Wang, L., Fu, Y., Song, H., Zhao, H., Deng, M., Zhang, J., and Fan, D. (2008). Preliminary investigation of effect of granulocyte colony stimulating factor on amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1–2.
Zhang, Y., Zhang, H., Fu, Y., Song, H., Wang, L., Zhang, J., and Fan, D. (2006). VEGF C2578A polymorphism does not contribute to amyotrophic lateral sclerosis susceptibility in sporadic Chinese patients. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 7, 119–122.
Zhao, L. R., Navalitloha, Y., Singhal, S., Mehta, J., Piao, C. S., Guo, W. P., Kessler, J. A., and Groothuis, D. R. (2007). Hematopoietic growth factors pass through the blood-brain barrier in intact rats. Exp. Neurol. 204, 569–573.
Zheng, C., Skold, M. K., Li, J., Nennesmo, I., Fadeel, B., and Henter, J. I. (2007). VEGF reduces astrogliosis and preserves neuromuscular junctions in ALS transgenic mice. Biochem. Biophys. Res. Commun. 363, 989–993.
Zhu, S., Stavrovskaya, I. G., Drozda, M., Kim, B. Y., Ona, V., Li, M., Sarang, S., Liu, A. S., Hartley, D. M., Wu, D. C., Gullans, S., Ferrante, R. J., Przedborski, S., Kristal, B. S., and Friedlander, R. M. (2002). Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 417, 74–78.
Keywords: amyotrophic lateral sclerosis, growth factors, therapy, clinical trial, animal model, motoneurons
Citation: Henriques A, Pitzer C and Schneider A (2010) Neurotrophic growth factors for the treatment of amyotrophic lateral sclerosis: where do we stand? Front. Neurosci. 4:32. doi: 10.3389/fnins.2010.00032
This article was submitted to Frontiers in Neuropharmacology, a specialty of Frontiers in Neuroscience.
Received: 14 February 2010;
Paper pending published: 11 March 2010;
Accepted: 07 May 2010;
Published online: 11 June 2010
Edited by:
Lucile Capuron, University Victor Segalen Bordeaux 2, FranceReviewed by:
Simon Lunn, University of Michigan, USAMassimo Corbo, Centro Clinico NeuroMuscular Omnicentre, Italy
Copyright: © 2010 Henriques, Pitzer and Schneider. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Armin Schneider, Department of Molecular Neurology, SYGNIS Bioscience, Im Neuenheimer Feld 515, 69120 Heidelberg, Germany. e-mail:c2NobmVpZGVyQHN5Z25pcy5kZQ==
Abbreviations: ALS, amyotrophic lateral sclerosis; ALSFRS, ALS Functional Rating Scale; BDNF, brain-derived neurotrophic factor; CNS, central nervous system; CNTF, ciliary neurotrophic factor; EPO, erythropoietin; FGF, fibroblast growth factor; FVC, forced vital capacity; G-CSF: granulocyte-colony stimulating factor; GDNF, glial-derived neurotrophic factor; IGF-1, insulin-like growth factor 1; MNs, motoneurons; MMT, manual muscle testing; NGF, nerve growth factor; NMJ, neuromuscular junction; NT-3, neurotrophin 3; NT-4/5, neurotrophin 4/5; SOD1: superoxide dismutase 1; TDI, therapy development institute; VEGF, vascular endothelial growth factor.