

- 1 Discipline of Medicine, Royal Adelaide Hospital, University of Adelaide, Adelaide, SA, Australia
- 2 NHMRC Centre of Clinical Research Excellence in Nutritional Physiology, Interactions and Outcomes, University of Adelaide, Adelaide, SA, Australia
Dietary fat interacts with receptors in both the oral cavity and the gastrointestinal (GI) tract to regulate fat and energy intake. This review discusses recent developments in our understanding of the mechanisms underlying the effects of fat, through its digestive products, fatty acids (FAs), on GI function and energy intake, the role of oral and intestinal FA receptors, and the implications that changes in oral and small intestinal sensitivity in response to ingested fat may have for the development of obesity.
Introduction
It is well established that the interaction of dietary fat, and specifically fatty acids (FAs), with the small intestine induces potent effects on gastrointestinal (GI) function (Heddle et al., 1988a; Herrmann et al., 1995; MacIntosh et al., 1999; Feinle-Bisset et al., 2005) that contribute to the suppression of hunger and energy intake (Seimon et al., 2010). Recently, evidence has also emerged for a sensory system that detects the presence of FAs in the oral cavity (Mattes, 1996; Gilbertson, 1998; Gilbertson et al., 1998), and this system may also play an important role in the regulation of fat and energy intake (Stewart et al., 2010a). However, despite the potent effects of small intestinal fat on GI function and energy intake suppression, there is a strong positive relationship between the intake of dietary fat with total energy intake and body weight (Golay and Bobbioni, 1997), and increasing evidence that the GI effects of fat are attenuated following consumption of a high-fat diet (Boyd et al., 2003; Park et al., 2007) and in obesity (Wisen and Johansson, 1992; French et al., 1993; Speechly and Buffenstein, 2000). Furthermore, a reduced ability to detect FAs in the oral cavity has been associated with higher energy and fat intakes, and higher body mass index (BMI) (Stewart et al., 2010a), suggesting that differences in, or impairment of, oral FA sensing mechanisms may contribute to overeating and obesity. This review will discuss current knowledge about the mechanisms underlying the effects of FAs on GI function and appetite, and the implications that changes in oral and small intestinal sensitivity to fat may have for the development of obesity. The review will have a particular focus on studies conducted in humans, however, most evidence for the mechanisms underlying the functional responses to fat ingestion comes from animal studies, and thus, animal data will be discussed where human data are lacking.
Effects of Fat Ingestion on Gastrointestinal Function, Appetite and Energy Intake
Gut-to-brain signaling, initiated by the sensing of nutrients in the small intestine, plays an important role in the regulation of GI function and energy intake (Schwartz, 2000). The interaction of nutrients, and most potently fat, with the small intestine results in feedback inhibition of gastric emptying (Cooke, 1977; Heddle et al., 1989), which serves to prolong gastric distension (Read et al., 1994), and regulate the rate at which nutrients enter the small intestine, allowing for their optimal digestion and absorption. The slowing of gastric emptying by fat is mediated by a relaxation of the proximal stomach (Azpiroz and Malagelada, 1985; Feinle et al., 1996), suppression of antral and duodenal pressures (Heddle et al., 1988a) and stimulation of tonic and phasic pyloric pressures (Heddle et al., 1988b). The presence of fat in the small intestine also stimulates the secretion of a number of GI hormones, including cholecystokinin (CCK), glucagon-like peptide-1 (GLP-1) and peptide YY (PYY), and suppresses ghrelin (Feinle et al., 2003; Feinle-Bisset et al., 2005). The important role of these peptides in the regulation of both GI function and energy intake is well described (Chaudhri et al., 2008). However, the mechanism(s) by which the small intestine senses fat are currently not well understood.
It is, nonetheless, well established that it is not fat (triacylglyceride) per se, but the digestive products of fat, FAs, that are sensed in the small intestine and are responsible for triggering the GI, and appetite-suppressant, effects of fat (O’Donovan et al., 2003; Feinle-Bisset et al., 2005). Ironically, much of the evidence for this was obtained using pharmacological agents, such as tetrahydrolipstatin, the active component of the anti-obesity drugs, Xenical® (Roche) and Alli® (GlaxoSmithKline), which were designed to decrease dietary fat absorption by inhibiting lipase activity, thereby, reducing total energy intake. The use of lipase inhibition in clinical studies has, however, revealed that the digestion of fat, and consequently the release of FAs in the small intestinal lumen, is essential for the effects of fat on gastric emptying, antropyloroduodenal motility, GI hormone secretion, appetite and energy intake (Schwizer et al., 1997; Borovicka et al., 2000; Matzinger et al., 2000; Feinle et al., 2001, 2003; O’Donovan et al., 2003; Pilichiewicz et al., 2003; Degen et al., 2006, 2007). Thus, weight loss strategies that compromise nutrient digestion and absorption interfere with those GI functions that are involved in the suppression of energy intake, most likely by impairing small intestinal nutrient sensing. In support of this, recent evidence indicates that rather than inhibiting fat digestion, strategies that enhance fat digestion and/or utilize the presence of small amounts of FAs in the small intestine may be more effective in modulating appetite and energy intake and the GI functions underlying it (Feltrin et al., 2004; Little et al., 2005, 2007; Marciani et al., 2007, 2009; Seimon et al., 2009).
Factors Modulating the Effects of Fat on Gastrointestinal Function and Energy Intake
Fat Droplet Size and Emulsification
It is conceivable that enhancing both the rate of fat digestion, and initial gastric emptying, increases the delivery of FAs to the small intestine, and as a result, potentiates the effects of fat on GI function and energy intake. One way of enhancing fat digestion is to reduce the size of fat droplets in an emulsion. This has a profound effect on lipolysis since it increases the surface area available for the binding of lipases. For example, duodenal lipolysis of a lipid emulsion containing fine droplets (0.7 μm in diameter) is ∼30% greater than that of a lipid emulsion containing coarse droplets (10 μm in diameter) (Armand et al., 1999). In a recent study in our laboratory, we were able to demonstrate that the smaller the droplet size of a lipid emulsion (0.26 μm vs. 30 μm vs. 170 μm, infused isocalorically directly into the duodenum), the greater the effect on the stimulation of pyloric pressures, CCK and PYY secretion, and the suppression of hunger (Seimon et al., 2009). Similarly, enhancing the acid stability of a fat emulsion results in an initially rapid phase of gastric emptying that increases the exposure of the small intestine to fat and its digestion products (Meyer et al., 1996, 1999). Acid-stable emulsions have been shown to increase plasma CCK secretion and fullness, and suppress hunger, more potently than acid-unstable emulsions, in which the oil rapidly layers above the aqueous components and, as a result, empties into the small intestine much more slowly (Little et al., 2007; Marciani et al., 2007, 2009). Taken together, these examples illustrate strategies that could be employed to structure fat-containing foods to enhance the liberation of FAs in the intestine, thereby resulting in greater modulation of both GI function and satiety.
Acyl Chain Length of Fatty Acids
Once FAs have been released in the process of digestion, their effects are critically dependent upon their acyl chain length, such that only FAs with a chain length of ≥12 carbon atoms notably slow gastric emptying, stimulate GI hormone release and suppress energy intake (Hunt and Knox, 1968; Meyer et al., 1998b; McLaughlin et al., 1999; Feltrin et al., 2004). For example, in humans, intraduodenal infusion of lauric acid (C12) resulted in a much more marked increase in plasma CCK concentrations than decanoic acid (C10) (McLaughlin et al., 1999; Feltrin et al., 2004). Similarly, intraduodenal infusion of sodium oleate (C18:1), but not sodium caprylate (C8), stimulated CCK secretion (Matzinger et al., 2000). A similar pattern of effect applies to other gut hormones, e.g., C12 caused a significant increase in plasma GLP-1 and PYY concentrations, and suppressed ghrelin, whilst C10 did not (Feltrin et al., 2004, 2006). Once fatty chain length exceeds 12 carbon atoms, FAs appear to exert comparable effects on GI function. For example, the effects of isocaloric intraduodenal loads of C12 and C18:1 on the stimulation of pyloric pressures and CCK secretion are comparable (Feltrin et al., 2008).
Given the important role of fatty acid chain length for the effects of FAs on GI motility and gut hormone secretion, it is not surprising that the effects of FAs on appetite and energy intake are also dependent upon an acyl chain length ≥12 (Matzinger et al., 2000; Feltrin et al., 2004). For example, Matzinger and colleagues reported that C18:1 (infused at 8 mmol/h, corresponding to 0.76 kcal/min), but not C8, suppressed energy intake (Matzinger et al., 2000). Furthermore, we found that infusion of C12 (at a load of 0.4 kcal/min) potently reduced hunger and desire to eat, and suppressed energy intake at a buffet meal by ∼680 kcal, when compared with control (Feltrin et al., 2004), confirming that the “cut-off” between C12 and C10, described for effects on GI motility and hormone release, also applies to the effects on energy intake. Within the group of FAs ≥C12, we evaluated the comparative effects of C12 and C18:1, and found that C12, but not C18:1, suppressed energy intake (Feltrin et al., 2008), with the lack of effect of C18:1 apparently in contrast to the findings of Matzinger et al. (2000). However, we infused at a much lower load of 0.4 kcal/min, indicating that: (1) the effects of C18:1 on energy intake are load-dependent, and the threshold load required for the suppression of energy intake by C18:1 is between 0.4 and 0.76 kcal/min, and (2) that, calorie for calorie, C12 is more potent. The mechanism(s) underlying this specific effect of C12 on energy intake is(are) unclear, however, medium chain fatty acids are more extensively oxidized in the liver to anorexigenic ketone bodies (Bach et al., 1996) and undergo conversion to malonyl CoA, which is an important signal of energy stores in the hypothalamus and has been implicated in energy intake regulation (Bouchard, 2000; Lane et al., 2005). Furthermore, since C12 is not highly prevalent in the western diet, the GI tract of populations that are not regularly exposed to this FA could display “hypersensitivity” to its effects.
High-Fat Diet Exposure
While, as discussed, fat has potent effects on GI function and energy intake, consumption of a high-fat diet has been implicated in the development of obesity (Lissner and Heitmann, 1995), and high-fat feeding is commonly used in animals studies to induce obesity (Woods et al., 2003). The mechanisms underlying the development of obesity in response to a high-fat diet are unclear, but are likely to be multi-factorial. There is increasing evidence that the effects of fat on GI function are attenuated following consumption of a high-fat diet (Cunningham et al., 1991; French et al., 1995; Covasa and Ritter, 1999, 2000; Boyd et al., 2003). Since both plasma CCK concentrations and pyloric pressures are independent predictors of acute energy intake (Seimon et al., 2010), it is conceivable that if the stimulatory effects of fat on these GI functions are attenuated by exposure to a high-fat diet, then this may also impact on energy intake. Indeed, in rats, exposure to a high-fat diet is associated with attenuation of the suppressive effects of small intestinal fat on gastric emptying (Covasa and Ritter, 2000) and food intake (Covasa and Ritter, 1999). Furthermore, although high-fat feeding has been reported to increase the CCK response to small intestinal oleate (Spannagel et al., 1996), the effects of intraperitoneal (i.p.) administration of CCK on gastric emptying (Covasa and Ritter, 2000), hindbrain activation (Covasa et al., 2000) and energy intake (Covasa and Ritter, 1998; Covasa et al., 2001) in rats are attenuated by high-fat feeding, indicating that sensitivity to CCK is reduced by excess dietary fat intake. Interestingly, CCK knock-out mice have recently been reported to be resistant to high-fat diet-induced obesity (DIO), an effect that was mediated by reduced fat absorption and increased energy expenditure in these animals compared with wild-type controls (Lo et al., 2010), indicating that CCK may also play an important role in regulating body fat and body weight. Consumption of a high-fat diet enriched with oleic acid has also been reported to result in decreased small intestinal oleoylethanolamine (OEA) levels in rats (Artmann et al., 2008), and it has been suggested that this may contribute to the increased energy intake associated with high-fat diets. Different strains of mice are resistant (diet-induced obesity-resistant, DIO-R) or prone (diet-induced obesity-prone, DIO-P) to high-fat diet-induced obesity, and these models may be useful for determining the mechanisms underlying the development of obesity. For example, it appears that while in DIO-P mice exposure to a high-fat diet attenuates the effects of lipid on gut-to-brain signaling (measured by the detection of c-fos in the nucleus tractus solitarius (NTS) of the brainstem), these changes do not occur in DIO-R mice (Donovan et al., 2009).
In humans, epidemiological evidence suggests a relationship between the incidence of overweight and obesity with increased dietary fat consumption (Rolls, 1995; Golay and Bobbioni, 1997). In the laboratory, covert manipulation of dietary fat content results in an ∼15% increase in total daily energy intake on a high-fat diet (45–50% fat), when compared with a medium-fat diet (30–35% fat), for 2 weeks, resulting in weight gain (Lissner et al., 1987). A small number of short-term studies have investigated the effects of a high-fat diet on GI function in humans, and the results are often conflicting (Cunningham et al., 1991; French et al., 1995; Castiglione et al., 2002; Boyd et al., 2003; Little et al., 2008), likely due to the different dietary approaches used, the short-term nature of the interventions (2–3 weeks), and issues with dietary compliance. Nonetheless, the patterns of responses in such studies suggest that sensitivity to the effects of fat on GI function and energy intake is reduced by a high-fat diet. For example, the slowing of gastric emptying by fat (Cunningham et al., 1991), and the stimulatory effects of intraduodenal lipid infusion on pyloric pressures (Boyd et al., 2003), have been reported to be attenuated following a high-fat diet. The underlying mechanisms are currently unknown. Since the stimulation of pyloric pressures is mediated, at least in part, by both CCK (Brennan et al., 2005) and GLP-1 (Schirra et al., 2000), the changes in GI motility in response to a high-fat diet may be due to changes in sensitivity to, or secretion of, these gut peptides. The observation that plasma CCK and GLP-1 concentrations in response to intraduodenal lipid did not differ following a high-fat, when compared with a low-fat, diet (Boyd et al., 2003), implicates a change in sensitivity. Nonetheless, both baseline plasma CCK concentrations (Little et al., 2008), and the plasma CCK response to a standardized breakfast (French et al., 1995), were increased following consumption of a high-fat diet, suggesting that changes in secretion may also occur. The effects of exogenous CCK-8 on GI motility and energy intake have been reported to be maintained following acute exposure to a high-fat diet, when compared with an isocaloric low-fat diet, for a period of 3 weeks (Little et al., 2008), suggesting that, in lean men, and in the absence of a change in body weight, the sensitivity to exogenous CCK-8 is not changed following a short period on a high-fat diet. These results appear to be contrast to the reports from animal studies (Covasa et al., 2001), however, it is unclear whether observations from studies in animals are applicable to humans. All of the studies conducted in humans were short-term and not associated with weight gain, hence, while differences in GI function and energy intake occur acutely in response to a high-fat diet, it remains unclear how these relate to real life, i.e., chronic consumption of a high-fat diet and the development of obesity. Much further work is needed to understand the relationships between high-fat diet exposure, intestinal fat sensing, energy intake and obesity. Interestingly, while, as discussed, there is a strong epidemiological link between dietary fat consumption and body weight, not all individuals who habitually consume a high-fat diet become overweight or obese (Blundell et al., 2005), and determining whether individuals who are resistant to weight gain maintain small intestinal sensitivity to fat, may provide important insights into the mechanisms underlying the development of obesity.
Fatty Acid Sensing Mechanisms in the Gastrointestinal Tract
The cellular and molecular mechanisms through which FAs are detected in the small intestine and induce signaling to regulate GI function and energy intake are not well understood, although research on the potential receptors involved has rapidly expanded over the last few years. A schematic drawing summarizing current understanding is shown in Figure 1. The recent identification of a number of G-protein coupled receptors (GPCRs), including GPR120 (Hirasawa et al., 2005; Tanaka et al., 2008), GPR40 (Edfalk et al., 2008) and GPR119 (Lan et al., 2009; Lauffer et al., 2009), which are activated by FAs ≥ C12, or their derivatives, such as n-acylethanolamines (e.g., OEA), provides strong evidence for the existence of a GI fatty acid sensing mechanism. The receptor, CD36, which plays an important role in fat absorption in the small intestine by transporting FAs across the cell membrane, is important for the production of OEA, which has been reported to suppress food intake (Lobo et al., 2001; Schwartz et al., 2008). Subsequently, post-absorptive processing of FAs with a chain length ≥ C12, including their packaging into chylomicrons, and the associated generation of apolipoprotein A-IV (apo A-IV), also appears to play an important role, stimulating the release of CCK, which in turn, activates vagal afferents to convey information about the small intestinal environment to the brain (Glatzle et al., 2002, 2004).
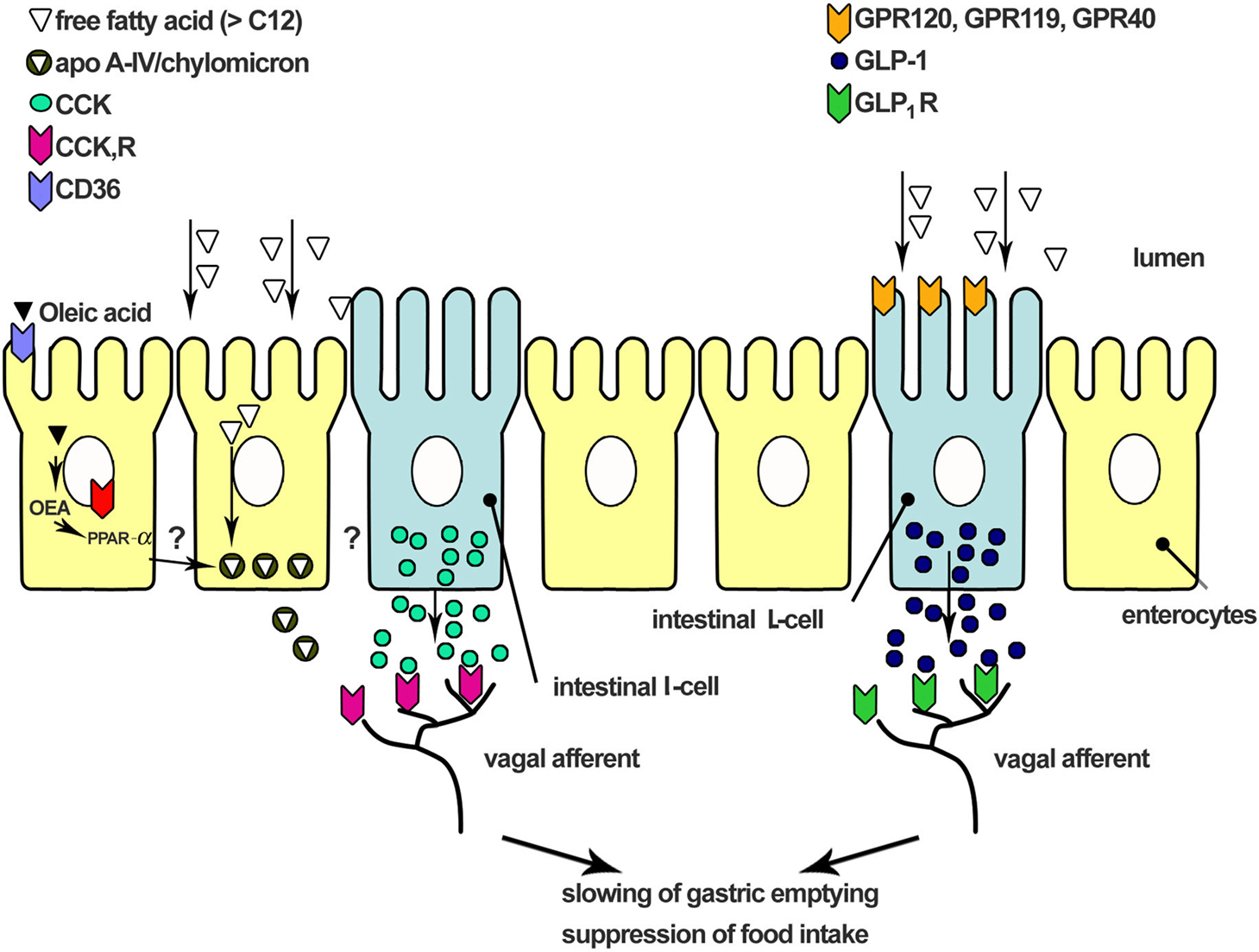
Figure 1. Schematic representation of the mechanisms involved in small intestinal fat sensing. Fatty acids are sensed by G-protein coupled receptors (e.g., GPR40, GPR120, and GPR119) located on enteroendocrine cells. These GPCRs appear to mediate gut peptide secretion induced by small intestinal fatty acids. Uptake of fatty acids into the enterocyte is mediated by the receptor CD36. This receptor is critical for the mobilization of oleoylethanolamine (OEA), which has been shown to reduce food intake and body weight via the activation of the nuclear receptor, peroxisome-proliferator-activated receptor-α (PPAR-α). Long chain fatty acids also stimulate chylomicron formation, and, in particular, the chylomicron component apolipoprotein A-IV (apo A-IV), is important in mediating CCK secretion and the effects of long-chain fatty acids on energy intake. The gut peptides released, such as CCK and GLP-1, interact with receptors on vagal afferent nerve terminals within the intestinal wall to convey signals to the brainstem and higher brain centers, resulting in changes in gastrointestinal function and the suppression of food intake.
Fatty Acid Detection
Intensive research has led to the identification of a number of GPCRs in the small intestine, dramatically advancing our understanding of the molecular mechanisms underlying the sensing of dietary fat. These GPCRs, including GPR120 (Hirasawa et al., 2005; Tanaka et al., 2008), GPR40 (Edfalk et al., 2008) and GPR119 (Lan et al., 2009; Lauffer et al., 2009), appear to be critical for the effects of fat on GI peptide secretion, although much of the evidence comes from cell line studies. The strongest evidence for a role in FA sensing exists for GPR120, which is activated by both saturated and unsaturated long-chain FAs (Miyauchi et al., 2010). GPR120 is expressed in the intestine of mouse and human, and in enteroendocrine cell lines (Hirasawa et al., 2005; Miyauchi et al., 2010), and in mice, is co-localized with endocrine L-cells in the large intestine (Hirasawa et al., 2005). In enteroendocrine cell lines, GPR120 is required for the fat-induced secretion of CCK (Tanaka et al., 2008) and GLP-1 (Hirasawa et al., 2005). Furthermore, GPR120 knock-out mice have impaired GLP-1 secretion in response to FA ingestion (Hirasawa et al., 2005). Thus, GPR120 appears important in the sensing of FAs by enteroendocrine cells. GPR40, which is activated by medium and long-chain saturated and unsaturated FAs (Briscoe et al., 2003), has also been reported to be expressed in enteroendocrine L cells, and GPR40−/− mice have an impaired GLP-1 and GIP response to oral fat ingestion (Edfalk et al., 2008). However, the role of GPR40 in the secretion of other gut peptides is unclear, since knockdown of GPR40 has no effect on CCK release from STC-1 cells (Tanaka et al., 2008). GPR119 is also expressed in L-cell models, including GLUTag cells, human NCI-H716 and primary rat intestinal cell lines (Lauffer et al., 2009). An endogenous ligand for GPR119 is OEA (Overton et al., 2006). GLP-1 is secreted in response to ileal perfusion of OEA in the rat, and knock-out of GPR119 abolishes the GLP-1 response to OEA in mGLUTag cells (Lauffer et al., 2009). These studies provide strong evidence for FA sensing mechanisms in the small intestinal lumen that mediate the effects of fat on GI peptide secretion. Much further work is needed to clarify the role of these receptors, and the precise underlying mechanisms, in fat sensing, how this relates to the regulation of energy intake in humans, and whether changes in the expression and/or sensitivity of these receptors underlie the development of obesity and/or the changes in GI function and energy intake observed following over-exposure to dietary fat.
Transduction of Fatty Acid-Induced Signaling: Release of Mediators and Vagal Transmission
Ingestion of fat triggers the production of OEA in the small intestine (Schwartz et al., 2008). In mice, peripheral administration of OEA has been reported to prolong the time taken to initiate the next meal and decrease meal frequency resulting in an overall reduction of food intake (Fu et al., 2008; Schwartz et al., 2008) and body weight (Fu et al., 2003, 2005). The production of OEA is dependent on the FA transporter, CD36, which mediates the uptake of FAs into the enterocytes. Mutant mice lacking CD36 have impaired production of OEA and fail to reduce their food intake in response to duodenal lipid (Schwartz et al., 2008). As discussed above, OEA has been shown to activate GPR119 (Lauffer et al., 2009), however, OEA is still able to reduce food intake in GPR119−/− mice, suggesting that this receptor does not mediate the anorexigenic effects of OEA. Instead, the effect of OEA to reduce food intake and body weight is mediated through the activation of the nuclear receptor, peroxisome-proliferator-activated receptor-α (PPAR-α) (Fu et al., 2003; Schwartz et al., 2008). The downstream target of PPAR-α is currently unclear, however, it may act by inducing apo A-IV release (Schwartz et al., 2008), a PPAR-α regulated protein that activates vagal afferents (Glatzle et al., 2004).
The uptake of FAs into the enterocyte is associated with the formation of chylomicrons and the stimulation of apo A-IV, processes that have both been demonstrated to be required for the signaling of information to the brain, mainly through the vagus nerve. For example, in rats, inhibiting chylomicron formation, using the “detergent” Pluronic L-81, attenuates the effects of lipid on the release of CCK (Raybould et al., 1998), activation of vagal afferents (Randich et al., 2000), and induction of c-fos like immunoreactivity in the NTS (Lo et al., 2007), and consequently blocks the gastric emptying (Raybould et al., 1998) and food intake (Meyer et al., 1998a) responses to fat ingestion. The induction of c-fos in the NTS by chylomicron formation and/or signaling is dependent on the CCK1 receptor (Lo et al., 2007). Thus, the formation of chylomicrons and the stimulation of apo A-IV appear to be critical for CCK release following ingestion of long-chain FAs. Activation of CCK1 receptors on vagal afferents, and locally on GI smooth muscle, then induces feedback effects on gastric emptying and food intake (Lo et al., 2007). In humans, the effects of FAs also appear dependent on the release of CCK. For example, the inhibitory effects of C12 on gastric emptying and the perception of intragastric volume (maximum volume tolerated) (Lal et al., 2004), and the inhibitory effects of C18:1 on energy intake (Matzinger et al., 2000), were attenuated by the CCK1 receptor antagonist, dexloxiglumide. A recent study in humans, using functional magnetic resonance imaging, has demonstrated that C12 increases activity in the brainstem and hypothalamus, and that this effect is dependent on the CCK1 receptor (Lassman et al., 2010), thus, in man, the sensing of lipid by the small intestine appears to also be signaled to the brainstem and hypothalamus via a CCK1 receptor mediated pathway. As the response was rapid (occurring within 2–4 min following C12 infusion), it is consistent with a vagally mediated response.
Taken together, it appears that small intestinal FAs are detected by GPCRs located on enteroendocrine cells to stimulate GI peptide release, and also by the mobilization of OEA, and chylomicron components, in particular apo A-IV, which in turn induce gut peptide secretion. These gut peptides interact with receptors on vagal afferent nerve terminals within the intestinal wall, conveying information about the small intestinal environment to the brainstem and higher brain centers, resulting in changes in GI function and behavioral responses, including the suppression of food intake.
Oral Fat “Taste”: Role of Oral Fatty Acid Detection in Dietary Preference and the Regulation of GI Function and Energy Intake
The taste of food is an important determinant of the amount consumed at a meal. Fat, in particular, increases the palatability of foods and can, therefore, lead to over-consumption. It has long been established that the presence of food in the oral cavity also induces physiological responses, including gastric and pancreatic secretion, as part of the so-called “cephalic phase”, which prepares the GI tract for nutrient exposure, thereby optimizing nutrient digestion and absorption. While it is well established that the human gustatory system can detect the taste qualities of sweet, sour, bitter, salty, and umami, more recently the existence of a sixth detection (“taste”) modality responsive to FAs has been revealed (Abumrad, 2005; Chale-Rush et al., 2007a,b; Gaillard et al., 2008; Mattes, 2009b). Dietary fats, i.e., triacylglycerols, do not appear to be effective stimuli per se; in analogy to intestinal fat sensing, the detection of fat in the oral cavity appears to be dependent on the presence of FAs. In humans, sensory detection of FAs occurs within the millimolar range (0.02–6.4 mM) (Stewart et al., 2010a). It has been argued that in humans the oral cavity would not be exposed to FAs as the presence of lingual lipase has been debated, however, this detection threshold is consistent with the concentrations of FAs naturally present in foods (0.76–3% w/v), and lipolytic activity in saliva has recently been shown to be sufficient to produce micromolar amounts of fatty acids within the detectable range (Stewart et al., 2010a). Importantly, the receptors implicated in oral FA detection have been characterized as analogous to those involved in small intestinal FA sensing. The following will discuss the mechanisms underlying oral FA detection, the physiological responses induced by the detection of FA in the oral cavity, and recent evidence that sensitivity to oral FAs is related to dietary fat preference and intake.
Oral Fatty Acid Detection
Oral detection of nutrients occurs as a result of the interaction of nutrients with receptors on the apical surface of taste receptor cells (TRCs). Animal studies have revealed the expression of a number of receptors, including CD36, delayed rectifying potassium channels, and a series of G-protein coupled receptors, including GPR40, GPR 41, GPR43, and GPR120 on TRCs (Mattes, 2009b). The detection of FAs by these mechanisms induces a signaling cascade activating gustatory nerves that transmit sensory information to the NTS in the brainstem (Gaillard et al., 2008), and from there to higher brain centers, such as the lateral hypothalamus and the nucleus accumbens, which play an important role in the regulation of food intake and in reward. CD36 appears to play a key role in oral fat detection; oral exposure to long-chain FAs increases [Ca2+] in TRCs, and c-fos expression in the NTS, an effect that is abolished in CD36 knockout animals (Gaillard et al., 2008). The role of GPCRs in fat taste signal transduction is less clear. However, GPR120 has been reported to be co-localized with phospholipase Cβ2 and α-gustducin in TRCs (Matsumura et al., 2009), both of which are involved in the transduction of other tastes, such as sweetness and bitterness.
Physiological Responses Induced by Oral Fatty Acids
Oral sensing of FAs appears to have two major roles: (i) mediation of cephalic phase responses and (ii) contribution to the regulation of fat and energy intake. Oral stimulation with fat, using modified sham-feeding techniques, has been reported in humans to stimulate gastric lipase secretion (Wojdemann et al., 1997), elevate serum triglycerides (Mattes, 2009a), suppress ghrelin (Heath et al., 2004), and to reduce appetite (Heath et al., 2004; Smeets and Westerterp-Plantenga, 2006; Smeets et al., 2009) and energy intake (Crystal and Teff, 2006).
Sensitivity to Oral Fatty Acids and Dietary Fat Preference and Intake
Heightened preference for, and increased consumption of, fatty foods amongst the obese suggests that fat intake is poorly regulated in this group (Mela and Sacchetti, 1991), raising the question of relationships between oral fat sensitivity, fat intake and obesity. In animals, FA sensitivity appears to vary across strains; e.g., DIO-P (Osborne–Mendel) rats have decreased oral FA acid sensitivity when compared with DIO-R (S5B/PL) rats (Gilbertson et al., 2005). The greater oral sensitivity in the DIO-R rats is associated with reduced fat intake, reduced preference for fat, and reduced predisposition for obesity when compared with DIO-P rats (Gilbertson et al., 1998). These observations provide compelling evidence that oral FA sensitivity contributes to dietary fat preference, energy intake and, consequently, body weight. Similarly, human studies have identified a large diversity in oral sensitivity to FAs, with detection thresholds ranging from 0.02–12 mM, although all subjects appeared able to detect FAs across this range (Stewart et al., 2010a,b). Those individuals who were defined as “hypersensitive” to C18:1 (those able to detect C18:1 at a concentration of 1.4 mM) had significantly lower habitual dietary energy and fat intakes and body weights than those defined as hyposensitive (those unable to detect the presence of 1.4 mM oleic acid) (Stewart et al., 2010a). In a further study (Stewart et al., 2010b), obese subjects, as a group, had significantly higher detection thresholds for C18:1, and intraduodenal infusion of C18:1 was associated with reduced stimulation of pyloric motility, when compared with lean individuals, indicating that the obese are less able to sense, and induce appropriate signaling responses to, both oral and intestinal fat (Figure 2).
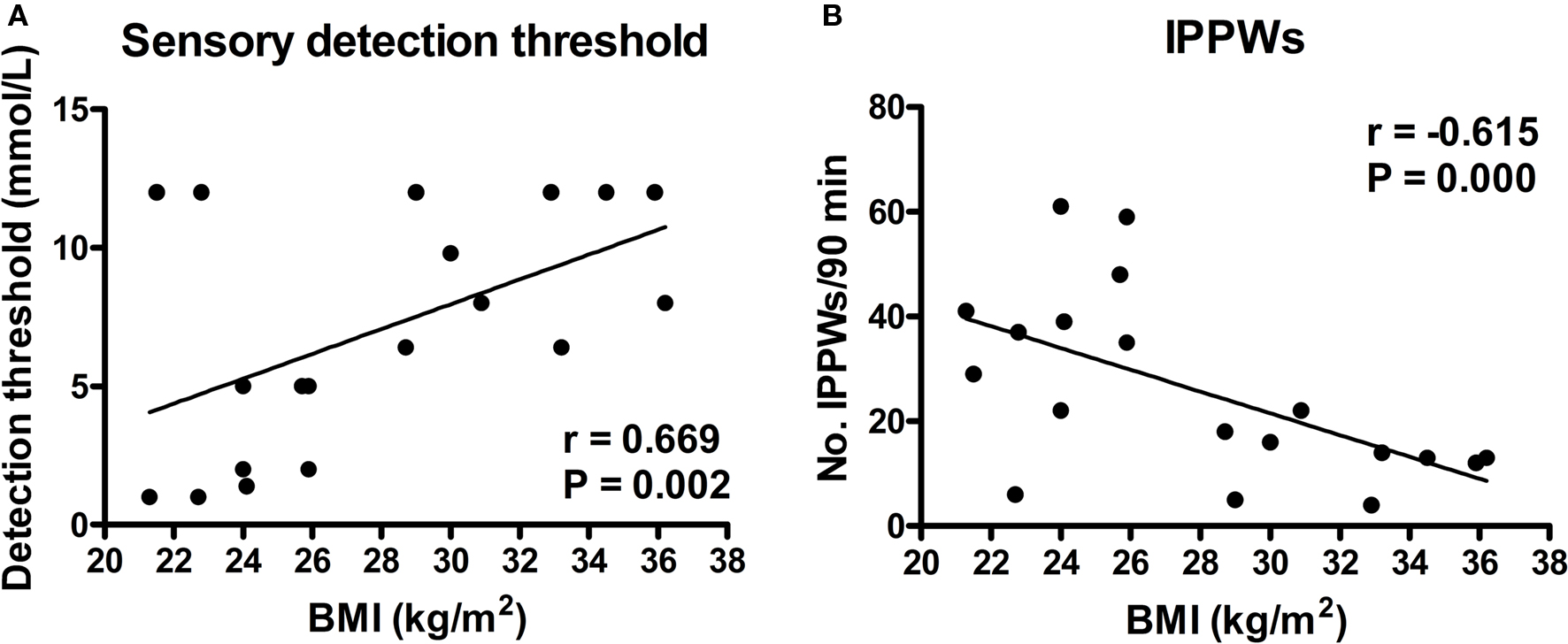
Figure 2. Relationship between (A) oral detection threshold for oleic acid (C18:1) and (B) the total number of isolated pyloric pressure waves (IPPWs) following intraduodenal infusion of C18:1 (0. 76 kcal/min for 90 min), with body mass index (BMI, kg/m2) in both lean (n = 10) and obese (n = 9) subjects (Stewart et al., 2010b).
Given the homology between the receptors involved in oral and intestinal fat sensing, this observation raises the possibility that receptor expression/sensitivity may be altered in obesity in both the oral cavity and the GI tract. Whether those individuals prone to obesity have altered expression of fat sensing receptors (i.e., decreased expression of receptors such as CD36 and GPR120 in the oral cavity and small intestine), or whether receptor expression/sensitivity is altered by factors that contribute to the development of obesity (i.e., induced by changes in environmental factors such as prolonged exposure to a high-fat diet), is currently unknown. However, given evidence of impaired small intestinal fat sensing following high-fat diet exposure, it is conceivable that dietary over-exposure to fat also modulates oral fat sensing. In order to understand the mechanisms underlying differences in oral fat detection, research is warranted to determine whether receptor expression/sensitivity can be modulated by energy/fat restriction to restore oral and intestinal signaling responses to fat.
Reward-Related Eating
Food is ingested not only to replenish energy stores, but also because it is enjoyable and rewarding, and this may particularly relate to fat-containing foods, due to their high palatability. Thus, dietary fat does not only influence homeostatic pathways, as discussed above, but there is increasing evidence that it can powerfully modify food intake by activating hedonic pathways, increasing the desire to consume foods that are highly palatable (Lutter and Nestler, 2009). For example, studies using fMRI have demonstrated in humans that oral exposure to dietary fat activates reward areas, such as the lateral hypothalamus, amygdala, insula, and anterior cingulate cortex (De Araujo and Rolls, 2004), and this may be enhanced in obesity (Rolls, 2010). It is currently not known how differences in the ability to detect fat in the oral cavity contribute to differences in food preference and reward-related eating and, thus, overall energy intake. Studies are warranted to evaluate whether these hedonic effects of fat are related to oral fat taste sensitivity, i.e., whether individuals with reduced sensitivity to oral fat need to consume more fat/energy to receive the same reward/satisfaction. Furthermore, since an individual’s perception of reward is also likely to be related to psychological factors, such as if they have experienced aversive responses to particular foods, or if certain foods were provided as a reward for good behavior when they were children, it will also be important to determine whether such psychological factors have the ability to influence oral sensitivity to FAs (Rolls, 2010).
Conclusions and Implications
This review has summarized our current understanding of the mechanisms involved in the sensing of fat both in the oral cavity and small intestinal lumen, and the potent effects elicited by fat on GI function, including changes in motor function and secretion of gut peptides, which lead to the suppression of appetite and energy intake. The knowledge of the cellular and molecular mechanisms underlying both oral and intestinal fat sensing has increased dramatically in recent years, however, much further work is needed to fully characterize these mechanisms, particularly in humans where data are currently scarce. It will be critical to determine the role of these mechanisms in the regulation of normal energy intake and reward, whether modulation of expression and/or sensitivity of these receptors predisposes to obesity, or mediates the development of obesity following dietary overconsumption (particularly of fat), and whether, for example, impaired oral and intestinal sensing of dietary fat, as observed in obesity, can be restored by energy restriction. Ultimately, these mechanisms may then provide exciting new targets for the treatment, or prevention, of obesity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Dr Tanya Little was supported by a National Health and Medical Research Council of Australia (NHMRC) Overseas Clinical Research Postdoctoral Training Fellowship (grant: 519349, 2008–2011) and A/Prof Feinle-Bisset by an NHMRC Senior Research Fellowship (grant: 627002, 2010–2014). We would like to thank Dr Richard Young, Nerve-Gut Research Laboratory, Royal Adelaide Hospital, for his help with the creation of Figure 1.
References
Abumrad, N. A. (2005). CD36 may determine our desire for dietary fats. J. Clin. Invest. 115, 2965–2967.
Armand, M., Pasquier, B., Andre, M., Borel, P., Senft, M., Peyrot, J., Salducci, J., Portugal, H., Jaussan, V., and Lairon, D. (1999). Digestion and absorption of 2 fat emulsions with different droplet sizes in the human digestive tract. Am. J. Clin. Nutr. 70, 1096–1106.
Artmann, A., Petersen, G., Hellgren, L. I., Boberg, J., Skonberg, C., Nellemann, C., Hansen, S. H., and Hansen, H. S. (2008). Influence of dietary fatty acids on endocannabinoid and N-acylethanolamine levels in rat brain, liver and small intestine. Biochim. Biophys. Acta 1781, 200–212.
Azpiroz, F., and Malagelada, J. R. (1985). Intestinal control of gastric tone. Am. J. Physiol. Gastrointest. Liver Physiol. 249, G501–G509.
Bach, A. C., Ingenbleek, Y., and Frey, A. (1996). The usefulness of dietary medium-chain triglycerides in body weight control: fact or fancy? J. Lipid Res. 37, 708–726.
Blundell, J. E., Stubbs, R. J., Golding, C., Croden, F., Alam, R., Whybrow, S., Le Noury, J., and Lawton, C. L. (2005). Resistance and susceptibility to weight gain: individual variability in response to a high-fat diet. Physiol. Behav. 86, 614–622.
Borovicka, J., Schwizer, W., Guttmann, G., Hartmann, D., Kosinski, M., Wastiel, C., Bischof-Delaloye, A., and Fried, M. (2000). Role of lipase in the regulation of postprandial gastric acid secretion and emptying of fat in humans: a study with orlistat, a highly specific lipase inhibitor. Gut 46, 774–781.
Bouchard, C. (2000). Inhibition of food intake by inhibitors of fatty acid synthase. N. Engl. J. Med. 343, 1888–1889.
Boyd, K. A., O’Donovan, D. G., Doran, S., Wishart, J., Chapman, I. M., Horowitz, M., and Feinle, C. (2003). High-fat diet effects on gut motility, hormone, and appetite responses to duodenal lipid in healthy men. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G188–G196.
Brennan, I. M., Feltrin, K. L., Horowitz, M., Smout, A. J., Meyer, J. H., Wishart, J., and Feinle-Bisset, C. (2005). Evaluation of interactions between CCK and GLP-1 in their effects on appetite, energy intake and antropyloroduodenal motility in healthy men. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R1477–R1485.
Briscoe, C. P., Tadayyon, M., Andrews, J. L., Benson, W. G., Chambers, J. K., Eilert, M. M. Ellis, C., Elshourbagy, N. A., Goetz, A. S., Minnick, D. T., Murdock, P. R., Sauls, H. R. Jr., Shabon, U., Spinage, L. D., Strum, J. C., Szekeres, P. G., Tan, K. B., Way, J. M., Ignar, D. M., Wilson, S., and Muir, A. I. (2003). The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 278, 11303–11311.
Castiglione, K. E., Read, N. W., and French, S. J. (2002). Adaptation to high-fat diet accelerates emptying of fat but not carbohydrate test meals in humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 282, R366–R371.
Chale-Rush, A., Burgess, J. R., and Mattes, R. D. (2007a). Evidence for human orosensory (taste?) sensitivity to free fatty acids. Chem. Senses 32, 423–431.
Chale-Rush, A., Burgess, J. R., and Mattes, R. D. (2007b). Multiple routes of chemosensitivity to free fatty acids in humans. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G1206–1212.
Chaudhri, O. B., Salem, V., Murphy, K. G., and Bloom, S. R. (2008). Gastrointestinal satiety signals. Annu. Rev. Physiol. 70, 239–255.
Cooke, A. R. (1977). Localization of receptors inhibiting gastric emptying in the gut. Gastroenterology 72, 875–880.
Covasa, M., Grahn, J., and Ritter, R. C. (2000). Reduced hindbrain and enteric neuronal response to intestinal oleate in rats maintained on high-fat diet. Auton. Neurosci. 84, 8–18.
Covasa, M., Marcuson, J. K., and Ritter, R. C. (2001). Diminished satiation in rats exposed to elevated levels of endogenous or exogenous cholecystokinin. Am. J. Physiol. Regul. Integr. Comp. Physiol. 280, R331–R337.
Covasa, M., and Ritter, R. C. (1998). Rats maintained on high-fat diets exhibit reduced satiety in response to CCK and bombesin. Peptides 19, 1407–1415.
Covasa, M., and Ritter, R. C. (1999). Reduced sensitivity to the satiation effect of intestinal oleate in rats adapted to high-fat diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 277, R279–R285.
Covasa, M., and Ritter, R. C. (2000). Adaptation to high-fat diet reduces inhibition of gastric emptying by CCK and intestinal oleate. Am. J. Physiol. Regul. Integr. Comp. Physiol. 278, R166–R170.
Crystal, S. R., and Teff, K. L. (2006). Tasting fat: cephalic phase hormonal responses and food intake in restrained and unrestrained eaters. Physiol. Behav. 89, 213–220.
Cunningham, K. M., Daly, J., Horowitz, M., and Read, N. W. (1991). Gastrointestinal adaptation to diets of differing fat composition in human volunteers. Gut 32, 483–486.
De Araujo, I. E., and Rolls, E. T. (2004). Representation in the human brain of food texture and oral fat. J. Neurosci. 24, 3086–3093.
Degen, L., Drewe, J., Piccoli, F., Grani, K., Oesch, S., Bunea, R., D’Amato, M., and Beglinger, C. (2007). Effect of CCK-1 receptor blockade on ghrelin and PYY secretion in man. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R1391–R1399.
Degen, L., Matzinger, D., Drewe, J., Nissle, S., Maecke, H., Lengsfeld, H., Hadvary, P., and Beglinger, C. (2006). Role of free Fatty acids in regulating gastric emptying and gallbladder contraction. Digestion 74, 131–139.
Donovan, M. J., Paulino, G., and Raybould, H. E. (2009). Activation of hindbrain neurons in response to gastrointestinal lipid is attenuated by high fat, high energy diets in mice prone to diet-induced obesity. Brain Res. 1248, 136–140.
Edfalk, S., Steneberg, P., and Edlund, H. (2008). GPR40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 57, 2280–2287.
Feinle, C., D’Amato, M., and Read, N. W. (1996). Cholecystokinin-A receptors modulate gastric sensory and motor responses to gastric distension and duodenal lipid. Gastroenterology 110, 1379–1385.
Feinle, C., O’Donovan, D. G., Doran, S., Andrews, J. M., Wishart, J., Chapman, I., and Horowitz, M. (2003). Effects of fat digestion on appetite, APD motility, and gut hormones in response to duodenal fat infusion in humans. Am. J. Physiol. Gastrointest. Liver Physiol. 284, G798–G807.
Feinle, C., Rades, T., Otto, B., and Fried, M. (2001). Fat digestion modulates gastrointestinal sensations induced by gastric distention and duodenal lipid in humans. Gastroenterology 120, 1100–1107.
Feinle-Bisset, C., Patterson, M., Ghatei, M. A., Bloom, S. R., and Horowitz, M. (2005). Fat digestion is required for suppression of ghrelin and stimulation of peptide YY and pancreatic polypeptide secretion by intraduodenal lipid. Am. J. Physiol. Endocrinol. Metab. 289, E948–E953.
Feltrin, K. L., Little, T. J., Meyer, J. H., Horowitz, M., Rades, T., Wishart, J., and Feinle-Bisset, C. (2008). Comparative effects of intraduodenal infusions of lauric and oleic acids on antropyloroduodenal motility, plasma cholecystokinin and peptide YY, appetite, and energy intake in healthy men. Am. J. Clin. Nutr. 87, 1181–1187.
Feltrin, K. L., Little, T. J., Meyer, J. H., Horowitz, M., Smout, A. J., Wishart, J., Pilichiewicz, A. N., Rades, T., Chapman, I. M., and Feinle-Bisset, C. (2004). Effects of intraduodenal fatty acids on appetite, antropyloroduodenal motility, and plasma CCK and GLP-1 in humans vary with their chain length. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R524–R533.
Feltrin, K. L., Patterson, M., Ghatei, M. A., Bloom, S. R., Meyer, J. H., Horowitz, M., and Feinle-Bisset, C. (2006). Effect of fatty acid chain length on suppression of ghrelin and stimulation of PYY, GLP-2 and PP secretion in healthy men. Peptides 27, 1638–1643.
French, S. J., Murray, B., Rumsey, R. D., Fadzlin, R., and Read, N. W. (1995). Adaptation to high-fat diets: effects on eating behaviour and plasma cholecystokinin. Br. J. Nutr. 73, 179–189.
French, S. J., Murray, B., Rumsey, R. D., Sepple, C. P., and Read, N. W. (1993). Preliminary studies on the gastrointestinal responses to fatty meals in obese people. Int. J. Obes. Relat. Metab. Disord. 17, 295–300.
Fu, J., Gaetani, S., Oveisi, F., Lo Verme, J., Serrano, A., Rodriguez De Fonseca, F., Rosengarth, A., Luecke, H., Di Giacomo, B., Tarzia, G., and Piomelli, D. (2003). Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature 425, 90–93.
Fu, J., Kim, J., Oveisi, F., Astarita, G., and Piomelli, D. (2008). Targeted enhancement of oleoylethanolamide production in proximal small intestine induces across-meal satiety in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R45–R50.
Fu, J., Oveisi, F., Gaetani, S., Lin, E., and Piomelli, D. (2005). Oleoylethanolamide, an endogenous PPAR-alpha agonist, lowers body weight and hyperlipidemia in obese rats. Neuropharmacology 48, 1147–1153.
Gaillard, D., Laugerette, F., Darcel, N., El-Yassimi, A., Passilly-Degrace, P., Hichami, A., Khan, N. A., Montmayeur, J. P., and Besnard, P. (2008). The gustatory pathway is involved in CD36-mediated orosensory perception of long-chain fatty acids in the mouse. FASEB J. 22, 1458–1468.
Gilbertson, T. A. (1998). Gustatory mechanisms for the detection of fat. Curr. Opin. Neurobiol. 8, 447–452.
Gilbertson, T. A., Liu, L., Kim, I., Burks, C. A., and Hansen, D. R. (2005). Fatty acid responses in taste cells from obesity-prone and -resistant rats. Physiol. Behav. 86, 681–690.
Gilbertson, T. A., Liu, L., York, D. A., and Bray, G. A. (1998). Dietary fat preferences are inversely correlated with peripheral gustatory fatty acid sensitivity. Ann. N. Y. Acad. Sci. 855, 165–168.
Glatzle, J., Darcel, N., Rechs, A. J., Kalogeris, T. J., Tso, P., and Raybould, H. E. (2004). Apolipoprotein (apo) A-IV stimulates duodenal vagal afferent fiber activity to inhibit gastric motility via a CCK-1 receptor dependent pathway. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R354–R359.
Glatzle, J., Kalogeris, T. J., Zittel, T. T., Guerrini, S., Tso, P., and Raybould, H. E. (2002). Chylomicron components mediate intestinal lipid-induced inhibition of gastric motor function. Am. J. Physiol. Gastrointest. Liver Physiol. 282, G86–G91.
Golay, A., and Bobbioni, E. (1997). The role of dietary fat in obesity. Int. J. Obes. Relat. Metab. Disord. 21(Suppl. 3), S2–S11.
Heath, R. B., Jones, R., Frayn, K. N., and Robertson, M. D. (2004). Vagal stimulation exaggerates the inhibitory ghrelin response to oral fat in humans. J. Endocrinol. 180, 273–281.
Heddle, R., Collins, P. J., Dent, J., Horowitz, M., Read, N. W., Chatterton, B., and Houghton, L. A. (1989). Motor mechanisms associated with slowing of the gastric emptying of a solid meal by an intraduodenal lipid infusion. J. Gastroenterol. Hepatol. 4, 437–447.
Heddle, R., Dent, J., Read, N. W., Houghton, L. A., Toouli, J., Horowitz, M., Maddern, G. J., and Downton, J. (1988a). Antropyloroduodenal motor responses to intraduodenal lipid infusion in healthy volunteers. Am. J. Physiol. Gastrointest. Liver Physiol. 254, G671–G679.
Heddle, R., Dent, J., Toouli, J., and Read, N. W. (1988b). Topography and measurement of pyloric pressure waves and tone in humans. Am. J. Physiol. Gastrointest. Liver Physiol. 255, G490–G497.
Herrmann, C., Goke, R., Richter, G., Fehmann, H. C., Arnold, R., and Goke, B. (1995). Glucagon-like peptide-1 and glucose-dependent insulin-releasing polypeptide plasma levels in response to nutrients. Digestion 56, 117–126.
Hirasawa, A., Tsumaya, K., Awaji, T., Katsuma, S., Adachi, T., Yamada, M., Sugimoto, Y., Miyazaki, S., and Tsujimoto, G. (2005). Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 11, 90–94.
Hunt, J. N., and Knox, M. T. (1968). A relation between the chain length of fatty acids and the slowing of gastric emptying. J. Physiol. (Lond) 194, 327–336.
Lal, S., McLaughlin, J., Barlow, J., D’Amato, M., Giacovelli, G., Varro, A., Dockray, G. J., and Thompson, D. G. (2004). Cholecystokinin pathways modulate sensations induced by gastric distension in man. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G72–G79.
Lan, H., Vassileva, G., Corona, A., Liu, L., Baker, H., Golovko, A., Abbondanzo, S. J., Hu, W., Yang, S., Ning, Y., Del Vecchio, R. A., Poulet, F., Laverty, M., Gustafson, E. L., Hedrick, J. A., and Kowalski, T. J. (2009). GPR119 is required for physiological regulation of glucagon-like peptide-1 secretion but not for metabolic homeostasis. J. Endocrinol. 201, 219–230.
Lane, M. D., Hu, Z., Cha, S. H., Dai, Y., Wolfgang, M., and Sidhaye, A. (2005). Role of malonyl-CoA in the hypothalamic control of food intake and energy expenditure. Biochem. Soc. Trans. 33, 1063–1067.
Lassman, D. J., McKie, S., Gregory, L. J., Lal, S., D’Amato, M., Steele, I., Varro, A., Dockray, G. J., Williams, S. C. R., and Thompson, D. G. (2010). Defining the role of CCK in the lipid induced human brain activation matrix. Gastroenterology 138, 1514–1524.
Lauffer, L. M., Iakoubov, R., and Brubaker, P. L. (2009). GPR119 is essential for oleoylethanolamide-induced glucagon-like peptide-1 secretion from the intestinal enteroendocrine L-cell. Diabetes 58, 1058–1066.
Lissner, L., and Heitmann, B. L. (1995). Dietary fat and obesity: evidence from epidemiology. Eur. J. Clin. Nutr. 49, 79–90.
Lissner, L., Levitsky, D. A., Strupp, B. J., Kalkwarf, H. J., and Roe, D. A. (1987). Dietary fat and the regulation of energy intake in human subjects. Am. J. Clin. Nutr. 46, 886–892.
Little, T. J., Feltrin, K. L., Horowitz, M., Meyer, J. H., Wishart, J., Chapman, I. M., and Feinle-Bisset, C. (2008). A high-fat diet raises fasting plasma CCK but does not affect upper gut motility, PYY, and ghrelin, or energy intake during CCK-8 infusion in lean men. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R45–R51.
Little, T. J., Feltrin, K. L., Horowitz, M., Smout, A. J., Rades, T., Meyer, J. H., Pilichiewicz, A. N., Wishart, J., and Feinle-Bisset, C. (2005). Dose-related effects of lauric acid on antropyloroduodenal motility, gastrointestinal hormone release, appetite, and energy intake in healthy men. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, R1090–R1098.
Little, T. J., Russo, A., Meyer, J. H., Horowitz, M., Smyth, D. R., Bellon, M., Wishart, J. M., Jones, K. L., and Feinle-Bisset, C. (2007). Free fatty acids have more potent effects on gastric emptying, gut hormones, and appetite than triacylglycerides. Gastroenterology 133, 1124–1131.
Lo, C. M., King, A., Samuelson, L. C., Kindel, T. L., Rider, T., Jandacek, R. J., Raybould, H. E., Woods, S. C., and Tso, P. (2010). Cholecystokinin knockout mice are resistant to high-fat diet-induced obesity. Gastroenterology 138, 1997–2005.
Lo, C. M., Ma, L., Zhang, D. M., Lee, R., Qin, A., Liu, M., Woods, S. C., Sakai, R. R., Raybould, H. E., and Tso, P. (2007). Mechanism of the induction of brain c-Fos-positive neurons by lipid absorption. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R268–R273.
Lobo, M. V., Huerta, L., Ruiz-Velasco, N., Teixeiro, E., de la Cueva, P., Celdran, A., Martin-Hidalgo, A., Vega, M. A., and Bragado, R. (2001). Localization of the lipid receptors CD36 and CLA-1/SR-BI in the human gastrointestinal tract: towards the identification of receptors mediating the intestinal absorption of dietary lipids. J. Histochem. Cytochem. 49, 1253–1260.
Lutter, M., and Nestler, E. J. (2009). Homeostatic and hedonic signals interact in the regulation of food intake. J. Nutr. 139, 629–632.
MacIntosh, C. G., Andrews, J. M., Jones, K. L., Wishart, J. M., Morris, H. A., Jansen, J. B., Morley, J. E., Horowitz, M., and Chapman, I. M. (1999). Effects of age on concentrations of plasma cholecystokinin, glucagon-like peptide-1, and peptide YY and their relation to appetite and pyloric motility. Am. J. Clin. Nutr. 69, 999–1006.
Marciani, L., Faulks, R., Wickham, M. S., Bush, D., Pick, B., Wright, J., Cox, E. F., Fillery-Travis, A., Gowland, P. A., and Spiller, R. C. (2009). Effect of intragastric acid stability of fat emulsions on gastric emptying, plasma lipid profile and postprandial satiety. Br. J. Nutr. 101, 919–928.
Marciani, L., Wickham, M., Singh, G., Bush, D., Pick, B., Cox, E., Fillery-Travis, A., Faulks, R., Marsden, C., Gowland, P. A., and Spiller, R. C. (2007). Enhancement of intragastric acid stability of a fat emulsion meal delays gastric emptying and increases cholecystokinin release and gallbladder contraction. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G1607–G1613.
Matsumura, S., Eguchi, A., Mizushige, T., Kitabayashi, N., Tsuzuki, S., Inoue, K., and Fushiki, T. (2009). Colocalization of GPR120 with phospholipase-Cbeta2 and alpha-gustducin in the taste bud cells in mice. Neurosci. Lett. 450, 186–190.
Mattes, R. D. (1996). Oral fat exposure alters postprandial lipid metabolism in humans. Am. J. Clin. Nutr. 63, 911–917.
Mattes, R. D. (2009a). Brief oral stimulation, but especially oral fat exposure, elevates serum triglycerides in humans. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G365–G371.
Matzinger, D., Degen, L., Drewe, J., Meuli, J., Duebendorfer, R., Ruckstuhl, N., D’Amato, M., Rovati, L., and Beglinger, C. (2000). The role of long chain fatty acids in regulating food intake and cholecystokinin release in humans. Gut 46, 688–693.
McLaughlin, J., Grazia-Luca, M., Jones, M. N., D’Amato, M., Dockray, G. J., and Thompson, D. G. (1999). Fatty acid chain length determines cholecystokinin secretion and effect on human gastric motility. Gastroenterology 116, 46–53.
Mela, D. J., and Sacchetti, D. A. (1991). Sensory preferences for fats: relationships with diet and body composition. Am. J. Clin. Nutr. 53, 908–915.
Meyer, J. H., Elashoff, J. D., and Lake, R. (1999). Gastric emptying of indigestible versus digestible oils and solid fats in normal humans. Dig. Dis. Sci. 44, 1076–1082.
Meyer, J. H., Hlinka, M., Kao, D., Lake, R., MacLaughlin, E., Graham, L. S., and Elashoff, J. D. (1996). Gastric emptying of oil from solid and liquid meals. Effect of human pancreatic insufficiency. Dig. Dis. Sci. 41, 1691–1699.
Meyer, J. H., Hlinka, M., Khatibi, A., Raybould, H. E., and Tso, P. (1998a). Role of small intestine in caloric compensations to oil premeals in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 275, R1320–R1333.
Meyer, J. H., Hlinka, M., Tabrizi, Y., DiMaso, N., and Raybould, H. E. (1998b). Chemical specificities and intestinal distributions of nutrient-driven satiety. Am. J. Physiol. Regul. Integr. Comp. Physiol. 275, R1293–R1307.
Miyauchi, S., Hirasawa, A., Ichimura, A., Hara, T., and Tsujimoto, G. (2010). New frontiers in gut nutrient sensor research: free fatty acid sensing in the gastrointestinal tract. J. Pharmacol. Sci. 112, 19–24.
O’Donovan, D., Feinle-Bisset, C., Wishart, J., and Horowitz, M. (2003). Lipase inhibition attenuates the acute effects of oral fat on food intake in healthy subjects. Br. J. Nutr. 90, 849–852.
Overton, H. A., Babbs, A. J., Doel, S. M., Fyfe, M. C., Gardner, L. S., Griffin, G., Jackson, H. C., Procter, M. J., Rasamison, C. M., Tang-Christensen, M., Widdowson, P. S., Williams, G. M., and Reynet, C. (2006). Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab. 3, 167–175.
Park, M. I., Camilleri, M., O’Connor, H., Oenning, L., Burton, D., Stephens, D., and Zinsmeister, A. R. (2007). Effect of different macronutrients in excess on gastric sensory and motor functions and appetite in normal-weight, overweight, and obese humans. Am. J. Clin. Nutr. 85, 411–418.
Pilichiewicz, A., O’Donovan, D., Feinle, C., Lei, Y., Wishart, J. M., Bryant, L., Meyer, J. H., Horowitz, M., and Jones, K. L. (2003). Effect of lipase inhibition on gastric emptying of, and the glycemic and incretin responses to, an oil/aqueous drink in type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 88, 3829–3834.
Randich, A., Tyler, W. J., Cox, J. E., Meller, S. T., Kelm, G. R., and Bharaj, S. S. (2000). Responses of celiac and cervical vagal afferents to infusions of lipids in the jejunum or ileum of the rat. Am. J. Physiol. 278, R34–R43.
Raybould, H. E., Meyer, J. H., Tabrizi, Y., Liddle, R. A., and Tso, P. (1998). Inhibition of gastric emptying in response to intestinal lipid is dependent on chylomicron formation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 274, R1834–R1838.
Read, N., French, S., and Cunningham, K. (1994). The role of the gut in regulating food intake in man. Nutr. Rev. 52, 1–10.
Rolls, E. T. (2010). Taste, olfactory and food texture reward processing in the brain and obesity. Int. J. Obes. (Lond). doi: 10.1038/ijo.2010.163.
Schirra, J., Houck, P., Wank, U., Arnold, R., Goke, B., and Katschinski, M. (2000). Effects of glucagon-like peptide-1(7-36)amide on antro-pyloro-duodenal motility in the interdigestive state and with duodenal lipid perfusion in humans. Gut 46, 622–631.
Schwartz, G. J. (2000). The role of gastrointestinal vagal afferents in the control of food intake: current prospects. Nutrition 16, 866–873.
Schwartz, G. J., Fu, J., Astarita, G., Li, X., Gaetani, S., Campolongo, P., Cuomo, V., and Piomelli, D. (2008). The lipid messenger OEA links dietary fat intake to satiety. Cell Metab. 8, 281–288.
Schwizer, W., Asal, K., Kreiss, C., Mettraux, C., Borovicka, J., Remy, B., Guzelhan, C., Hartmann, D., and Fried, M. (1997). Role of lipase in the regulation of upper gastrointestinal function in humans. Am. J. Physiol. Gastrointest. Liver Physiol. 273, G612–G620.
Seimon, R. V., Lange, K., Little, T. J., Brennan, I. M., Pilichiewicz, A., Feltrin, K. L., Smeets, A. J., Horowitz, M., and Feinle-Bisset, C. (2010). Pooled data analysis identifies pyloric pressures and plasma CCK concentrations as major determinants of acute energy intake in healthy lean males. Am. J. Clin. Nutr. 92, 61–68.
Seimon, R. V., Wooster, T., Otto, B., Golding, M., Day, L., Little, T. J., Horowitz, M., Clifton, P. M., and Feinle-Bisset, C. (2009). The droplet size of intraduodenal fat emulsions influences antropyloroduodenal motility, hormone release, and appetite in healthy males. Am. J. Clin. Nutr. 89, 1729–1736.
Smeets, A. J., Lejeune, M. P., and Westerterp-Plantenga, M. S. (2009). Effects of oral fat perception by modified sham feeding on energy expenditure, hormones and appetite profile in the postprandial state. Br. J. Nutr. 101, 1360–1368.
Smeets, A. J., and Westerterp-Plantenga, M. S. (2006). Satiety and substrate mobilization after oral fat stimulation. Br. J. Nutr. 95, 795–801.
Spannagel, A. W., Nakano, I., Tawil, T., Chey, W. Y., Liddle, R. A., and Green, G. M. (1996). Adaptation to fat markedly increases pancreatic secretory response to intraduodenal fat in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 270, G128–G135.
Speechly, D. P., and Buffenstein, R. (2000). Appetite dysfunction in obese males: evidence for role of hyperinsulinaemia in passive overconsumption with a high fat diet. Eur. J. Clin. Nutr. 54, 225–233.
Stewart, J. E., Feinle-Bisset, C., Golding, M., Delahunty, C., Clifton, P. M., and Keast, R. S. (2010a). Oral sensitivity to fatty acids, food consumption and BMI in human subjects. Br. J. Nutr. 104, 145–152.
Stewart, J. E., Seimon, R. V., Otto, B., Keast, R. S. J., Clifton, P. M., and Feinle-Bisset, C. (2010b). Differences in gustatory and gastrointestinal (GI) sensitivity to oleic acid between lean and obese. Neurogastroenterol. Motil. 22(Suppl. 1), 24.
Tanaka, T., Katsuma, S., Adachi, T., Koshimizu, T. A., Hirasawa, A., and Tsujimoto, G. (2008). Free fatty acids induce cholecystokinin secretion through GPR120. Naunyn Schmiedebergs Arch. Pharmacol. 377, 523–527.
Wisen, O., and Johansson, C. (1992). Gastrointestinal function in obesity: motility, secretion, and absorption following a liquid test meal. Metab. Clin. Exp. 41, 390–395.
Wojdemann, M., Olsen, O., Norregaard, P., Sternby, B., and Rehfeld, J. F. (1997). Gastric lipase secretion after sham feeding and cholinergic blockade. Dig. Dis. Sci. 42, 1070–1075.
Keywords: fatty acids, energy intake, gastrointestinal function, taste, fat sensing, high-fat diet, obesity
Citation: Little TJ and Feinle-Bisset C (2010) Oral and gastrointestinal sensing of dietary fat and appetite regulation in humans: modification by diet and obesity. Front. Neurosci. 4:178. doi: 10.3389/fnins.2010.00178
Received: 03 May 2010;
Paper pending published: 20 May 2010;
Accepted: 23 September 2010;
Published online: 19 October 2010
Edited by:
L. Ashley Blackshaw, University of Adelaide, AustraliaReviewed by:
Tobias Liebregts, University Hospital Essen, Medical School University DuisburgEssen, GermanyAmanda J. Page, Royal Adelaide Hospital, Australia
Copyright: © 2010 Little and Feinle-Bisset. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Tanya J. Little, Discipline of Medicine, Royal Adelaide Hospital, University of Adelaide, Level 6, Eleanor Harrald Building, North Terrace, Adelaide, SA 5000, Australia. e-mail:dGFueWEubGl0dGxlQGFkZWxhaWRlLmVkdS5hdQ==