


- Laboratory of Molecular Physiology, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, MD, USA
Expression of heterologous proteins in adult mammalian neurons is a valuable technique for the study of neuronal function. The post-mitotic nature of mature neurons prevents effective DNA transfection using simple, cationic lipid-based methods. Adequate heterologous protein expression is often only achievable using complex techniques that, in many cases, are associated with substantial toxicity. Here, a simple method for high efficiency transfection of mammalian primary neurons using in vitro transcribed mRNA and the cationic lipid transfection reagent Lipofectamine™ 2000 is described. Optimal transfection conditions were established in adult mouse dissociated dorsal root ganglion (DRG) neurons using a 96-well based luciferase activity assay. Using these conditions, a transfection efficiency of 25% was achieved in DRG neurons transfected with EGFP mRNA. High transfection efficiencies were also obtained in dissociated rat superior cervical ganglion (SCG) neurons and mouse cortical and hippocampal cultures. Endogenous Ca2+ currents in EGFP mRNA-transfected SCG neurons were not significantly different from untransfected neurons, which suggested that this technique is well suited for heterologous expression in patch clamp recording experiments. Functional expression of a cannabinoid receptor (CB1R), a G protein inwardly rectifying K+ channel (GIRK4) and a dominant-negative G protein α-subunit mutant (GoA G203T) indicate that the levels of heterologous protein expression attainable using mRNA transfection are suitable for most functional protein studies. This study demonstrates that mRNA transfection is a straightforward and effective method for heterologous expression in neurons and is likely to have many applications in neuroscience research.
Introduction
Heterologous protein expression in primary neurons is a valuable technique for the study of neuronal function, which allows the roles of specific proteins to be studied in a physiologically relevant environment. Heterologous expression from plasmid cDNA vectors using simple cationic lipid-based transfection methods are effective in dividing cells, but often result in very low transfection efficiencies in post-mitotic cells such as mature neurons. As a result, these methods are unsuitable for many studies and, when used, raise the question of how well the few successfully transfected neurons represent the population of neurons under investigation.
Other methods for heterologous expression in neurons have been developed but most have significant drawbacks. A modified electroporation method, “nucleofection,” can produce high transfection efficiency and minimal toxicity in dissociated hippocampal neuron cultures (Teruel et al., 1999), but, in our lab, it has been difficult to apply this technique to peripheral ganglion neurons without altering the electrophysiological properties of the cells (unpublished observations). Biolistic gene transfer is a simple and rapid technique for heterologous expression in neurons, but is associated with substantial cell death and has a relatively low transfection efficiency (Wellmann et al., 1999). Lentiviral vectors can efficiently transduce hippocampal slice cultures with low toxicity (Ehrengruber et al., 2001). Unfortunately, the relatively long period between transduction of neurons and sufficient transgene expression can be a problem: extended culture may be difficult for some types of neuron and often results in substantial neurite growth that makes reliable voltage-clamp experiments difficult to carry out. In addition, preparation of lentiviral vectors is time-consuming and appropriate laboratory safety considerations are necessary. Intranuclear microinjection of cDNA provides a reliable method for heterologous protein expression, which is suitable for single-cell assays such as electrophysiology and many imaging-based techniques (Lu et al., 2009), but this approach requires considerable equipment, user expertise and is a rather laborious task. The absence of a simple and effective method for heterologous protein expression in neurons restricts many experiments to other models such as cell lines, which may lack many of the characteristics of primary neurons. Therefore, development of such a method would be a valuable tool for neuroscience research.
The post-mitotic nature of neurons is likely to be the reason why cationic lipid-mediated DNA transfection is inefficient in neurons. Previous studies have shown that the nuclear envelope is the major barrier to successful of cationic lipid-mediated transfection (Zabner et al., 1995) and breakdown of nuclear envelope during mitosis is thought to be the primary route for entry of foreign DNA into the nucleus (Mortimer et al., 1999; Brunner et al., 2000). mRNA translation takes place in the cytosol and therefore protein expression from exogenous mRNA does not require entry into the nucleus. For this reason, mRNA transfection has the potential to be an efficient method for heterologous expression in post-mitotic cells such as primary neurons. While cationic lipid-mediated transfection of mRNA was developed some time ago (Malone et al., 1989), its potential for transfection of non-dividing cells has only recently been demonstrated (Van Tendeloo et al., 2007; Zou et al., 2010).
In these studies, we describe a simple method for high efficiency transfection of primary neurons using in vitro transcribed mRNA and the cationic lipid reagent, Lipofectamine™ 2000 (L2K). Using this mRNA transfection protocol, heterologous expression of functional receptors and channels capable of coupling to endogenous signaling pathways is shown. This method appears to be of low toxicity and levels of protein expression are shown to be sufficiently high to conduct a dominant-negative experiment.
Methods
Primary Neuron Cultures
All animals were sacrificed using methods in accordance with the NIH Guidelines for Animal Care and Use. To prepare dissociated DRG neurons, approximately 40 ganglia from 2- to 4-month-old Swiss Webster or C57BL/6 mice were removed from cervical, thoracic and lumbar levels of the spine. Ganglia were combined and dissociated enzymatically with 1 mg/ml collagenase type D and 0.3 mg/ml trypsin (both from Worthington Biochemical Corp, Lakewood, NJ, USA) in Earle’s Balanced Salt Solution (EBSS) saturated with 5% CO2/95% O2 in a shaking incubator at 37°C for 1 h. After this digestion step, neurons were dissociated by vigorous shaking of the flask by hand for 10 s. The cells were then washed and spun at 50 × g twice for 6 min, and resuspended in culture medium consisting of minimal essential medium (MEM; Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA) and 1 unit/ml penicillin, 1 μg/ml streptomycin (Life Technologies). For luciferase assays, DRG neurons from two mice were plated into 48 wells of a poly-l-lysine-coated 96-well plate (Corning, Lowell, MA, USA). For imaging experiments, DRG neurons from one mouse were plated on poly-l-lysine-coated glass-bottomed 35 mm Petri dishes (MatTek, Ashland, MA, USA).
Superior cervical ganglion neurons from male adult Wistar rats (200–250 g) were dissociated enzymatically according to the protocol described previously (Ikeda, 2004) with the following modifications: after the first wash and spin of dissociated neurons, the cells were resuspended in 10 ml of OptiMEM (Life Technologies) and then split into a 4-ml and a 6-ml aliquot. The cells were centrifuged a second time and the 4-ml aliquot (control cells) was resuspended in 400 μl of culture medium and plated in two poly-l-lysine-coated 35 mm Petri dishes. The second aliquot of cells (6 ml) was spun down and resuspended in 2 ml of the transfection mix (described below). The cell suspension was transferred to a 0.15% agar-coated 35 mm dish and incubated for 1 h at 37°C. After the incubation, 3 ml of the culture medium was added to the cells and the suspension was transferred to a 15 ml centrifuge tube. The neurons were spun at 50 × g, and resuspended in 600 μl of culture medium and plated in three poly-l-lysine-coated 35 mm Petri dishes. Neurons were cultured for approximately 16 h at 37°C prior to further experiments.
Hippocampal and cerebral cortical tissues were dissected from P0 to P1 Sprague Dawley rats and enzymatically dissociated using papain, as previously described (Thaler et al., 2009). Cells were plated on poly-l-lysine-coated glass-bottomed 35 mm dishes and incubated at 37°C for 5 days prior to transfection.
Molecular Biology
Standard PCR- and restriction-based cloning techniques were used to construct the cDNA plasmids (from which the mRNA was transcribed) in this study. Full details and plasmid vector maps are provided in the Supplementary Material. Briefly, cDNA was cloned into a custom-made pCI-based plasmid vector (Promega, Madison, WI, USA) containing an optimized T7 promoter and a tandem of human β-globin 3′-UTRs, as previously described (Williams et al., 2009). The following coding sequences were cloned into the vector: Renilla luciferase (hRluc, from phRG-TK) and firefly luciferase (+luc from pGL3-Control; both from Promega); human cannabinoid receptor 1 (CNR1 transcript variant 1; NM_016083); rat G protein-coupled inwardly rectifying K+ channel subtype 4 (GIRK4; Kcnj5; NM_017297) containing the S143T mutation, which is capable of forming homomeric channels (Vivaudou et al., 1997); human heterotrimeric G protein α-subunit GoA (GNAO1 transcript variant 1; NM_02098) containing the G203T “dominant-negative” mutation (Slepak et al., 1993).
mRNA was transcribed from linearized plasmid DNA using mMESSAGE mMACHINE T7 kit. mRNA was recovered from the transcription reaction using lithium chloride precipitation according to the manufacturer’s instructions (Ambion, Austin, TX, USA). mRNA were resuspended in RNase-free H2O, split into 10 μl aliquots and stored at −70°C. The integrity of the mRNA was assessed on a formaldehyde agarose gel. Concentrations were determined using a Biophotometer (Eppendorf, Hamburg, Germany) by measuring the absorbance at 260 nm.
Transfection
The transfection mix consisted of mRNA, Lipofectamine™ 2000 (L2K) and OptiMEM (both from Life Technologies, Carlsbad, CA, USA). For the luciferase assays, a total volume of 65 μl was used per transfection. For all other experiments (carried out in 35 mm dishes), a total volume of 2 ml was used for each transfection. The quantities of L2K and mRNA for each experiment are specified in the section “Results.” The transfection mix was prepared in two parts of equal volume: one part containing the RNA, the second part containing L2K. Each component was gently mixed, incubated at room temperature for 5 min before being combined, mixed again, and incubated at room temperature for a further 30 min. Cells were incubated with the transfection mix for the length of time described in the section “Results.” Following incubation, cells were washed twice with PBS and culture media added and incubated at 37°C, until experiments were performed. Protocols for transfection of adherent neurons and neurons in suspension in a 35 mm dish are provided in the Supplementary Material.
Luciferase Assay
Twenty-four hours after plating, cells were washed twice with PBS before being transfected in triplicate using the transfection conditions described in the section “Results.” Prior to lysis, neurons were washed twice with PBS. 80 μl of M-Per lysis buffer (Thermo Scientific, Rockford, IL, USA) was added to each well and thoroughly mixed using a multichannel pipette. After 15 min incubation at room temperature, the lysate was diluted fivefold in PBS, and the luciferase activity of 10 μl of the diluted lysate was assayed using 100 μl of 5 μM benzyl-coelenterazine (NanoLight Technology, Pinetop, AZ, USA) or Dual Luciferase Reporter Assay System (Promega) in an LB 960 microplate luminometer (Berthold Technologies, Oak Ridge, TN, USA). Luciferase activity was normalized to a control transfection condition or to the transfection condition that leads to maximum activity, as indicated in the section “Results.”
Imaging
Transfection efficiency calculations were carried out using images acquired on a TE2000-U microscope equipped with a Plan Fluor 10× (0.3 NA) objective, GFP filter cube (all Nikon Instruments, Melville, NY, USA), a 12-bit Orca-ER CCD camera (Hamamatsu, Bridgewater, NJ, USA), and an X-Cite series 120Q mercury arc lamp (EXFO, Mississauga, ON, Canada). Transfection efficiency was calculated using a phase contrast cell image and epifluorescence image, acquired using a 2 s exposure, of the same field. The neurons were identified and counted manually from the phase contrast image using the ImageJ program (NIH, http://rsb.info.nih.gov/ij) and the “Cell Counter” plug-in. Transfected cells were identified from the fluorescence image. Approximate neuron-sized regions of the fluorescence image with intensities above the background of untransfected neurons were identified using the particle analysis algorithm of ImageJ. The minimum intensity threshold was calculated from the intensities of fluorescence images measured from untransfected neurons, and a minimum particle area of 21 μm2 was used. A mask of regions of high fluorescence (created by the particle analysis algorithm) was overlaid onto the phase image (see Figure 1Biii), and neurons that were both identified in the initial total neuron count and covered by the mask were designated as transfected. The transfection efficiency was calculated by dividing the number of transfected neurons by the total number of neurons in the phase image. For each dish, the mean transfection efficiency was calculated from images from four fields of view. At least two dishes were imaged per experiment. For clarity, linear adjustment of the contrast of the fluorescence images presented in the figures has been made. The same contrast adjustment was applied to both control and experimental images.
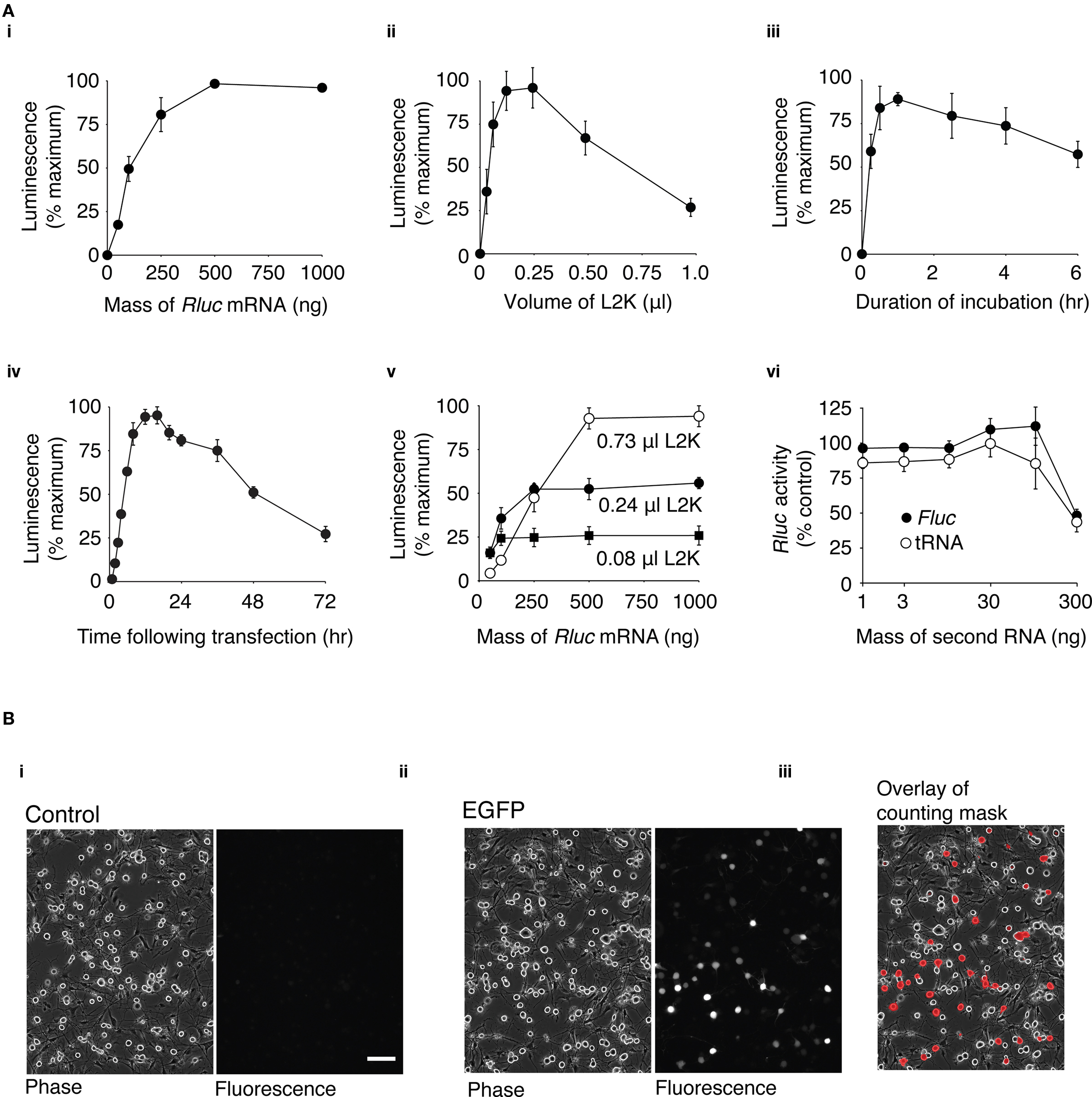
Figure 1. Optimization of mRNA transfection in dissociated DRG neurons. (A) Rluc mRNA transfection of DRG neurons using different transfection conditions was assessed by a luciferase assay of cell lysates. Unless indicated, the transfection mix contained 0.24 μl L2K and 100 ng Rluc mRNA. Neurons were incubated with the transfection mix for 1 h and luciferase activity was measured 16 h after transfection. Except for (vi), data were normalized to the condition within the experiment with the maximum luciferase activity. (i) Transfection mix containing 0–1000 ng of Rluc mRNA (n = 4) (ii) Transfection mix containing 0–1.0 μl of L2K (n = 8). (iii) Neurons incubated with transfection mix for a duration of 0–6 h (n = 4). (iv) Time course of luciferase activity following transfection. Luciferase activity was measured between 0 and 72 h following transfection (n = 4). (v) Luciferase expression using a transfection mix containing 30–1000 ng Rluc mRNA and 0.08 μl (■), 0.24 μl (●), or 0.73 μl (○) L2K (n = 3). (vi) Transfection mix containing 100 ng of Rluc mRNA and 1–300 ng of Fluc mRNA (●) or yeast tRNA (○). Data was normalized to the luciferase activity of cell lysates transfected with 100 ng Rluc mRNA alone (n = 3). The points on the graphs represent the mean luciferase activity ± s.e.m. (B) Transfection efficiency of DRG neurons. (i) Phase and fluorescence image of untransfected DRG neurons. (ii) Phase and fluorescence image of DRG neurons transfected with 3.1 μg GFP mRNA, 16 h after transfection (iii) Phase image with overlay of a mask created from the fluorescence image. The mask indicates regions of the image with fluorescence intensities over a threshold above which neurons were classified as transfected (see Methods). 100 μm scale bar (for all images).
Electrophysiological Recording
Currents were recorded using conventional whole-cell patch clamp techniques. Recordings were obtained using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA). Patch pipettes were fabricated from Corning 8250 borosilicate glass capillaries (King Precision Glass, Claremont, CA, USA) using a P-97 Flaming-Brown micropipette puller (Sutter Instruments, Novato, CA, USA). Pipettes were coated with Sylgard (Dow-Corning, Midland, MI, USA) and fire-polished to a final resistance of approximately 5 MΩ when filled with internal solution. Voltage protocol generation and data acquisition were carried out using custom designed software (S5) running on a Mac G4 computer (Apple, Cupertino, CA, USA). Currents were filtered at 10 kHz using a four-pole low-pass Bessel filter, and digitized at 10 kHz. Uncompensated series resistance was <6 MΩ and was electrically compensated to approximately 90%. For Ca2+ currents, the external solution contained (in mM): 145 tetraethylammonium hydroxide (TEA-OH), 140 methanesulfonate, 10 HEPES, 15 glucose, and 10 CaCl2, and 0.0003 tetrodotoxin (TTX). Internal pipette solution contained (in mM): 120 N-methyl-d-glucamine, 20 TEA-OH, 10 HEPES, 11 EGTA, 1 CaCl2, 14 Tris-creatine phosphate, 4 MgATP, and 0.3 Na2GTP. For GIRK currents, the external solution contained (in mM): 130 NaCl, 5.4 KCl, 10 HEPES, 0.8 MgCl2, 10 CaCl2, 15 glucose, 15 sucrose, and 0.0003 TTX. GIRK internal pipette solution contained (in mM): 135 KCl, 11 EGTA, 1 CaCl2, 2 MgCl2, 10 HEPES, 4 MgATP, and 0.3 Na2GTP. The pH of the solutions were adjusted to 7.4. All recordings were carried out at room temperature (20–22°C). Voltage protocols are described in the figure legends.
Drugs were applied to the cells using a gravity-driven perfusion system. 2-arachidonylglycerol (2-AG) was purchased from Tocris Bioscience, Inc. (Ellisville, MO, USA), and dissolved to a stock concentration of 10 mM in dimethyl sulfoxide. For experiments using 2-AG, fatty acid-free bovine serum albumin (0.5 mg/ml) was added to all external solutions. Norepinephrine (NE) bitartrate salt (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in H2O to a stock concentration of 10 mM. All drugs were diluted to their final concentrations in external solution just prior to the experiment.
Data Analysis and Statistics
Data were analyzed using IgorPro 6.0 (WaveMetrics, Lake Oswego, OR, USA) and Prism 5.0 (GraphPad Software, San Diego, CA, USA). Where possible, the distribution of data was assessed using D’Agostino and Pearson omnibus normality test. Normally distributed data were expressed as the mean ± s.e.m., and statistical comparisons were made using unpaired Student’s t-test. Non-Gaussian data were expressed as the median and the interquartile range (IQR), and statistical comparisons were made using the Mann–Whitney non-parametric test. P values <0.05 were considered significant.
Results
Optimization of mRNA Transfection Conditions for Dissociated DRG Neurons
Cationic lipid-mediated transfection of plasmid DNA results in very poor levels of heterologous expression in adult dissociated DRG neurons (see Supplementary Material and Figure S1) and therefore a simple alternative method of heterologous expression would be highly desirable. As described in the introduction, cationic lipid transfection of mRNA has potential advantages for heterologous expression in non-dividing cells. For this reason a series of experiments were undertaken to establish the optimal conditions for mRNA transfection of dissociated DRG neurons. This preparation provided a sufficient quantity of neurons (with a minimal amount of glial cells) for a “high-throughput” 96-well plate-based assay. Neurons were transfected with Rluc mRNA and luciferase expression was quantified from cell lysates using a luminescence-based luciferase activity assay.
First, experiments were carried out to determine the optimum amount of mRNA for transfection. In these experiments, a constant volume of L2K (0.24 μl) was used and the amount of Rluc mRNA was varied between 50 and 1000 ng per transfection. DRG neurons were incubated in the presence of the transfection mix for 1 h. The maximum expression of luciferase occurred at around 500 ng of Rluc mRNA and reached a plateau (Figure 1Ai). Transfection of 100 ng of Rluc mRNA resulted in 49 ± 7% (n = 4) of the maximum level of luciferase activity and was used in subsequent optimization experiments unless stated otherwise. This quantity of mRNA was chosen because it provided a large dynamic range for assessing luciferase expression. In addition, when the quantity of mRNA is scaled up to that required for transfection in a 35 mm dish, preparation of quantities of mRNA equivalent to >100 ng becomes impractical. To establish the optimum amount of L2K to use per transfection, neurons were transfected with 100 ng of Rluc mRNA and the amount of L2K was varied between 0.03 and 0.98 μl (Figure 1Aii). DRG neurons were incubated in the presence of the transfection mix for 1 h. The maximum amount of luciferase activity occurred using a transfection mix containing between 0.12 and 0.24 μl of L2K, and decreased appreciably with larger volumes of L2K. The optimum duration of incubation with the transfection mix was ascertained by incubating the neurons for 0.25–6 h with a transfection mix containing 0.24 μl L2K and 100 ng of Rluc (Figure 1Aiii). The peak luciferase activity occurred with an incubation duration of approximately 1 h. The time course of luciferase expression was determined by incubating neurons with a transfection mix containing 0.24 μl L2K and 100 ng of Rluc for 1 h, lysing the neurons and measuring luciferase activity at time-points between 0.5 and 72 h after transfection. As shown in Figure 1Aiv, the luciferase activity reached a peak 12–16 h after transfection.
To investigate the potential for a different combination of Rluc mRNA mass and L2K volume to provide a higher level of Rluc expression, neurons were transfected with transfection mix containing 50–1000 ng Rluc mRNA and either 0.08, 0.24, or 0.75 μl L2K. Cells were incubated with the transfection mix for 1 h and Rluc activity was measured 16 h after transfection. As shown in Figure 1Av, the maximum Rluc activity increased as the volume of L2K increased, but in transfections using less than 250 ng of Rluc mRNA, luciferase activity was lower using 0.74 μl than with either 0.24 μl or 0.08 μl of L2K per transfection.
Transfection Efficiency
Once the optimum transfection conditions were determined by the luciferase-based assay, transfection efficiency was calculated using an imaging-based method to quantify the proportion of DRG neurons that express (green fluorescent protein) GFP following transfection with GFP mRNA. DRG neurons were plated in 35 mm dishes and the quantities of the transfection reagents were scaled up to the appropriate volume: 3.1 μg of GFP mRNA, 3.75 μl L2K in a 2 ml volume. Neurons were incubated with the transfection mix for 1 h and the cells were imaged 16 h after transfection. As shown in Figure 1Bi, minimal fluorescence is visible in the untransfected control cells, while a range of GFP expression is clearly visible in the transfected cells (Figure 1Bii). The transfection efficiency was quantified using a mask of the fluorescence image overlaid onto the phase image (Figure 1Biii; see Methods for details). 26 ± 3% (n = 8) of DRG neurons expressed levels of GFP detectable by this method. A high threshold was chosen so that autofluorescence from DRG neurons did not complicate the transfection efficiency measurements. For this reason, neurons expressing very low levels of GFP were not considered “transfected” in our calculations.
Optimization of the mRNA Vector
It would be advantageous to maximize the level of protein expression from mRNA. The untranslated regions (UTRs) of mRNA are important for controlling levels of protein expression by influencing mRNA stability and translation efficiency (Pesole et al., 2001). For this reason, the effect of replacing the UTR of our Rluc mRNA with UTRs that have previously been shown to increase heterologous protein was investigated. Experiments were carried out using DRG neurons in a 96-well luciferase-based assay format, as described above. Neurons were transfected with 30, 100, or 300 ng of mRNA transcribed from templates containing the same Rluc sequence but with variations to the 5′-UTR or 3′-UTR (Figure 2Ai). The luciferase activity was normalized to the activity in lysates transfected with 100 ng of control Rluc mRNA (Figure 2Aii). A plasmid vector, pRNA2-(A)128, was constructed which contained a 128-base polyadenine [poly(a)] tract on the 3′-UTR (see Supplementary Material). Transcription using a pRNA2-(A)128 template yielded a mRNA with a fixed length of poly(a) tail, which did not require the enzymatic polyadenylation step which can lead to variable lengths of the poly(a) tail (Holtkamp et al., 2006; see Figure S2 in Supplementary Material). In cells in transfected with 100 ng Rluc mRNA containing the (A)128 tail, luciferase activity was 144 ± 4% (n = 3) of the control mRNA construct. Removal of the poly(a) tail led to a substantial reduction to 0.5 ± 0.1% (n = 3) of control luciferase activity, which demonstrates the importance of the poly(a) tract for efficient protein expression from mRNA.
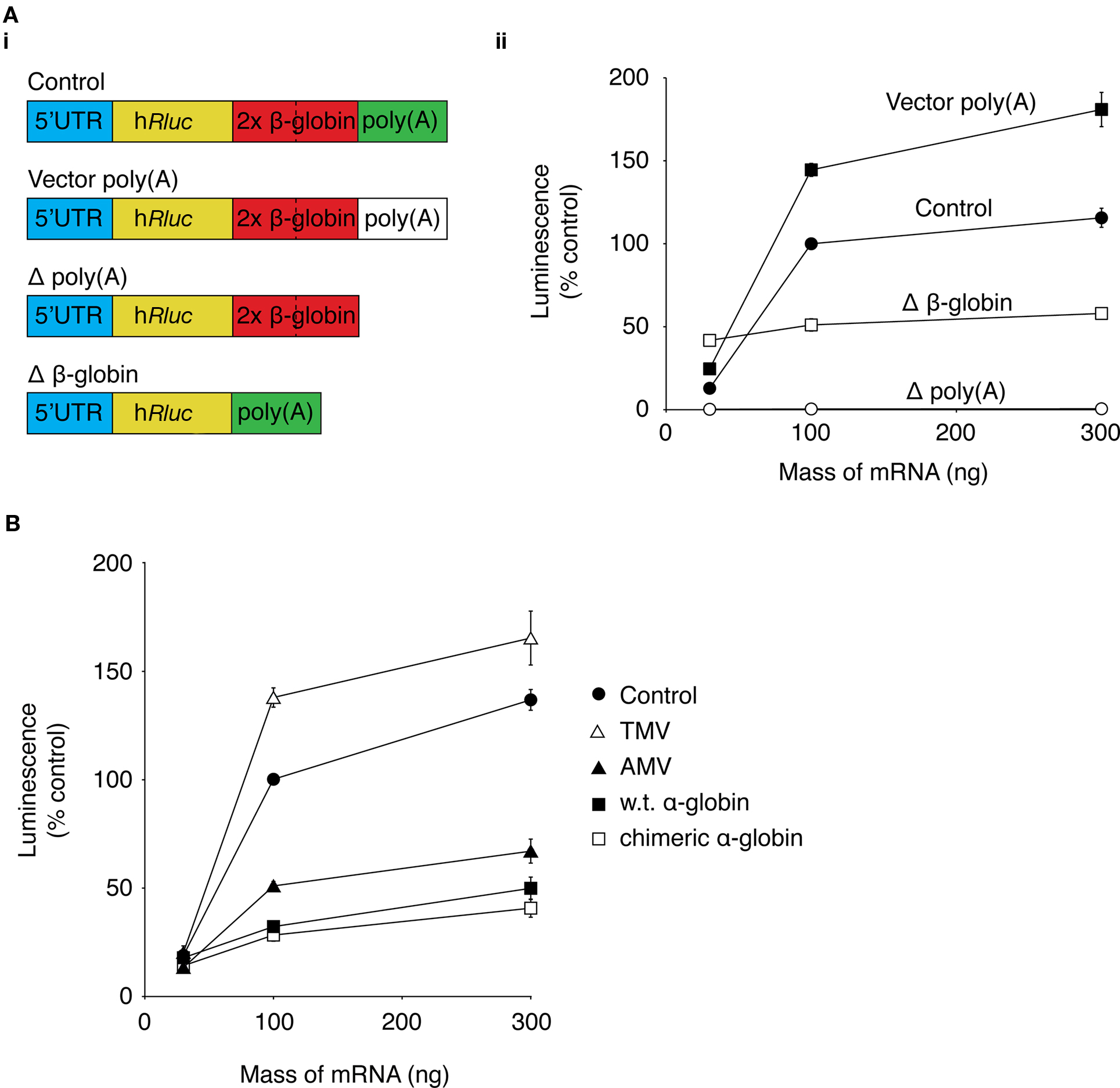
Figure 2. Optimization of mRNA UTRs for maximum protein expression. The effect of changes to the Rluc mRNA UTRs on expression of Rluc was investigated using a luciferase activity assay. (A) (i) a schematic of modifications to the 3′-UTR of Rluc mRNA. (ii) Graph showing luciferase activity of lysates from DRG neurons transfected with 30, 100, and 300 ng of control Rluc mRNA (●), Rluc mRNA containing vector poly(a) sequence (vector poly(A); ■), Rluc mRNA without β-globin sequences (Δ β-globin; □), or Rluc mRNA lacking a poly(a) sequence (Δ poly(a); ○). Data were normalized to luciferase activity of neurons transfected with 100 ng of control Rluc mRNA (n = 3). (B) Graph of luciferase activity of cell lysates from DRG neurons transfected with 30, 100, 300 ng of control Rluc mRNA (●) or with the control 5′-UTR replaced with the 5′-UTR from tobacco mosaic virus (TMV; ∆), alfalfa mosaic virus (AMV; ▲), rabbit wild type α-globin (■), or rabbit chimeric α-globin (□). Data were normalized to the luciferase activity of neurons transfected with 100 ng of control Rluc mRNA (n = 3).
The mRNA vector used in the current study contains a tandem repeat of two human β-globin 3′-UTR sequences, which has been shown to increase protein expression in dendritic cells (Holtkamp et al., 2006). Luciferase activity in neurons transfected with 100 ng of mRNA lacking the β-globin sequences (Δβ-globin; Figure 2Ai) was 44 ± 1% of luciferase activity of neurons transfected with 100 ng of the control construct (n = 3; Figure 3Aii). Interpretation of this modest change in Rluc activity is difficult. As described in the section “Results” in Supplementary Material (Figure S3 in Supplementary Material), transfection of different preparations of Rluc mRNA transcribed from the same template can result in a range of luciferase activities. Therefore, it is difficult to determine whether changes in luciferase activity arise from changes to protein expression as a result of alterations to the mRNA sequence or because of variability between mRNA preparations.
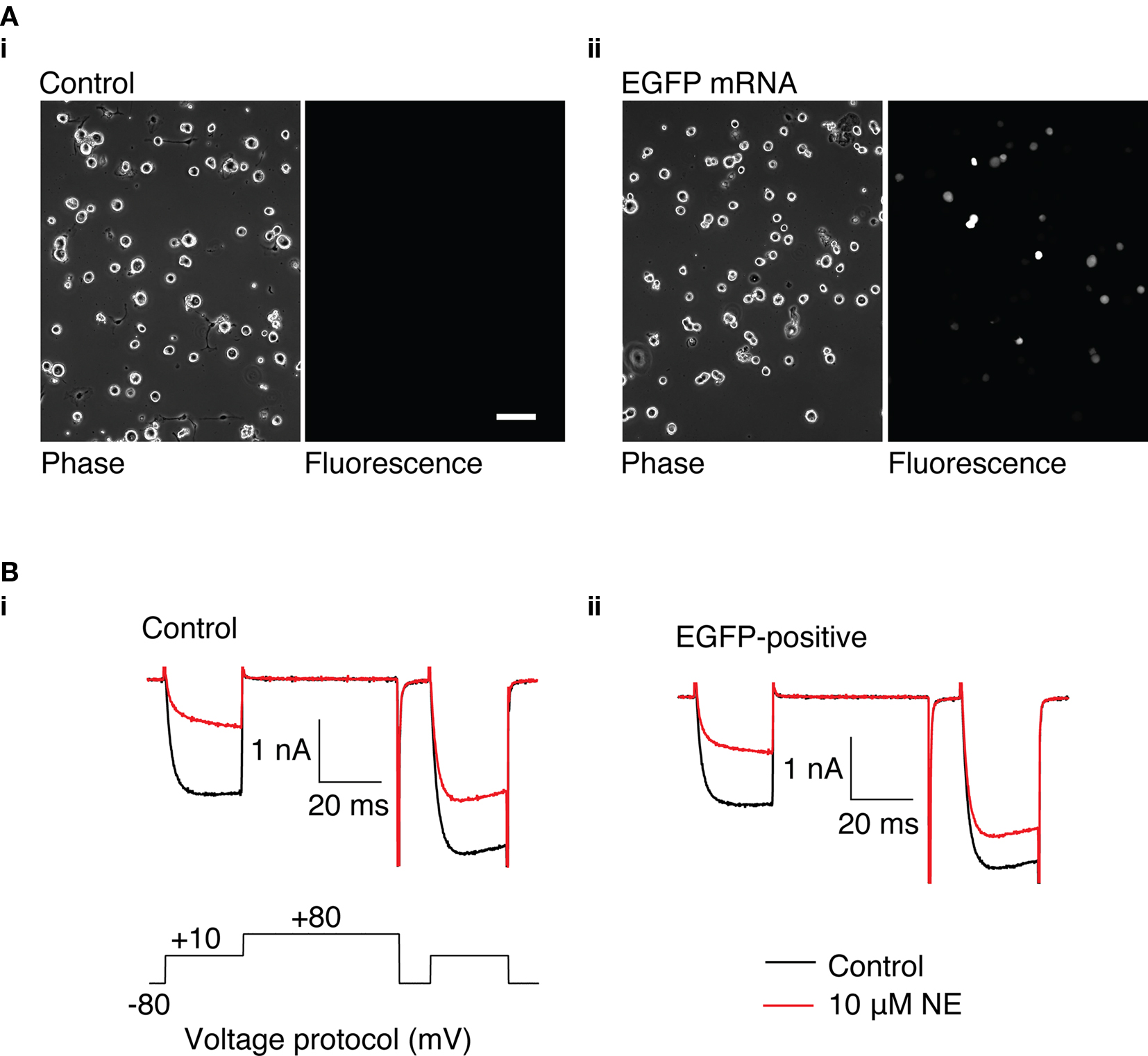
Figure 3. GFP expression in rat SCG neurons using a suspension-based mRNA transfection technique. (A) (i) Phase and fluorescence image of untransfected SCG neurons. (ii) Phase and fluorescence image of SCG neurons transfected with 3.1 μg GFP mRNA, 16 h after transfection. 100 μm scale bar. (B) Representative Ca2+ currents recorded from control SCG neurons (i) and neurons transfected with 3.1 μg GFP mRNA (ii). Currents were evoked using the voltage protocol shown in (i). Control currents are shown in black, and currents in the presence of 10 μM NE are shown in red.
The 5′-UTR sequence of the mRNA is an important determinant of translational efficiency (Pain, 1996). In an attempt to maximize protein expression from the mRNA, the 5′-UTR sequence of our mRNA was replaced with 5′-UTRs that have previously been demonstrated to confer high translational efficiency to the coding mRNA to which they are attached. So far, the 5′-UTR of the mRNA used in this study consists of the multiple cloning region of the cDNA plasmid template. This “control” 5′-UTR was replaced with 5′-UTRs from tobacco mosaic virus (TMV; Gallie et al., 1987a), alfalfa mosaic virus (AMV; Jobling and Gehrke, 1987), wild type rabbit α-globin, or a chimeric α-globin containing a region of β-globin 5′-UTR sequence (Kozak, 1994). Rluc activity of lysates from DRG neurons transfected with 30, 100, or 300 ng of the Rluc mRNA containing the alternative 5′-UTRs was normalized to the luciferase activity of lysates transfected with 100 ng of the control luciferase mRNA (Figure 2B). The Rluc activity of neurons transfected with 100 ng of TMV 5′-UTR mRNA was 138 ± 4% (n = 3) of control Rluc activity. In neurons transfected with 100 ng of the AMV 5′-UTR, the luciferase activity was 51 ± 2% of the control construct. Rluc activity from neurons transfected with 100 ng of wild type or chimeric α-globin 5′-UTR were 32 ± 2% (n = 3) and 28 ± 3% (n = 3) of the control Rluc construct, respectively. These result suggest that replacement of the control 5′-UTR was unable to produce a substantial increase in Rluc expression.
Transfection of Dissociated SCG Neurons
The ability to heterologously express proteins in neurons and carry out electrophysiological recordings is a valuable technique for neuroscience research. In our hands, reliable whole-cell patch clamp recordings of Ca2+ currents in SCG neurons using a method other than intranuclear microinjection for acute heterologous expression is difficult. To determine the suitability of mRNA transfection for patch clamp experiments, SCG neurons were transfected with GFP mRNA and endogenous Ca2+ currents were monitored using whole-cell patch recording. In preliminary experiments, in which the SCG were plated up to 6 h prior to transfection, the majority of neurons detached from the dish when the transfection mix was replaced with culture media. Reliable Ca2+ current recording from the remaining cells was problematic: GΩ seals were difficult to achieve, neurons were “leaky” (i.e., had low input resistance), and Ca2+ currents were smaller than in untransfected neurons (results not shown). A modified method of transfection was developed in which cells were transfected in suspension (see Methods). Using this technique, the neurons adhered well to the dish and GFP was expressed in neurons (Figure 3A) with similar efficiency to the DRG neuron experiments described above. Ca2+ currents were evoked by a double-pulse protocol consisting of two test pulses (“prepulse” and “postpulse”) to +10 mV separated by a large depolarizing conditioning pulse to +80 mV (FElmslie et al., 1990; igure 3B). A comparison of the amplitude of the prepulse Ca2+ current, the current density, and the basal facilitation of Ca2+ currents (the ratio of the postpulse to prepulse current amplitude in the absence of agonist), which has been shown to arise from a small amount of tonic G protein activation (Ikeda, 1991), shows there are no significant differences between GFP-transfected and untransfected neurons (Table 1). Application of 10 μM NE produced a voltage-dependent inhibition of Ca2+ currents that was similar in both GFP-transfected and untransfected neurons (Figure 3B and Table 1). The increase in the facilitation ratio during agonist application is characteristic of the G protein βγ-subunit-mediated inhibition of Ca2+ current that follows NE activation of α2-adrenoceptors (Herlitze et al., 1996; Ikeda, 1996). The similarity between the transfected and untransfected cells indicates that GFP transfection has little effect on endogenous Ca2+ currents and the modulation of Ca2+ currents that follows α2-adrenoceptor activation.
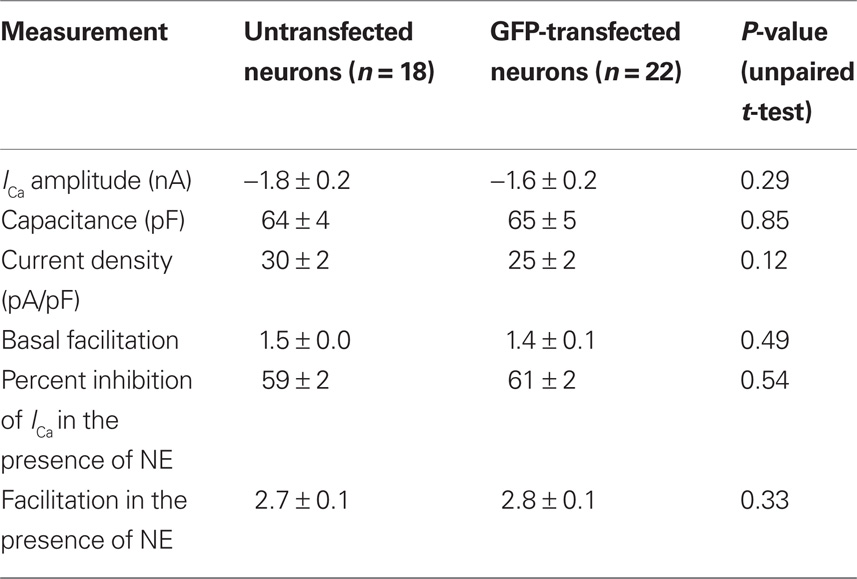
Table 1. The effect of GFP mRNA transfection on Ca2+ currents and NE-mediated Ca2+ current inhibition in SCG neurons. Characteristics of endogenous Ca2+ currents were measured in untransfected SCG neurons and neurons transfected with 3.1 μg of GFP. Facilitation was calculated from the ratio of postpulse Ca2+-current amplitude to the prepulse current amplitude.
Functional Heterologous Receptor, Channel and Dominant-Negative Expression from mRNA
The GFP transfection experiments suggest that mRNA transfection does not cause substantial changes to SCG neuron signaling and health. To determine if functional receptors could be expressed and couple to endogenous signaling pathways, neurons were cotransfected with 3.1 μg of each of the mRNAs encoding the transcript variant 1 of human CB1R and EGFP (as a marker to identify successfully transfected neurons). Ca2+ currents were evoked using the double-pulse protocol described above. In control neurons, in the presence of 10 μM of the CB1R agonist 2-AG, the mean change in amplitude of the prepulse current was 0 ± 1% (n = 17) of the current of in the absence of 2-AG (Figure 4A). In neurons transfected with CB1R and EGFP, the mean amplitude of the prepulse current during application of 2-AG was 51 ± 3% of the control amplitude (n = 18; P < 0.001). The inhibition displayed the hallmarks of Gβγ-mediated inhibition (described above) and is consistent with previous studies using intranuclear microinjection of cDNA to express CB1R in SCG neurons (Guo and Ikeda, 2004).
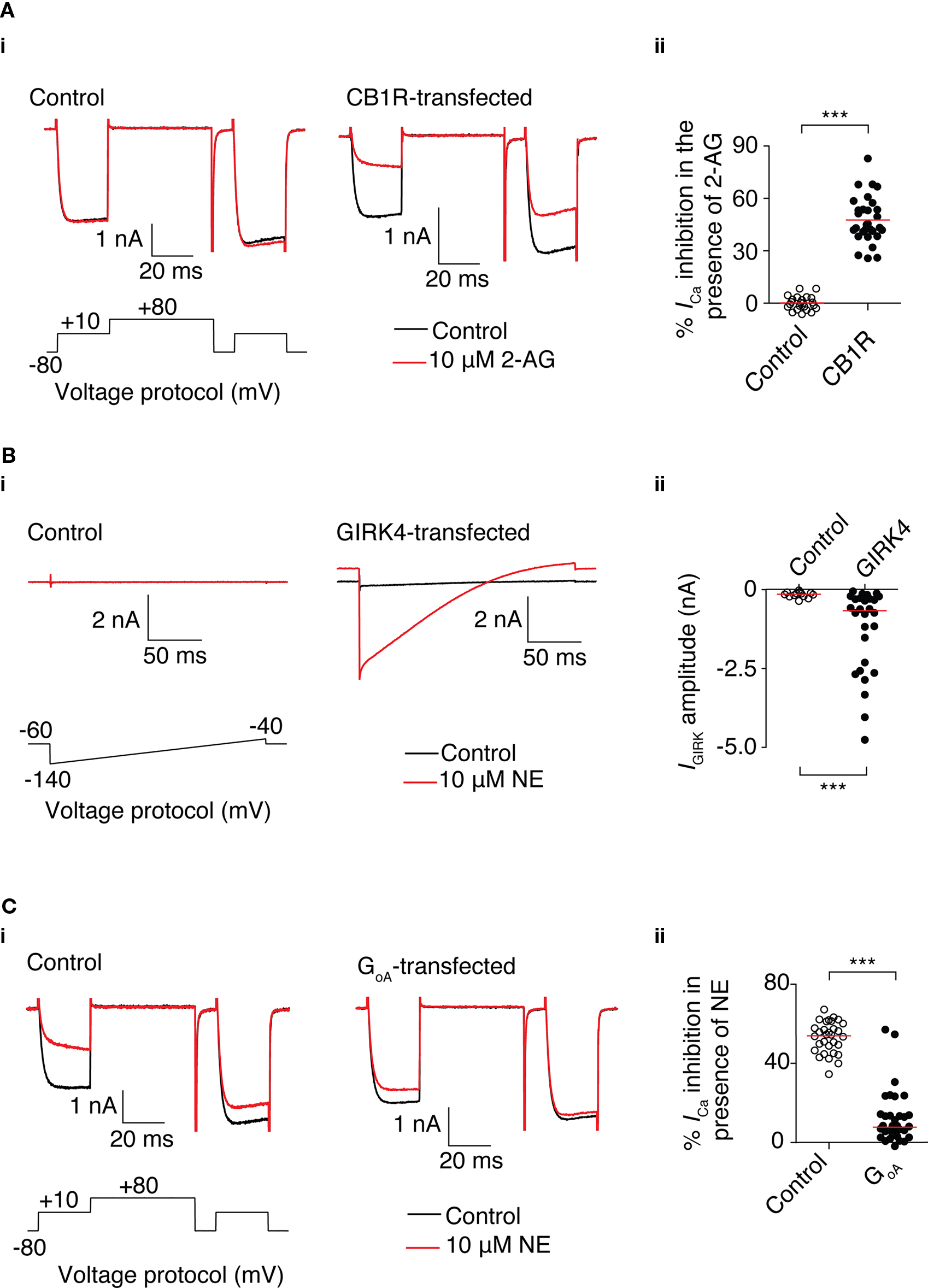
Figure 4. Functional heterologous expression of a receptor, channel, and dominant-negative GoA using mRNA transfection in SCG neurons. (A) 2-AG-mediated Ca2+ current inhibition in SCG neurons transfected with 3.1 μg CB1R mRNA. (i) Superimposed traces of Ca2+ currents in the absence (black) and presence of 10 μM 2-AG (red) in control neurons (left) and neurons transfected with CB1R mRNA (right). Currents were evoked using the voltage protocol shown. (ii) Scatter plot showing the inhibition of Ca2+ current by 2-AG in untransfected neurons (○) or neurons transfected with CB1R mRNA (●). % Inhibition was calculated from the amplitude of the prepulse current before and in the presence of 2-AG. Each data point represents a recording from a different neuron. The red line represents the mean value. ***Represents a P < 0.001, unpaired t-test. (B) NE-mediated GIRK currents in SCG neurons transfected with 3.1 μg GIRK4 S143T mRNA. (i) Superimposed traces of currents in the absence (black) and presence of 10 μM NE (red) in control neurons (left) and neurons transfected with GIRK4 mRNA (right). Currents were evoked using the voltage protocol shown. (ii) Scatter plot showing GIRK current by elicited by NE in untransfected neurons (○) or neurons transfected with GIRK4 mRNA (●). GIRK current was measured 40 ms after the start of the voltage ramp. Each data point represents a measurement from a different neuron. The red line represents the median value. ***Represents a P = 0.001, Mann–Whitney test. (C) Reduction of NE-mediated Ca2+ current inhibition in SCG neurons transfected with 3.1 μg GoA G203T. (i) Superimposed traces of Ca2+ currents in the absence (black) and presence of 10 μM NE (red) in control neurons (left) and neurons transfected with GoA mRNA (right). Currents were evoked using the voltage protocol shown. (ii) Scatter plot showing inhibition of Ca2+ current by 10 μM NE in untransfected neurons (○) or neurons transfected with GoA mRNA (●). % Inhibition was calculated from the amplitudes of the prepulse current before and in the presence of NE. Each data point represents a recording from different neuron. The red line represents the median value. ***Represents a P < 0.001, Mann–Whitney test.
To determine if functional ion channels could be expressed from mRNA in SCG neurons, neurons were cotransfected with 3.1 μg mRNA encoding the GIRK4 S143T mutant subunit and 3.1 μg of GFP mRNA. A GIRK channel was chosen because of the lack of native expression of GIRK subunits in SCG neurons (Ruiz-Velasco and Ikeda, 1998). GIRK currents were evoked by a 200 ms voltage ramp from −140 to −40 mV from a holding potential of −60 mV following activation of endogenous α2-adrenoceptors by 10 μM NE. In GFP-positive cells, NE elicited a clear inwardly rectifying current that was not present in untransfected cells (Figure 4Bi). The amplitude of the current was measured at 40 ms after the start of the ramp protocol (corresponding to a potential of approximately −120 mV) in the presence of 10 μM NE. In the GFP-positive cells, the median amplitude of the current was 670 pA (IQR: −270 to −2500 pA; n = 28) and in untransfected control cells, the median amplitude was 150 pA (IQR −10 to −210 pA; n = 13; P = 0.001; Figure 4Bii). These results demonstrate mRNA transfection is suitable for heterologous expression of channels that can couple to endogenous signaling pathways.
Heterologous expression of a “dominant-negative” protein provides a useful method to investigate the function of a specific protein (Herskowitz, 1987). This method often requires substantial levels of expression for a phenotype to become apparent. To determine whether sufficient expression can be achieved using mRNA transfection for a dominant-negative experiment, SCG neurons were transfected with mRNA encoding a GoA mutant. Intranuclear microinjection of a similar GoA mutant cDNA plasmid has previously been demonstrated to abolish the NE-mediated Ca2+ current inhibition in SCG neurons (Ikeda, 1996). Neurons were cotransfected with 3.1 μg GoA mRNA and 3.1 μg of GFP mRNA. Ca2+ currents were evoked using the double-pulse voltage protocol described earlier. As shown in Figure 4C, NE caused a median reduction of 54% (IQR 47–50%; n = 31) of prepulse Ca2+ current in untransfected neurons, whereas in neurons transfected with GoA mRNA, the median reduction in Ca2+ current was 8% (IQR 3.5–14%; n = 33; P < 0.001). These results suggest that mRNA transfection can provide sufficient GoA expression to act as a Gβγ sink, which reduces NE-mediated Ca2+ current inhibition in SCG neurons.
Transfection of Hippocampal and Cortical Neuronal Cultures
Heterologous expression in cultures of central neurons using cationic lipids and cDNA plasmid vectors is often of low efficiency (Washbourne and McAllister, 2002). In order to assess the suitability of mRNA transfection for heterologous expression in central neurons, cultures of hippocampal and cortical neurons from P0 to P1 rats were transfected with 2 ml transfection mix contain 3.1 μg of GFP mRNA and 3.75 μl of L2K per dish. Cultures were incubated in the presence of the transfection mix for 1 h at 37°C. After removal of the transfection mix, cultures were washed twice with PBS, the culture media was replaced and the cell incubated for approximately 16 h before imaging. As shown in Figure 5, a variety of cell types could be transfected and, judging by the gross morphology of the cells, transfection was not substantially toxic.
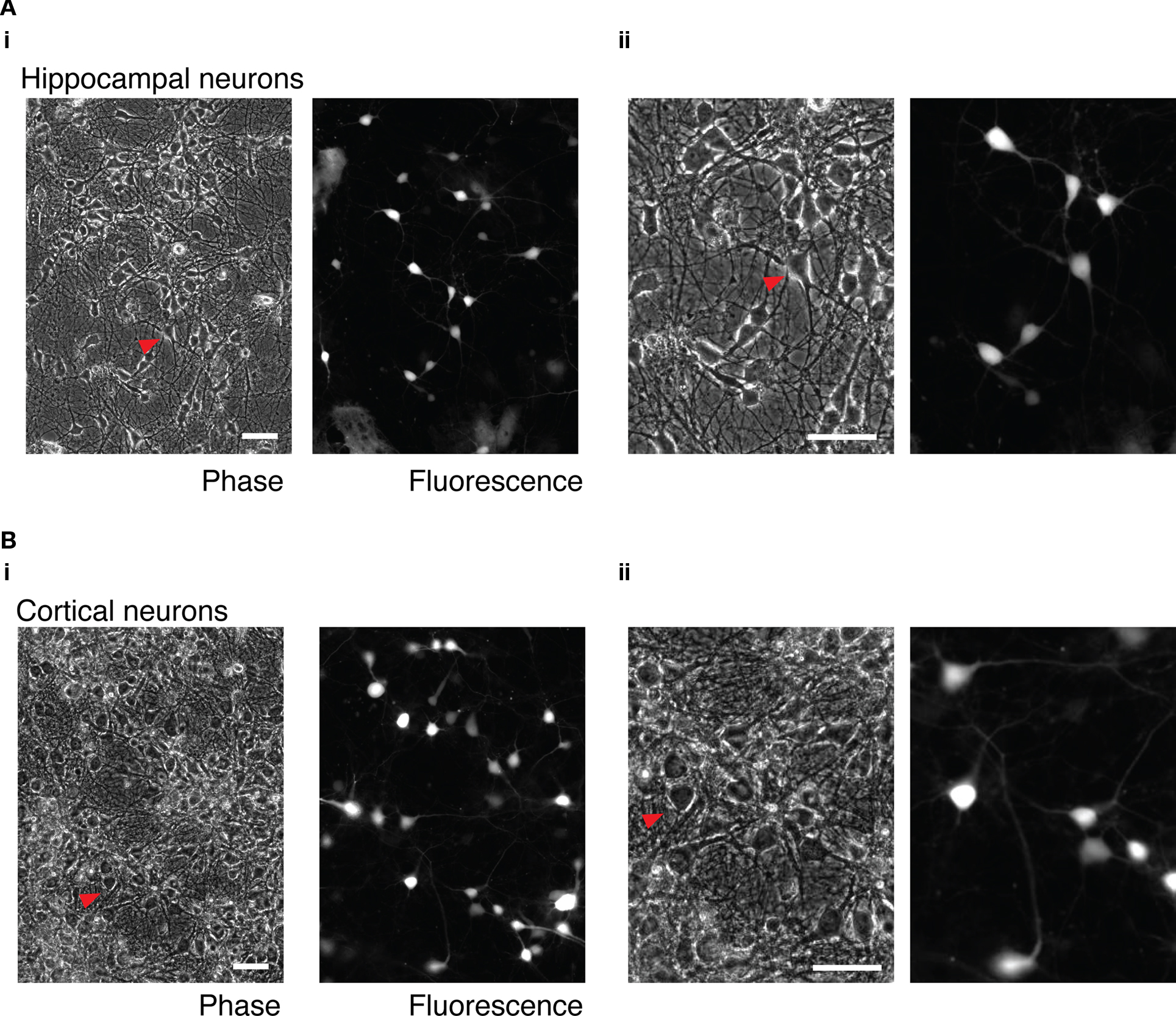
Figure 5. mRNA transfection of hippocampal and cortical neuron cultures. (A) Cultured, dissociated hippocampal neurons transfected with 3.1 μg GFP mRNA. (i) wide-field phase and fluorescence images. (ii) Enlarged detail of the same images. The red arrow indicates the same transfected neuron in both images. (B) Cultured dissociated cortical neurons. (i) Wide-field phase and fluorescence images. (ii) Enlarged detail of the same images. The red arrow indicates the same transfected cell in the wide-field and enlarged images. Phase and fluorescence images were acquired from the same field of view. 50 μm scale bar in all images.
Discussion
This study has shown that mRNA-cationic lipid complexes can be used as an efficient method for heterologous protein expression in a variety of neurons. mRNA transfection is well suited for acute expression of proteins for patch clamp recording experiments – a technique which is particularly difficult using other heterologous protein expression methods because of toxicity or the extended period of time in culture required for sufficient expression. The technique described is easy to perform and requires minimal equipment.
Functional Expression of Proteins at a High Transfection Efficiency
A transfection efficiency of 25% was measured in GFP-transfected DRG neurons. This figure is a conservative measurement because of the high threshold of fluorescence intensity by which successful transfection was judged. A much higher transfection efficiency would likely be obtained using a more sensitive assay such as such as β-galactosidase activity. However, when assessing a method of heterologous expression, it is important to consider the transfection efficiency in relation to the level of heterologous protein expression. A sufficient proportion of the cells should express enough protein to generate the phenotype under investigation so that one can be confident that the cells chosen for further study (electrophysiological recording, imaging, etc) are a good representation of the population of the cells under investigation. While the levels of expression obtained using the mRNA transfection method described here are substantially lower than levels of expression that typically result from intranuclear microinjection of cDNA, there sufficient expression to generate a clear phenotype for all the transgenes chosen in this study (Figure 3). This functional data is a good indication that mRNA transfection is suitable for functional expression of a variety of proteins at a high transfection efficiency.
Potential for Expression of High Levels of Heterologous Proteins
Inhibition of Ca2+ current by overexpression of Gα subunits is assumed to require a relatively high level of expression (Ikeda, 2004), and based on the inhibition of Ca2+ currents in the GoA overexpression experiment (Figure 4C), the levels of expression is likely to be suitable for most studies. If higher levels of expression are required, it may be possible to increase the amount of heterologous protein by increasing the amount of L2K and RNA. As shown in Figure 1Av, the level at which Rluc expression reached a plateau increased as the volume of L2K increased. These results suggest that the plateau in mRNA expression that occurs at higher RNA quantities (see also Figure 1Ai) is likely to be caused by a limiting amount of L2K, rather than saturation of the translation machinery. The hypothesis is supported by the experiments in which addition of increasing amounts of a second coding mRNA (Fluc) to the transfection mix was shown to have little effect on expression of the Rluc mRNA (Figure 1Avi). Reduced expression of Rluc is only apparent at when the transfection mix contained large amounts of Fluc mRNA. A similar reduction in Rluc expression also occurs when large amounts of untranslated RNA (tRNA) is added to the transfection mix, which suggests that there might be competition between the two RNA species for L2K to form L2K-RNA complexes.
There are likely to be drawbacks in simply using more mRNA/L2K as an approach to increasing the expression of a heterologous protein. In addition to the inconvenience and high cost of preparing large amounts of mRNA, increased exposure of neurons to L2K may be unfavorable. In experiments where neurons were exposure to high levels of L2K (Figures 1Aii, Av for quantities of mRNA <250 ng) or a long period of exposure to L2K was used (Figure 1Aiii), there was a decrease in luciferase activity. This reduction is consistent with previous studies showing that some cationic lipids are associated with toxicity (Farhood et al., 1992; Lappalainen et al., 1994; Jacobsen et al., 2009), and suggests that the amount of L2K should be kept as low as possible.
Preliminary experiments to assess the toxicity of mRNA transfection were carried out using a modified “MTT” assay (Denizot and Lang, 1986) (results not shown). Unfortunately, because of the relatively low number of cells used in our experiments, the assay was found to be too insensitive to be of much practical use. It is likely that treatment of cells with cationic lipids will result in a number of changes to the cell (Jacobsen et al., 2009) and appropriate control experiments should be run. However, in our experience, the ability to obtain robust whole-cell patch clamp recording from SCG neurons is very sensitive to cell health and, as shown in Table 1, recordings obtained from transfected neurons were not significantly different to untransfected neurons. These result suggest that mRNA transfection is a relatively gentle method of heterologous expression, and toxic effects are minimal.
Maximizing Expression from mRNA
Because of the disadvantages of increasing the amount of mRNA and L2K, alternative approaches to increase protein expression were attempted. There are a number of steps in transfection that are potentially limiting to the level of protein expression. Previous studies show that once mRNA-cationic lipid complexes enter the cell, only a small proportion of the mRNA is released from the complexes and available to be translated (Bettinger et al., 2001; Barreau et al., 2006). Alternative transfection reagents have the potential to be more effective at delivering mRNA for translation, but preliminary experiments using PEI or Lipofectamine LTX reagents were unsuccessful. For this reason, optimization of the transcript to ensure maximum translation provides an approach with greater potential to increase protein expression. At the level of the transcript, the amount of expression is likely to be determined by the stability and the translation efficiency of mRNA (Ross, 1995; Wilkie et al., 2003). In this study, a number of modifications were made to the UTRs of the mRNA transcript, which have previously been shown to increase the amount of protein expression from exogenous mRNA. In this investigation, many of changes caused by the modifications were fairly small (approximately ±50% of the control protein expression; Figure 2) and could be attributed to variability between mRNA preparations (Figure S3 in Supplementary Material). Only the reduction in expression that followed removal of the poly(a) sequence was of sufficient magnitude to be confident that it resulted from changes to the mRNA transcript.
Optimization of mRNA Stability
mRNAs are highly labile in the presence of ribonucleases that exist in the cytosol. The short half-life of mRNA and the resulting short window in which translation can occur is thought to be the cause of lower levels of protein expression when compared to expression from plasmid DNA (Wolff et al., 1990; Kariko et al., 1998; Bettinger and Read, 2001). The in vitro transcribed RNA transcript used in this study contains a poly(a) tail, a feature that is found in almost all mature eukaryotic mRNA transcripts. It is well established that the poly(a) tail is vital for stable mRNA (Marbaix et al., 1975) and is also required for protein translation (Sachs et al., 1997). The importance of the poly(a) tail was confirmed by the very large reduction luciferase expression from mRNA lacking a poly(a) tail (Figure 2A).
Previous studies have shown that enzymatic polyadenylation is variable and that changes to length of the poly(a) tail results in changes to mRNA stability and/or protein expression (Elango et al., 2005; Holtkamp et al., 2006). It is possible that the variability of enzymatic polyadenylation was the cause of the variability in transcript size (Figure S3ii in Supplementary Material) and protein expression (Figure S3iii in Supplementary Material) that occurred between different mRNAs transcribed from the same template. Transcription from a template containing a poly(a) sequence is likely to give a fixed length of poly(a) tail (Holtkamp et al., 2006). Given that the level of protein expression was adequate from both enzymatically polyadenylated mRNA or mRNA transcribed from pRNA2-(a)128 (Figure 2A), the template poly(a) may provide a method to avoid the variability in protein expression that occurs when enzymatically polyadenylated mRNA are used in transfections.
There are a number of sequences in the UTRs that affect mRNA stability (Ross, 1995). The majority of these sequences are destabilizing which reduce protein expression. Of the few sequences that increase mRNA stability, most are binding sites for proteins that prevent the access of an endonuclease to a specific target sequence (Chen et al., 2000; Tebo et al., 2000; Wang and Kiledjian, 2000) rather than stabilizing the mRNA per se. One of the few sequences to actively promote mRNA stability is the β-globin 3′-UTR. The stabilizing effect is mediated by the binding of a multisubunit complex to a sequence in the 3′-UTR which protects the tail from deadenylation and subsequent breakdown of the mRNA (Yu and Russell, 2001; Jiang et al., 2006). The addition of β-globin UTR to mRNA is a commonly used approach to maximize protein expression from in vitro transcribed mRNA; for example, addition of β-globin UTRs to Rluc mRNA has been shown to increase protein expression by ninefold in NIH/3T3 cells (Malone et al., 1989), and the addition of tandem β-globin 3′-UTRs to GFP mRNA leads to a twofold increase in GFP expression in dendritic cells (Holtkamp et al., 2006). In the current study, the control mRNA in the study contained a tandem repeat of the human β-globin 3′-UTR. Removal of the tandem sequence caused a 50% decrease in expression (Figure 2A). The cause of this reduction is difficult to determine; in addition to a reduction in mRNA stability and variations between mRNA preparations, removal of the 3′-UTR can decrease translation efficiency. Previous studies have showed that reducing the 3′-UTR from 27 to 7 bases (not including the poly(a) tail) causes a decrease in translation efficiency that results in a 35-fold reduction in protein expression from exogenous mRNA in CHO cells. In the current study, the 3′-UTR of the mRNA lacking the β-globin repeat is only 12 bases long, which is likely to have a substantial effect on translation efficiency.
Maximizing Translation Efficiency
Translation efficiency is mostly influenced by sequences in the 5′-UTR (Wilkie et al., 2003). Much like for mRNA stability, the majority of the identified sequences that influence translation efficiency reduce protein expression. These sequences form stable secondary structures or providing binding sites for regulatory proteins, which prevent movement of the translation machinery along the mRNA. Efficient translation appears to be dependent on the 5′-UTR being without significant secondary structure and of sufficient length (over 20 bases), rather than the presence of specific sequences (Kozak, 1991b).
In this study, four different 5′-UTRs that have previously been shown to enhance protein expression were tested: AMV, TMV, wild type α-globin, and a chimeric α-globin that contains a region of β-globin sequence. The unstructured sequences of the AMV and two α-globin 5′-UTRs are thought to enhance translation by a lack of secondary structure mechanism described above (Gehrke et al., 1983; Kozak, 1994). Previously, replacement of the control 5′-UTR sequence with the α-globin 5′-UTR sequences has been shown to increase expression in vitro and in cells approximately twofold (Kozak, 1994; Babendure et al., 2006). Exchanging a control 5′-UTR with the AMV 5′-UTR increases protein expression up to 35-fold in in vitro transcription systems (Jobling and Gehrke, 1987) and between two- and eightfold in cells, depending on the cell type (Gallie et al., 1987b). The TMV sequence represent one of the few 5′-UTR that actively enhances translation. In protoplasts, enhancement is dependent on binding of heat shock protein 101 to the 5′-UTR and involves the recruitment of the translation initiation factor eIF4F to the 5′ cap (Wells et al., 1998; Gallie, 2002). In plant cells the addition of a TMV 5′-UTR sequence has been shown to increase expression up to 30-fold (Gallie et al., 1987a) and increase two- to fourfold in mammalian cells (Gallie and Walbot, 1990; Gallie et al., 1991).
In this study, the different 5′-UTRs did not increase heterologous protein expression in comparison with the control construct. Perhaps, this absence of effect is to be expected: the increase in protein expression is dependent on the control construct, and the 5′-UTR of control construct used in this study may already contain the characteristics which confer optimal protein expression. For example, analysis of the thermodynamic stability of secondary structures in the control 5′-UTR (ΔG = −22 kcal/mol; Zuker, 2003) indicate that the sequence is unlikely to form a highly stable secondary structure. Any potential secondary structure formed is unlikely to be stable or in a position to substantially reduce translation efficiency (Kozak, 1986; Babendure et al., 2006). In addition, the control 5′-UTR is free from destabilizing sequences and sufficiently long (90 bases) to confer high efficiency translation, both of which lead to a high level of protein expression.
The 5′-UTR of TMV did not cause a substantial increase in protein expression. The large difference in enhancement of protein expression between plant cells and other cell types has lead to the hypothesis that different mechanisms of enhancement exist (Gallie et al., 1991). A search of mammalian genes reveals no HSP101 gene or obvious homolog, which suggests that mammalian cells do not possess the machinery required for the substantial enhancement of translation that occurs in plant cells. The increase in expression that has been demonstrated in previous studies may be caused by the relative inefficiency of protein expression from the control construct rather than an “active” enhancement. The control construct described in the aforementioned studies is short (17 bases), which may reduce translation efficiency (Kozak, 1991a). Replacement of this sequence with the TMV 5′-UTR, which is long (41 bases), free from destabilizing sequences and significant secondary structure (ΔG = −5 kcal/mol), is likely to cause an increase in expression. The similarity between the TMV and the control 5′-UTR used in the current study means that they are likely to influence translation to a similar degree, which is supported by the similar levels of protein expression obtained (Figure 2B).
Our results indicate suggest that there is limited potential to increase protein expression from our mRNA transcript by changing the UTR sequences. If high levels of heterologous protein are required, other approaches are necessary; for example, transfection of a self-replicating mRNA transcript has been shown to increase heterologous expression in cell lines (Johanning et al., 1995). Alternatively, delivery of “naked” mRNA has the potential to increase heterologous expression because the mRNA does not have to be released from a mRNA-cationic lipid complex before translation. It is possible that development of the “nucleofection” electroporation technique (Teruel et al., 1999) could provide an effective method for delivery of naked mRNA into neurons. Since mRNA only has to enter to the cytosol, less aggressive electroporation parameters than those used for cDNA plasmids are likely to be required. This more gentle process may be less detrimental to neuron health than cDNA plasmid electroporation.
Applications of Neuronal mRNA Transfection
As shown in this study, mRNA transfection is particularly well suited for single-cell assays such as voltage-clamp recording. In addition, the ability to express a genetically-encoded probe at high efficiencies could be particularly useful for identification of neuronal subtypes in cultures containing a mixed population. This technique also enables experiments that have previously been confined to cell lines to be carried out in neurons; for example; heterologous expression in neurons to be can be applied to “high-throughput”-style experiments and biochemical studies. These examples and the current study demonstrate that this technique has great potential and utility in many areas of neuroscience research.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the intramural program at the National Institutes of Health, National Institute on Alcohol Abuse and Alcoholism, Bethesda, MD, USA.
References
Babendure, J. R., Babendure, J. L., Ding, J. H., and Tsien, R. Y. (2006). Control of mammalian translation by mRNA structure near caps. RNA 12, 851–861.
Barreau, C., Dutertre, S., Paillard, L., and Osborne, H. B. (2006). Liposome-mediated RNA transfection should be used with caution. RNA 12, 1790–1793.
Bettinger, T., Carlisle, R. C., Read, M. L., Ogris, M., and Seymour, L. W. (2001). Peptide-mediated RNA delivery: a novel approach for enhanced transfection of primary and post-mitotic cells. Nucleic Acids Res. 29, 3882–3891.
Bettinger, T., and Read, M. L. (2001). Recent developments in RNA-based strategies for cancer gene therapy. Curr. Opin. Mol. Ther. 3, 116–124.
Brunner, S., Sauer, T., Carotta, S., Cotten, M., Saltik, M., and Wagner, E. (2000). Cell cycle dependence of gene transfer by lipoplex, polyplex and recombinant adenovirus. Gene Ther. 7, 401–407.
Chen, C. Y., Gherzi, R., Andersen, J. S., Gaietta, G., Jurchott, K., Royer, H. D., Mann, M., and Karin, M. (2000). Nucleolin and YB-1 are required for JNK-mediated interleukin-2 mRNA stabilization during T-cell activation. Genes Dev. 14, 1236–1248.
Denizot, F., and Lang, R. (1986). Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 89, 271–277.
Ehrengruber, M. U., Hennou, S., Bueler, H., Naim, H. Y., Deglon, N., and Lundstrom, K. (2001). Gene transfer into neurons from hippocampal slices: comparison of recombinant semliki forest virus, adenovirus, adeno-associated virus, lentivirus, and measles virus. Mol. Cell. Neurosci. 17, 855–871.
Elango, N., Elango, S., Shivshankar, P., and Katz, M. S. (2005). Optimized transfection of mRNA transcribed from a d(A/T)100 tail-containing vector. Biochem. Biophys. Res. Commun. 330, 958–966.
Elmslie, K. S., Zhou, W., and Jones, S. W. (1990). LHRH and GTP-gamma-S modify calcium current activation in bullfrog sympathetic neurons. Neuron 5, 75–80.
Farhood, H., Bottega, R., Epand, R. M., and Huang, L. (1992). Effect of cationic cholesterol derivatives on gene transfer and protein kinase C activity. Biochim. Biophys. Acta 1111, 239–246.
Gallie, D. R. (2002). The 5′-leader of tobacco mosaic virus promotes translation through enhanced recruitment of eIF4F. Nucleic Acids Res. 30, 3401–3411.
Gallie, D. R., Feder, J. N., Schimke, R. T., and Walbot, V. (1991). Post-transcriptional regulation in higher eukaryotes: the role of the reporter gene in controlling expression. Mol. Gen. Genet. 228, 258–264.
Gallie, D. R., Sleat, D. E., Watts, J. W., Turner, P. C., and Wilson, T. M. (1987a). The 5′-leader sequence of tobacco mosaic virus RNA enhances the expression of foreign gene transcripts in vitro and in vivo. Nucleic Acids Res. 15, 3257–3273.
Gallie, D. R., Sleat, D. E., Watts, J. W., Turner, P. C., and Wilson, T. M. (1987b). A comparison of eukaryotic viral 5′-leader sequences as enhancers of mRNA expression in vivo. Nucleic Acids Res. 15, 8693–8711.
Gallie, D. R., and Walbot, V. (1990). RNA pseudoknot domain of tobacco mosaic virus can functionally substitute for a poly(A) tail in plant and animal cells. Genes Dev. 4, 1149–1157.
Gehrke, L., Auron, P. E., Quigley, G. J., Rich, A., and Sonenberg, N. (1983). 5′-Conformation of capped alfalfa mosaic virus ribonucleic acid 4 may reflect its independence of the cap structure or of cap-binding protein for efficient translation. Biochemistry 22, 5157–5164.
Geiser, M., Cebe, R., Drewello, D., and Schmitz, R. (2001). Integration of PCR fragments at any specific site within cloning vectors without the use of restriction enzymes and DNA ligase. BioTechniques 31, 88–92.
Guo, J., and Ikeda, S. R. (2004). Endocannabinoids modulate N-type calcium channels and G-protein-coupled inwardly rectifying potassium channels via CB1 cannabinoid receptors heterologously expressed in mammalian neurons. Mol. Pharmacol. 65, 665–674.
Herlitze, S., Garcia, D. E., Mackie, K., Hille, B., Scheuer, T., and Catterall, W. A. (1996). Modulation of Ca2+ channels by G-protein βγ subunits. Nature 380, 258–262.
Herskowitz, I. (1987). Functional inactivation of genes by dominant negative mutations. Nature 329, 219–222.
Holtkamp, S., Kreiter, S., Selmi, A., Simon, P., Koslowski, M., Huber, C., Tureci, O., and Sahin, U. (2006). Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 108, 4009–4017.
Ikeda, S. R. (1991). Double-pulse calcium channel current facilitation in adult rat sympathetic neurones. J. Physiol. (Lond.) 439, 181–214.
Ikeda, S. R. (1996). Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature 380, 255–258.
Ikeda, S. R. (2004). Expression of G-protein signaling components in adult mammalian neurons by microinjection. Methods Mol. Biol. 259, 167–181.
Jacobsen, L., Calvin, S., and Lobenhofer, E. (2009). Transcriptional effects of transfection: the potential for misinterpretation of gene expression data generated from transiently transfected cells. BioTechniques 47, 617–624.
Jiang, Y., Xu, X. S., and Russell, J. E. (2006). A nucleolin-binding 3′ untranslated region element stabilizes β-globin mRNA in vivo. Mol. Cell. Biol. 26, 2419–2429.
Jobling, S. A., and Gehrke, L. (1987). Enhanced translation of chimaeric messenger RNAs containing a plant viral untranslated leader sequence. Nature 325, 622–625.
Johanning, F. W., Conry, R. M., LoBuglio, A. F., Wright, M., Sumerel, L. A., Pike, M. J., and Curiel, D. T. (1995). A sindbis virus mRNA polynucleotide vector achieves prolonged and high level heterologous gene expression in vivo. Nucleic Acids Res. 23, 1495–1501.
Kariko, K., Kuo, A., Barnathan, E. S., and Langer, D. J. (1998). Phosphate-enhanced transfection of cationic lipid-complexed mRNA and plasmid DNA. Biochim. Biophys. Acta 1369, 320–334.
Kozak, M. (1986). Influences of mRNA secondary structure on initiation by eukaryotic ribosomes. Proc. Natl. Acad. Sci. U.S.A. 83, 2850–2854.
Kozak, M. (1991a). A short leader sequence impairs the fidelity of initiation by eukaryotic ribosomes. Gene Expr. 1, 111–115.
Kozak, M. (1991b). Structural features in eukaryotic mRNAs that modulate the initiation of translation. J. Biol. Chem. 266, 19867–19870.
Kozak, M. (1994). Features in the 5′ non-coding sequences of rabbit a and β-globin mRNAs that affect translational efficiency. J. Mol. Biol. 235, 95–110.
Lappalainen, K., Jaaskelainen, I., Syrjanen, K., Urtti, A., and Syrjanen, S. (1994). Comparison of cell proliferation and toxicity assays using two cationic liposomes. Pharm. Res. 11, 1127–1131.
Lei, Q., Jones, M. B., Talley, E. M., Schrier, A. D., McIntire, W. E., Garrison, J. C., and Bayliss, D. A. (2000). Activation and inhibition of G protein-coupled inwardly rectifying potassium (Kir3) channels by G protein beta gamma subunits. Proc. Natl. Acad. Sci. USA. 97, 9771–9776.
Lu, V. B., Williams, D. J., Won, Y., and Ikeda, S. R. (2009). Intranuclear microinjection of DNA into dissociated adult mammalian neurons. J. Vis. Exp. 34, 1614.
Malone, R. W., Felgner, P. L., and Verma, I. M. (1989). Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. U.S.A. 86, 6077–6081.
Marbaix, G., Huez, G., Burny, A., Cleuter, Y., Hubert, E., Leclercq, M., Chantrenne, H., Soreq, H., Nudel, U., and Littauer, U. Z. (1975). Absence of polyadenylate segment in globin messenger RNA accelerates its degradation in Xenopus oocytes. Proc. Natl. Acad. Sci. U.S.A. 72, 3065–3067.
Mortimer, I., Tam, P., MacLachlan, I., Graham, R. W., Saravolac, E. G., and Joshi, P. B. (1999). Cationic lipid-mediated transfection of cells in culture requires mitotic activity. Gene Ther. 6, 403–411.
Pain, V. M. (1996). Initiation of protein synthesis in eukaryotic cells. Eur. J. Biochem. 236, 747–771.
Pesole, G., Mignone, F., Gissi, C., Grillo, G., Licciulli, F., and Liuni, S. (2001). Structural and functional features of eukaryotic mRNA untranslated regions. Gene 276, 73–81.
Ruiz-Velasco, V., and Ikeda, S. R. (1998). Heterologous expression and coupling of G protein-gated inwardly rectifying K+ channels in adult rat sympathetic neurons. J. Physiol. (Lond.) 513, 761–773.
Sachs, A. B., Sarnow, P., and Hentze, M. W. (1997). Starting at the beginning, middle, and end: translation initiation in eukaryotes. Cell 89, 831–838.
Slepak, V. Z., Wilkie, T. M., and Simon, M. I. (1993). Mutational analysis of G protein alpha subunit G(o) alpha expressed in Escherichia coli. J. Biol. Chem. 268, 1414–1423.
Tebo, J. M., Datta, S., Kishore, R., Kolosov, M., Major, J. A., Ohmori, Y., and Hamilton, T. A. (2000). Interleukin-1-mediated stabilization of mouse KC mRNA depends on sequences in both 5′- and 3′-untranslated regions. J. Biol. Chem. 275, 12987–12993.
Teruel, M. N., Blanpied, T. A., Shen, K., Augustine, G. J., and Meyer, T. (1999). A versatile microporation technique for the transfection of cultured CNS neurons. J. Neurosci. Methods 93, 37–48.
Thaler, C., Koushik, S. V., Puhl, H. L. 3rd, Blank, P. S., and Vogel, S. S. (2009). Structural rearrangement of CaMKIIα catalytic domains encodes activation. Proc. Natl. Acad. Sci. U.S.A. 106, 6369–6374.
Van Tendeloo, V. F., Ponsaerts, P., and Berneman, Z. N. (2007). mRNA-based gene transfer as a tool for gene and cell therapy. Curr. Opin. Mol. Ther. 9, 423–431.
Vivaudou, M., Chan, K. W., Sui, J. L., Jan, L. Y., Reuveny, E., and Logothetis, D. E. (1997). Probing the G-protein regulation of GIRK1 and GIRK4, the two subunits of the KACh channel, using functional homomeric mutants. J. Biol. Chem. 272, 31553–31560.
Wang, Z., and Kiledjian, M. (2000). The poly(A)-binding protein and an mRNA stability protein jointly regulate an endoribonuclease activity. Mol. Cell. Biol. 20, 6334–6341.
Washbourne, P., and McAllister, A. K. (2002). Techniques for gene transfer into neurons. Curr. Opin. Neurobiol. 12, 566–573.
Wellmann, H., Kaltschmidt, B., and Kaltschmidt, C. (1999). Optimized protocol for biolistic transfection of brain slices and dissociated cultured neurons with a hand-held gene gun. J. Neurosci. Methods 92, 55–64.
Wells, D. R., Tanguay, R. L., Le, H., and Gallie, D. R. (1998). HSP101 functions as a specific translational regulatory protein whose activity is regulated by nutrient status. Genes Dev. 12, 3236–3251.
Wilkie, G. S., Dickson, K. S., and Gray, N. K. (2003). Regulation of mRNA translation by 5′- and 3′-UTR-binding factors. Trends Biochem. Sci. 28, 182–188.
Williams, D. J., Puhl, H. L. 3rd, and Ikeda, S. R. (2009). Rapid modification of proteins using a rapamycin-inducible tobacco etch virus protease system. PLoS ONE 4, e7474. doi: 10.1371/journal.pone.0007474.
Wolff, J. A., Malone, R. W., Williams, P., Chong, W., Acsadi, G., Jani, A., and Felgner, P. L. (1990). Direct gene transfer into mouse muscle in vivo. Science 247, 1465–1468.
Yu, J., and Russell, J. E. (2001). Structural and functional analysis of an mRNP complex that mediates the high stability of human beta-globin mRNA. Mol. Cell. Biol. 21, 5879–5888.
Zabner, J., Fasbender, A. J., Moninger, T., Poellinger, K. A., and Welsh, M. J. (1995). Cellular and molecular barrieirs to gene-transfer by a cationic lipid. J. Biol. Chem. 270, 18997–19007.
Zou, S., Scarfo, K., Nantz, M. H., and Hecker, J. G. (2010). Lipid-mediated delivery of RNA is more efficient than delivery of DNA in non-dividing cells. Int. J. Pharm. 389, 232–243.
Keywords: peripheral neurons, voltage-clamp, untranslated regions, tobacco mosaic virus, alfalfa mosaic virus, globin
Citation: Williams DJ, Puhl HL III and Ikeda SR (2010) A simple, highly efficient method for heterologous expression in mammalian primary neurons using cationic lipid-mediated mRNA transfection. Front. Neurosci. 4:181. doi: 10.3389/fnins.2010.00181
Received: 12 April 2010;
Accepted: 06
October 2010;
Published online: 04 November 2010.
Edited by:
Karl Deisseroth, Stanford University, USAReviewed by:
Karl Deisseroth, Stanford University, USAJames S. Trimmer, University of California, USA
Copyright: © 2010 Williams, Puhl III and Ikeda. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Stephen R. Ikeda, Laboratory of Molecular Physiology, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, MD 20892, USA. e-mail:c2lrZWRhQG1haWwubmloLmdvdg==