 Maureen T. Cruz1
Maureen T. Cruz1 Michal Bajo2
Michal Bajo2 M. Elisabetta Maragnoli3 Boris Tabakoff3 George R. Siggins2
M. Elisabetta Maragnoli3 Boris Tabakoff3 George R. Siggins2 Marisa Roberto1*
Marisa Roberto1*
- 1 Committee on the Neurobiology of Addictive Disorders, The Scripps Research Institute, La Jolla, CA, USA
- 2 Molecular and Integrative Neurosciences Department, The Scripps Research Institute, La Jolla, CA, USA
- 3 Department of Pharmacology, School of Medicine, University of Colorado Health Sciences Center, Aurora, CO, USA
The GABAergic system in the central amygdala (CeA) plays a major role in ethanol dependence and in the anxiogenic response to ethanol withdrawal. Previously, we found that both ethanol and corticotropin releasing factor (CRF) increase GABAergic transmission in mouse and rat CeA neurons, in part by enhancing the release of GABA via activation of presynaptic CRF1 receptors. CRF1 receptors are coupled to the enzyme adenylyl cyclase (AC), which produces the second messenger cyclic AMP. There are nine isoforms of AC, but we recently found that CRF1 receptors in the pituitary were coupled to the Type 7 AC (AC7). Therefore, using an in vitro electrophysiological approach in brain slices, here we have investigated a possible role of the AC7 signaling pathway in ethanol and CRF effects on CeA GABAergic synapses of genetically modified mice with diminished brain Adcy7 activity (HET) compared to their littermate male wild-type (WT) mice. We found no significant differences in basal membrane properties, mean baseline amplitude of evoked GABAA receptor-mediated inhibitory postsynaptic potentials (IPSPs), or paired-pulse facilitation (PPF) of GABAA-IPSPs between HET and WT mice. In CeA neurons of WT mice, ethanol superfusion significantly augmented (by 39%) GABAA-IPSPs and decreased PPF (by 25%), suggesting increased presynaptic GABA release. However, these effects were absent in HET mice. CRF superfusion also significantly augmented IPSPs (by 38%) and decreased PPF (by 23%) in WT CeA neurons, and still elicited a significant but smaller (by 13%) increase of IPSP amplitude, but no effect on PPF, in HET mice. These electrophysiological data suggest that AC7 plays an important role in ethanol and CRF modulation of presynaptic GABA release in CeA and thus may underlie ethanol-related behaviors such as anxiety and dependence.
Introduction
Behavioral studies have demonstrated that the gamma-aminobutyric acid (GABA)ergic system in central nucleus of the amygdala (CeA) is linked to the reinforcing properties of ethanol (Koob, 2003, 2006, 2008; Roberto et al., 2010). Thus, microinjection of GABAA receptor antagonists into the rat CeA significantly reduces ethanol self-administration (Hyytia and Koob, 1995; Roberts et al., 1996) in rodents. Further, corticotropin releasing factor (CRF) and its receptors (CRF1 and CRF2) are also linked to alcohol dependence and anxiety-like behaviors in rodents (Heilig and Koob, 2007; Lowery and Thiele, 2010; Roberto et al., 2010). CRF1 may also contribute to alcohol abuse in humans, as evidenced by the association of CRF1 gene variants with specific drinking patterns in humans (Treutlein et al., 2006). However, the molecular mechanisms coupling CRF1, responses to alcohol, and negative emotional state related to ethanol have yet to be determined.
Our previous studies of the CeA demonstrated that acute ethanol application increases IPSC (inhibitory postsynaptic current) amplitudes via increased presynaptic GABA release in rat (Roberto et al., 2003) and mouse (Nie et al., 2004). Like ethanol, CRF also augments IPSCs in rat (Roberto et al., 2010) and mouse (Nie et al., 2004, 2009; Bajo et al., 2008) CeA. The ethanol- and CRF-induced increase in GABAergic transmission is mediated by CRF1 receptor activation, but the intracellular signaling mechanisms involved in this alteration in GABA transmission are still unknown. The CRF1 receptor is a G-protein-coupled receptor (GPCR) that is predominantly linked to the activation of adenylyl cyclase (AC), and also other second messenger systems such as inositol trisphosphate and protein kinase C (PKC; Grammatopoulos et al., 2001; Blank et al., 2003). AC produces the second messenger cyclic AMP, which can bind to the protein kinase A (PKA) regulatory subunit (Hanoune and Defer, 2001). There is evidence for PKA acting at neurotransmitter release machinery to regulate synaptic activity (Trudeau et al., 1996; Chheda et al., 2001; Seino and Shibasaki, 2005). It has been suggested that the type 7 isoform of adenylyl cyclase (AC7) might activate the cAMP-dependent signaling pathway downstream of CRF1 (Mons et al., 1998a). Antoni et al. (2003) demonstrated a coupling of CRF1 to AC9 and AC7. A switch in coupling from AC9 to AC7 results in a more robust cAMP signal when CRF binds to CRF1 (Antoni, 2000; Antoni et al., 2003). It should be emphasized that AC7 is localized both postsynaptically (striatum, hippocampus) and presynaptically (nucleus accumbens, amygdala; Mons et al., 1998a,b) and is anatomically positioned to receive signals from GPCRs on both dendrites and axon terminals. AC7 is also a target of ethanol in the brain. The activity of this enzyme is enhanced by ethanol, such that moderately intoxicating concentrations of ethanol significantly potentiate AC7-mediated activation of the cAMP signaling cascade (Mons et al., 1998b). The physiological consequences of this AC activation have been linked to the release of ACTH from the pituitary (Antoni et al., 2003; Pronko et al., 2010) and predisposition to depression in women (Hines et al., 2006); furthermore, recent studies indicate that acute ethanol exposure can increase neurotransmitter release via activation of AC (Kelm et al., 2008), providing another clue into the possible role of AC7 in brain.
Based on prior results (Pronko et al., 2010; Roberto et al., 2010), we investigated a physiological role for the AC7 signaling pathway in the ethanol and CRF effects on CeA GABAergic transmission by comparing HET (one copy of the ADCY7 gene disrupted) and wild-type (WT) mice. We previously noted that disruption of both copies of ADCY7 in mice results in fetal death on day 11 of gestation (Hines et al., 2006). Our current studies showed no significant differences in the mean baseline amplitude of evoked GABAA receptor-mediated inhibitory postsynaptic potentials (GABAA-IPSPs) in CeA of HET compared to WT mice. However, in CeA neurons of HET mice, the previously characterized ethanol-induced increase of GABAA-IPSPs was abolished. In addition, in CeA neurons from HET mice, the CRF-induced augmentation of IPSPs was also significantly decreased compared to those from WT mice. Thus, our data identify the involvement of the AC7 signaling pathway in both ethanol and CRF sensitivity of GABAergic synapses in CeA.
Materials and Methods
Electrophysiological Studies
Mouse breeding and care
We studied 14 male adenylyl cyclase type 7 heterozygous mice in which once copy of the ADCY7 gene was disrupted (HET), and their respective WT counterparts (WT; n = 14) generated by Hines et al. (2006). We conducted all mouse breeding and care procedures in accordance with the Institutional Animal Care and Use Committee (IACUC) of the University of Colorado at Denver and Health Science Center and The Scripps Research Institute IACUC policies.
Slice preparation
All electrophysiological experiments were also performed in accordance with The Scripps Research Institute IACUC and NIH guidelines on the care and use of laboratory animals. We prepared CeA slices as previously described (Roberto et al., 2003, 2004; Bajo et al., 2008), from male HET mice and their respective WT counterparts (4–5 months old at the time of experimentation) that were anesthetized with halothane (3%) and decapitated. We cut 400 μm thick transverse slices on a Vibratome Series 3000 (Technical Products International, St. Louis, MO, USA), incubated them in an interface configuration for about 30 min, and then completely submerged and continuously superfused (flow rate of 2–4 ml/min) them with warm (31°C), gassed ACSF of the following composition in mM: NaCl, 130; KCl, 3.5; NaH2PO4, 1.25; MgSO4·7H2O, 1.5; CaCl2, 2.0; NaHCO3, 24; glucose, 10. Drugs were added to the ACSF from stock solutions to obtain known concentrations in the superfusate.
Electrophysiology
We recorded from neurons principally in the medial subdivision of the CeA with sharp micropipettes filled with 3 M KCl (impedance 60–90 MΩ) using current-clamp mode. We held most neurons near their resting membrane potential (RMP). We evoked pharmacologically isolated GABAA-IPSPs by stimulating locally within the CeA through a bipolar stimulating electrode while superfusing the slices with the glutamate receptor blockers 6,7-dinitroquinoxaline-2,3-dione (DNQX; 20 μM) and DL-2-amino-5-phosphonovalerate (DL-APV; 30 μM), and the GABAB receptor antagonist (CGP 55845A; 1 μM). We often superfused 30 μM bicuculline or 50 μM picrotoxin at the end of the experiment to confirm the GABAA nature of the IPSPs. Data were acquired with an Axoclamp-2A preamplifier (Axon Instruments, Foster City, CA, USA) and stored for later analysis using pClamp software (Axon Instruments, Foster City, CA, USA). To determine the experimental IPSP response parameters for each cell, we performed an input–output protocol consisting of a range of five current stimulations (typically between 50 and 250 mA; 0.125 Hz), starting at the threshold current required to elicit an IPSP up to the strength required to elicit the maximum amplitude. The range of stimulation intensities was very comparable between the two groups. The I/O method helps minimize inter-slice variability and, when averaged over multiple cells or slices, reproducibly reveals significant changes in synaptic excitability. The half-maximal (half-max) stimulus strength was maintained throughout the entire duration of the experiment. We normalized the three middle (omitting threshold and maximal) stimulus intensities of five equal steps as 1–3X. Stability of IPSPs was established by stimulating for at least 15 min prior to beginning experiments. We quantified the synaptic responses by averaging two consecutive responses evoked by a half-max intensity (30 s apart, i.e., 1 data point/min) and calculating the IPSP amplitude with Clampfit software (Axon Instruments). We also applied hyperpolarizing and depolarizing current steps (200 pA increments, 750 ms duration) to generate voltage–current (V–I) curves.
To estimate changes in presynaptic effects in each neuron, we examined paired-pulse facilitation (PPF), a phenomenon whereby a secondary evoked synaptic response is increased by a preceding primary stimulation of equal intensity. In our studies, we used 50 ms inter-stimulus intervals. We calculated the paired-pulse ratio as the second IPSP amplitude over that of the first IPSP. All measures were taken before ethanol or CRF superfusion (control), during their superfusion (5–10 min), and following washout (20–30 min). To avoid tachyphylaxis, we superfused ethanol and CRF only once onto a single cell. We express all values as mean ± SEM, analyzed them with a between-subjects ANOVA or within-subject ANOVA with repeated measures and, when appropriate, with the Bonferroni post hoc test, with P < 0.05 considered statistically significant. We also used a Student’s paired or unpaired t-test for individual means comparisons.
Drugs
CGP 55845A was a gift from Novartis Pharma. We purchased DL-AP5, DNQX, picrotoxin, and bicuculline from Sigma (St. Louis, MO, USA), CRF from Chempacific Corp. (Baltimore, MD, USA) and ethanol from Remet (La Mirada, CA, USA).
Results
Basal GABAergic Transmission in CeA Neurons is Comparable between HET and WT Mice
We recorded from 78 CeA neurons from male HET and their respective (WT) littermate mice. The mean RMP was −77 ± 1.5 mV and mean input resistance was 106 ± 4 MΩ. The two groups did not show significant differences in these properties. In addition, there were no significant differences in the voltage–current relationship between HET and WT mice (not shown). We evoked pharmacologically isolated GABAA-IPSPs by stimulating locally within the CeA. Interestingly, baseline IPSP input–output curves generated by equivalent stimulus intensities were similar in CeA neurons from HET mice and in those from WT littermates (Figure 1A). We examined PPF of the IPSPs at 50 ms inter-stimulus intervals and did not find any difference in the basal PPF ratio of IPSPs between the two groups (Figure 1B).
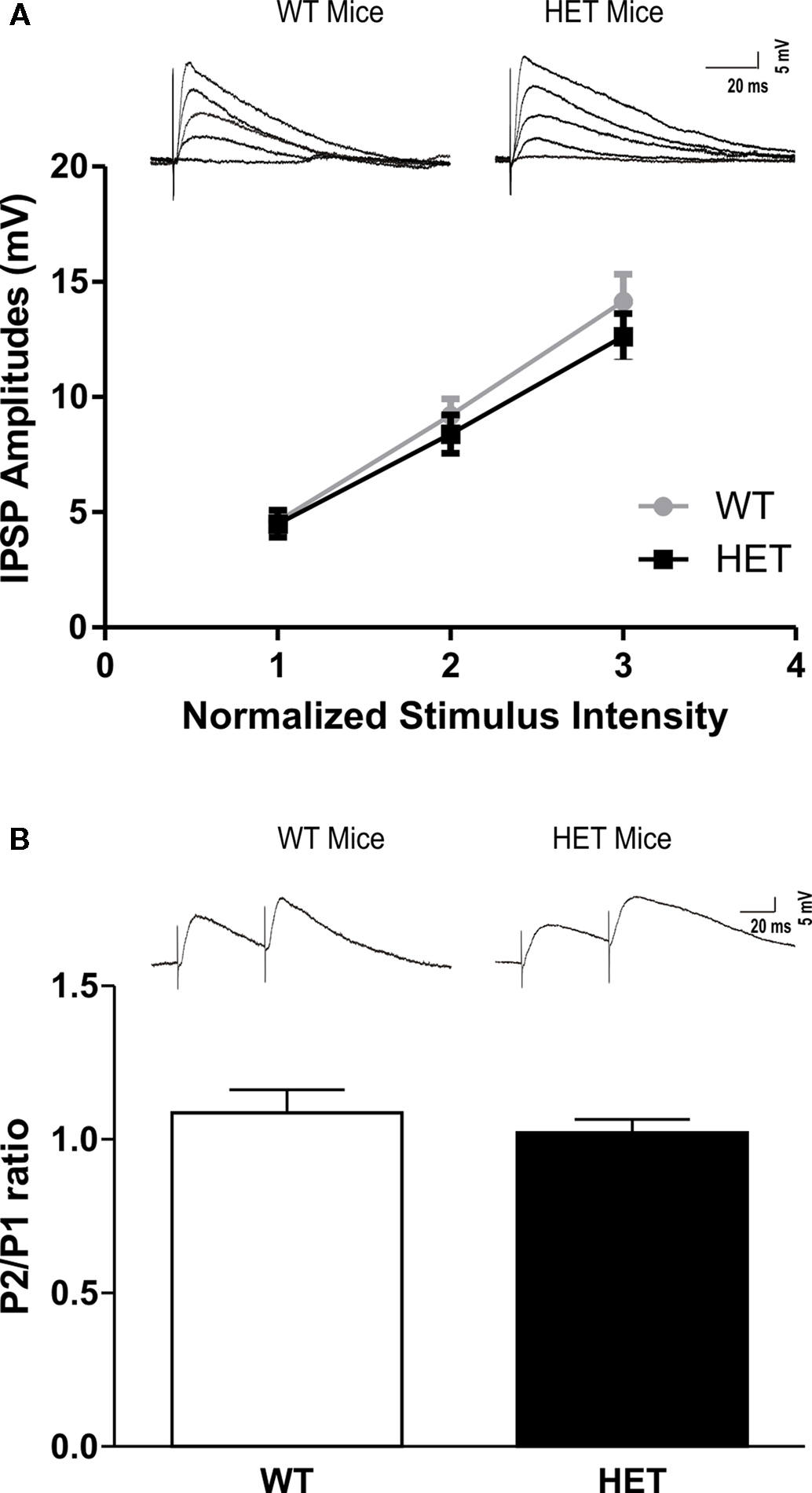
Figure 1. There is no significant difference in basal GABAergic transmission in the CeA between WT and HET mice. (A) Basal input/output curves of mean GABAA-IPSP amplitudes in the CeA were not altered in HET compared to WT mice (WT: n = 21 and HET: n = 22). (B) Paired-pulse facilitation protocol showed no differences in P2/P1 ratio between WT and HET mice (WT: n = 18 and HET: n = 22). The top panels in (A) and (B) show representative recordings from individual CeA neurons. The values are presented as mean IPSP amplitudes ± SEM. Significance was calculated by Student’s t-test and set at *P < 0.05.
Ethanol Enhancement of GABAergic Transmission is Absent in CeA Neurons of HET Mice
Previously we found that ethanol dose-dependently increased GABAergic transmission in CeA slices by increasing presynaptic vesicular GABA release (Roberto et al., 2003). In the present study, we investigated whether AC7 plays a role in ethanol-stimulated GABA release. In CeA slices from WT mice, 10 min superfusion of 44 mM ethanol significantly (P < 0.01) increased the mean amplitude of evoked IPSPs by 39 ± 3% at each stimulus strength tested (Figure 2A; n = 13). To test whether the site of ethanol action was pre or postsynaptic, we assessed PPF, a measure inversely related to neurotransmitter release (Andreasen and Hablitz, 1994; Bonci and Williams, 1997; Roberto et al., 2003). Ethanol significantly (P < 0.01) decreased the PPF of evoked IPSPs by 22 ± 5% in 10 of 11 the CeA neurons from WT mice (Figure 2C), suggesting that ethanol increases GABA release. In HET mice with diminished brain AC7 activity, the ethanol-induced augmentation of evoked IPSP amplitudes was completely absent (103 ± 6% of control, n = 15, P > 0.05; Figure 2B), as was the ethanol-induced reduction in PPF of IPSPs (100 ± 5% of control, n = 10, P > 0.05; Figure 2D). Ethanol had no significant effects on the membrane properties (V–I curves, RMPs, and input resistance) in either HET or WT mice (data not shown). These data suggest that AC7 plays a critical role in the effects of ethanol on GABAergic transmission in the CeA.
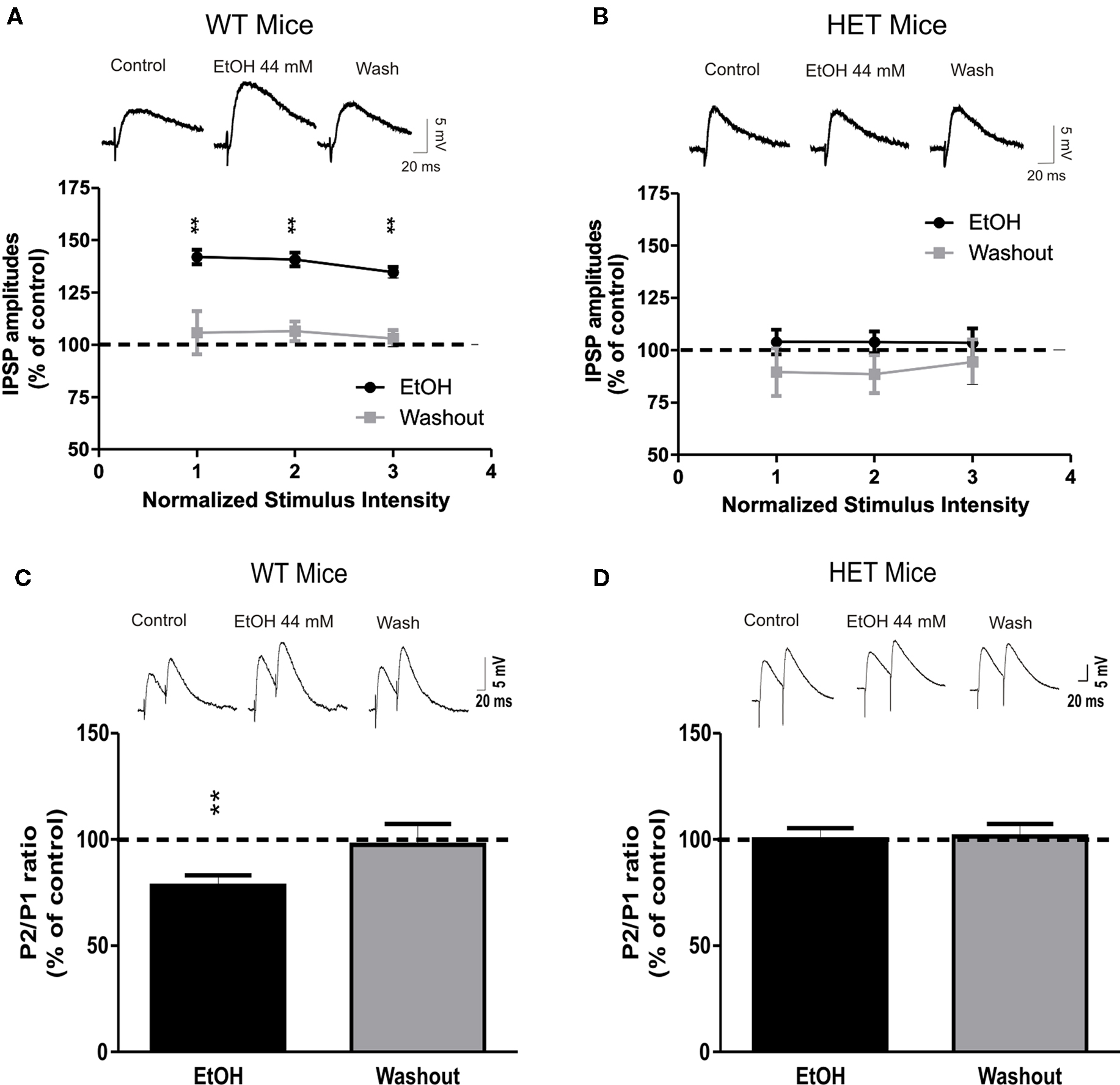
Figure 2. The ethanol effects on GABAergic transmission in the CeA are abolished in HET mice. (A) Superfusion of 44 mM ethanol increased IPSP amplitudes, with recovery on washout in CeA neurons of WT naïve (n = 13) but not of AC7 HET mice [n = 15; (B)]. Each point represents the mean ± SEM. Percent increase in IPSP amplitude. Top panel: representative recordings showing effect of ethanol on GABAA-IPSPs in the CeA neurons of WT and HET mice, respectively. (C) Pooled data of PPF against control (at 50 ms inter-stimulus interval). Ethanol significantly decreased PPF in CeA slices from WT (n = 10) but had no effect in HET (n = 10) mice (D). PPF is inversely related to transmitter release, suggesting that acute ethanol increases release of GABA in WT. Top panels of (C) and (D): representative recordings showing effect of ethanol on PPF in CeA neurons of WT and HET mice, respectively. Statistical significance of ethanol and CRF effects calculated by Student’s t-test and set at *P < 0.05 and **P < 0.01.
CRF Enhancement of Gabaergic Transmission is Reduced in CeA Neurons of HET Mice
We previously reported that CRF, via activation of CRF1s, presynaptically augments GABAergic transmission in CeA slices from C57BL6/J mice (Nie et al., 2004, 2009; Bajo et al., 2008). Recent data suggest that CRF1 activation increases the enzymatic activity of AC7 (Antoni et al., 2003; Pronko et al., 2010). Therefore, we explored the possible role of AC7 in CRF-dependent stimulation of GABA release in the CeA. In accord with our previous studies (Nie et al., 2004, 2009; Bajo et al., 2008), superfusion of 200 nM CRF for 10 min significantly (P < 0.01) increased the mean amplitude of evoked IPSPs of WT CeA by 38 ± 3% (Figure 3A; n = 9). The CRF-induced increase in the amplitudes of WT IPSPs was associated with a significant (P < 0.05) decrease in PPF by 20 ± 6% in eight of nine cells (Figure 3C), suggesting that CRF acts presynaptically to increase GABA release in CeA from WT mice. Although CRF significantly (P < 0.05) increased IPSP amplitude by 13 ± 5%, in HET neurons, this CRF effect represented a significant reduction (by 65%, P < 0.01) compared to the CRF effects in WT mice (Figure 3B; n = 13). Contrary to the effects seen in WT mice, CRF did not significantly (P > 0.05) alter the PPF ratio of IPSPs in HET mice (95 ± 6% of control, n = 11; Figure 3D). We did not observe any significant changes in the membrane properties of the CeA neurons induced by CRF in HET or WT mice (data not shown). Thus, our data suggest that both ethanol and CRF act mainly via presynaptic mechanisms. Because we are measuring only evoked (macroscopic) IPSPs of relatively short duration, and not tonic GABA release, changes in conductance and potential could be detected only during the IPSP itself, during either the baseline or treatment conditions.
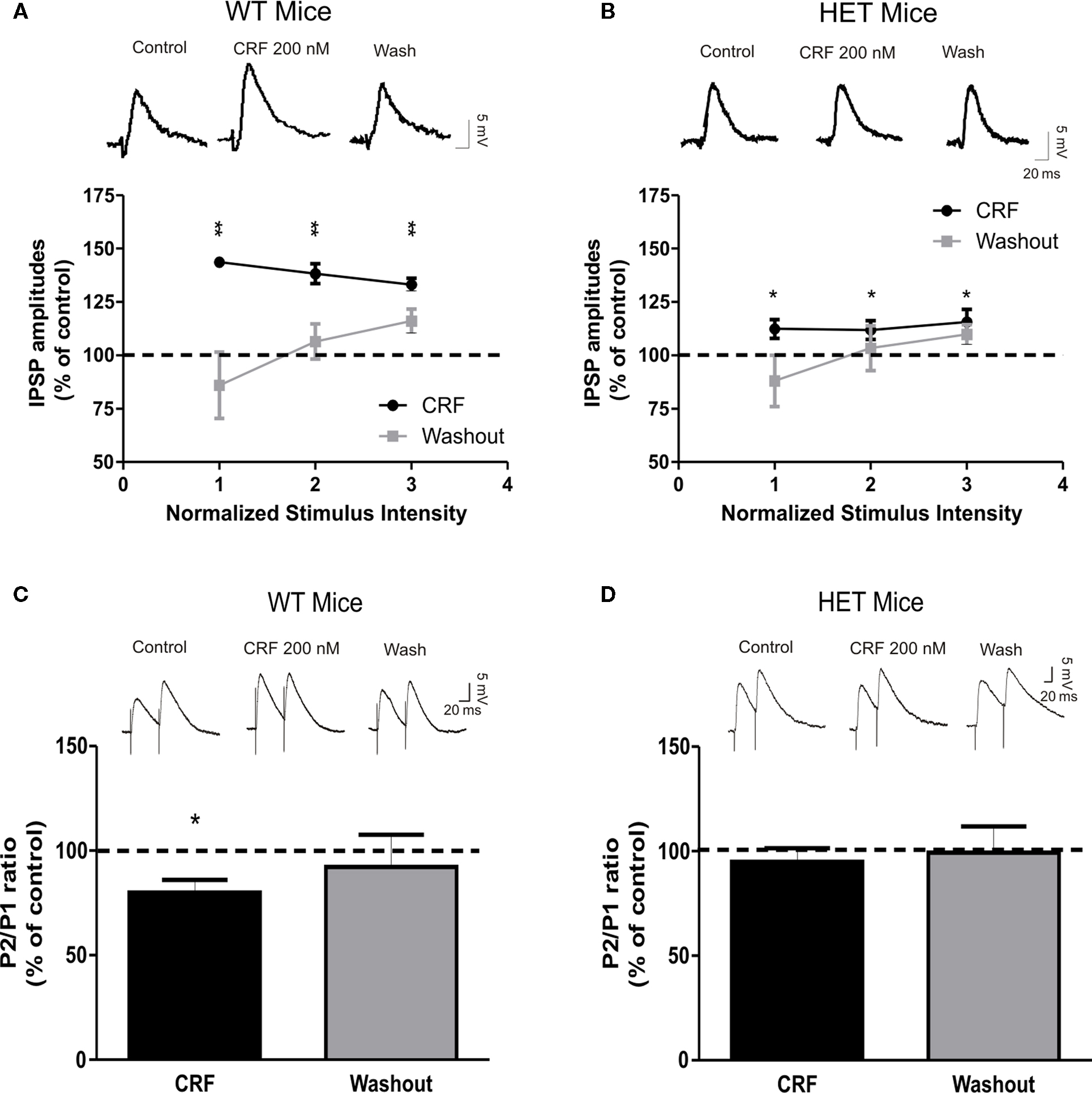
Figure 3. The presynaptic effect of CRF on GABAergic transmission in the CeA is reduced in HET mice. (A) Superfusion of 200 nM CRF significantly increased IPSP amplitudes (38 ± 3%) in CeA neurons of WT naïve (n = 9). Although still significant, the CRF-dependent increase in mean IPSP amplitude was significantly (P < 0.01) decreased (to 65%) in HET [n = 13; (B)] compared to WT mice. Upper panels show representative recordings from the CeA neurons of WT and HET mice. (C) Superfusion of CRF significantly decreased PPF in CeA slices from WT (n = 8) mice, but had no effect on PPF in HET mice [n = 11; (D)]. Top panels show representative recordings from PPF experiments. Data presented as mean ± SEM. Statistical significance calculated by Student’s t-test and set at *P < 0.05and **P < 0.01.
Discussion
The CeA plays a critical role in ethanol dependence and anxiety-related behaviors (Roberto et al., 2010). In the present study, we validated previous reports from our laboratory that ethanol and CRF increase GABA transmission in CeA neurons via increased presynaptic GABA release (Nie et al., 2009; Roberto et al., 2010). More importantly, we established that the AC7 pathway is involved in the alteration of GABA release by ethanol and CRF.
The effector molecules that translate signals from receptors (particularly GPCRs) to intracellular signaling pathways (e.g., PKA) are important mediators of ethanol’s synaptic effects. AC is one of the most ubiquitous signal transduction molecules and its activity in response to activation of GPCRs is altered by acute and chronic ethanol treatment (Tabakoff and Hoffman, 1998). Activation of many GPCRs stimulates AC activity to convert ATP to the intracellular second messenger, cAMP. Cyclic AMP, in turn, activates cAMP-dependent PKA, which modulates metabolism, and gene expression. It appears that the magnitude of ethanol’s actions on AC activity depends on the isoform of AC (Yoshimura and Tabakoff, 1995, 1999). Nine isoforms of the mammalian AC enzyme exist. The type VII isoform of AC (AC7) is distinguished by the fact that it is insensitive to inhibition by Giα, but is stimulated by the βγ subunits (Yoshimura et al., 1996). It is sensitive to calcium, and moderate concentrations of ethanol significantly enhance AC7-mediated activation of the cAMP signaling cascade via phosphorylation of AC7 by PKCdelta (Nelson et al., 2003), whereas AC 7 is not readily activated by forskolin (Yoshimura and Tabakoff, 1999).
Previously we found that the ethanol-induced increase in GABA release in CeA slices is mediated by a mechanism dependent on activation of CRF1s (Nie et al., 2004, 2009; Bajo et al., 2008). CRF1 is a potent mediator of stress- and anxiety-related behaviors (Baldwin et al., 1991; Rassnick et al., 1993; Pich et al., 1995) and CRF1s are abundantly expressed in the CeA. CRF1 is a GPCR that is positively linked to the activation of AC and other second messenger systems such as IP3 and PKC (Bajo et al., 2008). Several reports suggest that CRF1 couples to both the AC7 and AC9 isoforms (Antoni, 2000; Antoni et al., 2003). A switch in coupling from AC9 to AC7 results in increased cAMP signaling when CRF1 is activated by CRF. Disruption of CRF1 activation reduces negative affective states of opiate and alcohol withdrawal (Contarino and Papaleo, 2005; Gilpin et al., 2008), and pharmacological inhibition of CRF1 suppresses stress-mediated alcohol relapse in rats genetically selected for high alcohol preference (Valdez and Koob, 2004; Hansson et al., 2006). In a recent study (Bajo et al., 2008) we demonstrated that PKCε serves two roles in the CeA: (i) to reduce spontaneous baseline GABA release, and (ii) to mediate CRF- and ethanol-stimulated release of GABA. Based on the present data, we believe that the AC/cAMP and PKCε cascades are intimately involved and cross-communicate at some point in their cascades to regulate GABA release, and we speculate that the final target for both pathways is downstream to these cascades. Our previous and current studies suggest that components of the PKC (Bajo et al., 2008) and PKA signaling pathways are critical for CRF1-mediated effects in the CeA that could underlie anxiety-like and ethanol-related behaviors.
In the present study, we used male HET mice with diminished AC7 activity (Hines et al., 2006) to investigate a role of AC in ethanol- and CRF1-dependent effects on GABAergic transmission in the CeA. We found that despite similar baseline GABA transmission, CeA neurons from male HET mice did not demonstrate the increased GABA transmission (mediated by increased presynaptic GABA release) seen in their WT counterparts after treatment with ethanol or CRF. These data indicate that any imbalance in AC7 activity may have a profound effect on ethanol- and CRF-dependent modulation of the GABA system in the mouse CeA. However, CRF superfusion still elicited a significant but smaller increase of IPSP amplitude in the HET mice, suggesting a different contribution of AC7 pathway to CRF vs ethanol effects. Using Affymetrix gene expression arrays, we previously demonstrated that the only protein coding mRNA which was altered (diminished) in the brain of male HET mice was the ADCY7 product (Hines et al., 2006). Although we cannot rule out a possible postsynaptic location of AC7 in the CeA, our current data and the prior data of Mons et al. (1998b) suggest that AC7 is localized in presynaptic axon terminals of GABAergic neurons in several brain regions including the cerebellum (Mons et al., 1998b). Given the sensitivity of AC7 to ethanol and the coupling of AC7 to CRF1, our findings suggest that AC7 responds to ethanol and CRF in the CeA by increasing cAMP signaling and subsequent activation of the PKA cascade. The ethanol- and CRF1-dependent PKA activation, in turn, could modulate downstream effector molecules such as synaptotagmin and voltage sensitive Ca++ channels, resulting in an increase in presynaptic vesicular GABA release. However, in the long term, this enhanced signaling may initiate neuroadaptations in AC activity and response to neurotransmitters (Saito et al., 1987) that may be critical in inducing alcohol tolerance or dependence.
Based on genome-wide association studies, CRF1 and its downstream signaling effector AC7 have been considered to contribute to alcohol abuse in humans. The variants in the CRHR1 gene encoding the human CRF1 have recently been associated with binge drinking and high alcohol intake in two independent sample populations (Treutlein et al., 2006). These data further support and validate findings obtained from animal models of alcohol addiction that dysregulation of the CRF system is underlies the development of alcohol addiction in humans. Interestingly, a recent whole genome association study of a large German population sample demonstrated an association of polymorphisms in ADCY7 and alcoholism in females (S. Desrivieres et al., personal communication).
Overall, our study has added a new player in the complex signaling pathways mediating ethanol and CRF1 effects in the CeA. We identified AC7 as a critical signaling effector molecule involved in the ethanol and CRF1 effects on GABAergic transmission in the CeA. Previous studies indicated that in the CeA, ethanol acts through activation of CRF1 and its signaling pathways. Based on our current study, those signaling pathways include AC7 and subsequently also PKA. Identification of key players of intracellular signaling pathways involved in ethanol effects on neuronal systems is crucial for understanding mechanisms leading to development of ethanol dependence and for finding therapeutic targets for the treatment of alcoholism.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This is manuscript number 20989 from The Scripps Research Institute. This work was supported by the Pearson Center for Alcoholism and Addiction Research and NIAAA grants AA015566, AA06420, AA016985, AA013517, AA013498, AA013162, AA016663, AA017447, and Banbury Fund.
References
Andreasen, M., and Hablitz, J. J. (1994). Paired-pulse facilitation in the dentate gyrus: a patch-clamp study in rat hippocampus in vitro. J. Neurophysiol. 72, 326–336.
Antoni, F. A. (2000). Molecular diversity of cyclic AMP signalling. Front. Neuroendocrinol. 21, 103–132.
Antoni, F. A., Sosunov, A. A., Haunso, A., Paterson, J. M., and Simpson, J. (2003). Short-term plasticity of cyclic adenosine 3′,5′-monophosphate signaling in anterior pituitary corticotrope cells: the role of adenylyl cyclase isotypes. Mol. Endocrinol. 17, 692–703.
Bajo, M., Cruz, M. T., Siggins, G. R., Messing, R., and Roberto, M. (2008). Protein kinase C epsilon mediation of CRF- and ethanol-induced GABA release in central amygdala. Proc. Natl. Acad. Sci. U.S.A. 105, 8410–8415.
Baldwin, H. A., Rassnick, S., Rivier, J., Koob, G. F., and Britton, K. T. (1991). CRF antagonist reverses the “anxiogenic” response to ethanol withdrawal in the rat. Psychopharmacology (Berl.) 103, 227–232.
Blank, T., Nijholt, I., Grammatopoulos, D. K., Randeva, H. S., Hillhouse, E. W., and Spiess, J. (2003). Corticotropin-releasing factor receptors couple to multiple G-proteins to activate diverse intracellular signaling pathways in mouse hippocampus: role in neuronal excitability and associative learning. J. Neurosci. 23, 700–707.
Bonci, A., and Williams, J. T. (1997). Increased probability of GABA release during withdrawal from morphine. J. Neurosci. 17, 796–803.
Chheda, M. G., Ashery, U., Thakur, P., Rettig, J., and Sheng, Z. H. (2001). Phosphorylation of snapin by PKA modulates its interaction with the SNARE complex. Nat. Cell Biol. 3, 331–338.
Contarino, A., and Papaleo, F. (2005). The corticotropin-releasing factor receptor-1 pathway mediates the negative affective states of opiate withdrawal. Proc. Natl. Acad. Sci. U.S.A. 102, 18649–18654.
Gilpin, N. W., Richardson, H. N., and Koob, G. F. (2008). Effects of CRF1-receptor and opioid-receptor antagonists on dependence-induced increases in alcohol drinking by alcohol-preferring (P) rats. Alcohol. Clin. Exp. Res. 32, 1535–1542.
Grammatopoulos, D. K., Randeva, H. S., Levine, M. A., Kanellopoulou, K. A., and Hillhouse, E. W. (2001). Rat cerebral cortex corticotropin-releasing hormone receptors: evidence for receptor coupling to multiple G-proteins. J. Neurochem. 76, 509–519.
Hanoune, J., and Defer, N. (2001). Regulation and role of adenylyl cyclase isoforms. Annu. Rev. Pharmacol. Toxicol. 41, 145–174.
Hansson, A. C., Cippitelli, A., Sommer, W. H., Fedeli, A., Bjork, K., Soverchia, L., Terasmaa, A., Massi, M., Heilig, M., and Ciccocioppo, R. (2006). Variation at the rat Crhr1 locus and sensitivity to relapse into alcohol seeking induced by environmental stress. Proc. Natl. Acad. Sci. U.S.A. 103, 15236–15241.
Heilig, M., and Koob, G. F. (2007). A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci. 30, 399–406.
Hines, L. M., Hoffman, P. L., Bhave, S., Saba, L., Kaiser, A., Snell, L., Goncharov, I., LeGault, L., Dongier, M., Grant, B., Pronko, S., Martinez, L., Yoshimura, M., and Tabakoff, B. (2006). A sex-specific role of type VII adenylyl cyclase in depression. J. Neurosci. 26, 12609–12619.
Hyytia, P., and Koob, G. F. (1995). GABAA receptor antagonism in the extended amygdala decreases ethanol self-administration in rats. Eur. J. Pharmacol. 283, 151–159.
Kelm, M. K., Criswell, H. E., and Breese, G. R. (2008). The role of protein kinase A in the ethanol-induced increase in spontaneous GABA release onto cerebellar Purkinje neurons. J. Neurophysiol. 100, 3417–3428.
Koob, G. F. (2006). The Neurobiology of Addiction: A Hedonic Calvinist View. New York: Guilford Press.
Lowery, E. G., and Thiele, T. E. (2010). Pre-clinical evidence that corticotropin-releasing factor (CRF) receptor antagonists are promising targets for pharmacological treatment of alcoholism. CNS Neurol. Disord. Drug Targets 9, 77–86.
Mons, N., Decorte, L., Jaffard, R., and Cooper, D. M. (1998a). Ca2+-sensitive adenylyl cyclases, key integrators of cellular signalling. Life Sci. 62, 1647–1652.
Mons, N., Yoshimura, M., Ikeda, H., Hoffman, P. L., and Tabakoff, B. (1998b). Immunological assessment of the distribution of type VII adenylyl cyclase in brain. Brain Res. 788, 251–261.
Nelson, E. J., Hellevuo, K., Yoshimura, M., and Tabakoff, B. (2003). Ethanol-induced phosphorylation and potentiation of the activity of type 7 adenylyl cyclase. Involvement of protein kinase C delta. J. Biol. Chem. 278, 4552–4560.
Nie, Z., Schweitzer, P., Roberts, A. J., Madamba, S. G., Moore, S. D., and Siggins, G. R. (2004). Ethanol augments GABAergic transmission in the central amygdala via CRF1 receptors. Science 303, 1512–1514.
Nie, Z., Zorrilla, E. P., Madamba, S. G., Rice, K. C., Roberto, M., and Siggins, G. R. (2009). Presynaptic CRF1 receptors mediate the ethanol enhancement of GABAergic transmission in the mouse central amygdala. Scientific World J. 9, 68–85.
Pich, E. M., Lorang, M., Yeganeh, M., Rodriguez de Fonseca, F., Raber, J., Koob, G. F., and Weiss, F. (1995). Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J. Neurosci. 15, 5439–5447.
Pronko, S. P., Saba, L. M., Hoffman, P. L., and Tabakoff, B. (2010). Type 7 adenylyl cyclase-mediated hypothalamic–pituitary–adrenal axis responsiveness: influence of ethanol and sex. J. Pharmacol. Exp. Ther. 334, 44–52.
Rassnick, S., Heinrichs, S. C., Britton, K. T., and Koob, G. F. (1993). Microinjection of a corticotropin-releasing factor antagonist into the central nucleus of the amygdala reverses anxiogenic-like effects of ethanol withdrawal. Brain Res. 605, 25–32.
Roberto, M., Cruz, M. T., Gilpin, N. W., Sabino, V., Schweitzer, P., Bajo, M., Cottone, P., Madamba, S. G., Stouffer, D. G., Zorrilla, E. P., Koob, G. F., Siggins, G. R., and Parsons, L. H. (2010). Corticotropin releasing factor-induced amygdala gamma-aminobutyric acid release plays a key role in alcohol dependence. Biol. Psychiatry 67, 831–839.
Roberto, M., Madamba, S. G., Moore, S. D., Tallent, M. K., and Siggins, G. R. (2003). Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc. Natl. Acad. Sci. U.S.A. 100, 2053–2058.
Roberto, M., Madamba, S. G., Stouffer, D. G., Parsons, L. H., and Siggins, G. R. (2004). Increased GABA release in the central amygdala of ethanol-dependent rats. J. Neurosci. 24, 10159–10166.
Roberts, A. J., Cole, M., and Koob, G. F. (1996). Intra-amygdala muscimol decreases operant ethanol self-administration in dependent rats. Alcohol. Clin. Exp. Res. 20, 1289–1298.
Saito, T., Lee, J. M., Hoffman, P. L., and Tabakoff, B. (1987). Effects of chronic ethanol treatment on the beta-adrenergic receptor-coupled adenylate cyclase system of mouse cerebral cortex. J. Neurochem. 48, 1817–1822.
Seino, S., and Shibasaki, T. (2005). PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol. Rev. 85, 1303–1342.
Tabakoff, B., and Hoffman, P. L. (1998). Adenylyl cyclases and alcohol. Adv. Second Messenger Phosphoprotein Res. 32, 173–193.
Treutlein, J., Kissling, C., Frank, J., Wiemann, S., Dong, L., Depner, M., Saam, C., Lascorz, J., Soyka, M., Preuss, U. W., Rujescu, D., Skowronek, M. H., Rietschel, M., Spanagel, R., Heinz, A., Laucht, M., Mann, K., and Schumann, G. (2006). Genetic association of the human corticotropin releasing hormone receptor 1 (CRHR1) with binge drinking and alcohol intake patterns in two independent samples. Mol. Psychiatry 11, 594–602.
Trudeau, L. E., Emery, D. G., and Haydon, P. G. (1996). Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron 17, 789–797.
Valdez, G. R., and Koob, G. F. (2004). Allostasis and dysregulation of corticotropin-releasing factor and neuropeptide Y systems: implications for the development of alcoholism. Pharmacol. Biochem. Behav. 79, 671–689.
Yoshimura, M., Ikeda, H., and Tabakoff, B. (1996). mu-Opioid receptors inhibit dopamine-stimulated activity of type V adenylyl cyclase but enhance dopamine-stimulated activity of type VII adenylyl cyclase. Mol. Pharmacol. 50, 43–51.
Yoshimura, M., and Tabakoff, B. (1995). Selective effects of ethanol on the generation of cAMP by particular members of the adenylyl cyclase family. Alcohol. Clin. Exp. Res. 19, 1435–1440.
Keywords: alcohol, CRH, electrophysiology, IPSPs, paired-pulse facilitation, knock-down mouse
Citation: Cruz MT, Bajo M, Maragnoli ME, Tabakoff B, Siggins GR and Roberto M (2011) Type 7 adenylyl cyclase is involved in the ethanol and CRF sensitivity of GABAergic synapses in mouse central amygdala. Front. Neurosci. 4:207. doi: 10.3389/fnins.2010.00207
Received: 28 October 2010;
Accepted: 20 December 2010;
Published online: 14 January 2011.
Edited by:
A. Leslie Morrow, University of North Carolina School of Medicine, USAReviewed by:
Carlos Fernando Valenzuela, University of New Mexico Health Sciences Center, USAMark S. Brodie, University of Illinois at Chicago, USA
Copyright: © 2011 Cruz, Bajo, Maragnoli, Tabakoff, Siggins and Roberto. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Marisa Roberto, Committee on Neurobiology of Addictive Disorders, The Scripps Research Institute, SP30-1160, 10550 N. Torrey Pines Road, La Jolla, CA 92037, USA.e-mail:bXJvYmVydG9Ac2NyaXBwcy5lZHU=