 Anni-Maija Linden1
Anni-Maija Linden1 Ulrich Schmitt2 Elli Leppä1
Ulrich Schmitt2 Elli Leppä1 Peer Wulff3
Peer Wulff3 William Wisden4
William Wisden4 Hartmut Lüddens2*
Hartmut Lüddens2* Esa R. Korpi1*
Esa R. Korpi1*
- 1 Pharmacology, Institute of Biomedicine, University of Helsinki, Helsinki, Finland
- 2 Department of Psychiatry and Psychotherapy, University Medical Centre, Johannes Gutenberg University Mainz, Mainz, Germany
- 3 Institute of Medical Sciences, University of Aberdeen, Foresterhill, Aberdeen, UK
- 4 Division of Cell and Molecular Biology, Imperial College London, London, UK
Ethyl alcohol (ethanol) has many molecular targets in the nervous system, its potency at these sites being low compared to those of sedative drugs. This has made it difficult to discover ethanol’s binding site(s). There are two putative binding sites at γ-aminobutyric acid (GABA) type A receptor subtypes for the proposed ethanol antagonist Ro 15-4513, the established γ2 subunit-dependent benzodiazepine site and the recently reported δ subunit-dependent Ro 15-4513/ethanol binding site. Here, we aimed at clarifying the in vivo role of Ro 15-4513 at these two sites. We found that the antagonism of ethanol actions by Ro 15-4513 in wildtype mice was dependent on the test: an open field test showed that light sedation induced by 1.5–1.8 g/kg ethanol was sensitive to Ro 15-4513, whereas several tests for ethanol-induced anxiolytic effects showed that the ethanol-induced effects were insensitive to Ro 15-4513. Antagonism of ethanol-induced sedation by Ro 15-4513 was unaffected in GABAA receptor δ subunit knockout mice. By contrast, when testing the GABAA receptor γ2 subunit F77I knock-in mouse line (γ2I77 mice) with its strongly reduced affinity of the benzodiazepine sites for Ro 15-4513, we found that the ethanol-induced sedation was no longer antagonized by Ro 15-4513. Indeed, γ2I77 mice had only a small proportion of high-affinity binding of [3H]Ro 15-4513 left as compared to wildtype mice, especially in the caudate–putamen and septal areas, but these residual sites are apparently not involved in ethanol antagonism. In conclusion, we found that Ro 15-4513 abolished the sedative effect of ethanol by an action on γ2 subunit-dependent benzodiazepine sites.
Introduction
Ethanol (ethyl alcohol) is a poorly discriminating pharmacological compound targeting various proteins in the central nervous system. As the dose increases, ethanol causes anxiolysis, loss of exploration, muscle relaxation and ataxia, sedation, and finally loss of righting reflex and hypnosis (Deitrich et al., 1989; Spanagel, 2009). The costs of alcoholism to society are immense (e.g., Olesen and Leonardi, 2003; Nutt et al., 2010); and in Finland more alcohol-caused deaths occur yearly than casualties in traffic accidents (Causes of Death 2008, 2010); in the UK, alcohol is considered the drug which does the most harm to society (Nutt et al., 2010). Therefore, a potent and efficient alcohol antagonist would be important to develop. To that end, reports on ethyl-8-azido-5,6-dihydro-5-methyl-6-oxo-4H-imidazo-1,4-benzodiazepine-3-carboxylate (Ro 15-4513), an imidazobenzodiazepine compound acting via the benzodiazepine sites of γ-aminobutyric acid type A receptors (GABAA), initially provoked enormous interest as this compound seemed to inhibit ethanol’s actions in animal models (Suzdak et al., 1986, 1988; Bonetti et al., 1988). Although Ro 15-4513’s efficacy as an alcohol antagonist was later not found high enough for clinical development, the mechanisms of action of this compound are still of interest.
Ro 15-4513 binds to all known GABAA receptor γ2 subunit-dependent benzodiazepine binding sites irrespective of the α or β variants (Luddens and Wisden, 1991; Wisden et al., 1991; Korpi et al., 2002). It usually acts as a partial inverse agonist at GABAA receptors (Bonetti et al., 1988; Hadingham et al., 1993; Korpi et al., 2002), except for α4 and α6 subunit-containing ones in which it acts as an agonist (Hadingham et al., 1996; Knoflach et al., 1996; Wafford et al., 1996). Some reports suggested that Ro 15-4513 inhibits ethanol-induced potentiation of GABAA receptor function in brain membrane vesicles and cultured neurons (Suzdak et al., 1986; Mehta and Ticku, 1988), making this simple receptor antagonism the possible mode of action. More recently, ethanol has been suggested to competitively inhibit the binding of Ro 15-4513 to α4/6βδ subunit-containing GABAA receptors (Hanchar et al., 2006; Wallner et al., 2006). For some neuronal classes (cerebellar granule cells, thalamic relay neurons, dentate granule cells), these receptor subtypes make up a large portion of extrasynaptic receptors that are exceptionally sensitive to GABA, and which do not desensitize. Such α4βδ and α6βδ GABAA receptors produce tonic inhibitory currents (Brickley et al., 2001; Chandra et al., 2006). It is also important to note that the α4 and α6 subunits combine recombinantly and in vivo with the γ2 subunit, and not just with the δ subunit (Wisden et al., 1991). For example in the cerebellar granule cells there will be α6βδ and α6βγ2 and/or α1/6βγ2 receptors (Wisden et al., 1996) and in the hippocampal CA1 pyramidal cells there will be α4βγ2 receptors and probably in other forebrain cell types as well (Olsen and Sieghart, 2008).
In an alcohol- and benzodiazepine-sensitive selectively bred rat line (Korpi et al., 1993) and more recently in outbred Wistar rats (Hanchar et al., 2005) the α6 subunit gene was found to have a point mutation (R100Q) which increases affinity to benzodiazepine agonists. Whereas the experiments in Wistar rats suggested that some effects of alcohol are enhanced by the mutated α6Q100 subunit-containing GABAA receptor (Hanchar et al., 2005), the same conclusion could not be made for the selectively bred rats, since increased alcohol sensitivity was not genetically segregating with the point mutation in the α6 subunit in the F2 generations of the cross between alcohol-sensitive and alcohol-insensitive rat lines (Sarviharju and Korpi, 1993; Botta et al., 2007).
From previous work, the mechanism of the alcohol antagonistic action of Ro 15-4513 might then be either the general inverse agonism of GABAA receptors at the established non-α4/non-α6 benzodiazepine sites (e.g., α1βγ2, α2βγ2, α3βγ2, and α5βγ2) and/or the selective inhibition of ethanol binding to specific non-benzodiazepine Ro 15-4513 binding sites of GABAA receptors with δ subunits. Here, we have tested these two modes of action with two mouse lines. Firstly, we used the GABAA receptor δ subunit knockout mouse line (Mihalek et al., 1999), which should remove α4βδ- and α6βδ-dependent high-affinity ethanol sites while ethanol antagonism by Ro 15-4513 could still be mediated via inverse agonism of α1/2/3/5βγ2 subunit-containing receptors. Secondly, we used the γ2I77 knock-in mouse line (Cope et al., 2004, 2005; Leppa et al., 2005) that carry a point mutation engineered into their γ2 subunit genes at the amino acid position 77, replacing phenylalanine (F) with isoleucine (I) and thus globally altering benzodiazepine pharmacology in the brain by greatly decreasing the affinity to the agonist zolpidem and the inverse agonist DMCM (methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate) as well as to Ro 15-4513 (Buhr et al., 1997; Wingrove et al., 1997; Ogris et al., 2004; Cope et al., 2005). This renders γ2I77 animals ideal as a complimentary tool to the δ-deficient mouse line to test whether possible high-affinity ethanol sites dependent on the δ subunit are obligatory for the ethanol antagonism by Ro 15-4513.
Materials and Methods
Animals
Generation of the δ−/− mouse line has been described (Mihalek et al., 1999). The genetic background of the mice obtained from Gregg E. Homanics from the University of Pittsburgh was C57BL/6J × strain 129Sv/SvJ. In Mainz, heterozygous mice were bred to obtain age-matched male and female littermate wildtype controls (n = 16, aged 3–8 months, 23–36 g males, 20–27 g females) and homozygous δ−/− mice (n = 18, aged 3–8 months, 24–34 g males, 20–28 g females). Genotyping was performed as described (Mihalek et al., 1999). Male C57BL/6J mice for open arena experiments were obtained from central animal facility (ZVTE) of the University Medical Center Mainz (n = 45, aged 2–4 months, 25–30 g).
Generation of the γ2I77 mouse line has been described (Cope et al., 2004). In brief, the γ2I77 mouse line was generated by mating male chimeras (C57BL/6J × 129ola) containing γ2I77 mutation with wildtype C57BL/6J females (Charles River Deutschland, Sulzfeld, Germany) to obtain heterozygous γ2F77/γ2I77 founder pairs as described (Cope et al., 2004). The γ2I77 mouse line used in our experiments was backcrossed at least four times with C57BL/6 mice (Harlan Netherlands, Horst, Netherlands). Heterozygous breeding was used to obtain age-matched male and female littermate wildtype controls (n = 50, aged 3–4 months, 24–31 g males, 18–21 g females) and homozygous γ2I77 mice (n = 42, aged 3–4 months, 26–31 g males, 18–22 g females) for open arena and elevated plus-maze experiments. Homozygous breeding was used to obtain additional γ2I77 mice (n = 130, aged 3–5 months, 21–30 g males, 17–24 g females) for the elevated plus-maze tests. Genotyping was performed as described (Cope et al., 2004). To obtain brains for autoradiography six female homozygous γ2I77 mice and six wildtype littermates (five females, one male) from heterozygous breeding, aged about 2 months, were used.
The mice were maintained in same-sex groups of either 1–2 (small plastic cages, 20 cm × 27 cm × 13 cm) or 4–8 (bigger cages, 40 cm × 30 cm × 15 cm) with food pellets and tap water ad libitum at standard housing conditions (12 h light–dark cycle, lights on at 6:00 A.M.; temperature, 20–23°C; relative humidity, 50–60%; aspen chip beddings).
All animal tests were carried out in accordance to the European Communities Council Directive of 24 November 1986 (86/609/EEC) and approved by the Laboratory Animal Committee of the Southern Finland Provincial Government and the Landesuntersuchungsamt Rheinland-Pfalz, Koblenz. All efforts were made to minimize the number and suffering of the experimental animals.
Behavioral Assays
To test for exploratory locomotor activity and ethanol-induced sedation, the mice were individually placed on a novel arena for 5 min (Linden et al., 2006; Chandra et al., 2010). Total locomotor activity of mice were analyzed automatically by following the center of the animal’s surface area monitored from above using a video tracking system with EthoVision 3.0 software (Noldus Information Technology, Wageningen, Netherlands). The number of rearing events was recorded by the experimenter from a video monitor. Lighting was adjusted to obtain light level of approximately 175 lux in the center of the arena. The arena was cleaned after each animal with diluted sodium hypochlorite solution (active chlorine approximately 0.005%).
To examine anxiety-related behaviors the mice were tested in an elevated plus-maze as described (Linden et al., 2006). The elevated plus-maze was made of gray plastic and elevated 50 cm from the floor level. It consisted of a central platform (5 cm × 5 cm), two open arms (5 cm × 40 cm with a 0.2 cm margin), and two enclosed arms (5 cm × 40 cm × 20 cm; Lister, 1987b). The mice were placed individually on the central platform facing the open arm and allowed free exploration of the maze for 5 min. An arm entry was recorded when the center of the mouse entered the arm at least 2 cm distal from the central platform. This corresponds to the definition of an arm entry with all four legs on the arm. Lighting was adjusted to obtain light level of approximately 20 lux in the central platform for tests with γ2I77 mice. The maze was cleaned after each animal with diluted sodium hypochlorite solution or with water. Testing of γ2I77 mice was performed in a quiet room adjacent to housing rooms. In the elevated plus-maze testing of δ−/− mice, lighting level was higher (exact level not determined), the maze was cleaned with water, a radio needed to be on to standardize background noise level, and the testing room was separate from housing rooms.
Quantitative Ligand Autoradiography
Flumazenil-sensitive [3H]Ro 15-4513 binding was determined in various brain regions as described (Makela et al., 1997). In brief, horizontal sections using a Leica CM 3050 S cryostat were cut from six brains for both genotypes, thaw-mounted onto gelatin-coated object glasses (Menzel GmbH, Braunschweig, Germany) and preincubated for 15 min in an ice-water bath in incubation buffer [50 mM Tris–HCl, 120 mM NaCl (pH 7.4)]. The final incubation took place with 15 nM [3H]Ro 15-4513 (Perkin-Elmer, Boston, MA, USA) in fresh buffer in the dark at +4°C for 60 min in plastic slide mailers. The required amount of the radioisotope in ethanol was evaporated to dryness under vacuum and taken up into a similar volume of dimethyl sulfoxide. Effects of 1–100 mM ethanol (Altia, Rajamäki, Finland) on radioligand binding were determined. Non-specific binding was determined with 10 μM flumazenil (Ro 15-1788; Tocris Biosciences, Ellisville, MO, USA). After incubation, the sections were washed with ice-cold incubation buffer for 2 × 60 s, dipped in distilled water, and dried in air flow at room temperature. The sections were exposed to Kodak Biomax MR film (Eastman Kodak, Rochester, NY, USA) for 8 weeks with [3H]microscale standards (GE Healthcare, Little Chalfont, Buckinghamshire, UK). For quantification of binding densities, the films were first scanned with the standards and then relevant brain regions analyzed with MCID M5 image analysis devices and programs (Imaging Research Inc., St. Catharines, ON, Canada). Binding densities were converted to nCi/mg radioactivity values on the basis of the simultaneously exposed standards. Non-specific binding in the presence of flumazenil was at the background level in sections of both mouse lines, except for some faint binding left in the cerebellar granule cell layer of wildtype sections (Figure 8). This component was not subtracted from the total binding. Representative images were obtained by scanning the films using an Epson expression 1680 Pro scanner.
In Situ Hybridization
In situ hybridization with 35S-labeled oligonucleotide probes was as described (Wisden et al., 1992; Wisden and Morris, 2002). Brain cryostat sections from adult γ2I77 mice were used. Images were generated from 4 to 12 week exposures to Kodak Biomax MR X-ray films. To assess non-specific labeling of the sections, each labeled oligonucleotide was hybridized to brain sections with a 100-fold excess of unlabeled oligonucleotide. Oligonucleotide sequences were as follows:
GABAA receptor γ1 subunit,
5′-ATGCAAGGTTCCGTATTCCATGAGTGCTGCAAACACAAAAATGAA-3′; GABAA receptor γ2 subunit,
5′-GTGTCTGGAATCCAGATTTTCCCCACCATATTGCTATTCAAC-3′;
GABAA receptor γ3 subunit,
5′-AGAGGGTGCTTGAAGGCTTATTCGATCAGGAATCCATCTTGTTGA-3′.
Drugs
Ethanol (94% w/w) was diluted with saline to 10% w/v and injected at volumes of 10, 15, or 18 ml/kg for the doses of 1.0, 1.5, and 1.8 g/kg, respectively. Ro 15-4513 (Tocris) was first carefully dissolved in 100% Tween 80 (Sigma, St. Louis, MO, USA; final concentration 3%) and then brought to the final concentration with saline or if co-administered with ethanol with 10% ethanol. Thus, the final Ro 15-4513 concentration was 0.167 or 0.3 mg/ml in the tests with 1.8 or 1 g/kg doses of ethanol, respectively. When Ro 15-4513 was administered 15 min before 1.5 g/kg ethanol, the drug suspension in Tween 80 was brought to the final 0.3 mg/ml concentration with saline and injected at a volume of 10 ml/kg.
Statistical Testing
All data are given as means ± SEM. Three-way analysis of variance (ANOVA) was used to test main drug, genotype and sex effects (SPSS 15.0 for Windows, SPSS Inc., Chicago, IL, USA). If no significant sex effect was found, the data from males and females were combined for post hoc Newman–Keuls multiple comparison test, or for Student’s t-test or Mann–Whitney analyses (GraphPad Prism 5.00, San Diego, CA, USA). The limit for significance was p < 0.05.
Results
Ro 15-4513 Reversed Ethanol-Induced Sedation in GABAA Receptor δ Subunit-Deficient Mice
Male and female δ−/− and littermate wildtype δ+/+ mice were administered first 3 mg/kg Ro15-4513, then 15 min later 1.5 g/kg ethanol, and 10 min later they were individually placed in an open arena (59 cm × 59 cm) for a 5-min test of locomotor activity.
Three-way ANOVA revealed a significant drug effect (F2,22 = 6.8, p < 0.01) on the percent time moving, but no sex effect or interaction. Therefore, the data of males and females were pooled (drug effect F2,28 = 5.8, p < 0.01 in two-way ANOVA) for subsequent analysis (Figure 1). Ethanol appeared to slightly reduce total locomotor activity (i.e., it induced a sedative effect) in both wildtype and δ−/− mice. However, this reduction was significant (p < 0.05) only in the knockout mice (Figure 1). Importantly, pretreatment with Ro 15-4513 prevented this ethanol-induced reduction (Figure 1). Since no genotype effect was in locomotor activity, we pooled the wildtype and knockout values to analyze the effect of ethanol and its combination with Ro 15-4513. Ethanol alone significantly (p < 0.01, one-way ANOVA followed by Student’s t-test) reduced locomotor activity compared to vehicle treatment (47.7 ± 4.6 vs. 67.5 ± 4.2% time moving, n = 10–12, respectively), whereas ethanol + Ro 15-4513 (65.4 ± 4.1, n = 12) did not differ from vehicle values, but was significantly (p < 0.01) higher than the values for ethanol alone.
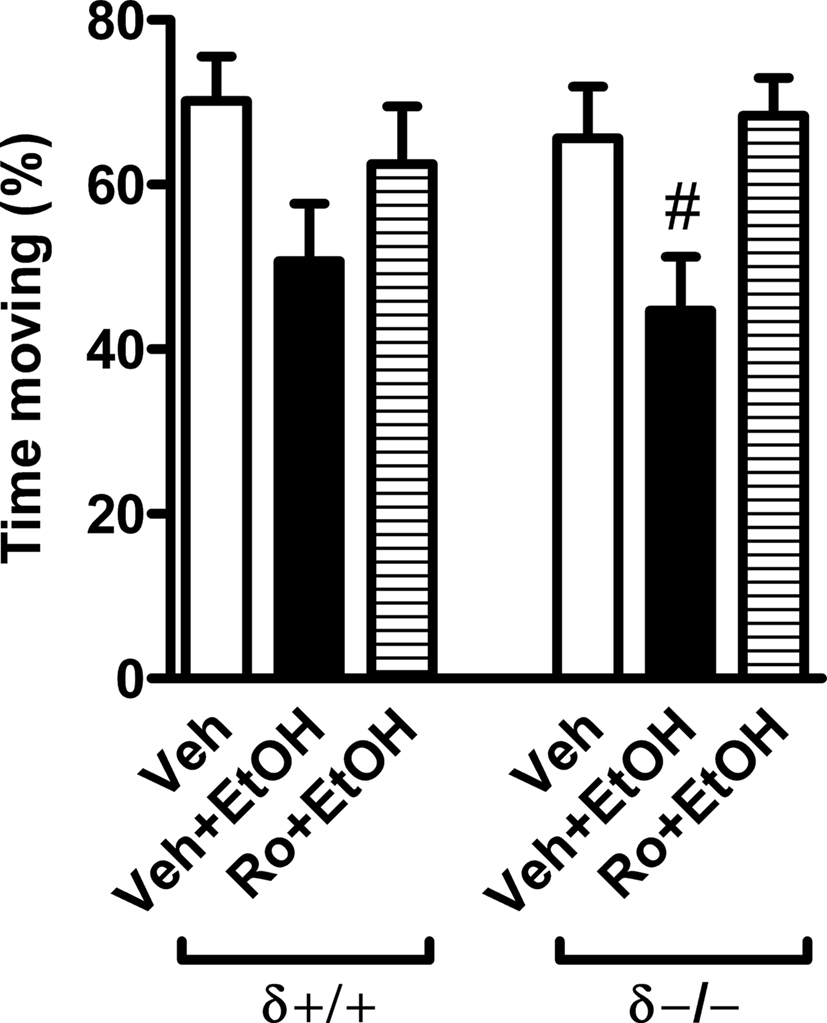
FIGURE 1. Ethanol-induced reduction in total locomotor activity was reversed by pretreatment with Ro 15-4513 in δ−/− mice. Male and female δ−/− and littermate wildtype δ+/+ mice were pretreated with 3 mg/kg Ro 15-4513 (i.p.) or vehicle (Veh) 15 min before administration of 1.5 g/kg ethanol (EtOH; i.p.) and tested 10 min later on a novel arena for 5 min for total locomotor activity analyzed as percent time moving of the total testing time. #p < 0.05 compared to vehicle controls within the same genotype (one-way ANOVA followed by Student’s t-test). The data of males and females were pooled. n = 5–7.
Antagonism of 1.8 g/Kg Ethanol-Induced Sedation by Ro 15-4513 in C57BL/6J Mice
Next, we tested whether Ro 15-4513 could inhibit sedation induced by a slightly higher dose (1.8 g/kg) of ethanol because the reduction in locomotor activity by the 1.5-g/kg dose barely reached statistical significance in the test described above. In C57BL/6J male mice, 1.8 g/kg ethanol significantly reduced locomotor activity in an open arena (59 cm × 59 cm) as verified in post hoc tests followed by one-way ANOVA (drug effects F3,41 > 16.5, p < 0.0001; Figures 2A,B). Importantly, Ro 15-4513 (3 mg/kg), co-administered at the same time as ethanol, completely inhibited the ethanol-induced reduction in total locomotor activity and partly the reduction in rearing (Figures 2A,B). Ro 15-4513 alone had no effect on locomotor activity.
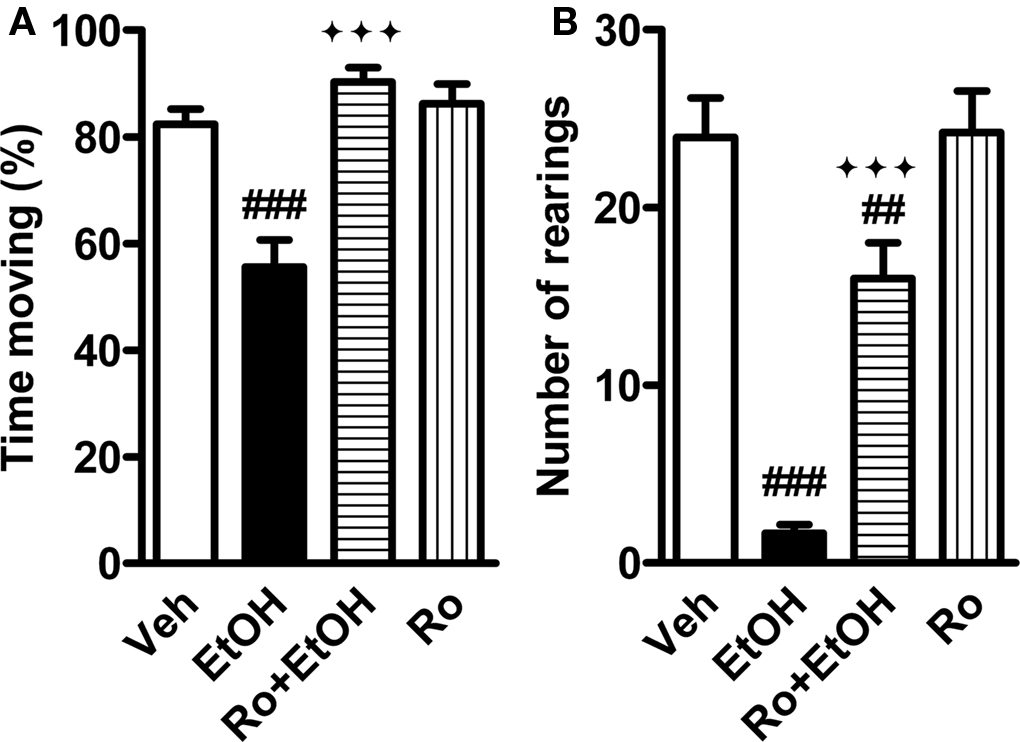
FIGURE 2. Reversal of ethanol-induced reduction in total locomotor activity by Ro 15-4513 in wildtype mice. Male C57BL/6J mice were co-administered with 1.8 g/kg ethanol (EtOH) and 3 mg/kg Ro15-4513 (or vehicle, Veh) 10 min before being tested on a novel arena for 5 min. (A) Co-treatment with Ro 15-4513 reversed ethanol-induced reduction in total locomotor activity. (B) Co-treatment with Ro 15-4513 attenuated ethanol-induced reduction in rearing. ##p < 0.01, ###p < 0.001 compared to vehicle treatment; ♦♦♦p < 0.001 compared to ethanol treatment (one-way ANOVA followed by Newman–Keuls test). n = 9–17.
Ro 15-4513 Failed to Reverse Ethanol-Induced Sedation in γ2I77 Mice
Ethanol (1.8 g/kg) was administered to male and female γ2I77 and littermate wildtype γ2F77 mice with or without 3 mg/kg Ro15-4513 and the mice were tested on an open arena (50 cm × 50 cm) 10 min later. Three-way ANOVA revealed no sex effect or interaction and therefore the data of males and females could be pooled without reservations for subsequent analysis. Two-way ANOVA showed that locomotor activity was significantly affected by drug treatments (F3,84 = 17.2, p < 0.001) and genotype (F1,84 = 12.3, p < 0.01) and that genotype × drug interaction (F3,84 = 5.6, p < 0.01) emerged. Post hoc comparisons verified that ethanol reduced locomotion in both wildtype and γ2I77 mice (Figure 3A), and that this reduction was completely prevented in the wildtype mice by co-administration with Ro 15-4513. In γ2I77 mice, Ro 15-4513 failed to prevent this effect of ethanol. Ro 15-4513 alone did not affect locomotion in either wildtype or γ2I77 mice (Figure 3A).
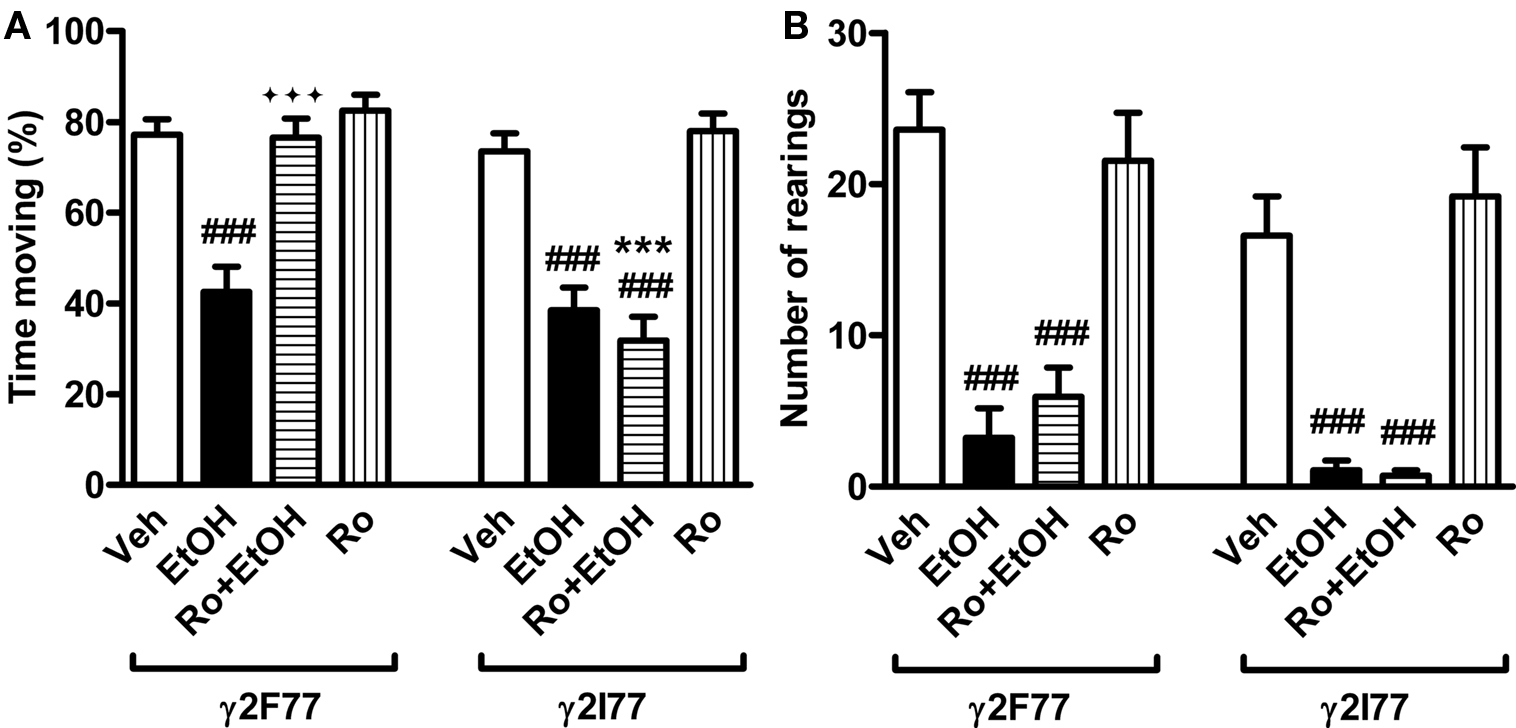
FIGURE 3. Ro 15-4513 reversed ethanol-induced reduction in total locomotor activity in wildtype γ2F77 mice, but not in γ2I77 knock-in mice. Male and female γ2I77 and littermate wildtype γ2F77 mice were co-administered with 1.8 g/kg ethanol and 3 mg/kg Ro15-4513 (or vehicle, Veh) 10 min before being tested on a novel arena for 5 min. (A) Ro 15-4513 reversed ethanol-induced reduction in total locomotor activity in the γ2F77, but not in γ2I77 mice. (B) Ro 15-4513 failed to reverse ethanol-induced reduction in rearing both in the γ2F77 and γ2I77 mice. Because three-way ANOVA revealed no sex effect or interaction the data of males and females were pooled. ***p < 0.001 compared to similarly treated γ2F77 mice; ###p < 0.001 compared to vehicle controls within the same genotype; ♦♦♦p < 0.001 compared to ethanol treatment within the same genotype (one-way ANOVA followed by Newman–Keuls multiple comparison test). n = 10–14.
Ethanol treatment reduced the number of rearings in both genotypes but this reduction was not prevented by Ro 15-4513 in either genotype (Figure 3B). Consistently, two-way ANOVA displayed drug (F3,84 = 40.6, p < 0.001) and genotype (F1,84 = 6.4, p < 0.05) effects without an interaction. The slightly lower number of rearing events in all treatment groups of γ2I77 mice compared to the corresponding wildtype values likely explains the significant genotype effect in two-way ANOVA.
Elevated Plus-Maze Behavior after Administration of Ethanol and Ro 15-4513 in the Wildtype and γ2I77 Mice
Ethanol (1 g/kg, i.p.) and Ro 15-4513 (3 mg/kg) were co-administered to male and female γ2I77 and wildtype littermate γ2F77 mice 10 min before the elevated plus-maze test. Three-way ANOVA revealed no sex effects in the behaviors, and, therefore, the male and female data were combined (Figure 4). The percent time spent on the open arms was significantly affected by drug treatment (F3,106 = 3.9, p < 0.05, two-way ANOVA), but not by genotype, and no genotype × drug interaction emerged. The only groups that were significantly different from the vehicle-treated mice in the post hoc comparisons were the ethanol-treated and Ro 15-4513 + ethanol-treated γ2I77 mouse groups. They spent more time on the open arms compared to vehicle-treated γ2I77 mice (Figure 4A). The number of open-arm entries was also significantly affected by drug treatment (F3,106 = 3.6, p < 0.05, two-way ANOVA). However, differences between the treatment groups were not significant in post hoc comparisons in either genotype (Figure 4B).
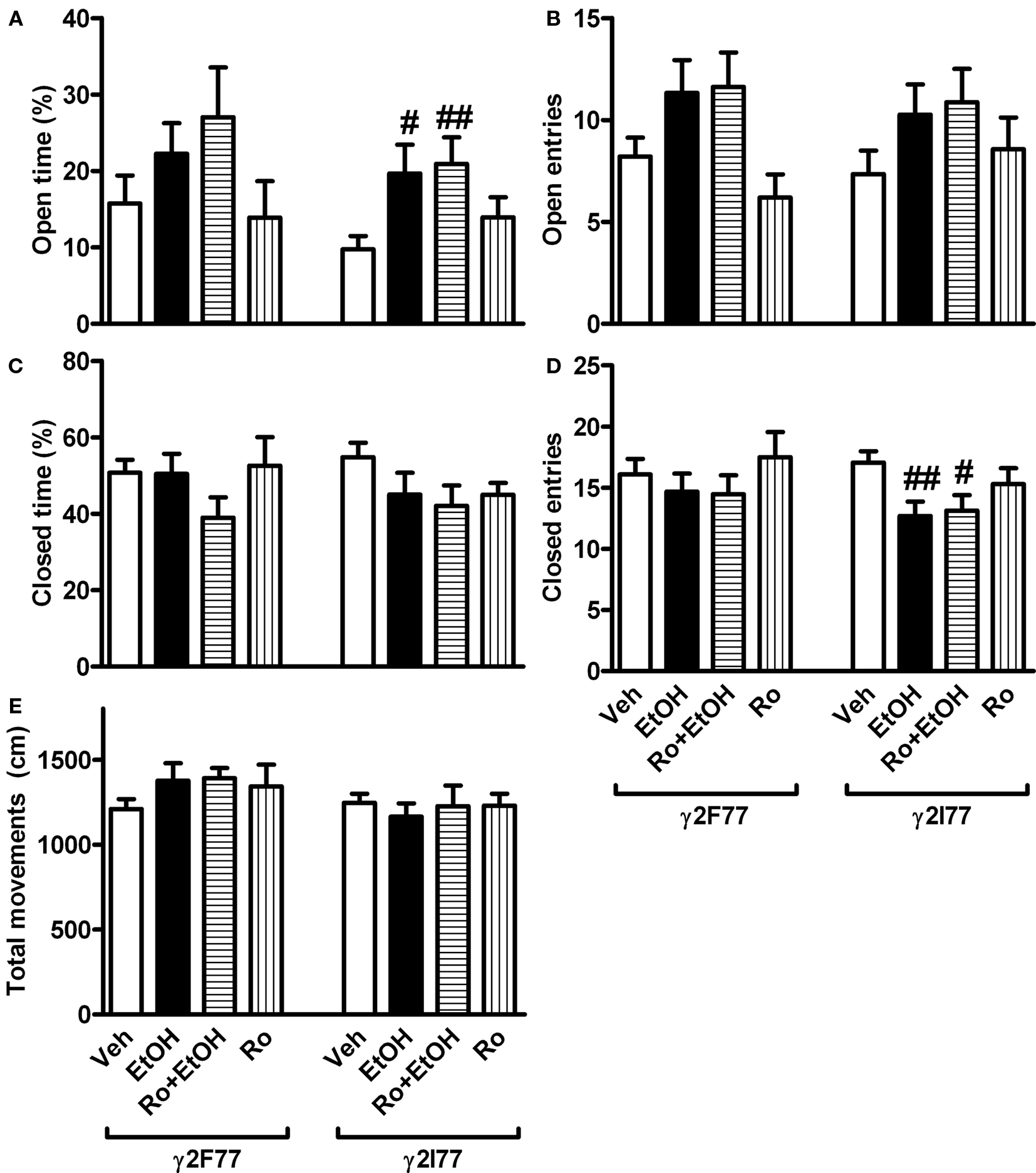
FIGURE 4. Effects of ethanol and its combination with Ro 15-4513 on elevated plus-maze behavior in wildtype γ2F77 and γ2I77 knock-in mice. Male and female γ2I77 and littermate wildtype γ2F77 mice were co-administered with 1 g/kg ethanol (EtOH) and 3 mg/kg Ro15-4513 (or vehicle, Veh) 10 min before being tested for anxiolytic-like behavior on an elevated plus-maze for 5 min. (A) The percent time spent in the open arms of the total testing time. (B) The number of open-arm entries. (C) The percent time spent in the closed arms of the total testing time. (D) The number of closed-arm entries. (E) Total movements. Because three-way ANOVA revealed no sex effect or interaction the data of males and females were pooled. #p < 0.05, ##p < 0.01 compared to vehicle-treated mice within the same genotype (one-way ANOVA followed by Student’s t-test). n = 10–19.
The percent time spent on the closed arms was not significantly affected by drug treatment or genotype (Figure 4C). However, the number of closed-arm entries was affected by drug treatment (F3,106 = 2.8, p < 0.05, two-way ANOVA) without genotype effect or genotype × drug interaction. Post hoc comparisons showed that the ethanol-treated and Ro 15-4513 + ethanol-treated γ2I77 mice visited significantly less frequently the closed arms than the vehicle-treated γ2I77 mice (Figure 4D). The dose of 1 g/kg ethanol or its combination with Ro 15-4513 (3 mg/kg) did not cause sedation as the amount of total movements was not significantly affected by drug treatment (p > 0.05, two-way ANOVA; Figure 4E).
It should be noted that we excluded from the results 5 (2 males and 3 females) out of 20 (25%) Ro 15-4513 + ethanol-treated γ2I77 mice; 1 (male) out of 14 (7.1%) Ro 15-4513 + ethanol-treated wildtype mice; and 1 (male) out of 13 (7.7%) ethanol-treated wildtype mice because they fell down from the maze during the test. Thus, the mean values of especially Ro 15-4513 + ethanol-treated γ2I77 mice may be biased by the inability of the most sensitive animals to perform the test.
In order to analyze the effects of ethanol on open-arm behaviors and the actions of Ro 15-4513 on them we pooled the data of γ2I77 and wildtype γ2F77 mice, since no genotype effect emerged in two-way ANOVA. Ethanol treatment increased significantly (p < 0.01, one-way ANOVA followed by Student’s t-test) the percent time spent in the open arms compared to vehicle treatment (20.7 ± 2.7 vs. 12.5 ± 2.0, n = 31, respectively). The open-arm time was also significantly (p < 0.01) longer in Ro 15-4513 + ethanol-treated mice (23.8 ± 3.5, n = 28). Similarly, ethanol treatment increased significantly (p < 0.05) the number of open-arm visits compared to vehicle treatment (10.7 ± 1.1 vs. 7.7 ± 0.7, n = 31, respectively), and they were also significantly increased (p < 0.05) in Ro 15-4513 + ethanol-treated mice (11.2 ± 1.2, n = 28).
In another elevated plus-maze experiment, the effect of co-administering Ro 15-4513 (3 mg/kg) and ethanol (1 g/kg) were tested in γ2I77 mice obtained from homozygous breeding. In this test the maze was cleaned with water, not with diluted sodium hypochlorite solution. We excluded 2 (females) out of 27 (7.4%) ethanol-treated mice because they fell down from the maze during the test. Two-way ANOVA revealed no sex effects. The Ro 15-4513 + ethanol-treated γ2I77 mice spent significantly more time on the open arms compared to the vehicle-treated mice (F3,92 = 2.8, p < 0.05, two-way ANOVA; Figure 5A). The number of open-arm entries was also significantly affected by drug treatment (F3,92 = 3.2, p < 0.05). However, differences between the treatment groups did not reach significance in post hoc comparisons (Figure 5B). Although, the drug treatments did not alter the time spent on the closed arms, the number of closed-arm entries was significantly affected by drug treatment (F3,92 = 4.6, p < 0.01), and the Ro 15-4513 + ethanol-treated γ2I77 mice visited significantly less frequently the closed arms than the vehicle-treated mice (Figure 5D). Total movements were not significantly affected by drug treatments (Figure 5E).
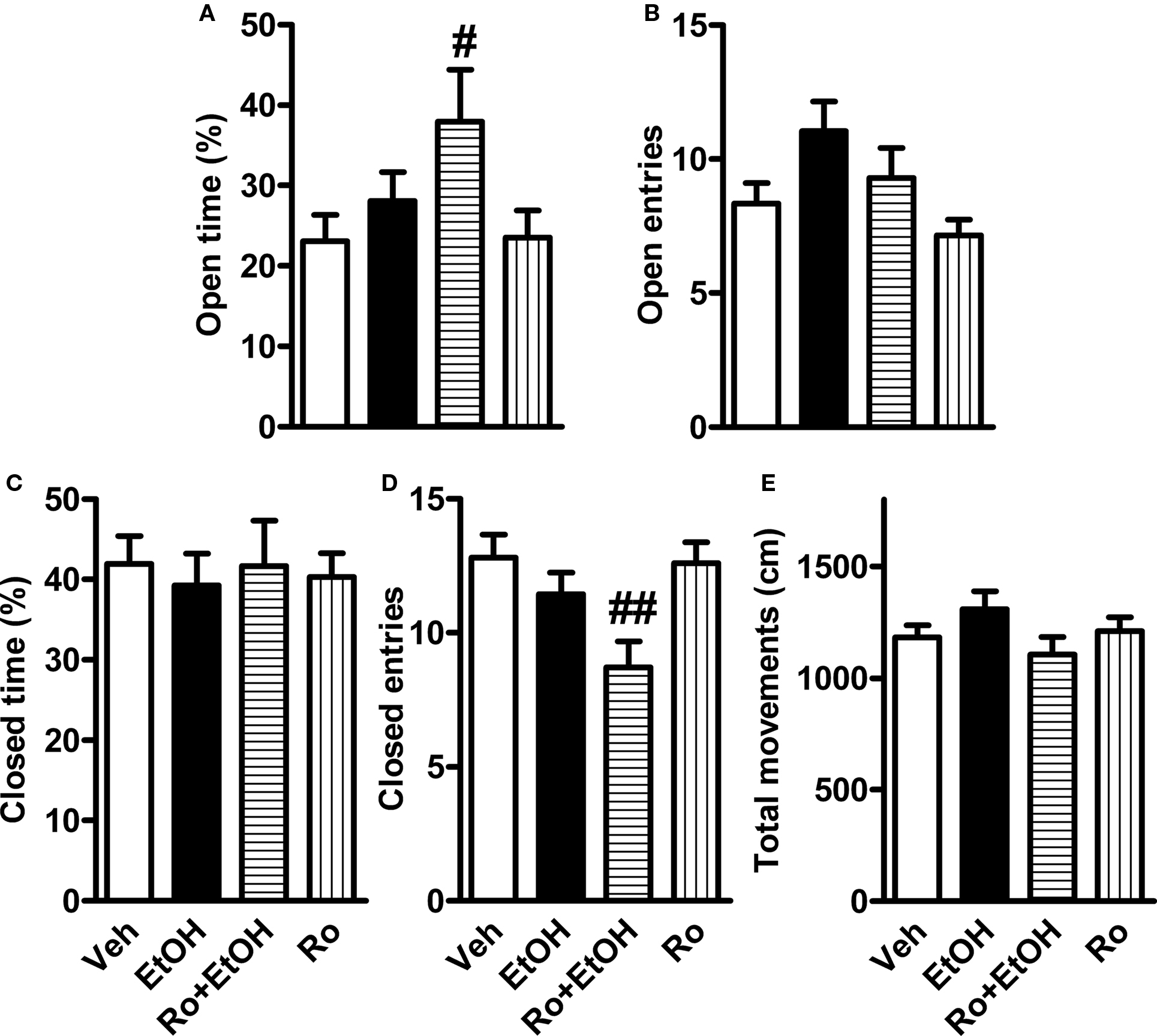
FIGURE 5. Effects of ethanol and its combination with Ro 15-4513 on elevated plus-maze behavior in γ2I77 knock-in mice. Male and female γ2I77 mice were co-administered with 1 g/kg ethanol (EtOH) and 3 mg/kg Ro 15-4513 (or vehicle, Veh) 10 min before being tested on the elevated plus-maze for 5 min. (A) The percent time spent in the open arms of the total testing time. (B) The number of open-arm entries. (C) The percent time spent in the closed arms of the total testing time. (D) The number of closed-arm entries. (E) Total movements. Because two-way ANOVA revealed no sex effect or interaction the data of males and females were pooled. #p < 0.05, ##p < 0.01 compared to vehicle-treated mice (one-way ANOVA followed by Student’s t-test). n = 24–26.
Elevated Plus-Maze Behavior after Ethanol and Ro 15-4513 Administration in the δ Subunit-Deficient Mice
Interactions of ethanol and Ro 15-4513 on elevated plus-maze behaviors were also analyzed in δ−/− and littermate δ+/+ mice (Figure 6). However, testing conditions differed considerably from those of γ2I77 mice (Figures 4 and 5) as explained in the Methods. Changes in experimental conditions probably explain the baseline differences in vehicle-treated wildtype mice in these tests performed in different laboratories. Ethanol (1 g/kg, i.p.) and Ro 15-4513 (3 mg/kg) were co-administered to male and female mice 10 min before the test. Three-way ANOVA revealed no sex effects in the behaviors, and, therefore, the male and female data were combined (Figure 6). The percent time spent on the open arms was not significantly affected by drug treatment or genotype, and no genotype × drug interaction emerged (two-way ANOVA; Figure 6A). However, the number of open-arm entries was significantly affected by drug treatment (F2,31 = 5.8, p < 0.01, two-way ANOVA). Differences between the treatment groups reached significance in post hoc comparisons in the wildtype mice only (Figure 6B). The percent time spent on the closed arms was significantly affected by drug treatment (F2,31 = 10.1, p < 0.01, two-way ANOVA), and the effect of ethanol was seen in both genotypes (Figure 6C). No sedation was observed as the amount of total movements was not significantly affected by drug treatments (Figure 6E).
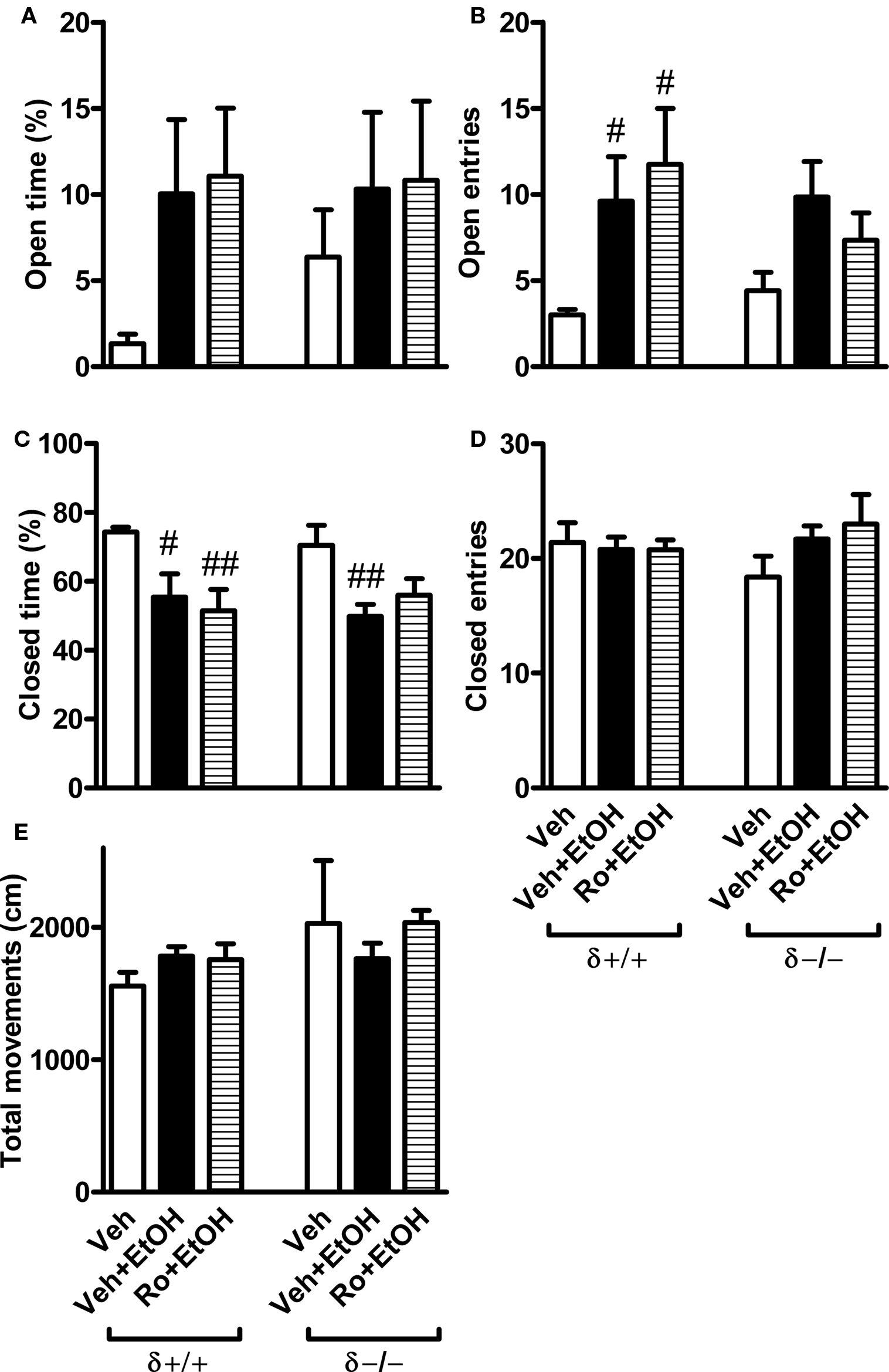
FIGURE 6. Effects of ethanol and its combination with Ro 15-4513 on elevated plus-maze behavior in δ+/+ and δ−/− mice. Male and female δ−/− and littermate wildtype δ+/+ mice were co-administered (i.p.) with 1 g/kg ethanol (EtOH) and 3 mg/kg Ro15-4513 (or vehicle, Veh) 10 min before being tested for anxiolytic-like behavior on an elevated plus-maze for 5 min. (A) The percent time spent in the open arms of the total testing time. (B) The number of open-arm entries. (C) The percent time spent in the closed arms of the total testing time. (D) The number of closed-arm entries. (E) Total movements. Because three-way ANOVA revealed no sex effect or interaction the data of males and females were pooled. #p < 0.05, ##p < 0.01 compared to vehicle-treated mice within the same genotype (one-way ANOVA followed by Student’s t-test). n = 4–7.
In order to analyze drug effects on open-arm behaviors we pooled the data of wildtype and knockout mice because no genotype effect emerged in two-way ANOVA. Open-arm time was not significantly affected. However, ethanol treatment increased significantly (p < 0.01, one-way ANOVA followed by Student’s t-test) the number of visits to the open arms compared to vehicle treatment (9.8 ± 1.5 vs. 3.7 ± 0.6, n = 10–12, respectively). The open-arm visits were also significantly (p < 0.01) increased in Ro 15-4513 + ethanol-treated mice (9.1 ± 1.7, n = 10).
Binding of [3H]Ro 15-4513 to Brain Sections from Wildtype and γ2I77 Mice: Effects of Ethanol
Horizontal brain sections from adult wildtype littermate γ2F77 animals showed a wide, but regionally varying distribution of [3H]Ro 15-4513 binding to flumazenil-sensitive benzodiazepine binding sites, in agreement with previous studies (Sieghart et al., 1987; Uusi-Oukari and Korpi, 1990; Turner et al., 1991; Figure 7A). The cerebral cortex and hippocampus had more than twofold more binding than the caudate–putamen, septal nuclei, and cerebellar granule cell layer (Table 1). In γ2I77 mouse brain sections, the binding was massively reduced throughout the brain, indicating a decreased affinity of [3H]Ro 15-4513 to point-mutated receptors (see Ogris et al., 2004 for pharmacological comparisons of wildtype and γ2I77 mutant brain membranes). Since we used [3H]Ro 15-4513 at the ligand concentration of 15 nM, which is more than twofold the Kd for native wildtype GABAA receptors (Uusi-Oukari and Korpi, 1990), the results should be indicative of the number of binding sites. Interestingly, clear residual binding was detectable in the septal nuclei and caudate–putamen (Figure 7A; Table 1), amounting to approximately 13% of the wildtype levels.
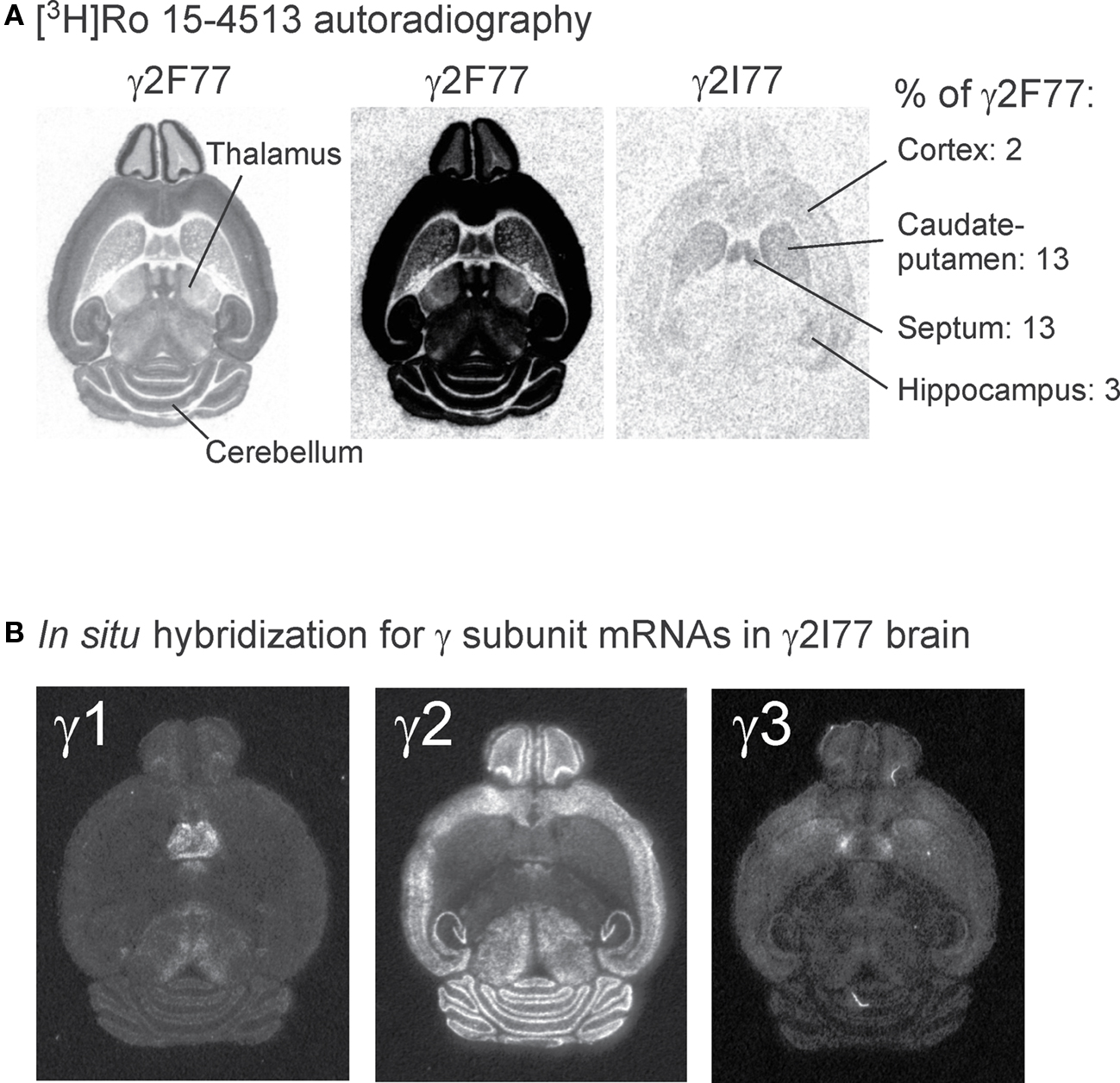
FIGURE 7. Residual benzodiazepine site binding of [3H]Ro 15-4513 to brain sections of γ2I77 knock-in mice as compared to the total binding in wildtype γ2F77 mice. (A) Total [3H]Ro 15-4513 binding to horizontal sections from adult wildtype γ2F77 and γ2I77 mice, showing on the left an optimized image for the γ2F77 section, on the right an optimized image for the residual binding to mutant mouse section, and in the middle the same γ2F77 image using the settings optimized for the mutant. The mean percentage values of the residual binding are given for those areas that show significant binding. (B) In situ hybridization signals for specific oligonucleotide probes for mRNAs of the three γ subunit genes in sections from adult γ2I77 mice.
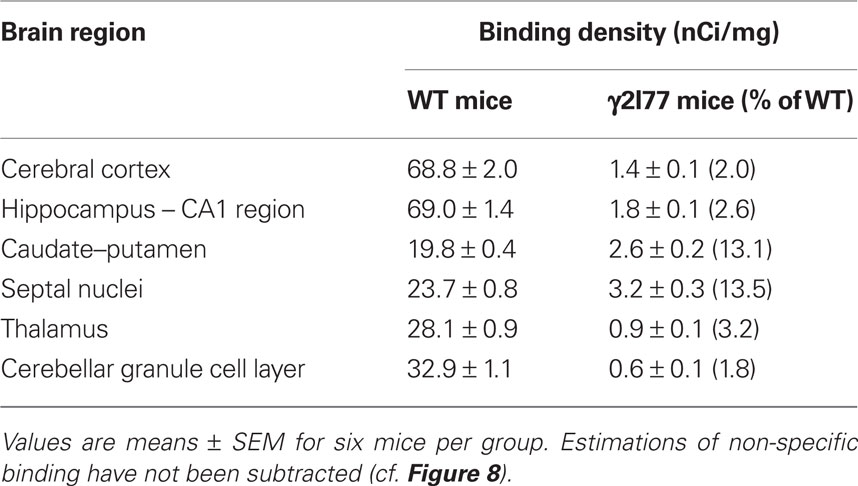
Table 1. Quantitative autoradiography of [3H]Ro 15-4513 binding to various brain regions from naïve γ2I77 and wildtype (WT) littermate γ2F77 mice.
Ethanol, at concentrations ranging from 1 to 100 mM only marginally, if at all, affected [3H]Ro 15-4513 binding (Figure 8) in wildtype and γ2I77 brain sections. The binding levels in the six brain regions analyzed (the same as in Table 1), were between 96–109% and 80–108% of the basal binding in the γ2F77 and γ2I77 brains, respectively (data not shown), thus contrasting with the proposed competitive inhibition of [3H]Ro 15-4513 binding by low millimolar concentrations of ethanol in native bovine GABAA receptors (Hanchar et al., 2006).
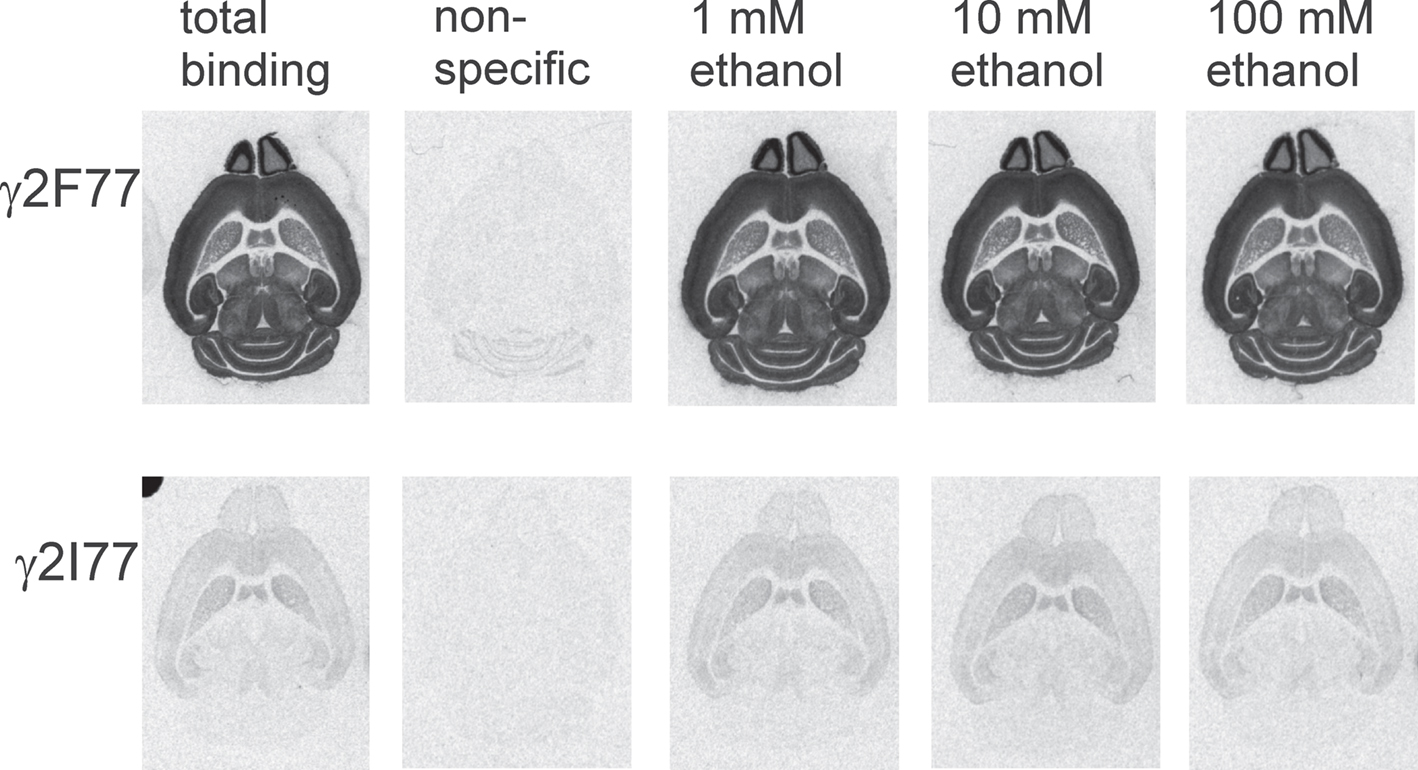
FIGURE 8. Effects of ethanol (1–100 mM) on [3H]Ro 15-4513 binding to horizontal sections from wildtype γ2F77 and γ2I77 knock-in mice. Representative images for both mouse lines have been prepared using identical imaging settings for contrast and brightness. No consistent inhibition of the binding by ethanol was observed. Non-specific binding in the presence of 10 μM flumazenil was negligible at the background level, except for the cerebellar granule cell layer of wildtype brains. In that area, it amounted to about 3% of total binding.
Figure 7B shows the expression patterns for the GABAA receptor γ1, γ2, and γ3 subunit genes in adult γ2I77 brain sections. Whereas the γ2 mRNA gene expression is the most widespread, the γ1 and γ3 genes do have discrete expressions in, for example, septal nuclei and the rostral caudate–putamen (γ3; Ymer et al., 1990; Herb et al., 1992; Wisden et al., 1992). Therefore, since the benzodiazepine receptor forms at the interface of the α and γ subunits (Sigel and Buhr, 1997), the residual flumazenil-sensitive [3H]Ro 15-4513 binding in γ2I77 mice probably originates from αβγ1 and/or αβγ3 subunit-containing GABAA receptors.
Discussion
Our main finding here was that Ro 15-4513 could reverse the sedative effect (i.e., the reduction of horizontal locomotor activity) produced by relatively low doses of ethanol (1.5–1.8 g/kg) in δ−/− knockout mice, but not in γ2I77 mice. Thus, the ability of Ro 15-4513 to antagonize this particular sedative effect of ethanol must depend on the intact benzodiazepine site formed by the γ2 subunit in αβγ2 receptors and only marginally or not at all on δ subunit-containing GABAA receptors.
The sedative ethanol doses we used produce moderate blood ethanol concentrations. For example, an ethanol dose of 1.6 g/kg (i.p.) in mice, produces approximately 40 and 35 mM blood ethanol concentrations at 5- and 15-min time points, respectively (Pastino et al., 1996), while 1 g/kg (i.p.) ethanol produces approximately 20 mM at the 20-min time point (Lallemand and de Witte, 2005). It could be argued that Ro 15-4513 only antagonizes very low ethanol concentrations, but it is difficult to test its efficacy in any behaving mouse model due to inconsistent behavioral effects of low ethanol doses. However, drugs that antagonize low alcohol doses are not needed in the clinic. In contrast, an efficient antagonist for alcohol poisoning (ethanol levels at or over 60–80 mM) is urgently needed, but this is an effect that Ro 15-4513 lacks (Nutt et al., 1988).
Our results with δ−/− mice indicate that δ subunit-containing GABAA receptors are dispensable for Ro 15-4513 to antagonize the light sedation produced by a 1.5-g/kg dose of ethanol. This is consistent with Ro 15-4513 shortening ethanol (3.5 g/kg)-induced loss of righting reflex in δ−/− mice similarly as for wildtype mice (Mihalek et al., 2001). Anxiolytic and hypnotic effects of ethanol were unaltered in δ−/− mice, whereas ethanol consumption, withdrawal responses, and anticonvulsant effects were affected (Mihalek et al., 2001).
We analyzed the effect of Ro 15-4513 on behaviors induced by a low dose of ethanol, and tested its effects on ethanol-induced anxiolytic-like behavior in the elevated plus-maze test. Ethanol alone at a dose of 1 g/kg produced a modest anxiolytic-like effect, i.e., open-arm activities of wildtype, γ2I77 as well as δ−/− mice in the elevated plus-maze test were slightly increased independently of the genotype. This anxiolytic effect was not antagonized by co-treatment with Ro 15-4513 in any of our experiments. In contrast to our results, there are reports demonstrating that Ro 15-4513 reverses anxiolytic effects of ethanol in several different anxiety tests including the elevated plus-maze test (Suzdak et al., 1986; Belzung et al., 1988; Misslin et al., 1988; Becker and Hale, 1991; Prunell et al., 1994). The lack of antagonism of ethanol’s anxiolytic-like action by Ro 15-4513 we observed may be related to the dosing regimen employed. Because we injected the drugs 10 min prior to testing, we may have analyzed the mice at a time point at which combining Ro 15-4513 and ethanol leads to some excitation. Indeed, Becker and Hale (1989) administered the drugs 5–6 min before testing and found that co-administration of ethanol and Ro 15-4513 increased locomotion although neither drug alone had any effect, and Weizman et al. (2001) have shown that Ro 15-4513 enhances the effects of low ethanol doses. Moreover, at the low doses of ethanol that elicit excitation, Ro 15-4513 has been shown to either potentiate (Becker and Hale, 1989; June and Lewis, 1989) or reverse (Lister, 1987a; Belzung et al., 1988; Misslin et al., 1988; June and Lewis, 1994) the effect depending on the experimental animals, conditions, and parameters measured.
We found inconsistent effects of Ro 15-4513 on rearing activity impaired by 1.8 g/kg dose of ethanol in different wildtype mice. Co-treatment with Ro 15-4513 partially reversed the reduction in rearing elicited by ethanol in C57BL/6J mice but not in the γ2F77 mice. The lack of effect in the latter mice may have been due to slightly stronger sedation in that particular experiment as indicated by more robust reduction in locomotor activity in γ2F77 mice (reduction of time moving on the average from 77 to 43%) compared to that in C57BL/6J mice (from 82 to 56%) after ethanol administration.
Finally, we showed that high-affinity binding of [3H]Ro 15-4513 requires the γ2 subunit as this binding was completely abolished in most forebrain regions (this study) and in the cerebellum (Korpi et al., 2007) of γ2I77 mice. Importantly, the residual binding detected in the γ2I77 brain sections was not localized in regions with high δ subunit expression levels such as the thalamus, dentate gyrus, or cerebellar granule cell layer (Wisden et al., 1992). Instead, the highest proportion of residual binding (approximately 13% of wildtype levels) was in the rostral caudate–putamen and septal nuclei suggesting that [3H]Ro 15-4513 binds to γ3 and/or γ1 subunit-containing receptors. It has to be noted that, while γ3 subunit-containing receptors bind [3H]Ro 15-4513 with high to moderate affinity (Hadingham et al., 1995; Lüddens and Korpi, 1995), γ1 subunit-containing receptors may not bind this ligand with high affinity at all (Benke et al., 1996) or have low efficacy (Wafford et al., 1993). The residual [3H]Ro 15-4513 binding was not displaced by ethanol ranging from 1 to 100 mM concentrations, which is in line with our earlier observations that [3H]Ro 15-4513 binding in the wildtype C57BL/6J mouse brain was unaffected by 1–100 mM ethanol (Korpi et al., 2007). Thus, we were unable to find evidence of γ2 subunit-independent, δ subunit-dependent Ro 15-4513/ethanol binding sites in the γ2I77 mouse brain, consistent with the lack of effect of ethanol on [3H]Ro 15-4513 binding to rat brain homogenates (Mehta et al., 2007). In addition, the δ and β3 subunit expression patterns are often mutually exclusive in areas such as many thalamic nuclei: instead the most frequent thalamic GABAA receptor is likely to be α4β2δ and/or α1α4β2γ2 (Wisden et al., 1991, 1992; Belelli et al., 2005). As the β3 subunit is claimed to be indispensable for the action of low concentrations of ethanol (Wallner et al., 2003), an in vivo effect at this site (via e.g., α4β3δ) seems unlikely.
In conclusion, our behavioral studies with δ−/− knockout mice and γ2I77 mice indicate that the antagonistic effect of Ro 15-4513 alcohol-induced sedation requires the classic and established γ2 benzodiazepine site: there, Ro 15-4513 exerts inverse agonistic actions leading to reversal of some behavioral effects induced by low/moderate ethanol concentrations. Competitive Ro 15-4513/ethanol binding sites could not be detected in native mouse brain.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors want to thank Chiara Procaccini for PCR genotyping, Katja Ojala for helping with behavioral tests on γ2I77 mice, Vera Marks for behavioral tests on C57BL/6J mice, and Cecile Malecot and Aira Säisä for carrying out autoradiography experiments. The study was supported by the Academy of Finland, the Sigrid Juselius Foundation, and the Finnish Foundation for Alcohol Studies (Esa R. Korpi).
References
Becker, H. C., and Hale, R. L. (1989). Ethanol-induced locomotor stimulation in C57BL/6 mice following RO15-4513 administration. Psychopharmacology (Berl.) 99, 333–336.
Becker, H. C., and Hale, R. L. (1991). RO15-4513 antagonizes the anxiolytic effects of ethanol in a nonshock conflict task at doses devoid of anxiogenic activity. Pharmacol. Biochem. Behav. 39, 803–807.
Belelli, D., Peden, D. R., Rosahl, T. W., Wafford, K. A., and Lambert, J. J. (2005). Extrasynaptic GABAA receptors of thalamocortical neurons: a molecular target for hypnotics. J. Neurosci. 25, 11513–11520.
Belzung, C., Misslin, R., and Vogel, E. (1988). Does RO 15-4513 reverse the anxiolytic effects of ethanol by its intrinsic properties? Pharmacol. Biochem. Behav. 30, 867–870.
Benke, D., Honer, M., Michel, C., and Mohler, H. (1996). GABAA receptor subtypes differentiated by their γ-subunit variants: prevalence, pharmacology and subunit architecture. Neuropharmacology 35, 1413–1423.
Bonetti, E. P., Burkard, W. P., Gabl, M., Hunkeler, W., Lorez, H. P., Martin, J. R., Moehler, H., Osterrieder, W., Pieri, L., Polc, P., Richards, J. G., Schaffner, R., Scherschlicht, R., Schoch, P., and Haefely, W. E. (1988). Ro 15-4513: partial inverse agonism at the BZR and interaction with ethanol. Pharmacol. Biochem. Behav. 31, 733–749.
Botta, P., Radcliffe, R. A., Carta, M., Mameli, M., Daly, E., Floyd, K. L., Deitrich, R. A., and Valenzuela, C. F. (2007). Modulation of GABAA receptors in cerebellar granule neurons by ethanol: a review of genetic and electrophysiological studies. Alcohol 41, 187–199.
Brickley, S. G., Revilla, V., Cull-Candy, S. G., Wisden, W., and Farrant, M. (2001). Adaptive regulation of neuronal excitability by a voltage-independent potassium conductance. Nature 409, 88–92.
Buhr, A., Baur, R., and Sigel, E. (1997). Subtle changes in residue 77 of the γ subunit of α1β2γ2 GABAA receptors drastically alter the affinity for ligands of the benzodiazepine binding site. J. Biol. Chem. 272, 11799–11804.
Causes of Death 2008. (2010). Statistics Finland, Health, Helsinki. Available at: http://www.stat.fi/til/kuol/index_en.html
Chandra, D., Halonen, L. M., Linden, A. M., Procaccini, C., Hellsten, K., Homanics, G. E., and Korpi, E. R. (2010). Prototypic GABAA receptor agonist muscimol acts preferentially through forebrain high-affinity binding sites. Neuropsychopharmacology 35, 999–1007.
Chandra, D., Jia, F., Liang, J., Peng, Z., Suryanarayanan, A., Werner, D. F., Spigelman, I., Houser, C. R., Olsen, R. W., Harrison, N. L., and Homanics, G. E. (2006). GABAA receptor α4 subunits mediate extrasynaptic inhibition in thalamus and dentate gyrus and the action of gaboxadol. Proc. Natl. Acad. Sci. U.S.A. 103, 15230–15235.
Cope, D. W., Halbsguth, C., Karayannis, T., Wulff, P., Ferraguti, F., Hoeger, H., Leppa, E., Linden, A. M., Oberto, A., Ogris, W., Korpi, E. R., Sieghart, W., Somogyi, P., Wisden, W., and Capogna, M. (2005). Loss of zolpidem efficacy in the hippocampus of mice with the GABAA receptor γ2 F77I point mutation. Eur. J. Neurosci. 21, 3002–3016.
Cope, D. W., Wulff, P., Oberto, A., Aller, M. I., Capogna, M., Ferraguti, F., Halbsguth, C., Hoeger, H., Jolin, H. E., Jones, A., McKenzie, A. N., Ogris, W., Poeltl, A., Sinkkonen, S. T., Vekovischeva, O. Y., Korpi, E. R., Sieghart, W., Sigel, E., Somogyi, P., and Wisden, W. (2004). Abolition of zolpidem sensitivity in mice with a point mutation in the GABAA receptor γ2 subunit. Neuropharmacology 47, 17–34.
Deitrich, R. A., Dunwiddie, T. V., Harris, R. A., and Erwin, V. G. (1989). Mechanism of action of ethanol: initial central nervous system actions. Pharmacol. Rev. 41, 489–537.
Hadingham, K. L., Garrett, E. M., Wafford, K. A., Bain, C., Heavens, R. P., Sirinathsinghji, D. J., and Whiting, P. J. (1996). Cloning of cDNAs encoding the human γ-aminobutyric acid type A receptor α6 subunit and characterization of the pharmacology of α6-containing receptors. Mol. Pharmacol. 49, 253–259.
Hadingham, K. L., Wafford, K. A., Thompson, S. A., Palmer, K. J., and Whiting, P. J. (1995). Expression and pharmacology of human GABAA receptors containing γ3 subunits. Eur. J. Pharmacol. 291, 301–309.
Hadingham, K. L., Wingrove, P., Le Bourdelles, B., Palmer, K. J., Ragan, C. I., and Whiting, P. J. (1993). Cloning of cDNA sequences encoding human α2 and α3 γ-aminobutyric acidA receptor subunits and characterization of the benzodiazepine pharmacology of recombinant α1-, α2-, α3-, and α5-containing human γ-aminobutyric acidA receptors. Mol. Pharmacol. 43, 970–975.
Hanchar, H. J., Chutsrinopkun, P., Meera, P., Supavilai, P., Sieghart, W., Wallner, M., and Olsen, R. W. (2006). Ethanol potently and competitively inhibits binding of the alcohol antagonist Ro15-4513 to α4/6β3δ GABAA receptors. Proc. Natl. Acad. Sci. U.S.A. 103, 8546–8551.
Hanchar, H. J., Dodson, P. D., Olsen, R. W., Otis, T. S., and Wallner, M. (2005). Alcohol-induced motor impairment caused by increased extrasynaptic GABAA receptor activity. Nat. Neurosci. 8, 339–345.
Herb, A., Wisden, W., Luddens, H., Puia, G., Vicini, S., and Seeburg, P. H. (1992). The third γ subunit of the γ-aminobutyric acid type A receptor family. Proc. Natl. Acad. Sci. U.S.A. 89, 1433–1437.
June, H. L., and Lewis, M. J. (1989). Ro15-4513 enhances and attenuates motor stimulant effects of ethanol in rats. Alcohol 6, 245–248.
June, H. L., and Lewis, M. J. (1994). Interactions of Ro15-4513, Ro15-1788 (flumazenil) and ethanol on measures of exploration and locomotion in rats. Psychopharmacology (Berl.) 116, 309–316.
Knoflach, F., Benke, D., Wang, Y., Scheurer, L., Luddens, H., Hamilton, B. J., Carter, D. B., Mohler, H., and Benson, J. A. (1996). Pharmacological modulation of the diazepam-insensitive recombinant γ-aminobutyric acidA receptors α4β2γ2 and α6β2γ2. Mol. Pharmacol. 50, 1253–1261.
Korpi, E. R., Debus, F., Linden, A. M., Malecot, C., Leppa, E., Vekovischeva, O., Rabe, H., Bohme, I., Aller, M. I., Wisden, W., and Luddens, H. (2007). Does ethanol act preferentially via selected brain GABAA receptor subtypes? The current evidence is ambiguous. Alcohol 41, 163–176.
Korpi, E. R., Grunder, G., and Luddens, H. (2002). Drug interactions at GABAA receptors. Prog. Neurobiol. 67, 113–159.
Korpi, E. R., Kleingoor, C., Kettenmann, H., and Seeburg, P. H. (1993). Benzodiazepine-induced motor impairment linked to point mutation in cerebellar GABAA receptor. Nature 361, 356–359.
Lallemand, F., and de Witte, P. (2005). Ethanol induces higher BEC in CB1 cannabinoid receptor knockout mice while decreasing ethanol preference. Alcohol Alcohol. 40, 54–62.
Leppa, E., Vekovischeva, O. Y., Linden, A. M., Wulff, P., Oberto, A., Wisden, W., and Korpi, E. R. (2005). Agonistic effects of the β-carboline DMCM revealed in GABAA receptor γ2 subunit F77I point-mutated mice. Neuropharmacology 48, 469–478.
Linden, A. M., Aller, M. I., Leppa, E., Vekovischeva, O., Aitta-Aho, T., Veale, E. L., Mathie, A., Rosenberg, P., Wisden, W., and Korpi, E. R. (2006). The in vivo contributions of TASK-1-containing channels to the actions of inhalation anesthetics, the α2 adrenergic sedative dexmedetomidine and cannabinoid agonists. J. Pharmacol. Exp. Ther. 317, 615–626.
Lister, R. G. (1987a). The benzodiazepine receptor inverse agonists FG 7142 and RO 15-4513 both reverse some of the behavioral effects of ethanol in a holeboard test. Life Sci. 41, 1481–1489.
Lister, R. G. (1987b). The use of a plus-maze to measure anxiety in the mouse. Psychopharmacology (Berl.) 92, 180–185.
Lüddens, H., and Korpi, E. R. (1995). GABA antagonists differentiate between recombinant GABAA/benzodiazepine receptor subtypes. J. Neurosci. 15, 6957–6962.
Luddens, H., and Wisden, W. (1991). Function and pharmacology of multiple GABAA receptor subunits. Trends Pharmacol. Sci. 12, 49–51.
Makela, R., Uusi-Oukari, M., Homanics, G. E., Quinlan, J. J., Firestone, L. L., Wisden, W., and Korpi, E. R. (1997). Cerebellar γ-aminobutyric acid type A receptors: pharmacological subtypes revealed by mutant mouse lines. Mol. Pharmacol. 52, 380–388.
Mehta, A. K., Marutha Ravindran, C. R., and Ticku, M. K. (2007). Low concentrations of ethanol do not affect radioligand binding to the δ-subunit-containing GABAA receptors in the rat brain. Brain Res. 1165, 15–20.
Mehta, A. K., and Ticku, M. K. (1988). Ethanol potentiation of GABAergic transmission in cultured spinal cord neurons involves γ-aminobutyric acidA-gated chloride channels. J. Pharmacol. Exp. Ther. 246, 558–564.
Mihalek, R. M., Banerjee, P. K., Korpi, E. R., Quinlan, J. J., Firestone, L. L., Mi, Z. P., Lagenaur, C., Tretter, V., Sieghart, W., Anagnostaras, S. G., Sage, J. R., Fanselow, M. S., Guidotti, A., Spigelman, I., Li, Z., DeLorey, T. M., Olsen, R. W., and Homanics, G. E. (1999). Attenuated sensitivity to neuroactive steroids in γ-aminobutyrate type A receptor δ subunit knockout mice. Proc. Natl. Acad. Sci. U.S.A. 96, 12905–12910.
Mihalek, R. M., Bowers, B. J., Wehner, J. M., Kralic, J. E., VanDoren, M. J., Morrow, A. L., and Homanics, G. E. (2001). GABAA-receptor δ subunit knockout mice have multiple defects in behavioral responses to ethanol. Alcohol. Clin. Exp. Res. 25, 1708–1718.
Misslin, R., Belzung, C., and Vogel, E. (1988). Interaction of RO 15-4513 and ethanol on the behaviour of mice: antagonistic or additive effects? Psychopharmacology (Berl.) 94, 392–396.
Nutt, D. J., King, L. A., and Phillips, L. D. (2010). Drug harms in the UK: a multicriteria decision analysis. Lancet 376, 1558–1565.
Nutt, D. J., Lister, R. G., Rusche, D., Bonetti, E. P., Reese, R. E., and Rufener, R. (1988). Ro 15-4513 does not protect rats against the lethal effects of ethanol. Eur. J. Pharmacol. 151, 127–129.
Ogris, W., Poltl, A., Hauer, B., Ernst, M., Oberto, A., Wulff, P., Hoger, H., Wisden, W., and Sieghart, W. (2004). Affinity of various benzodiazepine site ligands in mice with a point mutation in the GABAA receptor γ2 subunit. Biochem. Pharmacol. 68, 1621–1629.
Olesen, J., and Leonardi, M. (2003). The burden of brain diseases in Europe. Eur. J. Neurol. 10, 471–477.
Olsen, R. W., and Sieghart, W. (2008). International union of pharmacology. LXX. Subtypes of γ-aminobutyric acidA receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol. Rev. 60, 243–260.
Pastino, G. M., Sultatos, L. G., and Flynn, E. J. (1996). Development and application of a physiologically based pharmacokinetic model for ethanol in the mouse. Alcohol Alcohol. 31, 365–374.
Prunell, M., Escorihuela, R. M., Fernandez-Teruel, A., Nunez, J. F., and Tobena, A. (1994). Differential interactions between ethanol and Ro 15-4513 on two anxiety tests in rats. Pharmacol. Biochem. Behav. 47, 147–151.
Sarviharju, M., and Korpi, E. R. (1993). Ethanol sensitivity and consumption in F2 hybrid crosses of ANT and AT rats. Alcohol 10, 415–418.
Sieghart, W., Eichinger, A., Richards, J. G., and Mohler, H. (1987). Photoaffinity labeling of benzodiazepine receptor proteins with the partial inverse agonist [3H]Ro 15-4513: a biochemical and autoradiographic study. J. Neurochem. 48, 46–52.
Sigel, E., and Buhr, A. (1997). The benzodiazepine binding site of GABAA receptors. Trends Pharmacol. Sci. 18, 425–429.
Spanagel, R. (2009). Alcoholism: a systems approach from molecular physiology to addictive behavior. Physiol. Rev. 89, 649–705.
Suzdak, P. D., Glowa, J. R., Crawley, J. N., Schwartz, R. D., Skolnick, P., and Paul, S. M. (1986). A selective imidazobenzodiazepine antagonist of ethanol in the rat. Science 234, 1243–1247.
Suzdak, P. D., Paul, S. M., and Crawley, J. N. (1988). Effects of Ro15-4513 and other benzodiazepine receptor inverse agonists on alcohol-induced intoxication in the rat. J. Pharmacol. Exp. Ther. 245, 880–886.
Turner, D. M., Sapp, D. W., and Olsen, R. W. (1991). The benzodiazepine/alcohol antagonist Ro 15-4513: binding to a GABAA receptor subtype that is insensitive to diazepam. J. Pharmacol. Exp. Ther. 257, 1236–1242.
Uusi-Oukari, M., and Korpi, E. R. (1990). Diazepam sensitivity of the binding of an imidazobenzodiazepine, [3H]Ro 15-4513, in cerebellar membranes from two rat lines developed for high and low alcohol sensitivity. J. Neurochem. 54, 1980–1987.
Wafford, K. A., Bain, C. J., Whiting, P. J., and Kemp, J. A. (1993). Functional comparison of the role of γ subunits in recombinant human γ-aminobutyric acidA/benzodiazepine receptors. Mol. Pharmacol. 44, 437–442.
Wafford, K. A., Thompson, S. A., Thomas, D., Sikela, J., Wilcox, A. S., and Whiting, P. J. (1996). Functional characterization of human γ-aminobutyric acidA receptors containing the α4 subunit. Mol. Pharmacol. 50, 670–678.
Wallner, M., Hanchar, H. J., and Olsen, R. W. (2003). Ethanol enhances α4β3δ and α6β3δ γ-aminobutyric acid type A receptors at low concentrations known to affect humans. Proc. Natl. Acad. Sci. U.S.A. 100, 15218–15223.
Wallner, M., Hanchar, H. J., and Olsen, R. W. (2006). Low-dose alcohol actions on α4β3δ GABAA receptors are reversed by the behavioral alcohol antagonist Ro15-4513. Proc. Natl. Acad. Sci. U.S.A. 103, 8540–8545.
Weizman, R., Paz, L., Peter, Y., and Pick, C. G. (2001). Mice performance on the staircase test following acute ethanol administration. Pharmacol. Biochem. Behav. 68, 491–495.
Wingrove, P. B., Thompson, S. A., Wafford, K. A., and Whiting, P. J. (1997). Key amino acids in the γ subunit of the γ-aminobutyric acidA receptor that determine ligand binding and modulation at the benzodiazepine site. Mol. Pharmacol. 52, 874–881.
Wisden, W., Herb, A., Wieland, H., Keinänen, K., Lüddens, H., and Seeburg, P. H. (1991). Cloning, pharmacological characteristics and expression pattern of the rat GABAA receptor α4 subunit. FEBS Lett. 289, 227–230.
Wisden, W., Korpi, E. R., and Bahn, S. (1996). The cerebellum: a model system for studying GABAA receptor diversity. Neuropharmacology 35, 1139–1160.
Wisden, W., Laurie, D. J., Monyer, H., and Seeburg, P. H. (1992). The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. J. Neurosci. 12, 1040–1062.
Wisden, W., and Morris, B. J. (2002) In situ hybridization with oligonucleotide probes. Int. Rev. Neurobiol. 47, 3–59.
Keywords: ethanol, GABAA receptor, Ro 15-4513, alcohol antagonist, inverse agonist
Citation: Linden A-M, Schmitt U, Leppä E, Wulff P, Wisden W, Lüddens H and Korpi ER (2011) Ro 15-4513 antagonizes alcohol-induced sedation in mice through αβγ2-type GABAA receptors. Front. Neurosci. 5:3. doi: 10.3389/fnins.2011.00003
Received: 01 November 2010;
Accepted: 04 January 2011;
Published online: 20 January 2011.
Edited by:
A. Leslie Morrow, University of North Carolina School of Medicine, USAReviewed by:
Gregg E. Homanics, University of Pittsburgh, USARobert Adron Harris, University of Texas at Austin, USA
Copyright: © 2011 Linden, Schmitt, Leppä, Wulff, Wisden, Lüddens and Korpi. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Esa R. Korpi, Pharmacology, Institute of Biomedicine, University of Helsinki, POB 63 (Haartmaninkatu 8), FI-00014 Helsinki, Finland .e-mail:ZXNhLmtvcnBpQGhlbHNpbmtpLmZp; Hartmut Lüddens, Department of Psychiatry and Psychotherapy, University Medical Centre, Johannes Gutenberg University Mainz, Untere Zahlbacher Strasse 8, 55131 Mainz, Germany. e-mail:bHVlZGRlbnNAdW5pLW1haW56LmRl