
- Division of Gastroenterology and Hepatology, The University of Texas Medical Branch, Galveston, TX, USA
Nitric oxide (NO) is a functionally important neurotransmitter signaling molecule generated by mammalian and bacterial nitric oxide synthases (NOS), and by chemical conversion of dietary nitrite in the gastrointestinal (GI) tract. Neuronal NOS (nNOS) is the most abundant isoenzyme in the enteric nervous system, and targeted deletion in transgenic mice has clearly demonstrated its importance in normal gut function. Enteric neuropathy is also often associated with abnormal NO production, for example in achalasia and diabetic gastroparesis. Not surprisingly therefore, aberrant nNOS activity is widely implicated in enteric disease, and represents a potential molecular target for therapeutic intervention. One physiological signaling mechanism of NO bioactivity is through chemical reaction with the heme center of guanylyl cyclase, resulting in the conversion of cGMP from GTP. This second messenger nucleotide signal activates cGMP-dependent protein kinases, phosphodiesterases, and ion channels, and is implicated in the neuronal control of GI function. However, few studies in the GI tract have fully related NO bioactivity with specific molecular targets of NO-derived signals. In the central nervous system (CNS), it is now increasingly appreciated that NO bioactivity is often actively transduced via S-nitrosothiol (SNO) signals rather than via activation of guanylyl cyclase. Moreover, aberrant S-nitrosylation of specific molecular targets is implicated in CNS pathology. S-nitrosylation refers to the post-translational modification of a protein cysteine thiol by NO, forming an endogenous SNO. Because cysteine residues are often key regulators of protein function, S-nitrosylation represents a physiologically important signaling mechanism analogous to other post-translational modifications, such as O-phosphorylation. This article provides an overview of how neurotransmitter NO is produced by nNOS as this represents the most prominent and well defined source of SNO production in the enteric nervous system. Further, it provides a perspective of how S-nitrosylation signals derived from multiple diverse sources may potentially transduce NO bioactivity in the GI tract. Possible lessons that might be learnt from the CNS, such as SNO mediated auto-inhibition of nNOS activity and modulation of neuronal cell death, are also explored as these may have pathophysiological relevance in enteric neuropathy. Thus, S-nitrosylation may mediate previously underappreciated NO-derived signals in the enteric nervous system that regulate homeostatic gut functions and disease susceptibility.
Nitric oxide (NO)-related biology was first described in the 1980s with the demonstration of free NO gas release during cellular metabolism (Murad, 1986; Ignarro et al., 1987). Following these initial reports, NO has become one of the most intensely studied cellular signaling molecules with more than a 100,000 related publications reported to date. NO is a highly reactive free radical gas with diverse physiological functions that are mediated via intracellular and intercellular communications (Ignarro, 1996; Andrew and Mayer, 1999; Li and Poulos, 2005). Numerous disease processes are also associated with pathological NO production (Foster et al., 2009). One important cellular source of NO is from L-arginine synthesis by P-450-like nitric oxide synthases (NOS), in the presence of oxygen and nicotinamide adenine dinucleotide phosphate (NADPH) co-substrates (Moncada and Higgs, 1993). Several genetically distinct NOS isoenzymes exist and are designated as neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS), respectively. These proteins are expressed at various levels in neurons and glia of the central and enteric nervous systems. However, nNOS is the predominant physiological isoenzyme in these tissues (Jaffrey et al., 2001) and forms an active homodimer to generate free NO by a process that is regulated by several cofactors including heme, flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), and tetrahydrobiopterin (Wang et al., 1999; Gangula et al., 2007; Foster et al., 2009).
In the enteric nervous system, nNOS-derived NO is an important neurotransmitter regulator of motility, vascular tone, blood supply, mucosal secretion, and barrier function (Bornstein et al., 2010). An emerging concept in the NO signaling field is that second messenger S-nitrosothiols (SNOs) play a crucial regulatory role in this cellular signal transduction process (Benhar et al., 2009). Preliminary studies of nNOS signaling in the enteric nervous system support this view (Vanden Berghe, 2008), and possibly implicate S-nitrosylation as a major signaling mechanism of NO neurotransmitter activity in the gastrointestinal (GI) tract. Consequently, this article will focus on nNOS expression and activity as a cellular source of SNO in the enteric nervous system, and how these NO-derived signals are altered in disease states.
Regulation of nNOS Activity as a Cellular Source of SNO in the Enteric Nervous System
Neuronal NOS is abundantly expressed throughout the enteric nervous system where it represents the predominant source of NO neurotransmitter (Takahashi, 2003; Van Geldre and Lefebvre, 2004; Bornstein et al., 2010). Within the myenteric plexus, nNOS activity is also associated with a significant production of cellular SNO (Figure 1). Therefore, the regulated activity of nNOS likely plays an important functional role in the GI tract, as is indicated by circadian correlates in nNOS expression, NO production and rhythmic changes in colonic motility (Hoogerwerf, 2010). Several functionally distinct 5′ mRNA nNOS splice variants have been identified, reflecting differential expression from alternative promoters. Because of this differential first exon usage and alternative splicing, nNOS is classified as a complex gene (Boissel et al., 2003; Newton et al., 2003; Secondo et al., 2003). Specific splice variants encode for distinct nNOSα, nNOSβ, or nNOSγ transcripts (Figure 2), which show differences in developmental and cephalocaudal expression in the enteric nervous system (Huber et al., 1998; Saur et al., 2000; Vannucchi et al., 2002). For example, all nNOS splice variants are expressed in the human colon, whereas the nNOSγ isoform is not detected in the stomach or small intestine (Stricker et al., 1997; Saur et al., 2002).
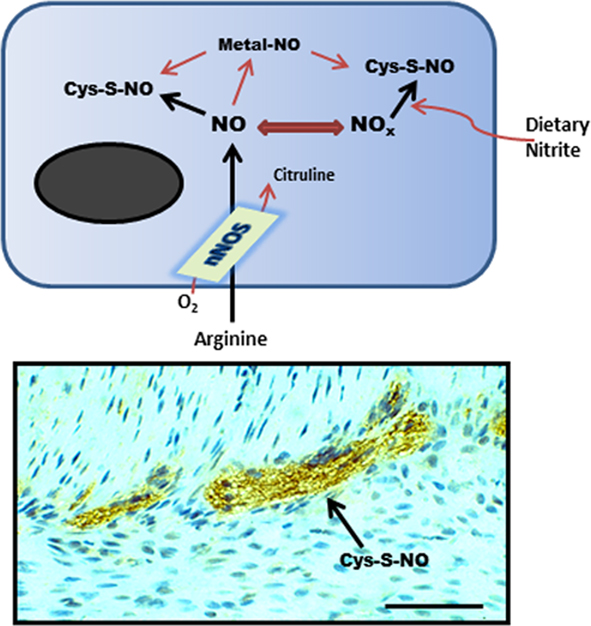
Figure 1. Schematic representation (top) of nNOS activity generating SNO from L-arginine or from exogenous nitrite. This synthesis step may involve direct protein–protein interactions with nNOS or another SNO-synthase, transfer of a coordinated NO from a transition metal center, or by chemical reaction with a reactive nitrosium ion intermediate. (Bottom) shows anti-SNO immunoreactivity in the mouse small intestinal myenteric plexus (scale bar = 40 μm).
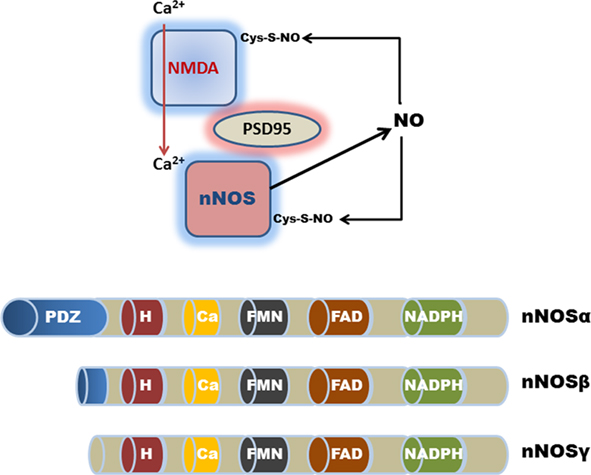
Figure 2. Schematic representation (top) of nNOSα PDZ domain-interactions with PSD-95 and the NMDA receptor in the CNS. Calcium ion entry activates nNOS production of NO. Excessive NO production results in the auto-S-nitrosylation and inhibition of nNOS and the NMDA receptor. Although auto-S-nitrosylation of nNOS represents a likely inhibitory mechanism in the GI tract, a physiological role for NMDA receptor signaling remains controversial. (Bottom) Shows the various nNOS protein isoforms; nNOSα contains a NH2-terminus PDZ/GLGF-domain (∼100 amino acids), that modulates interactions with other PDZ-containing proteins. nNOSβ and nNOSγ lack a PDZ domain; H, heme consensus site; Ca, calmodulin consensus site; FMN, flavin mononucleotide consensus site; and FAD, flavin adenine dinucleotide consensus site.
Transcriptional Regulation of nNOS
The genomic organization of nNOS is known and although canonical TATA and CCAAT boxes are absent in proximal regions upstream of the transcription start sites, the promoter region contains several putative transcription factor binding sites for Sp-1, AP-1, NFκB, E-box, and half sites of glucocorticoid response elements (Jeong et al., 2000; Molero et al., 2002; Garban et al., 2005). Although the absence of a TATA box does not necessarily affect transcription rates, it can result in promiscuous initiation of transcription by RNA polymerase II, resulting in multiple initiation sites. In several eukaryotic genes, it has been observed that promoters lacking a TATA box are recognized by Sp-1. In this regard, because the minimal promoter of nNOS exon 1c is GC-rich, TATA-less, and Sp-regulated, it resembles those for other constitutively expressed genes (including eNOS), where promoter regulation is poorly understood (Böhm et al., 1995; Wu, 2002). In addition, it has been shown that Sp-1 actively interacts with multiple components of the transcriptional machinery in GC-rich promoter regions that lack a canonical TATA box (Cook et al., 1999). Therefore Sp-1 plays a critical role in the assembly of the transcription initiation complex and although not yet demonstrated, it is also feasible that nNOS is transcriptionally regulated (possibly repressed) by SNO, because this is a major regulator of Sp-1 activity (Zaman et al., 2004; see nNOS-Derived SNO and Intestinal Disease). It is likely that the presence of multiple alternative promoters permits cell-, tissue-, and site-specific transcriptional regulation of nNOS activity under different physiological and pathological states.
nNOS Splice Variants are Functionally Distinct
Functionally, the nNOSα protein isoform is unique in that it contains a specific N-terminus PDZ domain, which is important for protein–protein interactions that are required for homodimerization into the active configuration (Brenman et al., 1996; Hashida-Okumura et al., 1999; Riefler and Firestein, 2001; Chanrion et al., 2007; Figure 2). By contrast, both nNOSβ and nNOSγ are N-terminus truncated proteins, and thus lack these protein–protein interaction domains. A recent study has highlighted the functional importance of this PDZ domain interaction in regulating nNOS activity in the stomach (Gangula et al., 2007). Reduced nNOSα dimerization, but not total nNOS expression, significantly altered gastric antrum nitrergic relaxation and solid gastric emptying resulting in gastroparesis. The lack of a dimerization domain in nNOSβ and nNOSγ suggests that these splice variants play different physiological roles to nNOSα (Figure 2). The catalytic activity of nNOSβ is approximately 80% of nNOSα, whereas nNOSγ is functionally inert and, as such, may function as a dominant-negative nNOS variant in the human colon (Brenman et al., 1996; Gangula et al., 2007). For example, nNOSα, but not nNOSβ appears to regulate pyloric sphincter relaxation and gastric stasis with delayed gastric emptying (Gangula et al., 2007). This view is supported by findings in nNOSα mutant mice where animals display a disease phenotype that closely resembles hypertrophic pyloric stenosis, with delayed gastric emptying of solids and fluids (Huang et al., 1993; Mashimo et al., 2000). Because of the continued expression of the alternative nNOSβ splice variant in nNOSα mutant mice, the disease pathology in other GI regions is less severe when compared for example with targeted deletion of NO-sensitive guanylyl cyclase in mice (Friebe et al., 2007). Differences in the N-terminus protein structure are therefore likely to confer functional distinctions between nNOSα, nNOSβ, and nNOSγ variants.
In the central nervous system (CNS), the PDZ domain in nNOSα also interacts with other functionally important regulatory proteins, such as the postsynaptic density protein PSD-95 which promotes cellular calcium ion entry by binding to the glutamate-activated N-methyl-D-aspartic acid (NMDA) receptor (Figure 2). nNOS (like eNOS) activity is promoted by elevated intracellular calcium ion concentrations. The functional regulation of nNOSα is complex as the N-terminus region also possesses a binding domain for nNOS protein inhibitor (PIN) and nitric oxide synthase interacting protein (NOSIP; Stricker et al., 1997). Moreover, these protein–protein interactions with nNOS are negatively regulated by auto-S-nitrosylation as a form of feed-back inhibition (Jaffrey et al., 2001; Benhar et al., 2009; Figure 2; see SNO Effector Signals in the Enteric Nervous System).
SNO Effector Signals in the Enteric Nervous System
The previous section detailed nNOS as a major (but not exclusive) enzymatic source for the generation of cellular SNO in the enteric nervous system. This section considers whether SNO may extend or potentiate nitrergic neurotransmission in the GI tract through the process of transnitrosylation. Endogenous SNO moieties are formed when NO reacts with the sulfur atom of cysteine residues on proteins or peptides resulting in an S–NO bond (Hogg, 2002; Gaston et al., 2003; Figure 1). S-nitrosylation may also occur following transfer of an NO moiety from cysteine-to-cysteine residues in a process known as transnitrosylation. S-nitrosylation is now known to post-translationally modify several hundreds of different cellular proteins (Stamler and Hess, 2010), and can regulate virtually all known cellular signaling cascades, ranging from inhibitory protein–DNA interactions (Li et al., 2007), to G-protein coupled receptor activation (Chvanov et al., 2006), and ion channel modulation (Foster et al., 2009). Small peptide SNOs, notably S-nitrosoglutathione (GSNO) is a potent endogenous transnitrosylating species, and preliminary studies in the GI tract have indicated that exogenous GSNO provides novel disease-attenuating signals in fulminant jejuno-ileitis resulting from functional ablation of the enteric nervous system (Savidge et al., 2007), burn-induced gut trauma (Costantini et al., 2010), and shigellosis (Flamant et al., 2010).
S-Nitrosylation Motif
Protein S-nitrosylation is not a random event, but is most often governed by an exposed consensus motif that encompasses the cysteine residue targeted for posttranslational modification. Mechanistically, this S-nitrosylation motif was initially thought to promote cysteine thiolate nucleophilicity to NO reactivity (Stamler et al., 1997), but it has more recently been demonstrated that the motif actively promotes catalytic protein–protein interactions in a manner similar to the reaction between phosphokinase enzymes and their protein target substrates (Doullas et al., 2010; Kornberg et al., 2010; Liu et al., 2010; Marino and Gladyshev, 2010; Stamler and Hess, 2010). This makes sense in light that NO is by itself a poor S-nitrosylating agent, although this reaction is favored following an electrophilic attack of a nitrosonium ion equivalent (NO+) on the thiolate anion (Figure 1). It is currently proposed that SNO– protein intermediates generated as a result of NOS activity may also function as specific SNO-synthases in cellular compartments that normally lack NOS activity, for example glyceraldehyde 3-phosphate dehydrogenase (GAPDH)–SNO in the nucleus specifically transnitrosylates deacetylating enzyme sirtuin-1 (SIRT-1), histone deacetylase-2 (HDAC2), and DNA-activated protein kinase (DNA-PK; Kornberg et al., 2010; Stamler and Hess, 2010). Because the SNO-synthases appear to target multiple protein substrates, intensive efforts are currently underway to fully characterize the complete SNO-synthase network, and this may now easily exceed the known repertoire of phosphokinases. Denitrosylating enzyme activities in the form of thioredoxin, Cu2+, Zn2+ superoxide dismutase, GSNO-reductase, and carbonyl reductase (Benhar et al., 2009; Staab et al., 2011) are more clearly defined, and it is perhaps not a coincidence that aberrant regulation of these enzymatic pathways is often associated with disease induction.
Structurally, the S-nitrosylation motif is encoded either into the primary amino acid sequence of a protein, or it may forma more integral part of the tertiary structure. For example, the ryanodine response calcium channel of skeletal muscle has many reduced cysteines, yet only one cysteine (Cys3635) forming part of an S-nitrosylation motif is selectively modified by calmodulin-dependent NO-mediated modulation of channel activity (Xu et al., 1998). The tertiary S-nitrosylation motif is often also allosterically regulated, thereby conferring an additional level of physiological control, for example via redox or ion-dependent reactions that modify the protein structure. An example is the S-nitrosylation of cysteine β93 on hemoglobin, which is allosterically regulated by oxygen tension, i.e., R versus T states. This allosteric mechanism facilitates carriage of vasodilatory NO to capillaries in oxygen deprived tissues (Foster et al., 2009). Our laboratory has recently demonstrated that allosteric S-nitrosylation reactions may also regulate toxin-induced neurogenic inflammation in the colon (unpublished data). As a specific example, Clostridium difficile toxins have evolved an elegant virulence mechanism to enter host cells by exploiting the allosteric cytosolic cofactor inositol hexakisphosphate (InsP6; Reineke et al., 2007). To combat this virulence mechanism, inflammation-induced SNO functions as a novel host defense mechanism by competitively inactivating a critical catalytic cysteine on the toxin during allosteric coupling by InsP6. Thus, elevated NOS activity in response to neurogenic inflammation promotes cellular SNO accumulation within the highly lipophilic plasma membrane microenvironment that subsequently inhibits toxin entry.
Location, Location, Location….also Applies to SNO-Regulation
Intracellular compartmentalization of target proteins is another important regulatory mechanism for S-nitrosylation (Iwakiri et al., 2006). For example, the mitochondrial microenvironment is enriched with inorganic electron acceptors, such as NAD+ and cytochrome c, which promote SNO formation and play an important regulatory role in caspase activation during apoptosis. Inactive pro-caspase 3 remains constitutively S-nitrosylated in the inner mitochondrial membrane. Once this protein is shuttled into the highly reducing microenvironment of the cytosol, it is rapidly denitrosylated into an active form (Rössig et al., 1999; Mannick et al., 2001). Therefore, reductive and enzymatic denitrosylation represent important physiological steps in NO signal transduction since the SNO-bond is otherwise a reasonably stable modification [Benhar et al., 2008; a theoretical half-life of seconds to years is evident for homolytic bond dissociation energies (22–32 kcal) for the S–N bond in SNOs]. However, not all proteins are rapidly denitrosylated in the cytosolic microenvironment, especially when the modified cysteine residue is located within a hydrophobic pocket. For example, extracellular signal-regulated kinase (ERK) remains S-nitrosylated even after prolonged exposure to powerful reducing agents (Paige et al., 2008).
SNO Modulation of Neuronal Plasticity and Signaling
A recent study has demonstrated that endogenous NO regulates inhibitory synaptic transmission and neuronal excitability in the ileal submucosal plexus (Bornstein et al., 2010). A significant component of this NO bioactivity appears to be cGMP-independent (J. Bornstein, personal communication), implicating S-nitrosylation as a potential postsynaptic regulatory mechanism of inhibitory synapticpotentials (IPSPs), slow excitatory synaptic potentials (EPSPs) and action potential firing in submucosal neurons. In the CNS, S-nitrosylation is known to regulate neuronal plasticity and signaling by directly modulating protein–protein interactions and neurotransmitter release, for example by S-nitrosylating actin (Lu et al., 2009). Neuronal plasticity is also modulated by SNO-regulation of membrane receptor and ion channel activity, such as the surface expression of glutamate-stimulated AMPA and NMDA receptors (Selvakumar et al., 2009). Because NMDA receptor expression has been identified on both CNS and enteric neurons (Liu et al., 1997) it is feasible that this constitutes a common regulatory mechanism for nNOS activity and neuronal plasticity, although this remains a contentious issue in the enteric nervous system (Ren et al., 2000). Notably, NMDA receptor stimulation of nNOS in the CNS results in S-nitrosylation of stargazin, a physiological regulator of AMPA receptor expression on the surface of neurons (Selvakumar et al., 2009). S-nitrosylation agents are also known to inhibit the activity of the epidermal growth factor receptor (EGFR), as well as several G-protein-coupled receptors, including the serotonin 5-HT2 receptor (Foster et al., 2009). Moreover, the activity of several voltage-gated, cation-activated, and receptor-coupled ion channels have been shown to be modulated by endogenous SNOs, although the regulatory cysteine residues that are functionally important in altering many of these channel activities remain to be fully characterized (Sun et al., 2006; Whalen et al., 2007). Neuromodulation by SNO’s may also target neuropeptides. For example, S-nitrosylation of vasointestinal peptide (VIP) augments the relaxations it evokes in smooth muscle cells (Jia and Stamler, 1999).
Target protein interaction with NOS or other SNO-synthases represents another important regulatory mechanism, for example activation of cyclooxygenase-2 is facilitated by the binding and subsequent S-nitrosylation by iNOS (Kim et al., 2005). In regard to neuronal NO signaling, PDZ domain-interactions appear to play a particularly important role in self-limiting nNOS enzymatic activity. Excessive NO production leads to auto-S-nitrosylation of nNOS, causing down-regulation of its enzymatic activity. S-nitrosylation of the NMDA receptor binding partner also occurs as a means of self-regulation in the CNS (Figure 2). Moreover, the neuronal NMDA receptor mediates the postsynaptic Ca2+ flux which is a regulatory requirement for nNOS activity (Selvakumar et al., 2009). The NMDA receptor is in turn S-nitrosylated on a single critical cysteine (Cys399) on the NR2A subunit, which inhibits the regulatory Ca2+ flux. Mechanistically, S-nitrosylation of the NMDA receptor by nNOS appears to inhibit its channel function by allosterically sensitizing the receptor complex to Zn2+ inhibitory modulators. In summary, it is possible that S-nitrosylation plays an important and largely unexplored role in transducing NO-derived signals in the GI tract, as has become increasingly evident in the CNS and other organs (Foster et al., 2009; Stamler and Hess, 2010).
nNOS-Derived SNO and Intestinal Disease
The previous sections have detailed nNOS activity as a major enzymatic source of NO and SNO in the enteric nervous system. Several reports have directly implicated aberrant NO activity in modulating GI disease (especially in motility disorders), although it is still not clearly understood how nNOS dysfunction contributes to the pathology (Takahashi, 2003; Pasricha et al., 2008; Gangula et al., 2010a,b). Because a precedent already exists for aberrant SNO signaling in CNS (Schonhoff et al., 2006), respiratory (Que et al., 2005), and cardiovascular disease (Lima et al., 2009), this section will consider abnormal S-nitrosylation reactions in gut pathophysiology.
Gut motility is controlled by contractile cholinergic and relaxant non-adrenergic non-cholinergic (NANC) neurons in the myenteric plexus (Lecci et al., 2002). nNOS-generated NO is a primary inhibitory NANC neurotransmitter, and aberrant NANC relaxation is implicated in the pathophysiology of functional GI motility disorders (Takahashi, 2003; Pasricha et al., 2008; Gangula et al., 2010a,b). The functional importance of NO signaling in gut motility has been clearly demonstrated following genetic ablation of NO-sensitive guanylyl cyclase in mice (Friebe et al., 2007). A subgroup of these transgenic pups develops fatal GI obstruction due to a grossly enlarged and dilated cecum. Thus, it seems plausible that aberrant nNOS activity could contribute to GI pathology, although other important transmitter molecules are also likely to orchestrate NANC inhibitory signals (Lecci et al., 2002; Vanden Berghe, 2008).
Aberrant nNOS Expression and Intestinal Pathology
Several transcription factors required for nNOS expression, notably Sp-1, NFκB, and glucocorticoid response elements, are also important regulators of intestinal disease and of the stress response. Not surprisingly therefore, aberrant nNOS expression is evident in several important clinical conditions, including achalasia (Mearin et al., 2006; Zarate et al., 2006) and diabetic gastroparesis (Watkins et al., 2000; Cellek et al., 2003; Surendran and Kondapaka, 2005; Iwasaki et al., 2006; Shotton et al., 2006). Altered NO signaling is also present in patients with functional dyspepsia, although it is unclear whether this is due to a reduction in NO release or nNOS expression in the gastric myenteric plexus (Tack et al., 2002; Walecka-Kapica et al., 2007). A deficiency in nNOS innervation may contribute to the pathogenesis of aperistalsis of the esophagus in patients with congenital esophageal stenosis. Infantile hypertrophic pyloric stenosis is also associated with decreased nNOS expression in the hypertrophied circular muscle sphincter (Takahashi, 2003). In addition, during pregnancy delayed gastric emptying may in part be due to abnormal NO activity (Shah et al., 2001). A pathogenic role has also been suggested in aging, where nNOS expression and activity decreases in the large intestine (Gabella, 1989). However, although aberrant nNOS activity is widely implicated in several important gut diseases, virtually nothing is known about whether this translates into altered SNO signals.
SNO Signals and S-Nitrosylation of Molecular Disease Targets
There is now an increasing appreciation that altered SNO homeostasis resulting in aberrant S-nitrosylation of regulatory proteins is sufficient to contribute to disease severity (Nakamura and Lipton, 2008; Stamler and Hess, 2010). For example, S-nitrosylation of Cys644 on dynamin-related protein 1 causes significant neuronal loss in Alzheimer’s disease (Cho et al., 2009). GAPDH (Hara et al., 2005), Parkin (Chung et al., 2004), peroxiredoxin-2 (Fang et al., 2007), and protein disulfide isomerase (Uehara et al., 2006) are other regulatory proteins that are aberrantly S-nitrosylated in Parkinson’s and Alzheimer’s disease. In amyotrophic lateral sclerosis, altered denitrosylating activity of mutant SOD-1 is implicated in the disease process (Johnson et al., 2001; Schonhoff et al., 2006). However, virtually nothing is known about endogenous SNO signals in the GI tract and whether altered SNO homeostasis may contribute to disease pathogenesis.
In the CNS, diminished SNO is evident in nNOSα deficient mice (Jaffrey et al., 2001). Using the biotin-switch assay as the gold standard method for identifying protein–SNO (Forrester et al., 2009), and by quantification of cysteine S-nitrosylation using fluorescence in a novel unbiased proteomic approach (SNOFlo) for characterizing the entire S-nitrosoproteome, we have demonstrated that SNO homeostasis is also altered in the gastric antrum of nNOSα deficient mice, with different protein populations being targeted for S-nitrosylation (unpublished findings). However, as significant quantities of protein–SNO are still measurable in the stomach of nNOSα deficient mice, regulation of the SNO pool appears to be more complex in the GI tract. This apparent disparity between nNOS activity and SNO accumulation may be due, in part, to bioactive SNO being generated and stored as stable intermediates following dietary nitrite conversion in the gut lumen (Suscheck et al., 2006; Figure 1). An estimated 20% of the total body NO production may occur from dietary nitrite reaction with gastric acid in the stomach Kuhnle et al., 2007; Milkowski et al., 2010), and the resulting stable SNO pool is sufficiently large to induce smooth muscle relaxation in the gastric fundus (Ogulener and Ergun, 2002). Bioactive SNO may also be generated from foods that are rich in heme-containing red meat which contains significant SNO-synthase activity. Dietary SNO has been implicated in colorectal cancer progression (Milkowski et al., 2010), but potentially may also play a significant role in regulating nNOS activity and neuronal cell death in the GI tract.
Diabetic autonomic neuropathy represents one interesting disease showcase because nNOS deficiency in diabetic gastropathy would be expected to slow gastric emptying, yet paradoxically this may also promote greater conversion of dietary nitrite into SNO. Diabetic gastropathy is thought to results from an impairment of nitrergic neurons that undergo a progressive loss of nNOS activity, followed by a degenerative process (Watkins et al., 2000; Cellek et al., 2003; Surendran and Kondapaka, 2005; Iwasaki et al., 2006; Shotton et al., 2006; Pasricha et al., 2008; Gangula et al., 2010a,b). In light of the evidence that elevated S-nitrosylation can impair insulin receptor β and insulin receptor substrate-1 signaling in diabetes (Carvalho-Filho et al., 2005; Kaneki et al., 2007), it is feasible that SNO-induced neurotoxicity may also contribute to neuronal degeneration. Mechanistically, progressive changes could occur at several levels: (i) by modulating nNOS gene expression, (ii) by auto-inhibiting nNOS activity, and (iii) by promoting preferential apoptosis of nitrergic neurons.
Transcriptional modulation by Sp-1 is known to be regulated by the endogenous S-nitrosylation agent GSNO (Zaman et al., 2004). Moderate GSNO concentrations augment Sp-1 and Sp-3 binding to GC-rich promoter Sp-binding sites, as described for nNOS in Section “Regulation of nNOS Activity as a Cellular Source of SNO in the Enteric Nervous System.” Further, transcriptional regulation by GSNO is not mediated by guanylyl cyclase activation, but appears to result from S-nitrosylation of cysteines in the zinc-finger binding domain of Sp-1 as it is fully reversible by reducing agents and by inhibition of cellular GSNO uptake. By contrast, excessive GSNO concentrations can inhibit this transcriptional activity, and it is possible that nNOS expression is similarly attenuated by excessive GSNO production. Elevated SNO signals may also inhibit nNOS activity by post-translational auto-inhibition, as described for CNS neurons in Section “SNO Effector Signals in the Enteric Nervous System,” or by desensitization of cGMP receptor signaling cascades (Mayer et al., 2009).
Several recent studies have described various SNO-dependent mechanisms that modulate neuronal susceptibility to cell death (He et al., 2007; Nakamura and Lipton, 2008). For example, excessive nNOS activation and intracellular SNO accumulation increases the susceptibility of neuronal cells to NO induced apoptosis by S-nitrosylation of housekeeping proteins, such as GAPDH or β-actin. GAPDH–SNO interacts with the E3 ubiquitin ligase, Siah, resulting in nuclear translocation where this SNO-synthase promotes cell death in part by transnitrosylating deacetylating enzyme SIRT-1, HDAC2, and DNA-PK (Hara et al., 2005; Kornberg et al., 2010). Conversely, a neuroprotective protein and competitor of Siah, GOSPEL, is also S-nitrosylated by nNOS, and a regulatory balance exists between these two proteins in deciding neuronal cell fate (Kornberg et al., 2010). SNO-regulation of mitochondrial fragmentation by dynamin-related protein-1 may also determine the fate of neuronal survival in response to cellular stress, including β-amyloid (Cho et al., 2009). Excessive NO production results in the S-nitrosylation and inhibition of dynamin-related protein-1, causing synaptic impairment and neuronal cell death. Future studies are needed to investigate whether similar signaling pathways are subject to SNO-regulation in the enteric nervous system, and to examine whether SNO accumulation in disease states is sufficient to impair nNOS activity and neuronal survival.
Summary and Perspectives
Neuronal NOS activity is located in several classes of neurons in the enteric nervous system, where NO production represents an important neurotransmitter regulator of GI function. Impairment of nNOS activity and NO signaling is important in the pathogenesis of GI disease, as is evident in mice with genetic deletions of nNOS and NO-sensitive guanylyl cyclase. Significant interest has therefore focused on the possibility of exploiting the nNOS system as a therapeutic tool to modulate NO signals in GI pathology (Dijkstra et al., 2004; Micci et al., 2005), although a better characterization of altered promoter usage, activation, or suppression by disease-specific transcription factors is required. Of particular relevance to this article, SNO second messengers are implicated in the signal transduction of NO bioactivity; in the regulated expression and activity of nNOS; and of the survival of enteric neurons in the GI tract. If excessive SNO can be shown to contribute to the loss of nitrergic neurons in the enteric nervous system, modulation of dietary SNO production may constitute an approach that is worthwhile investigating, for example by limiting dietary nitrite and/or meat intake, neutralizing gastric acid pH, or by increasing the consumption of SNO reducing agents such as ascorbic acid (Milkowski et al., 2010). Alternatively, since exogenous GSNO is known to confer protection against neuronal loss associated with jejuno-ileitis (Savidge et al., 2007), therapeutic approaches that modulate SNO bioavailability may represent possible interventions that regulate enteric nNOS activity and protect neuronal survival. Further studies are needed to evaluate the potential use of SNO-derived signals in GI pathology.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by NIDDK R21-DK078032-01, 1UL1RR029876-01, and John S. Dunn Gulf Coast Consortium for Chemical Genomics Robert A. Welch Collaborative Grant Program.
References
Andrew, P. J., and Mayer, B. (1999). Enzymatic function of nitric oxide synthases. Cardiovasc. Res. 43, 521–531.
Benhar, M., Forrester, M., T., Hess, D. T., and Stamler, J. S. (2008). Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science 320, 1050–1054.
Benhar, M., Forrester, M. T., and Stamler, J. S. (2009). Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat. Rev. Mol. Cell Biol. 10, 721–732.
Böhm, S. K., Gum, J. R. Jr., Erickson, R. H., Hicks, J. W., and Kim, Y. S. (1995). Human dipeptidyl peptidase IV gene promoter: tissue-specific regulation from a TATA-less GC-rich sequence characteristic of a housekeeping gene promoter. Biochem. J. 311, 835–843.
Boissel, J. P., Zelenka, M., Gödtel-Armbrust, U., Feuerstein, T. J., and Förstermann, U. (2003). Transcription of different exons 1 of the human neuronal nitric oxide synthase gene is dynamically regulated in a cell- and stimulus-specific manner. Biol. Chem. 384, 351–362.
Bornstein, J. C., Marks, K. A., Foong, J. P. P., Gwynne, R. M., and Wang, Z. H. (2010). Nitric oxide enhances inhibitory synaptic transmission and neuronal excitability in guinea-pig submucous plexus. Front. Neurosci. 4:30. doi: 10.3389/fnins.2010.00030
Brenman, J. E., Chao, D. S., Gee, S. H., McGee, A. W., Craven, S. E., Santillano, D. R., Wu, Z., Huang, F., Xia, H., Peters, M. F., Froehner, S. C., and Bredt, D. S. (1996). Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and α1-syntrophin mediated by PDZ domains. Cell 84, 757–767.
Carvalho-Filho, M. A., Ueno, M., Hirabara, S. M., Seabra, A. B., Carvalheira, J. B., de Oliveira, M. G., Velloso, L. A., Curi, R., and Saad, M. J. (2005). S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: a novel mechanism of insulin resistance. Diabetes 54, 959–967.
Cellek, S., Foxwell, N. A., and Moncada, S. (2003). Two phases of nitrergic neuropathy in streptozotocin-induced diabetic rats. Diabetes 52, 2353–2362.
Chanrion, B., Mannoury la Cour, C., Bertaso, F., Lerner-Natoli, M., Freissmuth, M., Millan, M. J., Bockaert, J., and Marin, P. (2007). Physical interaction between the serotonin transporter and neuronal nitric oxide synthase underlies reciprocal modulation of their activity. Proc. Natl. Acad. Sci. U.S.A. 104, 8119–8124.
Cho, D. H., Nakamura, T., Fang, J., Cieplak, P., Godzik, A., Gu, Z., and Lipton, S. A. (2009). S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science 324, 102–105.
Chung, K. K., Thomas, B., Li, X., Pletnikova, O., Troncoso, J. C., Marsh, L., Dawson, V. L., and Dawson, T. M. (2004). S-nitrosylation of Parkin regulates ubiquitination and compromises Parkin’s protective function. Science 304, 1328–1331.
Chvanov, M., Gerasimenko, O. V., Petersen, O. H., and Tepikin, A. V. (2006). Calcium-dependent release of NO from intracellular S-nitrosothiols. EMBO J. 25, 3024–3032.
Cook, T., Gebelein, B., and Urrutia, R. (1999). Sp1 and its likes: biochemical and functional predictions for a growing family of zinc finger transcription factors. Ann. N. Y. Acad. Sci. 880, 94–102.
Costantini, T. W., Bansal, V., Krzyzaniak, M. J., Putnam, J. G., Peterson, C. Y., Loomis, W. H., Wolf, P. L., Baird, A., Eliceiri, B. P., and Coimbra, R. (2010). Vagal nerve stimulation protects against burn-induced intestinal injury through activation of enteric glia cells. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G1308–G1318.
Dijkstra, G., van Goor, H., Jansen, P. L., and Moshage, H. (2004). Targeting nitric oxide in the gastrointestinal tract. Curr. Opin. Investig. Drugs 5, 529–536.
Doullas, P. T., Greene, J. L., Greco, T. M., Tenopoulou, M., Seeholzer, S. H., Dunbrack, R. L., and Ischiropoulos, H. (2010). Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc. Natl. Acad. Sci. U.S.A. 107, 16958–16963.
Fang, J., Nakamura, T., Cho, D. H., Gu, Z., and Lipton, S. A. (2007). S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 104, 18742–18747.
Flamant, M., Aubert, P., Rolli-Derkinderen, M., Bourreille, A., Neunlist, M. R., Mahé, M. M., Meurette, G., Marteyn, B., Savidge, T., Galmiche, J. P., Sansonetti, P. J., and Neunlist, M. (2010). Enteric glia protect against Shigella flexneri invasion in intestinal epithelial cells: a role for S-nitrosoglutathione. Gut doi: 10.1136/gut2010.229237. [Epub ahead of print].
Forrester, M. T., Foster, M. W., Benhar, M., and Stamler, J. S. (2009). Detection of protein S-nitrosylation with the biotin-switch technique. Free Radic. Biol. Med. 46, 119–126.
Foster, M. W., Hess, D. T., and Stamler, J. S. (2009). Protein S-nitrosylation in health and disease: a current perspective. Trends. Mol. Med. 15, 391–404.
Friebe, A., Mergia, E., Dangel, O., Lange, A., and Koesling, D. (2007). Fatal gastrointestinal obstruction and hypertension in mice lacking nitric oxide-sensitive guanylyl cyclase. Proc. Natl. Acad. Sci. U.S.A. 104, 7699–7704.
Gabella, B. (1989). Fall in the number of myenteric neurons in aging guinea pigs. Gastroenterology 96, 1487–1493.
Gangula, P. R., Maner, W. L., Micci, M. A., Garfield, R. E., and Pasricha, P. J. (2007). Diabetes induces sex-dependent changes in neuronal nitric oxide synthase dimerization and function in the rat gastric antrum. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G725–G733.
Gangula, P. R., Mukhopadhyay, S., Pasricha, P. J., and Ravella, K. (2010a). Sepiapterin reverses the changes in gastric nNOS dimerization and function in diabetic gastroparesis. Neurogastroenterol. Motil. 12, 1325–1331.
Gangula, P. R., Mukhopadhyay, S., Ravella, K., Cai, S., Channon, K. M., Garfield, R. E., and Pasricha, P. J. (2010b). Tetrahydrobiopterin (BH4), a cofactor for nNOS, restores gastric emptying and nNOS expression in female diabetic rats. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G692–G699.
Garban, H. J., Marquez-Garban, D. C., Pietras, R. J., and Ignarro, L. J. (2005). Rapid nitric oxide-mediated S-nitrosylation of estrogen receptor: regulation of estrogen-dependent gene transcription. Proc. Natl. Acad. Sci. U.S.A. 102, 2632–2636.
Gaston, B. M., Carver, J., Doctor, A., and Palmer, L. A. (2003). S-nitrosylation signaling in cell biology. Mol. Interv. 3, 253–263.
Hara, M. R., Agrawal, N., Kim, S. F., Cascio, M. B., Fujimuro, M., Ozeki, Y., Takahashi, M., Cheah, J. H., Tankou, S. K., Hester, L. D., Ferris, C. D., Hayward, S. D., Snyder, S. H., and Sawa, A. (2005). S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 7, 665–674.
Hashida-Okumura, A., Okumura, N., Iwamatsu, A., Buijs, R. M., Romijn, H. J., and Nagai, K. (1999). Interaction of neuronal nitric-oxide synthase with alpha1-syntrophin in rat brain. J. Biol. Chem. 274, 11736–11741.
He, J., Wang, T., Wang, P., Han, P., Yin, Q., and Chen, C. (2007). A novel mechanism underlying the susceptibility of neuronal cells to nitric oxide: the occurrence and regulation of protein S-nitrosylation is the checkpoint. J. Neurochem. 102, 1863–1874.
Hogg, N. (2002). The biochemistry and physiology of S-nitrosothiols. Annu. Rev. Pharmacol. Toxicol. 42, 585–600.
Hoogerwerf, W. A. (2010). Role of clock genes in gastrointestinal motility. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G549–G555.
Huang, P. L., Dawson, T. M., Bredt, D. S., Snyder, S. H., and Fishman, M. C. (1993). Targeted disruption of the neuronal nitric oxide synthase gene. Cell 75, 1273–1286.
Huber, A., Saur, D., Kurjak, M., Schusdziarra, V., and Allescher, H. D. (1998). Characterization and splice variants of neuronal nitric oxide synthase in rat small intestine. Am. J. Physiol. 275, G1146–G1156.
Ignarro, L. J. (1996). Physiology and pathophysiology of nitric oxide. Kidney Int. Suppl. 55, S2–S5.
Ignarro, L. J., Buga, G. M., Wood, K. S., Byrns, R. E., and Chaudhuri, G. (1987). Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 84, 9265–9269.
Iwakiri, Y., Satoh, A., Chatterjee, S., Toomre, D. K., Chalouni, C. M., Fulton, D., Groszmann, R. J., Shah, V. H., and Sessa, W. C. (2006). Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc. Natl. Acad. Sci. U.S.A. 103, 19777–19782.
Iwasaki, H., Kajimura, M., Osawa, S., Kanaoka, S., Furuta, T., Ikuma, M., and Hishida, A. (2006). A deficiency of gastric interstitial cells of Cajal accompanied by decreased expression of neuronal nitric oxide synthase and substance P in patients with type 2 diabetes mellitus. J. Gastroenterol. 41, 1076–1087.
Jaffrey, S. R., Erdjument-Bromage, H., Ferris, C. D., Tempst, P., and Snyder, S. H. (2001). Protein S-nitrosylation: a physiological signal for neural nitric oxide. Nat. Cell Biol. 3, 193–197.
Jeong, Y., Won, J., Kim, C., and Yim, J. (2000). 5′-Flanking sequence and promoter activity of the rabbit neuronal nitric oxide synthase (nNOS) gene. Mol. Cells 10, 566–574.
Jia, L., and Stamler, J. S. (1999). Dual actions of S-nitrosylated derivative of vasoactive intestinal peptide as a vasoactive intestinal peptide-like mediator and a nitric oxide carrier. Eur. J. Pharmacol. 366, 79–86.
Johnson, M. A., Macdonald, T. L., Mannick, J. B., Conaway, M. R., and Gaston, B. (2001). Accelerated S-nitrosothiol breakdown by amyotrophic lateral sclerosis mutant copper, zinc-superoxide dismutase. J. Biol. Chem. 276, 39872–39878.
Kaneki, M., Shimizu, N., Yamada, D., and Chang, K. (2007). Nitrosative stress and pathogenesis of insulin resistance. Antioxid. Redox Signal. 9, 319–329.
Kim, S. F., Huri, D. A., and Snyder, S. H. (2005). Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science 310, 1966–1970.
Kornberg, M. D., Sen, N., Hara, M. R., Juluri, K. R., Nguyen, J. V. K., Snowman, A. M., Law, L., Hester, L. D., and Snyder, S. H. (2010). GAPDH mediates nitrosylation of nuclear proteins. Nat. Cell Biol. 12, 1094–1100.
Kuhnle, G. G., Story, G. W., Reda, T., Mani, A. R., Moore, K. P., Lunn, J. C., and Bingham, S. A. (2007). Diet-induced endogenous formation of nitroso compounds in the GI tract. Free Radic. Biol. Med. 43, 1040–1047.
Lecci, A., Santicioli, P., and Maggi, C. A. (2002). Pharmacology of transmission to gastrointestinal muscle. Curr. Opin. Pharmacol. 2, 630–641.
Li, F., Sonveaux, P., Rabbani, Z. N., Liu, S., Yan, B., Huang, Q., Vujaskovic, Z., Dewhirst, M. W., and Li, C. Y. (2007). Regulation of HIF-1α stability through S-nitrosylation. Mol. Cell 26, 63–74.
Li, H., and Poulos, T. L. (2005). Structure-function studies on nitric oxide synthases. J. Inorg. Biochem. 99, 293–305.
Lima, B., Lam, G. K., Xie, L., Diesen, D. L., Villamizar, N., Nienaber, J., Messina, E., Bowles, D., Kontos, C. D., Hare, J. M., Stamler, J. S., and Rockman, H. A. (2009). Endogenous S-nitrosothiols protect against myocardial injury. Proc. Natl. Acad. Sci. U.S.A. 106, 6297–6302.
Liu, M., Hou, J., Huang, L., Huang, X., Heibeck, T. H., Zhao, R., Pasa-Tolic, L., Smith, R. D., Li, Y., Fu, K., Zhang, Z., Hinrichs, S. H., and Ding, S. J. (2010). Site-specific proteomics approach for study protein S-nitrosylation. Anal. Chem. 82, 7160–7168.
Liu, M. T., Rothstein, J. D., Gershon, M. D., and Kirchgessner, A. L. (1997). Glutamatergic enteric neurons. J. Neurosci. 17, 4764–4784.
Lu, J., Katano, T., Okuda-Ashitaka, E., Oishi, Y., Urade, Y., and Ito, S. (2009). Involvement of S-nitrosylation of actin in inhibition of neurotransmitter release by nitric oxide. Mol. Pain 5, 58.
Mannick, J. B., Schonhoff, C., Papeta, N., Ghafourifar, P., Szibor, M., Fang, K., and Gaston, B. (2001). S-nitrosylation of mitochondrial caspases. J. Cell Biol. 154, 1111–1116.
Marino, S. M., and Gladyshev, V. N. (2010). Structural analysis of cysteine S-nitrosylation: a modified acid-based motif and the emerging role of trans-nitrosylation. J. Mol. Biol. 395, 844–859.
Mashimo, H., Kjellin, A., and Goyal, R. K. (2000). Gastric stasis in neuronal nitric oxide synthase-deficient knockout mice. Gastroenterology 119, 766–773.
Mayer, B., Kleschyov, A. L., Stessel, H., Russwurm, M., Münzel, T., Koesling, D., and Schmidt, K. (2009). Inactivation of soluble guanylate cyclase by stoichiometric S-nitrosation. Mol. Pharmacol. 75, 886–891.
Mearin, F., García-González, M. A., Strunk, M., Zárate, N., Malagelada, J. R., and Lanas, A. (2006). Association between achalasia and nitric oxide synthase gene polymorphisms. Am. J. Gastroenterol. 101, 1979–1984.
Micci, M. A., Kahrig, K. M., Simmons, R. S., Sarna, S. K., Espejo-Navarro, M. R., and Pasricha, P. J. (2005). Neural stem cell transplantation in the stomach rescues gastric function in neuronal nitric oxide synthase-deficient mice. Gastroenterology 129, 1817–1824.
Milkowski, A., Garg, H. K., Coughlin, J. R., and Bryan, N. S. (2010). Nutritional epidemiology in the context of nitric oxide biology: a risk-benefit evaluation for dietary nitrite and nitrate. Nitric Oxide 22, 110–119.
Molero, L., García-Durán, M., Diaz-Recasens, J., Rico, L., Casado, S., and López-Farré, A. (2002). Expression of estrogen receptor subtypes and neuronal nitric oxide synthase in neutrophils from women and men: regulation by estrogen. Cardiovasc. Res. 56, 43–51.
Moncada, S., and Higgs, A. (1993). The l-arginine-nitric oxide pathway. N. Engl. J. Med. 329, 2002–2012.
Murad, F. (1986). Cyclic guanosine monophosphate as a mediator of vasodilation. J. Clin. Invest. 78, 1–5.
Nakamura, T., and Lipton, S. A. (2008). Emerging roles of S-nitrosylation in protein misfolding and neurodegenerative diseases. Antioxid. Redox Signal. 10, 87–101.
Newton, D. C., Bevan, S. C., Choi, S., Robb, G. B., Millar, A., Wang, Y., and Marsden, P. A. (2003). Translational regulation of human neuronal nitric-oxide synthase by an alternatively spliced 5'-untranslated region leader exon. J. Biol. Chem. 278, 636–644.
Ogulener, N., and Ergun, Y. (2002). A putative role for S-nitrosoglutathione as the source of nitric oxide in photorelaxation of the mouse gastric fundus. Eur. J. Pharmacol. 450, 267–275.
Paige, J, S., Xu, G., Stancevic, B., and Jaffrey, S. R. (2008). Nitrosothiol reactivity profiling identifies S-nitrosylated proteins with unexpected stability. Chem. Biol. 15, 1307–1316.
Pasricha, P. J., Pehlivanov, N. D., Gomez, G., Vittal, H., Lurken, M. S., and Farrugia, G. (2008). Changes in the gastric enteric nervous system and muscle: a case report on two patients with diabetic gastroparesis. BMC Gastroenterol. 8, 21. doi: 10.1186/1471-230X-8-21
Que, L. G., Liu, L., Yan, Y., Whitehead, G. S., Gavett, S. H., Schwartz, D. A., and Stamler, J. S. (2005). Protection from experimental asthma by an endogenous bronchodilator. Science 308, 1618–1621.
Reineke, J., Tenzer, S., Rupnik, M., Koschinski, A., Hasselmayer, O., Schrattenholz, A., Schild, H., and von Eichel-Streiber, C. (2007). Autocatalytic cleavage of Clostridium difficile toxin B. Nature 446, 415–419.
Ren, J., Hu, H. Z., Liu, S., Xia, Y., and Wood, J. D. (2000). Glutamate receptors in the enteric nervous system: ionotropic or metabolotropic. Neurogastroenterol. Motil. 12, 257–164.
Riefler, G. M., and Firestein, B. L. (2001). Binding of neuronal nitric-oxide synthase (nNOS) to carboxyl-terminal-binding protein (CtBP) changes the localization of CtBP from the nucleus to the cytosol: a novel function for targeting by the PDZ domain of nNOS. J. Biol. Chem. 276, 48262–48268.
Rössig, L., Fichtlscherer, B., Breitschopf, K., Haendeler, J., Zeiher, A. M., Mülsch, A., and Dimmeler, S. (1999). Nitric oxide inhibits caspase-3 by S-nitrosation in vivo. J. Biol. Chem. 274; 6823–6826.
Saur, D., Neuhuber, W. L., Gengenbach, B., Huber, A., Schusdziarra, V., and Allescher, H. D. (2002). Site-specific gene expression of nNOS variants in distinct functional regions of rat gastrointestinal tract. Am. J. Physiol. Gastrointest. Liver Physiol. 282, G349–58.
Saur, D., Paehge, H., Schusdziarra, V., and Allescher, H. D. (2000). Distinct expression of splice variants of neuronal nitric oxide synthase in the human gastrointestinal tract. Gastroenterology 118, 849–958.
Savidge, T. C., Newman, P., Pothoulakis, C., Ruhl, A., Neunlist, M., Bourreille, A., Hurst, R., and Sofroniew, M. V. (2007). Enteric glia regulate intestinal barrier function and inflammation via release of S-nitrosoglutathione. Gastroenterology 132, 1344–1358.
Schonhoff, C. M., Matsuoka, M., Tummala, H., Johnson, M. A., Estevez, A. G., Wu, R., Kamaid, A., Ricart, K. C., Hashimoto, Y., Gaston, B., Macdonald, T. L., Xu, Z., and Mannick, J. B. (2006). S-nitrosothiol depletion in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 103, 2404–2409.
Secondo, A., Sirabella, R., Formisano, L., D’Alessio, A., Castaldo, P., Amoroso, S., Ingleton, P., Di Renzo, G., and Annunziato, L. (2003). Involvement of PI3′-K, mitogen-activated protein kinase and protein kinase B in the up-regulation of the expression of nNOSalpha and nNOSbeta splicing variants induced by PRL-receptor activation in GH3 cells. J. Neurochem. 84, 1367–1377.
Selvakumar, B., Huganir, R. L., and Snyder, S. H. (2009). S-nitrosylation of stargazin regulates surface expression of AMPA-glutamate neurotransmitter receptors. Proc. Natl. Acad. Sci. U.S.A. 106, 16440–16445.
Shah, S., Nathan, L., Singh, R., Fu, Y. S., and Chaudhuri, G. (2001). E2 and not P4 increases NO release from NANC nerves of the gastrointestinal tract: implications in pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 280, R1546–R1554.
Shotton, H. R., Adams, A., and Lincoln, J. (2006). Diabetes only affects nitric oxide synthase-containing myenteric neurons that do not contain heme oxygenase 2. Brain Res. 1068, 248–256.
Staab, C. A., Hartmanová, T., El-Hawari, Y., Ebert, B., Kisiela, M., Wsol, V., Martin, H. J., and Maser, E. (2011). Studies on reduction of S-nitrosoglutathione by human carbonyl reductases 1 and 3. Chem. Biol. Interact. doi:10.106/j.cbi.2011.01.016. [Epub ahead of print].
Stamler, J. S., Toone, E. J., Lipton, S. A., and Sucher, N. J. (1997). (S)NO signals: translocation, regulation, and a consensus motif. Neuron 18, 691–696.
Stricker, N. L., Christopherson, K. S., Yi, B. A., Schatz, P. J., Raab, R. W., Dawes, G., Bassett, D. E. Jr., Bredt, D. S., and Li, M. (1997). PDZ domain of neuronal nitric oxide synthase recognizes novel C-terminal peptide sequences. Nat. Biotechnol. 15, 336–342.
Sun, J., Picht, E., Ginsburg, K. S., Bers, D. M., Steenbergen, C., and Murphy, E. (2006). Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel α subunit and reduced ischemia/reperfusion injury. Circ. Res. 98, 403–411.
Surendran, S., and Kondapaka, S. B. (2005). Altered expression of neuronal nitric oxide synthase in the duodenum longitudinal muscle-myenteric plexus of obesity induced diabetes mouse: implications on enteric neurodegeneration. Biochem. Biophys. Res. Commun. 338, 919–922.
Suscheck, C. V., Schewe, T., Sies, H., and Kröncke, K. D. (2006). Nitrite, a naturally occurring precursor of nitric oxide that acts like a “prodrug.” Biol. Chem. 387, 499–506.
Tack, J., Demedts, I., Dehondt, G., Caenepeel, P., Fischler, B., Zandecki, M., and Janssens, J. (2002). Clinical and pathophysiological characteristics of acute-onset functional dyspepsia. Gastroenterology 122, 1738–1747.
Takahashi, T. (2003). Pathophysiological significance of neuronal nitric oxide synthase in the gastrointestinal tract. J. Gastroenterol. 38, 421–430.
Uehara, T., Nakamura, T., Yao, D., Shi, Z. Q., Gu, Z., Ma, Y., Masliah, E., Nomura, Y., and Lipton, S. A. (2006). S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441, 513–517.
Van Geldre, L. A., and Lefebvre, R. A. (2004). Interaction of NO and VIP in gastrointestinal smooth muscle relaxation. Curr. Pharm. Des. 10, 2483–2497.
Vanden Berghe, P. (2008). Electrochemical detection of neurotransmitters in the gut wall. Neurogastroenterol. Motil. 20, 1185–1188.
Vannucchi, M. G., Corsani, L., Bani, D., and Faussone-Pellegrini, M. S. (2002). Myenteric neurons and interstitial cells of Cajal of mouse colon express several nitric oxide synthase isoforms. Neurosci. Lett. 326, 191–195.
Walecka-Kapica, E., Klupińska, G., Harasiuk, A., Felicka, E., Foryš, S., and Chojnacki, C. (2007). The influence of melatonin on concentration of nitric oxide metabolites in gastric juice in subjects with functional dyspepsia. Pol. Merkur. Lekarski 22, 332–335.
Wang, Y., Newton, D. C., and Marsden, P. A. (1999). Neuronal NOS: gene structure, mRNA diversity, and functional relevance. Crit. Rev. Neurobiol. 13, 21–43.
Watkins, C. C., Sawa, A., Jaffrey, S., Blackshaw, S., Barrow, R. K., Snyder, S. H., and Ferris, C. D. (2000). Insulin restores neuronal nitric oxide synthase expression and function that is lost in diabetic gastropathy. J. Clin. Invest. 106, 373–384.
Whalen, E. J., Foster, M. W., Matsumoto, A., Ozawa, K., Violin, J. D., Que, L. G., Nelson, C. D., Benhar, M., Keys, J. R., Rockman, H. A., Koch, W. J., Daaka, Y., Lefkowitz, R. J., and Stamler, J. S. (2007). Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell 129, 511–522.
Wu, K. K. (2002). Regulation of endothelial nitric oxide synthase activity and gene expression. Ann. N. Y. Acad. Sci. 962, 122–130.
Xu, L., Eu, J. P., Meissner, G., and Stamler, J. S. (1998). Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279, 234–237.
Zaman, K., Palmer, L. A., Doctor, A., Hunt, J. F., and Gaston, B. (2004). Concentration-dependent effects of endogenous S-nitrosoglutathione on gene regulation by specificity proteins Sp3 and Sp1. Biochem. J. 380, 67–74.
Keywords: S-nitrosothiol, nitric oxide, S-nitrosoglutathione, nNOS, enteric neuropathy, enteric glia and neurons
Citation: Savidge TC (2011) S-nitrosothiol signals in the enteric nervous system: lessons learnt from big brother. Front. Neurosci. 5:31. doi: 10.3389/fnins.2011.00031
Received: 11 October 2010;
Paper pending published: 02 January 2011;
Accepted: 28 February 2011;
Published online: 09 March 2011.
Edited by:
L. Ashley Blackshaw, University of Adelaide, AustraliaReviewed by:
Nicholas Spencer, Flinders University, AustraliaSimon J. H. Brookes, Flinders University, Australia
Copyright: © 2011 Savidge. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Tor C. Savidge, Division of Gastroenterology, The University of Texas Medical Branch, Galveston, TX 77555, USA. e-mail:dGNzYXZpZGdAdXRtYi5lZHU=