 Erzsebet Kovesdi1
Erzsebet Kovesdi1 Andrea B. Gyorgy2
Andrea B. Gyorgy2 Sook-Kyung C. Kwon2 Daniel L. Wingo2
Sook-Kyung C. Kwon2 Daniel L. Wingo2 Alaa Kamnaksh2 Joseph B. Long3
Alaa Kamnaksh2 Joseph B. Long3 Christine E. Kasper1
Christine E. Kasper1 Denes V. Agoston2*
Denes V. Agoston2*
- 1 U.S. Department of Veterans Affairs, Veterans Affairs Central Office, Washington, DC, USA
- 2 Department of Anatomy, Physiology and Genetics, School of Medicine, Uniformed Services University, Bethesda, MD, USA
- 3 Division of Military Casualty Research, Walter Reed Army Institute of Research, Silver Spring, MD, USA
De novo hippocampal neurogenesis contributes to functional recovery following traumatic brain injury (TBI). Enriched environment (EEN) can improve the outcome of TBI by positively affecting neurogenesis. Blast induced traumatic brain injury (bTBI) characterized by memory impairment and increased anxiety levels, is a leading cause of chronic disability among soldiers. Using a rodent model of bTBI we asked: (a) whether long-term exposure to EEN after injury can ameliorate behavioral abnormalities and (b) what the effects of EEN are at the molecular and cellular levels and on de novo neurogenesis. We found that housing injured animals in EEN resulted in significantly improved spatial memory while animals in normal housing (NH) showed persistent memory impairment. VEGF and Tau protein but not Interleukin-6 (IL-6) levels were normalized in the dorsal hippocampus (DHC) of EEN rats while all three markers remained elevated in NH rats. Interestingly, after peaking at 6 weeks post-injury, anxiety returned to normal levels at 2 months independent of housing conditions. Housing animals in EEN had no significant effect on VEGF and Tau protein levels in the ventral hippocampus (VHC) and the amygdala (AD). We also found that EEN reduced IL-6 and IFNγ levels in the VHC; these markers remained elevated following NH. We observed an increase in GFAP and DCX immunoreactivities in the VHC of NH animals at 2 months post-injury. Conversely, injured animals housed in EEN showed no increase in GFAP or DCX immunoreactivity in their VHC. In summary, long-term exposure of injured animals to EEN appears to play a positive role in the restoration of memory functions but not on anxiety, which returned to normal levels after a significant period of time. Cellular and molecular changes in response to EEN appear to be a part of neurogenesis-independent as well as dependent recovery processes triggered by bTBI.
Introduction
Traumatic brain injury (TBI) is one of the leading causes of chronic disability worldwide (Bruns and Hauser, 2003; Tagliaferri et al., 2006). The main neurobehavioral symptoms of survivable TBI include memory problems, increased anxiety, and depression (Gentilini et al., 1985; Schoenhuber and Gentilini, 1988; Ponsford et al., 1995). Blast induced traumatic brain injury (bTBI) caused by explosive devices has become a signature injury of the recent conflicts in Iraq and in Afghanistan (Warden and French, 2005; Taber et al., 2006; Warden, 2006). Although bTBI shares some of the clinical features of the closed head and the penetrating TBI models, bTBI appears to be a different form of neurotrauma (Ling et al., 2009). The secondary injury process after either form of TBI includes metabolic changes, hypoxia, edema, neuroinflammation, and gliosis (Kaur et al., 1997; Mayorga, 1997; Cernak et al., 2001; Taber et al., 2006). As epidemiological and experimental data show, even moderate and mild forms of bTBI can cause long-term behavioral changes (Ryan and Warden, 2003; Okie, 2005; Cernak et al., 2010). The observed changes include impaired memory and increased anxiety, implicating damage to the hippocampus (HC) and the prefrontal cortex (PFC).
Exposure to severe blast causes vascular, neuronal, and glial damage that results in cerebral edema, vasospasm, and the loss of fiber tracts (Kaur et al., 1997; Mayorga, 1997; Cernak et al., 2001; Taber et al., 2006). Moderate and mild blast result in gliosis and neuroinflammation characterized by elevated levels of proinflammatory molecules like Interleukin-6 (IL-6; Vallieres et al., 2002; Kwon et al., 2011). Inflammation can cause neuronal cell death as well as increase vulnerability to noxious factors like neurotoxins (Cacci et al., 2005; Agoston et al., 2009). IL-6 is also a potent inhibitor of hippocampal de novo neurogenesis (Vallieres et al., 2002; Barkho et al., 2006).
Intact de novo hippocampal neurogenesis in the adult brain has been shown to play an important role in maintaining normal neurobehavioral functions (Kempermann, 2002b; Kempermann and Kronenberg, 2003; Kempermann et al., 2004; Sahay and Hen, 2007; Aimone et al., 2010; Deng et al., 2010). Neurogenesis is a part of the innate regenerative processes triggered by various insults to the brain including TBI (Dash et al., 2001; Hallbergson et al., 2003; Parent, 2003). Following TBI, the rate of de novo neurogenesis is substantially upregulated (Dash et al., 2001; Chirumamilla et al., 2002; Lee and Agoston, 2009), which contributes to the amelioration of the neurobehavioral consequences of TBI (Sun et al., 2007). Neurogenesis is a complex process that is not restricted to the proliferation of neural stem (progenitor) cells; it includes neuronal fate determination, differentiation of immature neurons, migration, survival, and functional integration into neuronal circuits (Kempermann, 2002a; Kozorovitskiy and Gould, 2003; Abrous et al., 2005; Hagg, 2005; Ming and Song, 2005; Zhao et al., 2008).
Previous studies indicated that socially and physically enriched environments (EEN) can significantly improve working memory, especially spatial learning along with other beneficial neurobehavioral effects (Kempermann et al., 1997; Pham et al., 1999; Van Praag et al., 2000; Dhanushkodi et al., 2007). The positive effects of EEN have been described in young intact animals (Tees et al., 1990) after various forms of brain lesions (Einon et al., 1980; Whishaw et al., 1984) like TBI, spinal cord injury (Berrocal et al., 2007; Fischer and Peduzzi, 2007; Kline et al., 2007; Hoffman et al., 2008), and ischemia (Johansson and Ohlsson, 1996; Dahlqvist et al., 2004; Buchhold et al., 2007; Briones et al., 2009). Kempermann et al. (1997) found that mice housed in EEN had more new granule cells in the dentate gyrus than control mice. The combination of physical and social interactions in EEN enhances hippocampal neurogenesis, synaptic efficacy, learning, and memory functions (Van Praag et al., 2000; Cao et al., 2004; Tashiro et al., 2007; Wright and Conrad, 2008). Therefore, the demonstrated benefits of EEN may offer an easily implementable “treatment” option to improve neurobehavior following brain insults.
In this study, we aimed to identify the effect of EEN on behavioral outcome as well as cellular and molecular alterations ensuing bTBI. The observed cellular and molecular changes suggest that recovery after bTBI involves complex processes like de novo neurogenesis and that EEN may modulate the temporal characteristics of these processes.
Materials and Methods
Animals and Housing Conditions
Sprague-Dawley male rats (245–265 g, n = 23; Charles River Laboratories, Wilmington, MA, USA) were housed in cages with free access to food and water in a reverse 12–12 h light-dark cycle. After 7 days of acclimation and handling, animals were exposed to a single blast (or sham) injury and then placed into the separate housing conditions until the end of the study. The experimental schedule is depicted in Figure 1. All animals were handled according to protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the Uniformed Services University of the Health Sciences (USUHS). All behavioral tests were performed during animals’ dark cycle.
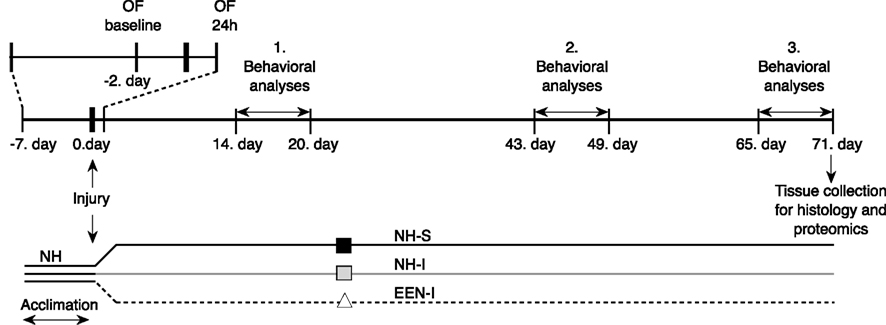
Figure 1. Outline of the experimental schedule.
Injury
On the day of the injury all rats (injury weight: ∼300 g) were transferred to Walter Reed Army Institute of Research (Silver Spring, MD, USA) where blast was generated using a compression-driven shock tube (Long et al., 2009). Before the induction of injury, rats were anesthetized in a bell jar for 6 min with 4% isoflurane (Forane, Baxter Healthcare Corporation, Deerfield, IL, USA). Anesthetized rats in chest protection were then placed in a holder in a transverse prone position and exposed to whole body blast injury at 20.63 psi pressure. Immediately after injury rats were moved back to their home cages and transported back to the USUHS animal facility. Sham-injured animals underwent the same procedure without being exposed to blast overpressure.
Housing Conditions After Injury
Following bTBI (n = 15) or sham injury (n = 8), rats were assigned to the following groups: normal housed-injured (NH-I; n = 8), normal housed-sham (NH-S; n = 8), and enriched environment-injured (EEN-I; n = 7). Rats under NH conditions were housed on hardwood chip bedding without any toys, two rats/standard rat cage. Rats under EEN conditions were housed in a four level metal cage (dimensions: 36′′L × 25′′W × 62 1/2′′H; MIDWEST Ferret Nation Double Unit Home with Stand, PetSmart) that contained different kinds of toys like activity wheels, tunnels, balls, and rodent igloos (Figure A1 in Appendix). Toys (PetSmart) were replaced with new ones weekly.
Behavioral Tests
All rats underwent a series of behavioral evaluations; basic motor function and anxiety were measured by elevated plus maze (EPM), and spatial learning and memory by Barnes maze (BM). Behavioral assessments were performed on separate days starting at 15, 44, and 66 days after injury (Figure 1).
Elevated plus maze
The EPM is a widely used, ethologically relevant test that assesses anxiety states in rodents (Carobrez and Bertoglio, 2005; Salzberg et al., 2007; Walf and Frye, 2007). The maze is an elevated structure (1 m above ground) consisting of four intersecting arms. The arms of the maze are 50 cm long and 10 cm wide; the closed arms have walls on three sides that are 40 cm high while the open arms have none. The lighting in the middle of the maze was set at 90 lux. On testing days rats were placed one by one in the center of the maze facing one of the open arms; each animal was allowed to explore freely for 5 min while its movement was video-tracked. Total distance traveled (meters) was used to measure changes in basic motor function. Time spent in the closed and open arms (seconds) was used to measure anxiety levels. All data from the maze were recorded using ANY-maze 4.2 Software (Stoelting Company, Wood Dale, IL, USA). The maze was cleaned with a 30% ethanol solution between each rat.
Barnes maze
Barnes maze represents an approved and less stressful alternative to the commonly used water maze test (Barnes, 1979; Maegele et al., 2005; Doll et al., 2009; Harrison et al., 2009). The maze is a circular platform (1.2 m in diameter) with 18 evenly spaced holes around the periphery. One of the holes is the entrance to a darkened escape box that is not visible from the surface of the board. Each rat was tested twice per day for five consecutive days to find the escape box (only day 1 of the first BM session had three trials). In each trial latency to locate and enter the escape box was measured (ANY-maze 4.2 Software, Stoelting Company, Wood Dale, IL, USA). During the first (teaching) trial of the first BM session, animals were trained to locate the escape chamber. Each animal was placed in the escape box and covered for 30 s; the escape box was then removed with the animal inside and moved to the center of the maze. The rat was removed from the box and allowed to explore the maze for a few seconds, after which the rat was returned to its home cage. No latency times were recorded for the teaching trials. The escape box and the maze were cleaned with a 30% ethanol solution between each trial. In the second trial the same rat was placed under a start box in the center of the maze for 30 s; the start box was then removed and the rat was allowed to explore freely to find the escape chamber. Training sessions ended after the animal had entered the escape box or when a pre-determined time (240 s) had elapsed. If the animal had not found the escape box during the given time period, it was placed in the escape box for 1 min at the end of the trial.
Tissue Collection and Processing
At the conclusion of the last behavioral testing session (day 71 post-blast or sham injury), animals were placed inside an induction chamber saturated with isoflurane and deeply anesthetized. For proteomics and ELISA assays (NH-S: n = 5; NH-I: n = 5; and EEN-I: n = 4), rats were decapitated and brains were immediately removed and placed on ice. The amygdala (AD), PFC, dorsal (DHC), and ventral hippocampus (VHC) were dissected, frozen, and stored at −80°C. For histology (NH-S: n = 3; NH-I: n = 3; and EEN-I: n = 3), rats were placed under deep isoflurane anesthesia and transcardially perfused with cold phosphate-buffered saline (PBS) followed by 4% paraformaldehyde solution. Fixed brains were then immersed in cold 15 and 30% sucrose in 1 × PBS (consecutively) for cryoprotection. Frozen brains were sectioned coronally at a 40-μm thickness using a cryostat (Cryocut 1800; Leica Microsystems, Bannockburn, IL, USA) and sections containing the DHC and the VHC were kept at −80°C until use.
Proteomics
Preparation of samples
Sample preparation, printing, scanning, and data analysis were performed as described in detail (Gyorgy et al., 2010). Briefly, flash frozen brain samples were pulverized in liquid nitrogen; 200 mg of the powder was transferred into 1 ml of T-per lysis buffer (Thermo Fisher, Waltham, MA, USA) with protease and phosphatase inhibitors (Thermo Fisher) and then sonicated. Samples were centrifuged for 15 min at 4°C and the supernatants were aliquoted and stored at −80°C. Tissue samples were diluted in print buffer (10% glycerol, 0.05% SDS, 50 mM DTT in 1 × TBS) to a final protein concentration of 1 mg/ml. Samples were then subjected to an 11-point serial 1:2 dilution and transferred into Genetix 384-well plates (X7022, Fisher Scientific, Pittsburg, PA, USA) using a JANUS Varispan Integrator and Expanded Platform Workstation (PerkinElmer, Waltham, MA, USA). Plates were transferred into an Aushon 2470 Arrayer (Aushon Biosytems, Billerica, MA, USA) to be printed on ONCYTE Avid (tissue samples) or ONCYTE Nova (serum samples) single-pad nitrocellulose coated glass slides (Grace Bio-Labs, Bend, OR, USA).
Printing parameters
The Aushon Arrayer was programmed to use 16 pins (4 × 4 pattern). Each sample was printed in 12 dilutions (12 rows) and in triplicate (3 columns), resulting in a block of 3 × 12 dots per sample. The Spot Diameter was set to 250 nm with a spacing of 500 nm between dots on the x-axis and 375 nm on the y-axis. Wash time was set at 2 s without delays. The printer was programmed for a single deposition per dot for printing serum and tissue extracts.
Immunochemical detection
Primary antibodies were diluted to 10× the optimal Western analysis concentration in antibody incubation buffer [0.1% bovine serum albumin (BSA), protease inhibitors (EDTA-free Halt protease and phosphatase inhibitor cocktail, Thermo Fisher, Waltham, MA, USA), 1 × TBS, 0.5%Tween 20]. Primary antibodies were used in the following dilutions for reverse phase protein microarray (RPPA): VEGF 1:100 (Abcam ab-53465) and Tau protein 1:20 (Santa Cruz sc-1995). The primary antibody solution was incubated overnight at 4°C with a cover slip (Nunc* mSeries LifterSlips, Fisher Scientific, Pittsburg, PA, USA). The following day slides were washed and then incubated with an Alexa Fluor® 635 goat anti-mouse (Cat# A-31574), goat anti-rabbit (Cat# A-31576), or rabbit anti-goat IgG (H + L; Cat# A-21086) secondary antibodies from Invitrogen at 1:6000 dilution in antibody incubation buffer for 1 h at room temperature (RT). After washing and drying, the fluorescent signals were measured by a Scan Array Express HT microarray scanner (Perkin Elmer, Waltham, MA, USA) using a 633-nm wavelength laser and a 647-nm filter; data were imported into a bioinformatics program.
Data analysis and bioinformatics
Data from the scanned images were imported into a Microsoft Excel-based bioinformatics program developed in house for analysis (Gyorgy et al., 2010). The tool calculates total net intensity after local background subtraction for each spot. The intensity data from the dilution series of each sample were then plotted against dilution on a log–log graph. The linear regression of the log–log data was calculated after the removal of flagged data, which include signal to noise ratios of less than 2, spot intensities in the saturation range or noise range, or high variability between duplicate spots (>10−15%). The total amount of antigen is determined by the Y-axis intercept (Gyorgy et al., 2010).
Interleukin-6 and Interferon Gamma Elisa
Interferon gamma (IFNγ) and IL-6 levels were measured from brain tissues using the Rat IFNγ ELISA and the Rat IL-6 ELISA kits (both are from Thermo Fisher, Waltham, MA, USA). The IL-6 ELISA kit required a 1:5 dilution using the supplied dilution buffer in order to avoid saturation in the wells. After the dilution of brain samples, the assay was performed according to the manufacturer’s instructions.
Histology
Immunohistochemistry
Sections from the PFC, DHC, and VHC were identified based on z-axis and morphology as described in the Chemoerchitectonic Atlas of The Rat Forebrain (Paxinos et al., 1999). The 1st and 10th sections were mounted on positively charged glass slides two sections per slide. Three slides per animal were selected per brain region for each immunostaining. Sections were equilibrated at RT and hydrated with 1 × PBS for 30 min. Antigen retrieval was performed by incubating in 10 mM citrate buffer (pH 6.0) at 80°C for 30 min followed by cooling down to RT. After rehydration with 1 × PBS, sections were permeabilized with 0.5% Triton X-100 in PBS for 1 h and blocked in 1 × PBS containing 5% normal goat serum (NGS), 5% BSA, 0.1% Sodium Azide, and 0.5% Triton X-100 for 1 h. The same solution used for blocking, with the exception of NGS, was used to dilute the primary antibodies. Primary antibodies include mouse anti-GFAP (Millipore, Temecula, CA, USA) and rabbit anti-Doublecortin (DCX; Cell Signaling Technology, Beverly, MA, USA). Sections were incubated with the primary antibodies overnight at 4°C. After washing with 1 × PBS, secondary antibody (Alexa Fluor 555 goat anti-mouse IgG or 488 goat anti-rabbit IgG was applied for 1 h at RT; 1 μg/ml of Hoechst 33342 (Molecular Probes, Eugene, OR, USA) was applied for 2 min for nuclear counterstaining and sections were coverslipped with anti-fading media (Vectashield, Vector Laboratories, Burlingame, CA, USA). Histological sections were visualized in an Olympus IX-71 microscope and images were collected using a SPOT digital camera (Diagnostic Instruments Inc., Sterling Heights, MI, USA). Immunofluorescent staining was acquired using the appropriate filters. The collected images were colored using TIFFany3rr Software.
Statistical Analysis
Statistical analysis was performed using Graph Pad Instat software. Behavioral test results were analyzed with ANOVA and Tukey post hoc tests. Differences were considered significant with a p value of 0.05. Proteomics data results were followed up with a one-way ANOVA (Gyorgy et al., 2010; Kwon et al., 2011) a significance level of 0.05 was used throughout. No adjustment was made for multiple comparisons, thus, significant findings should be considered exploratory and in need of confirmation by further studies. IL-6 and IFNγ ELISA results were analyzed with Student t-test. Data are reported as the average ± SE of the mean. For each of our numerical measurements, we determined statistical significance among experimental groups by (p < 0.05 depicted by one star; and p < 0.01 by two).
Results
Behavioral Changes
Anxiety
We found that general locomotion decreased over the period of 2 months in all experimental groups. Animals traveled less distances on day 66 post-injury (or sham) than day 15 at the beginning of the behavioral tests (Figure 2A). Among all experimental groups, NH-I animals traveled the least at all time points measured, however the values were only statistically significant at day 44 post-injury. EEN-I rats traveled greater distances than NH-I animals but the difference was not statistically significant.
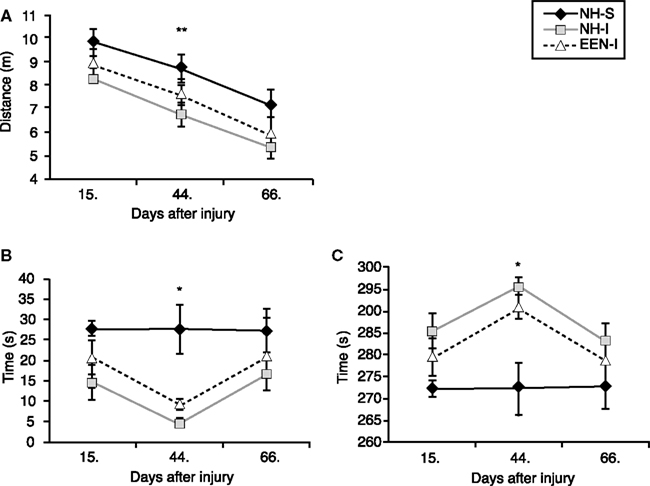
Figure 2. Anxiety and basic motor function of animals in the various experimental groups. Elevated plus maze was used to measure changes in basic motor function and anxiety levels. Total distance traveled (meter) (A), duration of time spent (seconds) in open arms (B) and duration of time spent (second) in closed arms (C) were measured at 15, 44, and 66 days after mbTBI. Housing rats in EEN after mbTBI mildly improved motor function and reduced anxiety. *p < 0.05 and **p < 0.01 for NH-I versus NH-S. Data are presented as mean ± SEM. (NH-I, normal housed-injured; NH-S, normal housed-sham; EEN-I, enriched environment-injured).
At 15 days post-injury, injured animals (regardless of housing) spent less time in the open arms and more time in the closed arms of the EPM compared to NH-S animals; the differences were statistically insignificant (Figures 2B,C). The difference in anxiety levels of NH-S and injured animals was the greatest at 44 days post-injury; animals in both EEN-I and NH-I groups spent substantially more time in the open arms and less time in the closed arms compared to the non-injured controls (Figures 2B,C).
At 66 days post-injury there were no significant differences among the experimental groups. All animals spent roughly the same amount of time in the closed arms as well as in the open arms of the maze independent of housing conditions or injury (Figures 2A–C).
Spatial learning and memory
The first testing session was performed between days 16 and 20 post-injury or sham. We found that NH-I animals had significant difficulty in learning the task on the first day of BM compared to animals in the NH-S or EEN-I groups (Figure 3A). Rats in the EEN-I and NH-S groups performed similarly and significantly better than NH-I rats.
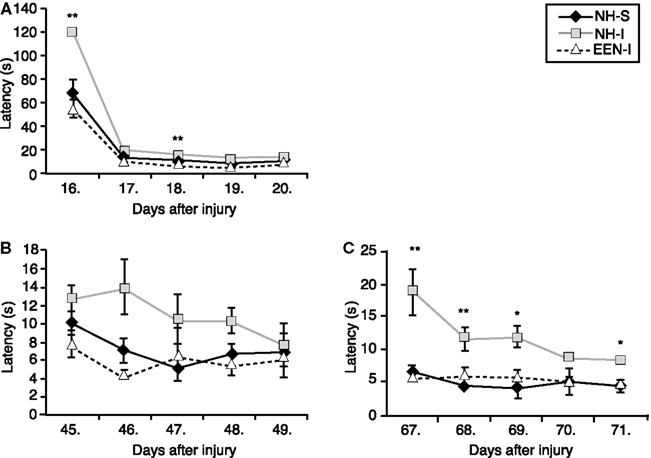
Figure 3. Spatial learning and memory of animals in the various experimental groups. Barnes maze was used to determine spatial learning and memory performance. Latency times (second) to find the escape box were measured on days 16–20 (A), days 45–49 (B), and days 67–71 (C) after mbTBI. Housing rats in EEN after mbTBI significantly improved spatial memory. *p < 0.05 and **p < 0.01 for NH-I versus NH-S. Data are presented as mean ± SEM. (NH-I, normal housed-injured; NH-S, normal housed-sham; EEN-I, enriched environment-injured).
The second BM session was performed between days 45 and 49 post-injury (or sham). Again, EEN-I rats performed similar to NH-S rats on every day of the testing period (Figure 3B). In contrast, rats in the NH-I group required more time to find the escape box every day of the testing session, however the difference was statistically insignificant.
The last BM testing session was performed between days 67 and 71 post-injury. The performance of EEN-I animals was practically identical to NH-S animals (Figure 3C). NH-I animals required significantly longer times to locate the escape box on days 67, 68, 69, and 71 post-injury. Interestingly, the behavior of NH-I animals in the last BM session was similar to the one measured during the first testing session. On the first day of BM, NH-I animals required the longest period of time to find the escape box.
Protein Changes
At the end of the last behavioral session, we analyzed changes in the expression of selected proteins in the AD, PFC, DHC, and VHC of animals in the various experimental groups using RPPA. NH-I animals had significantly elevated levels of Tau protein, a marker of axonal degeneration, in all brain regions compared to levels detected in the NH-S group (Figure 4). Importantly, housing injured animals in EEN resulted in a significant decrease in Tau levels in the DHC compared to those measured in the DHC of NH-S animals; no such decrease was detected in the DHC of NH-I animals. A similar decrease in Tau levels was observed in the PFC of EEN-I animals; no such effect of EEN was seen in the VHC or the AD.
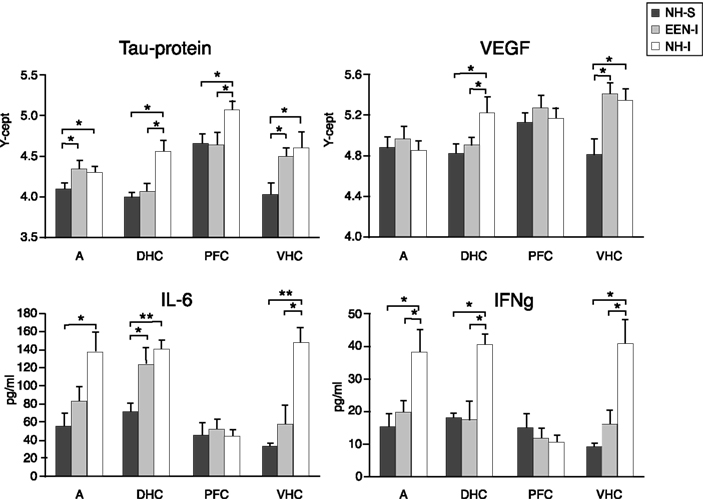
Figure 4. Protein markers in the AD, PFC, DHC, and the VHC of animals in the various experimental groups. Tissue extracts were prepared from dissected brain regions of NH-S, NH-I, and EEN-I rats. Tissue levels of selected protein markers were assayed using either RPPM or ELISA. The Y-axis intercept (Y-cept) and pg/ml (IL-6 and IFNγ) indicate measured protein levels. *p < 0.05 and **p < 0.01; data are presented as mean ± SEM. (NH-I, normal housed-injured; NH-S, normal housed-sham; EEN-I, enriched environment-injured).
Housing animals in EEN after injury had a similar effect on VEGF levels. EEN-I animals had decreased VEGF levels in the DHC but not in the VHC. In NH-I animals, VEGF levels increased in both the DHC and the VHC, but not in the AD or the PFC (Figure 4). Importantly, neither injury nor housing conditions had any significant effect on VEGF levels in the AD or the PFC (Figure 4).
To assess the potential inflammatory response to injury and housing conditions, we measured IL-6 and IFNγ, molecules associated with neuroinflammation in the same brain regions. Injury increased IL-6 concentrations in all brain regions except in the PFC of NH-I animals (Figure 4). Interestingly, housing injured animals in EEN restored IL-6 levels to NH-S levels in the VHC but not in the DHC. A similar trend was seen in the AD but the differences between EEN-I and NH-S animals were not statistically significant. Importantly, housing conditions had no effect on IL-6 values in the DHC; IL-6 remained elevated regardless of post-injury housing conditions compared to values measured in NH-S animals.
Injury had a similar effect on IFNγ levels; we measured elevated values in all brain regions except in the PFC (Figure 4). Interestingly, housing animals in EEN after injury restored IFNγ to control levels not only in the VHC and the AD, but also in the DHC.
Cellular Changes
Astrogliosis
We analyzed the DHC and the VHC at 71 days post-injury for GFAP expression to identify some of the cellular responses to injury and the effects of EEN. We found increased GFAP immunoreactivity in the VHC of NH-I animals compared to NH-S (Figures 5A–F). GFAP immunoreactive cells were located in the hilus of the VHC but many GFAP positive cells were also present in the dentate gyrus; these cells resembled reactive astrocytes with a stellar appearance and elaborate processes. Importantly, housing animals in EEN resulted in GFAP immunoreactivity similar to that of the DHC of NH-S animals, suggesting that EEN may mitigate the effect of injury on GFAP immunoreactivity.
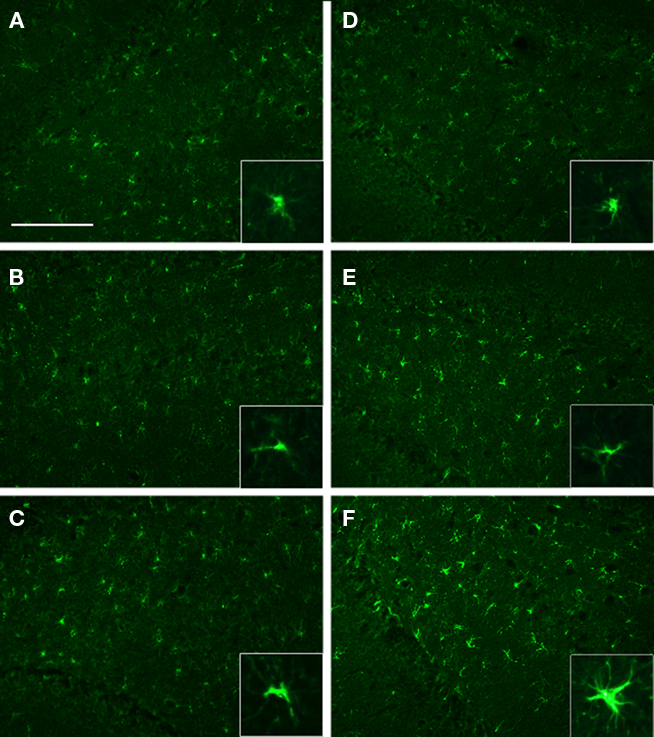
Figure 5. GFAP immunoreactivity in selected brain regions of animals in the various experimental groups. Frozen coronal sections containing the DHC and the VHC were cut and processed for immunohistochemistry using a GFAP antibody. DHC of NH-S (A), NH-I (B), and EEN-I (C) rats; VHC of NH-S (D), NH-I (E), and EEN-I (F) rats. Inlays magnify GFAP morphology. Scale bar (A–F) = 100 μm.
Neurogenesis
To identify the potential effects of injury and EEN on de novo neurogenesis, we analyzed the DHC and the VHC for DCX immunoreactivity. We found similar DCX immunoreactivity in the DHC and the VHC of NH-S animals (Figures 6A,D). We also found an apparent increase in DCX immunoreactivity in the VHC of NH-I animals (Figure 6F). Many of these DCX immunoreactive cells displayed elaborate, branching processes (Figure 6C, insert). Interestingly, we observed the lowest DCX immunoreactivity in the DHC and the VHC of EEN-I animals. Conversely, the highest DCX immunoreactivity was in the DHC and the VHC of NH-I animals (compare Figures 6A–F).
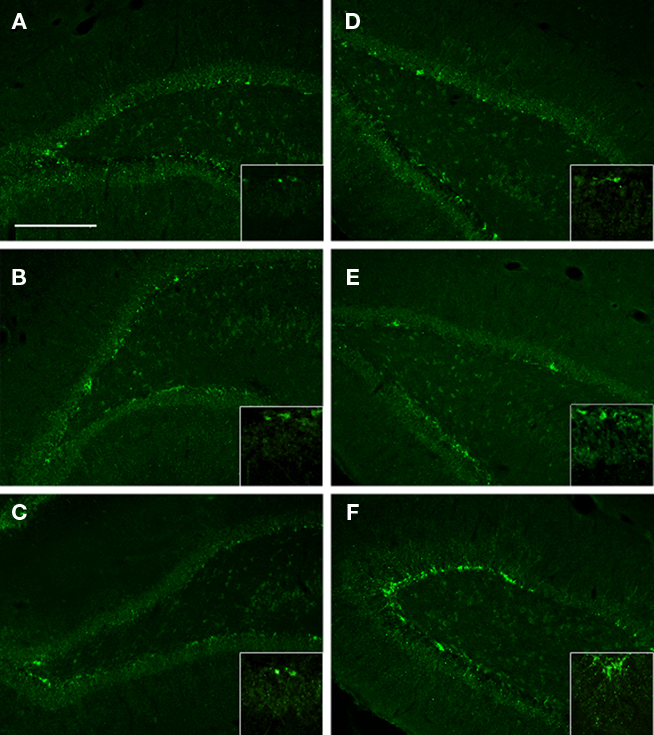
Figure 6. The DCX immunoreactivity in selected brain regions of animals in the various experimental groups. Frozen coronal sections containing the DHC and the VHC were cut and processed for immunohistochemistry using a DCX antibody. DHC of (A) NH-S, (B) NH-I, and (C) EEN-I rats; VHC of (D) NH-S, (E) NH-I, and (F) EEN-I rats. Inlays magnify DCX cell body and process. Scale bar (A–F) = 200 μm.
Discussion
Our study shows that the normalization of anxiety in a rodent model of bTBI can take more than 2 months after the insult, the process is independent of housing conditions and most likely involves de novo neurogenesis. We found that anxiety was high at 15 days and further increased at 44 days post-injury. Importantly, at 66 days the anxiety of animals was rather similar irrespective of housing conditions or injury. To our knowledge, our study is one of the very few that conducted behavioral assessments at various time points after injury over a 2-month period of time. Two months in rodent life roughly translates into a multi-year period of time for humans (Quinn, 2005). Thus, our findings may have implications for conducting longitudinal human clinical studies.
Our work also implicates studies that only measure the short-term effects of TBI in that they may not be able to provide insight into the full effect of the insult and/or the various treatments tested. Therefore, our findings underline the importance of monitoring behavior at several time points post-injury in the same sets of animals. There are very few studies that examine neurobehavior following TBI for longer than 2 weeks and even less that assess behavior at several time points post-injury. A recent study (Liu et al., 2010b) using the lateral fluid percussion injury model in rats monitored behavior and performed in vivo imaging at 1, 3, and 6 months after injury. The study showed that a single insult is capable of triggering behavioral and morphological changes that evolve over several months. Using the same blast overpressure TBI model in an independent study, we found that anxiety increased within the first 2 days after injury, reached a maximum at 1 month, and returned to normal levels at 2 months after bTBI (Kwon et al., 2011). This illustrates that the pathophysiology as well as the recovery process after bTBI changes significantly over a longer period of time than previously thought.
Our findings indicate that increased anxiety resulting from bTBI should dissipate over time. However, epidemiological studies demonstrate that some individuals develop a chronic condition (Woon et al., 2010) with symptoms similar to post-traumatic stress disorder (PTSD; Gross and Hen, 2004; Yehuda and Ledoux, 2007). Previous studies have shown that hippocampal abnormalities can play a role in individuals’ vulnerability to the long-term effects of stress. According to human imaging studies, reduced hippocampal volume increases pathologic vulnerability to psychological trauma and the development of PTSD (Tischler et al., 2006). An in vivo imaging study indicates that CA3/DG volume is significantly reduced in veterans with combat-related PTSD (Wang et al., 2010). The abovementioned long-term animal study also showed significant morphological changes in the HC 6 months after injury (Liu et al., 2010a). Neuroanatomical substrates mediating anxiety include the HC in addition to the medial PFC and the AD (Bremner, 2007; Liberzon and Sripada, 2008); anxiety possesses neurogenesis dependent and independent components (Sahay and Hen, 2007).
While the VHC is predominantly involved in mediating anxiety-related functions, the DHC mediates learning and memory (Henke, 1990; Moser et al., 1995). As opposed to the housing-independent normalization of anxiety levels, the recovery of spatial memory after bTBI was clearly EEN dependent. The conception of EEN comes from Hebb’s (1947, 1949) research ; EEN is an experimental model that provides multiple possibilities for physical and social interactions (Rosenzweig and Bennett, 1996; Van Praag et al., 2000; Puurunen and Sivenius, 2002; Mora et al., 2007). In EEN, rats are usually housed in big multi-level cages containing a variety of toys, which are changed daily to provide opportunities for sensory and physical interaction (Van Praag et al., 2000). The average rat number in EEN is 8–12 rats per cage for social interaction purposes. EENs impart various learning experiences to animals such as motor learning through the exploration of new toys or objects in the cage, and spatial learning as they learn the place of water bottles, food, and toys. In addition to other social learning mechanisms as the rats interact with each other. EEN improved motor performance on a beam-walk task in adult rats after sensorimotor cortex lesions (Held et al., 1985; Gentile et al., 1987; Rose et al., 1987). Rats housed in larger EEN cages demonstrated better recovery of motor function than standard housed rats (Johansson, 1996), largely due to the space available for exploration and other activities. EEN improved spatial learning in a water maze task after moderate (Hamm et al., 1996) and severe fluid percussion brain injury (Passineau et al., 2001). EEN housing also enhanced cell genesis and microglia proliferation in adult murine neocortices (Ehninger and Kempermann, 2003).
Interestingly, the learning ability of EEN-I rats was significantly better than NH-I rats after 2 weeks only; day 16 was the first day of BM and rats had no pre-training before bTBI. Moreover, the performance of EEN-I rats was slightly better than that of control rats (NH-S). The positive effect of EEN was observed throughout the entire length of the study. This early improvement in spatial memory performance suggests that the positive effect of EEN did not involve de novo neurogenesis but rather the mitigation of the pathological processes induced by bTBI. Housing animals in EEN results in other complex molecular, morphological, and functional changes in the brain; these include increased synaptic plasticity and neuronal survival (Will et al., 2004). EEN can also induce the expression of various trophic factors and mitigate neuroinflammation (Shum et al., 2007) in addition to increasing the number of synapses per neuron and dendritic branching in the HC (Juraska et al., 1985; Juraska and Kopcik, 1988).
Some of our proteomics and immunohistochemical findings are consistent with the conclusion that EEN can exert its positive effects in a neurogenesis-independent manner. In our experiment, EEN normalized tau protein levels in the DHC but not in the VHC. Tau protein is a microtubule-associated protein that is involved in microtubule assembly and stabilization and is predominantly present in neurons and axons (Wilhelmsen, 1999); damaged axons may be an important source of amyloid-beta (Aβ) and Tau proteins following TBI (Marklund et al., 2009). Tau can become toxic and in turn lead to neuronal cell death. Therefore, increased Tau is a pathological hallmark of many neurodegenerative disorders (Lasagna-Reeves et al., 2010). The normalization of Tau levels by EEN suggests that EEN has a positive effect on axonal damage, a hallmark of bTBI (Buki and Povlishock, 2006; Farkas and Povlishock, 2007). Axonal damage can then result in neuronal malfunction and increased neuronal cell death.
Enriched environment specifically normalized VEGF levels in the DHC but not in the VHC. VEGF is a signaling protein that promotes the development of new blood vessels (Neufeld et al., 1999), regulates vascular permeability (Kaur and Ling, 2008), is associated with areas of growth or healing (Krum and Khaibullina, 2003), and acts as a positive regulator of adult de novo hippocampal neurogenesis (Rosenstein and Krum, 2004; Yasuhara et al., 2004). VEGF is significantly upregulated by TBI (Jin et al., 2002; Lee and Agoston, 2009, 2010). We previously found that VEGF promotes survival in the injured brain by blocking apoptotic cell death of de novo hippocampal neurons rather than a proliferative factor (Lee and Agoston, 2009). Due to its complex role in de novo neurogenesis and in other aspects of the post-injury process, changes in VEGF levels in the DHC compared to the VHC are difficult to interpret without additional studies.
Independent of housing conditions, IL-6 levels remained elevated after injury in the DHC, but were significantly lowered in the VHC of EEN animals. We found that IFNγ was elevated in the NH-I group, but decreased to control levels in the EEN-I group. These inflammatory molecules can have direct or indirect effects like increased neurotoxicity (Minagar et al., 2002). IL-6 is also a potent inhibitor of hippocampal de novo neurogenesis (Vallieres et al., 2002). The role of inflammation, including the role of microglia in the post-injury recovery process is rather complex (Barron, 1995; Kempermann and Neumann, 2003). Thus, whether elevated IL-6 levels contribute to the suppression of de novo neurogenesis in the DHC after bTBI remains to be established.
Our histological data showed brain region specific changes in GFAP immunoreactivity. Interestingly, housing conditions had no effect on GFAP immunoreactivity in the DHC, the VHC, or the AD at 71 days post-injury. However, housing injured animals in EEN resulted in an apparent decrease of GFAP positive cells in the VHC. Increased numbers of reactive astrocytes and elevated GFAP expression are hallmarks of CNS neurotrauma (Ridet et al., 1997; Dihne et al., 2001; Kernie et al., 2001). The role of elevated GFAP immunoreactivity and the presence of stellar astroglia in CNS injury is rather complex. Astrocytic response can either reflect reparative or pathological processes depending on the time elapsed after injury (Eng and Ghirnikar, 1994; Mueller et al., 2005). There is evidence indicating a protective role of astrogliosis in reducing the toxic effects of extracellular glutamate and enabling barrier reconstruction (Buffo et al., 2010). Elevated GFAP immunoreactivity in the VHC of injured animals may reflect reparative mechanisms that may involve de novo neurogenesis for the restoration of anxiety levels (Alvarez-Buylla and Garcia-Verdugo, 2002).
The full maturation and functional integration of de novo granule cells requires ∼6–8 weeks in the adult rodent brain (Kempermann et al., 2004). Newborn neurons, marked by DCX expression, migrate from the subgranular layer (SGL) to the granular cell layer (GCL) where many of the surviving neurons differentiate into granule cells (Altman and Das, 1965; Cameron et al., 1993). Some of the differentiated granule cells are then integrated into the existing hippocampal circuitry where they contribute to hippocampal function in the normal adult brain (Shors et al., 2001; Saxe et al., 2006). The rate of de novo hippocampal neurogenesis can be significantly upregulated by environmental conditions like EEN (Kempermann et al., 1997).
The DCX has been shown to be a specific, reliable marker that reflects adult neurogenesis levels and its modulation (Couillard-Despres et al., 2005). Importantly, the analysis of neurogenesis through the detection of DCX does not require in vivo labeling of proliferating cells. Even though it has been accepted that an increase in DCX+ cells indicate increased de novo hippocampal neurogenesis, DCX is only transiently expressed by de novo neurons (Kempermann and Gage, 2000; Kempermann, 2002a). Our finding that DCX immunoreactivity was elevated in the VHC of NH-I animals can be interpreted as increased de novo neurogenesis in NH-I animals compared to EEN-I animals. However, an alternative interpretation can be that in EEN animals the neurogenetic process already passed the DCX positive phase. Thus, a more detailed BrdU/Prox1 double immunohistochemical and stereological quantification of the histology results is required to determine changes in de novo neurogenesis after EEN and blast.
In summary, our data suggests that complex neurogenesis dependent and independent processes are involved in functional recovery after bTBI. EEN appears to have a positive and relatively fast-acting effect on the restoration of spatial memory after bTBI probably through a neurogenesis-independent process. In contrast, post-injury normalization of anxiety levels seems to be independent of EEN but likely involves de novo neurogenesis. The latter being much longer than previously thought and investigated. Additional histological and proteomics analyses at multiple post-injury time points as well as the use of additional markers (e.g., quantitative BrdU/Prox1 double label immunohistochemistry) would clarify the precise involvement of de novo neurogenesis in the various aspects of the regenerative process following bTBI.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Neurotrauma Team (WRAIR) for their technical help during the blast exposures; Dr. David Jacobowitz (USU) for his help in brain micropunch dissections; Ms. Cara Olsen (USU) for her help in statistical analysis; Drs. Neil Grunberg and John Wu (USU) for their input in designing and interpreting the behavioral experiments. The work was supported by VA grant # B5044R.
References
Abrous, D. N., Koehl, M., and Le Moal, M. (2005). Adult neurogenesis: from precursors to network and physiology. Physiol. Rev. 85, 523–569.
Agoston, D. V., Gyorgy, A., Eidelman, O., and Pollard, H. B. (2009). Proteomic biomarkers for blast neurotrauma: targeting cerebral edema, inflammation, and neuronal death cascades. J. Neurotrauma 26, 901–911.
Aimone, J. B., Deng, W., and Gage, F. H. (2010). Adult neurogenesis: integrating theories and separating functions. Trends Cogn. Sci. 14, 325–337.
Altman, J., and Das, G. D. (1965). Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J. Comp. Neurol. 124, 319–335.
Alvarez-Buylla, A., and Garcia-Verdugo, J. M. (2002). Neurogenesis in adult subventricular zone. J. Neurosci. 22, 629–634.
Barkho, B. Z., Song, H., Aimone, J. B., Smrt, R. D., Kuwabara, T., Nakashima, K., Gage, F. H., and Zhao, X. (2006). Identification of astrocyte-expressed factors that modulate neural stem/progenitor cell differentiation. Stem Cells Dev. 15, 407–421.
Barnes, C. A. (1979). Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J. Comp. Physiol. Psychol. 93, 74–104.
Berrocal, Y., Pearse, D. D., Singh, A., Andrade, C. M., Mcbroom, J. S., Puentes, R., and Eaton, M. J. (2007). Social and environmental enrichment improves sensory and motor recovery after severe contusive spinal cord injury in the rat. J. Neurotrauma 24, 1761–1772.
Bremner, J. D. (2007). Functional neuroimaging in post-traumatic stress disorder. Expert Rev. Neurother. 7, 393–405.
Briones, T. L., Rogozinska, M., and Woods, J. (2009). Environmental experience modulates ischemia-induced amyloidogenesis and enhances functional recovery. J. Neurotrauma 26, 613–625.
Bruns, J. Jr., and Hauser, W. A. (2003). The epidemiology of traumatic brain injury: a review. Epilepsia 44(Suppl.), 2.
Buchhold, B., Mogoanta, L., Suofu, Y., Hamm, A., Walker, L., Kessler, C., and Popa-Wagner, A. (2007). Environmental enrichment improves functional and neuropathological indices following stroke in young and aged rats. Restor. Neurol. Neurosci. 25, 467–484.
Buffo, A., Rolando, C., and Ceruti, S. (2010). Astrocytes in the damaged brain: molecular and cellular insights into their reactive response and healing potential. Biochem. Pharmacol. 79, 77–89.
Buki, A., and Povlishock, J. T. (2006). All roads lead to disconnection? – Traumatic axonal injury revisited. Acta Neurochir. (Wien) 148, 181–193. [Discussion 193–184].
Cacci, E., Claasen, J. H., and Kokaia, Z. (2005). Microglia-derived tumor necrosis factor-alpha exaggerates death of newborn hippocampal progenitor cells in vitro. J. Neurosci. Res. 80, 789–797.
Cameron, H. A., Woolley, C. S., Mcewen, B. S., and Gould, E. (1993). Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. Neuroscience 56, 337–344.
Cao, L., Jiao, X., Zuzga, D. S., Liu, Y., Fong, D. M., Young, D., and During, M. J. (2004). VEGF links hippocampal activity with neurogenesis, learning and memory. Nat. Genet. 36, 827–835.
Carobrez, A. P., and Bertoglio, L. J. (2005). Ethological and temporal analyses of anxiety-like behavior: the elevated plus-maze model 20 years on. Neurosci. Biobehav. Rev. 29, 1193–1205.
Cernak, I., Merkle, A. C., Koliatsos, V. E., Bilik, J. M., Luong, Q. T., Mahota, T. M., Xu, L., Slack, N., Windle, D., and Ahmed, F. A. (2010). The pathobiology of blast injuries and blast-induced neurotrauma as identified using a new experimental model of injury in mice. Neurobiol. Dis. 41, 538–551.
Cernak, I., Wang, Z., Jiang, J., Bian, X., and Savic, J. (2001). Ultrastructural and functional characteristics of blast injury-induced neurotrauma. J. Trauma. 50, 695–706.
Chirumamilla, S., Sun, D., Bullock, M. R., and Colello, R. J. (2002). Traumatic brain injury induced cell proliferation in the adult mammalian central nervous system. J. Neurotrauma 19, 693–703.
Couillard-Despres, S., Winner, B., Schaubeck, S., Aigner, R., Vroemen, M., Weidner, N., Bogdahn, U., Winkler, J., Kuhn, H. G., and Aigner, L. (2005). Doublecortin expression levels in adult brain reflect neurogenesis. Eur. J. Neurosci. 21, 1–14.
Dahlqvist, P., Ronnback, A., Bergstrom, S. A., Soderstrom, I., and Olsson, T. (2004). Environmental enrichment reverses learning impairment in the Morris water maze after focal cerebral ischemia in rats. Eur. J. Neurosci. 19, 2288–2298.
Dash, P. K., Mach, S. A., and Moore, A. N. (2001). Enhanced neurogenesis in the rodent hippocampus following traumatic brain injury. J. Neurosci. Res. 63, 313–319.
Deng, W., Aimone, J. B., and Gage, F. H. (2010). New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat. Rev. Neurosci. 11, 339–350.
Dhanushkodi, A., Bindu, B., Raju, T. R., and Kutty, B. M. (2007). Exposure to enriched environment improves spatial learning performances and enhances cell density but not choline acetyltransferase activity in the hippocampus of ventral subicular-lesioned rats. Behav. Neurosci. 121, 491–500.
Dihne, M., Block, F., Korr, H., and Topper, R. (2001). Time course of glial proliferation and glial apoptosis following excitotoxic CNS injury. Brain Res. 902, 178–189.
Doll, H., Truebel, H., Kipfmueller, F., Schaefer, U., Neugebauer, E. A., Wirth, S., and Maegele, M. (2009). Pharyngeal selective brain cooling improves neurofunctional and neurocognitive outcome after fluid percussion brain injury in rats. J. Neurotrauma 26, 235–242.
Ehninger, D., and Kempermann, G. (2003). Regional effects of wheel running and environmental enrichment on cell genesis and microglia proliferation in the adult murine neocortex. Cereb. Cortex 13, 845–851.
Einon, D. F., Morgan, M. J., and Will, B. E. (1980). Effects of post-operative environment on recovery from dorsal hippocampal lesions in young rats: tests of spatial memory and motor transfer. Q. J. Exp. Psychol. 32, 137–148.
Farkas, O., and Povlishock, J. T. (2007). Cellular and subcellular change evoked by diffuse traumatic brain injury: a complex web of change extending far beyond focal damage. Prog. Brain Res. 161, 43–59.
Fischer, F. R., and Peduzzi, J. D. (2007). Functional recovery in rats with chronic spinal cord injuries after exposure to an enriched environment. J. Spinal. Cord Med. 30, 147–155.
Gentile, A. M., Beheshti, Z., and Held, J. M. (1987). Enrichment versus exercise effects on motor impairments following cortical removals in rats. Behav. Neural Biol. 47, 321–332.
Gentilini, M., Nichelli, P., Schoenhuber, R., Bortolotti, P., Tonelli, L., Falasca, A., and Merli, G. A. (1985). Neuropsychological evaluation of mild head injury. J. Neurol. Neurosurg. Psychiatr. 48, 137–140.
Gross, C., and Hen, R. (2004). Genetic and environmental factors interact to influence anxiety. Neurotox. Res. 6, 493–501.
Gyorgy, A. B., Walker, J., Wingo, D., Eidelman, O., Pollard, H. B., Molnar, A., and Agoston, D. V. (2010). Reverse phase protein microarray technology in traumatic brain injury. J. Neurosci. Methods. 192, 96–101.
Hagg, T. (2005). Molecular regulation of adult CNS neurogenesis: an integrated view. Trends Neurosci. 28, 589–595.
Hallbergson, A. F., Gnatenco, C., and Peterson, D. A. (2003). Neurogenesis and brain injury: managing a renewable resource for repair. J. Clin. Invest. 112, 1128–1133.
Hamm, R. J., Temple, M. D., O’dell, D. M., Pike, B. R., and Lyeth, B. G. (1996). Exposure to environmental complexity promotes recovery of cognitive function after traumatic brain injury. J. Neurotrauma 13, 41–47.
Harrison, F. E., Hosseini, A. H., and Mcdonald, M. P. (2009). Endogenous anxiety and stress responses in water maze and Barnes maze spatial memory tasks. Behav. Brain Res. 198, 247–251.
Hebb, D. O. (1947). The effects of early experience on problem-solving at maturity. Am. Psychol. 2, 306–307.
Held, J. M., Gordon, J., and Gentile, A. M. (1985). Environmental influences on locomotor recovery following cortical lesions in rats. Behav. Neurosci. 99, 678–690.
Henke, P. G. (1990). Hippocampal pathway to the amygdala and stress ulcer development. Brain Res. Bull. 25, 691–695.
Hoffman, A. N., Malena, R. R., Westergom, B. P., Luthra, P., Cheng, J. P., Aslam, H. A., Zafonte, R. D., and Kline, A. E. (2008). Environmental enrichment-mediated functional improvement after experimental traumatic brain injury is contingent on task-specific neurobehavioral experience. Neurosci. Lett. 431, 226–230.
Jin, K., Zhu, Y., Sun, Y., Mao, X. O., Xie, L., and Greenberg, D. A. (2002). Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 99, 11946–11950.
Johansson, B. B. (1996). Functional outcome in rats transferred to an enriched environment 15 days after focal brain ischemia. Stroke 27, 324–326.
Johansson, B. B., and Ohlsson, A. L. (1996). Environment, social interaction, and physical activity as determinants of functional outcome after cerebral infarction in the rat. Exp. Neurol. 139, 322–327.
Juraska, J. M., Fitch, J. M., Henderson, C., and Rivers, N. (1985). Sex differences in the dendritic branching of dentate granule cells following differential experience. Brain Res. 333, 73–80.
Juraska, J. M., and Kopcik, J. R. (1988). Sex and environmental influences on the size and ultrastructure of the rat corpus callosum. Brain Res. 450, 1–8.
Kaur, C., and Ling, E. A. (2008). Blood brain barrier in hypoxic-ischemic conditions. Curr. Neurovasc. Res. 5, 71–81.
Kaur, C., Singh, J., Lim, M. K., Ng, B. L., Yap, E. P., and Ling, E. A. (1997). Ultrastructural changes of macroglial cells in the rat brain following an exposure to a non-penetrative blast. Ann. Acad. Med. Singapore 26, 27–29.
Kempermann, G. (2002a). Regulation of adult hippocampal neurogenesis – implications for novel theories of major depression. Bipolar Disord. 4, 17–33.
Kempermann, G. (2002b). Why new neurons? Possible functions for adult hippocampal neurogenesis. J. Neurosci. 22, 635–638.
Kempermann, G., and Gage, F. H. (2000). Neurogenesis in the adult hippocampus. Novartis Found. Symp. 231, 220–235. [discussion 235–241, 302–226].
Kempermann, G., and Kronenberg, G. (2003). Depressed new neurons – adult hippocampal neurogenesis and a cellular plasticity hypothesis of major depression. Biol. Psychiatry 54, 499–503.
Kempermann, G., Kuhn, H. G., and Gage, F. H. (1997). More hippocampal neurons in adult mice living in an enriched environment. Nature 386, 493–495.
Kempermann, G., and Neumann, H. (2003). Neuroscience. Microglia: the enemy within? Science 302, 1689–1690.
Kempermann, G., Wiskott, L., and Gage, F. H. (2004). Functional significance of adult neurogenesis. Curr. Opin. Neurobiol. 14, 186–191.
Kernie, S. G., Erwin, T. M., and Parada, L. F. (2001). Brain remodeling due to neuronal and astrocytic proliferation after controlled cortical injury in mice. J. Neurosci. Res. 66, 317–326.
Kline, A. E., Wagner, A. K., Westergom, B. P., Malena, R. R., Zafonte, R. D., Olsen, A. S., Sozda, C. N., Luthra, P., Panda, M., Cheng, J. P., and Aslam, H. A. (2007). Acute treatment with the 5-HT(1A) receptor agonist 8-OH-DPAT and chronic environmental enrichment confer neurobehavioral benefit after experimental brain trauma. Behav. Brain Res. 177, 186–194.
Kozorovitskiy, Y., and Gould, E. (2003). Adult neurogenesis: a mechanism for brain repair? J. Clin. Exp. Neuropsychol. 25, 721–732.
Krum, J. M., and Khaibullina, A. (2003). Inhibition of endogenous VEGF impedes revascularization and astroglial proliferation: roles for VEGF in brain repair. Exp. Neurol. 181, 241–257.
Kwon, S.-K. C., Kovesdi, E., Gyorgy, A. B., Wingo, D., Kamnaksh, A., Walker, J., Long, J. B., and Agoston, D. V. (2011). Stress and traumatic brain injury; a behavioral, proteomics and histological study. Front. Neurol. 2:12. doi: 10.3389/fneur.2011.00012
Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Guerrero-Muoz, M. J., Jackson, G. R., and Kayed, R. (2010). Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49, 10039–10041.
Lee, C., and Agoston, D. V. (2009). Inhibition of VEGF receptor 2 increased cell death of dentate hilar neurons after traumatic brain injury. Exp. Neurol. 220, 400–403.
Lee, C., and Agoston, D. V. (2010). Vascular endothelial growth factor is involved in mediating increased de novo hippocampal neurogenesis in response to traumatic brain injury. J. Neurotrauma 27, 541–553.
Liberzon, I., and Sripada, C. S. (2008). The functional neuroanatomy of PTSD: a critical review. Prog. Brain Res. 167, 151–169.
Ling, G., Bandak, F., Armonda, R., Grant, G., and Ecklund, J. (2009). Explosive blast neurotrauma. J. Neurotrauma 26, 815–825.
Liu, Q., Li, B., Zhu, H. Y., Wang, Y. Q., Yu, J., and Wu, G. C. (2010a). Glia atrophy in the hippocampus of chronic unpredictable stress-induced depression model rats is reversed by electroacupuncture treatment. J. Affect. Disord. 128, 309–313.
Liu, Y. R., Cardamone, L., Hogan, R. E., Gregoire, M. C., Williams, J. P., Hicks, R. J., Binns, D., Koe, A., Jones, N. C., Myers, D. E., O’brien, T. J., and Bouilleret, V. (2010b). Progressive metabolic and structural cerebral perturbations after traumatic brain injury: an in vivo imaging study in the rat. J. Nucl. Med. 51, 1788–1795.
Long, J. B., Bentley, T. L., Wessner, K. A., Cerone, C., Sweeney, S., and Bauman, R. A. (2009). Blast overpressure in rats: recreating a battlefield injury in the laboratory. J. Neurotrauma 26, 827–840.
Maegele, M., Lippert-Gruener, M., Ester-Bode, T., Sauerland, S., Schafer, U., Molcany, M., Lefering, R., Bouillon, B., Neiss, W. F., Angelov, D. N., Klug, N., Mcintosh, T. K., and Neugebauer, E. A. (2005). Reversal of neuromotor and cognitive dysfunction in an enriched environment combined with multimodal early onset stimulation after traumatic brain injury in rats. J. Neurotrauma 22, 772–782.
Marklund, N., Blennow, K., Zetterberg, H., Ronne-Engstrom, E., Enblad, P., and Hillered, L. (2009). Monitoring of brain interstitial total tau and beta amyloid proteins by microdialysis in patients with traumatic brain injury. J. Neurosurg. 110, 1227–1237.
Minagar, A., Shapshak, P., Fujimura, R., Ownby, R., Heyes, M., and Eisdorfer, C. (2002). The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J. Neurol. Sci. 202, 13–23.
Ming, G. L., and Song, H. (2005). Adult neurogenesis in the mammalian central nervous system. Annu. Rev. Neurosci. 28, 223–250.
Mora, F., Segovia, G., and Del Arco, A. (2007). Aging, plasticity and environmental enrichment: structural changes and neurotransmitter dynamics in several areas of the brain. Brain Res. Rev. 55, 78–88.
Moser, M. B., Moser, E. I., Forrest, E., Andersen, P., and Morris, R. G. (1995). Spatial learning with a minislab in the dorsal hippocampus. Proc. Natl. Acad. Sci. U.S.A. 92, 9697–9701.
Mueller, F. J., Mckercher, S. R., Imitola, J., Loring, J. F., Yip, S., Khoury, S. J., and Snyder, E. Y. (2005). At the interface of the immune system and the nervous system: how neuroinflammation modulates the fate of neural progenitors in vivo. Ernst Schering Res. Found. Workshop 53, 83–114.
Neufeld, G., Cohen, T., Gengrinovitch, S., and Poltorak, Z. (1999). Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 13, 9–22.
Parent, J. M. (2003). Injury-induced neurogenesis in the adult mammalian brain. Neuroscientist 9, 261–272.
Passineau, M. J., Green, E. J., and Dietrich, W. D. (2001). Therapeutic effects of environmental enrichment on cognitive function and tissue integrity following severe traumatic brain injury in rats. Exp. Neurol. 168, 373–384.
Paxinos, G., Kus, L., Ashwell, K. W. S., and Watson, C. (1999). Chemoarchitectonic Atlas of the Rat Forebrain. San Diego: Academic Press.
Pham, T. M., Soderstrom, S., Winblad, B., and Mohammed, A. H. (1999). Effects of environmental enrichment on cognitive function and hippocampal NGF in the non-handled rats. Behav. Brain Res. 103, 63–70.
Ponsford, J. L., Olver, J. H., and Curran, C. (1995). A profile of outcome: 2 years after traumatic brain injury. Brain Inj. 9, 1–10.
Puurunen, K., and Sivenius, J. (2002). Influence of enriched environment on spatial learning following cerebral insult. Rev. Neurosci. 13, 347–364.
Quinn, R. (2005). Comparing rat’s to human’s age: how old is my rat in people years? Nutrition 21, 775–777.
Ridet, J. L., Malhotra, S. K., Privat, A., and Gage, F. H. (1997). Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 20, 570–577.
Rose, F. D., Davey, M. J., Love, S., and Dell, P. A. (1987). Environmental enrichment and recovery from contralateral sensory neglect in rats with large unilateral neocortical lesions. Behav. Brain Res. 24, 195–202.
Rosenstein, J. M., and Krum, J. M. (2004). New roles for VEGF in nervous tissue – beyond blood vessels. Exp. Neurol. 187, 246–253.
Rosenzweig, M. R., and Bennett, E. L. (1996). Psychobiology of plasticity: effects of training and experience on brain and behavior. Behav. Brain Res. 78, 57–65.
Sahay, A., and Hen, R. (2007). Adult hippocampal neurogenesis in depression. Nat. Neurosci. 10, 1110–1115.
Salzberg, M., Kumar, G., Supit, L., Jones, N. C., Morris, M. J., Rees, S., and O’Brien, T. J. (2007). Early postnatal stress confers enduring vulnerability to limbic epileptogenesis. Epilepsia 48, 2079–2085.
Saxe, M. D., Battaglia, F., Wang, J. W., Malleret, G., David, D. J., Monckton, J. E., Garcia, A. D., Sofroniew, M. V., Kandel, E. R., Santarelli, L., Hen, R., and Drew, M. R. (2006). Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc. Natl. Acad. Sci. U.S.A. 103, 17501–17506.
Schoenhuber, R., and Gentilini, M. (1988). Anxiety and depression after mild head injury: a case control study. J. Neurol. Neurosurg. Psychiatr. 51, 722–724.
Shors, T. J., Miesegaes, G., Beylin, A., Zhao, M., Rydel, T., and Gould, E. (2001). Neurogenesis in the adult is involved in the formation of trace memories. Nature 410, 372–376.
Shum, F. W., Wu, L. J., Zhao, M. G., Toyoda, H., Xu, H., Ren, M., Pinaud, R., Ko, S. W., Lee, Y. S., Kaang, B. K., and Zhuo, M. (2007). Alteration of cingulate long-term plasticity and behavioral sensitization to inflammation by environmental enrichment. Learn. Mem. 14, 304–312.
Sun, D., Mcginn, M. J., Zhou, Z., Harvey, H. B., Bullock, M. R., and Colello, R. J. (2007). Anatomical integration of newly generated dentate granule neurons following traumatic brain injury in adult rats and its association to cognitive recovery. Exp. Neurol. 204, 264–272.
Taber, K. H., Warden, D. L., and Hurley, R. A. (2006). Blast-related traumatic brain injury: what is known? J. Neuropsychiatry Clin. Neurosci. 18, 141–145.
Tagliaferri, F., Compagnone, C., Korsic, M., Servadei, F., and Kraus, J. (2006). A systematic review of brain injury epidemiology in Europe. Acta Neurochir. (Wien) 148, 255–256.
Tashiro, A., Makino, H., and Gage, F. H. (2007). Experience-specific functional modification of the dentate gyrus through adult neurogenesis: a critical period during an immature stage. J. Neurosci. 27, 3252–3259.
Tees, R. C., Buhrmann, K., and Hanley, J. (1990). The effect of early experience on water maze spatial learning and memory in rats. Dev. Psychobiol. 23, 427–439.
Tischler, L., Brand, S. R., Stavitsky, K., Labinsky, E., Newmark, R., Grossman, R., Buchsbaum, M. S., and Yehuda, R. (2006). The relationship between hippocampal volume and declarative memory in a population of combat veterans with and without PTSD. Ann. N. Y. Acad. Sci. 1071, 405–409.
Vallieres, L., Campbell, I. L., Gage, F. H., and Sawchenko, P. E. (2002). Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J. Neurosci. 22, 486–492.
Van Praag, H., Kempermann, G., and Gage, F. H. (2000). Neural consequences of environmental enrichment. Nat. Rev. Neurosci. 1, 191–198.
Walf, A. A., and Frye, C. A. (2007). The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat. Protoc. 2, 322–328.
Wang, Z., Neylan, T. C., Mueller, S. G., Lenoci, M., Truran, D., Marmar, C. R., Weiner, M. W., and Schuff, N. (2010). Magnetic resonance imaging of hippocampal subfields in posttraumatic stress disorder. Arch. Gen. Psychiatry 67, 296–303.
Warden, D. (2006). Military TBI during the Iraq and Afghanistan wars. J. Head Trauma Rehabil. 21, 398–402.
Warden, D. L., and French, L. (2005). Traumatic brain injury in the war zone. N. Engl. J. Med. 353, 633–634.
Whishaw, I. Q., Zaborowski, J. A., and Kolb, B. (1984). Postsurgical enrichment aids adult hemidecorticate rats on a spatial navigation task. Behav. Neural Biol. 42, 183–190.
Will, B., Galani, R., Kelche, C., and Rosenzweig, M. R. (2004). Recovery from brain injury in animals: relative efficacy of environmental enrichment, physical exercise or formal training (1990–2002). Prog. Neurobiol. 72, 167–182.
Woon, F. L., Sood, S., and Hedges, D. W. (2010). Hippocampal volume deficits associated with exposure to psychological trauma and posttraumatic stress disorder in adults: a meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 34, 1181–1188.
Wright, R. L., and Conrad, C. D. (2008). Enriched environment prevents chronic stress-induced spatial learning and memory deficits. Behav. Brain Res. 187, 41–47.
Yasuhara, T., Shingo, T., Kobayashi, K., Takeuchi, A., Yano, A., Muraoka, K., Matsui, T., Miyoshi, Y., Hamada, H., and Date, I. (2004). Neuroprotective effects of vascular endothelial growth factor (VEGF) upon dopaminergic neurons in a rat model of Parkinson’s disease. Eur. J. Neurosci. 19, 1494–1504.
Yehuda, R., and Ledoux, J. (2007). Response variation following trauma: a translational neuroscience approach to understanding PTSD. Neuron 56, 19–32.
Keywords: neurogenesis, enriched environment, traumatic brain injury, memory, anxiety, hippocampus, histology, proteomics
Citation: Kovesdi E, Gyorgy AB, Kwon S-KC, Wingo DL, Kamnaksh A, Long JB, Kasper CE and Agoston DV (2011) The effect of enriched environment on the outcome of traumatic brain injury; a behavioral, proteomics, and histological study. Front. Neurosci. 5:42. doi:10.3389/fnins.2011.00042
Received: 31 December 2010;
Accepted: 15 March 2011;
Published online: 01 April 2011.
Edited by:
Gerd Kempermann, Center for Regenerative Therapies, GermanyReviewed by:
Brian R. Christie, University of British Columbia, CanadaDong Sun, Virginia Commonwealth University, USA
Copyright: © 2011 Kovesdi, Gyorgy, Kwon, Wingo, Kamnaksh, Long, Kasper and Agoston. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Denes V. Agoston, Department of Anatomy, Physiology and Genetics, School of Medicine, Uniformed Services University, 4301 Jones Bridge Road, Bethesda, MD 20814, USA. e-mail:dmFnb3N0b25AdXN1aHMuZWR1