 Phyllis Pugh
Phyllis Pugh Elena Adlaf Chuan-Sheng Zhao
Elena Adlaf Chuan-Sheng Zhao Sean Markwardt Cristin Gavin Jacques Wadiche
Sean Markwardt Cristin Gavin Jacques Wadiche Linda Overstreet-Wadiche*
Linda Overstreet-Wadiche*
- McKnight Brain Institute and Department of Neurobiology, University of Alabama at Birmingham, Birmingham, AL, USA
The production and integration of adult-generated neurons in the dentate gyrus is dramatically perturbed by a variety of pathological insults, including repetitive seizures and hypoxia/ischemia. Less is known about how insults affect early postnatal neurogenesis, during the developmental period when the majority of dentate neurons are produced. Here we tested how single episodes of hypoxia or chemically induced seizure activity in postnatal day 10 mice alter granule cell production and integration. Although neither insult was sufficient to alter the number of newborn neurons nor the population of proliferating cells, both treatments increased the dendritic complexity of newborn granule cells that were born around the time of the insult. Surprisingly, only the dendritic enhancement caused by hypoxia was associated with increased synaptic integration. These results suggest that alterations in dendritic integration can be dissociated from altered neural production and that integration appears to have a lower threshold for perturbation. Furthermore, newborn neurons in adult mice that experienced neonatal hypoxia had reduced dendritic length while having no alterations in number. Together these results suggest that single insults during the neonatal period can have both long- and short-term consequences for neuronal maturation.
Introduction
In the mammalian dentate gyrus (DG), neurogenesis persists through adulthood. Although their precise role in hippocampal-dependent functions is not clear, postnatal-generated granule cells (GCs) undergo a prolonged period of maturation during which they exhibit enhanced excitability and synaptic plasticity compared to mature GCs (Snyder et al., 2001; Schmidt-Hieber et al., 2004; Overstreet-Wadiche et al., 2006a; Ge et al., 2007). These properties could endow immature GCs with a distinct role in hippocampal network activity compared with the much larger population of mature GCs. Interestingly, the production and development of new GCs appears highly regulated by both physiological and pathological stimuli, supporting the idea that neurogenesis enables adaptive network plasticity in response to changing demands (Deisseroth et al., 2004). In fact, a variety of pathological conditions profoundly affect the production and development of adult-generated GCs, and considerable effort is underway to understand the functional consequences of these changes. Less is known about the regulation of neural production and integration of newborn GCs in the developing brain.
Epileptic seizures are strong triggers of neural proliferation in adult rodents (Parent et al., 1997). In chemically induced epilepsy models, acute enhancement of neurogenesis is accompanied by abnormalities in maturation of newborn neurons, including ectopic migration and aberrant dendritic structure, as well as accelerated synaptic integration (reviewed in Shapiro et al., 2008; Zhao and Overstreet-Wadiche, 2008). In the chronic phase of experimentally induced epilepsy, neurogenesis is sharply reduced (Hattiangady and Shetty, 2008). Abnormal neurogenesis in these models is thus predicted to contribute to cognitive impairments associated with epilepsy and perhaps to the establishment of network hyperexcitability (Scharfman, 2004). Brief global ischemia also triggers a robust transient increase in proliferation in the DG (Liu et al., 1998; reviewed in Kokaia and Lindvall, 2003), although abnormalities in the maturation of ischemia-induced new GCs have not been reported (Tanaka et al., 2004). Despite the large relative increase in neural production in these models, newborn GCs remain only a fraction of the total number of dentate GCs.
The majority of GCs are generated during the neonatal period, during a time when seizure susceptibility in rodents and humans is high and hypoxic injury is common (Jensen, 2009; Lawrence and Inder, 2010). In contrast to adult rodents, seizures, and hypoxia in young rodents can either reduce or enhance progenitor proliferation, depending on the age and paradigm (Sankar et al., 2000; McCabe et al., 2001; Fagel et al., 2006; Xiu-Yu et al., 2007). The basis of such differential regulation is not known, although one potential difference is the extent of neural injury associated with the insult. In adults, chemically induced prolonged seizures (status epilepticus) produce significant cell death in sensitive brain regions like the hippocampus, whereas little or no injury is produced by such seizures during the first two postnatal weeks when newborn neurons comprise a larger proportion of total cells (Holmes, 2002).
Here we examined how presumably mild neonatal insults affect the morphology and synaptic connectivity of newborn GCs, using a brief hypoxia paradigm that is reported to generate seizure activity in rats (Jensen et al., 1991b) as well as a single episode of kainate-induced status epilepticus. Our results demonstrate that such neonatal insults can alter the structural and functional integration of newborn GCs without affecting neuronal production.
Materials and Methods
Animals
Experiments were performed in heterozygous −13/+8 pro-opiomelanocortin (POMC)–EGFP mice (Cowley et al., 2001; Overstreet et al., 2004) maintained by out-breeding homozygous males with wild-type C57BL/6J females. Litters were housed with their dam until weaning at P21. All protocols were performed following review and approval by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham and were in accordance with NIH guidelines on the ethical use of experimental animals. All animals were kept under a constant 12 h light/dark cycle with ad libitum access to food and water.
Neonatal Insults
We used a model developed for hypoxia-induced seizures in rats (Jensen et al., 1991b; Koh et al., 2004; Sanchez et al., 2007). Briefly, pups at P10 were placed in an airtight chamber on heating pads. The O2 concentration in the chamber was lowered to 7–8% for 4 min, 6–7% for 8 min, then lowered by 1% per minute until the animals became apneic for 30 s. The chamber was then uncovered and exposed to room air. If animals became apneic before 5–7% O2 was achieved, pups were removed from the chamber for a 2-min recovery period, then returned to the chamber until the total time exposed to hypoxia was 14–16 min. Control animals were placed in the same chamber for a similar amount of time, but not subjected to the hypoxia treatment. Following treatment, control and experimental animals were returned to their dam.
For kainate-induced seizure induction, pups at P10 were injected subcutaneously with a single dose of kainic acid (1.45 mg/kg in saline, Ascent Scientific, UK). Seizures were scored using the Racine scale. All kainate-injected pups had behavioral seizures that lasted approximately 2 h. Age-matched control mice received 0.9% NaCl injections. None of the control mice displayed seizure activity.
Tissue Processing and Immunohistochemistry
At either P17 or P70, mice were anesthetized with 2,2,2-tribromoethanol (Avertin; Aldrich, St.Louis, MO, USA) and perfused transcardially with 0.9% NaCl followed by 4% paraformaldehyde in PBS. The brains were removed and post-fixed overnight. Horizontal sections through the hippocampus (50 μm) were cut on a vibratome and stored at −20°C in cryoprotection buffer (30% ethylene glycol, 20% glycerin, and 0.05 M NaPO4). All processing and analysis was performed by an investigator blinded to treatment conditions.
For GFP and Ki67 staining, free-floating sections were washed three times in TBS then incubated in a blocking buffer [TBS, 0.4% TritonX-100, 1% bovine serum albumin (BSA), and 10% normal goat serum] for 90 min. Sections were incubated in a primary antibody (rabbit polyclonal anti-Ki67, 1:300; overnight at 4°C; Abcam, Cambridge, MA, USA), then incubated in secondary antibody (goat anti-rabbit IgG Alexa 568, 1:200; overnight at 4°C; Invitrogen, Carlsbad, CA, USA). Following a brief wash, sections were incubated with Alexa 488-conjugated anti-GFP polyclonal antibody (1:1000; overnight at 4°C, Invitrogen). For propidium iodide staining, free-floating sections were washed three times in PBS then soaked in propidium iodide (1.5 mg/ml stock, 1:1000 in PBS) for 10 min. After another round of washing, sections were mounted on slides with Prolong Antifade (Invitrogen) and imaged with an Olympus FluoView 300 confocal microscope.
Stereological Quantification
Transverse sections (50 μm) were taken from the mid- to ventral-region of the DG consistent between each animal. After choosing a random start, every sixth slice was quantified. The number of GFP+ cells, proliferating cells, and the total number of neurons in the granule cell layer (GCL) were determined for each experimental group using the optical fractionator method (Stereo Investigator, MicroBrightField; West et al., 1991). Counting was performed on an Olympus BX-51 with a 100X oil-immersion lens (1.4 NA). Parameters for the set were determined that yielded a coefficient of error (CE) of less than 0.10 for each sample.
Dendrite Analysis
Neuronal morphology was traced from confocal image stacks using Neurolucida (v. 7, MicroBrightField, Inc., Williston, VT, USA). Cells with obvious truncations were excluded from analysis. Measurements included total dendrite length (TDL) and Sholl analyses of length, nodes, and intersections (at 5 μm intervals). Furthest extent of dendritic projections was determined by the furthest Sholl radius containing measurable dendrite length (i.e., rounded to the nearest 5 μm). To determine the location of GC somata, images were divided into five layers (hilus, inner GCL, middle GCL, outer GCL, molecular layer), and the location of each soma was scored for each image. Where possible, TDL, nodes, and dendritic extents were compared by one-way ANOVA with Bonferroni post-tests (Prism, Graph Pad Software, La Jolla, CA, USA), and their distributions were compared using Kolmogorov–Smirnov tests (performed at http://www.physics.csbsju.edu/stats/KS-test.html). Sholl analyses were compared using a two-way ANOVA with Bonferroni post-tests. For comparisons between P17 and P70 animals, unpaired t-tests, with Welch’s correction if necessary, were used.
Acute Hippocampal Slice Preparation
Acute hippocampal slices were prepared from P16–18 pups that were sacrificed by decapitation under isoflurane anesthesia. Brains were quickly removed and placed in cutting solution containing (in mM): 110 choline chloride, 26 D-glucose, 2.5 MgCl2, 2.5 KCl, 1.25 Na2PO4, 0.5 CaCl2, 1.3 Na-ascorbate, 3 Na-pyruvate, and 25 NaHCO3, bubbled with 95% O2/5% CO2 (4°C). Horizontal sections (350 μm) were prepared using a vibratome (3000EP, Vibratome, Bannockburn, IL, USA). Slices were incubated at 37°C for ∼30 min in recording ACSF solution containing (in mM): 125 NaCl, 2.5 KCl, 1.25 Na2PO4, 2 CaCl2, 1 MgCl2, 25 NaHCO3, and 25 D-glucose bubbled with 95%O2/5%CO2, then transferred to room temperature in the same solution.Electrophysiological Recordings
Slices were placed in a submersion chamber and superfused with oxygenated recording ACSF at room temperature. All recordings were performed by an investigator blinded to treatment conditions. Cells were visualized with custom-made Dodt Optics on an Olympus BS51W1 microscope and fluorescence illumination provided by a mercury lamp and GFP filter set (GFP-A Basic; Semrock). Whole-cell patch clamp recordings were acquired from either newborn POMC–GFP labeled GCs or unlabeled mature GCs (Markwardt et al., 2009). For recordings of spontaneous GABAA receptor-mediated postsynaptic currents (PSCs) at −70 mV in the DG, the internal solution contained (in mM): CsCl 129, MgCl2 2, EGTA 10, HEPES 10, Na-ATP 3, GTP 0.3 (pH 7.2). The input resistance and capacitance of newborn GCs, determined by current transients in response to 5 mV voltage steps, were not different across treatment groups. For recordings of IPSCs in CA1 pyramidal cells at +10 mV, pipettes contained (in mM): Cs-gluconate 123, NaCl 4, MgCl2 2, EGTA 10, HEPES 10, Na-ATP 4, GTP 0.3 (pH 7.2). Filled pipette resistance varied from 2–4 MΩ (mature cells) to 4–6 MΩ (newborn cells). All recordings were done at 22°C. Currents were filtered at 2 kHz and sampled at 10 kHz (MultiClamp 700B; Molecular Devices, Union City, CA, USA). Series resistance was uncompensated (10–25 MΩ), and experiments were discarded if substantial changes (> 20%) were observed. Voltages were not corrected for junction potentials. Spontaneous IPSCs (sIPSCs) and miniature IPSCs (mIPSCs) were detected and analyzed using the event detection package with a variable amplitude template in AxoGraph X (Axograph Scientific). We compared the average parameter (frequency, amplitude) from each cell within each treatment group, such that n = no. of cells. Mean values across treatment groups were compared with a Student’s t-test (two groups, unpaired) or ANOVA (three groups) with Bonferroni post hoc test, with p < 0.05.
NBQX (1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide, 10 μM), APV (DL-2-amino-5-phosphonovaleric acid, 100 μM), picrotoxin (PTX, 100 mM), tetrodotoxin (TTX, 0.5 μM), and 4-aminopyridine (4-AP, 100 μM) were diluted from stock solutions to final concentrations in ACSF. All drugs were purchased from Sigma-Aldrich (St. Louis, MO, USA) or Tocris Bioscience (Ellisville, MO, USA).
Results
We used transgenic mice with EGFP expression controlled by regulatory elements of the POMC gene to identify newborn GCs in the DG (Overstreet et al., 2004). Transient expression of POMC–GFP results in a loss of labeling in individual cells as their maturation proceeds, allowing identification of GCs that are ∼1–3 weeks postmitotic throughout the lifespan of the mouse. The immature neuronal morphology and physiological properties of POMC–GFP labeled cells indicate that they are postmitotic neurons rather than progenitors, consistent with a lack of co-labeling with BrdU at 1-day after administration and co-expression of the transcription factor that drives granule cell differentiation, NeuroD1 (Overstreet-Wadiche et al., 2006a). Thus, POMC–GFP labeling allows selective identification of newborn postmitotic GCs.
Neonatal Insults Enhance Dendritic Structure of Newborn GCs
Insults such as repetitive seizure activity in adult mice alter the migration, morphology, and synaptic integration of newly generated GCs (Zhao and Overstreet-Wadiche, 2008). To determine whether insults early in life similarly alter the development of newborn GCs, we measured the dendritic length and complexity of POMC–GFP labeled GCs in P17 mice that received insults at P10. We found that both hypoxia- and kainate-induced seizures increased the TDL of newborn GCs by 15 and 20%, respectively (Figures 1A–C). The most obvious change in dendrite structure was an increase in the extent of the dendrites through the molecular layer (Figures 1A,B,D). The mean furthest extent was 87 ± 3 μm in control (n = 27 cells from seven mice), 101 ± 4 μm in hypoxia treatment (n = 21 cells from six mice), and 105 ± 3 μm in kainate treatment (n = 30 cells from five mice, Figure 1D). Because the extent of the dendritic arbor was measured relative to the soma, it did not stem from changes in position of the cell bodies. In all three conditions, approximately 80% of the somata were located within the inner GC layer (control, 78.7%; hypoxia, 74.0%; KA, 80.6%; p = 0.13 by two-way ANOVA). Sholl analysis revealed that the difference in dendrite structure resulted primarily from longer dendrites located 75–105 μm from the cell body (Figure 1E). KA-treated animals exhibited a slight increase in cross-sectional soma area (86.8 ± 4.3 μm2, p < 0.05, ANOVA) compared to control animals (71.2 ± 2.8 μm2), an effect not seen in hypoxia-treated animals (72.8 ± 3.3 μm2). These results appear qualitatively similar to, although less dramatic than, the enhanced TDL observed in newborn GCs following pilocarpine-induced seizures in adults (Overstreet-Wadiche et al., 2006b) and suggest that insults alter the maturation of newborn GCs during an ontological period when a large number of GCs are being produced.
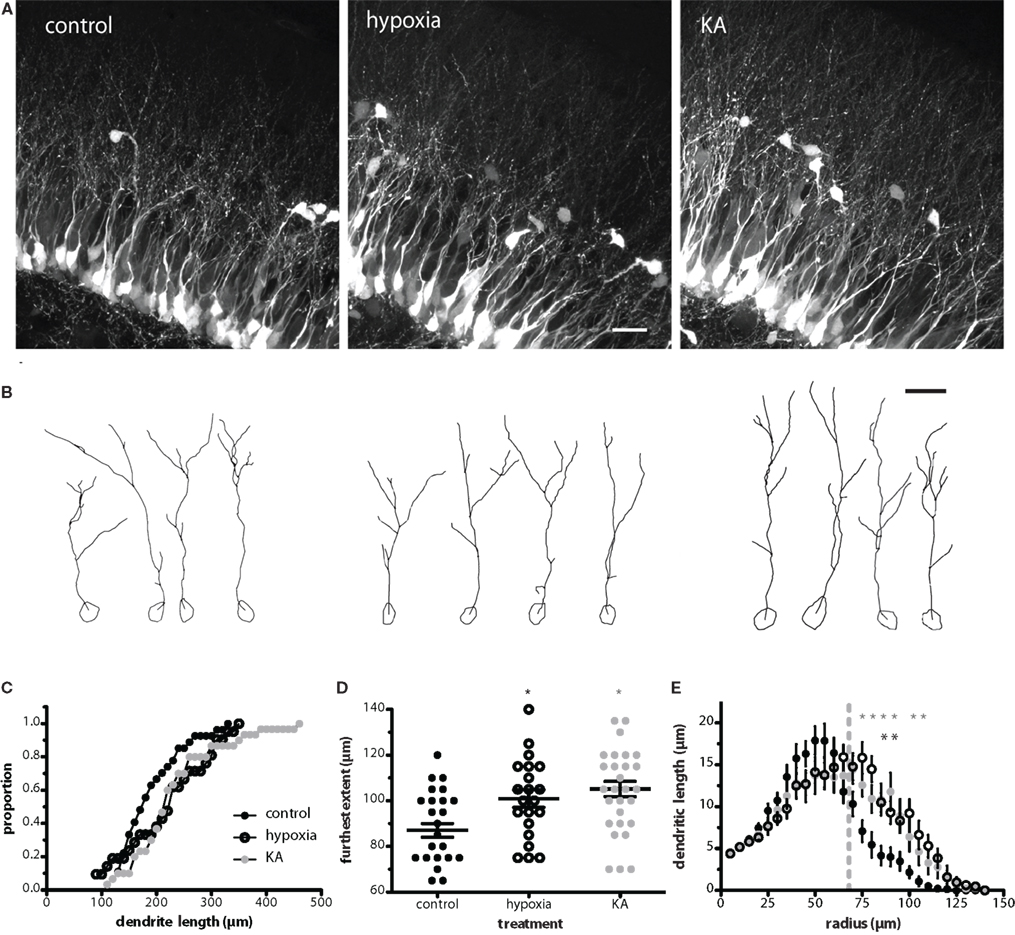
Figure 1. Neonatal insults enhance dendritic structure of newborn GCs. (A) Confocal images of newborn GCs at P17 illustrate the enhanced dendritic structure of newborn GCs following hypoxia and KA insults on P10. Images are maximum z-projections of 28 images taken at 0.5 μm intervals. Scale bar, 20 μm. (B) Examples of individual cells from Neurolucida reconstructions. The high density of labeled cells made it difficult to follow small branches, so quantification was repeated by two additional blinded reviewers, both whom acquired similar results. Scale bar, 20 μm. (C) Cumulative proportion of total dendrite lengths for control, hypoxia-, and KA-treated mice on P17. The TDL of the KA group was significantly increased over control (p < 0.05; Kolmogorov–Smirnov test). (D) The furthest extent of newborn neuron dendrites into the molecular layer was enhanced by both hypoxia and KA treatment (p < 0.05 and 0.001, respectively; one-way ANOVA with Bonferroni post-test). (E) Sholl analysis revealed that the difference in dendrite structure resulted primarily from longer dendrites located 75–105 μm from the cell body. Asterisks indicate significant differences from control; p < 0.05, two-way ANOVA with Bonferroni post-test). Gray line indicates the (average) depth of the GC layer.
Altered Synaptic Input to Newborn GCs
To determine whether enhanced dendrite length was associated with altered synaptic connectivity, we compared spontaneous synaptic input to newborn GCs in acute hippocampal slices from P16–18 mice representing the different treatment groups. GCs at the early developmental stage when POMC–GFP is expressed exhibit exclusively GABAergic synaptic input. Consistent with our previous studies in older and younger mice (Overstreet Wadiche et al., 2005; Overstreet-Wadiche et al., 2006a), newborn GCs in juvenile mice displayed a low frequency of spontaneous GABAergic synaptic currents that were blocked by the GABAA receptor antagonist, picrotoxin (see Figure 2A bottom traces). In slices from control mice, the fraction of cells with spontaneous events during the recording period (5/8 cells) and the overall frequency of events (0.003 ± 0.001 Hz, n = 8) was increased when network activity was enhanced by the K+ channel blocker 4-AP (100 μM; 8/8 cells, 0.028 ± 0.008 Hz), with no change in the amplitude of individual events (14.4 ± 0.3 pA, n = 5, vs. 12.5 ± 1.0 pA, n = 8). Newborn GCs from hypoxia-treated mice had an increased frequency of spontaneous events in control conditions (0.024 ± 0.009, n = 7; events detected in 6/7 cells, Figure 2A, right) with the same amplitude as in control mice (16.0 ± 1.1 pA, n = 6). In hypoxia-treated mice, 4-AP similarly enhanced the frequency of spontaneous events to 0.28 ± 0.09 (Figure 2C), with no significant change in amplitude (24 ± 4.7 pA, n = 7). Thus, the enhanced dendritic structure of newborn GCs in mice treated with hypoxia was associated with an elevated frequency of individual spontaneous synaptic currents under basal conditions as well as when network activity was enhanced with 4-AP (Figures 2B,C). Consistent with previously described synchronized activation of gap-junction-coupled interneuron networks (Michelson and Wong, 1994; Zsiros et al., 2007; Markwardt et al., 2009), 4-AP-induced large and long-lasting compound events that were blocked by picrotoxin in newborn GCs from both control and hypoxia-treated mice. Furthermore, these compound events in hypoxia-treated mice had the same frequency and amplitude (0.03 ± 0.004 Hz and 240 ± 65 pA, n = 9) as the compound events in control mice (0.03 ± 0.004 Hz and 237 ± 77 pA, n = 8).
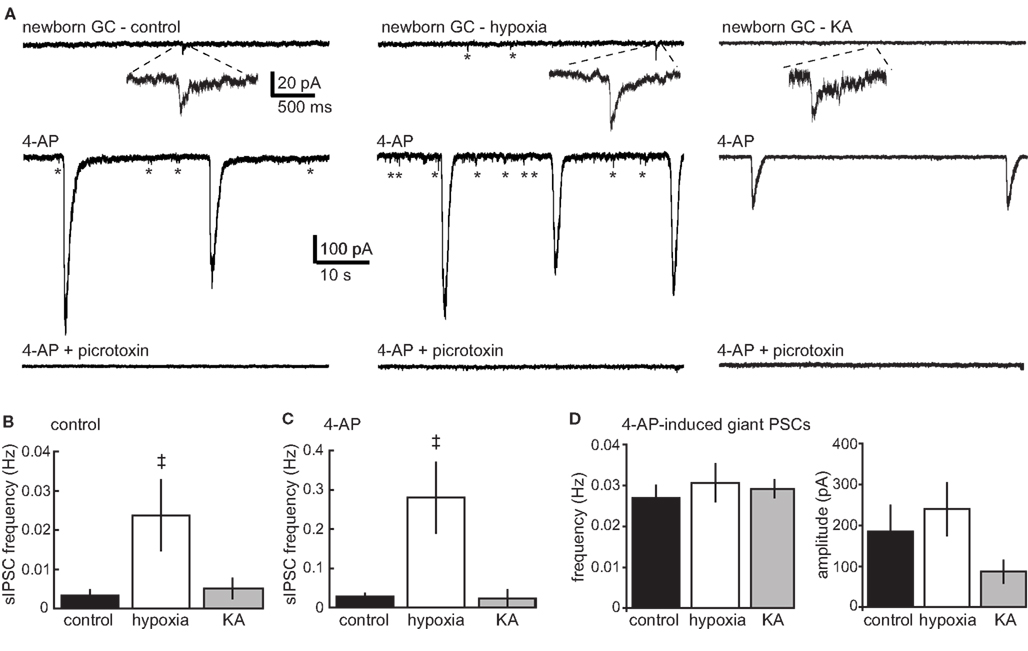
Figure 2. Enhanced GABAergic input to newborn GCs after hypoxia but not KA treatment. (A) Current traces from newborn GCs in control, hypoxia-, and KA-treated mice recorded on P17.Top traces show spontaneous synaptic events during a control period in the absence of receptor antagonists. Addition of the K+ channel blocker 4-AP (100 μM; middle traces) increased the frequency of spontaneous events (asterisks) in GCs from control and hypoxia-treated mice, but not in GCs from KA-treated mice. 4-AP also triggered large compound events in GCs from all groups. All spontaneous synaptic currents were blocked by picrotoxin (bottom traces). (B,C) The frequency of spontaneous events was increased in newborn GCs from hypoxia-treated mice. The double dagger (‡) indicates significantly different from control (ANOVA with Bonferroni post-test). The same results were obtained when cells with no detected events in the control condition were excluded from analysis (not shown). The frequency of spontaneous events was not increased in newborn GCs from KA-treated mice. (D) There was no difference in the frequency or amplitude of 4-AP-induced compound events (ANOVA, p = 0.18).
In contrast to the results following hypoxia treatment, the frequency of spontaneous synaptic currents in newborn GCs following KA seizures remained low. In KA-treated mice, only 4/10 cells displayed events resulting in an overall frequency of 0.005 ± 0.003 Hz (n = 10) that was not significantly different from the sIPSC frequency from control mice (Figure 2B), even if non-responding newborn GCs were excluded (0.013 ± 0.004 Hz, n = 4). The amplitude of individual events was similar to that in control and hypoxia-treated mice (13.7 ± 3.4 pA, n = 4). Furthermore, unlike control and hypoxia-treated mice, 4-AP did not enhance the frequency of individual sIPSCs in newborn GCs from KA-treated mice (to 0.024 ± 0.016 Hz, n = 10; p = 0.3; Wilcoxon paired; Figure 2C), although 4-AP did trigger large compound events with comparable amplitudes (87 ± 28 pA), and frequencies (0.03 ± 0.002 Hz, n = 9, Figures 2A,D). Even when comparing only cells exhibiting individual synaptic currents under basal conditions (0.013 ± 0.004 Hz, n = 4), 4-AP did not significantly enhance the frequency of events in KA-treated mice (0.048 ± 0.039, p = 0.3). Thus, 4-AP-induced synchronized network activity was similar in all groups, but there was an increase in the frequency of sIPSCs in newborn GCs from hypoxia-treated mice that was not seen in KA-treated mice, both under basal conditions and when network activity was enhanced with 4-AP.
Gabaergic Synaptic Input to Mature GCs Following Hypoxia
We reasoned that enhanced spontaneous GABAergic input to newborn GCs observed after hypoxia could result from either enhanced activity of GABAergic interneurons or enhanced dendritic structure of newborn neurons resulting in greater integration into the network. To differentiate between these possibilities, we recorded sIPSCs in neighboring unlabeled (mature) GCs in control and hypoxia-treated mice at P16–18. There was no difference in the amplitude and frequency of pharmacologically isolated sIPSCs or mIPSCs in mature GCs from hypoxia-treated mice (Figures 3A,B; Table 1). This suggests that enhanced synaptic input to newborn GCs after hypoxia treatment results from their enhanced integration into the network rather than from enhanced GABAergic activity within the network. However, we cannot rule out the possibility that the interneuron subtype(s) that project to newborn GCs (Markwardt et al., 2009) mediates only a small fraction of spontaneous events in mature GCs, making any alteration in these inputs to mature GCs difficult to detect. To further examine potential changes in network activity, we also examined sIPSCs in CA1 pyramidal cells in control and hypoxia-treated mice. Febrile or hypoxia-induced seizures in P10 rats can increase or decrease, respectively, the frequency of sIPSCs in CA1 pyramidal cells 1 week following seizure induction (Chen et al., 1999; Sanchez et al., 2007). However, using depolarized recording conditions that allow detection of sIPSCs without blocking glutamatergic transmission, we found no difference in sIPSCs after our hypoxia treatment in mice (Figure 3C). The amplitude of sIPSCs was 27 ± 4.4 pA in control (n = 5) compared to 32 ± 5.3 after hypoxia (n = 4, p > 0.05) and the frequency was 5.5 ± 1.3 Hz in control compared to 5.2 ± 0.4 Hz after hypoxia, with no changes in the rise and decay phases of the individual events (not shown). Together these results suggest that the hypoxia paradigm we used did not alter synaptic inhibition throughout the hippocampus, but rather selectively enhanced GABAergic input to newborn GCs, evidence of their enhanced synaptic integration. Surprisingly, KA treatment did not increase the synaptic integration of newborn GCs despite their enhanced dendritic structure.
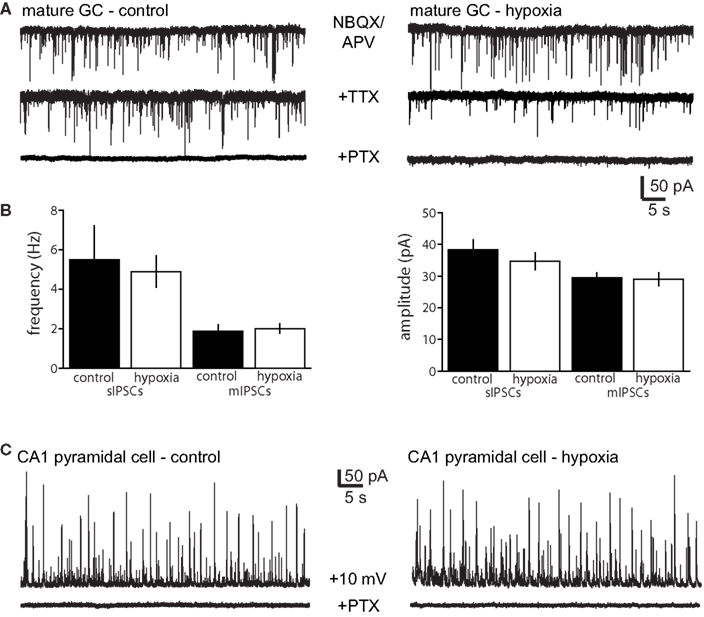
Figure 3. No change in spontaneous IPSCs in mature neurons after hypoxia. (A) Examples of spontaneous GABAergic synaptic activity in mature granule cells (top) from control (left) and hypoxia-treated (right) mice. Glutamate receptors were blocked with NBQX and APV in all recordings. Miniature IPSCs were recorded with the addition of TTX (middle traces), and all events were blocked by picrotoxin (PTX, bottom). (B) There was no difference in the frequency and amplitude of sIPSCs and mIPSCs in mature GCs from control and hypoxia-treated mice (five control mice and three hypoxia-treated mice, Table 1). (C) Example of sIPSCs from CA1 pyramidal cells in control (left, n = 5 cells from three mice) and hypoxia-treated mice (n = 4 cells from two mice). There was also no difference in the frequency or amplitude of sIPSCs. Recordings were made at +10 mV to allow identification of outward IPSCs in the absence of glutamate receptor blockers.
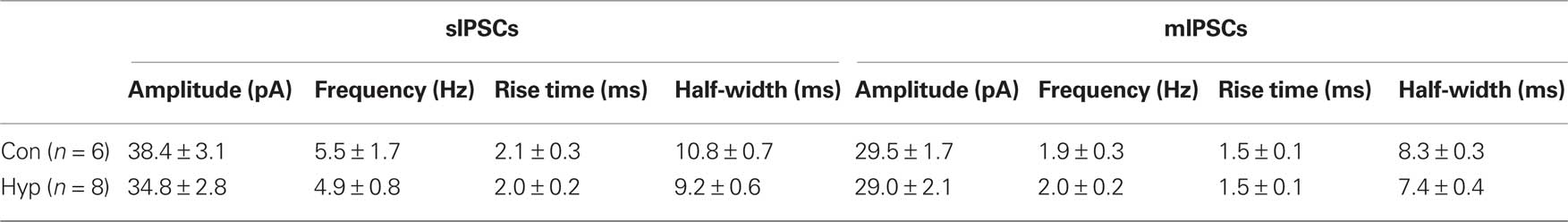
Table 1. Properties of sIPSCs and mIPSCs in mature GCs.
Cell Number is Unaffected
Previous work has shown that seizures and chronic hypoxia during the neonatal period can decrease or enhance neural proliferation, depending on the age of the rodent and paradigm (reviewed by Porter, 2008). To determine whether the paradigms we used altered progenitor proliferation and neural production, we quantified stage-specific granule cell populations at P17. Proliferating cells were identified with Ki67, newborn neurons were identified with POMC–GFP and the total number of cells in the GCL was quantified with the nuclear stain propidium iodide (PI). We found that the sizes of these cell populations were not altered by either insult (Figure 4). In addition, the ratio of newborn neurons to total cells (POMC–GFP+ cells/PI+ cells) was similar across treatment groups (9.7, 10.2, and 9.0% for control, hypoxia, and KA, respectively) and the ratio of proliferating cells to total cells (Ki67+ cells/PI+ cells) was similar across treatment groups (4.7, 5.8, and 4.7% for control, hypoxia, and KA, respectively). Although we cannot rule out the possibility that proliferation was affected at earlier or later time points, these results suggest that seizure-induced alterations in morphology can be dissociated from changes in neural production and progenitor proliferation.
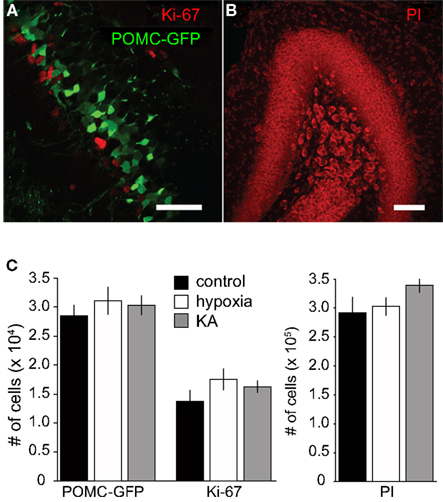
Figure 4. Insults on P10 did not alter neural production. (A) A confocal image showing proliferating progenitors expressing Ki67 (red) and newborn GCs expressing POMC–GFP (green) from a P17 mouse. There was no co-localization between Ki67 and POMC–GFP in any condition, consistent with the idea that POMC–GFP labels postmitotic newborn neurons. Scale bar, 50 μm. (B) Propidium iodide (PI) was used as a non-specific nuclear stain to quantify the total number of cells in the granule cell layer. Scale bar, 100 μm. (C) Stereological quantification revealed no differences in the number of POMC–GFP-, Ki67-, or PI-labeled cells on P17 between treatment groups (p > 0.05; ANOVA). For POMC–GFP labeling, n = 8, 9, and 9 mice per group, respectively. For Ki67 labeling, n = 3, 4, and 4, respectively.
Long-Term Consequences of Neonatal Seizures
Previous work using BrdU labeling has identified biphasic responses of neural proliferation following repetitive neonatal insults, with acute reduced proliferation followed by enhanced proliferation in the weeks following insults (Fagel et al., 2006; Xiu-Yu et al., 2007). To determine whether neonatal insults altered the number of newborn and mature GCs over a longer time scale, we quantified the number of GFP labeled newborn GCs in mice 60 days following seizure induction. At this time point, there was no difference in the number of newborn GCs across groups, with 5726 ± 346 cells in control (n = 7), 6946 ± 720 cells in hypoxia-treated mice (n = 5), and 6773 ± 641 cells in KA-treated mice (n = 6; Figures 5A,C). There was also no difference in the total number of cells in the GCL assessed by PI staining at P70 (control: 218,870 ± 8,205, n = 6; hypoxia: 222,288 ± 4,221, n = 5; KA: 213,198 ± 2,977, n = 5; Figures 5B,C). Together these results suggest that if our seizure paradigms altered the number of neurons produced at time points we did not test, they did not have a cumulative effect on the total number of dentate GCs nor the number of newborn GCs in young adulthood.
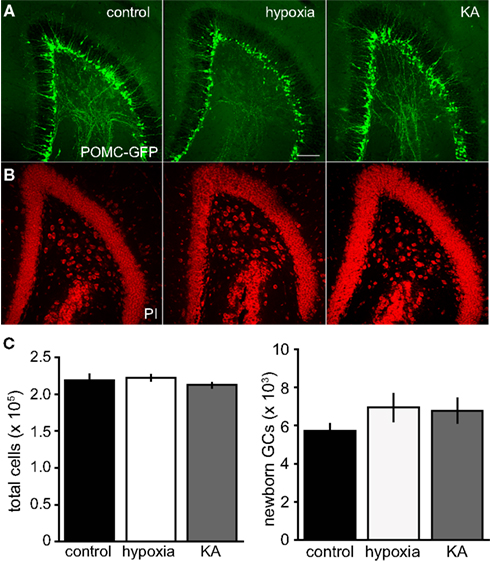
Figure 5. Insults on P10 did not alter neural populations at P70. (A,B) Confocal images of newborn GCs expressing POMC–GFP (A) and total cells stained with PI (B) in P70 mice. Scale bar, 100 μm. (C) Stereological quantification revealed no significant differences in the number of newborn GCs (left, p = 0.25, ANOVA) or total number of cells (right, p = 0.5, ANOVA).
To test the persistence of the enhanced dendritic structure of newborn GCs seen at P17, we also quantified the morphology of POMC–GFP labeled newborn GCs at P70. By P70 the depth of the GCL in control animals expanded approximately 50% (68 ± 2 μm at P17, n = 10, compared to 90 ± 3 μm at P70, n = 20, p < 0.0001; Figure 6A), presumably due to the addition of new GCs after P17. The increased depth of the GCL was associated with similar increases in both TDL (50 ± 11%, Figure 6B) and furthest extent (42 ± 6%, Figure 6C) of newborn GC dendrites. In addition, by P70, more of the dendritic arbors of newborn GCs exist in the inner molecular layer (IML, 34 ± 2%, n = 62, vs. 20 ± 2%, n = 27, Figure 6D). This suggests that later-developing GCs not only reach the IML during the newborn phase, but also expand more of their dendritic arbor within it. Whether this age-related difference results from changes in the extracellular environment, the slower maturational rate of adult-generated neurons (Overstreet-Wadiche et al., 2006a; Zhao et al., 2006) or altered POMC–GFP expression remains to be determined.
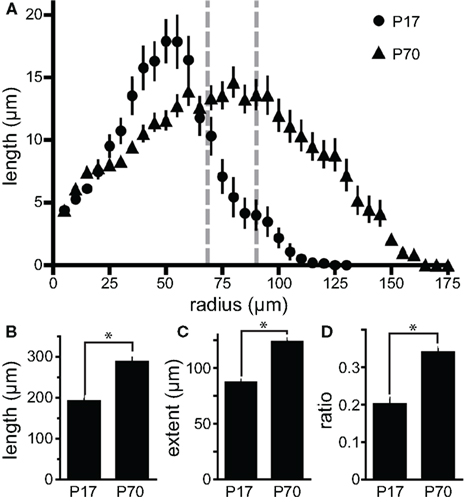
Figure 6. Developmental alterations in dendritic structure. (A) Measurements of apical dendrite length in various Sholl shells indicate that peak dendritic density for newborn GCs shifts outward from P17 (circles) to P70 (triangles). In both cases the majority of dendritic length is contained within the granule cell layer as indicated by the gray dashed lines [GC layer depth at P17: 68 ± 2 μm (n = 10) to P70: 90 ± 3 μm (n = 20)]. (B, C) Newborn granule cells at P70 exhibit both (B) greater TDL (n = 27 and n = 62 for P17 and P70, respectively) and (C) further reach than those at P17. (D) At P70, a greater proportion of the dendritic architecture of newborn neurons lies outside the GC layer. Bars represent the proportion of TDL beyond the average depth of the GCL layer. Asterisks indicate p < 0.0001.
Surprisingly, at P70 there was a selective decrease in the TDL of newborn GCs in hypoxia-treated mice, with no difference in the KA-treated animals (control: 288 ± 15 μm, n = 62; hypoxia: 239 ± 12 μm, n = 58; KA: 294 ± 18 μm, n = 51; Figures 7A–C). The alteration in the length appeared to derive from a reduction of dendritic length in the area between 45 and 60 μm from the soma, e.g., within the GCL (Figure 7D). There was no difference in the location of cell bodies between treatment groups, with approximately 90% of the somata positioned within the inner GC layer (control, 91.4%; hypoxia, 87.0%; KA, 84.0%; p = 0.36 by two-way ANOVA). The decrease in TDL was accompanied by a decrease in dendritic complexity as total nodes were selectively decreased in hypoxia-treated mice (control: 6.3 ± 0.4, n = 62; hypoxia: 4.8 ± 0.3, n = 58, KA: 6.6 ± 0.5, n = 43; Figure 7E, p < 0.05 for control vs. hypoxia, ANOVA), with the most striking difference occurring between 20 and 60 μm from the soma (data not shown). These results suggest that while hypoxia treatment does not alter the total number of newborn GCs or total neurons, effects on the patterns of dendritic outgrowth are persistent in subsequent generations of newborn neurons.
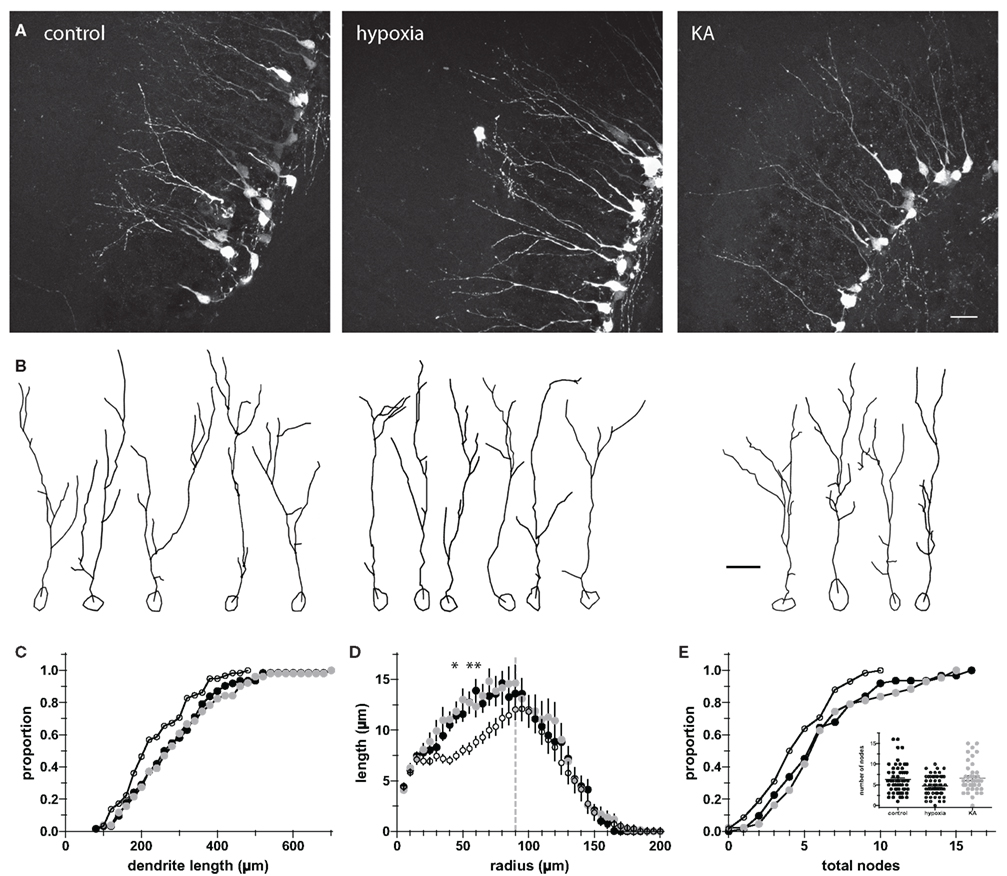
Figure 7. Dendritic structure alterations remain at P70. (A) Confocal images (maximum z-projections) of newborn GCs at P70 illustrate the dendritic structure of newborn GCs following hypoxia and KA insults on P10. Scale bar, 20 μm. (B) Examples of individual dendrite tracings. Scale bar, 20 μm. (C, D) Hypoxia-treated animals (o) had a reduction in total dendrite length [(C), p < 0.05 for control vs. hypoxia, Kolmogorov–Smirnov test], resulting primarily from shorter dendrites located 45–60 μm from the cell body (D). Asterisks indicate significant differences from control; p < 0.05, ANOVA. (E) Dendritic structure of newborn GCs in hypoxia-treated animals exhibits reduced complexity at P70. Inset, On average, hypoxia-treated animals had 1.5 fewer nodes than their control (•) or KA-treated (•) counterparts (p < 0.05).
Discussion
Here we examined the development of postnatal-generated GCs following neonatal insults. Both hypoxia treatment and KA-induced seizures on P10 resulted in an increase in dendritic development 1 week later that, in the case of hypoxia, was accompanied by an increase in synaptic input. The number of newborn GCs and proliferating cells 1 week after the insults was unaffected, suggesting that morphological and functional changes can be triggered in response to insults that are insufficient to alter proliferation and neural production. In addition, hypoxia had long-lasting consequences, such that newborn GCs generated in adult mice that experienced hypoxia at P10 had reduced dendritic structure 2 months following the insult. Together, these results demonstrate that single insults during the neonatal period can have both long- and short-term consequences for neuronal maturation.
Newly generated GCs are particularly susceptible to undergoing abnormal dendritic development following insults. In adult rodents, seizures induce dendritic abnormalities selectively in GCs that are born during a ∼1-month window surrounding the seizure event, whereas older GCs are essentially unaffected (Walter et al., 2007; Kron et al., 2010). Newborn GCs labeled with POMC–GFP are within this vulnerable developmental period. In adult mice, POMC–GFP labeled GCs are less than 3 weeks postmitotic, and in young mice, when maturation is accelerated, POMC–GFP labeled cells are less than 2 weeks postmitotic (Overstreet-Wadiche et al., 2006a). Here we examined the effect of insults on POMC–GFP labeled newborn GCs during an early ontological period when many GCs are at an immature stage. Whereas newborn GCs constitute <3% of the total GC population in young adult rodents, in the first postnatal week they constitute ∼30% of the total GC population (Overstreet-Wadiche et al., 2006a). Thus insults occurring early in life have the potential to affect the development of a large cohort of GCs.
We interpret the increased dendrite length measured 1 week after the insults as an acceleration of maturation. POMC–GFP is a useful indicator of the rate of neural maturation because it provides an exogenous reporter of neuronal morphology that is not associated with endogenous protein expression (Overstreet et al., 2004). Similar increases in dendrite length following electroconvulsant treatment were reported using a retroviral labeling method that is also independent of endogenous protein expression (Ma et al., 2009). We previously observed dramatic outgrowth of POMC–GFP newborn GC dendrites in adult mice after pilocarpine-induced seizures without alterations in BrdU co-labeling, indicating enhanced maturation rather than altered timing of GFP expression (Overstreet-Wadiche et al., 2006b). We speculate that accelerated maturation may be a consequence of enhanced neural activity in the DG that promotes granule cell development by both cell-autonomous and non-cell-autonomous mechanisms (Tozuka et al., 2005; Ge et al., 2006; Overstreet-Wadiche et al., 2006a; Ma et al., 2009). Whether accelerated maturation results in persistent alterations in the function of newborn GCs is not yet clear. In support of this idea, seizures in the adult brain have also been associated with long-term alterations in synaptic input to 6- to 8-week-old adult-generated GCs (Jakubs et al., 2006). Identifying persistent alterations in the function of developing GCs following neonatal insults will require methods to track the fate of newborn GCs across time.
The dendritic alterations seen at P70 for the hypoxia-treated animals are also intriguing. The localized loss of dendritic growth and branching within the GCL suggests that long-lasting changes have occurred such that dendritic elongation is favored over branching. Whether this change results from lessened synaptic activity onto newborn cells within the GC layer (Tsigankov and Koulakov, 2009) or other changes in the extracellular environment impinging on the developing dendrites (Hely et al., 2001) remains open to investigation. Alternatively, hypoxia-induced alterations in postnatal stem cells/progenitors that persist to generate subsequent generations of GCs (Mathews et al., 2010) could contribute to long-term dendritic dysregulation via cell-autonomous mechanisms.
Our findings that single episodes of neonatal seizure or hypoxia are insufficient to alter the production of newborn GCs are consistent with previous reports that multiple episodes of seizures are necessary to suppress neurogenesis in neonate rodents (McCabe et al., 2001; Liu et al., 2003; Xiu-Yu et al., 2007). Thus it appears that seizure-induced alterations in progenitor proliferation and the maturation of newborn GCs are dissociable processes (Plumpe et al., 2006) and that maturation has a lower threshold for perturbation compared to proliferation.
Even a single seizure episode induced by KA in neonates can produce long-term impairments in hippocampal-based cognitive function, including impairments in short- and long-term spatial memory and retrieval (Lynch et al., 2000; Sayin et al., 2004). Such behavioral outcomes are associated with alterations in synaptic transmission (Lynch et al., 2000; Cornejo et al., 2007). It is possible that abnormal GC development shown here, and/or alterations in synaptic transmission throughout cortical structures, contributes to long-term behavioral outcomes (Cornejo et al., 2008). Additionally, our finding that the enhanced dendrite structure of newborn GCs after KA is not associated with enhanced GABAergic input suggests that GABAergic signaling within the DG may be impaired. Indeed, previous studies have shown reduced GABAergic signaling in the hippocampus following experimentally induced seizures (Cossart et al., 2001; Kobayashi and Buckmaster, 2003). Although we did not detect differences in spontaneous GABAergic input to mature GCs 1 week after KA treatment (not shown), reduced GABA release by specific interneuron subtype(s) (Cossart et al., 2001; Kobayashi and Buckmaster, 2003) may be difficult to detect if those interneurons mediate only a fraction of the total inputs to mature cells. Alternatively, the absence of enhanced sIPSCs following KA seizures could result from selective impairment of GABAergic synaptogenesis in newborn GCs. These possibilities merit further investigation.
Brief hypoxia in P10–12 rats has been developed as a model of hypoxic encephalopathy that is a major cause of seizures in human neonates (Jensen et al., 1991b). Similar to the KA rat model, the neonatal hypoxia seizure model in rats is associated with long-term alterations in cellular excitability and seizure susceptibility (Jensen et al., 1991a, 1998), as well as cognitive impairment (Yang et al., 2004). Although hypoxia in P10–12 rats induces acute behavioral seizure activity (Jensen et al., 1991b; Yang et al., 2004), we did not observe behavioral seizures in hypoxia-treated mouse pups. Furthermore, we did not detect a reduction in sIPSCs in CA1 pyramidal cells as was reported in neonatal rats following hypoxia-induced seizures (Sanchez et al., 2007). Species-specific differences in development or seizure response could underlie these discrepancies. However, dendritic alterations in newborn GCs in the rat model have been reported (Sanchez et al., 2008) and the newborn GC dendrite phenotype was similar in both KA- and hypoxia-treated mice, suggesting that newborn GCs can respond to disparate insults in a similar manner.
In conclusion, our results demonstrate that alterations in newborn neuron synaptic integration can occur in response to neonatal insults that are below the threshold for affecting the number of newly generated neurons. Our results also provide a cautionary illustration that enhanced dendrite structure is not always accompanied by increased functional (synaptic) integration. Although the long-term consequence of our observations are not yet known, they add to the growing list of functional alterations triggered by neonatal insults that may contribute to long-term abnormalities in injury models that are devoid of neuron loss or major structural reorganization (Dube et al., 2009; Rakhade and Jensen, 2009).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Wadiche labs for helpful comments and Swati Gupta for participating in preliminary experiments. This work was supported by the Epilepsy Foundation (Linda Overstreet-Wadiche), the Civitan International Research Center (Chuan-Sheng Zhao and Linda Overstreet-Wadiche), NIH RO1NS064025 (Linda Overstreet-Wadiche), and NIH Neuroscience Blueprint Core Grant NS57098.
References
Chen, K., Baram, T. Z., and Soltesz, I. (1999). Febrile seizures in the developing brain result in persistent modification of neuronal excitability in limbic circuits. Nat. Med. 5, 888–894.
Cornejo, B. J., Mesches, M. J., and Benke, T. A. (2008). A single early-life seizure impairs short-term memory but does not alter spatial learning, recognition memory, or anxiety. Epilepsy Behav. 13, 585–592.
Cornejo, B. J., Mesches, M. J., Coultrap, S., Browning, M. D., and Benke, T. A. (2007). A single episode of neonatal seizures permanently alters glutamatergic synapses. Ann. Neurol. 61, 411–426.
Cossart, R., Dinocourt, C., Hirsch, J. C., Merchan-Perez, A., De Felipe, J., Ben-Ari, Y., Esclapez, M., and Bernard, C. (2001). Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat. Neurosci. 4, 52–62.
Cowley, M. A., Smart, J. L., Rubinstein, M., Cerdan, M. G., Diano, S., Horvath, T. L., Cone, R. D., and Low, M. J. (2001). Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411, 480–484.
Deisseroth, K., Singla, S., Toda, H., Monje, M., Palmer, T. D., and Malenka, R. C. (2004). Excitation-neurogenesis coupling in adult neural stem/progenitor cells. Neuron 42, 535–552.
Dube, C. M., Brewster, A. L., and Baram, T. Z. (2009). Febrile seizures: mechanisms and relationship to epilepsy. Brain Dev. 31, 366–371.
Fagel, D. M., Ganat, Y., Silbereis, J., Ebbitt, T., Stewart, W., Zhang, H., Ment, L. R., and Vaccarino, F. M. (2006). Cortical neurogenesis enhanced by chronic perinatal hypoxia. Exp. Neurol. 199, 77–91.
Ge, S., Goh, E. L., Sailor, K. A., Kitabatake, Y., Ming, G. L., and Song, H. (2006). GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 439, 589–593.
Ge, S., Yang, C. H., Hsu, K. S., Ming, G. L., and Song, H. (2007). A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain. Neuron 54, 559–566.
Hattiangady, B., and Shetty, A. K. (2008). Implications of decreased hippocampal neurogenesis in chronic temporal lobe epilepsy. Epilepsia 49(Suppl. 5), 26–41.
Hely, T. A., Graham, B., and van Ooyen, A. (2001). A computational model of dendrite elongation and branching based on MAP2 phosphorylation. J. Theor. Biol. 210, 375–84.
Jakubs, K., Nanobashvili, A., Bonde, S., Ekdahl, C. T., Kokaia, Z., Kokaia, M., and Lindvall, O. (2006). Environment matters: synaptic properties of neurons born in the epileptic adult brain develop to reduce excitability. Neuron 52, 1047–1059.
Jensen, F. E. (2009). Neonatal seizures: An update on mechanisms and management. Clin. Perinatol. 30, 881–900.
Jensen, F. E., Applegate, C., Burchfiel, J., and Lombroso, C. T. (1991a). Differential effects of perinatal hypoxia and anoxia on long term seizure susceptibility in the rat. Life Sci. 49, 399–407.
Jensen, F. E., Applegate, C. D., Holtzman, D., Belin, T. R., and Burchfiel, J. L. (1991b). Epileptogenic effect of hypoxia in the immature rodent brain. Ann. Neurol. 29, 629–637.
Jensen, F. E., Wang, C., Stafstrom, C. E., Liu, Z., Geary, C., and Stevens, M. C. (1998). Acute and chronic increases in excitability in rat hippocampal slices after perinatal hypoxiain vivo. J. Neurophysiol. 79, 73–81.
Kobayashi, M., and Buckmaster, P. S. (2003). Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J. Neurosci. 23, 2440–2452.
Koh, S., Tibayan, F. D., Simpson, J. N., and Jensen, F. E. (2004). NBQX or topiramate treatment after perinatal hypoxia-induced seizures prevents later increases in seizure-induced neuronal injury. Epilepsia 45, 569–575.
Kokaia, Z., and Lindvall, O. (2003). Neurogenesis after ischaemic brain insults. Curr. Opin. Neurobiol. 13, 127–132.
Kron, M. M., Zhang, H., and Parent, J. M. (2010). The developmental stage of dentate granule cells dictates their contribution to seizure-induced plasticity. J. Neurosci. 30, 2051–2059.
Lawrence, R., and Inder, T. (2010). Neonatal status epilepticus. Semin. Pediatr. Neurol. 17, 163–168.
Liu, H., Kaur, J., Dashtipour, K., Kinyamu, R., Ribak, C. E., and Friedman, L. K. (2003). Suppression of hippocampal neurogenesis is associated with developmental stage, number of perinatal seizure episodes, and glucocorticosteroid level. Exp. Neurol. 184, 196–213.
Liu, J., Solway, K., Messing, R. O., and Sharp, F. R. (1998). Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J. Neurosci. 18, 7768–7778.
Lynch, M., Sayin, U., Bownds, J., Janumpalli, S., and Sutula, T. (2000). Long-term consequences of early postnatal seizures on hippocampal learning and plasticity. Eur. J. Neurosci. 12, 2252–2264.
Ma, D. K., Jang, M. H., Guo, J. U., Kitabatake, Y., Chang, M. L., Pow-Anpongkul, N., Flavell, R. A., Lu, B., Ming, G. L., and Song, H. (2009). Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 323, 1074–1077.
Markwardt, S. J., Wadiche, J. I., and Overstreet-Wadiche, L. S. (2009). Input-specific GABAergic signaling to newborn neurons in adult dentate gyrus. J. Neurosci. 29, 15063–15072.
Mathews, E. A., Morgenstern, N. A., Piatti, V. C., Zhao, C., Jessberger, S., Schinder, A. F., and Gage, F. H. (2010). A distinctive layering pattern of mouse dentate granule cells is generated by developmental and adult neurogenesis. J. Comp. Neurol. 518, 4479–4490.
McCabe, B. K., Silveira, D. C., Cilio, M. R., Cha, B. H., Liu, X., Sogawa, Y., and Holmes, G. L. (2001). Reduced neurogenesis after neonatal seizures. J. Neurosci. 21, 2094–2103.
Michelson, H. B., and Wong, R. K. (1994). Synchronization of inhibitory neurones in the guinea-pig hippocampus in vitro. J. Physiol. 477, 35–45.
Overstreet, L. S., Hentges, S. T., Bumaschny, V. F., de Souza, F. S., Smart, J. L., Santangelo, A. M., Low, M. J., Westbrook, G. L., and Rubinstein, M. (2004). A transgenic marker for newly born granule cells in dentate gyrus. J. Neurosci. 24, 3251–3259.
Overstreet Wadiche, L., Bromberg, D. A., Bensen, A. L., and Westbrook, G. L. (2005). GABAergic signaling to newborn neurons in dentate gyrus. J. Neurophysiol. 94, 4528–4532.
Overstreet-Wadiche, L. S., Bensen, A. L., and Westbrook, G. L. (2006a). Delayed development of adult-generated granule cells in dentate gyrus. J. Neurosci. 26, 2326–2334.
Overstreet-Wadiche, L. S., Bromberg, D. A., Bensen, A. L., and Westbrook, G. L. (2006b). Seizures accelerate functional integration of adult-generated granule cells. J. Neurosci. 26, 4095–4103.
Parent, J. M., Yu, T. W., Leibowitz, R. T., Geschwind, D. H., Sloviter, R. S., and Lowenstein, D. H. (1997). Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J. Neurosci. 17, 3727–3738.
Plumpe, T., Ehninger, D., Steiner, B., Klempin, F., Jessberger, S., Brandt, M., Romer, B., Rodriguez, G. R., Kronenberg, G., and Kempermann, G. (2006). Variability of doublecortin-associated dendrite maturation in adult hippocampal neurogenesis is independent of the regulation of precursor cell proliferation. BMC Neurosci. 7, 77. doi: 10.1186/1471-2202-7-77
Porter, B. E. (2008). Neurogenesis and epilepsy in the developing brain. Epilepsia 49(Suppl. 5), 50–54.
Rakhade, S. N., and Jensen, F. E. (2009). Epileptogenesis in the immature brain: emerging mechanisms. Nat. Rev. Neurol. 5, 380–391.
Sanchez, R. M., Justice, J. A., and Zhang, K. (2007). Persistently decreased basal synaptic inhibition of hippocampal CA1 pyramidal neurons after neonatal hypoxia-induced seizures. Dev. Neurosci. 29, 159–167.
Sanchez, R. M., Zhang, K., and Shapiro, L. A. (2008). “Neuroanatomical alterations to the immature dentate gyrus following seizure-inducing hypoxia in neonatal rat,” in Neuroscience Meeting Planner, Program No. 448.8 (Washington, DC: Society for Neuroscience). [Online].
Sankar, R., Shin, D., Liu, H., Katsumori, H., and Wasterlain, C. G. (2000). Granule cell neurogenesis after status epilepticus in the immature rat brain. Epilepsia 41(Suppl. 6), 53–56.
Sayin, U., Sutula, T. P., and Stafstrom, C. E. (2004). Seizures in the developing brain cause adverse long-term effects on spatial learning and anxiety. Epilepsia 45, 1539–1548.
Scharfman, H. E. (2004). Functional implications of seizure-induced neurogenesis. Adv. Exp. Med. Biol. 548, 192–212.
Schmidt-Hieber, C., Jonas, P., and Bischofberger, J. (2004). Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature 429, 184–187.
Shapiro, L. A., Ribak, C. E., and Jessberger, S. (2008). Structural changes for adult-born dentate granule cells after status epilepticus. Epilepsia 49(Suppl. 5), 13–18.
Snyder, J. S., Kee, N., and Wojtowicz, J. M. (2001). Effects of adult neurogenesis on synaptic plasticity in the rat dentate gyrus. J. Neurophysiol. 85, 2423–2431.
Tanaka, R., Yamashiro, K., Mochizuki, H., Cho, N., Onodera, M., Mizuno, Y., and Urabe, T. (2004). Neurogenesis after transient global ischemia in the adult hippocampus visualized by improved retroviral vector. Stroke 35, 1454–1459.
Tozuka, Y., Fukuda, S., Namba, T., Seki, T., and Hisatsune, T. (2005). GABAergic excitation promotes neuronal differentiation in adult hippocampal progenitor cells. Neuron 47, 803–815.
Tsigankov, D., and Koulakov, A. (2009). Optimal axonal and dendritic branching strategies during the development of neural circuitry. Front. Neural Circuits 3:18. doi: 10.3389/neuro.04.018.2009
Walter, C., Murphy, B. L., Pun, R. Y., Spieles-Engemann, A. L., and Danzer, S. C. (2007). Pilocarpine-induced seizures cause selective time-dependent changes to adult-generated hippocampal dentate granule cells. J. Neurosci. 27, 7541–7552.
West, M. J., Slomiana, L., and Gunderson, H. J. (1991). Unbiased stereological estimation of the total number neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat. Rec. 231, 482–497.
Xiu-Yu, S., Ruo-Peng, S., and Ji-Wen, W. (2007). Consequences of pilocarpine-induced recurrent seizures in neonatal rats. Brain Dev. 29, 157–163.
Yang, S. N., Huang, C. B., Yang, C. H., Lai, M. C., Chen, W. F., Wang, C. L., Wu, C. L., and Huang, L. T. (2004). Impaired SynGAP expression and long-term spatial learning and memory in hippocampal CA1 area from rats previously exposed to perinatal hypoxia-induced insults: beneficial effects of A68930. Neurosci. Lett. 371, 73–78.
Zhao, C., Teng, E. M., Summers, R. G. Jr., Ming, G. L., and Gage, F. H. (2006). Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus. J. Neurosci. 26, 3–11.
Zhao, C. S., and Overstreet-Wadiche, L. (2008). Integration of adult generated neurons during epileptogenesis. Epilepsia 49(Suppl. 5), 3–12.
Zsiros, V., Aradi, I., and Maccaferri, G. (2007). Propagation of postsynaptic currents and potentials via gap junctions in GABAergic networks of the rat hippocampus. J. Physiol. 578, 527–544.
Keywords: kainate, hypoxia, postnatal neurogenesis, dentate gyrus, synaptic, seizure, granule cell, neonate
Citation: Pugh P, Adlaf E, Zhao C-S, Markwardt S, Gavin C, Wadiche J and Overstreet-Wadiche L (2011) Enhanced integration of newborn neurons after neonatal insults. Front. Neurosci. 5:45. doi: 10.3389/fnins.2011.00045
Received: 24 January 2011;
Paper pending published: 15 February 2011;
Accepted: 17 March 2011;
Published online: 30 March 2011.
Edited by:
Joseph Loturco, University of Connecticut, USAReviewed by:
Joseph Loturco, University of Connecticut, USAJack M. Parent, University of Michigan, USA
Copyright: © 2011 Pugh, Adlaf, Zhao, Markwardt, Gavin, Wadiche and Overstreet-Wadiche. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Linda Overstreet-Wadiche, Department of Neurobiology, University of Alabama at Birmingham, 1825 University Boulevard, Birmingham, AL 35294, USA. e-mail:bHdhZGljaGVAdWFiLmVkdQ==