 Dawn Béraud1
Dawn Béraud1 Margaret Twomey1
Margaret Twomey1 Benjamin Bloom1
Benjamin Bloom1 Andrew Mittereder1 Vy Ton1 Katherine Neitzke1 Sergey Chasovskikh2
Andrew Mittereder1 Vy Ton1 Katherine Neitzke1 Sergey Chasovskikh2 Timothy R. Mhyre1
Timothy R. Mhyre1 Kathleen A. Maguire-Zeiss1*
Kathleen A. Maguire-Zeiss1*- 1 Interdisciplinary Program in Neuroscience, Department of Neuroscience, Georgetown University Medical Center, Washington, DC, USA
- 2 Lombardi Comprehensive Cancer Center; Georgetown University Medical Center, Washington, DC, USA
Parkinson’s disease, an age-related neurodegenerative disorder, is characterized by the loss of dopamine neurons in the substantia nigra, the accumulation of α-synuclein in Lewy bodies and neurites, and neuroinflammation. While the exact etiology of sporadic Parkinson’s disease remains elusive, a growing body of evidence suggests that misfolded α-synuclein promotes inflammation and oxidative stress resulting in neurodegeneration. α-Synuclein has been directly linked to microglial activation in vitro and increased numbers of activated microglia have been reported in an α-synuclein overexpressing mouse model prior to neuronal loss. However, the mechanism by which α-synuclein incites microglial activation has not been fully described. Microglial activation is governed in part, by pattern recognition receptors that detect foreign material and additionally recognize changes in homeostatic cellular conditions. Upon proinflammatory pathway initiation, activated microglia contribute to oxidative stress through release of cytokines, nitric oxide, and other reactive oxygen species, which may adversely impact adjacent neurons. Here we show that microglia are directly activated by α-synuclein in a classical activation pathway that includes alterations in the expression of toll-like receptors. These data suggest that α-synuclein can act as a danger-associated molecular pattern.
Introduction
Parkinson’s disease is the second most common neurodegenerative disorder, affecting approximately five million people worldwide. Sporadic as well as familial forms of this disease are typified by the loss of substantia nigra pars compacta (SNpc) dopamine neurons, dystrophic projections to the striatum, increased oxidation of proteins, lipids and DNA, increased numbers of activated microglia, and intracytoplasmic proteinaceous inclusions in the surviving SNpc dopamine neurons, called Lewy bodies (Duvoisin, 1992; Forno, 1996). One major component of Lewy bodies is fibrillar α-synuclein, a conformation purported to be toxic to neurons (Spillantini et al., 1997; Giasson et al., 1999; Masliah et al., 2000; Lee et al., 2001; Song et al., 2004; Periquet et al., 2007; Parihar et al., 2008; Feng et al., 2010). In addition to its presence in Lewy bodies, α-synuclein is further implicated in Parkinson’s disease since point mutations and overexpression of the α-synuclein gene, SNCA, are associated with familial forms of this disorder (Polymeropoulos et al., 1996, 1997; Kruger et al., 1998; Singleton et al., 2003, 2004). Moreover, genome-wide association studies (GWAS) have linked SNCA polymorphisms with an increased risk for developing sporadic Parkinson’s disease (Satake et al., 2009; Simon-Sanchez et al., 2009; Hamza et al., 2010). While the normal function of α-synuclein is not completely understood, genetic and pathological evidence suggests that Parkinson’s disease pathogenesis is closely linked with a toxic gain-of-function of misfolded α-synuclein.
Whereas native α-synuclein maintains a random coil structure, this protein exhibits a propensity to misfold into protofibrils and higher-order oligomers following changes in pH and ionic strength, increases in molecular crowding, and interactions with lipid membranes as well as secondary modification such as dopamine adduction, nitrosylation, and phosphorylation (Conway et al., 1998, 2000, 2001; Hashimoto et al., 1999; Kowall et al., 2000; Vila et al., 2000; Perrin et al., 2001; Volles et al., 2001; Ding et al., 2002; Shtilerman et al., 2002; Volles and Lansbury, 2002; Sharon et al., 2003; Fink, 2006; Tsigelny et al., 2008b). Multiple lines of evidence suggest that the pathological role of α-synuclein is linked to this ability to misfold and self-assemble into higher-order structures. In cell culture models, α-synuclein-induced cell death has been associated with the formation of oligomeric α-synuclein, increased cell membrane conductance, mitochondrial, lysosomal and proteasomal dysfunction, and microglial activation (Biasini et al., 2004; Zhang et al., 2005; Reynolds et al., 2008; Su et al., 2008, 2009; Xilouri et al., 2009; Feng et al., 2010). One consequence of these synuclein-driven perturbations is an overall increase in oxidative stress which can result from neuronal production of reactive oxygen species (ROS), decreased antioxidant responses as well as ROS emanating from surrounding activated microglia (Hsu et al., 2000; Parihar et al., 2008, 2009). Importantly, studies using α-synuclein transgenic models also support that α-synuclein is associated with the aforementioned cellular changes, resulting in neuronal dysfunction and degeneration, microglial activation, and increased oxidative stress (Feany and Bender, 2000; He et al., 2001; Dawson et al., 2002; Su et al., 2008; Kim et al., 2011).
It is not surprising that Parkinson’s disease patients, most of whom have already exhibited a reduction in dopamine content due to presynaptic terminal loss, demonstrate an over six-fold increase in activated microglia compared to control patients (Ouchi et al., 2005, 2009; Bartels and Leenders, 2007). While these immune surveillance cells phagocytose cell debris emanating from dying cells and dystrophic neurites, the evidence that microglia are activated in mouse, rat, and non-human primate models of Parkinson’s disease prior to frank neuron death is compelling (Czlonkowska et al., 1996; Kohutnicka et al., 1998; Cicchetti et al., 2002; Depino et al., 2003; Sugama et al., 2003; Wu et al., 2005; Zhang et al., 2005; Cho et al., 2006; Liu, 2006; Qian et al., 2006; Sawada et al., 2006; Su et al., 2008, 2009). Also noteworthy are the results from a recent GWAS, identifying an association between sporadic Parkinson’s disease and a major histocompatibility complex cell surface receptor region on chromosome 6, supporting a role for inflammation in the pathogenesis of Parkinson’s disease (Hamza et al., 2010). Importantly, α-synuclein leads to increased numbers of activated microglia in mouse models of α-synuclein overexpression prior to SNpc dopamine neuron death and has a direct effect on microglial activation in cell culture experiments (Zhang et al., 2005; Su et al., 2008, 2009; Theodore et al., 2008; Lee et al., 2010). Although these studies demonstrate a direct effect of α-synuclein on microglia, the mechanism and type of activation awaits delineation.
Microglia continuously monitor and react to their microenvironment and activation can be mediated by pattern recognition receptors (PRRs) that are specific for pathogen-associated molecular patterns (PAMPs) such as bacterial- and viral-derived carbohydrates, nucleic acids, and lipoproteins (Hu et al., 1996; Muzio et al., 2000; Lee and Lee, 2002; Block et al., 2007). These receptors are localized to microglial membranes and intracellular compartments and include families of scavenger receptors and toll-like receptors (TLRs). Once engaged by ligands (e.g., PAMPs), a cascade of molecular events ensues which can result in the production and release of proinflammatory cytokines (e.g., tumor necrosis factor-α, TNF-α and interleukin-1β, IL-1β), nitric oxide, and superoxide: a classical activation pathway. Alternatively, microglia can be activated to produce anti-inflammatory cytokines (e.g., arginase-1 and transforming growth factor-β) demonstrating the ability of these cells to regulate inflammation and allow for repair (Colton and Wilcock, 2010). In addition to the typical PAMPs, researchers have characterized sterile, non-pathogen related forms of inflammation in which endogenous, disease-related signals, “danger/damage-associated molecular patterns” (DAMPs), are recognized by microglia via PRRs and result in activation (Halle et al., 2008; Chen and Nunez, 2010; Duewell et al., 2010; Stewart et al., 2010).
One study suggests that α-synuclein activates microglia through a mechanism that involves CD36; however it is likely that other PRRs are also required for this activation since microglia derived from CD36 knockout mice are still activated following exposure to α-synuclein, albeit to a lesser extent (Su et al., 2008, 2009). Importantly, the identification of PRRs involved in microglial activation directed by α-synuclein could provide clinically relevant therapeutics. In this study, using a murine microglia cell line (BV-2) and mouse primary microglia, we determined whether human wild-type α-synuclein-mediated microglial activation altered the expression of TLRs. Here we report that α-synuclein caused direct microglial activation with classical cytokine upregulation, increased expression of antioxidant response enzymes and demonstrate for the first time changes in TLR gene expression.
Materials and Methods
Chemicals and Reagents
Dulbecco’s modified Eagle medium (DMEM) and minimum essential medium (MEM) were obtained from Cellgro (St. Louis, MO, USA). Fetal bovine serum was purchased from Hyclone (Logan, UT, USA). All other reagents for cell culture and general use, if not indicated, were obtained from Invitrogen (Carlsbad, CA, USA) or Sigma-Aldrich (St. Louis, MO, USA).
Expression, Purification, and Manipulation of α-Synuclein
The bacterial expression vector pRK172 containing wild-type human α-synuclein cDNA was a kind gift of Dr. Giasson (Giasson et al., 1999). α-Synuclein was bacterially expressed in Escherichia coli BL21 (DE3), purified as previously described, followed by lyophilization and storage at −20°C until use (Maguire-Zeiss et al., 2006). The lyophilized protein was resuspended by sonication at 20 Hz (2 × 10 s bursts with a 10-s rest between bursts) and diluted to 1 mg/ml in TEN buffer (10 mM Tris–HCl, pH 7.5, 1 mM EDTA, 20 mM NaCl) followed by incubation for 5 days at 33–37°C with rotation at 1000 rpm (SYNTR, Labnet Orbit M60 shaker; Labnet International, Edison, NJ, USA). TEN buffer was incubated in the same manner and used as the buffer control for all treatments (BufferTR). Endotoxin contamination of the SYNTR and BufferTR was evaluated using an E-TOXATE test kit following the manufacturer’s instructions (Sigma-Aldrich). The detection limit of the kit was 0.13 endotoxin units (EU)/ml (10 EU = 1 ng).
Characterization of α-Synuclein
Western blot analysis
One microgram of protein was added to denaturing sample buffer [62.5 mM Tris, pH 6.8, 10% (v/v) glycerol, 2% (w/v) SDS, 5% (v/v) β-mercaptoethanol, and 1% (w/v) bromophenol blue], boiled for 90 s, and subjected to polyacrylamide gradient (4–20%) gel electrophoresis under denaturing conditions followed by transfer to polyvinylidene difluoride (PVDF) membranes (PerkinElmer, Waltham, MA, USA). Membranes were blocked in TBST/NFDM [20 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.1% (v/v) Tween, 5% (w/v) non-fat dry milk]. Mouse anti-α-synuclein primary antibody (1:1000; BD Biosciences, San Jose, CA, USA) was used to probe for α-synuclein conformers. Immune complexes were visualized on film following incubation with HRP-conjugated goat anti-mouse secondary antibody (1:2000; Chemicon, Temecula, CA, USA) and Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific, Waltham, MA, USA).
Thioflavin T assay
Forty micromolar Thioflavin T in 10 mM Tris–HCl pH 8.0 (80 μl of 50 μM stock) and 12.5 μM SYNTR (20 μl of 62.5 μM stock) or 20 μl of the appropriate buffer control (BufferTR) were incubated in a 96-well black clear bottom plate. Fluorescent measurements were obtained using a Fluoromark™ microplate fluorometer (BioRad) with an excitation of 450 nm and an emission of 490 nm (Naiki et al., 1989; LeVine, 1999). All measurements were performed in triplicate in three separate experimental replicates.
Atomic force microscopy
Freshly cleaved muscovite mica was incubated in a mixture of 1-(3-aminopropyl) silatrane (APS) solution for 30 min to prepare APS-mica. SYNTR or BufferTR was added to the APS-mica and allowed to adhere for 2 min, washed with de-ionized water, and dried with nitrogen gas (Shlyakhtenko et al., 2000, 2003). The mica was attached to a metal disk with double-sided tape for imaging. Images were acquired in tapping mode, using silicon tapping mode probes and a Multimode SPM Nanoscope IIIa system (Veeco/Digital Instruments, Santa Barbara, CA, USA). Nominal spring constants of 60 N/m and a resonant frequency of 245 Hz were used.
Cell Culture
BV-2 microglia treatment
BV-2 murine microglial cells (BV-2 cells) were plated at a density of 5 × 105 cells per well (6-well plates) in 2 ml of DMEM supplemented with 5% fetal bovine serum and allowed to adhere for 24 h (Blasi et al., 1990; Horvath et al., 2008; Henn et al., 2009). One hour prior to treatment, serum-containing media was replaced with serum-free DMEM. Cells were subsequently treated with 50 nM SYNTR or BufferTR in DMEM for various time points as indicated in the figure legends. Following treatment, media were collected, centrifuged at 1000 rpm for 2 min and stored at −20°C until assayed. All treatments were preformed in triplicate in three separate biological replicates.
TNF-α secretion
Tumor necrosis factor-α levels in the media of treated microglia were measured by an enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN, USA). All measurements were performed in triplicate in three separate biological replicates.
Nitric oxide release
Nitric oxide (NO) release into the media of treated microglia was determined by measuring the stable NO metabolite, nitrite, using a Greiss reagent assay kit according to the manufacturer’s instructions (Invitrogen). All measurements were performed in triplicate in three separate biological replicates.
Preparation and treatment of primary microglia
Primary microglia cultures were prepared from P1 to P3 C57Bl/6 mouse cortices as previously described (Su et al., 2008) except that microglia were isolated from mixed glial cultures (∼DIV 14) by shaking at 125 rpm for 5 h at 37°C on a rotary shaker and collecting the microglia-enriched medium. Microglia were plated at a density of 5 × 105 cells per well (6-well plates) in 2 ml of MEM supplemented with 0.01% pyruvate, 0.6% glucose, and 5% fetal bovine serum (microglia growth media) and allowed to adhere for 24 h. Cells were subsequently treated with 50 nM SYNTR or BufferTR in microglia growth media for 24 h. All treatments were preformed in triplicate in two separate biological replicates. Animals were maintained and treated in accordance with the regulatory standards of the Animal Welfare Act and approved for use by the Georgetown University Animal Care and Use Committee.
RNA extraction
Following treatment, RNA was harvested from cultured cells using an RNeasy mini kit and on-column DNase I digestion according to the manufacturer’s instructions (Qiagen, Valencia, CA, USA). RNA purity was assessed using an Agilent 2100 bioanalyzer (Santa Clara, CA, USA) and concentration measured using a NanoDrop 1000 spectrophotometer (Thermo Scientific).
qRT-PCR
RNA was reverse transcribed in a 20 μl reaction using a High-Capacity cDNA Archive Kit (Applied Biosystems, Carlsbad, CA, USA). The quality of cDNA was verified following RT-PCR for β-actin expression. cDNA samples (10 μl) were then added to 90 μl of TaqMan® Universal PCR master mix and loaded onto TaqMan® low density arrays (TLDA) preloaded with probes and primers for various targets and one endogenous control (see figure legends and Tables 1 and 2). Additional gene targets were assayed in a 96-well plate format, where 2.5 μl of cDNA from each sample was added to 17.5 μl of master mix containing the appropriate primer/probe pairs and TaqMan® Universal PCR master mix. All real-time PCR were run using the ABI Prism 7900HT Sequence Detection System (Applied Biosystems). The results were analyzed using the relative quantification ΔΔCt method, normalizing samples to endogenous controls, either 18S rRNA (TLDA) or GAPDH (individual targets) followed by normalization to the appropriate BufferTR treated controls. All measurements were performed in triplicate in three separate biological replicates. (Primers/Probes used: TNF-α Mm00443258_m1, IL-1β Mm0434228_m1, NfkB1 Mm0047361_m1, Aif1 Mm00479862_g1, TLR1 Mm0120884_m1, TLR2 Mm00442346_m1, TLR3 Mm00446577_g1, TLR4 Mm00445273_m1, TLR6 Mm01208943_s1, TLR7 Mm00446590_m1, TLR9 Mm00446193, Myd88 Mm01351743, Scarb1 Mm00450234_m1, Hmox1 Mm00516004_m1, Prdx1 Mm01621996_s1, 18S rRNA Hs99999901_s1, and GAPDH 4352339E.) Statistical analysis was performed by Student’s t-test on ΔCt values and the significance level was set at P < 0.05. Gene expression changes are graphically represented as fold change (2−ΔΔCt).
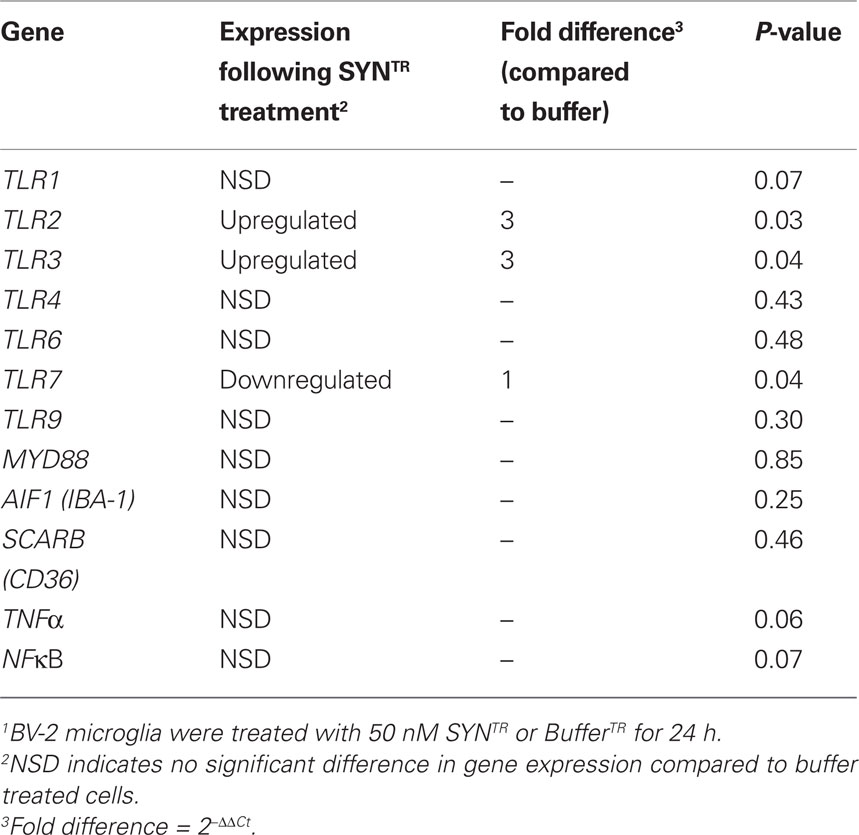
Table 1. Gene expression changes following SYNTR treatment of BV-2 microglia1.
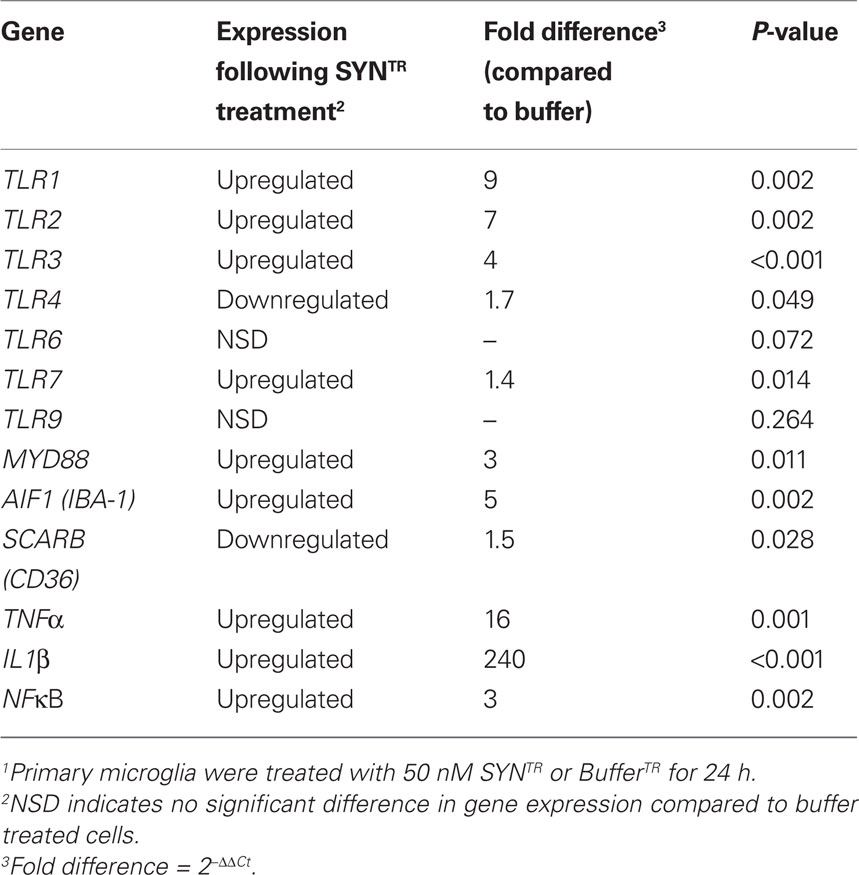
Table 2. Gene expression changes following SYNTR treatment of Primary microglia1.
Western blot analysis
BV-2 cells were treated as described above. Following treatment, cells were washed with ice-cold PBS and lysed on ice in modified RIPA buffer (50 mM Tris–HCl pH 7.4, 1% (v/v) NP-40, 0.25% (w/v) sodium deoxycholate, 150 mM NaCl) supplemented with Protease Inhibitor Cocktail (for mammalian cells; Sigma-Aldrich). Cell lysates were subjected to gentle rotation for 20 min at 4°C then sonicated at 18 Hz (3 × 10 s bursts with 30 s rest between bursts). Lysates were cleared by centrifugation at 17,000 rpm for 10 min at 4°C. Cleared lysates (25 μg) were treated as described in above and subjected to denaturing polyacrylamide gel electrophoresis (10%) followed by transfer to PVDF membranes (PerkinElmer). Membranes were blocked in TBST/NFDM [20 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.1% (v/v) Tween, 5% (w/v) non-fat dry milk] followed by incubation with mouse anti-heme oxygenase-1 primary antibody (HO-1; 1:1000; Abcam, Cambridge, MA, USA). Immune complexes were visualized on film following incubation with HRP-conjugated goat anti-mouse secondary antibody (1:2000; Chemicon) and Super Signal West Pico Chemiluminescent Substrate. Membranes were reprobed with α-tubulin (1:1000; Abcam), which served as the loading control. Densitometric analysis of digitized images was completed using the EC3 Imaging System (UVP, Upland, CA, USA).
Statistical Analysis
All statistical analyses were carried out using Graphpad Prism 5 (Graphpad Software Inc., La Jolla, CA, USA). An ANOVA was performed followed by Bonferroni’s post hoc test or Student’s t-test where appropriate. All data are reported as mean ± SD. P-values ≤0.05 were considered significant.
Results
Characterization of Manipulated α-Synuclein
The mechanism by which α-synuclein activates microglia is unknown, but purported to be involved in Parkinson’s disease pathogenesis (Zhang et al., 2005; Thomas et al., 2007; Su et al., 2008, 2009). In order to investigate the response of microglia to α-synuclein, we prepared recombinant human wild-type α-synuclein which was subsequently incubated for 5 days at 33–37°C (T) with mechanical rotation at 1000 rpm (R). The resultant α-synuclein, SYNTR, was tested for endotoxin contamination, which was found to be below the detectable limit of the assay (<0.013 ng/ml; data not shown). First, to determine whether our preparation included SDS-stable oligomers, SYNTR was subjected to polyacrylamide gel electrophoresis under denaturing conditions, followed by Western blot analysis. As shown in Figure 1A, SYNTR contains monomeric as well as SDS-stable oligomers of α-synuclein (arrows). We next determined whether our preparation contained amyloid fibrils by measuring the excitation/emission spectra following incubation with Thioflavin T. We show in Figure 1B enhanced Thioflavin T fluorescence following incubation with SYNTR, suggesting the presence of amyloid fibrils (P ≤ 0.05; Figure 1B). Lastly, we characterized SYNTR under non-denaturing conditions using atomic force microscopy (AFM) to visualize the height and distribution of α-synuclein conformers (Figures 1C,D). The largest proportion of SYNTR had a height of <5 nm, likely representing monomeric α-synuclein, but amorphous aggregates of >10 nm were also present. Together, these data establish that SYNTR contains monomers, SDS-stable oligomers, amorphous aggregates and amyloid structures.
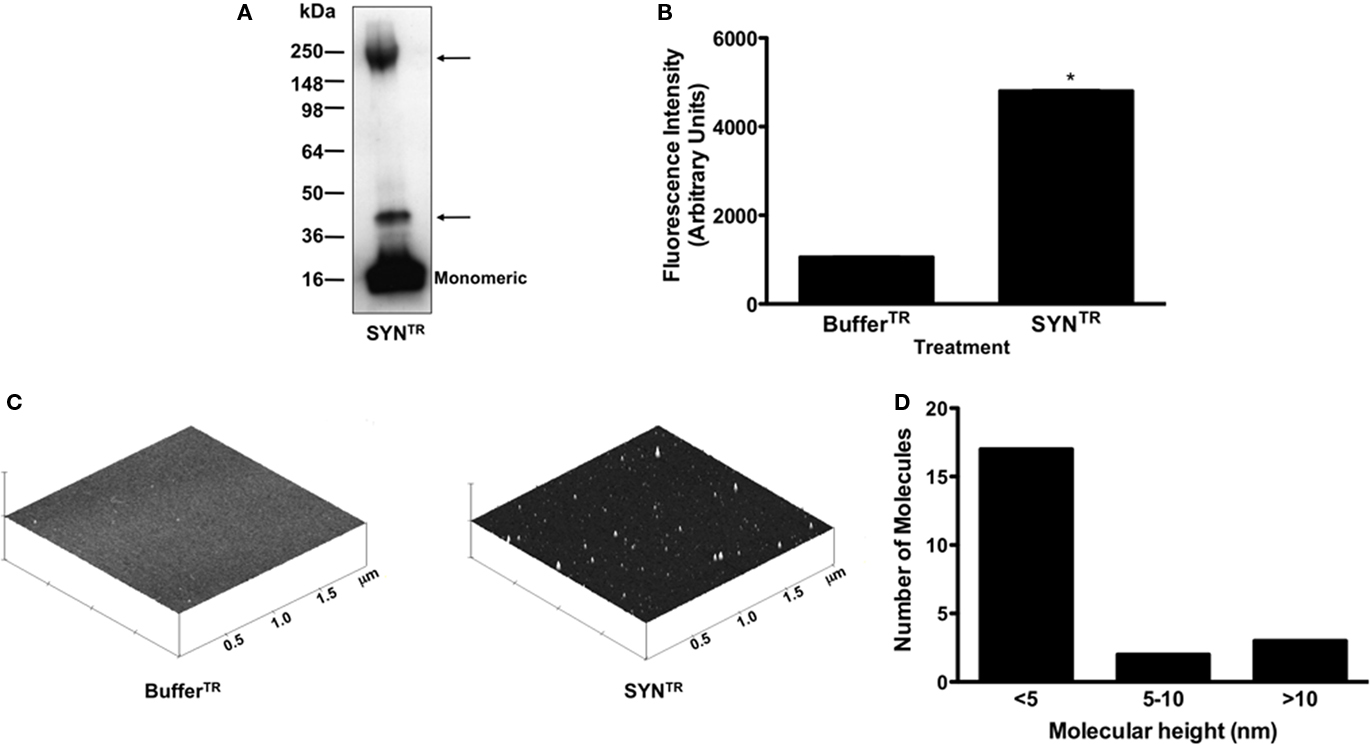
Figure 1. Manipulation of α-synuclein results in the formation of aggregates. (A) α-Synuclein prepared in TEN buffer or TEN buffer alone was incubated at 33–37°C with mechanical rotation (1000 rpm) for 5 days (SYNTR; BufferTR). SYNTR was then subjected to anti-synuclein Western blot analysis following separation on a 4–20% Tris–glycine polyacrylamide gel under denaturing conditions. Arrows indicate SDS-stable α-synuclein aggregates. (B) Thioflavin T fluorescence measurements were obtained using an excitation wavelength of 450 nm and an emission wavelength of 490 nm. SYNTR contains amyloid fibrils (*P < 0.05; each data point represents mean ± SD, n = 3). (C) SYNTR and BufferTR were subjected to AFM. This analysis demonstrated the formation of α-synuclein aggregates in the SYNTR samples. AFM height images of SYNTRand similarly manipulated buffer are shown with the height of aggregates displayed along the z-axis. Height images shown at 2 μm × 2 μm × 2 μm. (D) AFM height measurements of SYNTR. The largest proportion of SYNTR had a height of <5 nm, likely representing monomeric α-synuclein, but aggregates of >10 nm were also present.
SYNTR Induces Classical Microglial Activation
To determine whether SYNTR directly activates microglia through the classical activation pathway we treated a microglia cell line, BV-2 cells, with SYNTR and assayed for markers of inflammation. When classically activated, microglia adapt morphological changes and secrete proinflammatory mediators such as NO, TNF-α, and IL-1β (Banati et al., 1993; Combs et al., 2001; Lee and Lee, 2002; Kim and Joh, 2006; Colton and Wilcock, 2010). Previous studies have demonstrated that the secretion of proinflammatory mediators from activated microglia is a time-dependent process; therefore, we examined the temporal response of BV-2 cells to SYNTR treatment (Lee and Lee, 2002; Zhang et al., 2005; Su et al., 2008, 2009). Microglia were treated with SYNTR (50 nM) or BufferTR for 2–36 h and the amount of nitrite, a stable NO metabolite, was quantified in the conditioned media (Figure 2; a dose response curve was used to identify this concentration of SYNTR; data not shown). NO secretion was first detected 6 h post-SYNTR treatment with maximum production observed at 24 h (P ≤ 0.05). We therefore chose 24 h post-treatment for all subsequent analyses. To further characterize BV-2 activation in response to α-synuclein, we examined release of the prototypical proinflammatory mediator TNF-α. Twenty-four hours after SYNTR treatment there was a significant increase in TNF-α released into the BV-2 cell conditioned media compared to media from BufferTR treated cells (Figure 3A; P ≤ 0.05). Although the release of TNF-α protein was increased in SYNTR-treated BV-2 cells there was not a significant increase in the gene expression for this target (Table 1). However, using qRT-PCR we determined that the gene expression level of a second prototypical proinflammatory molecule, IL-1β, was significantly upregulated compared to control conditions following SYNTR treatment (Figure 3B; P ≤ 0.05). Taken together, these data establish that α-synuclein stimulates the production and release of classical proinflammatory molecules from BV-2 cells.
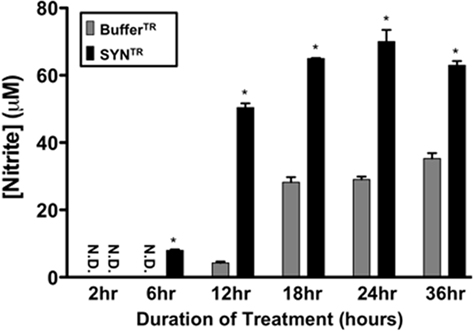
Figure 2. SYNTR treatment stimulates nitric oxide (NO) release from microglia. BV-2 cells were treated with 50 nM of SYNTR or BufferTR for 2–36 h. Following treatment, a Greiss reagent assay was performed on the conditioned media to determine NO release and subsequent nitrite production. NO release was significantly higher in cells treated with SYNTR than buffer control beginning at 6 h of treatment (*P < 0.05, n = 3). NO release reached a maximum by 24 h of treatment. Nitrite concentrations are represented as mean ± SD; ND indicates none detected.
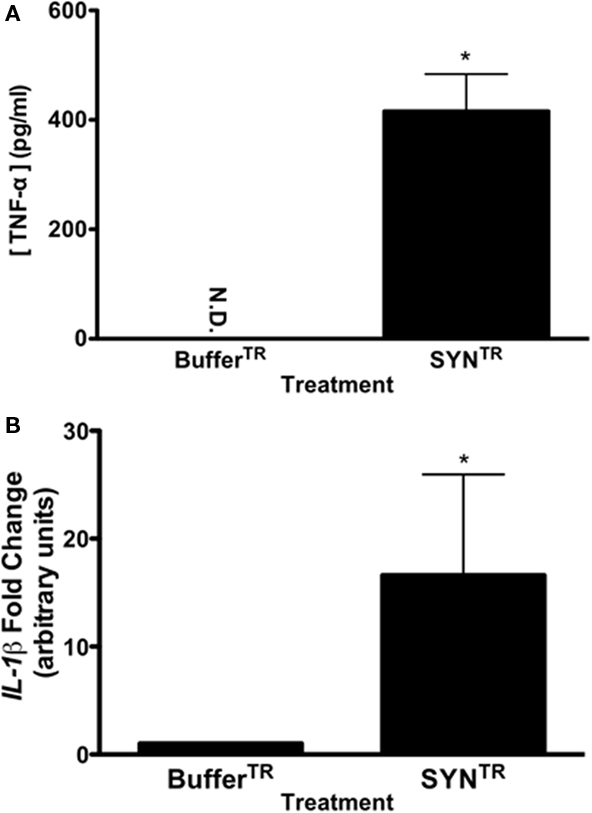
Figure 3. SYNTR treatment increases the expression and release of proinflammatory molecules. (A) BV-2 cells were treated with 50 nM of SYNTR or BufferTR for 24 h. Following treatment, the conditioned media were evaluated for TNF-α protein secretion using an ELISA. Cells treated with SYNTR released significantly more TNF-α than BufferTR treated microglia (ND indicates none detected; *P < 0.05, n = 3). (B) Quantitative RT-PCR for IL1β was performed on cDNA from SYNTR and buffer treated cells. Microglia treated with SYNTR had significantly higher expression of IL1β than buffer treated cells (*P < 0.05; n = 3). Expression values were normalized to 18S rRNA as an internal control. Statistics were performed on ΔCt values.
SYNTR Upregulates Antioxidant Enzymes
One consequence of NO and proinflammatory molecule production in glia is an overall increase in oxidative stress. Microglia are capable of responding to oxidative stress by increasing the expression of antioxidant response genes that are regulated by the transcription factor Nrf-2 [Nuclear factor (erythroid-derived 2)-like 2; Chowdhury et al., 2009; Bast et al., 2010]. The upregulation of these phase II detoxification enzymes provides cellular protection from oxidative stress. Therefore, we investigated whether two Nrf-2 regulated enzymes, peroxiredoxin-1 (PRDX-1), and heme oxygenase-1 (HO-1), both previously associated with activated microglia, were altered following SYNTR or BufferTR treatment (Kitamura et al., 1998a,b; Tanaka et al., 2006; Bast et al., 2010). cDNA was prepared from SYNTR and BufferTR treated BV-2 cells and qRT-PCR performed to interrogate PRDX1 and HMOX1 expression levels. As predicted, the expression levels of both antioxidant response genes were significantly upregulated in SYNTR-treated BV-2 cells compared with BufferTR treated cells (Figures 4A,B; P ≤ 0.05). To validate the gene expression findings, HO-1 protein levels were analyzed in lysates from SYNTR-treated BV-2 cells by Western blot analysis. SYNTR treatment of these cells caused a significant upregulation of HO-1 protein, confirming the qRT-PCR results (Figures 4C,D; P ≤ 0.05). These data demonstrate a robust BV-2 cell derived antioxidant response following treatment with SYNTR.
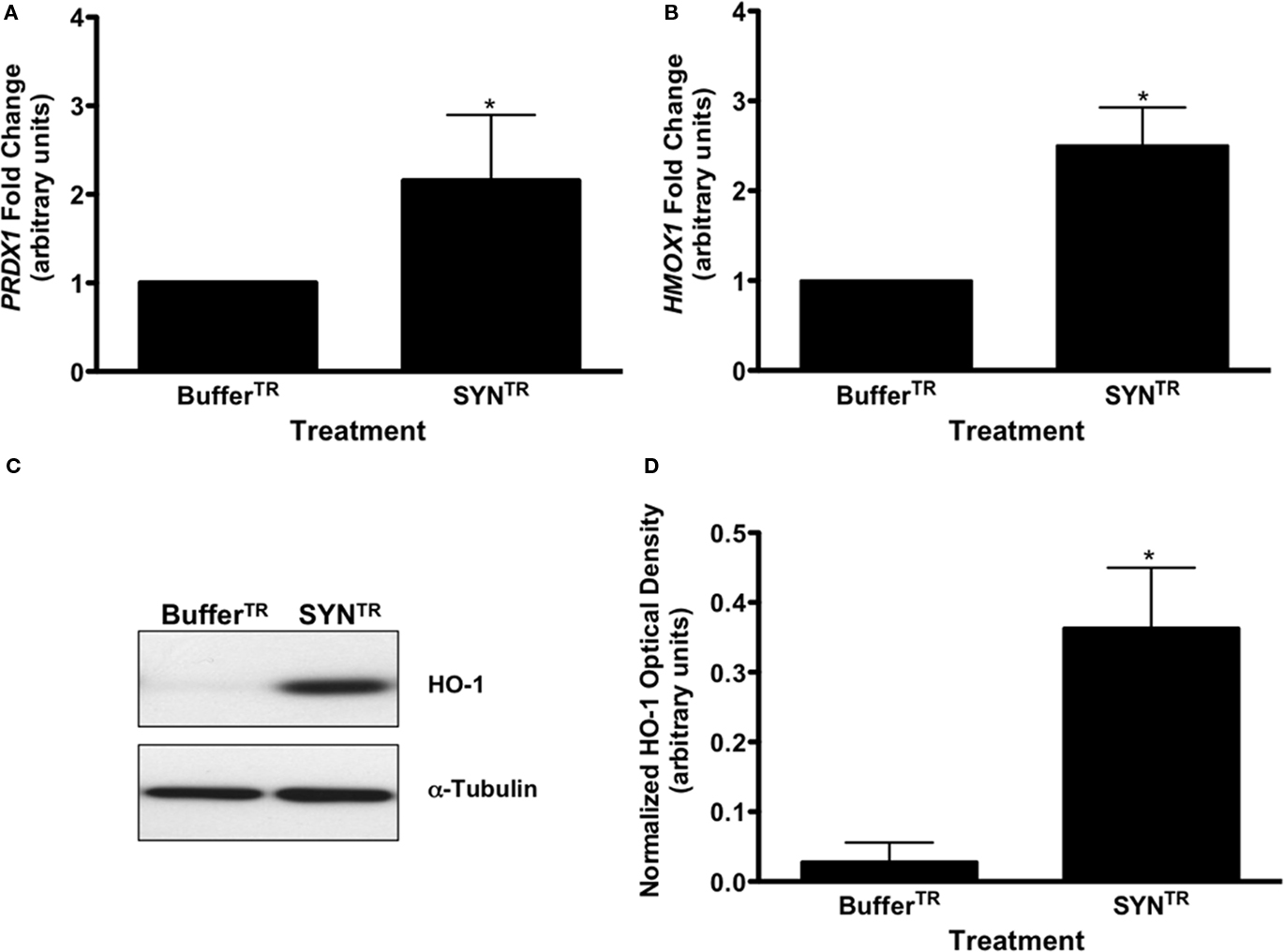
Figure 4. SYNTR treatment increases the expression of antioxidant response genes. (A) Quantitative RT-PCR for (A) peroxiredoxin-1 (PRDX1) and (B) heme oxygenase-1 (HMOX1) was performed on cDNA from BufferTR- or SYNTR-treated BV-2 cells. Cells treated with SYNTR (50 nM) had a significantly higher expression of PRDX1 and HMOX1 compared to buffer treated cells (BufferTR; *P < 0.05). Expression values were normalized to GAPDH as an internal control. Statistics were performed on ΔCt values. (C) Representative HO-1 Western blot analysis of BV-2 cell lysates following treatment. BV-2 cells were treated with 50 nM SYNTR or BufferTR for 24 h. Protein lysates were prepared and subjected to 10% SDS-polyacrylamide gel electrophoresis and immunoblotted for HO-1. Blots were reprobed for α-tubulin as a loading control. (D) Immunocomplexes were quantified by densitometric analysis and normalized to the loading control. BV-2 cells treated with SYNTR had significantly higher levels of HO-1 protein than buffer treated cells (BufferTR; *P < 0.05; n = 3).
SYNTR-Directed Microglial Activation Alters TLR Gene Expression
Thus far we have established that SYNTR directly activates BV-2 cells inciting the expression and release of proinflammatory molecules and the upregulation of antioxidant response enzymes. Since microglial activation is in part mediated through the engagement of DAMPs we asked whether the expression of classic PRRs was altered following exposure to SYNTR. BV-2 cells were treated with BufferTR or SYNTR, 24 h later RNA was prepared and gene expression levels for PRRs, co-receptors, and proinflammatory molecules were quantified (Table 1). Here, we show for the first time, a significant upregulation of TLR2 and TLR3 expression with a concomitant downregulation of TLR7 expression following SYNTR treatment of BV-2 cells (Table 1). The other targets were not significantly changed. Next we asked whether primary microglia derived from mouse cortices demonstrate similar SYNTR-mediated changes in gene expression. Similar to BV-2 cells, primary microglia demonstrate a significant upregulation of TLR2 and TLR3 expression following SYNTR treatment (Table 2). In addition, the expression of TLR1, TLR7, MYD88, IBA-1, NFκB, TNFα, and IL1β were all significantly increased while the expression of other gene targets were either unchanged or decreased (TLR4, TLR6, TLR9, CD36). These data support and extend previous observations that α-synuclein directly activates primary microglia through a classical activation pathway which we now show includes a role for TLRs.
Discussion
The novel finding of this paper is that α-synuclein activates BV-2 and primary microglia through a classical pathway that includes the upregulation of TLR expression. We also show that α-synuclein mediates a robust increase in the expression of antioxidant response genes. Together these results support a role for α-synuclein as a DAMP capable of activating microglial PRRs and inciting oxidative stress.
Specifically, we demonstrate that BV-2 cells exposed to SYNTR show increased release of TNF-α protein and NO as well as increased IL1β gene expression, all indicative of a classical activation pathway (Colton, 2009). Importantly, SYNTR also increases prototypical proinflammatory molecule gene expression in primary microglia (TNFα, IL1β). The upregulation and release of proinflammatory molecules following treatment of microglia with wild-type, mutant, or aggregated α-synuclein has been previously shown, but the activation mechanism is poorly understood (Zhang et al., 2005; Su et al., 2008, 2009). One study demonstrated that primary microglia derived from CD36 knockout mice had a reduced response to exogenous α-synuclein treatment, suggesting a function for this scavenger receptor in activation (Su et al., 2008). Here we confirm previous findings regarding α-synuclein’s ability to stimulate microglia through a classical pathway and extend our understanding of this activation by demonstrating a role for the TLR family of PRRs.
Pattern recognition receptors are associated both with membranes on the cell surface and organelles within microglia, and respond to specific ligands by altering downstream molecular pathways leading to altered transcription of NFκB-, C-Jun-, IRF7-, and CREB-regulated genes (Ajmone-Cat et al., 2003; Ousman et al., 2005; Waetzig et al., 2005; Colton, 2009). The pattern of transcriptional activation represents a specific microglial activation state classified as classical activation, alternative activation and acquired deactivation (Colton, 2009). Our data suggest that SYNTR induces a classical activation pathway since the prototypical proinflammatory molecules, NO, TNF-α, and IL-1β, are increased. It is relevant that these proinflammatory molecules are also elevated in Parkinson’s disease patients (Blum-Degen et al., 1995; Knott et al., 2000; Nagatsu et al., 2000). Additionally, we demonstrate a robust increase in HO-1 following treatment of BV-2 cells with SYNTR. HO-1 is a member of the inducible phase II detoxification enzyme family of genes with transcription regulated by the antioxidant response element binding protein Nrf-2 (Moi et al., 1994; Alam et al., 1999; Kensler et al., 2007; Paine et al., 2010). Under cellular stress, HO-1 helps to maintain antioxidant/oxidant homeostasis by degrading free heme into carbon monoxide, biliverdin, and free iron (Paine et al., 2010). In Parkinson’s disease patients there is an increase in HO-1 in substantia nigra dopamine neurons and in Lewy bodies (Castellani et al., 1996; Schipper et al., 1998; Schipper, 2004). In our reduced system we are examining the direct role of SYNTR on BV-2 cells and find that increased oxidative stress augments HO-1 expression.
Most notably, we have shown for the first time that SYNTR-mediated microglial activation modulates TLR expression. We demonstrate that these changes in TLR expression occur in both the BV-2 cell line and primary microglia derived from mouse cortices. It is interesting that the primary microglia have a more robust response to SYNTR with greater-fold increases in TLR and proinflammatory gene expression than BV-2 cells. TLR1 and TLR2 belong to a subfamily of TLRs that can bind to and be activated by hydrophobic ligands. These TLRs are found on the surface of microglia where they can heterodimerize with each other or with other PRRs to facilitate downstream signaling events (Jin and Lee, 2008; O’Neill et al., 2009). TLR1 and TLR2 are associated with a wide range of fungal, microbial, and endogenous ligands including other misfolded proteins (El Khoury et al., 2003; Jana et al., 2008; Jin et al., 2008; Reed-Geaghan et al., 2009; Stewart et al., 2010). In contrast, TLR3 and TLR7 belong to a single-domain family of TLRs, are localized to endosomes and are associated with the recognition of nucleic acids (O’Neill et al., 2009). Both are potent anti-viral TLRs responding to dsRNA and subsequently activating innate immune responses. The exact role of TLRs in α-synuclein-mediated microglial activation remains to be determined. However, another pathogenic protein, fibrillar β-amyloid, acts as a DAMP activating microglia via TLR2, TLR4, and TLR6 as well as the scavenger receptors, CD14 and CD36 supporting the dual role of TLRs as mediators of glial activation by both exogenous and endogenous ligands (El Khoury et al., 2003; Jana et al., 2008; Jin et al., 2008; Reed-Geaghan et al., 2009; Stewart et al., 2010). In this study we demonstrate a direct activation of microglia by α-synuclein and we hypothesize that α-synuclein also acts as a TLR ligand. First, due to its hydrophobic nature, α-synuclein may bind directly to TLRs since molecules with high hydrophobicity have been shown to contribute to proinflammatory events through interaction with TLRs (Seong and Matzinger, 2004). Second, Lee et al. (2008) have shown that fibrillar α-synuclein, once internalized, is trafficked along the endosomal pathway before it is eventually degraded in the lysosome. Therefore, it is conceivable that α-synuclein is available to directly bind both TLR complexes on the extracellular membrane of microglia and internally on endosomes. Further studies are needed to address this important issue.
In summary, we have demonstrated that α-synuclein activates microglia and alters the expression of TLRs in the process. We have utilized a reduced system, which allowed us to determine the direct effect of SYNTR on microglia. In vivo and cell culture experiments show that α-synuclein is available as a released protein and on cell membranes to activate microglia. Specifically, this protein is found in neuronal intracytoplasmic inclusions called Lewy bodies, on neuronal membranes and neurites, as well as in cerebrospinal fluid and plasma of Parkinson’s disease patients (Spillantini et al., 1997; Borghi et al., 2000; El-Agnaf et al., 2003). Relevant to our work, α-synuclein is released from neurons in an activity dependent manner and from cultured cells that overexpress this protein (Lee et al., 2005, 2010; Su et al., 2008; Emmanouilidou et al., 2010; Feng et al., 2010; Jang et al., 2010). Additionally, α-synuclein is localized to the cell membranes where it would be in juxtaposition to activate surveying microglia (McLean et al., 2000; Tsigelny et al., 2008a; Feng et al., 2010). Important issues remain: what is the structure of α-synuclein that is released from cells, does this structure regulate the type or extent of microglial activation and can we mimic this structure in vitro? α-Synuclein is an intrinsically disordered protein that adapts different conformations depending on the local cellular environment and toxicity has been ascribed to different aggregation states (Uversky, 2010). In vitro it is possible to affect the structure of α-synuclein as shown here and elsewhere, but obtaining a homogenous population of α-synuclein with one specific and stable conformation is a challenge (Uversky and Eliezer, 2009). Herein we show that a mixture of α-synuclein structures (amyloid, monomer, SDS-stable oligomers) activate both BV-2 cells and primary microglia in a manner that includes changes in the expression of TLRs. We do not yet know which if any particular conformation is required for this activation but are currently undertaking those studies. Taken together our results suggest that α-synuclein plays a role in danger/damage-associated molecular inflammation. This work provides key knowledge for future mechanistic studies involving α-synuclein-directed microglial activation and the possible development of anti-inflammatory therapies that act through TLR antagonism.
Conclusion
α-Synuclein incites microglial activation resulting in the expression and release of classical proinflammatory molecules. The novel finding in this paper is that α-synuclein treatment alters TLR expression suggesting that this protein acts as a DAMP. Further work is underway to determine whether specific structural conformations of α-synuclein exhibit preferential TLR binding.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIEHS (R01ES014470; Kathleen A. Maguire-Zeiss).
References
Ajmone-Cat, M. A., De Simone, R., Nicolini, A., and Minghetti, L. (2003). Effects of phosphatidylserine on p38 mitogen activated protein kinase, cyclic AMP responding element binding protein and nuclear factor-kappaB activation in resting and activated microglial cells. J. Neurochem. 84, 413–416.
Alam, J., Stewart, D., Touchard, C., Boinapally, S., Choi, A. M., and Cook, J. L. (1999). Nrf2, a Cap‘n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 274, 26071–26078.
Banati, R. B., Gehrmann, J., Schubert, P., and Kreutzberg, G. W. (1993). Cytotoxicity of microglia. Glia 7, 111–118.
Bartels, A. L., and Leenders, K. L. (2007). Neuroinflammation in the pathophysiology of Parkinson’s disease: evidence from animal models to human in vivo studies with [11C]-PK11195 PET. Mov. Disord. 22, 1852–1856.
Bast, A., Erttmann, S. F., Walther, R., and Steinmetz, I. (2010). Influence of iNOS and COX on peroxiredoxin gene expression in primary macrophages. Free Radic. Biol. Med. 49, 1881–1891.
Biasini, E., Fioriti, L., Ceglia, I., Invernizzi, R., Bertoli, A., Chiesa, R., and Forloni, G. (2004). Proteasome inhibition and aggregation in Parkinson’s disease: a comparative study in untransfected and transfected cells. J. Neurochem. 88, 545–553.
Blasi, E., Barluzzi, R., Bocchini, V., Mazzolla, R., and Bistoni, F. (1990). Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J. Neuroimmunol. 27, 229–237.
Block, M. L., Zecca, L., and Hong, J. S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69.
Blum-Degen, D., Muller, T., Kuhn, W., Gerlach, M., Przuntek, H., and Riederer, P. (1995). Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci. Lett. 202, 17–20.
Borghi, R., Marchese, R., Negro, A., Marinelli, L., Forloni, G., Zaccheo, D., Abbruzzese, G., and Tabaton, M. (2000). Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci. Lett. 287, 65–67.
Castellani, R., Smith, M. A., Richey, P. L., and Perry, G. (1996). Glycoxidation and oxidative stress in Parkinson disease and diffuse Lewy body disease. Brain Res. 737, 195–200.
Chen, G. Y., and Nunez, G. (2010). Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 10, 826–837.
Cho, B. P., Song, D. Y., Sugama, S., Shin, D. H., Shimizu, Y., Kim, S. S., Kim, Y. S., and Joh, T. H. (2006). Pathological dynamics of activated microglia following medial forebrain bundle transection. Glia 53, 92–102.
Chowdhury, I., Mo, Y., Gao, L., Kazi, A., Fisher, A. B., and Feinstein, S. I. (2009). Oxidant stress stimulates expression of the human peroxiredoxin 6 gene by a transcriptional mechanism involving an antioxidant response element. Free Radic. Biol. Med. 46, 146–153.
Cicchetti, F., Brownell, A. L., Williams, K., Chen, Y. I., Livni, E., and Isacson, O. (2002). Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur. J. Neurosci. 15, 991–998.
Colton, C. A. (2009). Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. 4, 399–418.
Colton, C. A., and Wilcock, D. M. (2010). Assessing activation states in microglia. CNS Neurol. Disord. Drug Targets 9, 174–191.
Combs, C. K., Karlo, J. C., Kao, S. C., and Landreth, G. E. (2001). Beta-amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 21, 1179–1188.
Conway, K. A., Harper, J. D., and Lansbury, P. T. (1998). Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat. Med. 4, 1318–1320.
Conway, K. A., Harper, J. D., and Lansbury, P. T. Jr. (2000). Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 39, 2552–2563.
Conway, K. A., Rochet, J. C., Bieganski, R. M., and Lansbury, P. T. Jr. (2001). Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science 294, 1346–1349.
Czlonkowska, A., Kohutnicka, M., Kurkowska-Jastrzebska, I., and Czlonkowski, A. (1996). Microglial reaction in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced Parkinson’s disease mice model. Neurodegeneration 5, 137–143.
Dawson, T., Mandir, A., and Lee, M. (2002). Animal models of PD: pieces of the same puzzle? Neuron 35, 219–222.
Depino, A. M., Earl, C., Kaczmarczyk, E., Ferrari, C., Besedovsky, H., Del Rey, A., Pitossi, F. J., and Oertel, W. H. (2003). Microglial activation with atypical proinflammatory cytokine expression in a rat model of Parkinson’s disease. Eur. J. Neurosci. 18, 2731–2742.
Ding, T. T., Lee, S. J., Rochet, J. C., and Lansbury, P. T. Jr. (2002). Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry 41, 10209–10217.
Duewell, P., Kono, H., Rayner, K. J., Sirois, C. M., Vladimer, G., Bauernfeind, F. G., Abela, G. S., Franchi, L., Nunez, G., Schnurr, M., Espevik, T., Lien, E., Fitzgerald, K. A., Rock, K. L., Moore, K. J., Wright, S. D., Hornung, V., and Latz, E. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361.
El-Agnaf, O. M., Salem, S. A., Paleologou, K. E., Cooper, L. J., Fullwood, N. J., Gibson, M. J., Curran, M. D., Court, J. A., Mann, D. M., Ikeda, S., Cookson, M. R., Hardy, J., and Allsop, D. (2003). Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 17, 1945–1947.
El Khoury, J. B., Moore, K. J., Means, T. K., Leung, J., Terada, K., Toft, M., Freeman, M. W., and Luster, A. D. (2003). CD36 mediates the innate host response to beta-amyloid. J. Exp. Med. 197, 1657–1666.
Emmanouilidou, E., Melachroinou, K., Roumeliotis, T., Garbis, S. D., Ntzouni, M., Margaritis, L. H., Stefanis, L., and Vekrellis, K. (2010). Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 30, 6838–6851.
Feany, M. B., and Bender, W. W. (2000). A Drosophila model of Parkinson’s disease. Nature 404, 394–398.
Feng, L. R., Federoff, H. J., Vicini, S., and Maguire-Zeiss, K. A. (2010). Alpha-synuclein mediates alterations in membrane conductance: a potential role for alpha-synuclein oligomers in cell vulnerability. Eur. J. Neurosci. 32, 10–17.
Fink, A. L. (2006). The aggregation and fibrillation of alpha-synuclein. Acc. Chem. Res. 39, 628–634.
Forno, L. S. (1996). Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 55, 259–272.
Giasson, B. I., Uryu, K., Trojanowski, J. Q., and Lee, V. M. (1999). Mutant and wild type human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. J. Biol. Chem. 274, 7619–7622.
Halle, A., Hornung, V., Petzold, G. C., Stewart, C. R., Monks, B. G., Reinheckel, T., Fitzgerald, K. A., Latz, E., Moore, K. J., and Golenbock, D. T. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 9, 857–865.
Hamza, T. H., Zabetian, C. P., Tenesa, A., Laederach, A., Montimurro, J., Yearout, D., Kay, D. M., Doheny, K. F., Paschall, J., Pugh, E., Kusel, V. I., Collura, R., Roberts, J., Griffith, A., Samii, A., Scott, W. K., Nutt, J., Factor, S. A., and Payami, H. (2010). Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat. Genet. 42, 781–785.
Hashimoto, M., Hsu, L. J., Xia, Y., Takeda, A., Sisk, A., Sundsmo, M., and Masliah, E. (1999). Oxidative stress induces amyloid-like aggregate formation of NACP/alpha-synuclein in vitro. Neuroreport 10, 717–721.
He, Y., Appel, S., and Le, W. (2001). Minocycline inhibits microglial activation and protects nigral cells after 6-hydroxydopamine injection into mouse striatum. Brain Res. 909, 187–193.
Henn, A., Lund, S., Hedtjarn, M., Schrattenholz, A., Porzgen, P., and Leist, M. (2009). The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX 26, 83–94.
Horvath, R. J., Nutile-Mcmenemy, N., Alkaitis, M. S., and Deleo, J. A. (2008). Differential migration, LPS-induced cytokine, chemokine, and NO expression in immortalized BV-2 and HAPI cell lines and primary microglial cultures. J. Neurochem. 107, 557–569.
Hsu, L. J., Sagara, Y., Arroyo, A., Rockenstein, E., Sisk, A., Mallory, M., Wong, J., Takenouchi, T., Hashimoto, M., and Masliah, E. (2000). Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 157, 401–410.
Hu, S., Chao, C. C., Khanna, K. V., Gekker, G., Peterson, P. K., and Molitor, T. W. (1996). Cytokine and free radical production by porcine microglia. Clin. Immunol. Immunopathol. 78, 93–96.
Jana, M., Palencia, C. A., and Pahan, K. (2008). Fibrillar amyloid-beta peptides activate microglia via TLR2: implications for Alzheimer’s disease. J. Immunol. 181, 7254–7262.
Jang, A., Lee, H. J., Suk, J. E., Jung, J. W., Kim, K. P., and Lee, S. J. (2010). Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 113, 1263–1274.
Jin, J. J., Kim, H. D., Maxwell, J. A., Li, L., and Fukuchi, K. (2008). Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer’s disease. J. Neuroinflammation 5, 23.
Jin, M. S., and Lee, J. O. (2008). Structures of the toll-like receptor family and its ligand complexes. Immunity 29, 182–191.
Kensler, T. W., Wakabayashi, N., and Biswal, S. (2007). Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 47, 89–116.
Kim, Y. H., Lussier, S., Rane, A., Choi, S. W., and Andersen, J. K. (2011). Inducible dopaminergic glutathione depletion in an alpha-synuclein transgenic mouse model results in age-related olfactory dysfunction. Neuroscience 172, 379–386.
Kim, Y. S., and Joh, T. H. (2006). Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson’s disease. Exp. Mol. Med. 38, 333–347.
Kitamura, Y., Furukawa, M., Matsuoka, Y., Tooyama, I., Kimura, H., Nomura, Y., and Taniguchi, T. (1998a). In vitro and in vivo induction of heme oxygenase-1 in rat glial cells: possible involvement of nitric oxide production from inducible nitric oxide synthase. Glia 22, 138–148.
Kitamura, Y., Matsuoka, Y., Nomura, Y., and Taniguchi, T. (1998b). Induction of inducible nitric oxide synthase and heme oxygenase-1 in rat glial cells. Life Sci. 62, 1717–1721.
Knott, C., Stern, G., and Wilkin, G. P. (2000). Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol. Cell. Neurosci. 16, 724–739.
Kohutnicka, M., Lewandowska, E., Kurkowska-Jastrzebska, I., Czlonkowski, A., and Czlonkowska, A. (1998). Microglial and astrocytic involvement in a murine model of Parkinson’s disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Immunopharmacology 39, 167–180.
Kowall, N. W., Hantraye, P., Brouillet, E., Beal, M. F., Mckee, A. C., and Ferrante, R. J. (2000). MPTP induces alpha-synuclein aggregation in the substantia nigra of baboons. Neuroreport 11, 211–213.
Kruger, R., Kuhn, W., Muller, T., Woitalla, D., Graeber, M., Kosel, S., Przuntek, H., Epplen, J. T., Schols, L., and Riess, O. (1998). Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108.
Lee, E. J., Woo, M. S., Moon, P. G., Baek, M. C., Choi, I. Y., Kim, W. K., Junn, E., and Kim, H. S. (2010). Alpha-synuclein activates microglia by inducing the expressions of matrix metalloproteinases and the subsequent activation of protease-activated receptor-1. J. Immunol. 185, 615–623.
Lee, F. J., Liu, F., Pristupa, Z. B., and Niznik, H. B. (2001). Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J. 15, 916–926.
Lee, H. J., Patel, S., and Lee, S. J. (2005). Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 25, 6016–6024.
Lee, H. J., Suk, J. E., Bae, E. J., Lee, J. H., Paik, S. R., and Lee, S. J. (2008). Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell Biol. 40, 1835–1849.
Lee, S. J., and Lee, S. (2002). Toll-like receptors and inflammation in the CNS. Curr. Drug Targets Inflamm. Allergy 1, 181–191.
LeVine, H. III. (1999). Quantification of beta-sheet amyloid fibril structures with thioflavin T. Meth. Enzymol. 309, 274–284.
Liu, B. (2006). Modulation of microglial pro-inflammatory and neurotoxic activity for the treatment of Parkinson’s disease. AAPS J. 8, E606–E621.
Maguire-Zeiss, K. A., Wang, C. I., Yehling, E., Sullivan, M. A., Short, D. W., Su, X., Gouzer, G., Henricksen, L. A., Wuertzer, C. A., and Federoff, H. J. (2006). Identification of human alpha-synuclein specific single chain antibodies. Biochem. Biophys. Res. Commun. 349, 1198–1205.
Masliah, E., Rockenstein, E., Veinbergs, I., Mallory, M., Hashimoto, M., Takeda, A., Sagara, Y., Sisk, A., and Mucke, L. (2000). Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science 287, 1265–1269.
McLean, P. J., Kawamata, H., Ribich, S., and Hyman, B. T. (2000). Membrane association and protein conformation of alpha-synuclein in intact neurons. Effect of Parkinson’s disease-linked mutations. J. Biol. Chem. 275, 8812–8816.
Moi, P., Chan, K., Asunis, I., Cao, A., and Kan, Y. W. (1994). Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. U.S.A. 91, 9926–9930.
Muzio, M., Polentarutti, N., Bosisio, D., Prahladan, M. K., and Mantovani, A. (2000). Toll-like receptors: a growing family of immune receptors that are differentially expressed and regulated by different leukocytes. J. Leukoc. Biol. 67, 450–456.
Nagatsu, T., Mogi, M., Ichinose, H., and Togari, A. (2000). Cytokines in Parkinson’s disease. J. Neural Transm. Suppl. 143–151.
Naiki, H., Higuchi, K., Hosokawa, M., and Takeda, T. (1989). Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Anal. Biochem. 177, 244–249.
O’Neill, L. A., Bryant, C. E., and Doyle, S. L. (2009). Therapeutic targeting of Toll-like receptors for infectious and inflammatory diseases and cancer. Pharmacol. Rev. 61, 177–197.
Ouchi, Y., Yagi, S., Yokokura, M., and Sakamoto, M. (2009). Neuroinflammation in the living brain of Parkinson’s disease. Parkinsonism Relat. Disord. 15(Suppl. 3), S200–S204.
Ouchi, Y., Yoshikawa, E., Sekine, Y., Futatsubashi, M., Kanno, T., Ogusu, T., and Torizuka, T. (2005). Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 57, 168–175.
Ousman, S. S., Wang, J., and Campbell, I. L. (2005). Differential regulation of interferon regulatory factor (IRF)-7 and IRF-9 gene expression in the central nervous system during viral infection. J. Virol. 79, 7514–7527.
Paine, A., Eiz-Vesper, B., Blasczyk, R., and Immenschuh, S. (2010). Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem. Pharmacol. 80, 1895–1903.
Parihar, M. S., Parihar, A., Fujita, M., Hashimoto, M., and Ghafourifar, P. (2008). Mitochondrial association of alpha-synuclein causes oxidative stress. Cell. Mol. Life Sci. 65, 1272–1284.
Parihar, M. S., Parihar, A., Fujita, M., Hashimoto, M., and Ghafourifar, P. (2009). Alpha-synuclein overexpression and aggregation exacerbates impairment of mitochondrial functions by augmenting oxidative stress in human neuroblastoma cells. Int. J. Biochem. Cell Biol. 41, 2015–2024.
Periquet, M., Fulga, T., Myllykangas, L., Schlossmacher, M. G., and Feany, M. B. (2007). Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J. Neurosci. 27, 3338–3346.
Perrin, R. J., Woods, W. S., Clayton, D. F., and George, J. M. (2001). Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J. Biol. Chem. 276, 41958–41962.
Polymeropoulos, M. H., Higgins, J. J., Golbe, L. I., Johnson, W. G., Ide, S. E., Di Iorio, G., Sanges, G., Stenroos, E. S., Pho, L. T., Schaffer, A. A., Lazzarini, A. M., Nussbaum, R. L., and Duvoisin, R. C. (1996). Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science 274, 1197–1199.
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., Pike, B., Root, H., Rubenstein, J., Boyer, R., Stenroos, E. S., Chandrasekharappa, S., Athanassiadou, A., Papapetropoulos, T., Johnson, W. G., Lazzarini, A. M., Duvoisin, R. C., Di Iorio, G., Golbe, L. I., and Nussbaum, R. L. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047.
Qian, L., Hong, J. S., and Flood, P. M. (2006). Role of microglia in inflammation-mediated degeneration of dopaminergic neurons: neuroprotective effect of interleukin 10. J. Neural Transm. Suppl. 367–371.
Reed-Geaghan, E. G., Savage, J. C., Hise, A. G., and Landreth, G. E. (2009). CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}stimulated microglial activation. J. Neurosci. 29, 11982–11992.
Reynolds, A. D., Kadiu, I., Garg, S. K., Glanzer, J. G., Nordgren, T., Ciborowski, P., Banerjee, R., and Gendelman, H. E. (2008). Nitrated alpha-synuclein and microglial neuroregulatory activities. J. Neuroimmune Pharmacol. 3, 59–74.
Satake, W., Nakabayashi, Y., Mizuta, I., Hirota, Y., Ito, C., Kubo, M., Kawaguchi, T., Tsunoda, T., Watanabe, M., Takeda, A., Tomiyama, H., Nakashima, K., Hasegawa, K., Obata, F., Yoshikawa, T., Kawakami, H., Sakoda, S., Yamamoto, M., Hattori, N., Murata, M., Nakamura, Y., and Toda, T. (2009). Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 41, 1303–1307.
Sawada, M., Imamura, K., and Nagatsu, T. (2006). Role of cytokines in inflammatory process in Parkinson’s disease. J. Neural Transm. Suppl. 373–381.
Schipper, H. M. (2004). Heme oxygenase expression in human central nervous system disorders. Free Radic. Biol. Med. 37, 1995–2011.
Schipper, H. M., Liberman, A., and Stopa, E. G. (1998). Neural heme oxygenase-1 expression in idiopathic Parkinson’s disease. Exp. Neurol. 150, 60–68.
Seong, S. Y., and Matzinger, P. (2004). Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat. Rev. Immunol. 4, 469–478.
Sharon, R., Bar-Joseph, I., Frosch, M. P., Walsh, D. M., Hamilton, J. A., and Selkoe, D. J. (2003). The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron 37, 583–595.
Shlyakhtenko, L. S., Gall, A. A., Filonov, A., Cerovac, Z., Lushnikov, A., and Lyubchenko, Y. L. (2003). Silatrane-based surface chemistry for immobilization of DNA, protein-DNA complexes and other biological materials. Ultramicroscopy 97, 279–287.
Shlyakhtenko, L. S., Potaman, V. N., Sinden, R. R., Gall, A. A., and Lyubchenko, Y. L. (2000). Structure and dynamics of three-way DNA junctions: atomic force microscopy studies. Nucleic Acids Res. 28, 3472–3477.
Shtilerman, M. D., Ding, T. T., and Lansbury, P. T. Jr. (2002). Molecular crowding accelerates fibrillization of alpha-synuclein: could an increase in the cytoplasmic protein concentration induce Parkinson’s disease? Biochemistry 41, 3855–3860.
Simon-Sanchez, J., Schulte, C., Bras, J. M., Sharma, M., Gibbs, J. R., Berg, D., Paisan-Ruiz, C., Lichtner, P., Scholz, S. W., Hernandez, D. G., Kruger, R., Federoff, M., Klein, C., Goate, A., Perlmutter, J., Bonin, M., Nalls, M. A., Illig, T., Gieger, C., Houlden, H., Steffens, M., Okun, M. S., Racette, B. A., Cookson, M. R., Foote, K. D., Fernandez, H. H., Traynor, B. J., Schreiber, S., Arepalli, S., Zonozi, R., Gwinn, K., Van Der Brug, M., Lopez, G., Chanock, S. J., Schatzkin, A., Park, Y., Hollenbeck, A., Gao, J., Huang, X., Wood, N. W., Lorenz, D., Deuschl, G., Chen, H., Riess, O., Hardy, J. A., Singleton, A. B., and Gasser, T. (2009). Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 41, 1308–1312.
Singleton, A., Gwinn-Hardy, K., Sharabi, Y., Li, S. T., Holmes, C., Dendi, R., Hardy, J., Crawley, A., and Goldstein, D. S. (2004). Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain 127, 768–772.
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., Hulihan, M., Peuralinna, T., Dutra, A., Nussbaum, R., Lincoln, S., Crawley, A., Hanson, M., Maraganore, D., Adler, C., Cookson, M. R., Muenter, M., Baptista, M., Miller, D., Blancato, J., Hardy, J., and Gwinn-Hardy, K. (2003). Alpha-synuclein locus triplication causes Parkinson’s disease. Science 302, 841.
Song, D. D., Shults, C. W., Sisk, A., Rockenstein, E., and Masliah, E. (2004). Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp. Neurol. 186, 158–172.
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840.
Stewart, C. R., Stuart, L. M., Wilkinson, K., Van Gils, J. M., Deng, J., Halle, A., Rayner, K. J., Boyer, L., Zhong, R., Frazier, W. A., Lacy-Hulbert, A., El Khoury, J., Golenbock, D. T., and Moore, K. J. (2010). CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11, 155–161.
Su, X., Federoff, H. J., and Maguire-Zeiss, K. A. (2009). Mutant alpha-synuclein overexpression mediates early proinflammatory activity. Neurotox. Res. 16, 238–254.
Su, X., Maguire-Zeiss, K. A., Giuliano, R., Prifti, L., Venkatesh, K., and Federoff, H. J. (2008). Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 29, 1690–1701.
Sugama, S., Yang, L., Cho, B. P., Degiorgio, L. A., Lorenzl, S., Albers, D. S., Beal, M. F., Volpe, B. T., and Joh, T. H. (2003). Age-related microglial activation in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurodegeneration in C57BL/6 mice. Brain Res. 964, 288–294.
Tanaka, S., Ide, M., Shibutani, T., Ohtaki, H., Numazawa, S., Shioda, S., and Yoshida, T. (2006). Lipopolysaccharide-induced microglial activation induces learning and memory deficits without neuronal cell death in rats. J. Neurosci. Res. 83, 557–566.
Theodore, S., Cao, S., Mclean, P. J., and Standaert, D. G. (2008). Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol. 67, 1149–1158.
Thomas, M. P., Chartrand, K., Reynolds, A., Vitvitsky, V., Banerjee, R., and Gendelman, H. E. (2007). Ion channel blockade attenuates aggregated alpha synuclein induction of microglial reactive oxygen species: relevance for the pathogenesis of Parkinson’s disease. J. Neurochem. 100, 503–519.
Tsigelny, I. F., Crews, L., Desplats, P., Shaked, G. M., Sharikov, Y., Mizuno, H., Spencer, B., Rockenstein, E., Trejo, M., Platoshyn, O., Yuan, J. X., and Masliah, E. (2008a). Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer’s and Parkinson’s diseases. PLoS ONE 3, e3135. doi: 10.1371/journal.pone.0003135
Tsigelny, I. F., Sharikov, Y., Miller, M. A., and Masliah, E. (2008b). Mechanism of alpha-synuclein oligomerization and membrane interaction: theoretical approach to unstructured proteins studies. Nanomedicine 4, 350–357.
Uversky, V. N. (2010). Mysterious oligomerization of the amyloidogenic proteins. FEBS J. 277, 2940–2953.
Uversky, V. N., and Eliezer, D. (2009). Biophysics of Parkinson’s disease: structure and aggregation of alpha-synuclein. Curr. Protein Pept. Sci. 10, 483–499.
Vila, M., Vukosavic, S., Jackson-Lewis, V., Neystat, M., Jakowec, M., and Przedborski, S. (2000). Alpha-synuclein up-regulation in substantia nigra dopaminergic neurons following administration of the parkinsonian toxin MPTP. J. Neurochem. 74, 721–729.
Volles, M. J., and Lansbury, P. T. Jr. (2002). Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 41, 4595–4602.
Volles, M. J., Lee, S. J., Rochet, J. C., Shtilerman, M. D., Ding, T. T., Kessler, J. C., and Lansbury, P. T. Jr. (2001). Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry 40, 7812–7819.
Waetzig, V., Czeloth, K., Hidding, U., Mielke, K., Kanzow, M., Brecht, S., Goetz, M., Lucius, R., Herdegen, T., and Hanisch, U. K. (2005). c-Jun N-terminal kinases (JNKs) mediate pro-inflammatory actions of microglia. Glia 50, 235–246.
Wu, X. F., Block, M. L., Zhang, W., Qin, L., Wilson, B., Zhang, W. Q., Veronesi, B., and Hong, J. S. (2005). The role of microglia in paraquat-induced dopaminergic neurotoxicity. Antioxid. Redox Signal. 7, 654–661.
Xilouri, M., Vogiatzi, T., Vekrellis, K., Park, D., and Stefanis, L. (2009). Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE 4, e5515. doi: 10.1371/journal.pone.0005515
Keywords: microglial activation, pattern recognition receptors, DAMP, inflammation, Parkinson’s disease
Citation: Béraud D, Twomey M, Bloom B, Mittereder A, Ton V, Neitzke K, Chasovskikh S, Mhyre TR and Maguire-Zeiss KA (2011) α-Synuclein alters toll-like receptor expression. Front. Neurosci. 5:80. doi: 10.3389/fnins.2011.00080
Received: 04 March 2011;
Accepted: 06 June 2011;
Published online: 29 June 2011.
Edited by:
Iliya Lefterov, University of Pittsburg, USAReviewed by:
Iliya Lefterov, University of Pittsburg, USASpaska Angelova Stanilova, Trakia University, Bulgaria
Nicholas Fitz, University of Pittsburgh, USA
Copyright: © 2011 Béraud, Twomey, Bloom, Mittereder, Ton, Neitzke, Chasovskikh, Mhyre and Maguire-Zeiss. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Kathleen A. Maguire-Zeiss, Department of Neuroscience, Georgetown University Medical Center, NRB EP08, 3970 Reservoir Road NW, Washington, DC 20057, USA. e-mail:a200NDVAZ2VvcmdldG93bi5lZHU=