 Harish Babu1
Harish Babu1 Jan-Hendrik Claasen2,3,4
Jan-Hendrik Claasen2,3,4 Suresh Kannan2
Suresh Kannan2 Annette E. Rünker2
Annette E. Rünker2 Theo Palmer1
Theo Palmer1 Gerd Kempermann2,3*
Gerd Kempermann2,3*- 1 Institute for Stem Cell Biology and Regenerative Medicine, Stanford University, Stanford, CA, USA
- 2 CRTD, Center for Regenerative Therapies Dresden, DFG-Research Center for Regenerative Therapies Dresden, Dresden, Germany
- 3 DZNE, German Center for Neurodegenerative Diseases, Dresden, Germany
- 4 Department of Neurology, Medizinische Fakultät Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
In vitro assays are valuable tools to study the characteristics of adult neural precursor cells under controlled conditions with a defined set of parameters. We here present a detailed protocol based on our previous original publication (Babu et al., 2007) to isolate neural precursor cells from the hippocampus of adult mice and maintain and propagate them as adherent monolayer cultures. The strategy is based on the use of Percoll density gradient centrifugation to enrich precursor cells from the micro-dissected dentate gyrus. Based on the expression of Nestin and Sox2, a culture-purity of more than 98% can be achieved. The cultures are expanded under serum-free conditions in Neurobasal A medium with addition of the mitogens Epidermal growth factor and Fibroblast growth factor 2 as well as the supplements Glutamax-1 and B27. Under differentiation conditions, the precursor cells reliably generate approximately 30% neurons with appropriate morphological, molecular, and electrophysiological characteristics that might reflect granule cell properties as their in vivo counterpart. We also highlight potential modifications to the protocol.
Introduction
In vitro cultures of adult hippocampal neural precursor cells and their differentiation into granule cell-like neurons is a key tool for deciphering the cellular and molecular mechanisms of adult neurogenesis. The sequence of adult neural precursor cell proliferation, neuronal differentiation, and subsequent integration into pre-existing neuronal circuitries occurs in the mature mammalian central nervous system throughout life (Kuhn et al., 1996; Ben Abdallah et al., 2010; Knoth et al., 2010).
Adult neural precursor cells reside in two distinct permissive microenvironments (neurogenic niches), the subventricular zone lining the lateral ventricle’s wall and the subgranular zone of the hippocampal dentate gyrus (Reynolds and Weiss, 1992; Palmer et al., 1995). Adult neurogenesis represents a highly complex interaction among the many cellular and molecular components of the unique niche with the genetic setting of the precursor cells. On one hand, in vitro studies of neural precursor cells allow analyzing precursor cells independently of niche signals from their former neighboring cells. On the other hand, the culture conditions have to replace the niche at least to the degree required to maintain the cells. It does not pose considerable problems to extract neural precursor cells and bring them into culture, but it is difficult to maintain and expand them in a way preserving their intrinsic properties and allow differentiation into defined cellular phenotypes.
Generally, adult neural precursor cells can be expanded in vitro using two different culture forms: as neurospheres, non-adherent spherical clusters of cells, or as adherent monolayer cultures.
Neurospheres have several undisputed advantages, most notably their stunning ease of use. The cytoarchitecture within the spheres is suggested to provide a microenvironment that might be advantageous for the precursor cells to survive in non-physiological conditions in vitro (Bez et al., 2003). However this cellular organization is disadvantageous with respect to in vitro expansion of the “stem cells.” Precursor cells inside the neurosphere have the tendency to differentiate resulting in increasing levels of cellular heterogeneity. With growing size neurospheres contain a more heterogeneous population of precursors with an external rim of rapidly dividing precursor cells and a core of differentiated postmitotic cells (Reynolds and Rietze, 2005) making frequent sphere dissociation inevitable. A reduced diffusion of growth factors into the sphere and a direct influence of already differentiated cells on the undifferentiated progeny may contribute to this layering effect. Other issues are the low efficiency of secondary sphere formation from dissociated single cells, and a tendency of floating cells to aggregate, making single-cell clonal analyses difficult to interpret. These and other caveats have been discussed in several critical comments and original publications (Jensen and Parmar, 2006; Singec et al., 2006; Jessberger et al., 2007; Marshall et al., 2007). Consequently, important modifications to the protocol have been suggested (Reynolds and Rietze, 2005; Rietze and Reynolds, 2006). Regardless, this culture form does have utility and has been the method of choice for multiple research projects including a side-by-side analysis of hippocampal cultures from rats and mice (Ray and Gage, 2006). For example, neurospheres might be rather faithful in vitro representations of the situation in the neurogenic niche and allowing to study the interaction of different cell types during differentiation (Imbeault et al., 2009). The point is not that neurospheres are per se inferior to monolayers as precursor cell model but that both have their individual pros and cons and that for certain questions monolayers are clearly preferable.
Adherent monolayer cultures circumvent some of the problems associated with neurospheres because they represent a more homogeneous undifferentiated population of precursor cells. Cells are uniformly exposed to growth factors in culture medium that supports expansion by symmetrical cell division and inhibits spontaneous differentiation, a finding that was described for neural stem cells derived from pluripotent mouse embryonic stem (ES) cells and cortical neural stem cell lines from mouse fetuses (E16.5) by Conti and colleagues in 2005 (Conti and Cattaneo, 2005).
A second advantage of the monolayer culture is that cells can be directly monitored and investigated. The arrangement in one single layer facilitates appreciation of the morphology and size of the individual cell and the change over time, e.g., with videomicroscopy. This aspect becomes central in functional assays, e.g., based on conditional luciferase expression. The opportunity to monitor the appearance of undifferentiated and unimpaired cells is also important in order to choose comparable vital cultures for experiments. In contrast, the densely packed cellular clusters of neurospheres limit the experimental manipulation and analysis of individual cells. It needs to be noted, however, that in vivo precursor cells are never isolated and without contact to other cells. The situation in the culture dish is thus highly artificial and represents a highly reductionist setting. The advantages are paid for with limitations that require critical judgment of all results obtained with such model.
Despite the fact that the process of adult hippocampal neurogenesis has been found in almost all mammals studied including rats (Cameron et al., 1993), monkeys (Kornack and Rakic, 1999), humans (Eriksson et al., 1998), the vast majority of studies are done in mice. The first protocol to instruct the generation of a serum-free monolayer culture of neural precursor cells from the micro-dissected mouse dentate gyrus was developed in our laboratory by Babu et al. (2007), a method that was based on monolayer cultures originally described for rat hippocampal precursor cells (Palmer et al., 1995, 1997, 1999). Using micro-dissection and enrichment protocols, we could show that precursor cells from the mouse dentate gyrus were self-renewing and that upon differentiation their progeny developed into neurons with properties of hippocampal granule cells (Babu et al., 2007).
Although highly artificial, monolayer culture systems preserve the criteria of “stemness” in these cells. This is a virtually unlimited capacity to replicate (self-renewal) and the ability to generate the three principal cell types of the nervous system (multipotency), including neurons as indicated by immunocytochemistry and electrophysiology. The monolayer method has proven useful to study intrinsic regulatory mechanisms of neural precursor cells and details are provided here on the conditions that are required to establish a monolayer cell culture for long-term expansion. However, prolonged expansion of neural precursor cells might carry the risk of genomic instability and subsequent chromosomal aberration. We did not detect overt changes in our cultures during long-term passage with regard to morphology, self-renewal, or molecular profiling. This observation is in accordance with Foroni et al. (2007) who did not detect any cellular transformation or genetic instability, when propagating subventricular zone adult neural precursor for up to 1 year. Nevertheless, if late passages are to be used, karyotyping might be advisable.
Although the methods described here depict the direct isolation of neural stem and progenitor cells from the dentate gyrus and their subsequent cultivation, we found these methods also useful to transfer neurospheres to monolayer culture and for isolating and expanding precursor cells from other brain regions. The here-described protocol is essentially based on our original publication from 2007, but in addition to the original method considers a dissection method for dentate gyrus tissue that does not require micro-dissections from vibratome slices (compare Hagihara et al., 2009). The only major change from the original publication consists of the improved cell separation with one single Percoll gradient centrifugation step. A small change is the use of medium instead of artificial CSF to store cells during the preparation, because artificial CSF has no advantages.
For more than 3 years our protocol has been successfully used in a wide range of our research projects and has been taught to cooperating research groups and visiting scientists. The here-described protocol was used in recent publications by us (Babu et al., 2009) and our collaborators (Ramirez-Rodriguez et al., 2009), as well as independent researchers in original (Wong Po Foo et al., 2009) or slightly modified form (Boku et al., 2009; Liu et al., 2010; Luo et al., 2010a,b).
Overview of the Procedure
Our neural precursor cell extraction protocol is based on Percoll gradient centrifugation that enables an enrichment of precursor cells for optimal culture conditions (Palmer et al., 1999).
The workflow is depicted in Figure 1. After separation of the hippocampus from the rest of the brain, the dentate gyrus is further isolated by micro-dissection from the adjacent walls of the lateral ventricles and cornu ammonis of the hippocampus to avoid a commixture of hippocampal neural precursor cells with other precursor cell populations. The excised tissue containing the neurogenic subgranular zone is then further dissociated into a single-cell suspension using an enzymatic solution supported by mechanical trituration steps with fire-polished Pasteur pipettes. In the ensuing step, neural precursor cells are separated from differentiated cells, myelin, and extracellular matrix using density gradient separation by a centrifugation step with 22% Percoll. Pelleted cells are separated from supernatant, washed, resuspended in chemically defined culture medium (Neurobasal A with supplements B27 and Glutamax-1 and growth factors EGF and FGF2) and plated on coated culture dishes. Two days after isolation neural precursor cells that have not yet attached to the surface are collected, centrifuged, and replated into the same culture dish with fresh medium. This enrichment step is necessary to ensure an optimal density for further cell expansion. From day 4 after cell extraction onwards, 75% of the culture medium is substituted every second day. In about 9–12 days 70–80% of the culture dish surface is populated and the culture is ready for the first passage.
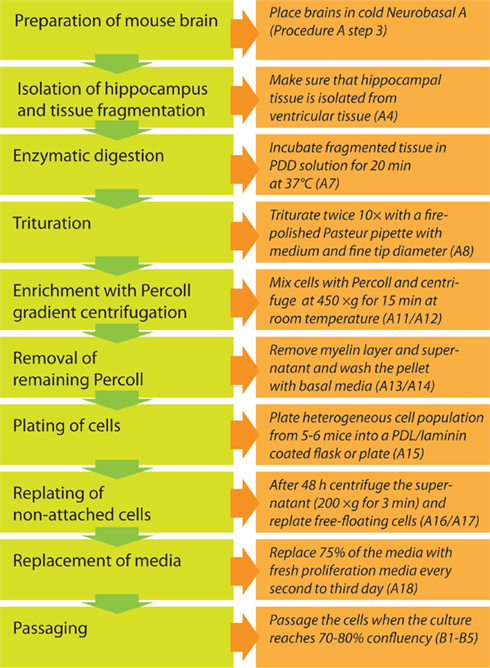
Figure 1. Workflow diagram for the experimental procedure.
The isolated and expanded cells display the typical morphology of adult neural precursor cells Figures 2A–C and express the typical neural precursor cell markers Nestin Figure 3C, or vimentin Figure 3D. Many cells also express the radial glia marker BLBP Figure 3B or transcription factor Id1 involved in self-renewal Figure 3F. In contrast, only few cells appear further committed to the neuronal lineage (expressing βIII-tubulin Figure 3E), and there are no identifiable mature astrocytes (expressing S100β Figure 3F). Virtually all cells express Ki67, a cellular marker for proliferation Figure 3A. Similarly, incubation with BrdU, a thymidine analog that is incorporated during DNA synthesis, labels almost all cells, indicating robust proliferation of the cultured neural precursor cells (not shown here, but see figures in Babu et al., 2007). Under differentiation conditions, neural precursor cells gradually acquire the morphology of postmitotic neurons and astrocytes (Figures2 D–F).
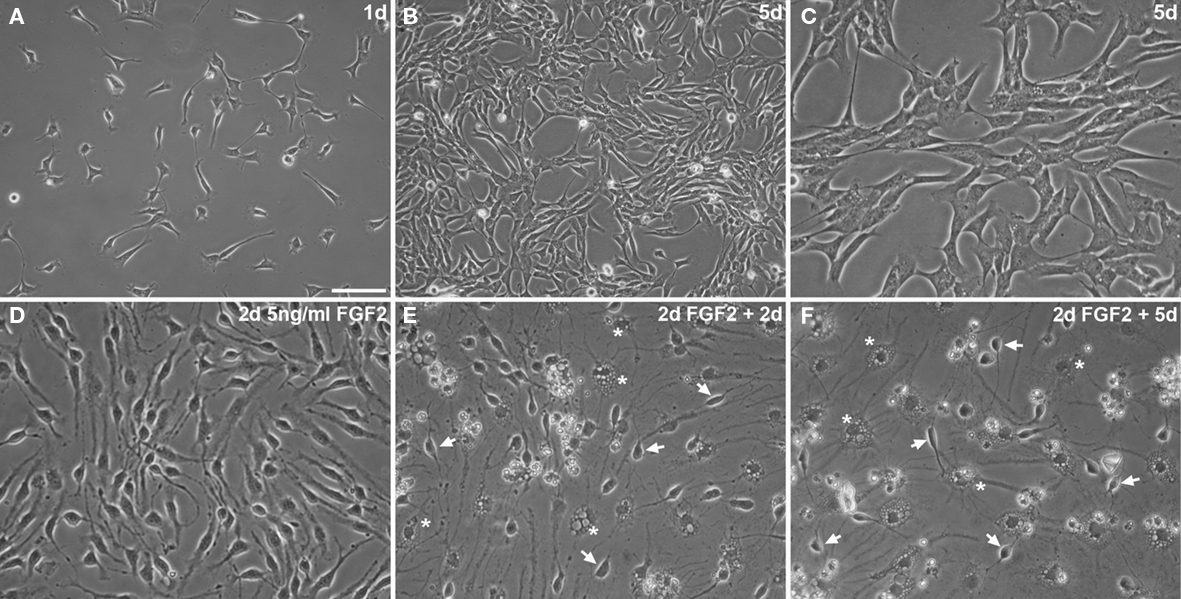
Figure 2. Highly enriched neural precursor cells propagated as adherent monolayer cultures (A–C). Adherent precursor cells cultures under routine proliferation conditions 1 day (A) and 5 days after seeding [(B) about 80% confluent; higher magnification in (C)]. (D–F) Upon differentiation with 2 days of 5 ng/ml FGF2 (D) and subsequent complete growth factor depletion (E,F), neural precursor cells change their morphology showing a progressively postmitotic and mature appearance of neuronal (arrows) and glial (asterisks) cells. Scale bar in (A): 100 μm for (A,B); 50 μm for (C–F).
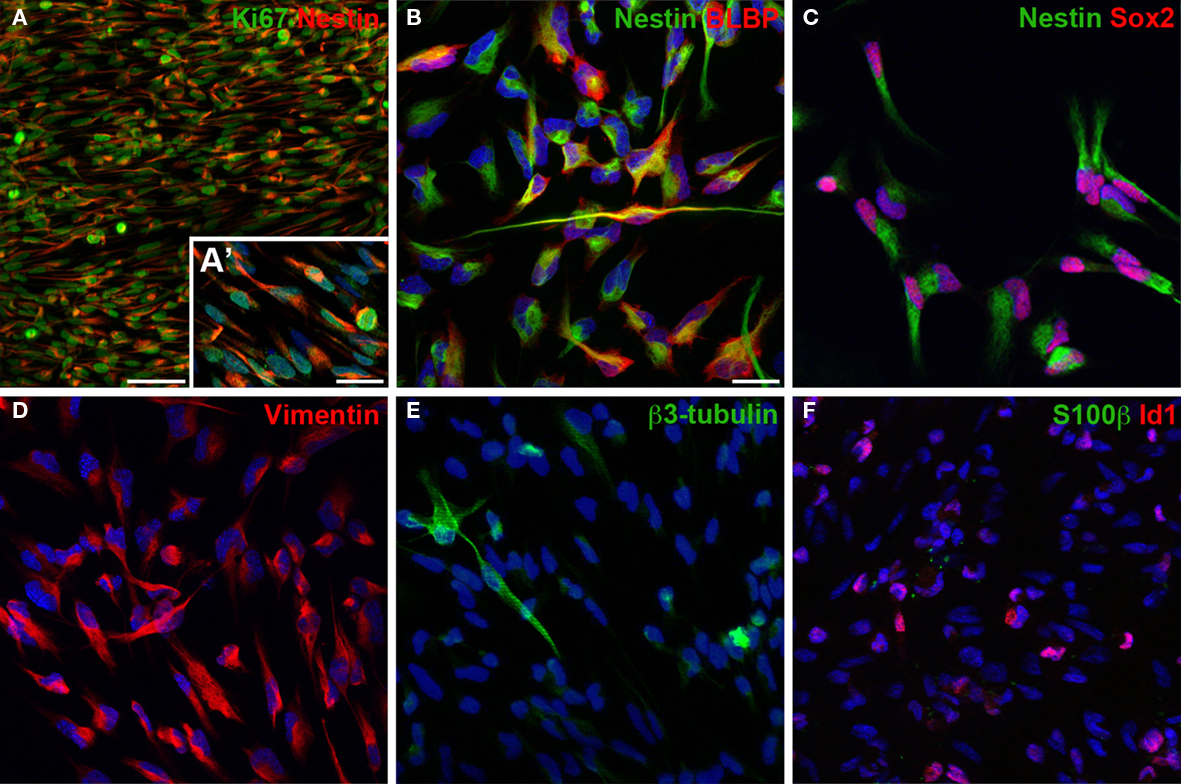
Figure 3. In cultures of dentate gyrus neural precursor cells virtually all cells express cellular marker for proliferation, Ki67 (A), and for progenitor cells of the nervous system, such as Nestin (A–C), Sox2 (C) or vimentin (D). Many cells express also the radial glia marker BLBP (B) and Id1 (F), a dominant-negative bHLH transcriptional regulator expressed in a fraction of adult neural stem cells important for their self-renewal capacity. Only few cells express β3-tubulin (E) indicating neuronally committed cells. No cells were positive for S100β (F), a marker that labels mature astrocytes. Cells were grown until about 80% confluent (3–5 days) before fixing and staining. Blue stain in (A–F) is the nucleic dye DAPI. Scale bar in (A): 50 μm, in (A′) and (B) for (A′–F): 20 μm.
Material
Animals
• C57BL6/J (The Jackson Laboratory) female and/or male mice with an age of 7–9 weeks are used. Five to six mice of this strain are sufficient to establish a primary adult neural precursor cell culture. All animals are kept under standard housing conditions with a 12-h light/dark cycle and free access to food and water.
IMPORTANT! Experiments involving live animals must conform to experimenter’s national and institutional rules and regulation.
3.2 Reagents
Culture medium
• Neurobasal A (Gibco, cat. no. 10888) or DMEM/F12 (Gibco, cat. no. 31330) for culture dish coating and cell separation procedure
• B27 Supplement (Gibco, cat. no. 17504)
• Glutamax-1 Supplement (Gibco, cat. no. 35050)
• Recombinant human Fibroblast growth factor 2 (FGF2; Peprotech, cat. no. 100-18B-B)
• Recombinant human Epidermal growth factor (EGF; Peprotech, cat. no. 100-15)
• Recombinant human Brain Derived Neurotrophic Factor (BDNF; Peprotech, cat. no. 450-02)
• Bovine serum albumin (BSA; Sigma, cat. no. A2153)
• D-Glucose (Sigma, cat. no. G7021)
• Retinoic acid (Sigma, cat. no. R2625)
Cell detachment
• Accutase (PAA, cat. no. L11-007)
• Ovomucoid trypsin inhibitor (Worthington, cat. no. 3085)
• Trypsin EDTA 10× (Gibco, cat. no. 15400054)
Cell extraction
• Papain (Worthington, cat. no. 3126)
• Dispase (Roche, cat. no. 10-165-859-001)
• DNase (Worthington, cat. no. 2139)
• Percoll (GE Healthcare, cat. no. 17-0891-01)
• PBS 10× (Gibco, cat. no. 70011)
Coating
• Laminin (Roche, cat. no. 11-243-217-001)
• Poly-D-Lysine (PDL; Sigma, cat. no. P1024)
• Ethanol 70% vol/vol
• PBS (Gibco, cat. no. 10010-023)
• H2SO4 (EMD, cat. no. SX1244-6)
• ddH2O
Cell banking
• Dimethyl sulfoxide (DMSO; Sigma Aldrich, cat. no. D8418)
Immunocytochemistry
• Paraformaldehyde (PFA; Electron Microscopy Sciences, cat. no. 19210)
• Triton-X100 (Sigma, cat. no. T8787)
• Normal donkey serum (Jackson Immunoresearch, cat. no. 017-000-121)
• Hoechst 33258 (Gibco, cat. no. H3569)
• Mouse anti-Map2ab antibody (Sigma, cat. no. M1406; 1:500)
• Rabbit anti-GFAP antibody (Dako, cat. no. Z0334; 1:500)
• Mouse anti-O4 antibody (R&D Systems, cat. no. MAB1326; 1:100)
• Mouse anti-Nestin antibody (BD Pharmingen, cat. no. 611658; 1:200)
• Rabbit anti-Sox2 antibody (Chemicon, cat. no. AB5603; 1:500)
• Rabbit anti-Ki67 antibody (Novocastra, cat. no. NCL-Ki67p; 1:400)
• Rabbit anti-BLBP antibody (Abcam, cat. no. ab32423; 1:400)
• Goat anti-Vimentin antibody (Sigma, cat. no. V4630; 1:500)
• Rabbit anti-Id1 antibody (Biocheck, cat. no. BCH-1/37-2; 1:100)
• Mouse anti-β3-tubulin antibody (Promega, cat. no. G712A; 1:1000)
• Rabbit anti-S100b antibody (Swant, cat. no. 37A; 1:1000)
Equipment
• Inverted microscope
• Dissecting microscope
• Water bath (37°C)
• Horizontal laminar flow hood for sterile cell culture work
• Dissection instruments: micro-dissection scissors (FST, cat. no. 15010-11 or cat. no. 15000-10), fine forceps (FST, cat. no. 11251-20), spatula (FST, cat. no. 10093-13), fine scissors (FST, cat. no. 14090-09), surgical scissors (FST, cat. no. 14010-15), disposable scalpel
• Standard cell culture incubator (37°C, 5% CO2, ≥95% humidity)
• Table top centrifuge with rotor capacity for 15 ml tubes
• Hemocytometer for cell counting
• Filter 0.22 μm (Millipore, cat. no. SE1M179M6)
• Polystyrene cell culture dishes: flasks with 0.2 μm vented plug seal, petri dishes, multiwell plates (BD Falcon)
• Pipettes, tubes, tips (BD Falcon)
• Cryogenic vial 1.2 ml (Corning, cat. no. 430487)
• Cryo 1°C freezing container (Nalgene, cat. no. 5100-0001)
• Coverglass slips 12 mm (Fisher, cat. no. 12-545-82)
• Pasteur pipettes (Fisher, cat. no. FB50623)
• Strainer 40 μm (BD Falcon, cat. no. 352340)
Set-Up
Material
Maintain strict sterile tissue culture practice!
BSA 0.1% wt/vol Dissolve BSA to a final concentration of 0.1% wt/vol in PBS and sterilize using a 0.22-μm filter.
Epidermal growth factor (EGF stock; 100 μg/ml) Dissolve EGF to a final concentration of 100 μg/ml in sterile PBS with 0.1% wt/vol BSA.
Fibroblast growth factor 2 (FGF2 stock; 100 μg/ml) Dissolve FGF2 to a final concentration of 100 μg/ml in sterile PBS with 0.1% wt/vol BSA. Store it in aliquots and avoid repeated freeze thaw cycles.
IMPORTANT! Stocks for BDNF, EGF, and FGF2 are stored in aliquots at −20°C for up to 6 months. After thawing, aliquots are kept at 4°C and used for maximal 2 weeks.
Basal growth medium Combine Neurobasal A (97 ml), B27 Supplement (2 ml), and Glutamax-1 (1 ml). This mixture is stable for 2 weeks at 4°C. We found that B27 supplemented with Vitamin A leads to higher cell viability than B27 without Vitamin A. Hence we recommend the use of B27 supplement including Vitamin A in standard media formulations. We have empirically determined that antibiotic-free medium is better at giving consistent results in our assays. To prevent the effect of penicillin and streptomycin on neural precursor cells beyond their traditional role as bactericidal agents we avoid using antibiotics in our culture medium. However using antibiotic-free media increases the risk of contaminations. It is up to the experimenter to calculate this risk.
Proliferation medium Add FGF2 (final concentration of 20 ng/ml) and EGF (final concentration of 20 ng/ml) to the basal growth medium just before use. Repeated freezing and thawing of the medium or growth factors can lead to a reduced effectiveness of the components. This severely effects the efficient derivation of differentiated cells from neural precursor cells. Cell lines can also be maintained with 10 ng/ml FGF2 and 10 ng/ml EGF in order to reduce consumption of the expensive growth factors. The activity of recombinant FGF2 can vary from lot to lot and a titration of FGF2 for peak mitogenesis on previously established cultures can be useful for determining the minimum dose required to maintain cells in an undifferentiated and rapidly dividing state.
IMPORTANT! Prolonged storage of medium with EGF and FGF2 leads to loss of mitogenic activity of growth factors within the medium. We recommend adding growth factors to the medium immediately before use.
Papain, Dispase and DNase mix (PDD) Prepare an enzymatic mixture of Papain (2.5 U/ml), Dispase (1 U/ml), and DNase (250 U/ml) in 100 ml Neurobasal A. Incubate the dilution for 30 min on a shaker at room temperature to get a clear solution. Filter sterilize by passing through a 0.22-μm filter. Freeze as single use aliquots.
Percoll buffer solution (Percoll-PBS) Dilute 9 ml of Percoll stock solution with 1 ml of 10× PBS. This constitutes the 100% Percoll buffer solution.
Glucose 2M Dissolve 36 g of d-Glucose in 100 ml ddH2O. Filter sterilize the solution with a 0.22-μm filter. This solution can be stored at 4°C for 2 months.
Phosphate buffered saline with Glucose (PBS-G) Add 1.5 ml of 2M Glucose to 100 ml of PBS to get a final concentration of 30 mM Glucose. Warm this solution to 37°C before use. This solution can be preserved at 4°C for 2 months.
Dimethyl sulfoxide (DMSO) 10% vol/vol Add 1 ml of DMSO to 9 ml of cold proliferation medium. Prepare fresh each time before use. Mix DMSO well with the medium. DMSO is heavier and thus settles down if not mixed appropriately.
IMPORTANT! DMSO is harmful to health and can easily penetrate the skin. Therefore avoid any contact with this substance and wear the appropriate glove type.
Poly-D-Lysine (PDL) stock Prepare a 1000× stock by reconstituting Poly-d-Lysine at 10 mg/ml in ddH2O. Prepare 500 μl aliquots in Eppendorf tubes and store at −20°C for max. 6–8 months. Tubes once thawed are maintained in 4°C and should not be refrozen.
H2SO4 2M Add 5.6 ml of concentrated H2SO4 to 44.4 ml of ddH2O.
Equipment
Disinfect and clean the coverglass slips by incubating in a rotatory shaker for 2 days in 2M H2SO4 followed by multiple rinses in ddH2O. Thoroughly dry by leaving the coverglass under the laminar flow hood for 1 day.
Fire-polish Pasteur pipettes by exposing the tip of the pipette to flame while rotating the pipette between hands. Round the sharp edges of the tip and prepare medium and fine tip diameters.
Poly-D-Lysine (PDL) coating of tissue culture surfaces Prepare a 10-μg/ml of PDL working solution in ddH2O. Cover the required surface with this solution. Leave under the laminar flow hood overnight at room temperature. The following day, remove PDL and wash the surface three times with ddH2O. Leave the surface to dry in the laminar flow hood. The dry coated surface can be stored at room temperature for future use and is stable for up to 6 months.
IMPORTANT! Avoid disinfection procedure of the laminar flow hood with UV-light during the coating process.
Laminin coating of tissue culture surfaces Prepare a working solution of 10 μg/ml of Laminin in Neurobasal A. Add the required amount to cover the PDL-coated surface and leave it in cell culture incubator at 37°C overnight. Use the plate next day for seeding cells. Laminin coated dishes can be stored at −20°C for up to 6 months.
IMPORTANT! Both Laminin and Neurobasal A have to be kept at 4°C when mixing and before adding to the PDL-coated surface. Laminin polymerizes at temperatures higher than 4°C, which would lead to uneven coating.
Detailed Protocol of the Procedure
A. Isolation of Neural Precursor Cells from Mouse Dentate Gyrus (3–3.5 h)
(1) Euthanize the mice with an appropriate method (e.g., overdose with anesthetics) approved by your institutional and national regulations.
(2) Disinfect head and neck of the mouse with 70% vol/vol ethanol. Decapitate the mouse with surgical scissors. Cut the skin sagittally along the midline till an arbitrary point between the eyes. Expose the skull free of overlying skin and subcutaneous tissue.
(3) Cut the skull mid-sagittally into two halves by cutting along the midline using fine scissors. This is done by incising the skull starting occipitally at the vertebral canal and then going forward toward the olfactory bulbs. The tip of the scissors should be kept close to the skull and away from the brain tissue to avoid damage. Further cross sections are made on either side from the vertebral canal laterally to the external auditory meatus. Reflect the overlying skull and expose the intact brain underneath. Carefully scoop out the brain and separate it from the rest of the calvarium. Place the brain in a 6-cm petri dish with cold Neurobasal A and remove the brains from remaining mice. Change to a clean 6 cm petri dish with fresh cold medium if the dish becomes bloody.
(4) Maintaining sterile working conditions, separate the cerebrum from the cerebellum, remove the meninges from the brain tissue and continue with dentate gyrus dissection:
(i) First option: dentate gyrus dissection from vibratome slices (this method has been used in the original publication Babu et al., 2007):
Section the brains coronally with a vibratome into 300 μm thick slices and collect those containing hippocampal tissue in cold Neurobasal A. Continue with dentate gyrus dissection using fine forceps under a dissecting microscope. Place an incision between the dentate gyrus and the ventricular wall of the lateral ventricle to avoid intermixture with subventricular zone neural precursor cells. As a next step transect along the hippocampal fissure and between the dentate gyrus and the CA3 region to free the dentate gyrus from the hippocampus (illustrated in Babu et al., 2007). Repeat with the remaining slices.
(ii) Second option: dentate gyrus dissection from entire hippocampus (this method is advantageous in our eyes but represents a change from the original publication Babu et al., 2007):
Cut the brain along the longitudinal fissure to divide both hemispheres. Continue the next steps under a dissection microscope. Remove the diencephalon, place the medial side of the hemisphere up, and resect the exposed hippocampus away from the neocortex. With the hippocampus isolated from the rest of the brain, separate the dentate gyrus from the hippocampus. Blood vessels running along the hippocampal fissure can be identified in the majority of animals. Place a longitudinal incision along the hippocampal fissure using these blood vessels as an anatomical landmark and mobilize the dentate gyrus. Free the knee-side of the dentate gyrus with an incision at the septal side of the hippocampal axis and liberate the dentate gyrus from CA1 and the lateral ventricle. Position the last cut almost at the temporal end of the hippocampal fissure and separate the hilar opening from CA3.
Watch the micro-dissection process from entire hippocampus in the movie provided as Supplementary File!
IMPORTANT! The dissection step carries the greatest risk of contamination and is thus ideally done with the microscope placed under a laminar flow hood. With adequate sterile work environment, however, these steps can also be performed outside the laminar flow hood. Dentate gyrus dissection from entire hippocampus demands greater technical skills compared to the technique using vibratome slices. However when performed accurately this technique is less time consuming and thus warrants a higher cell yield.
(5) Place the tissue in a 15-ml tube with cold Neurobasal A while dissecting the other animals. Avoid contact of the dissected tissue with blood-containing medium to prevent contamination with hematopoetic cells.
(6) With the pooled tissue in Neurobasal A accomplish the next steps in a sterile laminar flow hood. Chop the obtained tissue with a scalpel into small fragments.
(7) Incubate the dissected tissue in pre-warmed PDD enzyme mix for 20 min at 37°C. Add 1 ml of PDD per animal. The proteolytic enzymes digest the extracellular matrix and the intercellular adhesion proteins during the incubation step. The tissue still appears in one piece.
IMPORTANT! Do not use serum-containing medium to inhibit PDD as the proliferative ability of the recovered neural precursor cells significantly decreases in the presence of serum.
(8) Triturate carefully to obtain a single-cell suspension. Triturate first for 10 times with medium sized tip and then 10 times with fine sized tip fire-polished Pasteur pipettes.
IMPORTANT! Avoid frothing and bubbling. Entrapment of cells in air bubbles impairs their viability. Triturating with fire-polished Pasteur pipettes gently breaks the tissue and avoids sticking of cells to the tip.
(9) Plate the heterogeneous cell population from five to six mice in 1 ml proliferation medium into a PDL/Laminin coated 24-well plate.
(10) Add 3.9 ml of Neurobasal A equilibrated at room temperature to the recovered cell pellet.
(11) Add 1.1 ml of the Percoll buffer solution (Percoll-PBS) to the cell suspension for a final concentration of 22% vol/vol Percoll.
IMPORTANT! Make sure that the Percoll is well mixed with the cells and evenly distributed in the tube.
(12) Centrifuge for 15 min in a swinging bucket rotor at 450 × g at room temperature.
(13) After centrifugation a cell pellet at the bottom and a milky white layer on top should be visible. The supernatant contains buoyant non-dividing cells (astrocytes, neurons etc.) as well as myelin debris. Carefully discard the supernatant without disturbing the pellet containing the enriched neural precursor cell fraction.
IMPORTANT! Gently remove the supernatant, as the pellet may not be firmly attached to the bottom of the tube. If using vacuum suction, use at low intensity.
(14) Wash the cells three times with Neurobasal A. The medium allows for a better removal of the Percoll.
IMPORTANT! Thorough washing is important to completely remove Percoll. Residual Percoll results in poor attachment and survival of neural precursor cells after plating.
(15) Plate the heterogeneous cell population from five to six mice in 1 ml proliferation medium into a PDL/Laminin coated 24-well plate.
IMPORTANT! Do not let the Laminin coated surface dry between removing the Laminin containing solution and plating the cells. Drying alters the Laminin coating and hinders adhesion of the freshly plated precursor cells to vital extracellular matrix attachment proteins.
(16) Forty-eight hours after the initial isolation of the precursor cells, collect the medium with all floating cells that have not firmly attached to the surface yet.
IMPORTANT! Cover the PDL/Laminin coated surface with proliferation medium to avoid drying.
(17) Centrifuge the cell suspension at 200 × g for 3 min at room temperature in a swinging bucket rotor. Discard the supernatant and add fresh proliferation medium. Plate the cells back into the same culture dish. The final volume is about 200–250 μl/cm2. Add FGF2 and EGF to a final concentration of 20 ng/ml each.
(18) On day 4 after isolation and plating, remove 75% of the medium and replace it with fresh proliferation medium. By now the precursor cells are firmly attached to the surface. To the total volume of the medium (100%) add fresh FGF2 and EGF to attain a final concentration of 20 ng/ml. Change 75% of medium every 2–3 days with fresh medium.
IMPORTANT! Monitor the color of the medium daily. Feed cells as frequently as required to avoid low pH.
(19) About 4–5 days after isolation and plating, the precursor cells will start to populate the tissue culture surface. At about 9–12 days the culture will produce colonies of adherent cells separated by areas devoid of cells. Secondary cultures that are derived from the first passage of the initially isolated cells will show uniform growth without time lag.
IMPORTANT! Do not allow the culture to grow to more than 80% confluence. A fully confluent culture will loose the precursor cell characteristics and undergo crisis with ensuing cell death.
B. Passaging (30 Min)
(1) Discard the medium from the culture dish or flask and rinse once with warm PBS-G.
IMPORTANT! Do not let the cells incubate in PBS-G. Prolonged incubation can lead to cell detachment and thus loss of cells.
(2) For cell detachment we recommend two options:
(i) First option (Accutase):
Flood the surface of adherent neural precursor cells with Accutase. Return the culture back to the 37°C incubator and allow the cells to detach for 3 min. Accutase is a sterile ready-to-use solution especially developed for gentle cell dissociation. Three minutes of incubation is sufficient to detach the cells from the surface.
Immediately stop the digestive activity by diluting Accutase with 10 ml basal medium. Because Accutase digests itself, no additional enzyme inhibitors are required.
(ii) Second option (Trypsin):
Dilute 1 ml of 10× Trypsin EDTA (0.5% vol/vol) with 9 ml Neurobasal A to the final concentration of 0.05% vol/vol Trypsin and evenly cover cells with the solution. Return culture back to 37°C incubator and allow the cells to detach for 1 min.
Add the double amount of Ovomucoid Trypsin inhibitor stock solution directly to the cells to neutralize Trypsin. Gently swirl the culture dish to evenly mix the Trypsin inhibitor with the cell suspension. Avoid triturating the cells at this point or detaching them from the surface with mechanical force.
IMPORTANT! Do not prolong this incubation step beyond 1 min. Trypsin will digest membrane proteins leading to a poor recovery of the cells.
Results at this step are highly dependent on personal experience, skills, and preference. It is recommended that the reader experiments with different protocols to find the optimal conditions.
(3) Collect cells into a 15-ml conical tube and fill up to 10 ml with Neurobasal A. Centrifuge at 200 × g for 2 min at room temperature to pellet the dissociated cells.
IMPORTANT! It is essential to free the cells from Accutase and Trypsin. Residual amounts of these substances can interfere with attachment and maintenance of the precursor cells and their subsequent efficient differentiation into neurons and glia cells.
(4) After the final centrifugation step discard the supernatant and add fresh proliferation medium with 20 ng/ml of EGF and FGF2 to the pellet. Count the number of viable cells using a hemocytometer and Trypan blue.
(5) Plate 104 cells/cm2 into culture dishes coated with PDL and Laminin for routine growth and maintenance of neural precursor cells. Disperse the cells evenly to prevent cell clumping and uneven intercellular interactions that may influence growth, proliferation, and preservation of their multipotential characteristics.
C. Cryopreservation of Neural Precursor Cells (30 Min)
(1) Collect the cells by using Accutase or Trypsin as described in B steps 1–3.
(2) Resuspend the pellet carefully in cold 10% vol/vol DMSO freezing solution with gentle agitation. Add enough DMSO freezing solution to obtain a final cell suspension of 106 cells ml.
(3) Immediately aliquot 500 μl of this cell suspension into 1.2 ml cryogenic vials. Place the vials in the Cryo 1°C freezing container and place it in −80°C for 2 days.
(4) Remove the vials from the −80°C freezer and place them into a liquid nitrogen tank for long-term storage.
D. Thawing Frozen Neural Precursor Cells (20 Min)
(1) For thawing frozen neural precursor cells transfer the vial from liquid nitrogen tank immediately into a 37°C water bath. After 1 min remove the vial from the water bath and dilute the content with 9 ml warm basal growth medium in a 15-ml centrifugation tube. Mix well to evenly resuspend the cells.
IMPORTANT! It is important to immediately add the thawn cells to warm medium (37°C) for efficient recovery. In general, mammalian cells are cryo-preserved by slow freezing while recovery of the cells is done by flash thawing at 37°C.
(2) Centrifuge the cell suspension at 200 × g for 2 min at room temperature. A pellet of the recovered cells should be visible on the bottom of the tube.
(3) Discard the supernatant and add fresh proliferation medium. Plate the cells on a PDL/Laminin coated culture dish at the appropriate density.
(4) Change medium the next day. Approximately 95% of the cells recover, when the protocol is rigorously followed. The cells can be used for any subsequent experimental assay as desired.
E. Differentiation of Adherent Monolayer Neural Precursor Cells (6–10 Days)
(1) Select flasks or dishes with neural precursor cells that are about 80% confluent. Detach cells with Accutase or Trypsin as indicated in B steps 1–4 and count the cells using a hemocytometer and Trypan blue.
(2) Plate 2 × 104 cells/cm2 in proliferation medium onto PDL/Laminin coated 12 mm glass coverslips placed in a 24 multiwell plate. Place the cells in the humidified 37°C cell culture incubator.
IMPORTANT! Make sure that cell density is constant when comparing differentiation experiments. The fraction of neurons decreases with higher cell density suggesting that cell–cell interaction might influence fate programming of the precursor cells.
(3) After 48 h (differentiation day 0) discard the medium leaving the adhered neural precursor cells on the glass coverslips. Do not let dry. Add basal growth medium without growth factors (first option) or 5 ng/ml FGF2 (second option) to the culture and place the cells back into the incubator. At this stage, the cells can be fixed for immunocytochemistry and detection of neural precursor cell markers (e.g., Nestin, Sox2).
(4) Two days later (differentiation day 2) discard half of the medium (first option) or complete medium (second option) from the well and add basal growth medium without growth factors to the cells. Place cells back into the incubator.
(5) After 96 h (differentiation day 4) discard half of the medium and replace with equal amount of basal growth medium without growth factors.
(6) After 6 days from the start of the differentiation (8 days after plating) the cells can be fixed to assess differentiated progenies from the neural precursor cells. Neurons are visualized by immunocytochemistry for microtubule associated protein (Map2ab), astrocytes for glial fibrillary acidic protein (GFAP) and oligodendrocytes for O4.
IMPORTANT! Do not use β3-tubulin as a sole indicator of mature neuronal phenotypes since it can be expressed at the precursor cell stage. Complex neurite arborization and cell morphology are also useful indicators of neuronal differentiation.
(7) Further differentiation can be maintained by replacing 50% of the medium with basal growth medium every second day.
IMPORTANT! The here demonstrated “naïve” differentiation protocol was developed to manipulate as little as possible the intrinsic fate properties of neural precursor cells. Induction of the differentiation process of neural precursor cells in vitro by growth factor withdrawal is generally a stressful situation and accompanied by cell loss. We have empirically determined that the extent of cell death can be reduced by gradual reduction of FGF2. However this strategy needs further validation. The fraction of neuronal cells from the differentiating precursor cell pool can be further increased using BDNF (100 ng/ml) and retinoic acid (0.5 μM). Note that no side-by-side comparisons of the different protocols have been published, thus the exact impact of the different procedures on differentiation remains largely undetermined.
F. Immunocytochemistry (2 Days)
(1) Remove the medium and fix the cells with 4% paraformaldehyde. Incubate for 20 min at room temperature.
(2) Wash 3× with PBS for 5 min each.
(3) Block non-specific sites by incubating the cells in PBS with 3% vol/vol donkey serum and 0.1% vol/vol Triton-X100 for 30 min at room temperature.
(4) Incubate cells in PBS with 3% vol/vol donkey serum and the appropriate concentration of primary antibody overnight with gentle shaking at 4°C.
(5) The following day remove the antibody mix and wash three times with PBS for 5 min each to remove excess antibody.
(6) After the final wash add donkey secondary antibodies conjugated to fluorophores diluted in PBS with 3% vol/vol donkey serum in appropriate dilution.
(7) Incubate for 1 h at room temperature with gentle shaking.
(8) Remove the secondary antibody and wash twice with PBS for 5 min.
(9) Incubate cells with Hoechst 33258 diluted in PBS (1:3000) for 10 min.
(10) Remove the dye and wash twice with PBS for 10 min.
(11) Mount the cover glass and visualize under the microscope.
Timing
(A) Dissection and plating of adult dentate gyrus neural precursor cells: 3–3.5 h/12 days
Steps 1–5 Dentate gyrus dissection: 1 h
Steps 6–15 Isolation and plating of neural precursor cells: 2–2.5 h
Steps 16–19 Expansion of neural precursor cell culture: ≈12 days
(B) Passaging and replating of neural precursor cells:
Step 1–5: 30 min
(C) Cryopreservation of cells:
Steps 1–4: 30 min
(D) Thawing of frozen cells:
Steps 1–4: 20 min
(E) Differentiation:
Steps 1–7: 6–10 days
(F) Immunocytochemistry:
Steps 1–11: 2 days
Expected Results
When appropriately cultured, a 75-cm2 flask that is ∼ 80% confluent will contain about 7–8 × 106 cells. The typical appearance of adherent neural precursor cells under ideal culture conditions is shown in Figures 2A–C. Note that the neural precursor cells are phase bright with two to three processes. Cells in active mitosis are rounded and brighter than cells that are not in mitosis. Precursor cells of the murine dentate gyrus have a cell cycle length of about 20–21 h in culture. If maintained as suggested, the neural precursor cells can be passaged for extended periods (≈25 passages) without loss of multipotency and other precursor cell characteristics. However, extended passaging can be accompanied by increased chromosome aberration and reduced differentiation capacity.
Neurons generated from cultured adult hippocampal neural precursor cells using the present protocols typically display a resting membrane potential of −59 ± 4 mV and generate tetrodotoxin sensitive action potentials consistent with the expression of neuronal sodium channels (Babu et al., 2007).
An example of a neural precursor cell culture differentiated for 7 days is shown in Figure 2F. Immunocytochemically, neurons are detected by Map2ab, astrocytes by GFAP, and oligodendrocytes by O4 expression (Figure 4). O4-positive oligodendrocytes generated from adult neural precursor cells appear after 10 days of differentiation. Table 1 depicts the estimated fraction of cells that are derived upon routine differentiation method described in this protocol.
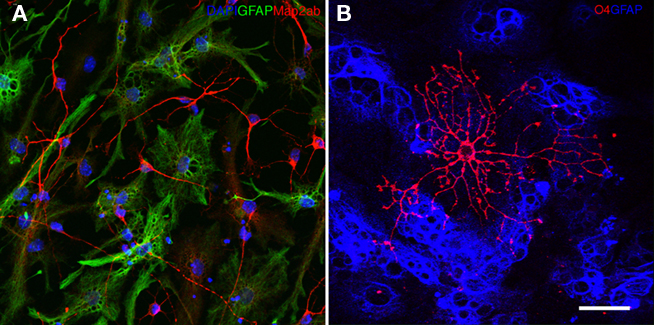
Figure 4. (A) Neural precursor cells upon 6 days of differentiation generate Map2ab (red) positive neurons and GFAP positive (green) glial cells. The nuclei are stained with DAPI (blue). (B) Ten days of differentiation generates O4 (red) positive oligodendrocytes. GFAP positive glial cells are depicted in blue.

Table 1. Cultured adult hippocampal neural precursor cells differentiate (10 days of growth factor withdrawal) into the three principal cell types of the nervous system demonstrating their multipotent properties.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/neurogenesis/10.3389/fnins.2011.00089/abstract
Movie S1. Micro-dissection and isolation of mouse dentate gyrus: the mouse brain is separated from the calvarium. The hippocampus that lies beneath the neocortex is dissected free from the rest of the brain. In the next step the dentate gyrus is isolated from the cornu ammonis by drawing a wedge through the hippocampal fissure.
References
Babu, H., Cheung, G., Kettenmann, H., Palmer, T. D., and Kempermann, G. (2007). Enriched monolayer precursor cell cultures from micro-dissected adult mouse dentate gyrus yield functional granule cell-like neurons. PLoS ONE 2, e388. doi:10.1371/journal.pone.0000388
Babu, H., Ramirez-Rodriguez, G., Fabel, K., Bischofberger, J., and Kempermann, G. (2009). Synaptic network activity induces neuronal differentiation of adult hippocampal precursor cells through BDNF signaling. Front. Neurosci. 3:49. doi: 10.3389/neuro.22.001.2009
Ben Abdallah, N., Slomianka, L., Vyssotski, A. L., and Lipp, H. P. (2010). Early age-related changes in adult hippocampal neurogenesis in C57 mice. Neurobiol. Aging 31, 151–161.
Bez, A., Corsini, E., Curti, D., Biggiogera, M., Colombo, A., Nicosia, R. F., Pagano, S. F., and Parati, E. A. (2003). Neurosphere and neurosphere-forming cells: morphological and ultrastructural characterization. Brain Res. 993, 18–29.
Boku, S., Nakagawa, S., Masuda, T., Nishikawa, H., Kato, A., Kitaichi, Y., Inoue, T., and Koyama, T. (2009). Glucocorticoids and lithium reciprocally regulate the proliferation of adult dentate gyrus-derived neural precursor cells through GSK-3beta and beta-catenin/TCF pathway. Neuropsychopharmacology 34, 805–815.
Cameron, H. A., Woolley, C. S., McEwen, B. S., and Gould, E. (1993). Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. Neuroscience 56, 337–344.
Conti, L., and Cattaneo, E. (2005). Controlling neural stem cell division within the adult subventricular zone: an APPealing job. Trends Neurosci. 28, 57–59.
Eriksson, P. S., Perfilieva, E., Bjork-Eriksson, T., Alborn, A. M., Nordborg, C., Peterson, D. A., and Gage, F. H. (1998). Neurogenesis in the adult human hippocampus. Nat. Med. 4, 1313–1317.
Foroni, C., Galli, R., Cipelletti, B., Caumo, A., Alberti, S., Fiocco, R., and Vescovi, A. (2007). Resilience to transformation and inherent genetic and functional stability of adult neural stem cells ex vivo. Cancer Res. 67, 3725–3733.
Hagihara, H., Toyama, K., Yamasaki, N., and Miyakawa, T. (2009). Dissection of hippocampal dentate gyrus from adult mouse. J. Vis. Exp. 3000, 1–6.
Imbeault, S., Gauvin, L. G., Toeg, H. D., Pettit, A., Sorbara, C. D., Migahed, L., DesRoches, R., Menzies, A. S., Nishii, K., Paul, D. L., Simon, A. M., and Bennett, S. A. (2009). The extracellular matrix controls gap junction protein expression and function in postnatal hippocampal neural progenitor cells. BMC Neurosci. 10, 13. doi: 10.1186/1471-2202-10-13
Jensen, J. B., and Parmar, M. (2006). Strengths and limitations of the neurosphere culture system. Mol. Neurobiol. 34, 153–161.
Jessberger, S., Clemenson, G. D. Jr., and Gage, F. H. (2007). Spontaneous fusion and nonclonal non-clonal growth of adult neural stem cells. Stem Cells 25, 871–874.
Knoth, R., Singec, I., Ditter, M., Pantazis, G., Capetian, P., Meyer, R. P., Horvat, V., Volk, B., and Kempermann, G. (2010). Murine features of neurogenesis in the human hippocampus across the lifespan from 0 to 100 Years. PLoS ONE 5, e8809. doi: 10.1371/journal.pone.0008809
Kornack, D. R., and Rakic, P. (1999). Continuation of neurogenesis in the hippocampus of the macaque monkey. Proc. Natl. Acad. Sci. U.S.A. 96, 5768–5773.
Kuhn, H. G., Dickinson-Anson, H., and Gage, F. H. (1996). Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J. Neurosci. 16, 2027–2033.
Liu, C., Teng, Z. Q., Santistevan, N. J., Szulwach, K. E., Guo, W., Jin, P., and Zhao, X. (2010). Epigenetic regulation of miR-184 by MBD1 governs neural stem cell proliferation and differentiation. Cell Stem Cell 6, 433–444.
Luo, C. X., Jin, X., Cao, C. C., Zhu, M. M., Wang, B., Chang, L., Zhou, Q. G., Wu, H. Y., and Zhu, D. Y. (2010a). Bidirectional regulation of neurogenesis by neuronal nitric oxide synthase derived from neurons and neural stem cells. Stem Cells 28, 2041–2052.
Luo, Y., Shan, G., Guo, W., Smrt, R. D., Johnson, E. B., Li, X., Pfeiffer, R. L., Szulwach, K. E., Duan, R., Barkho, B. Z., Li, W., Liu, C., Jin, P., and Zhao, X. (2010b). Fragile x mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet. 6, e1000898. doi: 10.1371/journal.pgen.1000898
Marshall, G. P. II, Reynolds, B. A., and Laywell, E. D. (2007). Using the neurosphere assay to quantify neural stem cells in vivo. Curr. Pharm. Biotechnol. 8, 141–145.
Palmer, T. D., Markakis, E. A., Willhoite, A. R., Safar, F., and Gage, F. H. (1999). Fibroblast growth factor-2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. J. Neurosci. 19, 8487–8497.
Palmer, T. D., Ray, J., and Gage, F. H. (1995). FGF-2-responsive neuronal progenitors reside in proliferative and quiescent regions of the adult rodent brain. Mol. Cell. Neurosci. 6, 474–486.
Palmer, T. D., Takahashi, J., and Gage, F. H. (1997). The adult rat hippocampus contains premordial neural stem cells. Mol. Cell. Neurosci. 8, 389–404.
Ramirez-Rodriguez, G., Klempin, F., Babu, H., Benitez-King, G., and Kempermann, G. (2009). Melatonin modulates cell survival of new neurons in the hippocampus of adult mice. Neuropsychopharmacology 34, 2180–2191.
Ray, J., and Gage, F. H. (2006). Differential properties of adult rat and mouse brain-derived neural stem/progenitor cells. Mol. Cell. Neurosci. 31, 560–573.
Reynolds, B. A., and Rietze, R. L. (2005). Neural stem cells and neurospheres – re-evaluating the relationship. Nat. Methods 2, 333–336.
Reynolds, B. A., and Weiss, S. (1992). Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255, 1707–1710.
Rietze, R. L., and Reynolds, B. A. (2006). Neural stem cell isolation and characterization. Meth. Enzymol. 419, 3–23.
Singec, I., Knoth, R., Meyer, R. P., Maciaczyk, J., Volk, B., Nikkhah, G., Frotscher, M., and Snyder, E. Y. (2006). Defining the actual sensitivity and specificity of the neurosphere assay in stem cell biology. Nat. Methods 3, 801–806.
Keywords: hippocampus, progenitor cell, precursor cell, adult neurogenesis, in vitro
Citation: Babu H, Claasen J-H, Kannan S, Rünker AE, Palmer T and Kempermann G (2011) A protocol for isolation and enriched monolayer cultivation of neural precursor cells from mouse dentate gyrus. Front. Neurosci. 5:89. doi: 10.3389/fnins.2011.00089
Received: 11 March 2011; Accepted: 28 June 2011;
Published online: 14 July 2011.
Edited by:
Silvia De Marchis, University of Turin, ItalyReviewed by:
Silvia De Marchis, University of Turin, ItalyD. Chichung Lie, Helmholtz Zentrum Munchen German Research Center for Environmental Health, Germany
Copyright: © 2011 Babu, Claasen, Kannan, Rünker, Palmer and Kempermann. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Gerd Kempermann, DFG Research Center for Regenerative Therapies Dresden, Center for Regenerative Therapies Dresden, Tatzberg 47–49, 01307 Dresden, Germany. e-mail:Z2VyZC5rZW1wZXJtYW5uQGNydC1kcmVzZGVuLmRl