 Jean M. Hébert1,2*
Jean M. Hébert1,2*- 1 Department of Neuroscience, Albert Einstein College of Medicine, Bronx, NY, USA
- 2 Department of Genetics, Albert Einstein College of Medicine, Bronx, NY, USA
From neurulation to postnatal processes, the requirements for FGF signaling in many aspects of neural precursor cell biology have been well documented. However, identifying a requirement for FGFs in a particular neurogenic process provides only an initial and superficial understanding of what FGF signaling is doing. How FGFs specify cell types in one instance, yet promote cell survival, proliferation, migration, or differentiation in other instances remains largely unknown and is key to understanding how they function. This review describes what we have learned primarily from in vivo vertebrate studies about the roles of FGF signaling in neurulation, anterior–posterior patterning of the neural plate, brain patterning from local signaling centers, and finally neocortex development as an example of continued roles for FGFs within the same brain area. The potential explanations for the diverse functions of FGFs through differential interactions with cell intrinsic and extrinsic factors is then discussed with an emphasis on how little we know about the modulation of FGF signaling in vivo. A clearer picture of the mechanisms involved is nevertheless essential to understand the behavior of neural precursor cells and to potentially guide their fates for therapeutic purposes.
Introduction
FGFs were first identified as factors derived from bovine pituitary and brain that were mitogenic for fibroblasts (Armelin, 1973; Gospodarowicz, 1974, 1975). The first two FGFs that were purified, cloned, and sequenced were acidic and basic FGF (aFGF, bFGF, now FGF1, and FGF2; Abraham et al., 1986a,b). FGFs can be found throughout metazoan species and the number of genes encoding them significantly expanded in early vertebrate evolution (Itoh and Ornitz, 2004, 2011; Popovici et al., 2005). In mammals there are 22 genes that encode FGFs. Since their identification as mitogenic factors for fibroblasts, the list of biological processes in which they are known to play crucial roles has grown to a surprising length – and continues to grow (Beenken and Mohammadi, 2009; Dorey and Amaya, 2010; Itoh and Ornitz, 2011). Nowhere is this more obvious than in the regulation of neural development where FGFs are found to be required for an increasing number of processes.
FGF genes can be divided into subfamilies based on their sequence and the mode of action of their respective peptides (Itoh and Ornitz, 2011). FGFs, except those from two subfamilies, are thought to act in a paracrine fashion. The hormone-like FGFs (FGF15/19, 21, 23) instead act in an endocrine manner (Beenken and Mohammadi, 2009), while the intracellular FGFs (FGF11–14) are thought to function in an FGF receptor-independent manner (Goldfarb, 2005). Although some members of all the subfamilies are likely to influence CNS development and/or function (see below; Mason, 2007; Iwata and Hevner, 2009; Umemori, 2009; Vaccarino et al., 2009; Guillemot and Zimmer, 2011), many of their roles may still remain unknown. This is in part because although almost all FGF genes have been knocked out individually (Itoh and Ornitz, 2011), those with overlapping expression patterns may functionally compensate for each other and mask the other’s role at different developmental times and in different tissues.
In contrast to the large number of genes encoding ligands, only four genes, Fgfr1–4, encode FGF receptors. This is a more manageable number of genes to work with in order to overcome compensation when disrupting FGF signaling. The extracellular portion of FGF receptors is comprised of three immunoglobulin-like domains and an acid box while the intracellular portion contains a split tyrosine kinase domain. A fifth receptor gene, Fgfrl1, also exists, but does not encode an intracellular domain and is not yet known to have a function in the CNS. Alternative splicing of Fgfr1–4 transcripts leads to receptors with differing extracellular domains, but most alternatively spliced forms contain the intracellular kinase domain (Johnson and Williams, 1993). Upon binding FGF ligand, receptors dimerize, autophosphorylate, and phosphorylate one or more of several immediate intracellular mediators described in sections below.
FGFs play essential roles in the induction and anterior–posterior (A–P) patterning of the neural plate, in the local patterning of several developing brain regions, in several steps in neurogenesis, and in establishing functional neural networks. Moreover, at the level of the cell, FGFs within the developing CNS are in some cases required for cell survival, fate specification, proliferation, migration, differentiation, or axon pathfinding. What can account for such diverse functions? Here, rather than providing a comprehensive review of FGFs in neurodevelopment, select examples of the different functions of FGFs in the developing neural plate and neocortex are provided to illustrate the broad spectrum of FGF functions. In addition, the differences in FGF ligands, receptors, intracellular and extracellular modulators, and signal transduction pathways are discussed as potential explanations for the wide range of FGF effects (Figure 1).
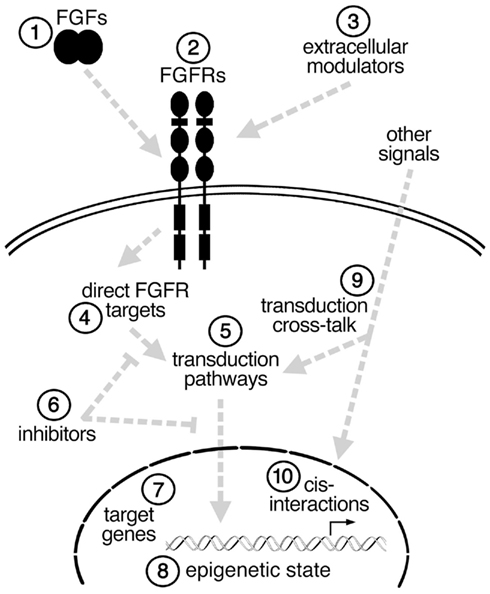
Figure 1. Simplified schema illustrating some of the possible steps (1–10) at which an FGF signal could be modulated in different cell types to affect cell fate. See text for details.
FGFs are Required for Early Patterning Processes in CNS Development
FGFs play roles in the earliest steps of CNS development (Table 1). To start with, FGF signaling participates in neural induction, although there is still some controversy as to the extent of its involvement in each species. In zebrafish, chick, and ascidian embryos FGF signaling is necessary and sufficient to initiate the acquisition of a neural fate (Rodriguez-Gallardo et al., 1997; Inazawa et al., 1998; Streit et al., 2000; Wilson et al., 2000; Hudson and Lemaire, 2001; Bertrand et al., 2003; Sheng et al., 2003; Kudoh et al., 2004), with perhaps a greater role in inducing posterior neural fates (Alvarez et al., 1998; Storey et al., 1998; Rentzsch et al., 2004; Londin et al., 2005; Takemoto et al., 2006). In Xenopus, even though some studies suggest no role for FGFs in neural induction (Kroll and Amaya, 1996; Wills et al., 2010), other studies indicate that it plays either a role in establishing posterior fates or in inducing anterior versus posterior fates in a time-dependent manner (Kengaku and Okamoto, 1995; Lamb and Harland, 1995; Launay et al., 1996; Sasai et al., 1996; Pera et al., 2003; Delaune et al., 2005; Marchal et al., 2009). A role for FGFs in neural induction in mammals remains to be clearly demonstrated.
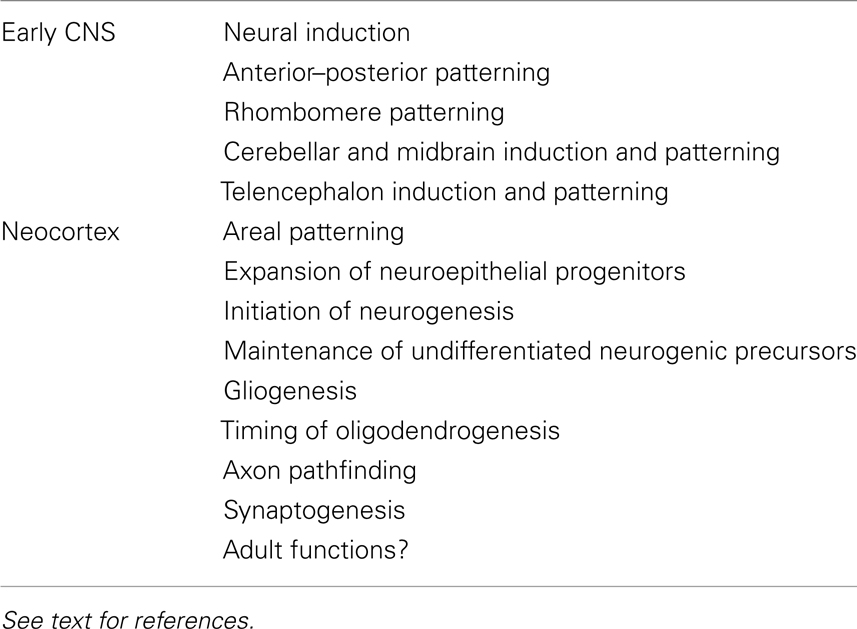
Table 1. Examples of functions for FGF signaling in the nascent central nervous system and developing neocortex.
Perhaps as an extension of its role in neural induction, FGF signaling is also essential for patterning the neural plate along its A–P axis as part of the overall A–P patterning of the embryo (Amaya et al., 1991; Isaacs et al., 1992; Griffin et al., 1995; Draper et al., 2003). Studies using chick, zebrafish, and Xenopus embryos show that graded FGF signaling, with high posterior and low anterior levels, patterns the neural plate at least in part by regulating the expression of Hox genes, which in turn determine the positional identities of neurons along the developing spinal cord (Isaacs et al., 1994; Cox and Hemmati-Brivanlou, 1995; Kengaku and Okamoto, 1995; Lamb and Harland, 1995; Pownall et al., 1998; Holowacz and Sokol, 1999; Ribisi et al., 2000; Liu et al., 2001; Bel-Vialar et al., 2002; Kudoh et al., 2002; Dasen et al., 2003). In mammals, FGFs are also likely required in A–P patterning of the neural plate given that FGFs (in particular Fgf8) are expressed in a high posterior to low anterior gradient in the neurulating embryo, ectopic FGF4 can suppress anterior development, and certain mutations in Fgfr1 cause general A–P patterning defects (Partanen et al., 1998; Davidson et al., 2000). Of course other factors, WNTs, retinoic acid, BMPs, and BMP antagonists, are likely to participate with FGFs in patterning the neural plate (Dorey and Amaya, 2010).
As development proceeds, signaling centers that express FGFs emerge in discrete areas of the developing brain. At least three signaling centers have been characterized. One is rhombomere 4 (zebrafish) or 5 and 6 (mouse or chick) in the hindbrain. In zebrafish, FGF3 and FGF8 emanating from rhombomere 4 are necessary and sufficient to promote development of adjacent rhombomeres 5 and 6 by regulating the expression of transcription factors including Krox20 (Maves et al., 2002; Walshe et al., 2002; Waskiewicz et al., 2002; Wiellette and Sive, 2003; Hernandez et al., 2004; Labalette et al., 2011). Similar FGF-dependent patterning mechanisms are likely to operate in the developing hindbrain of other vertebrates since FGFs also regulate Krox20 expression in chicks (Aragon et al., 2005). Moreover, FGF signaling also plays a role later in rhombomere development: FGF20a in zebrafish is essential during neurogenesis for maintaining precursor cells in segment centers in an undifferentiated state (Gonzalez-Quevedo et al., 2010).
A second signaling center that expresses FGFs is the isthmus at the mid–hindbrain boundary. Here FGF8, and to some extent FGF17 and 18, mediate the isthmus’ organizing activity for inducing and patterning the midbrain and cerebellum. An ectopic source of FGF8 in the caudal diencephalon is sufficient to induce the formation of a normally organized, ectopic, mirror-image midbrain whereas deletion of Fgf8 results in the loss of most or all midbrain and cerebellar structures due to cell death of the early precursors (Crossley et al., 1996; Martinez et al., 1999; Chi et al., 2003; Liu et al., 2003; Trokovic et al., 2003; Basson et al., 2008). FGF signaling is required concurrently and in a dose-dependent manner for both survival and patterning of cerebellar and midbrain precursors (Chi et al., 2003; Basson et al., 2008; Chen et al., 2009). As for rhombomere development, FGFs in the cerebellum play other roles later in development, for example as attractants for trochlear axons and in synaptogenesis (Irving et al., 2002; Umemori, 2004; Partanen, 2007; Yaguchi et al., 2009).
Finally, a third signaling center in which FGFs are required is the anterior neural ridge (ANR) at the anterior-most tip of the embryo between the neuroectoderm and underlying ectoderm. At least five FGFs, FGF3, 8, 15, 17, and 18, are expressed in the ANR or its early derivatives and three receptors FGFR1, FGFR2, and FGFR3 are expressed broadly in the anterior neuroectoderm. Similar to their function in mid–hindbrain development, FGFs emanate from the ANR to induce broad telencephalic cell types in a dose-dependent manner and to maintain the survival of telencephalic precursor cells (Shimamura and Rubenstein, 1997; Shanmugalingam et al., 2000; Shinya et al., 2001; Gunhaga et al., 2003; Walshe and Mason, 2003; Gutin et al., 2006; Storm et al., 2006; Theil et al., 2008; Paek et al., 2009). FGFs are also critical for several later steps of telencephalon development. The known requirements for FGFs in the developing neocortex, which comprises the dorsal part of the telencephalon, are described below as an example of the continued and varied functions for FGFs within one part of the brain.
Requirements for FGFs in the Developing Neocortex
In addition to inducing cell fates and maintaining cell survival in the early telencephalon, FGFs play key roles in the developing neocortex (Table 1; Iwata and Hevner, 2009). Early on, FGFs pattern and expand the neocortex. FGF8 acts as a morphogen and is necessary and sufficient to assign positional identities to cortical precursors along the anterior–lateral to posterior–medial axis in a dose-dependent manner (Fukuchi-Shimogori and Grove, 2001; Garel et al., 2003; Storm et al., 2006; Toyoda et al., 2010). FGF17, meanwhile, is required for patterning the frontal neocortex (Cholfin and Rubenstein, 2007). In addition to patterning, FGFs are required for expanding the pool of neuroepithelial precursor cells by extending the number of precursor cell divisions prior to generating neurons. For example, in the absence of FGF2, there are fewer precursor cells resulting in fewer neurons, especially in the anterior neocortex (Vaccarino et al., 1999; Raballo et al., 2000; Korada et al., 2002).
FGFs are also essential for the onset of cortical neurogenesis: FGF10 is required in rostral areas to promote the transition of neuroepithelial cells to radial glial cells, which in turn generate intermediate progenitors and neurons, marking the start of neurogenesis (Sahara and O’Leary, 2009). FGF8, meanwhile, is required to generate rostral Cajal–Retzius cells, one of the earliest born type of cortical neuron (Zimmer et al., 2010). During neurogenesis itself, if FGF signaling is disrupted by deleting one or more Fgfr genes, then the radial glial precursors fail to maintain their undifferentiated state and prematurely differentiate (Kang et al., 2009; Stevens et al., 2010). In this case, FGF18, which is expressed in neurons, is likely one of the ligands that feeds back to radial glia to maintain their precursor state (Hasegawa et al., 2004).
In addition to its roles in cortical patterning and neurogenesis, FGF signaling regulates the production of the other major classes of neural cell types, astrocytes, and oligodendrocytes. Toward the end of neurogenesis, FGFR1 and FGFR2, possibly responding to FGF9, are required for radial glia to transition from generating neurons to generating astrocytes (Smith et al., 2006; Tole et al., 2006; Seuntjens et al., 2009). And postnatally, FGFR3 is required for the timely onset of oligodendrocyte production (Oh et al., 2003). Hence FGF signaling plays continuous, yet distinct, roles throughout cortical development.
Not surprisingly, disruption of FGF signaling during cortical patterning, neurogenesis, and gliogenesis are associated with defects in axon pathfinding and behavioral anomalies in adult mice. The loss of midline glial cell types that result from disruption of Fgfr1 leads to a failure of callosal and other axons to cross the midline and connect the neocortex of both hemispheres (Shanmugalingam et al., 2000; Walshe and Mason, 2003; Smith et al., 2006; Tole et al., 2006). Connections within each hemisphere of the cortex can also be disrupted, as shown in Fgf8 hypomorphs (Huffman et al., 2004). Moreover, early ectopic sources of FGF8 can cause misrouting of thalamocortical axons (Shimogori and Grove, 2005). Imbalances in early cortical cell fate specification due to disruption of FGF signaling can also lead to behavioral anomalies such as abnormal social behaviors or hyperactivity, although the mechanism for the latter, whether due to a reduction in glutamatergic or a reduction in GABAergic neurons in the cortex, remains unclear (Shin et al., 2004; Muller-Smith et al., 2008; Scearce-Levie et al., 2008). Interestingly, FGF22 and FGF7 are required for the formation of glutamatergic and GABAergic synapses, respectively, in hippocampal CA3 neurons (Terauchi et al., 2010).
Finally, the roles for FGFs in adult neurogenesis, physiology, and homeostasis are largely unexplored. One or more of the intracellular FGFs (the FGFR-independent FGFs11–14) are likely to directly regulate the excitability of neocortical neurons as they do in the hippocampus and cerebellum (Goldfarb et al., 2007; Xiao et al., 2007; Shakkottai et al., 2009). The secreted FGFs are also likely to have functions in the adult cortex as diverse as during development, but these functions have yet to be determined in detail in vivo.
FGF–FGFR Signaling, Differing Levels Versus Different Mechanisms
The genetic disruption of individual FGF ligand and receptor genes can clearly result in distinct phenotypes in the developing CNS and embryo (e.g., above; Beenken and Mohammadi, 2009). Much of these phenotypic differences are likely due to the individual patterns and levels of expression for each gene, which does not explain how FGF signaling promotes cell survival in one case, or proliferation, differentiation, or the adoption of one of many cell identities in other cases.
Two broad, non-mutually exclusive possibilities exist to explain the varied effects of FGFs on cells: the first is that the individual ligands and/or receptors in fact transmit different signals to cells (for example, some FGF–FGFR combinations activate one transduction pathway whereas another combination induces a different pathway); and the second is that individual target cell types and their environments modulate and interpret an FGF signal differently (for example, one cell is predisposed to respond to FGFs by activating one intracellular pathway whereas another cell type responds by activating a different pathway). Although both of these possibilities will undoubtedly be found to affect FGF function in vivo, the little evidence that exists to date suggests that mechanistic differences in ligand–receptor signaling will be less substantial than the inherent modulation of the signal by the target cell type and its environment.
Although alternatively spliced forms of both ligands and receptors exist and these can affect binding specificities and levels of signaling as measured in cultured cells, binding of the FGF ligands to their receptors remains rather promiscuous (Ornitz et al., 1996; Zhang et al., 2006). Moreover, there is little evidence that distinct FGFs binding to the same receptors illicit distinct cellular responses. For example, the different effects of FGF8a and FGF8b on midbrain and cerebellar cell fates appear to simply reflect their different potencies (i.e., levels of activity; Sato and Nakamura, 2004).
Moreover, the existing evidence suggests that the receptors themselves transmit signal via the same intracellular pathways for a given cell type. For example, activation of FGFR1 promotes proliferation in some cell types, but activation of the intracellular domain of this receptor in chondrocytes leads to suppression of proliferation in a manner similar to FGFR3, the receptor that is normally expressed in these cells (Wang et al., 2001). Similarly, in zebrafish, constitutively active forms of several FGFRs each caused dorsalization, brain caudalization, and secondary axis formation, suggesting that signal transduction is similar among FGFRs for several embryonic cell types (Ota et al., 2009).
In addition, when the three Fgfr genes, Fgfr1, Fgfr2, and Fgfr3, that are expressed in the embryonic neuroepithelium are deleted in the early mid–hindbrain region, the telencephalon, or the neocortex, the cell death, patterning, and differentiation phenotypes obtained are dramatically more severe than loss of one or two receptors alone (Saarimaki-Vire et al., 2007; Kang et al., 2009; Paek et al., 2009). Hence the receptors can largely compensate for each other indicating that they likely signal through the same intracellular pathways for a given cell type.
This does not mean the receptors are equivalent and interchangeable since the efficiencies with which they activate downstream mediators may differ, leading to quantitative or qualitative differences in a cell’s response. There might even be cases in which individual FGF ligands, which have different receptor binding preferences, illicit different responses on the same cells. For example, Fgf15 and Fgf8 appear to have opposite effects on telencephalic precursors (Borello et al., 2008; Danjo et al., 2011). In cases in which phenotypic differences are associated with different ligands, it becomes of interest to decipher the underlying mechanism, whether it is the activation of different receptors, the duration or strength of receptor activation, and/or the recruitment of different intracellular signal transduction components.
Extracellular Modulators of FGF Signaling
Extracellular molecules that interact directly with FGFs or their receptors may affect both the levels of signaling and the cellular responses. Perhaps the best characterized molecule that interacts with FGFs is heparan sulfate (HS) as part of heparan sulfate proteoglycans. HS is widely believed to be a required component of FGF signaling (Dailey et al., 2005; Mason, 2007; Beenken and Mohammadi, 2009; Krejci et al., 2009; Umemori, 2009; Itoh and Ornitz, 2011). For example, disruption of the Ndst1 gene, which encodes an HS modifying enzyme, results in cerebral hyperplasia and mimics aspects of the forebrain phenotypes of FGF and SHH mutants (Grobe et al., 2005).
However, a strict requirement for heparan sulfate for FGF signaling has yet to be demonstrated in vivo. Expression of the Exostoses 1 (Ext1) gene is essential for heparan sulfate biosynthesis in mice and without it no heparan sulfate can be detected (Lin et al., 2000). Yet, the Ext1 null mutant survives later than the earliest FGF-related lethal phenotype, the Fgf4 null phenotype (Feldman et al., 1995; Lin et al., 2000). Similarly, the phenotype of a CNS specific deletion of Ext1 using a Nestin-Cre driver only recapitulates defects obtained with Fgf8 hypomorphs or partial loss of Fgfr expression (Inatani et al., 2003). These results cast doubt on a strict requirement for heparan sulfate, although they do not rule it out due to other potential explanations for the discrepancies between Ext1 associated phenotypes and the most severe FGF-associated phenotypes (such as strain differences, timing of recombination, heparan sulfate perdurance, or simultaneous loss in the Ext1 mutants of other signals that act antagonistically to FGFs and therefore partially rescue loss of FGF signaling). The protein moieties of the heparan sulfate proteoglycans involved in FGF signaling within the developing CNS are largely uncharacterized, although Glypican-1 plays an important role during neurogenesis (Jen et al., 2009). Nevertheless, it remains likely that the association of heparan sulfates with FGF ligands and receptors promotes higher levels of signaling rather than affecting the nature of the intracellular response.
FGF ligands and receptors can also interact with a variety of other extracellular molecules, including NCAM, cadherins, integrins, fibronectin, Klotho, anosmin-1, EphA4, and others (Polanska et al., 2009). These could potentially regulate levels of signaling and cellular responses to FGFs. However, the importance of these proteins to FGF signaling and neurodevelopment remains unclear.
Intracellular FGF Signal Transduction
Proteins that can interact directly with the intracellular domain of FGF receptors and that may mediate signal transduction have been identified. Potential mediators include FGF receptor substrate (FRS) 2 and 3, Phospholipase-Cγ (PLCγ), CRK, growth factor receptor bound (GRB) 14, Src homology domain-2 containing protein B (SHB), and possibly a complex comprised of GRB2, SH2-containing transforming protein C (SHC), and son of sevenless (SOS; Mohammadi et al., 1991; Wang et al., 1996; Kanai et al., 1997; Kouhara et al., 1997; Curto et al., 1998; Xu et al., 1998; Larrson et al., 1999; Ong et al., 2000; Reilly et al., 2000; Cross et al., 2002). FGF receptors can also translocate to the nucleus and interact directly with nuclear components (Wiedlocha and Sorensen, 2004; Bryant and Stow, 2005; Stachowiak et al., 2007). Importantly, the role for each protein that potentially interacts physically with the FGF receptors in mediating signaling remains unclear for any neurodevelopmental process in vivo.
The FRS proteins, in particular FRS2, are generally accepted as key mediators of FGF signaling (Wang et al., 1996; Kouhara et al., 1997; Xu et al., 1998; Ong et al., 2000; Hadari et al., 2001; Mason, 2007; Turner and Grose, 2010). FRS2 is a docking protein that appears constitutively bound to the juxtamembrane region of FGF receptors and becomes phosphorylated in the presence of FGFs. Upon phosphorylation, FRS2 is thought to recruit Grb2 and SHP2 leading to activation of the PI3K → AKT and Sos → Ras → Raf → MEK → MAPK pathways. These findings are based mainly on biochemical and cell culture data. Few studies have addressed the functional requirement for the interaction between FGF receptors and FRS proteins directly during neurodevelopment.
An Fgfr1 mutant that specifically lacks the ability to bind FRS2 and FRS3 was directly compared to an Fgfr1 null mutant in the same strain background. Whereas the Fgfr1 null mutant exhibits failures in mesoderm migration during gastrulation, somitogenesis, and neural tube closure, leading to small, truncated embryos that die shortly after gastrulation, the Fgfr1 mutant that cannot bind FRS undergoes normal gastrulation and somitogenesis, but exhibits neural tube, tail bud, and pharyngeal arch deficits (Deng et al., 1994; Yamaguchi et al., 1994; Hoch and Soriano, 2006). This suggests at the very least that FRS adaptor proteins are not the exclusive effectors of FGFR1 signal transduction in vivo. Likewise, an Fgfr2 mutant that lacks the binding site for FRS2 and FRS3, can rescue the effects of a gain-of-function cis mutation in Fgfr2 (Eswarakumar et al., 2006). However, no phenotype was reported for the FRS-binding mutation itself, yielding little insight on the normal requirement for the interaction between FGFR2 and FRS.
The phenotype of Frs2 null mice themselves does not clarify the role of this molecule in mediating FGF signaling. Frs2 null embryos lack normal extraembryonic tissue and die at ∼E8 (Gotoh et al., 2005), a phenotype that may overlap but does not recapitulate any FGF-associated phenotype. The interpretation of this result is confounded by the possibilities that there is some compensation by FRS3 and that FRS2 signaling is not specific to the FGF pathway (e.g., FRS2 can mediate neurotrophin signaling as well). Frs2 mutants that lack the binding residues for the tyrosine phosphatase SHP2 fail to maintain intermediate progenitor cells during cortical neurogenesis (Yamamoto et al., 2005). However, intermediate progenitors do not express detectable levels of Fgfr genes and mutants in which FGF signaling is abolished during cortical neurogenesis fail to maintain the radial glial stem cells rather than the intermediate progenitors, which are unaffected (Kang et al., 2009), suggesting that FRS2 may be acting downstream of neurotrophins rather than FGFs in this process. Note that no knockout or knockdown studies for Frs3 have been reported. Interestingly, the function of FRS-like proteins as direct targets of FGF receptors is not conserved in flies and worms (Wilson et al., 2004; Lo et al., 2010). Hence, in mammals there is still uncertainty as to where, when, and whether FRS is required in vivo for FGF signaling and how it might differentially affect cellular responses.
Similarly, PLCγ is postulated to be an important immediate target of FGFR phosphorylation (Mason, 2007; Turner and Grose, 2010), and regulation of this interaction could influence a cell’s response. However, in vivo evidence is still sparse. PLCγ activates the IP3 → Ca++ and DAG → PKC pathways. A direct comparison of Fgfr1 hypomorphic embryos to ones carrying an Fgfr1 point mutation that abolishes the interaction with PLCγ revealed that both exhibit homeotic vertebral transformations, but in some cases in opposite directions along the A–P axis, leading to the suggestion that signaling through PLCγ may negatively feedback on FGF function (Partanen et al., 1998). However, the overall requirement for the FGFR–PLCγ interaction in vivo remains largely obscure. In addition, the requirements for potential immediate targets of FGFRs other than PLCγ and FRS2, for example CRK, SHB, and GRB, are also not understood and need to be explored. Nevertheless, the use of different FGFR targets is likely to account, in part, for different cellular responses.
The immediate targets of activated FGF receptors will in turn activate one or more downstream effectors that include components of the Src, STAT, Shc, PI3K, and MAPK pathways (Mason, 2007; Beenken and Mohammadi, 2009; Turner and Grose, 2010; Guillemot and Zimmer, 2011). There is evidence that some of these pathways are differentially activated in different neural cell types in response to FGF signals, providing part of the explanation for how FGFs induce a variety of cellular responses. Activation of the Ras–Erk pathway has been examined most closely and has been implicated in mediating FGF signals, for example, in neural induction and patterning in Xenopus (Ribisi et al., 2000; Delaune et al., 2005), in patterning rhombomeres and promoting ventral forebrain development (Shinya et al., 2001; Hernandez et al., 2004; Aragon and Pujades, 2009), and in generating cerebellar, but not midbrain, cell fates at the mid–hindbrain boundary in the chick (Sato and Nakamura, 2004). On the other hand, conditional deletion of Erk2 during cortical neurogenesis appears to affect primarily the proliferation of intermediate progenitors (Samuels et al., 2008), a cell type in which FGF signaling appears not to play a significant role (Kang et al., 2009). The roles of the other intracellular pathways that are potentially activated by FGFs remain unclear.
In addition to mediators of FGF signaling, there are a number of important inhibitors. The functions of some of these have begun to be characterized in CNS development and include members of the Sprouty (Spry) family, inhibitors of the Ras–ERK pathway (Mason et al., 2006). Spry genes are typically induced by FGF signals and negatively feedback to restrict the amount of signaling. For example, Spry2 misexpression reduces FGF signaling and disrupts mid–hindbrain development in mice (Basson et al., 2008) and spry4 constrains FGF activity in patterning the hindbrain and expanding the telencephalon in zebrafish (Furthauer et al., 2001; Labalette et al., 2011). There are also negative feedback regulators of FGF signaling other than the Spry proteins, including Sef, MapK phosphatases (MKPs, also know as Dusps), and factors promoting receptor turnover (e.g., Furthauer et al., 2002; Tsang and Dawid, 2004; Echevarria et al., 2005; Ron et al., 2008). These are likely important in regulating neurogenic processes. However, their functions in the CNS have just begun to be explored.
In different parts of the CNS and at different stages of development, FGF signaling promotes and inhibits different sets of target genes. Target genes induced by FGFs usually include genes that encode feedback inhibitors (e.g., Spry) and members of the Ets family of transcription factors (including Pea3, Erm, Er81, and others) suggesting that these are immediate targets. The transcription complexes downstream of FGF signaling that regulate expression of these genes are largely uncharacterized. Cell type specific activation of transcription factors and target genes is likely due in part to the use of different intracellular signal transducers and inhibitors. Although little characterized, the epigenetic states of the target cell types will also be instrumental in determining which genes are activated by FGFs and what cell fates are adopted.
Integration of Multiple Extracellular Signals
In addition to intracellular modulators of FGF signaling affecting cellular responses, the presence of other secreted factors in the environment of a cell is bound to affect its response to FGFs as it tries to integrate the multiple signals. How a cell integrates multiple concurrent signals to adopt an appropriate response remains a fundamental question in developmental biology. Integration of multiple signals can potentially occur at several levels. For instance, cross-talk can occur between the different transduction pathways activated by different extracellular factors. In addition, the regulatory sequences of target genes can act as sites at which transduction pathways converge and integrate to affect gene expression and ultimately a cell’s fate.
Although few to date, there are some studies that demonstrate such interactions between FGFs and other signals that affect a cell’s response and its fate in the developing nervous system. For example, at the level of cross-talk between transduction pathways, BMP, WNT, FGF, and/or IGF signals during neural induction can be integrated via phosphorylation of alternate sites on SMAD1 (Pera et al., 2003; Fuentealba et al., 2007). Also, in patterning the posterior neural tube in Xenopus, FGF, WNT, and antagonistic BMP signals converge on regulatory elements of the Xcad gene (Haremaki et al., 2003). Finally, at the earliest stages of telencephalon development in the mouse, loss of either FGF or WNT signaling leads to the death of the neuroectoderm, whereas loss of Smad4 can rescue this phenotype (Paek et al., 2009, 2011). In this case, integration of FGF, WNT, and TGFβ signals occurs at several levels including the regulation of Cdkn1a expression via direct binding of SMAD/FOX and MYC complexes to its cis regulatory elements (Seoane et al., 2004; Paek et al., 2011). However, the few examples to date provide only limited mechanistic insights into the interactions of FGFs with other signals in regulating neural cell fates.
Summary and Perspective
In humans, there are at least 70 nucleotide substitutions that affect the amino acid sequences of the FGFR genes (Wilkie, 2005; Beenken and Mohammadi, 2009). Mutations in ligands also exist and some of these are strongly associated with disease. Before we can devise effective treatments to remedy or cure neural or FGF-related disorders, it might be useful, if not essential, to know what intracellular pathways transduce FGF signaling in each target cell type and how signaling is affected by intracellular and extracellular modulators. Despite our progress in identifying the many requirements for FGFs in neurodevelopment and neurogenesis, our understanding of how FGFs fulfill so many functions remains preliminary and superficial. This is because we do not yet have a firm grasp of the mechanisms for how an FGF signal at the cell surface is transduced and how combined with other signals it leads to changes in gene expression that ultimately affect a cell’s fate.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author is grateful to his laboratory members for helpful discussions and comments on the manuscript. Jean M. Hébert is supported by NIMH, NINDS, the Feinberg Foundation, and the Hirschl/Weill-Caulier Foundation.
References
Abraham, J. A., Mergia, A., Whang, J. L., Tumolo, A., Friedman, J., Hjerrild, K. A., Gospodarowicz, D., and Fiddes, J. C. (1986a). Nucleotide sequence of a bovine clone encoding the angiogenic protein, basic fibroblast growth factor. Science 233, 545–548.
Abraham, J. A., Whang, J. L., Tumolo, A., Mergia, A., Friedman, J., Gospodarowicz, D., and Fiddes, J. C. (1986b). Human basic fibroblast growth factor: nucleotide sequence and genomic organization. EMBO J. 5, 2523–2528.
Alvarez, I. S., Araujo, M., and Nieto, M. A. (1998). Neural induction in whole chick embryo cultures by FGF. Dev. Biol. 199, 42–54.
Amaya, E., Musci, T. J., and Kirschner, M. W. (1991). Expression of a dominant negative mutant of the FGF receptor disrupts mesoderm formation in Xenopus embryos. Cell 66, 257–270.
Aragon, F., and Pujades, C. (2009). FGF signaling controls caudal hindbrain specification through Ras-ERK1/2 pathway. BMC Dev. Biol. 9, 61. doi:10.1186/1471-213X-9-61
Aragon, F., Vazquez-Echeverria, C., Ulloa, E., Reber, M., Cereghini, S., Alsina, B., Giraldez, F., and Pujades, C. (2005). vHnf1 regulates specification of caudal rhombomere identity in the chick hindbrain. Dev. Dyn. 234, 567–576.
Armelin, H. A. (1973). Pituitary extracts and steroid hormones in the control of 3T3 cell growth. Proc. Natl. Acad. Sci. U.S.A. 70, 2702–2706.
Basson, M. A., Echevarria, D., Ahn, C. P., Sudarov, A., Joyner, A. L., Mason, I. J., Martinez, S., and Martin, G. R. (2008). Specific regions within the embryonic midbrain and cerebellum require different levels of FGF signaling during development. Development 135, 889–898.
Beenken, A., and Mohammadi, M. (2009). The FGF family: biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 8, 235–253.
Bel-Vialar, S., Itasaki, N., and Krumlauf, R. (2002). Initiating Hox gene expression: in the early chick neural tube differential sensitivity to FGF and RA signaling subdivides the HoxB genes in two distinct groups. Development 129, 5103–5115.
Bertrand, V., Hudson, C., Caillol, D., Popovici, C., and Lemaire, P. (2003). Neural tissue in ascidian embryos is induced by FGF9/16/20, acting via a combination of maternal GATA and Ets transcription factors. Cell 115, 615–627.
Borello, U., Cobos, I., Long, J. E., McWhirter, J. R., Murre, C., and Rubenstein, J. L. (2008). FGF15 promotes neurogenesis and opposes FGF8 function during neocortical development. Neural Dev. 3, 17.
Bryant, D. M., and Stow, J. L. (2005). Nuclear translocation of cell-surface receptors: lessons from fibroblast growth factor. Traffic 6, 947–954.
Chen, Y., Mohammadi, M., and Flanagan, J. G. (2009). Graded levels of FGF protein span the midbrain and can instruct graded induction and repression of neural mapping labels. Neuron 62, 773–780.
Chi, C. L., Martinez, S., Wurst, W., and Martin, G. R. (2003). The isthmic organizer signal FGF8 is required for cell survival in the prospective midbrain and cerebellum. Development 130, 2633–2644.
Cholfin, J. A., and Rubenstein, J. L. (2007). Patterning of frontal cortex subdivisions by Fgf17. Proc. Natl. Acad. Sci. U.S.A. 104, 7652–7657.
Cox, W. G., and Hemmati-Brivanlou, A. (1995). Caudalization of neural fate by tissue recombination and bFGF. Development 121, 4349–4358.
Cross, M. J., Lu, L., Magnusson, P., Nyqvist, D., Holmqvist, K., Welsh, M., and Claesson-Welsh, L. (2002). The Shb adaptor protein binds to tyrosine 766 in the FGFR-1 and regulates the Ras/MEK/MAPK pathway via FRS2 phosphorylation in endothelial cells. Mol. Biol. Cell 13, 2881–2893.
Crossley, P. H., Martinez, S., and Martin, G. R. (1996). Midbrain development induced by FGF8 in the chick embryo. Nature 380, 66–68.
Curto, M., Frankel, P., Carrero, A., and Foster, D. A. (1998). Novel recruitment of Shc, Grb2, and Sos by fibroblast growth factor receptor-1 in v-Src-transformed cells. Biochem. Biophys. Res. Commun. 243, 555–560.
Dailey, L., Ambrosetti, D., Mansukhani, A., and Basilico, C. (2005). Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 16, 233–247.
Danjo, T., Eiraku, M., Muguruma, K., Watanabe, K., Kawada, M., Yanagawa, Y., Rubenstein, J. L. R., and Sasai, Y. (2011). Subregional specification of embryonic stem cell-derived ventral telencephalic tissues by timed and combinatory treatment with extrinsic signals. J. Neurosci. 31, 1919–1933.
Dasen, J. S., Liu, J. P., and Jessell, T. M. (2003). Motor neuron columnar fate imposed by sequential phases of Hox-c activity. Nature 425, 926–933.
Davidson, B. P., Cheng, L., Kinder, S. J., and Tam, P. P. (2000). Exogenous FGF-4 can suppress anterior development in the mouse embryo during neurulation and early organogenesis. Dev. Biol. 221, 41–52.
Delaune, E., Lemaire, P., and Kodjabachian, L. (2005). Neural induction in Xenopus requires early FGF signalling in addition to BMP inhibition. Development 132, 299–310.
Deng, C. X., Wynshaw-Boris, A., Shen, M. M., Daugherty, C., Ornitz, D. M., and Leder, P. (1994). Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev. 8, 3045–3057.
Dorey, K., and Amaya, E. (2010). FGF signalling: diverse roles during early vertebrate embryogenesis. Development 137, 3731–3742.
Draper, B. W., Stock, D. W., and Kimmel, C. B. (2003). Zebrafish fgf24 functions with fgf8 to promote posterior mesodermal development. Development 130, 4639–4654.
Echevarria, D., Martinez, S., Marques, S., Lucas-Teixeira, V., and Belo, J. A. (2005). Mkp3 is a negative feedback modulator of Fgf8 signaling in the mammalian isthmic organizer. Dev. Biol. 277, 114–128.
Eswarakumar, V. P., Ozcan, F., Lew, E. D., Bae, J. H., Tomé, F., Booth, C. J., Adams, D. J., Lax, I., and Schlessinger, J. (2006). Attenuation of signaling pathways stimulated by pathologically activated FGF-receptor 2 mutants prevents craniosynostosis. Proc. Natl. Acad. Sci. U.S.A. 103, 18603–18608.
Feldman, B., Poueymirou, W., Papaioannou, V. E., DeChiara, T. M., and Goldfarb, M. (1995). Requirement of FGF-4 for postimplantation mouse development. Science 267, 246–249.
Fuentealba, L. C., Eivers, E., Ikeda, A., Hurtado, C., Kuroda, H., Pera, E. M., and De Robertis, E. M. (2007). Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell 131, 980–993.
Fukuchi-Shimogori, T., and Grove, E. A. (2001). Neocortex patterning by the secreted signaling molecule FGF8. Science 294, 1071–1074.
Furthauer, M., Lin, W., Ang, S. L., Thisse, B., and Thisse, C. (2002). Sef is a feedback-induced antagonist of Ras/MAPK-mediated FGF signalling. Nat. Cell Biol. 4, 170–174.
Furthauer, M., Reifers, F., Brand, M., Thisse, B., and Thisse, C. (2001). Sprouty4 acts in vivo as a feedback-induced antagonist of FGF signaling in zebrafish. Development 128, 2175–2186.
Garel, S., Huffman, K. J., and Rubenstein, J. L. (2003). Molecular regionalization of the neocortex is disrupted in Fgf8 hypomorphic mutants. Development 130, 1903–1914.
Goldfarb, M. (2005). Fibroblast growth factor homologous factors: evolution, structure, and function. Cytokine Growth Factor Rev. 16, 215–220.
Goldfarb, M., Schoorlemmer, J., Williams, A., Diwakar, S., Wang, Q., Huang, X., Giza, J., Tchetchik, D., Kelley, K., Vega, A., Matthews, G., Rossi, P., Ornitz, D. M., and D’Angelo, E. (2007). Fibroblast growth factor homologous factors control neuronal excitability through modulation of voltage-gated sodium channels. Neuron 55, 449–463.
Gonzalez-Quevedo, R., Lee, Y., Poss, K. D., and Wilkinson, D. G. (2010). Neuronal regulation of the spatial patterning of neurogenesis. Dev. Cell 18, 136–147.
Gospodarowicz, D. (1974). Localization of a fibroblast growth factor and its effect alone and with hydrocortisone on 3T3 cell growth. Nature 249, 123–127.
Gospodarowicz, D. (1975). Purification of a fibroblast growth factor from bovine pituitary. J. Biol. Chem. 250, 2515–2520.
Gotoh, N., Manova, K., Tanaka, S., Murohashi, M., Hadari, Y., Lee, A., Hamada, Y., Hiroe, T., Ito, M., Kurihara, T., Nakazato, H., Shibuya, M., Lax, I., Lacy, E., and Schlessinger, J. (2005). The docking protein FRS2alpha is an essential component of multiple fibroblast growth factor responses during early mouse development. Mol. Cell. Biol. 25, 4105–4116.
Griffin, K., Patient, R., and Holder, N. (1995). Analysis of FGF function in normal and no tail zebrafish embryos reveals separate mechanisms for formation of the trunk and the tail. Development 121, 2983–2994.
Grobe, K., Inatani, M., Pallerla, S. R., Castagnola, J., Yamaguchi, Y., and Esko, J. D. (2005). Cerebral hypoplasia and craniofacial defects in mice lacking heparan sulfate Ndst1 gene function. Development 132, 3777–3786.
Guillemot, F., and Zimmer, C. (2011). From cradle to grave: the multiple roles of fibroblast growth factors in neural development. Neuron 71, 574–588.
Gunhaga, L., Marklund, M., Sjodal, M., Hsieh, J. C., Jessell, T. M., and Edlund, T. (2003). Specification of dorsal telencephalic character by sequential Wnt and FGF signaling. Nat. Neurosci. 6, 701–707.
Gutin, G., Fernandes, M., Pallazolo, L., Paek, H., Kai, Y., Ornitz, D., McConnell, S. K., and Hebert, J. M. (2006). FGF acts independently of SHH to generate ventral telencephalic cells. Development 133, 2937–2946.
Hadari, Y. R., Gotoh, N., Kouhara, H., Lax, I., and Schlessinger, J. (2001). Critical role for the docking-protein FRS2a in FGF receptor-mediated signal transduction pathways. Proc. Natl. Acad. Sci. U.S.A. 98, 8578–8583.
Haremaki, T., Tanaka, Y., Hongo, I., Yuge, M., and Okamoto, H. (2003). Integration of multiple signal transducing pathways on Fgf response elements of the Xenopus caudal homologue Xcad3. Development 130, 4907–4917.
Hasegawa, H., Ashigaki, S., Takamatsu, M., Suzuki-Migishima, R., Ohbayashi, N., Itoh, N., Takada, S., and Tanabe, Y. (2004). Laminar patterning in the developing neocortex by temporally coordinated fibroblast growth factor signaling. J. Neurosci. 24, 8711–8719.
Hernandez, R. E., Rikhof, H. A., Bachmann, R., and Moens, C. B. (2004). vhnf1 integrates global RA patterning and local FGF signals to direct posterior hindbrain development in zebrafish. Development 131, 4511–4520.
Hoch, R. V., and Soriano, P. (2006). Context-specific requirements for Fgfr1 signaling through Frs2 and Frs3 during mouse development. Development 133, 663–673.
Holowacz, T., and Sokol, S. (1999). FGF is required for posterior neural patterning but not for neural induction. Dev. Biol. 205, 296–308.
Hudson, C., and Lemaire, P. (2001). Induction of anterior neural fates in the ascidian Ciona intestinalis. Mech. Dev. 100, 189–203.
Huffman, K. J., Garel, S., and Rubenstein, J. L. (2004). Fgf8 regulates the development of intra-neocortical projections. J. Neurosci. 24, 8917–8923.
Inatani, M., Fumitoshi, I., Plump, A. S., Tessier-Lavigne, M., and Yamaguchi, Y. (2003). Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science 302, 1044–1046.
Inazawa, T., Okamura, Y., and Takahashi, K. (1998). Basic fibroblast growth factor induction of neuronal ion channel expression in ascidian ectodermal blastomeres. J. Physiol. (Lond.) 511, 347–359.
Irving, C., Malhas, A., Guthrie, S., and Mason, I. (2002). Establishing the trochlear motor axon trajectory: role of the isthmic organiser and Fgf8. Development 129, 5389–5398.
Isaacs, H. V., Pownall, M. E., and Slack, J. M. (1994). eFGF regulates Xbra expression during Xenopus gastrulation. EMBO J. 13, 4469–4481.
Isaacs, H. V., Tannahill, D., and Slack, J. M. (1992). Expression of a novel FGF in the Xenopus embryo. A new candidate inducing factor for mesoderm formation and anteroposterior specification. Development 114, 711–720.
Itoh, N., and Ornitz, D. M. (2004). Evolution of the Fgf and Fgfr gene families. Trends Genet. 20, 563–569.
Itoh, N., and Ornitz, D. M. (2011). Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J. Biochem. 149, 121–130.
Iwata, T., and Hevner, R. F. (2009). Fibroblast growth factor signaling in development of the cerebral cortex. Dev. Growth Differ. 51, 299–323.
Jen, Y. H., Musacchio, M., and Lander, A. D. (2009). Glypican-1 controls brain size through regulation of fibroblast growth factor signaling in early neurogenesis. Neural Dev. 4, 33.
Johnson, D. E., and Williams, L. T. (1993). Structural and functional diversity in the FGF receptor multigene family. Adv. Cancer Res. 60, 1–41.
Kanai, M., Goke, M., Tsunekawa, S., and Podolsky, D. K. (1997). Signal transduction pathway of human fibroblast growth factor receptor 3. Identification of a novel 66-kDa phosphoprotein. J. Biol. Chem. 272, 6621–6628.
Kang, W., Wong, L. C., Shi, S., and Hébert, J. M. (2009). The transition from radial glial to intermediate progenitor cell is inhibited by FGF signaling during corticogenesis. J. Neurosci. 29, 14571–14580.
Kengaku, M., and Okamoto, H. (1995). bFGF as a possible morphogen for the anteroposterior axis of the central nervous system in Xenopus. Development 121, 3121–3130.
Korada, S., Zheng, W., Basilico, C., Schwartz, M. L., and Vaccarino, F. M. (2002). Fibroblast growth factor 2 is necessary for the growth of glutamate projection neurons in the anterior neocortex. J. Neurosci. 22, 863–875.
Kouhara, H., Hadari, Y. R., Spivak-Kroizman, T., Schilling, J., Bar-Sagi, D., Lax, I., and Schlessinger, J. (1997). A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK pathway. Cell 89, 693–702.
Krejci, P., Prochazkova, J., Bryja, V., Kozubik, A., and Wilcox, W. R. (2009). Molecular pathology of the fibroblast growth factor family. Hum. Mutat. 30, 1245–1255.
Kroll, K. L., and Amaya, E. (1996). Transgenic Xenopus embryos from sperm nuclear transplantations reveal FGF signaling requirements during gastrulation. Development 122, 3173–3183.
Kudoh, T., Concha, M. L., Houart, C., Dawid, I. B., and Wilson, S. W. (2004). Combinatorial Fgf and Bmp signalling patterns the gastrula ectoderm into prospective neural and epidermal domains. Development 131, 3581–3592.
Kudoh, T., Wilson, S. W., and Dawid, I. B. (2002). Distinct roles for Fgf, Wnt and retinoic acid in posteriorizing the neural ectoderm. Development 129, 4335–4346.
Labalette, C., Bouchoucha, Y. X., Wassef, M. A., Gongal, P. A., Le Men, J., Becker, T., Gilardi-Hebenstreit, P., and Charnay, P. (2011). Hindbrain patterning requires fine-tuning of early krox20 transcription by Sprouty 4. Development 138, 317–326.
Lamb, T. M., and Harland, R. M. (1995). Fibroblast growth factor is a direct neuralinducer, which combined with noggin generates anterior-posterior neural pattern. Development 121, 3627–3636.
Larrson, H., Klint, P., Landgren, E., and Claesson-Welsh, L. (1999). Fibroblast growth factor receptor-1 mediated endothelial cell proliferation is dependent on the src homology (SH2/SH3) domain-containing adaptor protein Crk. J. Biol. Chem. 274, 25726–25734.
Launay, C., Fromentoux, V., Shi, D. L., and Boucaut, J. C. (1996). A truncated FGF receptor blocks neural induction by endogenous Xenopus inducers. Development 122, 869–880.
Lin, X., Wei, G., Shi, Z., Dryer, L., Esko, J. D., Wells, D. E., and Matzuk, M. M. (2000). Disruption of gastrulation and heparan sulfate biosynthesis in EXT1-deficient mice. Dev. Biol. 224, 299–311.
Liu, A., Li, J. Y., Bromleigh, C., Lao, Z., Niswander, L. A., and Joyner, A. L. (2003). FGF17b and FGF18 have different midbrain regulatory properties from FGF8b or activated FGF receptors. Development 130, 6175–6185.
Liu, J. P., Laufer, E., and Jessell, T. M. (2001). Assigning the positional identity of spinal motor neurons: rostrocaudal patterning of Hox-c expression by FGFs, Gdf11, and retinoids. Neuron 32, 997–1012.
Lo, T. W., Bennett, D. C., Goodman, S. J., and Stern, M. J. (2010). Caenorhabditis elegans fibroblast growth factor receptor signaling can occur independently of the multi-substrate adaptor FRS2. Genetics 185, 537–547.
Londin, E. R., Niemiec, J., and Sirotkin, H. I. (2005). Chordin, FGF signaling, and mesodermal factors cooperate in zebrafish neural induction. Dev. Biol. 279, 1–19.
Marchal, L., Luxardi, G., Thome, V., and Kodjabachian, L. (2009). BMP inhibition initiates neural induction via FGF signaling and Zic genes. Proc. Natl. Acad. Sci. U.S.A. 106, 17437–17442.
Martinez, S., Crossley, P. H., Cobos, I., Rubenstein, J. L., and Martin, G. R. (1999). FGF8 induces formation of an ectopic isthmic organizer and isthmocerebellar development via a repressive effect on Otx2 expression. Development 126, 1189–1200.
Mason, I. (2007). Initiation to end point: the multiple roles of fibroblast growth factors in neural development. Nat. Rev. Neurosci. 8, 583–596.
Mason, J. M., Morrison, D. J., Basson, M. A., and Licht, J. D. (2006). Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 16, 45–54.
Maves, L., Jackman, W., and Kimmel, C. B. (2002). FGF3 and FGF8 mediate a rhombomere 4 signaling activity in the zebrafish hindbrain. Development 129, 3825–3837.
Mohammadi, M., Honegger, A. M., Rotin, D., Fischer, R., Bellot, F., Li, W., Dionne, C. A., Jaye, M., Rubinstein, M., and Schlessinger, J. (1991). A tyrosine-phosphorylated carboxy-terminal peptide of the fibroblast growth factor receptor (Flg) is a binding site for the SH2 domain of phospholipase C-g1. Mol. Cell. Biol. 11, 5068–5078.
Muller-Smith, K., Fagel, D. M., Stevens, H. E., Rabenstein, R. L., Maragnoli, M. E., Ohkubo, Y., Picciotto, M. R., Schwartz, M. L., and Vaccarino, F. M. (2008). Deficiency in inhibitory cortical interneurons associates with hyperactivity in fibroblast growth factor receptor 1 mutant mice. Biol. Psychiatry 63, 953–962.
Oh, L. Y., Denninger, A., Colvin, J. S., Vyas, A., Tole, S., Ornitz, D. M., and Bansal, R. (2003). Fibroblast growth factor receptor 3 signaling regulates the onset of oligodendrocyte terminal differentiation. J. Neurosci. 23, 883–894.
Ong, S. H., Guy, G. R., Hadari, Y. R., Laks, S., Gotoh, N., Schlessinger, J., and Lax, I. (2000). FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors. Mol. Cell. Biol. 20, 979–989.
Ornitz, D. M., Xu, J., Colvin, J. S., McEwen, D. G., MacArthur, C. A., Coulier, F., Gao, G., and Goldfarb, M. (1996). Receptor specificity of the fibroblast growth factor family. J. Biol. Chem. 271, 15292–15297.
Ota, S., Tonou-Fujimori, N., and Yamasu, K. (2009). The roles of the FGF signal in zebrafish embryos analyzed using constitutive activation and dominant-negative suppression of different FGF receptors. Mech. Dev. 126, 1–17.
Paek, H., Gutin, G., and Hebert, J. M. (2009). FGF signaling is strictly required to maintain early telencephalic precursor cell survival. Development 136, 2457–2465.
Paek, H., Hwang, J. Y., Zukin, R. S., and Hebert, J. M. (2011). β-catenin-dependent FGF signaling sustains cell survival in the anterior embryonic head by countering Smad4. Dev. Cell 20, 689–699.
Partanen, J. (2007). FGF signalling pathways in development of the midbrain and anterior hindbrain. J. Neurochem. 101, 1185–1193.
Partanen, J., Schwartz, L., and Rossant, J. (1998). Opposite phenotypes of hypomorphic and Y766 phosphorylation site mutations reveal a function for Fgfr1 in anteroposterior patterning of mouse embryos. Genes Dev. 12, 2332–2344.
Pera, E. M., Ikeda, A., Eivers, E., and De Robertis, E. M. (2003). Integration of IGF, FGF, and anti-BMP signals via Smad1 phosphorylation in neural induction. Genes Dev. 17, 3023–3028.
Polanska, U. M., Fernig, D. G., and Kinnunen, T. (2009). Extracellular interactome of the FGF receptor-ligand system: complexities and the relative simplicity of the worm. Dev. Dyn. 238, 277–293.
Popovici, C., Roubin, R., Coulier, F., and Birnbaum, D. (2005). An evolutionary history of the FGF superfamily. Bioessays 27, 849–857.
Pownall, M. E., Isaacs, H. V., and Slack, J. M. (1998). Two phases of Hox gene regulation during early Xenopus development. Curr. Biol. 8, 673–676.
Raballo, R., Rhee, J., Lyn-Cook, R., Leckman, J. F., Schwartz, M. L., and Vaccarino, F. M. (2000). Basic fibroblast growth factor (Fgf2) is necessary for cell proliferation and neurogenesis in the developing cerebral cortex. J. Neurosci. 20, 5012–5023.
Reilly, J. F., Mickey, G., and Maher, P. A. (2000). Association of fibroblast growth factor receptor 1 with the adaptor protein Grb14. J. Biol. Chem. 275, 7771–7778.
Rentzsch, F., Bakkers, J., Kramer, C., and Hammerschmidt, M. (2004). Fgf signaling induces posterior neuroectoderm independently of Bmp signaling inhibition. Dev. Dyn. 231, 750–757.
Ribisi, S. Jr., Mariani, F. V., Aamar, E., Lamb, T. M., Frank, D., and Harland, R. M. (2000). Ras-mediated FGF signaling is required for the formation of posterior but not anterior neural tissue in Xenopus laevis. Dev. Biol. 227, 183–196.
Rodriguez-Gallardo, L., Climent, V., Garcia-Martinez, V., Schoenwolf, G. C., and Alvarez, I. S. (1997). Targeted over-expression of FGF in chick embryos induces formation of ectopic neural cells. Int. J. Dev. Biol. 41, 715–723.
Ron, D., Fuchs, Y., and Chorev, D. S. (2008). Know thy Sef: a novel class of feedback antagonists of receptor tyrosine kinase signaling. Int. J. Biochem. Cell Biol. 40, 2040–2052.
Saarimaki-Vire, J., Peltopuro, P., Lahti, L., Naserke, T., Blak, A. A., Vogt Weisenhorn, D. M., Yu, K., Ornitz, D. M., Wurst, W., and Partanen, J. (2007). Fibroblast growth factor receptors cooperate to regulate neural progenitor properties in the developing midbrain, and hindbrain. J. Neurosci. 27, 8581–8592.
Sahara, S., and O’Leary, D. M. (2009). Fgf10 regulates transition period of cortical stem cell differentiation to radial glia controlling generation of neurons and basal progenitors. Neuron 63, 48–62.
Samuels, I. S., Karlo, J. C., Faruzzi, A. N., Pickering, K., Herrup, K., Sweatt, J. D., Saitta, S. C., and Landreth, G. E. (2008). Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J. Neurosci. 28, 6983–6995.
Sasai, Y., Lu, B., Piccolo, S., and De Robertis, E. M. (1996). Endoderm induction by the organizer-secreted factors chordin and noggin in Xenopus animal caps. EMBO J. 15, 4547–4555.
Sato, T., and Nakamura, H. (2004). The Fgf8 signal causes cerebellar differentiation by activating Ras-ERK signaling pathway. Development 131, 4275–4285.
Scearce-Levie, K., Roberson, E. D., Gerstein, H., Cholfin, J. A., Mandiyan, V. S., Shah, N. M., Rubenstein, J. L., and Mucke, L. (2008). Abnormal social behaviors in mice lacking Fgf17. Genes Brain Behav. 7, 344–354.
Seoane, J., Le, H. V., Shen, L., Anderson, S. A., and Massague, J. (2004). Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 117, 211–223.
Seuntjens, E., Nityanandam, A., Miquelajauregui, A., Debruyn, J., Stryjewska, A., Goebbels, S., Nave, K. A., Huylebroeck, D., and Tarabykin, V. (2009). Sip1 regulates sequential fate decisions by feedback signaling from postmitotic neurons to progenitors. Nat. Neurosci. 12, 1373–1380.
Shakkottai, V. G., Xiao, M., Xu, L., Wong, M., Nerbonne, J. M., Ornitz, D. M., and Yamada, K. A. (2009). FGF14 regulates the intrinsic excitability of cerebellar Purkinje neurons. Neurobiol. Dis. 33, 81–88.
Shanmugalingam, S., Houart, C., Picker, A., Reifers, F., Macdonald, R., Barth, A., Griffin, K., Brand, M., and Wilson, S. W. (2000). Ace/Fgf8 is required for forebrain commissure formation and patterning of the telencephalon. Development 127, 2549–2561.
Sheng, G., dos Reis, M., and Stern, C. D. (2003). Churchill, a zinc finger transcriptional activator, regulates the transition between gastrulation and neurulation. Cell 115, 603–613.
Shimamura, K., and Rubenstein, J. L. (1997). Inductive interactions direct early regionalization of the mouse forebrain. Development 124, 2709–2718.
Shimogori, T., and Grove, E. A. (2005). Fibroblast growth factor 8 regulates neocortical guidance of area-specific thalamic innervation. J. Neurosci. 25, 6550–6560.
Shin, D. M., Korada, S., Raballo, R., Shashikant, C. S., Simeone, A., Taylor, J. R., and Vaccarino, F. (2004). Loss of glutamatergic pyramidal neurons in frontal and temporal cortex resulting from attenuation of FGFR1 signaling is associated with spontaneous hyperactivity in mice. J. Neurosci. 24, 2247–2258.
Shinya, M., Koshida, S., Sawada, A., Kuroiwa, A., and Takeda, H. (2001). Fgf signalling through MAPK cascade is required for development of the subpallial telencephalon in zebrafish embryos. Development 128, 4153–4164.
Smith, K. M., Ohkubo, Y., Maragnoli, M. E., Rasin, M. R., Schwartz, M. L., Sestan, N., and Vaccarino, F. M. (2006). Midline radial glia translocation and corpus callosum formation require FGF signaling. Nat. Neurosci. 9, 787–797.
Stachowiak, M. K., Maher, P. A., and Stachowiak, E. K. (2007). Integrative nuclear signaling in cell development – a role for FGF receptor-1. DNA Cell Biol. 26, 811–826.
Stevens, H. E., Smith, K. M., Maragnoli, M. E., Fagel, D., Borok, E., Shanabrough, M., Horvath, T. L., and Vaccarino, F. M. (2010). Fgfr2 is required for the development of the medial prefrontal cortex and its connections with limbic circuits. J. Neurosci. 30, 5590–5602.
Storey, K. G., Goriely, A., Sargent, C. M., Brown, J. M., Burns, H. D., Abud, H. M., and Heath, J. K. (1998). Early posterior neural tissue is induced by FGF in the chick embryo. Development 125, 473–484.
Storm, E., Garel, S., Borello, U., Hébert, J. M., Martinez, S., McConnell, S. K., Martin, G. R., and Rubenstein, J. L. R. (2006). Dosage dependent functions of Fgf8 in regulating telencephalic patterning centers. Development 133, 1831–1844.
Streit, A., Berliner, A. J., Papanayotou, C., Sirulnik, A., and Stern, C. D. (2000). Initiation of neural induction by FGF signalling before gastrulation. Nature 406, 74–78.
Takemoto, T., Uchikawa, M., Kamachi, Y., and Kondoh, H. (2006). Convergence of Wnt and FGF signals in the genesis of posterior neural plate through activation of the Sox2 enhancer N-1. Development 133, 297–306.
Terauchi, A., Johnson-Venkatesh, E. M., Toth, A. B., Javed, D., Sutton, M. A., and Umemori, H. (2010). Distinct FGFs promote differentiation of excitatory and inhibitory synapses. Nature 465, 783–787.
Theil, T., Dominguez-Frutos, E., and Schimmang, T. (2008). Differential requirements for Fgf3 and Fgf8 during mouse forebrain development. Dev. Dyn. 237, 3417–3423.
Tole, S., Gutin, G., Remedios, R., Bhatnagar, L., and Hebert, J. M. (2006). Development of midline cell types and commissural axon tracts requires Fgfr1 in the cerebrum. Dev. Biol. 289, 141–151.
Toyoda, R., Assimacopoulos, S., Wilcoxon, J., Taylor, A., Feldman, P., Suzuki-Hirano, A., Shimogori, T., and Grove, E. A. (2010). FGF8 acts as a classic diffusible morphogen to pattern the neocortex. Development 137, 3439–3448.
Trokovic, R., Trokovic, N., Hernesniemi, S., Pirvola, U., Vogt Weisenhorn, D. M., Rossant, J., McMahon, A. P., Wurst, W., and Partanen, J. (2003). FGFR1 is independently required in both developing mid- and hindbrain for sustained response to isthmic signals. EMBO J. 22, 1811–1823.
Tsang, M., and Dawid, I. B. (2004). Promotion and attenuation of FGF signaling through the Ras-MAPK pathway. Sci. STKE 228, pe17.
Turner, N., and Grose, R. (2010). Fibroblast growth factor signalling from development to cancer. Nat. Rev. Cancer 10, 116–129.
Umemori, H. (2009). Weaving the neuronal net with target-derived fibroblast growth factors. Dev. Growth Differ. 51, 263–270.
Umemori, H., Linhoff, M. W., Ornitz, D. M., and Sanes, J. R. (2004). FGF22 and its close relatives are presynaptic organizing molecules in the mammalian brain. Cell 118, 257–270.
Vaccarino, F. M., Grigorenko, E. L., Smith, K. M., and Stevens, H. E. (2009). Regulation of cerebral cortical size and neuron number by fibroblast growth factors: implications for autism. J. Autism Dev. Disord. 39, 511–520.
Vaccarino, F. M., Schwartz, M. L., Raballo, R., Nilsen, J., Rhee, J., Zhou, M., Doetschman, T., Coffin, J. D., Wyland, J. J., and Hung, Y. T. (1999). Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat. Neurosci. 2:848.
Walshe, J., Maroon, H., McGonnell, I. M., Dickson, C., and Mason, I. (2002). Establishment of hindbrain segmental identity requires signaling by FGF3 and FGF8. Curr. Biol. 12, 1117–1123.
Walshe, J., and Mason, I. (2003). Unique and combinatorial functions of Fgf3 and Fgf8 during zebrafish forebrain development. Development 130, 4337–4349.
Wang, J.-K., Xu, H., Li, H.-C., and Goldfarb, M. (1996). Broadly expressed SNTlike proteins link FGF receptor stimulation to activators of the Ras pathway. Oncogene 13, 721–729.
Wang, Q., Green, R. P., Zhao, G., and Ornitz, D. M. (2001). Differential regulation of endochondral bone growth and joint development by FGFR1 and FGFR3 tyrosine kinase domains. Development 128, 3867–3876.
Waskiewicz, A. J., Rikhof, H. A., and Moens, C. B. (2002). Eliminating zebrafish pbx proteins reveals a hindbrain ground state. Dev. Cell 3, 723–733.
Wiedlocha, A., and Sorensen, V. (2004). Signaling, internalization, and intracellular activity of fibroblast growth factor. Curr. Top. Microbiol. Immunol. 286, 45–79.
Wiellette, E. L., and Sive, H. (2003). vhnf1 and Fgf signals synergize to specify rhombomere identity in the zebrafish hindbrain. Development 130, 3821–3829.
Wilkie, A. O. (2005). Bad bones, absent smell, selfish testes: the pleiotropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev. 16, 187–203.
Wills, A. E., Choi, V. M., Bennett, M. J., Khokha, M. K., and Harland, R. M. (2010). BMP antagonists and FGF signaling contribute to different domains of the neural plate in Xenopus. Dev. Biol. 337, 335–350.
Wilson, R., Battersby, A., Csiszar, A., Vogelsang, E., and Leptin, M. (2004). A functional domain of Dof that is required for fibroblast growth factor signaling. Mol. Cell. Biol. 24, 2263–2276.
Wilson, S. I., Graziano, E., Harland, R., Jessell, T. M., and Edlund, T. (2000). An early requirement for FGF signalling in the acquisition of neural cell fate in the chick embryo. Curr. Biol. 10, 421–429.
Xiao, M., Xu, L., Laezza, F., Yamada, K., Feng, S., and Ornitz, D. M. (2007). Impaired hippocampal synaptic transmission and plasticity in mice lacking fibroblast growth factor 14. Mol. Cell. Neurosci. 34, 366–377.
Xu, H., Lee, K. W., and Goldfarb, M. (1998). Novel recognition motif on fibroblast growth factor receptor mediates direct association and activation of SNT adapter proteins. J. Biol. Chem. 273, 17987–17990.
Yaguchi, Y., Yu, T., Ahmed, M. U., Berry, M., Mason, I., and Basson, M. A. (2009). Fibroblast growth factor (FGF) gene expression in the developing cerebellum suggests multiple roles for FGF signaling during cerebellar morphogenesis and development. Dev. Dyn. 238, 2058–2072.
Yamaguchi, T. P., Harpal, K., Henkemeyer, M., and Rossant, J. (1994). fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev. 8, 3032–3044.
Yamamoto, S., Yoshino, I., Shimazaki, T., Murohashi, M., Hevner, R. F., Lax, I., Okano, H., Shibuya, M., Schlessinger, J., and Gotoh, N. (2005). Proc. Natl. Acad. Sci. U.S.A. 102, 15983–15988.
Zhang, X., Ibrahimi, O. A., Olsen, S. K., Umemori, H., Mohammadi, M., and Ornitz, D. M. (2006). Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J. Biol. Chem. 281, 15694–15700.
Keywords: FGF, neural patterning, cell survival, neurogenesis, telencephalon, neocortex
Citation: Hébert JM (2011) FGFs: neurodevelopment’s Jack-of-all-trades – how do they do it? Front. Neurosci. 5:133. doi: 10.3389/fnins.2011.00133
Received: 05 June 2011; Accepted: 18 November 2011;
Published online: 05 December 2011.
Edited by:
Nicholas Gaiano, Johns Hopkins School of Medicine, USAReviewed by:
Maria J. Donoghue, Georgetown University, USAUmberto Di Porzio, Institute of Genetics and Biophysics, Italy
Copyright: © 2011 Hébert. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jean M. Hébert, Department of Neuroscience, Albert Einstein College of Medicine, Kennedy Building, 1410 Pelham Parkway South, Bronx, NY 10461, USA. e-mail:amVhbi5oZWJlcnRAZWluc3RlaW4ueXUuZWR1