 Marvin R. Diaz1
Marvin R. Diaz1


- 1 Department of Neurosciences, University of New Mexico Health Sciences Center, Albuquerque, NM, USA
- 2 Department of Neurosciences, Medical University of South Carolina, Charleston, SC, USA
The granule cell layer of the cerebellum functions in spatio-temporal encoding of information. Granule cells (GCs) are tonically inhibited by spillover of GABA released from Golgi cells and this tonic inhibition is facilitated by acute ethanol. Recently, it was demonstrated that a specialized Ca2+-activated anion-channel, bestrophin1 (Best1), found on glial cells, can release GABA that contributes up to 50–75% of the tonic GABAergic current. However, it is unknown if ethanol has any actions on Best1 function. Using whole-cell electrophysiology, we found that recombinant Best1 channels expressed in HEK-293 cells were insensitive to 40 and 80 mM ethanol. We attempted to measure the Best1-mediated component of the tonic current in slices using 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB). We confirmed that this agent blocks recombinant Best1 channels. Unexpectedly, we found that NPPB significantly potentiated the tonic current and the area and decay of GABAA-mediated spontaneous inhibitory post-synaptic currents (IPSCs) in GCs in rodent slices under two different recording conditions. To better isolate the Best1-dependent tonic current component, we blocked the Golgi cell component of the tonic current with tetrodotoxin and found that NPPB similarly and significantly potentiated the tonic current amplitude and decay time of miniature IPSCs. Two other Cl−-channel blockers were also tested: 4′-diisothiocyanatostilbene-2,2′-disulfonic acid disodium salt hydrate (DIDS) showed no effect on GABAergic transmission, while niflumic acid (NFA) significantly suppressed the tonic current noise, as well as the mIPSC frequency, amplitude, and area. These data suggest that acute ethanol exposure does not modulate Best1 channels and these findings serve to challenge recent data indicating that these channels participate in the generation of tonic GABAergic currents in cerebellar GCs.
Introduction
The cerebellum controls motor coordination, cognitive function, and emotion. Its unique and well organized neuro-architecture is comprised of numerous layers each containing specific neuron-types that process incoming excitatory information from the brain stem and spinal cord. Mossy fibers, one of the two major excitatory inputs into the cerebellar cortex, synapse onto cerebellar granule cells (GCs) whose axons form parallel fibers that transfer information to Purkinje cells, the main output of the cerebellar cortex. GCs function as the gateway for information into the cerebellar cortex as their excitability is controlled by activation of synaptic GABAA receptors (GABAARs) via phasic GABA release from a specialized interneuron, the Golgi cell. In addition, spillover of GABA released from Golgi cells tonically activates extrasynaptic GABAARs that help regulate the excitability of GCs.
In cerebellar GCs, tonic currents are mediated by extrasynaptic GABAARs containing α6βδ subunits. These extrasynaptic GABAARs have a have high affinity for GABA, and are easily activated by ambient levels of neurotransmitter (Brickley et al., 2001; Hamann et al., 2002) as glial GABA transporters are unable to fully eliminate GABA from Golgi cell-to-GC synapses (Attwell et al., 1993). In addition, a glomerulus encases these synapses and slows diffusion, thus facilitating the activation of extrasynaptic receptors by GABA (Wall and Usowicz, 1997; Rossi et al., 2003). Numerous studies have characterized tonic currents in the cerebellum and have shown that these are sensitive to GABAA antagonists (Brickley et al., 1996; Carta et al., 2004; Bright et al., 2011). They are also mediated in part by action potential-dependent mechanisms, as tetrodotoxin (TTX) can significantly reduce both synaptic GABA events driven by spontaneous firing of Golgi cells and the tonic current mediated by extrasynaptic GABAARs (Kaneda et al., 1995; Brickley et al., 1996; Wall and Usowicz, 1997; Carta et al., 2004).
Studies examining the effects of acute ethanol on tonic currents in GCs have revealed that ethanol potentiates these currents, at least in part, by increasing Golgi cell firing via slight inhibition of the Na+/K+ ATPase (Carta et al., 2004; Botta et al., 2010). The facilitation of the tonic current by ethanol was abolished by TTX (Carta et al., 2004), consistent with the hypothesis that the effects of ethanol on the tonic current are predominantly mediated through changes in Golgi cell excitability. It has also been proposed that ethanol increases the tonic current via direct potentiation of extrasynaptic GABAARs (Hanchar et al., 2005), but these findings are controversial (Botta et al., 2007a,b). Alternative mechanisms responsible for the actions of ethanol on tonic GABAergic currents in GCs have yet to be investigated.
Bestrophin channels have recently been suggested as a mechanism providing the predominant source of GABA for tonic currents in GCs. These are Ca2+-activated anion-channels that have been linked to human eye diseases [reviewed in (Marmorstein et al., 2009)], but have also been shown to exist in the brain (Petrukhin et al., 1998). Specifically, bestrophin1 (Best1) channels are abundantly expressed in hippocampal astrocytes where they conduct anions and glutamate (Park et al., 2009). A recent study reported that cerebellar astrocytes and Bergmann glia express Best1 channels that were found to be permeable to GABA (Lee et al., 2010). Importantly, this study showed that GABA release from Best1 contributes up to 50–75% of the tonic current in GCs as three different Cl−-channel blockers, 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB) with the largest effect, 4′-diisothiocyanatostilbene-2,2′-disulfonic acid disodium salt hydrate (DIDS), and niflumic acid (NFA) significantly attenuated tonic GABAergic currents by blocking Best1. In addition, shRNA-mediated down regulation of Best1 eliminated the effect of NPPB on the tonic current. Lee et al. (2010) also demonstrated that these agents blocked Cl− and GABA conductances in recombinant Best1 channels, but did not act on recombinant GABA receptors, further suggesting that the observed inhibition of the tonic current by these antagonists was solely mediated by blockade of GABA release through glial Best1 channels. Based on these findings, it can be concluded that extrasynaptic GABAARs in GCs are activated by three pools of GABA: (1) GABA released by glial cells via Best1; (2) spillover of GABA synaptically released from Golgi cells; and (3) ambient GABA levels.
As mentioned above, we previously showed that potentiation of tonic currents by ethanol in GCs is not observed under conditions where the Golgi cell-dependent component of the tonic current is blocked (Carta et al., 2004). According to the findings of Lee et al. (2010) the majority of the Golgi cell-independent component of the tonic current is mediated by Best1 channel-mediated GABA release from glia cells. We therefore hypothesized that the Best1-dependent component of the tonic current is insensitive to acute ethanol exposure. We tested this hypothesis using patch-clamp electrophysiological techniques on human recombinant Best1 channels expressed in human embryonic kidney (HEK)-293 cells and acute cerebellar slices from rodents.
Materials and Methods
Studies with Recombinant Best1
Unless indicated, all chemicals were purchased from Sigma Chemical Company (St. Louis, MO, USA). The wild-type human Best1 cDNA was kindly provided by Dr. J. Nathans (Baltimore, MD, USA). Site-directed mutagenesis was performed using the Quik-Change mutagenesis kit (Invitrogen, Carlsbad, CA, USA) and mutants were confirmed by DNA sequencing. HEK-293 cells were obtained from American Type Culture Collection (Manassas, VA, USA) and were maintained in serum-supplemented Dulbecco’s Modified Eagle Media in a humidified incubator supplied with 5% CO2 (Xu and Woodward, 2006). For recordings, cells were plated onto poly ornithine coated 35 mm dishes and transfected with plasmids encoding Best1, Best1 (W93C), or enhanced green fluorescent protein (eGFP) using Lipofectamine 2000 (Invitrogen Inc., Carlsbad, CA, USA) according to the manufacturer’s recommendation. The Best1 cDNA contained an Internal Ribosome Entry Site–eGFP domain that allowed for detection of transfected cells. Following transfection, cells were maintained in the incubator for 24–72 h prior to use.
Dishes containing transfected cells were mounted on the stage of an Olympus IX50 inverted microscope and perfused with extracellular recording solution at 1–2 ml/min. The recording solution contained (in mM): 135 NaCl, 5.4 KCl, 1.8 CaCl2, 5 HEPES, 10 glucose (pH adjusted to 7.4 and osmolarity adjusted to 310–325 mOsm with sucrose). Patch pipettes (2–5 MΩ) were pulled from borosilicate glass (1.5 mm × 0.86 mm) and filled with internal solution containing (in mM): 133 CsCl, 4 MgCl2, 3.5 CaCl2, 5 EGTA, 10 HEPES, 3.1 Na-ATP, and 0.42 Na-GTP (pH adjusted to 7.1 with CsOH). Transfected cells were identified by eGFP fluorescence and whole-cell voltage-clamp recordings were carried out at room temperature using an Axopatch 200B amplifier (Molecular Devices, Union City, CA, USA). Cells were held at −70 mV to monitor seal breakthrough and then stepped to 0 mV. Series resistance was monitored over the course of the experiment and cells with unstable holding currents or significant changes in series resistance were not used for analysis. Best1 currents were evoked using a ramp protocol in which cells were held at 0 mV and then jumped to −100 mV followed by a 1.3 s ramp to 100 mV and a return step to 0 mV. Control ramps obtained in normal recording solution were interleaved with those in which the cells were exposed to 80 mM ethanol delivered using a Warner Faststep multi-barrel perfusion system (Hamden, CT, USA). In another set of experiments, Best1-expressing HEK-293 cells were subject to the previously mentioned protocol using 40 mM ethanol or NPPB (100 μM). Data were filtered at 1–2 kHz and acquired at 5 kHz using an Instrutech ITC-16 digital interface (Instrutech Corp., Port Washington, NY, USA) controlled by AxographX software (Axograph, Sydney, Australia). Data were analyzed offline using AxographX software (Axograph, Sydney, NSW, Australia).
Statistical analyses were done with Prism (GraphPad, San Diego, CA, USA) using linear regressions followed by an unpaired t-test to compare slopes. A P < 0.05 was considered statistically significant.
Cerebellar Slice Electrophysiology
We utilized male Sprague-Dawley rats at postnatal day (P) 23–30 from Harlan (Indianapolis, IN, USA) and C57/B6 mice (P28–33) from Charles River (Wilmington, MA, USA). Animals were group-housed and received food and water ad libitum until the day of the experiment. All animal procedures were approved by the UNM-Health Sciences Center Institutional Animal Care and Use Committee and conformed to National Institutes of Health Guidelines. Animals were sacrificed by rapid decapitation under deep anesthesia with ketamine (250 mg/kg i.p.). For most experiments, brains were quickly removed and submerged for 2 min in cold sucrose artificial cerebral spinal fluid (aCSF) containing (in mM): 220 sucrose, 2 KCl, 1.25 NaH2PO4, 26 NaHCO3, 12 MgSO4, 10 glucose, 0.2 CaCl2, and 0.43 ketamine, pre-equilibrated with 95% O2/5% CO2. The vermis of the cerebellum was sliced in the sucrose aCSF at 200 μm using a vibrating tissue slicer (Leica Microsystems, Bannockburn, IL, USA). Immediately following this procedure, slices were transferred to a chamber containing normal aCSF and allowed to recover for 40 min at 35–36°C. This normal aCSF contained (in mM): 126 NaCl, 2 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 glucose, 1 MgSO4, 2 CaCl2, and 0.4 ascorbic acid and was continuously bubbled with 95% O2/5% CO2. When indicated, we used the procedures described by Lee et al. (2010); please refer to Table 1 for more details.
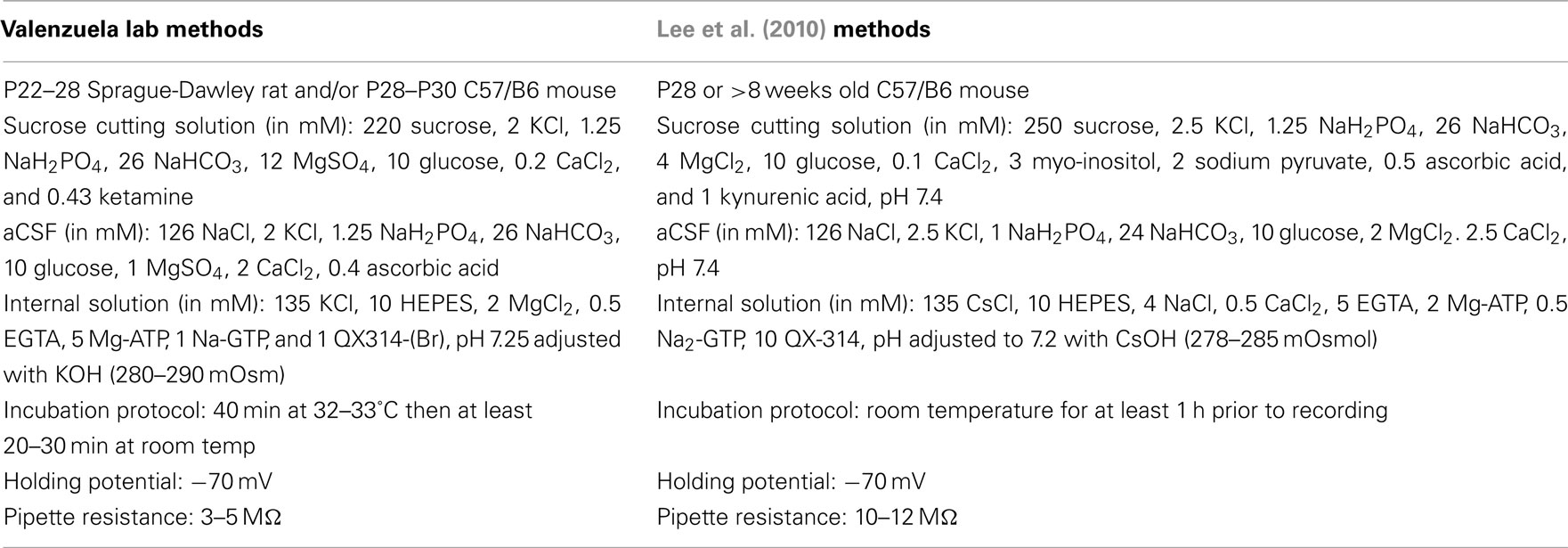
Table 1. Methodological differences in slice preparation and electrophysiological recording conditions.
Whole-cell patch-clamp techniques were used to record tonic currents and spontaneous activity. Recordings were performed in a chamber perfused with aCSF at a rate of 2–3 ml/min and maintained at 32–33°C. Neurons were visualized using infrared-differential interference contrast microscopy and recordings were performed with an Axopatch 200B amplifier. GCs were identified on the basis of their location in the GC layer, morphology (small and round sized), and capacitance = ∼2–5 pF. Patch pipettes (tip resistance = 3–5 MΩ) were filled with an internal solution containing (in mM): 135 KCl, 10 HEPES, 2 MgCl2, 0.5 EGTA, 5 Mg-ATP, 1 Na-GTP, and 1 N-(2,6-Dimethylphenylcarbamoylmethyl)triethylammonium) bromide (QX314-Br), pH 7.25, osmolarity 280–290 mOsm. The holding potential was −70 mV. Inclusion criteria for analysis was that access resistance did not change >20% throughout the duration of the experiment. GABAergic synaptic transmission was isolated by blocking AMPA and NMDA receptors using kynurenic acid (1 mM) and DL-APV (50 μM), respectively. During application of glutamate antagonists, neurons were allowed to equilibrate (∼5 min) prior to beginning an experiment. Data were acquired in gap-free mode at 10 kHz and filtered at 2 kHz.
Data were analyzed with Clampfit-10 (Molecular Devices) and Mini Analysis 6.0.3 (Synaptosoft, Decatur, GA, USA). As previously shown, the tonic current amplitude and noise were calculated by fitting a Gaussian distribution to an all-point histogram for every minute of the recording, constraining the fit to eliminate a contribution of spontaneous synaptic events (Botta et al., 2007b). Tonic current amplitude and noise were defined as the mean current and the standard deviation, respectively, recorded in the absence minus that recorded in the presence of gabazine (10 μM). Data were initially analyzed with the D’Agostino and Pearson omnibus normality tests. If data followed a normal distribution, these were analyzed using parametric tests. If this was not the case, then non-parametric tests were used. Pooled data were statistically analyzed with Prism and are presented as mean ± SEM. A P < 0.05 was considered to be statistically significant.
Ethanol was purchased from Aaper Alcohol and Chemical Company (Shelbyville, KY, USA). QX-314 Br and gabazine were obtained from Tocris (Ellisville, MO, USA or Bristol, UK), TTX was obtained from Calbiochem (Darmstadt, Germany) while all other chemicals were obtained from Sigma Chemical Company (St. Louis, MO, USA).
Results
Effect of Ethanol on Recombinant Best1 Channels
We initially investigated whether recombinant Best1 channels were sensitive to acute ethanol exposure. These channels were transiently transfected into HEK-293 cells and activated using a ramp protocol. Figure 1 shows a current density–voltage relationship in two different sets of Best1-expressing HEK-293 cells demonstrating that the recombinant Best1 channels easily pass current in and out of the cell with a reversal potential of 0 mV under our recording conditions (n = 9 for both experiments). Using a high Cl− and Ca2+-based internal solution, these channels do not show any voltage dependent activation, consistent with what has been previously shown (Park et al., 2009). We found that application of 80 mM ethanol (n = 8) did not alter current density at any given membrane voltage potential, suggesting no effect of this ethanol concentration on Best1 conductance (Figure 1A, slope: Best1 = 0.1742 ± 0.0053; Best1 + 80 mM; EtOH = 0.1634 ± 0.0035). To confirm that currents were mediated by Best1 channels, we transfected cells with Best1 (W93C), in which a mutation in the pore significantly reduces Ca2+-activated anion-channel currents (Park et al., 2009). As expected, currents were largely eliminated in cells expressing Best1 (W93C) channels (n = 3) and this was significantly different from Best1 (Figure 1A, slope: Best1 (W93C) = 0.0425 ± 0.0009; P < 0.0001 vs. Best1). Using another set of Best1-transfected HEK-293 cells, we also assessed a lower concentration of ethanol and found that application of 40 mM ethanol (n = 7) also did not alter current density at any of the membrane potentials tested (Figure 1B, slope: Best1 = 0.3421 ± 0.0129; Best1 + 40 mM; EtOH = 0.3267 ± 0.0117). We also tested the putative Best1 antagonist, NPPB, on Best1 functionality and found that the currents were significantly attenuated (Figure 1B; slope: Best1 + NPPB = 0.1423 ± 0.0027, n = 8; P < 0.0001; vs. Best1) similar to currents measured in non-transfected cells (Figure 1B; slope: non-transfected = 0.0887 ± 0.0041, n = 6; P < 0.0001 vs. Best1). Together, these data suggest that Best1 is insensitive to acute ethanol exposure and that NPPB blocks conductances in recombinant Best1 channels. These findings are in agreement with a previous report indicating that NPPB blocked recombinant Best1 channels (Park et al., 2009).
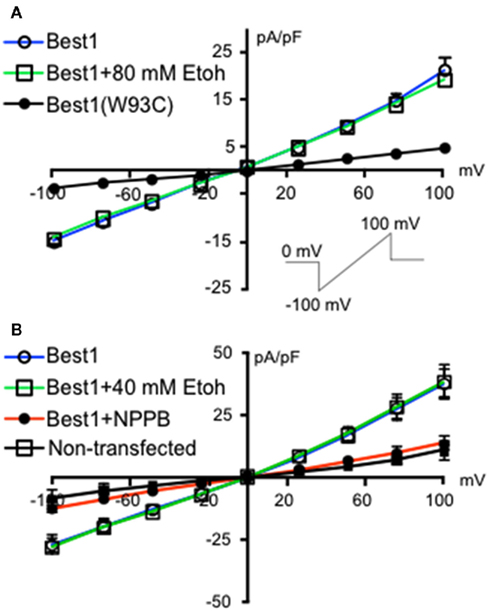
Figure 1. Ethanol does not change Best1 currents in HEK-293 cells. Non-transfected HEK-293 cells and HEK-293 cells transfected with Best1 or Best1 (W93C) were voltage-clamped at 0 mV and then stepped to −100 mV. A 1.3 s voltage ramp from −100 to +100 mV was then run before returning cells to 0 mV [(A), inset]. Ramps were repeated in the presence of (A) 80 mM ethanol (EtOH), (B) 40 mM EtOH, or NPPB (100 μM). Data are shown as mean current density expressed as pA/pF (±SEM; n = 9 for Best1 (top and bottom graphs), n = 8 for Best1 + 80 mM Ethanol, n = 3 for Best1(W93C), n = 7 for Best1 + 40 mM EtOH, n = 8 for Best1 + NPPB, n = 6 for non-transfected).
Effect of NPPB on GABAergic Currents in GCs
NPPB was shown to decrease tonic GABAergic currents in GCs in cerebellar slices from mice (Lee et al., 2010). We first tested whether this effect could be reproduced in rat GCs. In contrast to the findings of Lee et al. (2010) and in agreement with a previous report (Rossi et al., 2003), we found that NPPB increases the tonic current amplitude, with no effect on the tonic current noise (Figure 2A1). To test if the Best1 contribution to the tonic current was species specific, we performed the same experiment in slices from C57/B6 mice and again found that NPPB facilitates the tonic current amplitude with no effect on the noise under our recording conditions (Figure 2A2). Given that there were several methodological differences between our study and the Lee et al. (2010) study, we repeated the experiments using conditions that more closely matched those described in that study (Table 1). Under these conditions, we also found robust potentiation of the tonic current amplitude by NPPB with no effect on the tonic current noise (Figure 2A3). Given that we found similar results under all of the conditions, we combined the data obtained with rat and mouse slices and this yielded an NPPB-induced increase in tonic current amplitude of 21.20 ± 5.59 pA (P < 0.05 by Wilcoxon Signed Rank Test compared to 0; Figure 2B1) or 232.2 ± 118.6% (P < 0.05 by Wilcoxon Signed Rank Test compared to 0; n = 7) and there was a non-significant change in tonic current noise of 0.14 ± 0.3 pA or 21.2 ± 15.9%; n = 7 (Figure 2B2). These experiments clearly demonstrate that NPPB is not a selective Cl−-channel/Best1 antagonist, as it potentiates tonic GABAergic currents in rodent GCs.
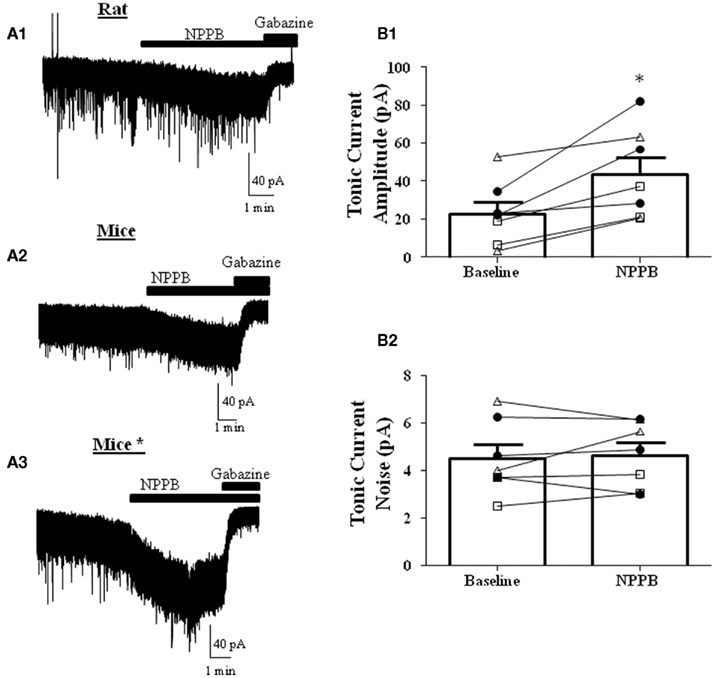
Figure 2. NPPB potentiates tonic currents in the absence of TTX. Sample traces of tonic currents with application of NPPB (100 μM) followed by gabazine (10 μM) in (A1) rats, (A2) mice, or (A3) mice* (using the methods from Lee et al., 2010; see Table 1). Effect of NPPB on (B1) tonic current amplitude and (B2) noise of individual cells (open triangles-rat, open squares-mice, dark circles-mice using the methods of Lee et al. (2010). NPPB significantly increased the tonic current amplitude while having no effect on the tonic current noise. *P < 0.05, paired t-test; n = 7.
We also examined whether NPPB altered sIPSCs in GCs, as it has been previously suggested that NPPB may act to increase GABA release (Rossi et al., 2003). Analysis of sIPSCs (Figure 3) from rat and mice slices (combined, n = 7) revealed no significant effect of NPPB (shown as % change from baseline) on frequency (3.09 ± 32.97%) or amplitude (11.30 ± 10.91%). In contrast, NPPB significantly increased the total current charge (change from control in sIPSC area = 98.93 ± 33.56%, P < 0.05, one sample t-test vs. 0) and decay time (change from control = 81.78 ± 26.86%, P < 0.05, Wilcoxon Signed Rank Test vs. 0).
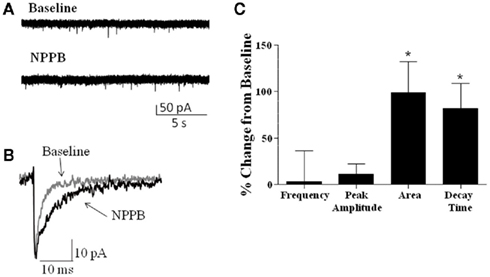
Figure 3. Effect of NPPB on sIPSCs. (A) Sample traces of sIPSCs recorded before and during application of NPPB (100 μM). (B) Exemplar average traces illustrating that NPPB decreased sIPSC decay and area. (C) Summary of the effect of NPPB on sIPSC frequency, amplitude, area, and decay time in slices from rat and mice (pooled). *P < 0.05, Wilcoxon test compared to 0; n = 7.
Effect of Other Best1 Channel Antagonists on Tonic Currents in Rat GCs in Presence of TTX
To further characterize the effect of Best1 antagonists on tonic GABAergic currents in rat slices, we blocked the Golgi cell-dependent component of these currents using TTX. This agent significantly decreased the tonic current (change in tonic current amplitude = −28.66 ± 10.38%; change in tonic current noise = −27.23 ± 8.89%; n = 20; P < 0.05 by Wilcoxon Signed Rank Test vs. 0), in addition to reducing sIPSCs (change in sIPSC frequency = −82.71 ± 4.22%; P < 0.0001 by Wilcoxon Signed Rank Test vs. 0). In the presence of TTX, NPPB significantly potentiated tonic current amplitude [Figure 4A, tonic current amplitude, TTX = 8.51 ± 2.48 pA, TTX + NPPB = 27.28 ± 4.11 pA (P < 0.05, paired t-test)] with a non-significant effect on tonic current noise [TTX = 2.41 ± 0.33 pA, TTX + NPPB = 4.52 ± 0.93 pA (P > 0.05, paired t-test; n = 5)]. We next tested the effect of two chemically distinct Cl−-channel blockers (DIDS and NFA) that were previously shown to inhibit Best1 channels and reduce the tonic current in GCs (Lee et al., 2010). In contrast to NPPB and the results of Lee et al. (2010), DIDS did not significantly modulate tonic currents [Figure 4B, tonic current amplitude, TTX = 12.41 ± 2.60 pA, TTX + DIDS = 11.29 ± 2.66 pA (P > 0.05 paired t-test); tonic current noise, TTX = 2.36 ± 0.43 pA, TTX + DIDS = 2.65 ± 0.42 pA (P > 0.05 paired t-test); n = 5]. On average, we found that NFA had no effect on the tonic current amplitude, but significantly decreased the tonic current noise [Figure 4C, tonic current amplitude, TTX = 14.30 ± 3.32 pA, TTX + NFA = 12.18 ± 2.17 pA (P > 0.05 paired t-test); tonic current noise, TTX = 2.66 ± 0.44 pA, TTX + NFA = 1.89 ± 0.23 pA (P < 0.05 paired t-test); n = 24].
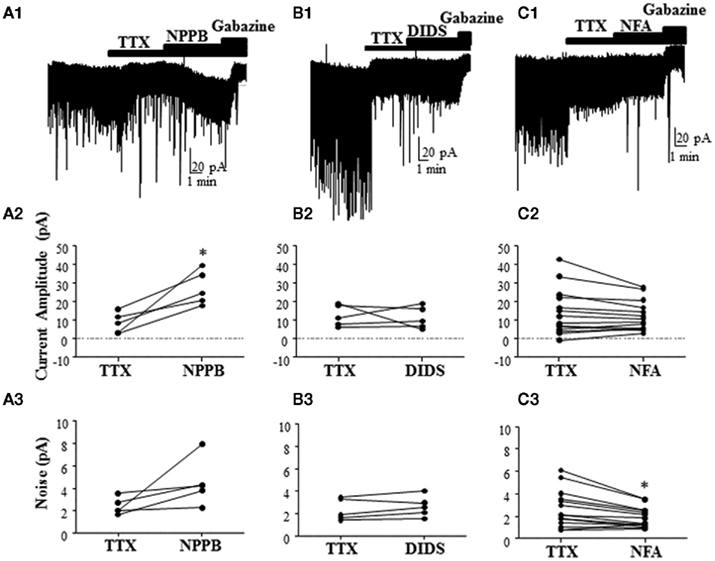
Figure 4. Effect of different Best1 antagonists on tonic currents in presence of TTX in rat slices. (A1,B1,C1) Sample traces showing the effect of Best1 channel antagonists on tonic currents in the presence of TTX. NPPB significantly increased the tonic current amplitude by 387.1 ± 180.6% (A2), but had no effect on the tonic current noise [101.7 ± 52.02%; (A3); n = 5]. DIDS had no effect on the tonic current amplitude [3.79 ± 23.31%; (B2); n = 5] or noise [15.02 ± 7.89%; (B3)]. NFA had no effect on the tonic current amplitude [−37.27 ± 36.87%; (C2); n = 24], but significantly decreased the tonic current noise by −18.34 ± 8.08% (C3). *P < 0.05, paired t-test.
We also determined whether the Best1 antagonists altered the properties of spontaneous synaptic events recorded in the presence of TTX (i.e., mIPSCs). Similar to our findings on sIPSCs, NPPB (Figure 5A) had no significant effect on mIPSC frequency (change from control = 18.62 ± 39.92%) or amplitude (change from control = 20.32 ± 22.44%). Although NPPB did not significantly increase the area of mIPSCs (change from control = 55.96 ± 23.53%) it did significantly increase the decay time (Figure 5A, change from control = 50.83 ± 17.73%, P < 0.05, one sample t-test vs. 0). DIDS (Figure 5B) did not significantly alter mIPSC frequency (change from control = −17.25 ± 13.49%), amplitude (change from control = −15.26 ± 7.92%), area (change from control = 10.97 ± 25.55%), or decay time (change from control = 18.56 ± 20.51%). Interestingly, NFA (Figure 5C) significantly decreased the frequency, amplitude, and area (change from control = −32.35 ± 1.93, −16.94 ± 3.73, and −21.74 ± 9.23%, respectively, P < 0.05 one sample t-test vs. 0) with no effect on the decay time (change from control = −0.02 ± 11.59%).
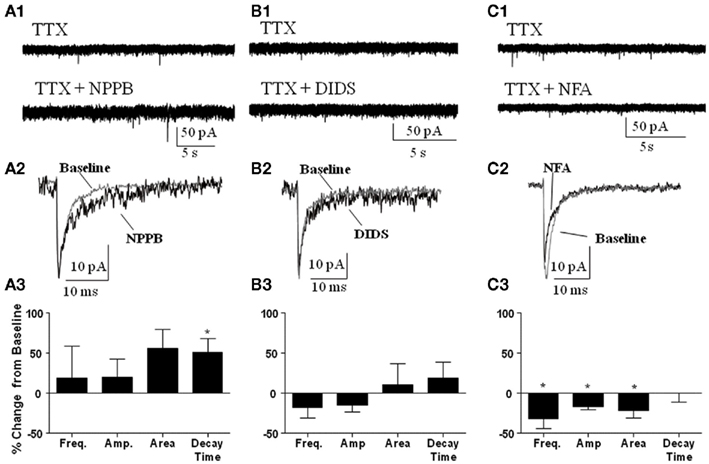
Figure 5. Effect of Best1 antagonists on mIPSCs. Sample traces of mIPSCs during baseline and after application of (A1) NPPB, (B1) DIDS, and (C1) NFA. Exemplar traces of averaged mIPSCs illustrating the effect of NPPB (A2), DIDS (B2), and NFA (C2). Summary graphs illustrating that NPPB significantly increased mIPSC decay time only [(A3); n = 3], DIDS had no effect on mIPSCs [(B3); n = 3], and NFA significantly decreased frequency, amplitude, and area of mIPSCs [(C3); n = 3]. *P < 0.05, one sample t-test compared to 0.
To determine the effect of the Best1 antagonists on resting conductances in GCs, we tested their actions in the presence of the GABAAR antagonist gabazine. NPPB minimally, but significantly, increased the residual current (not shown; current amplitude, gabazine (10 μM) = 1.92 ± 2.17 pA, gabazine + NPPB = 4.47 ± 2.65 pA, P < 0.05 paired t-test; n = 3). DIDS did not significantly affect the residual current amplitude (not shown; current amplitude, gabazine = −3.20 ± 2.87 pA, gabazine + DIDS = 1.10 ± 0.70; n = 3, P > 0.05 paired t-test). Likewise, NFA did not alter the residual current (not shown; current amplitude, gabazine = 1.88 ± 3.35 pA, gabazine + NFA = 0.02 ± 3.33 pA; n = 3, P > 0.05 by paired t-test).
Discussion
Understanding the mechanism by which ethanol modulates the tonic GABAAR current in GCs is important given the role of the cerebellar GC layer on spatio-temporal encoding (see review by D’Angelo and De Zeeuw, 2009). In the current study, we tested whether ethanol’s effects on the tonic current were, at least in part, mediated by Best1, an anion-channel found on glia that has recently been shown to mediate release of GABA from these cells and robustly contribute to the tonic current in cerebellar slices (Lee et al., 2010). We initially assessed the effect of acute ethanol on recombinant Best1 channels expressed in HEK-293 cells and found that it had no significant effect on currents carried by these channels. Unexpectedly, in cerebellar brain slices we were unable to detect theBest1-mediated contribution to the tonic current with several anion-channel blockers (regardless of the recording condition or the animal species) precluding assessment of the effect of ethanol on native Best1 function in this preparation. Based on these findings, we conclude that the acute effects of ethanol on the tonic current in cerebellar slices are independent of any action on Best1.
Acute Ethanol Does not Modulate Recombinant Best1 Channels
We successfully expressed recombinant Best1 channels in HEK-293 cells that could be blocked with NPPB. As expected, cells transfected with a functionally inactivated Best1 channel (Best1 W93C) had very small conductances over the membrane potentials tested, similar to non-transfected cells. We found that acute exposure to 40 and 80 mM ethanol had no effect on the functional conductance of recombinant Best1 at increasing or decreasing membrane potentials, supporting the hypothesis that these channels are insensitive to acute ethanol exposure. Moreover, these data support our previous findings indicating that the facilitatory effect of ethanol on tonic GABAergic currents in GCs is due to an increase in Golgi cell firing, with direct potentiation of extrasynaptic GABAARs perhaps contributing to the mechanism of action of ethanol under some experimental conditions (Carta et al., 2004; Hanchar et al., 2005; Botta et al., 2010), but see (Botta et al., 2007a). It is worth noting that although the experiments with recombinant Best1 suggest no effect of ethanol on this channel, it is still possible that it may have an effect on Best1 in a native system. Future studies in which Best1-mediated currents are directly recorded from glial cells, similar to the studies done by Lee et al. (2010) should be pursued to address this issue. It is also possible that ethanol could affect native Best channels expressed in other cell types such as epithelial cells, underlying certain actions of ethanol in these tissues. In fact, it has been shown that ethanol can inhibit Ca2+-activated Cl−-channels (Sanna et al., 1994; Clayton and Woodward, 2000), which Best1 channels are (Kunzelmann et al., 2011), suggesting that some Best channel sub-types may show some sensitivity to ethanol. Additional studies should investigate this possibility as well as the chronic actions of ethanol on these channels.
NPPB Potentiates Tonic and Phasic GABAA Receptor-Mediated Currents in GCs.
We attempted to block Best1-mediated tonic GABAergic currents using NPPB, an anion-channel blocker that was shown to inhibit the tonic current in GCs (Lee et al., 2010). Unexpectedly, we consistently found that this agent induces significant potentiation of the tonic current under a variety of experimental conditions, despite the fact that NPPB produced the expected block of recombinant Best1 channel conductances. We thought that differences in animal species or slice preparation/recording methodology could explain the differences between our results and those of Lee et al. (2010). However, we ruled out some of these factors by measuring GABAergic currents in slices from both rats and mice, and also by closely following the methodology used by Lee et al. (2010). NPPB-mediated potentiation of tonic GABAergic currents in GCs was reported in a previous paper, where it was suggested that NPPB could produce this effect by increasing GABA release from Golgi cells (Rossi et al., 2003). Our sIPSC and mIPSC recordings suggest that NPPB does not increase action potential-dependent or -independent GABA release, as the frequency of these events was not significantly affected by NPPB. Moreover, NPPB increased tonic GABAergic currents to a similar extent in the absence and presence of TTX, suggesting that it does not potentiate these currents by increasing spontaneous Golgi cell firing. This finding is consistent with direct potentiation of extrasynaptic GABAARs by NPPB. Our experiments indicate that NPPB also modulates synaptic GABAARs, as the area and decay times of sIPSCs were significantly increased. The Lee et al. (2010) study reported NPPB did not affect the function of GABAC receptors expressed in HEK-293 T cells, but the effect of this agent on GABAARs was not evaluated. In addition, that study also reported that NPPB slightly increased currents in GCs induced by local application of GABA, but the authors interpreted this finding as a consequence of blockade of the tonic current noise, leading to enhancement of synaptic events. The results from the current study argue against this mechanism, suggesting that NPPB directly potentiates GABAARs expressed in GCs. Clearly, this agent lacks selectivity for Best1 channels and should not be used to characterize the role of these channels on GABAergic transmission.
Different Anion–Channel Blockers Failed to Reduce Tonic GABAergic Currents.
Tonic GABAergic currents are, in part, mediated by action potential-dependent GABA release from Golgi cells, while Best1 channels have been suggested to mediate the remaining portion of the tonic current (Lee et al., 2010). However, when we eliminated the Golgi cell component using TTX and isolated the (presumed) Best1-mediated component, we were still unable to significantly reduce tonic currents with any of the Best1 antagonists used in the study of Lee et al. (2010). As mentioned above, NPPB potentiated the tonic current to the same extent as in experiments done without TTX. In addition, analysis of mIPSCs showed that NPPB significantly lengthened the decay time, providing further evidence for a post-synaptic effect on GABAARs. DIDS and NFA had no significant effect on the Golgi-independent component of the tonic current, although NFA reduced the tonic current in some cells. DIDS did not alter mIPSCs, while NFA significantly decreased frequency, amplitude, and area of mIPSCs, suggesting a pre- and post-synaptic effect of NFA on phasic GABA transmission, with the post-synaptic effect being consistent with NFA’s affinity for GABAA receptors (Sinkkonen et al., 2003).
Although we were unable to replicate the findings of Lee et al. (2010) using anion-channel blockers in a slice preparation, it should be noted that these investigators demonstrated a Best1-mediated component in the GC tonic current by knocking down Best1 using shRNA techniques. A potential explanation for the discrepancies between our study and the work of Lee et al. (2010) is that Best1 channels do mediate GABA release from astrocytes, but only under certain conditions; for example, when slice oxygenation is not optimal. Indeed, an increase in tonic GABAergic currents was demonstrated in the frontal cortex of an animal model of stroke (Clarkson et al., 2010), and ambient GABA levels were shown to increase following ischemic-like insults in the hippocampus (Allen and Attwell, 2004; Allen et al., 2004; Ransom et al., 2010). Consistent with this possibility, our studies revealed an average gabazine-sensitive tonic current of 17.6 ± 2.5 pA (n = 29 cells), in contrast to the 35.7 ± 0.1 pA tonic current amplitude previously reported (Lee et al., 2010). Moreover, the tonic current sample traces included in the paper of Lee et al. (2010) do not show the presence of sIPSCs, indicating that Golgi cells were not active under their recording conditions, perhaps due to compromised slice health. Clearly, studies should further investigate the possibility that Best1 channel function depends on the metabolic status of GCs in the acute slice preparation, as well as the intact cerebellum.
Conclusion
In the current study, we sought to investigate the effect of ethanol on Best1 channels and the contribution of these channels to the ethanol-induced potentiation of tonic GABAergic inhibition in cerebellar GCs. In a recombinant system, we found that acute ethanol did not alter NPPB-sensitive Best1 channel conductances. Using three chemically distinct antagonists of Best1 channels, we were unable to detect a Best1-dependent component in the tonic currents in a slice preparation. The results of this study are consistent with the model that the ethanol-induced potentiation of the tonic GABAergic current in cerebellar GCs is, at least in part, a consequence of an increase in Golgi cell firing and GABA spill-over.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Allen, N. J., and Attwell, D. (2004). The effect of simulated ischemia on spontaneous GABA release in area CA1 of the juvenile rat hippocampus. J. Physiol. (Lond.) 561, 485–498.
Allen, N. J., Rossi, D. J., and Attwell, D. (2004). Sequential release of GABA by exocytosis and reversed uptake leads to neuronal swelling in simulated ischemia of hippocampal slices. J. Neurosci. 24, 3837–3849.
Attwell, D., Barbour, B., and Szatkowski, M. (1993). Nonvesicular release of neurotransmitter. Neuron 11, 401–407.
Botta, P., De Souza, F. M., Sangrey, T., De Schutter, E., and Valenzuela, C. F. (2010). Alcohol excites cerebellar Golgi cells by inhibiting the Na+/K+ ATPase. Neuropsychopharmacology 35, 1984–1996.
Botta, P., Mameli, M., Floyd, K. L., Radcliffe, R. A., and Valenzuela, C. F. (2007a). Ethanol sensitivity of GABAergic currents in cerebellar granule neurons is not increased by a single amino acid change (R100Q) in the alpha6 GABAA receptor subunit. J. Pharmacol. Exp. Ther. 323, 684–691.
Botta, P., Radcliffe, R. A., Carta, M., Mameli, M., Daly, E., Floyd, K. L., Deitrich, R. A., and Valenzuela, C. F. (2007b). Modulation of GABAA receptors in cerebellar granule neurons by ethanol: a review of genetic and electrophysiological studies. Alcohol 41, 187–199.
Brickley, S. G., Cull-Candy, S. G., and Farrant, M. (1996). Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J. Physiol. (Lond.) 497(Pt 3), 753–759.
Brickley, S. G., Revilla, V., Cull-Candy, S. G., Wisden, W., and Farrant, M. (2001). Adaptive regulation of neuronal excitability by a voltage-independent potassium conductance. Nature 409, 88–92.
Bright, D. P., Renzi, M., Bartram, J., Mcgee, T. P., Mackenzie, G., Hosie, A. M., Farrant, M., and Brickley, S. G. (2011). Profound desensitization by ambient GABA limits activation of delta-containing GABAA receptors during spillover. J. Neurosci. 31, 753–763.
Carta, M., Mameli, M., and Valenzuela, C. F. (2004). Alcohol enhances GABAergic transmission to cerebellar granule cells via an increase in Golgi cell excitability. J. Neurosci. 24, 3746–3751.
Clarkson, A. N., Huang, B. S., Macisaac, S. E., Mody, I., and Carmichael, S. T. (2010). Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature 468, 305–309.
Clayton, R., and Woodward, J. J. (2000). Effects of ethanol on three endogenous membrane conductances present in Xenopus laevis oocytes. Neurochem. Int. 36, 67–74.
D’Angelo, E., and De Zeeuw, C. I. (2009). Timing and plasticity in the cerebellum: focus on the granular layer. Trends Neurosci. 32, 30–40.
Hamann, M., Rossi, D. J., and Attwell, D. (2002). Tonic and spillover inhibition of granule cells control information flow through cerebellar cortex. Neuron 33, 625–633.
Hanchar, H. J., Dodson, P. D., Olsen, R. W., Otis, T. S., and Wallner, M. (2005). Alcohol-induced motor impairment caused by increased extrasynaptic GABA(A) receptor activity. Nat. Neurosci. 8, 339–345.
Kaneda, M., Farrant, M., and Cull-Candy, S. G. (1995). Whole-cell and single-channel currents activated by GABA and glycine in granule cells of the rat cerebellum. J. Physiol. (Lond.) 485(Pt 2), 419–435.
Kunzelmann, K., Kongsuphol, P., Chootip, K., Toledo, C., Martins, J. R., Almaca, J., Tian, Y., Witzgall, R., Ousingsawat, J., and Schreiber, R. (2011). Role of the Ca2+-activated Cl-channels bestrophin and anoctamin in epithelial cells. Biol. Chem. 392, 125–134.
Lee, S., Yoon, B. E., Berglund, K., Oh, S. J., Park, H., Shin, H. S., Augustine, G. J., and Lee, C. J. (2010). Channel-mediated tonic GABA release from glia. Science 330, 790–796.
Marmorstein, A. D., Cross, H. E., and Peachey, N. S. (2009). Functional roles of bestrophins in ocular epithelia. Prog. Retin. Eye Res. 28, 206–226.
Park, H., Oh, S. J., Han, K. S., Woo, D. H., Mannaioni, G., Traynelis, S. F., and Lee, C. J. (2009). Bestrophin-1 encodes for the Ca2+-activated anion channel in hippocampal astrocytes. J. Neurosci. 29, 13063–13073.
Petrukhin, K., Koisti, M. J., Bakall, B., Li, W., Xie, G., Marknell, T., Sandgren, O., Forsman, K., Holmgren, G., Andreasson, S., Vujic, M., Bergen, A. A., Mcgarty-Dugan, V., Figueroa, D., Austin, C. P., Metzker, M. L., Caskey, C. T., and Wadelius, C. (1998). Identification of the gene responsible for best macular dystrophy. Nat. Genet. 19, 241–247.
Ransom, C. B., Wu, Y., and Richerson, G. B. (2010). Postdepolarization potentiation of GABAA receptors: a novel mechanism regulating tonic conductance in hippocampal neurons. J. Neurosci. 30, 7672–7684.
Rossi, D. J., Hamann, M., and Attwell, D. (2003). Multiple modes of GABAergic inhibition of rat cerebellar granule cells. J. Physiol. (Lond.) 548, 97–110.
Sanna, E., Dildy-Mayfield, J. E., and Harris, R. A. (1994). Ethanol inhibits the function of 5-hydroxytryptamine type 1c and muscarinic M1 G protein-linked receptors in Xenopus oocytes expressing brain mRNA: role of protein kinase C. Mol. Pharmacol. 45, 1004–1012.
Sinkkonen, S. T., Mansikkamaki, S., Moykkynen, T., Luddens, H., Uusi-Oukari, M., and Korpi, E. R. (2003). Receptor subtype-dependent positive and negative modulation of GABA(A) receptor function by niflumic acid, a nonsteroidal anti-inflammatory drug. Mol. Pharmacol. 64, 753–763.
Wall, M. J., and Usowicz, M. M. (1997). Development of action potential-dependent and independent spontaneous GABAA receptor-mediated currents in granule cells of postnatal rat cerebellum. Eur. J. Neurosci. 9, 533–548.
Keywords: bestrophin, tonic current, GABA, cerebellum, ethanol, NPPB, DIDS, NFA
Citation: Diaz MR, Wadleigh A, Hughes BA, Woodward JJ and Valenzuela CF (2012) Bestrophin1 channels are insensitive to ethanol and do not mediate tonic GABAergic currents in cerebellar granule cells. Front. Neurosci. 5:148. doi: 10.3389/fnins.2011.00148
Received: 05 November 2011;
Accepted: 27 December 2011;
Published online: 11 January 2012.
Edited by:
A. Leslie Morrow, University of North Carolina School of Medicine, USAReviewed by:
Jason B. Wu, Cedars-Sinai Medical Center, USAThomas Louis Kash, University of North Carolina Chapel Hill, USA
Copyright: © 2012 Diaz, Wadleigh, Hughes, Woodward and Valenzuela. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: C. Fernando Valenzuela, Department of Neurosciences, School of Medicine, 1 University of New Mexico, MSC08 4740, Albuquerque, NM 87131-0001, USA. e-mail:ZnZhbGVuenVlbGFAc2FsdWQudW5tLmVkdQ==