
 Michael Heide1
Michael Heide1
- 1 Department of Neuroanatomy, Institute of Anatomy and Cell Biology, University of Heidelberg, Heidelberg, Germany
- 2 Institute of Reconstructive Neurobiology, Life and Brain Center, University of Bonn, Bonn, Germany
Sonic hedgehog (Shh) signaling is essential for thalamic development. The Gli transcription factors act downstream of Shh – while Gli2 is the major activator (GliA), Gli3 acts primarily as a repressor (GliR). The thalamus is remarkable among dorsal structures because of its proximity to the mid-diencephalic organizer, a unique dorsal Shh source. This lends complexity to the interactions between Shh, Gli2, and Gli3, suggesting the presence of a dorsal Gli activator which elsewhere is found only ventrally, and making the dissection of thalamic Gli functions particularly interesting. A current model based on mutant phenotypes in telencephalon and midbrain postulates a degree of reciprocal antagonism of Shh and Gli3 in dorsal brain regions. To approach the role of Gli factors in thalamic specification we first analyzed mice deficient in Gli2 or Gli3. In Gli2 mutants, the thalamus is small and poorly differentiated with the exception of the medial and intralaminar nuclei which, in contrast, are specifically and severely affected by Gli3 inactivation. Gbx2 expression is very reduced in the Gli3 mutant. Most thalamic nuclei are present in both mutants, although incompletely differentiated, as reflected by the loss of specific markers. The ventral posterior group, revealed by novel specific marker Hes1, is present in both mutants and extends axons to the telencephalon. To test the Gli3/Shh interaction we generated a novel mutant deficient in Gli3 and neuroepithelial Shh. The thalamus of the n-Shh/Gli3 double mutants is very large and very poorly differentiated except for a broad domain of Gbx2, Lhx2, and Calb2 expression. In utero electroporation experiments on wild type embryos suggest that a stage-specific factor acting early is responsible for this prepattern. We show that, in the thalamus, GliA acts downstream of Shh to specify pattern and size of the thalamic nuclei to the exception of the medial and intralaminar groups. Gli3A can partially substitute for Gli2A in the Gli2 mutant. GliR is essential for specification and growth of the medial and intralaminar nuclei, contributes to the specification of other thalamic nuclei and reduces thalamic size. GliA (from neuroepithelial Shh signaling) and GliR do not show reciprocal antagonism in the thalamus, and their joint abolition does not rescue the wild type phenotype.
Introduction
How different molecules and signaling pathways work together to build the intricate and precisely patterned structure of the brain is not yet clear. The thalamus is a complex of neuronal nuclei relaying sensory input to the cortex. It develops from the neuroepithelium of the caudal diencephalon also known as alar portion of p2 (Puelles et al., 1987; Puelles and Rubenstein, 1993), which generates five masses of tissue or pronuclei which further differentiate into thalamic nuclei gradually building a complex nested structure (Rose, 1942; Jones, 2007). Its development is a model of the formation of an intricate, three-dimensional brain region from a two-dimensional neuroepithelium.
The Sonic hedgehog (Shh) signaling pathway is essential for thalamic development (Kiecker and Lumsden, 2004; Vieira et al., 2005; Scholpp and Lumsden, 2010). Shh is a secreted morphogen conferring fate specification through a concentration gradient (Ericson et al., 1997; Jessell, 2000; Dessaud et al., 2008).
The Shh pathway is very complex, including several steps – posttranslational modifications, release of Shh to the extracellular space through a unique mechanism, regulation of Shh spread, and gradient formation through binding to several membrane-bound proteins and to a multipart receptor system as well as intricate intracellular signal transduction; each of these steps depends on the cooperation of a large number of ancillary proteins (Ingham and McMahon, 2001; Ingham and Placzek, 2006; Dessaud et al., 2008).
The Shh protein gradient is interpreted by the progenitor cells through regulation of the activity of zinc-finger transcription factor cubitus interruptus (Ci) in Drosophila (Methot and Basler, 2001) or its vertebrate homologs, the Gli transcription factors Gli1, Gli2, and Gli3 (Hui et al., 1994; Lee et al., 1997; Ruiz i Altaba, 1998). While Ci can act as a transcriptional activator or, after proteolytic cleavage, as a repressor (reviewed in Jacob and Briscoe, 2003), the functions of each of the three Gli as activator or repressor vary between species and between CNS regions inside the same species.
In particular, Gli1 and Gli2 exert some overlapping functions but also play some distinctive roles depending on animal species and organ. For instance, in the mouse, Gli2 is an Hh-dependent activator of the Hh response in the nervous system and in the limbs as well as in the lungs; it is also essential for early development (Mo et al., 1997; Ding et al., 1998; Matise et al., 1998; Motoyama et al., 1998). In contrast, although transcription of Gli1 is indeed upregulated by Hh (and Gli1 can activate the Hh response), it is not required for neural patterning or early development (Park et al., 2000). Then again, Gli1 and Gli2 have overlapping activator roles as indicated by the fact that Gli1-mediated activation can rescue the Gli2-deficient phenotype in mouse (Bai and Joyner, 2001).
In the zebrafish, as opposed to the mouse, Gli1 is the major activator downstream of Hh while Gli2 shows activator and repressor roles in different regions of the embryo (Karlstrom et al., 2003). While zebrafish deficient in Gli1 show an important neural phenotype, Gli2 deficiency causes only small alterations in this species (Karlstrom et al., 2003). In agreement, experiments overexpressing Gli proteins in Xenopus embryos also suggest that Gli1 is a transcriptional activator while Gli2 can behave as both activator and repressor (Ruiz i Altaba, 1998; Mullor et al., 2001).
Whereas the roles of Gli1 and Gli2 are phylogenetically divergent, the dual role of Gli3 as repressor as well as weak activator, however, seems conserved in fish and mouse (Tyurina et al., 2005). Finally, although the functions carried out by individual Gli proteins differ between species, within a given species there is significant functional equivalency between Gli family members.
In the mouse, Gli2 and Gli3 are activated directly by Shh signaling, and they in turn activate transcription of Gli1 (Dai et al., 1999; Sasaki et al., 1999; Park et al., 2000). Gli2 is the major activator downstream of Shh, Gli1 is also an activator, and Gli3 exists as activator (Gli3A) and, after proteasome-mediated truncation, as repressor (Gli3R); the formation of Gli3R is negatively regulated by Shh (Bai and Joyner, 2001; Bai et al., 2002, 2004; Persson et al., 2002; Nguyen et al., 2005; Stamataki et al., 2005; Tyurina et al., 2005). Since Shh attenuates the formation of Gli3R, the Shh gradient results in an opposite gradient of Gli3 repressor activity (Persson et al., 2002; Bai et al., 2004). In this way, the outcome of the activation of the Shh pathway is the induction in the developing neural tube of two gradients of transcriptional regulators with opposite polarities – activator (high to low from ventral or medial to dorsal or lateral) and repressor (high to low from dorsal or lateral to ventral or medial; Jacob and Briscoe, 2003; Ruiz i Altaba et al., 2007).
Shh acts on the thalamic primordium after being secreted by two neuroepithelial domains – the zona limitans and the basal midbrain (Shimamura et al., 1995), each with essential and specific effects (Scholpp et al., 2006; Vieira and Martinez, 2006; Jeong et al., 2011). In particular, the positioning of the zona limitans is under tight regulation (Scholpp et al., 2007). Activation of the Shh pathway in the thalamic progenitors leads to their appropriate specification and therefore to correct differentiation of thalamic nuclei (Szabo et al., 2009b; Vue et al., 2009).
The Gli factors cooperate to integrate Shh and possibly other essential thalamic pathways (Zhou et al., 2004; Kataoka and Shimogori, 2008) in a region- and stage-specific manner, resulting in the specification of cell number and identity and ultimately of the size and morphology of brain regions (reviewed in Stecca and Ruiz, 2010). Despite numerous original contributions and reviews investigating the role of Shh in the development of the thalamus in different species, the Gli code has not been explored yet in this structure. The quantitative relation between Gli2 and Gli3 and its effects on thalamic development, however, have been approached before (Hashimoto-Torii et al., 2003).
Double mutants lacking Shh signaling as well as Gli3 partially rescue the Shh null mutant phenotype in the spinal cord and ventral telencephalon (Litingtung and Chiang, 2000; Aoto et al., 2002; Persson et al., 2002; Rallu et al., 2002; Wijgerde et al., 2002), and so it has been postulated that Shh and Gli3R cooperate in repressing and modifying a prepattern resulting from yet unknown factors (reviewed in Ruiz i Altaba et al., 2003). The effects of removing both Shh and Gli3 in the thalamus have not been analyzed yet.
Here we begin to assess the role of Gli factors in thalamic development by analyzing thalamic nuclear differentiation in mouse embryos deficient either in Gli2 (Mo et al., 1997) or in Gli3 (the Extra-toes mutant; Hui and Joyner, 1993; Maynard et al., 2002; Genestine et al., 2007). Furthermore, we approach the question of the reciprocally antagonistic action of Shh and Gli3 by analyzing the thalamus of a novel double mutant deficient in neuroepithelial Shh and in Gli3 (termed n-Shh/Gli3 mutant) and by loss-of-function and gain-of-function experiments in mouse embryos.
Our results suggest a combinatorial specification of each thalamic nuclear group by Gli2 and Gli3. An important exception is represented by the medial and intralaminar thalamic nuclei, which develop independently of Shh signaling but are Gli3R-dependent. Finally, simultaneous deficiency in neuroepithelial Shh and Gli3 reveals a prepattern defined by Gbx2 and Lhx2 expression and partial specification of the medial and intralaminar nuclei, as well as an increase in size. In utero electroporation experiments suggest that the effects of abolishing all Gli-mediated transcriptional regulation in the thalamus are nucleus- and stage-specific.
Materials and Methods
Mouse Lines
Animals were housed and handled in ways that minimize pain and discomfort, in accordance with German animal welfare regulations and in agreement with the European Communities Council Directive (2010/63/EU). The authorization for the experiments was granted by the Regierungspräsidium Karlsruhe (state authorities) and the experiments were performed under surveillance of the Animal Welfare Officer responsible for the Institute of Anatomy and Cell Biology. To obtain embryos, timed-pregnant females of the appropriate crossings were sacrificed by cervical dislocation; the embryos were decapitated.
Gli2zfd (Gli2 zinc finger-deleted) mice
This Gli2 null mutant mouse line was generated (Mo et al., 1997) by replacing the exons encoding for zinc fingers 3 to 5. The deletion leads to an out-of-frame mutation causing disrupted transcription from the deletion site to the 3′ end of the Gli2 gene. This results in translation of a truncated protein unable to bind to DNA, since the zinc fingers 4 and 5 are essential for DNA binding (Pavletich and Pabo, 1993).
Gli3Xt/+ (extra-toes) mouse line
Breeding pairs of heterozygotes were a kind gift from Dr. Thomas Theil (University of Edinburgh). This line carries a large deletion that inactivates the Gli3 locus but does not affect flanking genes (Hui and Joyner, 1993; Maynard et al., 2002; Genestine et al., 2007).
n-Shh/Gli3 mutants
The double homozygous mutants for neuroepithelial Shh and Gli3 were generated by crossings between Foxb1Cre/+; ShhloxP/+ mice; and Gli3Xt/+ mice. The double mutants (Foxb1Cre/+; ShhloxP/loxP; Gli3Xt/Xt, termed n-Shh/Gli3 mutants) do not survive beyond birth. The Foxb1Cre/+ mice show a normal phenotype (Zhao et al., 2007, 2008), Foxb1Cre/Cre homozygotes were never used in this study. The ShhloxP/+ conditional mutants were generated in the laboratory of Dr. Andrew McMahon (University of Harvard) and we obtained them through Jackson Labs (www.jax.org). The n-Shh mutants lack all Shh expression in the forebrain neuroepithelium (Foxb1Cre/+; ShhloxP/loxP; Szabo et al., 2009a,b).
In situ Hybridization
Embryos or embryonic brains were dissected, fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned at 14 μm. RNA in situ hybridization was performed as described (Blaess et al., 2011).
Immunohistochemistry
Immunofluorescent staining on sections was performed according to standard protocols. Primary antibodies were: mouse anti-Calretinin (Chemicon-Millipore, Billerica, MA, USA; MAB1568, 1:20, no unmasking, 2 days of incubation), mouse anti-neurofilament antibody 2H3 (DSHB, Iowa City; 1:100), rabbit anti-Hes1 (Chemicon AB5702; 1:100), mouse anti-NeuN (Chemicon MAB377; 1:100), rabbit anti-glial fibrillary acidic protein (GFAP) polyclonal (Chemicon AB5804; 1:100). Secondary antibodies: horse anti-mouse biotin (Vector BA2000; 1:200), Streptavidin-HRP (GE Healthcare RPN1231V; 1:100), anti-mouse IgG Alexa Fluor 488 (Invitrogen; 1:300), anti-rabbit IgG Alexa 594 (Invitrogen; 1:300).
DNA Constructs for Transfection
We used an expression vector driven by pCAGGS (Niwa et al., 1991) and inserted, upstream of an internal ribosomal entry site (IRES) and either EmGFP (kind gift of Dr. Boris Fehse, University of Hamburg; Weber et al., 2010) or tdTomato (kind gift of Dr. Roger Y. Tsien, UCSD) as reporters, one of the following cDNAs:
(1) A mutated form of human PTCH1 in which we deleted part (between MfeI and NsiI) of the second large extracellular loop (PTCH1Δ-loop2).
(2) A mutated form of mouse Smoothened (mSmo) in which we caused a G-to-T substitution by directed mutagenesis changing codon 539 from Trp to Leu (SmoM2; Xie et al., 1998).
(3) An ATG plus Kozak consensus sequence (Kozak, 1987) followed by the sequence encoding amino acids 471–645 of the mouse Gli3 zinc-finger (mGli-ZnF).
Similar constructs have been published (Hynes et al., 2000; Briscoe et al., 2001; Cayuso et al., 2006).
In utero Electroporation
This procedure was carried out as described (Saito and Nakatsuji, 2001; Saito, 2006) with the caveats for thalamus targeting (Kataoka and Shimogori, 2008; Vue et al., 2009; Matsui et al., 2011). In short, pregnant mice at E12.5 were anesthetized with a mixture of Halothane (Isoflurane, Baxter) and oxygen (0.5 l/min) administered with a Komesaroff Anaesthetic Machine. The uterus was exposed through laparotomy and the DNA solution (1 μg/μl) was injected with a glass micropipette in the ventricular system of the brain of each embryo through the uterine wall. Electric pulses were administered with a CUY21 electroporator (Nepagene, Japan; 5 square-wave pulses, 50 V, 50 ms on/950 ms off) and 5 mm tweezer electrodes. After the surgery, the embryos were allowed to develop in utero for 6 days and collected at E18.5 for analysis. The embryonic brains were dissected out, sectioned into 75 μm thick sections with a vibrating microtome (Compresstome VF-300, Precisionary Instruments Inc., Greenville, NC, USA) and observed and photographed with a Zeiss confocal microscope.
Statistics
Statistical assessment of the electroporation data was performed with Prism 5 software (GraphPad Software, San Diego, CA, USA).
Results
Regionalization Defects in the Gli Mutants
Whole mount detection of Gli3 mRNA on wild type embryos at E9.5 shows robust expression in the thalamic primordium (arrowhead in Figure 1A). Expression of Shh on E12.5 embryos (Figures 1B,C) shows an elongated zona limitans in the Gli3Xt/Xt mutant, suggesting an expansion of the thalamic primordium along the dorso-ventral axis. Expression of Pax6, a marker gene of the prethalamus (Stoykova and Gruss, 1994; Stoykova et al., 1996), confirms the presence of this region in the Gli3Xt/Xt mutant, although it appears dorsally elongated (Figures 1D,E) in keeping with the expansion of the zona limitans (Figures 1B,C). Ptch1, a diagnostic marker gene for activation of the Shh pathway (reviewed in Lewis et al., 2001), was present (arrowheads in Figures 1F,G), consistent with the normal expression of Shh in the mutant zona limitans (Figures 1B,C). To further explore thalamic regionalization in the Gli3Xt/Xt mutant, we examined specific thalamic regional marker gene Dbx1 (Shoji et al., 1996; Ishibashi and McMahon, 2002), which was appropriately expressed in the Gli3Xt/Xt mutant thalamus (Figures 1I,J). A second thalamic marker gene, however, Gbx2 (Miyashita-Lin et al., 1999; Hevner et al., 2002), was indeed expressed in the mutants but the expression was very reduced in extension and intensity (Figures 1L,M).
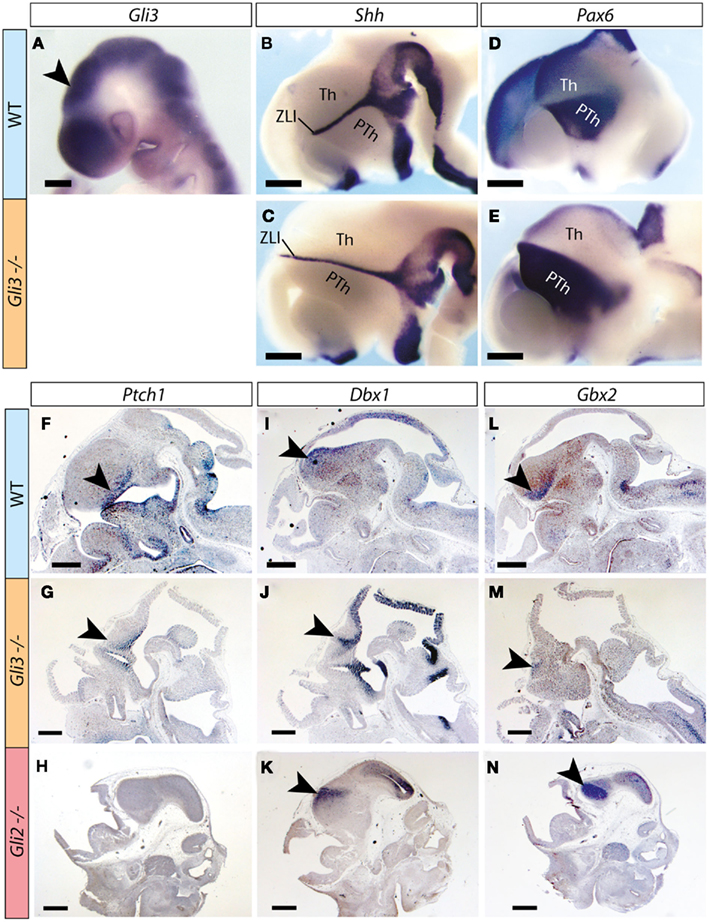
Figure 1. Thalamic regionalization in the Gli3Xt/Xt mutant. (A–E) Whole mount in situ detection of markers in E9.5 (A) and E12.5 (B–E) mouse embryos, markers, and genotypes as indicated. Rostral to the left. Arrowhead in (A) points to the thalamic primordium. (F–N) In situ detection of marker expression on sections of E12.5 mouse embryos, markers, and genotypes as indicated. Arrowheads point to diencephalic expression. PTh, prethalamus; Th, thalamus; ZLI, zona limitans interthalamica. The brown precipitate in (F,I,L) corresponds to background staining. Scale bar in (A), 200 μm; in (B–N), 500 μm.
Upon examination of the Gli2 mutant embryo at the same age, however, we found severely downregulated or undetectable expression of Ptch1 in the thalamus (Figure 1H), although the mRNAs of Dbx1 and Gbx2 were readily detected (Figures 1K,N).
We concluded that thalamic structures and expression of essential thalamic markers are induced in both Gli3Xt/Xt and Gli2zfd/zfd mutants
Evaluation of Thalamic Differentiation in the Gli3Xt/Xt and the Gli2zfd/zfd Mutant Thalami
To assess thalamic differentiation in the Gli3Xt/Xt and in the Gli2zfd/zfd mutants we performed an in situ analysis of specific marker expression (Figures 2 and 3; Table 1). Both mutants survive until E18.5, which was chosen as age of analysis. We used as markers several genes previously mapped to different thalamic nuclei and whose usefulness for phenotype assessment has been demonstrated (Szabo et al., 2009b): Calb2 (Calretinin; Arai et al., 1992, 1994; Winsky et al., 1992; Frassoni et al., 1998), Gbx2 (Jones and Rubenstein, 2004), Lhx2 and Ngn2 (Nakagawa and O’Leary, 2001), and Cdh6 (Jones and Rubenstein, 2004). These results are summarized in Table 1. To these markers we add here expression of Hes1 (hairy and enhancer of split 1), a transcription factor in the Notch pathway (Kageyama et al., 2000), as novel marker for the ventral posterior nuclear group of the thalamus at E18.5 (Figures 4 and 6; Lein et al., 2007). Our list is certainly not exhaustive, and the search for marker genes specific for different thalamic regions and nuclei is ongoing (Suzuki-Hirano et al., 2011; Yuge et al., 2011).
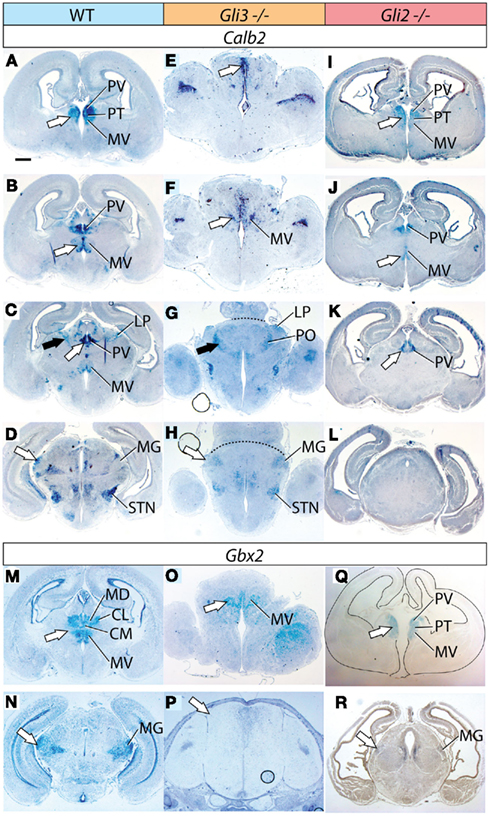
Figure 2. Thalamic differentiation in the Gli3Xt/Xt and Gli2 mutants (1). In situ detection of Calb2 (A–L) and Gbx2 (M–R) expression on transverse sections of wild type (A–D,M,N), Gli3Xt/Xt (E–H,O,P), and Gli2zfd/zfd (I–L,Q,R) E18.5 mouse brains. Comparable rostro-caudal thalamic levels are represented side-by-side. White and black arrowheads point at comparable structures across genotypes. The dotted line in (G) and (H) delimits the mutant thalamus from more caudal structures. The outline of the section has been delineated in (Q). Scale bar, 500 μm.
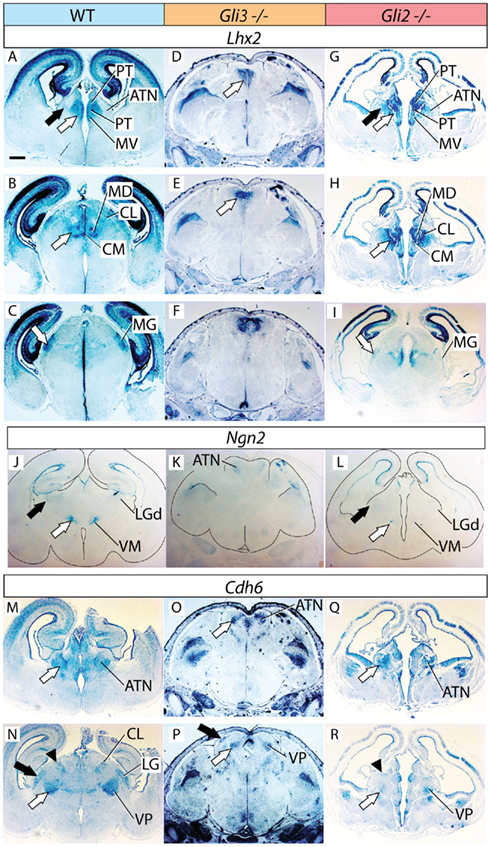
Figure 3. Thalamic differentiation in the Gli3Xt/Xt and Gli2 mutants (2). RNA in situ detection of Lhx2 (A–I), Ngn2 (J–L), and Cdh6 (M–R) expression on transverse sections of wild type (A–C,J,M,N), Gli3Xt/Xt (D–F,K,O,P), and Gli2zfd/zfd (G–I,L,Q,R) E18.5 mouse brains. Comparable rostro-caudal thalamic levels are represented side-by-side. White and black arrows point at comparable structures at comparable rostro-caudal levels across genotypes. The outline of the sections has been delineated in (J–L). Scale bar, 500 μm.
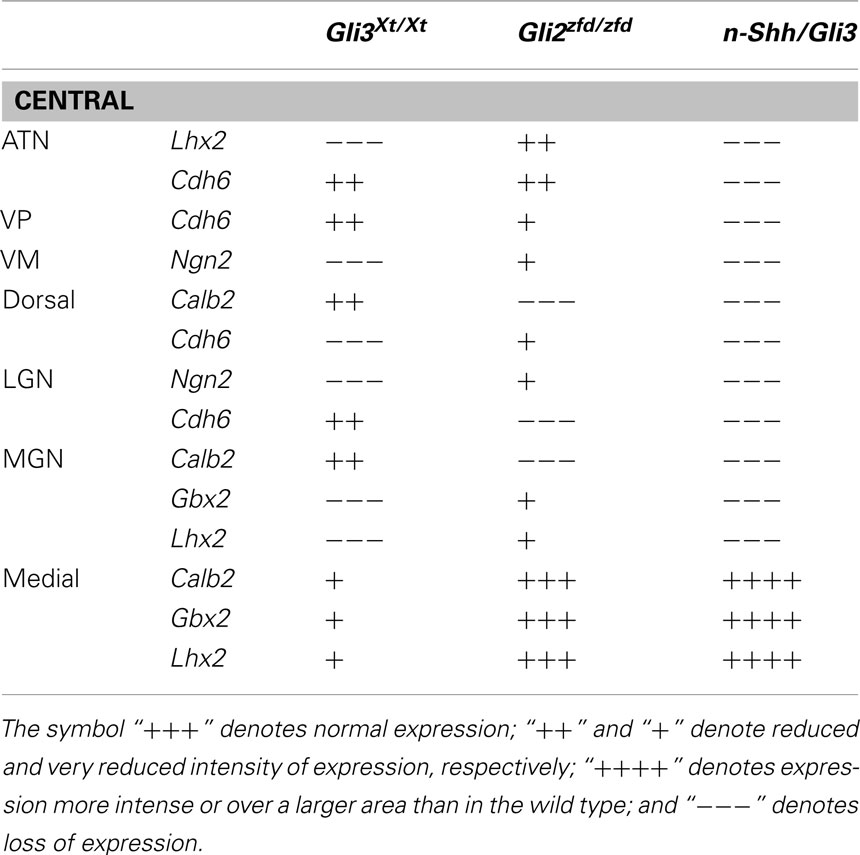
Table 1. Markers and nuclei in the wild type and Gli3Xt/Xt and Gli2zfd/zfd thalamus.
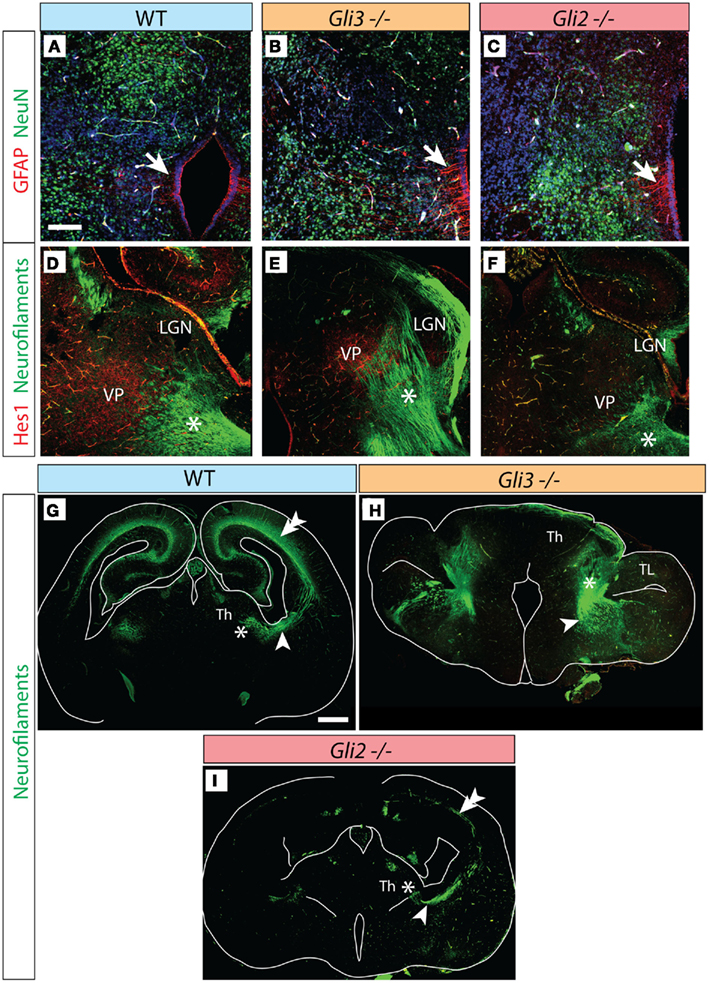
Figure 4. Thalamocortical axons in the Gli3Xt/Xt and Gli2 mutants. (A–C) Antibody detection of GFAP (red) and NeuN (green) on transverse sections of E18.5 wild type (A), Gli3Xt/Xt (B), and Gli2zfd/zfd (C) thalamus. (D–F) Antibody detection of Hes1 (red) and neurofilaments (green) on transverse sections of E18.5 wild type (D), Gli3Xt/Xt (E), and Gli2zfd/zfd (F) thalamus. (G–I) Confocal mosaic images of transverse sections through the E18.5 wild type (E), Gli3Xt/Xt (B), and Gli2zfd/zfd (C) brain showing antibody detection of neurofilaments, green. Scale bars, 100 μm (A–F) and 500 μm (G–I).
The developing thalamic mantle layer forms five neuronal masses or pronuclei out of which the adult thalamic nuclei gradually differentiate. The pronuclei and their derivatives are: medial pronucleus (medial and intralaminar nuclei), central pronucleus (anterior and ventral nuclear groups), dorsal pronucleus (lateral and posterior nuclear groups), lateral geniculate pronucleus, and medial geniculate pronucleus (Rose, 1942; Jones, 2007). We decided to use this classification for our analysis of the mutant thalamic phenotypes.
The Midline and Intralaminar Nuclei are Severely Affected in the Gli3Xt/Xt but Not in the Gli2zfd/zfd Mutant Thalamus
First, we sought to define the derivatives of the medial pronuclear group, including the medial nuclei [mediodorsal, medioventral (or reuniens), paraventricular, paratenial, and rhomboid] and the intralaminar nuclei (particularly the centromedial, paracentral, and centrolateral) by detecting the expression of marker genes Calb2, Gbx2, and Lhx2. Calb2 shows a complex and characteristic expression pattern in this structure (Arai et al., 1992, 1994; Winsky et al., 1992; Frassoni et al., 1998), which makes it useful for a general assessment of differentiation (Szabo et al., 2009b; Figures 2A–D).
The Gli3Xt/Xt mutant shows a gross morphological alteration of the di-telencephalic junction (Theil et al., 1999; Fotaki et al., 2006; Rash and Grove, 2011) affecting all rostral and dorsal structures (Figures 2E,F). In contrast, Calb2 expression was conserved in the midline (white arrows in Figures 2E,F) and recognizable on the basis of position and morphological traits in the medioventral nucleus (MV in Figure 2F), in the dorsal group (LP and PO, black arrows in Figures 2C,G) and in the medial geniculate nucleus (MG, white arrow in Figure 2H).
In the Gli2 mutant, the thalamus seemed of normal morphology but of reduced size and the size reduction was most prominent in caudal sections (Figures 2I–L). Calb2 was expressed in the medial group rostrally following a somewhat reduced normal pattern (Figures 2I,J). This marker however disappeared at more caudal levels and was not found in the dorsal group or in the medial geniculate (Figures 2K,L). We confirmed these results by antibody staining for calretinin (not shown).
Gbx2 labels the medial and intralaminar nuclei very specifically in the wild type (Figure 2M). Its expression is severely reduced but can still be detected in a midline domain in the Gli3Xt/Xt mutant (arrow in Figure 2O). In the Gli2zfd/zfd mutant (arrow in Figure 2Q), on the contrary, Gbx2 expression is strong and specific in this nuclear group.
Lhx2 is also expressed characteristically by the derivatives of the medial pronucleus (white arrows in Figures 3A,B). Its expression is present only in a small, compact, medial and dorsal structure in the Gli3Xt/Xt mutant (arrows in Figures 3D,E). The Gli2zfd/zfd mutant shows robust and appropriate expression of Lhx2 (white arrows in Figures 3G,H) indicating complete preservation of the medial and intralaminar nuclei.
We concluded that derivatives of the medial pronucleus (midline and intralaminar nuclei), are drastically altered in the Gli3Xt/Xt mutant but show essentially normal marker expression and size in the Gli2zfd/zfd mutant.
The Central and Dorsal Pronuclei are Differentially Affected in the Gli3Xt/Xt and the Gli2zfd/zfd Mutants
We used the markers Lhx2, Ngn2, and Cdh6 in order to explore the anterior and ventral thalamic nuclear groups, derivatives of the central pronucleus. The anterior thalamic nuclei were labeled by Lhx2 (black arrow in Figure 3A) and by Cdh6 (white arrow in Figure 3M). In the Gli3Xt/Xt mutant, they lost Lhx2 expression (Figure 3D) but are still labeled by Cdh6 (white arrow in Figure 3O). The Gli2zfd/zfd mutant showed expression of the two markers in a rostral structure clearly identifiable as part of the anterior thalamic complex (Lhx2, black arrow in Figure 3G; Cdh6, white arrow in Figure 3Q).
Specific Ngn2 expression in the ventromedial nucleus (white arrow in Figure 3J) was absent from the Gli3Xt/Xt mutant (Figure 3K) but present, very reduced in extension and intensity, in the Gli2zfd/zfd mutant (white arrow in Figure 3L). Finally, we detected Cdh6, a specific marker of the ventral posterior nuclei (white arrow in Figure 3N), in both mutants (white arrows in Figures 3P,R).
Derivatives of the dorsal pronucleus (lateral and posterior thalamic nuclei) can be labeled by Calb2 (black arrow in Figure 2C) or Cdh6 (arrowhead in Figure 3N) expression. The Gli3Xt/Xt mutant showed Calb2 expression in structures reasonably identifiable as the lateral and posterior nuclei (LP, PO, black arrow in Figure 2G), but no Cdh6 expression in the appropriate position (Figure 3P). The Gli2zfd/zfd mutant showed only expression of Cdh6 in the possible lateral group (arrowhead in Figure 3R).
Both mutants express some, but not all of the specific markers for thalamic nuclei derived from the central and dorsal pronuclei. We concluded that differentiation of the central and dorsal pronuclei is altered in both mutants, albeit with differences in the effects and severity.
The Lateral and Medial Geniculate Nuclei
To investigate the differentiation of the lateral geniculate nucleus (dorsal) we used markers Ngn2 (black arrow in Figure 3J) and Cdh6 (black arrow in Figure 3N). We were able to detect only Cdh6 in the lateral geniculate of the Gli3Xt/Xt mutant (black arrow in Figure 3P). The Gli2zfd/zfd mutant, on the contrary, expressed Ngn2, although at very low intensity, in the lateral geniculate (black arrow in Figure 3L) but no trace of Cdh6 (Figure 3R).
The medial geniculate is labeled by expression of Calb2 (MG, white arrow in Figure 2D), Gbx2 (white arrow in Figure 2N), Lhx2 (white arrow in Figure 3C), and Cdh6 (Szabo et al., 2009b). We discarded Cdh6 as a useful marker for this structure since it labels also the lateral geniculate nucleus, which is contiguous to the medial along the antero-posterior axis and could cause confusion. Only Calb2 was expressed in the appropriate caudal level of the Gli3Xt/Xt mutant thalamus (white arrow in Figure 2H). The Gli2zfd/zfd mutant showed no Calb2 expression in the appropriate region. Gbx2 (white arrow in Figure 2R) and Lhx2 (white arrow in Figure 3I) were however present in the medial geniculate (although very reduced in extension and intensity).
In summary, the medial and intralaminar nuclei (medial pronucleus) showed a remarkable degree of preservation in the Gli2zfd/zfd mutant but were severely affected in the Gli3Xt/Xt mutant. Each of the other pronuclei (dorsal, central, lateral geniculate, medial geniculate) expressed at least one of its characteristic markers in both mutants. The loss or preservation of specific marker expression was often complementary between the Gli2zfd/zfd and the Gli3Xt/Xt mutant (Table 1). Finally, the Gli2zfd/zfd mutant thalamus was of reduced size and, the Gli3Xt/Xt mutant seemed to be larger than in the wild type, in agreement with previous morphometric work (Aoto et al., 2002).
Thalamocortical Efferents in the Gli Mutants
In addition to our marker analysis with in situ hybridization we used antibodies against pan-neuronal marker protein NeuN (Wolf et al., 1996; green in Figures 4A–C) as well as against glial marker GFAP (arrow, red labeling in Figures 4A–C) in order to ascertain that the Gli3Xt/Xt as well as the Gli2zfd/zfd mutant thalamus are able to appropriately express general differentiation markers, as was the case (Figures 4A–C).
Essential to thalamic function are the thalamocortical efferents, whose development is a complex process depending on the appropriate fate specification of thalamic neurons by a cascade of key transcription factor genes (Lopez-Bendito and Molnar, 2003). Exploring the status of these efferents can provide important information about the degree of thalamic differentiation.
The thalamocortical axons of the somatosensory pathway originate from the ventral posterior complex (ventral postero-medial and ventral postero-lateral nuclei). At E18.5, transcription factor gene Hes1 is expressed in a single, well-delimited thalamic region (Lein et al., 2007) which we surmised, because of its localization, could be the ventral posterior group. Antibody detection of Hes1 on wild type E18.5 thalamus showed staining of the ventral posterior complex (red in Figure 4D), and neurofilament co-detection demonstrated as expected thalamocortical axons streaming out of the complex (asterisk in Figure 4D). In the Gli3Xt/Xt mutant, Hes1 was also expressed by a nuclear group (Figure 4E) extending very numerous axons into the direction of the telencephalon (asterisk in Figure 4E). The Gli2zfd/zfd mutant, however, showed weak and spatially reduced Hes1 expression in the region of the ventral posterior complex. A reduced number of axons originated in this area (asterisk in Figure 4F).
We then used stitching of multiple confocal images to analyze the broad pattern of axons connecting thalamus and telencephalon in Gli2 and Gli3 null mutants (Figures 4G–I). Thalamocortical axons were revealed as much in the wild type brain (arrowhead in Figure 4G) as in the Gli3Xt/Xt (arrowhead in Figure 4H) and the Gli2zfd/zfd (arrowhead in Figure 4I) mutants. However there were clear differences between the mutants and between each mutant and the wild type. In the wild type, the thalamocortical axons form a well-developed layer in the cortex (double arrowhead in Figure 4G). The Gli3Xt/Xt mutant seems to send very large numbers of axons in the general direction of the telencephalon, but at E18.5 they had not been able to proceed further into this region (Figure 4H), consistent with the severe defects in the cortex of the Gli3Xt/Xt (Theil et al., 1999; Theil, 2005). The Gli2zfd/zfd mutant thalamus sends only very sparse thalamocortical axons, resulting in a thin labeled axonal layer in the cortex (double arrowhead in Figure 4I).
The Thalamus of the Double Shh-Gli3Xt/Xt Mutant
Gli3R and Shh have reciprocally antagonistic functions (Litingtung and Chiang, 2000), and there is evidence that, in the telencephalon, deficit in both of them results in a phenotype closer to the wild type than that resulting from the mutation of each gene alone (Rallu et al., 2002). The implication is that other factors, acting earlier, are able to provide the telencephalon with a pattern (the so-called “prepattern”). During normal development, the combined action of Shh and Gli3 would later eliminate this prepattern and impose a new pattern (i.e., the normal thalamic pattern). Only when both Shh and Gli3 are experimentally inactivated is the prepattern uncovered (Ruiz i Altaba et al., 2003; Gutin et al., 2006; Hebert and Fishell, 2008).
Here we attempted for the first time to uncover such a possible “prepattern” in the thalamus by generating mice lacking Shh expression in the forebrain neuroepithelium as well as Gli3 expression. In previous work, we used a Cre-lox strategy to generate a mouse mutant lacking Shh expression in the forebrain neuroepithelium (the n-Shh mutant mouse; Szabo et al., 2009a,b). Here we crossed the Foxb1Cre/+, the ShhloxP/loxP, and the Gli3Xt/+ mouse lines to obtain homozygous null mutant embryos for neural Shh as well as Gli3 (Foxb1Cre/+; ShhloxP/loxP; Gli3Xt/Xt, in what follows called “double mutants” or n-Shh/Gli3 mutants; see Materials and Methods for details). These embryos are not born alive but can usually be collected at E18.5. At this age, the double mutant shows (Figure 5) a large thalamic primordium lacking characteristic dorsal structures (diencephalic roof and epithalamus-habenula). The lumen of the third ventricle, widened dorsally, correlates with the absence of the prethalamus (which is also absent in the n-Shh mutant; Szabo et al., 2009b).
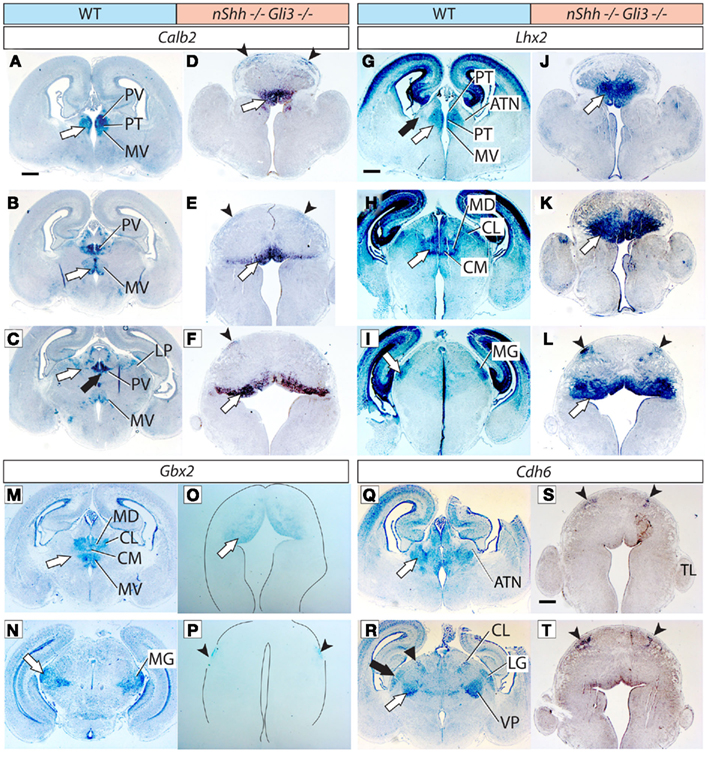
Figure 5. Thalamic differentiation in the double n-Shh/Gli3 mutant. In situ detection of Calb2 (A–F), Lhx2 (G–L), Gbx2 (M–P), and Cdh6 (Q–T) expression on transverse sections of wild type (A–C,G–I,M,N,Q,R) and n-Shh/Gli3 (D–F,J–L,O,P,S,T) E18.5 mouse brains. Comparable rostro-caudal thalamic levels are represented side-by-side. White arrows and black arrows point at comparable structures across genotypes. The outline of the section has been delineated in (O,P). Scale bar, 500 μm.
We then explored thalamic differentiation in the wild type and double mutant by using as markers Calb2 (Figures 5A–F), Lhx2 (Figures 5G–L), Gbx2 (Figures 5M–P), and Cdh6 (Figures 5Q–T). As for Ngn2, we were not able to detect expression of this gene in the thalamus of the double mutant at all (not shown). A striking finding was a wide ventral band of Calb2 (white arrows in Figures 5D–F), Lhx2 (white arrows in Figures 5J–L), and Gbx2 (white arrow in Figure 5O) expression extending along most of the rostro-caudal length of the double mutant thalamic primordium. Since these three markers are characteristic for the medial pronucleus (medial and intralaminar nuclei), and given the anatomical localization of the expression area, this result suggests that at least the ventral part of the medial pronucleus is present, and largely expanded, in the double mutant.
In addition, we noticed the presence of two small, paired, dorsal domains of weak marker expression that should correspond to two dorsal stripes visible with each of the four probes used, Calb2 (arrowheads in Figures 5D–F), Lhx2 (arrowheads in Figure 5L), Gbx2 (Figure 5P), and Cdh6 (arrowheads in Figures 5S,T). No other thalamic nucleus or structure could be recognized in the double mutant thalamus.
Furthermore, we determined that the double mutant thalamus expresses NeuN, a general marker of neurons, as well as GFAP, a marker of radial glia (Figure 6A)
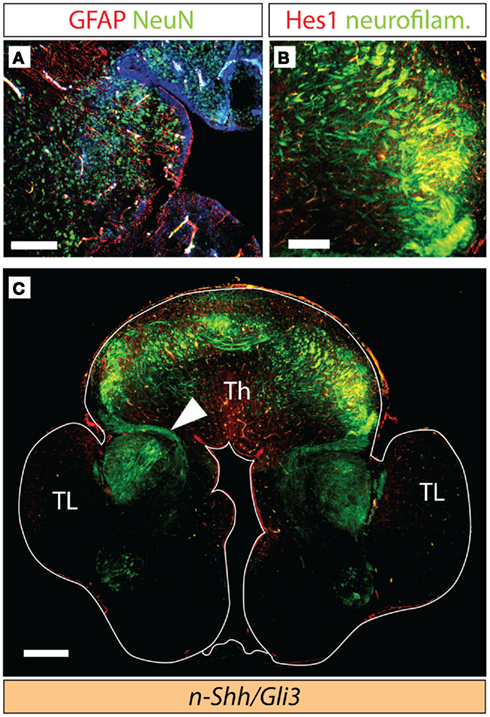
Figure 6. Axonal tracts in the double n-Shh/Gli3Xt/Xt mutant. Antibody detection of markers on transverse sections through the diencephalon of the n-Shh/Gli3Xt/Xt double mutant brain at E18.5. (A) NeuN (green) and GFAP (red). (B,C) Neurofilaments [green in (B,C)] and Hes1 [red in (B)]. (C) Shows a confocal mosaic, (B) shows a detail of (C) under higher magnification. Scale bars 100 μm (A), 200 μm (B), and 500 μm (C).
Thalamocortical Axons in the n-Shh/Gli3 Double Mutant
Next, we investigated the development of thalamocortical axons in the double-mutant through detection of neurofilaments and of Hes1 protein (see above, Figures 2 and 3). Hes1 was weakly expressed in the region giving rise to the presumed thalamocortical axons (Figure 6B). We were able to detect axons, although not very numerous, extending from the thalamic primordium in the direction of the telencephalon (arrowhead in Figure 6C). The telencephalon (including the cortex) of the double mutants showed a very abnormal phenotype, and the axons seemed unable to enter the telencephalic primordium (Figure 6C).
These results provide evidence that, in the absence of Gli3 expression and neuroepithelial Shh expression there is some appropriate thalamic differentiation.
An Approach to the Shh vs Gli3 Reciprocal Antagonism Through in utero Manipulation of the Shh Pathway
The increase in size obvious in the double mutant thalamus indicated that an abnormal surge in proliferation, independent from neuroepithelial Shh, had been unleashed by the simultaneous deficiencies in neuroepithelial Shh and in Gli3. The most striking size increase corresponded to the most differentiated part of the double mutant thalamus, a wide ventral domain which could correspond to some of the nuclei of the medial group (Figure 6). Since we could not recognize any other thalamic nucleus, we reasoned that a surge in proliferation activity could specifically affect the progenitors of the medial group at an early stage, when progenitor pools are still expanding, rather than later, when the actual neurons are generated (E12.7–E14.4; Bayer and Altman, 1995; Clancy et al., 2007). Therefore, we wanted to know if a late abolition of neuroepithelial Shh and Gli3 activity would also cause an increase in proliferation.
We approached this question by experimentally altering the Shh pathway in the progenitors of the dorsal lateral geniculate nucleus (which develops early) on wild type embryos developing in utero. The lateral geniculate nucleus was chosen as a candidate since it belongs to a different pronuclear group developing comparatively early (E11.9–E12.7; Bayer and Altman, 1995; Clancy et al., 2007) but still late enough that it can be experimentally approached in utero.
In order to establish a basis for the interpretation of our results, we first performed experiments transfecting DNA constructs that either block or constitutively activate the Shh pathway activity cell autonomously (see below). Since we transfect a 2:1 mixture of green (control):red (experimental) constructs, the effect of any experimental construct on proliferation can be measured by the ratio between green fluorescent cells and red fluorescent cells that result from the transfection. If the experimental construct has no effect at all, 100% of labeled cells will express the green reporter and 50% of them would express additionally the red reporter (since they are transfected with a 2:1 ratio); since green plus red fluorescence will appear yellow, we would expect 50% of all labeled cells to be green fluorescent (expressing only EmGFP) and 50% to be yellow fluorescent (expressing EmGFP- and tdTomato). Any increase or decrease in the proportion of red reporter-expressing cells (yellow) vs only green reporter-expressing cells will indicate increase or decrease in proliferation induced by the corresponding experimental construct.
Blocking Shh Signaling in the Developing Lateral Geniculate Nucleus
We first used in utero electroporation to transfect a 2:1 mixture of plasmids carrying either the reporter gene EmGFP (green) or a mutated form of the Shh receptor Ptch1 (Ptch1Δloop2) plus reporter gene tdTomato (red) into the thalamus of E12.5 embryos. Both constructs included a powerful constitutive promoter (see Materials and Methods for details). Transfection of this mutated form of Ptch1 blocks Shh signaling cell-autonomously (Briscoe et al., 2001). The transfected embryonic brains were collected and sectioned after 6 days survival in utero (E18.5) and labeled cells were counted in the region of the lateral geniculate nucleus (five embryos, nine sections; Figures 7A–C).
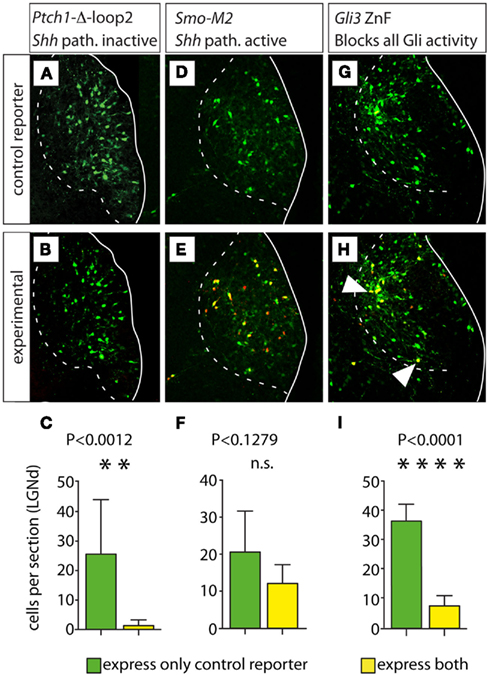
Figure 7. Loss of all Gli activity reduces proliferation of lateral geniculate progenitors. (A,B,D,E,G,H) Confocal images of the E18.5 lateral geniculate nucleus in representative transverse Compresstome sections showing expression of green fluorescent reporter (top row) or both green and red fluorescent reporter (middle row) after in utero electroporation transfection at E12.5 of plasmid constructs as indicated. Cells expressing both the control and experimental construct are yellow in (B,E,H). White arrowheads in (H) indicate double-labeled (yellow) cells. (C,F,I) Cell counting histograms (mean ± SD) after the corresponding transfections; n.s., non-significant.
This experiment yielded ca. 92.40% green cells (carrying the control construct expressing EmGFP) and only 5.60% yellow cells (carrying both the EmGFP control construct plus the Ptch1Δloop2 construct with red reporter tdTomato) in the lateral geniculate nucleus (a small percent of the labeled cells could not be unequivocally attributed red or yellow color). This result confirms that cell-autonomous inactivation of the Shh pathway leads to decreased proliferation (Ishibashi and McMahon, 2002; Szabo et al., 2009b). Additionally, an increase in Gli3R in absence of Shh signaling could increase cell death.
Constitutive Activation of Shh Signaling in the Developing Lateral Geniculate Nucleus
Next, we performed similar experiments using as experimental plasmid a DNA construct carrying SmoM2, a mutated form of the gene encoding Smoothened, a transmembrane protein essential for Shh signaling (Xie et al., 1998; Hynes et al., 2000). Expression of SmoM2 activates the Shh pathway constitutively (Hynes et al., 2000). Transfection of SmoM2 (four embryos, six sections) resulted in non-significant difference between the number of cells expressing only the control reporter (green cells) and the number of cells expressing both the control reporter and the red (experimental) reporter (yellow cells; Figures 7D–F). This is the result that we would expect if constitutive activation of the Shh pathway at E12.5 had no effect on progenitor proliferation (see above).
Blocking All Transcriptional Regulation by Gli Proteins in the Developing Lateral Geniculate Nucleus
In order to mimic the effect of the double deficiency in neuroepithelial Shh and in Gli3, we generated DNA constructs carrying a mutated form of Gli3 containing only the DNA-binding zinc-finger-domain (Gli-ZnF; Cayuso et al., 2006). Expression of Gli-ZnF results in the synthesis of a truncated Gli protein such that it specifically binds to DNA blocking all Gli-mediated transcriptional activation and repression; therefore, it can be considered the equivalent of a simultaneous loss of Shh and Gli3 (Cayuso et al., 2006).
This experiment (two embryos, five sections) resulted in 83.0% of green cells and 16.0% of yellow cells (Figures 7G–I). This indicates that blocking all GliA and GliR activity at E12.5 in the lateral geniculate (dorsal) does not increase proliferation at all (as would be expected on the basis of the n-Shh/Gli3 mutant phenotype), but rather decreases it significantly.
Discussion
We have attempted to clarify the Gli code as it applies to thalamic development in the mouse. Our conclusions are based on the analysis of mutant phenotypes, among them a novel double mutant, and on in utero experiments on wild type background. Although this approach is straightforward, several caveats apply to our interpretation of the data.
First of all, we use gene marker expression to determine if a phenotype is altered. One possible concern here is that we use only a tiny fraction of all existing specific marker genes. A full description of all specific nuclear markers for the thalamus is not yet available; it is also possible that the number of markers would be too large to be of practical interest. All things considered, though, the fact that the expression of some specific genes is altered is enough to state that the development of a specific nucleus is altered. If some nuclei are more altered than others, or if other markers would have shown a more complex phenotype, are valid questions that we cannot approach in this contribution.
Additionally, in our paper we do not address which genes or cellular processes act downstream of the Gli proteins in order to control thalamic development. While this is a very interesting field of inquiry for the future, it is obviously beyond the scope of this study. Finally, since the Gli3Xt/Xt mutants and the n-Shh/Gli3 double mutants die before birth, here we do not analyze the adult thalamus – however, it is in principle possible that the defective phenotypes we describe could have been compensated somehow later in development.
Functions of Gli Factors Downstream of Shh in the Thalamus
Expression of Ptch1 is upregulated in cells that receive high levels of Shh and in which GliA is induced, and it is considered diagnostic of activation of the Shh pathway (reviewed in Lewis et al., 2001). We have not been able to detect Ptch1 expression in the Gli2zfd/zfd mutant mice at E12.5, indicating that Ptch1 expression is not induced or that expression levels are too low to be detected (Figure 1). We are not aware of other publications analyzing Ptch1 expression in the Gli2zfd/zfd thalamus. In any case, the fact that Ptch1 is downregulated only in the Gli2zfd/zfd but not in the Gli3Xt/Xt thalamus indicates that Gli2 is the main activator downstream of Shh in this structure. This suggests that the main functions of the elements of the Shh pathway are the same in the thalamus as the ones classically described in the spinal cord (Matise et al., 1998; Litingtung and Chiang, 2000).
Intriguingly, Gli1 expression, another readout for Shh signaling, is very reduced but still detectable in the Gli2zfd/zfd mutants in the dorsal diencephalon at E10.5 (S. Blaess, unpublished results). This is remarkable, since Ptch1 and Gli1 are both readouts for high levels of Shh signaling. It is possible that the loss of Gli2 can be compensated by Gli3A only at early embryonic stages (before E12.5) but not late, or perhaps Gli1 is expressed at slightly higher levels of in the mutants, that are easier to detect.
The Gli2zfd/zfd Thalamic Phenotype
The Gli2zfd/zfd mutant thalamus is remarkably reduced in size and has a reduced marker expression. This agrees with the function of Gli2 as the major activator downstream of Shh. The fact that the thalamus is only reduced and not completely absent in the Gli2zfd/zfd mutants is most likely due to a compensatory role of Gli3A. In the absence of Gli2, the activator form of Gli3 can be induced by high levels of Shh signaling, and in combination with Gli1A, Gli3 can rescue at least partially the Gli2 deficiency (Bai et al., 2004). However, Gli3A is only a weak activator (Dai et al., 1999; Shin et al., 1999; Litingtung and Chiang, 2000; Motoyama et al., 2003; Bai et al., 2004) and cannot completely restore proliferation or differentiation. This is confirmed by the poor development of the thalamocortical projection in the Gli2zfd/zfd thalamus.
The thalamic primordium is exposed to Shh secreted by a large neuroepithelial domain comprised by a rostral, transverse portion (the zona limitans) and a ventral, longitudinal portion (the basal midbrain; Shimamura et al., 1995). Both portions are important to create a zone of very high Shh signaling ensuring proper specification of thalamic progenitors (Jeong et al., 2011). The curved shape of this rostral-plus-ventral domain is also observed in the immediately adjacent domain of Ptch1 expression that is an indicator of high levels of Shh signaling and Gli2A activity (Figures 1F,G; see also Lein et al., 2007). Therefore, we would expect the most severe Gli2zfd/zfd phenotype rostro-ventrally, i.e., in the area of highest Shh signaling. This is however masked by the good preservation of the derivatives of the medial pronucleus, i.e., the medial and intralaminar nuclei, whose development is not affected in mutants deficient in neuroepithelial Shh either (Szabo et al., 2009b; see below). These nuclei are of normal size in the Gli2zfd/zfd mutant and express their characteristic markers Calb2, Gbx2, and Lhx2. Since they occupy the central part and the rostral 2/3 of the thalamus, their good preservation in the mutant causes the misleading impression that the Gli2zfd/zfd thalamus is particularly small caudally.
The Gli3Xt/Xt Thalamic Phenotype
The Gli3Xt/Xt mutant thalamus shows alterations in size and AP patterning. Specifically, the rostral–dorsal part of the mutant thalamus is severely altered by the loss of di-telencephalic junction (Theil et al., 1999; Fotaki et al., 2006; Quinn et al., 2009; Rash and Grove, 2011). The thalamus of this mutant is larger than the wild type thalamus (Aoto et al., 2002), suggesting that in this brain region Gli3 represses proliferation or survival. In the dorsal midbrain region, Gli3R is essential for restricting growth to a normal level and for correct AP patterning in the mid-hindbrain region (Blaess et al., 2008). The effect of Gli3 on growth is very much context dependent (Yu et al., 2009a) and, for example, the cerebellum is reduced in size in Gli3Xt/Xt mutants (Blaess et al., 2008).
Additionally, there are alterations of nuclear specification: the role of Gli3R in thalamic differentiation affects specific genes in specific nuclei (Table 1). This, together with the often complementary loss of specific markers in the Gli3Xt/Xt and the Gli2zfd/zfd thalamus is consistent with a “GliA/R code” in the thalamus which specifies every nucleus on the basis of GliA/R ratios determined by local Shh signaling levels (Hashimoto-Torii et al., 2003). That the thalamocortical axons do not enter the telencephalon can also be due to specific alteration in telencephalic differentiation in the absence of Gli3 (Magnani et al., 2010).
The alterations in the Gli3Xt/Xt thalamus are likely due to loss of Gli3R rather than loss of Gli3A, since at the high levels of Shh signaling that result in Gli3A formation, Gli2A is also strongly upregulated and could compensate for the loss of Gli3A (Bai et al., 2004). This is also evident in the Ptch1 expression in the Gli3Xt/Xt thalamus, which is not reduced in the mutants. Other studies emphasize the importance of the repressor function of Gli3 – in mouse mutants with reduced production of Gli3R (not Gli3A), cortical defects ensue similar to those found in the Gli3Xt/Xt mutant (Willaredt et al., 2008; Besse et al., 2011; Wilson et al., 2011). Furthermore, Gli3R is required to specify V0 interneurons in the ventral spinal cord (Persson et al., 2002) as well as for fate specification in cortical neurons (Wang et al., 2011) and for cerebellar foliation (Blaess et al., 2008; Kim et al., 2011). The Gli3 repressor function is also essential for limb (Hill et al., 2009), kidney (Cain et al., 2009), and thymus specification (Hager-Theodorides et al., 2009).
The Gli3 Repressor Function is Essential for the Development of the Medial and Intralaminar Nuclei
We have previously shown that the derivatives of the medial pronucleus (the medial and intralaminar nuclei) are specifically lost in Gbx2 mutants (Szabo et al., 2009b). Early Gbx2 expression defines the entire thalamic neuroepithelium (Martinez-de-la-Torre et al., 2002; Hashimoto-Torii et al., 2003), and the whole thalamus is derived from the Gbx2 lineage (Chen et al., 2009). Around midgestation, however, Gbx2 expression becomes restricted to a thalamic region including mostly the neurons of the medial and intralaminar group (Lein et al., 2007). We show that Gbx2 expression is severely reduced in the Gli3Xt/Xt mutant at E12.5. In keeping with this reduction, the medial and intralaminar nuclei appear to be almost absent in the Gli3Xt/Xt mutant thalamus. It would also be possible that the development of the medial and intralaminar nuclei is rescued in the Gli2zfd/zfd mutant by Gli3A. However, since other rostral thalamic nuclei are affected in the Gli2zfd/zfd mutant, and the medial and intralaminar nuclei are not affected in mutants deficient in neuroepithelial Shh (Szabo et al., 2009b), we do not think that a Gli3 activator function is at work here.
These data indicate that thalamic specification depends not only on the Shh pathway but rather on the interplay between this and other signaling systems like the Wnt and Fgf8 pathways (Zhou et al., 2004; Kataoka and Shimogori, 2008; Bluske et al., 2009; Martinez-Ferre and Martinez, 2009; Quinlan et al., 2009), possibly even in cooperation (Rash and Grove, 2011). Since, as we show, expression of transcriptional repressor Hes1, a Notch effector, specifically labels a thalamic nuclear group at E18.5 (see also Lein et al., 2007), the Notch pathway is also a candidate to be involved in thalamic specification and differentiation. Additionally, all or some of these pathways could act through regulating the activity of proneural genes (Scholpp et al., 2009).
Shh vs Gli3 in the Dorsal Diencephalon: A Thalamic Prepattern?
Shh downregulates Gli3R by preventing the processing of full-length Gli3A by the proteasome into a Gli3R form (Marigo et al., 1996; Ruiz i Altaba, 1998; Wang et al., 2000; Kise et al., 2009). In mutants deficient in both Shh signaling and Gli3, ventral patterning defects observed in Shh null mutants are partially rescued in the spinal cord and in the telencephalon (Litingtung and Chiang, 2000; Rallu et al., 2002; Wijgerde et al., 2002), indicating that one important function of Shh is to antagonize the formation of Gli3R. The concept of a “prepattern” has emerged, i.e., a pattern of the neural tube independent of Shh and Gli3 (Ruiz i Altaba et al., 2003).
Deleting Shh in a Gli3Xt/Xt background, however, does not fully rescue the Gli3Xt/Xt phenotype in the telencephalon (Aoto et al., 2002; Persson et al., 2002; Rash and Grove, 2007; Yu et al., 2009b). Moreover, in the midbrain or cerebellum, mutants with inactive Gli3 and inactive Shh signaling have a phenotype that is almost indistinguishable from Gli3 mutants, suggesting that the antagonistic function of Shh and Gli3 is not reciprocal (Blaess et al., 2008). Here we show that in the dorsal diencephalon the removal of Shh from the neuroepithelium in Gli3 mutants does not rescue the Gli3Xt/Xt phenotype, and even results in a much more severe phenotype. If we consider that the double mutant allows us to define for the first time the prepattern of the thalamus, that is, a pattern existing before any influence of Gli3 or neuroepithelial Shh, this prepattern would only include a rough rostro-caudal and dorso-ventral axis specification, overgrowth and partial specification of the medial pronuclear group as well as thalamocortical axonal extension.
The fact that the ventral component of the medial group (expressing Gbx2, Lhx2, and Calb2) seems to be not only present but is abnormally large in the double mutant, suggests that neuroepithelial Shh and Gli3 restrict the expansion of a specific progenitor pool in the medial region of the thalamic neuroepithelium (see below). In general, the increased size of the double mutant suggests that Gli3 and neuroepithelial Shh cooperatively reduce the growth of the thalamic region. This is in agreement with the increase in proliferation in the spinal cord of the Gli2/Gli3 double mutants that has been linked to changes in the cell cycle (Bai et al., 2004).
The electroporation experiments blocking Shh and Gli3 at E12.5 on a wild type background show a reduction in cell number, i.e., the opposite effect as in the mutant. Therefore, blocking all Gli-mediated transcriptional regulation per se does not result in increased growth. This suggests that the factor or factors responsible for the prepattern act would act early, in a defined time-window. Their effect is therefore stage-specific.
Shh from the prechordal plate, which is intact in the n-Shh/Gli3 double mutants (since it is intact in the n-Shh mutants;Szabo et al., 2009a) could be responsible for the patterning of the n-Shh/Gli3 mutant thalamus, as well as the Fgf8, Wnt and maybe Notch pathways (see above).
Conclusion
We have analyzed for the first time the relation between Shh, GliA, and GliR in a dorsal portion of the mouse nervous system, the thalamus. We show that Shh acts upstream of GliA in the specification of pattern and size of most thalamic nuclei (except the medial and intralaminar groups). In a complementary manner, GliR is essential for specification and growth of the medial and intralaminar nuclei (although it also contributes to the specification of other thalamic nuclei and to restrict thalamic size). Additionally, we have found that if Gli2A is abolished, Gli3A can partially substitute for it in the thalamus. Finally, and against what has been established in ventral regions of the neural tube, GliA (from neuroepithelial Shh signaling) and GliR are not reciprocally antagonistic in the thalamus.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Extra-toes mice are a kind gift of Dr. Thomas Theil (University of Edinburgh). Dr. Boris Fehse (University of Hamburg) and Dr. Roger Y. Tsien (University of California, San Diego) contributed reporter plasmids EmGFP and tdTomato. Beate Quenzer helped with the in situ hybridization. The 2H3 antineurofilament antibody, developed by Drs. T. M. Jessell and J. Dodd, was obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the NICHD and maintained by the Department of Biology, The University of Iowa, Iowa City, IA 52242, USA. Roberta Haddad-Tóvolli is the recipient of a fellowship from the Deutscher Akademischer Austausch Dienst (DAAD). This study was supported by a Research Award from the Ministry of Science and Research of North Rhine Westphalia (Programm zur Förderung der Rückkehr des wissenschaftlichen Spitzennachwuchses aus dem Ausland, to Sandra Blaess) and by the Deutsche Forschungsgemeinschaft (AL603/2-1 to Gonzalo Alvarez-Bolado).
Abbreviations
ATN, anterior thalamic nuclei (central pronucleus); CL, centrolateral nucleus (medial pronucleus); CM, centromedial nucleus (medial pronucleus); LG/LGN, lateral geniculate nucleus; LGd/LGNd, lateral geniculate nucleus (dorsal); LP, lateral posterior nucleus (dorsal pronucleus); MD, mediodorsal nucleus (medial pronucleus); MG/MGN, medial geniculate nucleus; MV, medioventral nucleus (reuniens; medial pronucleus); PO, posterior nucleus (dorsal pronucleus); PT, paratenial nucleus (medial pronucleus); PTh, prethalamus; PV, thalamic paraventricular nucleus; STN, subthalamic nucleus; Th, thalamus; TL, telencephalon; VM, ventromedial nucleus (central pronucleus); VP, ventral posterior nucleus (central pronucleus); ZLI, zona limitans interthalamica.
References
Aoto, K., Nishimura, T., Eto, K., and Motoyama, J. (2002). Mouse GLI3 regulates Fgf8 expression and apoptosis in the developing neural tube, face, and limb bud. Dev. Biol. 251, 320–332.
Arai, M., Arai, R., Kani, K., and Jacobowitz, D. M. (1992). Immunohistochemical localization of calretinin in the rat lateral geniculate nucleus and its retino-geniculate projection. Brain Res. 596, 215–222.
Arai, R., Jacobowitz, D. M., and Deura, S. (1994). Distribution of calretinin, calbindin-D28k, and parvalbumin in the rat thalamus. Brain Res. Bull. 33, 595–614.
Bai, C. B., Auerbach, W., Lee, J. S., Stephen, D., and Joyner, A. L. (2002). Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 129, 4753–4761.
Bai, C. B., and Joyner, A. L. (2001). Gli1 can rescue the in vivo function of Gli2. Development 128, 5161–5172.
Bai, C. B., Stephen, D., and Joyner, A. L. (2004). All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev. Cell 6, 103–115.
Bayer, S.A., and Altman, J. (1995). “Neurogenesis and neuronal migration,” in The Rat Nervous System, ed. G. Paxinos (San Diego: Academic Press), 1079–1098.
Besse, L., Neti, M., Anselme, I., Gerhardt, C., Ruther, U., Laclef, C., and Schneider-Maunoury, S. (2011). Primary cilia control telencephalic patterning and morphogenesis via Gli3 proteolytic processing. Development 138, 2079–2088.
Blaess, S., Bodea, G. O., Kabanova, A., Chanet, S., Mugniery, E., Derouiche, A., Stephen, D., and Joyner, A. L. (2011). Temporal–spatial changes in Sonic hedgehog expression and signaling reveal different potentials of ventral mesencephalic progenitors to populate distinct ventral midbrain nuclei. Neural Dev. 6, 29.
Blaess, S., Stephen, D., and Joyner, A. L. (2008). Gli3 coordinates three-dimensional patterning and growth of the tectum and cerebellum by integrating Shh and Fgf8 signaling. Development 135, 2093–2103.
Bluske, K. K., Kawakami, Y., Koyano-Nakagawa, N., and Nakagawa, Y. (2009). Differential activity of Wnt/beta-catenin signaling in the embryonic mouse thalamus. Dev. Dyn. 238, 3297–3309.
Briscoe, J., Chen, Y., Jessell, T. M., and Struhl, G. (2001). A hedgehog-insensitive form of patched provides evidence for direct long-range morphogen activity of sonic hedgehog in the neural tube. Mol. Cell 7, 1279–1291.
Cain, J. E., Islam, E., Haxho, F., Chen, L., Bridgewater, D., Nieuwenhuis, E., Hui, C. C., and Rosenblum, N. D. (2009). GLI3 repressor controls nephron number via regulation of Wnt11 and Ret in ureteric tip cells. PLoS ONE 4, e7313. doi: 10.1371/journal.pone.0007313
Cayuso, J., Ulloa, F., Cox, B., Briscoe, J., and Marti, E. (2006). The Sonic hedgehog pathway independently controls the patterning, proliferation and survival of neuroepithelial cells by regulating Gli activity. Development 133, 517–528.
Chen, L., Guo, Q., and Li, J. Y. (2009). Transcription factor Gbx2 acts cell-nonautonomously to regulate the formation of lineage-restriction boundaries of the thalamus. Development 136, 1317–1326.
Clancy, B., Kersh, B., Hyde, J., Darlington, R. B., Anand, K. J., and Finlay, B. L. (2007). Web-based method for translating neurodevelopment from laboratory species to humans. Neuroinformatics 5, 79–94.
Dai, P., Akimaru, H., Tanaka, Y., Maekawa, T., Nakafuku, M., and Ishii, S. (1999). Sonic hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J. Biol. Chem. 274, 8143–8152.
Dessaud, E., McMahon, A. P., and Briscoe, J. (2008). Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development 135, 2489–2503.
Ding, Q., Motoyama, J., Gasca, S., Mo, R., Sasaki, H., Rossant, J., and Hui, C. C. (1998). Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development 125, 2533–2543.
Ericson, J., Briscoe, J., Rashbass, P., van Heyningen, V., and Jessell, T. M. (1997). Graded sonic hedgehog signaling and the specification of cell fate in the ventral neural tube. Cold Spring Harb. Symp. Quant. Biol. 62, 451–466.
Fotaki, V., Yu, T., Zaki, P. A., Mason, J. O., and Price, D. J. (2006). Abnormal positioning of diencephalic cell types in neocortical tissue in the dorsal telencephalon of mice lacking functional Gli3. J. Neurosci. 26, 9282–9292.
Frassoni, C., Arcelli, P., Selvaggio, M., and Spreafico, R. (1998). Calretinin immunoreactivity in the developing thalamus of the rat: a marker of early generated thalamic cells. Neuroscience 83, 1203–1214.
Genestine, M., Robert, B., and Lallemand, Y. (2007). High-resolution mapping of the Gli3 deletion in the mouse extra-toesH mutant. Genesis 45, 107–112.
Gutin, G., Fernandes, M., Palazzolo, L., Paek, H., Yu, K., Ornitz, D. M., McConnell, S. K., and Hebert, J. M. (2006). FGF signalling generates ventral telencephalic cells independently of SHH. Development 133, 2937–2946.
Hager-Theodorides, A. L., Furmanski, A. L., Ross, S. E., Outram, S. V., Rowbotham, N. J., and Crompton, T. (2009). The Gli3 transcription factor expressed in the thymus stroma controls thymocyte negative selection via hedgehog-dependent and -independent mechanisms. J. Immunol. 183, 3023–3032.
Hashimoto-Torii, K., Motoyama, J., Hui, C. C., Kuroiwa, A., Nakafuku, M., and Shimamura, K. (2003). Differential activities of Sonic hedgehog mediated by Gli transcription factors define distinct neuronal subtypes in the dorsal thalamus. Mech. Dev. 120, 1097–1111.
Hebert, J. M., and Fishell, G. (2008). The genetics of early telencephalon patterning: some assembly required. Nat. Rev. Neurosci. 9, 678–685.
Hevner, R. F., Miyashita-Lin, E., and Rubenstein, J. L. (2002). Cortical and thalamic axon pathfinding defects in Tbr1, Gbx2, and Pax6 mutant mice: evidence that cortical and thalamic axons interact and guide each other. J. Comp. Neurol. 447, 8–17.
Hill, P., Gotz, K., and Ruther, U. (2009). A SHH-independent regulation of Gli3 is a significant determinant of anteroposterior patterning of the limb bud. Dev. Biol. 328, 506–516.
Hui, C. C., and Joyner, A. L. (1993). A mouse model of greig cephalopolysyndactyly syndrome: the extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat. Genet. 3, 241–246.
Hui, C. C., Slusarski, D., Platt, K. A., Holmgren, R., and Joyner, A. L. (1994). Expression of three mouse homologs of the Drosophila segment polarity gene cubitus interruptus, Gli, Gli-2, and Gli-3, in ectoderm- and mesoderm-derived tissues suggests multiple roles during postimplantation development. Dev. Biol. 162, 402–413.
Hynes, M., Ye, W., Wang, K., Stone, D., Murone, M., Sauvage, F., and Rosenthal, A. (2000). The seven-transmembrane receptor smoothened cell-autonomously induces multiple ventral cell types. Nat. Neurosci. 3, 41–46.
Ingham, P. W., and McMahon, A. P. (2001). Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15, 3059–3087.
Ingham, P. W., and Placzek, M. (2006). Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nat. Rev. Genet. 7, 841–850.
Ishibashi, M., and McMahon, A. P. (2002). A sonic hedgehog-dependent signaling relay regulates growth of diencephalic and mesencephalic primordia in the early mouse embryo. Development 129, 4807–4819.
Jacob, J., and Briscoe, J. (2003). Gli proteins and the control of spinal-cord patterning. EMBO Rep. 4, 761–765.
Jeong, Y., Dolson, D. K., Waclaw, R. R., Matise, M. P., Sussel, L., Campbell, K., Kaestner, K. H., and Epstein, D. J. (2011). Spatial and temporal requirements for sonic hedgehog in the regulation of thalamic interneuron identity. Development 138, 531–541.
Jessell, T. M. (2000). Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat. Rev. Genet. 1, 20–29.
Jones, E. G., and Rubenstein, J. L. (2004). Expression of regulatory genes during differentiation of thalamic nuclei in mouse and monkey. J. Comp. Neurol. 477, 55–80.
Kageyama, R., Ohtsuka, T., and Tomita, K. (2000). The bHLH gene Hes1 regulates differentiation of multiple cell types. Mol. Cells 10, 1–7.
Karlstrom, R. O., Tyurina, O. V., Kawakami, A., Nishioka, N., Talbot, W. S., Sasaki, H., and Schier, A. F. (2003). Genetic analysis of zebrafish gli1 and gli2 reveals divergent requirements for gli genes in vertebrate development. Development 130, 1549–1564.
Kataoka, A., and Shimogori, T. (2008). Fgf8 controls regional identity in the developing thalamus. Development 135, 2873–2881.
Kiecker, C., and Lumsden, A. (2004). Hedgehog signaling from the ZLI regulates diencephalic regional identity. Nat. Neurosci. 7, 1242–1249.
Kim, J. J., Gill, P. S., Rotin, L., van Eede, M., Henkelman, R. M., Hui, C. C., and Rosenblum, N. D. (2011). Suppressor of fused controls mid-hindbrain patterning and cerebellar morphogenesis via GLI3 repressor. J. Neurosci. 31, 1825–1836.
Kise, Y., Morinaka, A., Teglund, S., and Miki, H. (2009). Sufu recruits GSK3beta for efficient processing of Gli3. Biochem. Biophys. Res. Commun. 387, 569–574.
Kozak, M. (1987). An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 15, 8125–8148.
Lee, J., Platt, K. A., Censullo, P., and Ruiz i Altaba, A. (1997). Gli1 is a target of Sonic hedgehog that induces ventral neural tube development. Development 124, 2537–2552.
Lein, E. S., Hawrylycz, M. J., Ao, N., Ayres, M., Bensinger, A., Bernard, A., Boe, A. F., Boguski, M. S., Brockway, K. S., Byrnes, E. J., Chen, L., Chen, T. M., Chin, M. C., Chong, J., Crook, B. E., Czaplinska, A., Dang, C. N., Datta, S., Dee, N. R., Desaki, A. L., Desta, T., Diep, E., Dolbeare, T. A., Donelan, M. J., Dong, H. W., Dougherty, J. G., Duncan, B. J., Ebbert, A. J., Eichele, G., Estin, L. K., Faber, C., Facer, B. A., Fields, R., Fischer, S. R., Fliss, T. P., Frensley, C., Gates, S. N., Glattfelder, K. J., Halverson, K. R., Hart, M. R., Hohmann, J. G., Howell, M. P., Jeung, D. P., Johnson, R. A., Karr, P. T., Kawal, R., Kidney, J. M., Knapik, R. H., Kuan, C. L., Lake, J. H., Laramee, A. R., Larsen, K. D., Lau, C., Lemon, T. A., Liang, A. J., Liu, Y., Luong, L. T., Michaels, J., Morgan, J. J., Morgan, R. J., Mortrud, M. T., Mosqueda, N. F., Ng, L. L., Ng, R., Orta, G. J., Overly, C. C., Pak, T. H., Parry, S. E., Pathak, S. D., Pearson, O. C., Puchalski, R. B., Riley, Z. L., Rockett, H. R., Rowland, S. A., Royall, J. J., Ruiz, M. J., Sarno, N. R., Schaffnit, K., Shapovalova, N. V., Sivisay, T., Slaughterbeck, C. R., Smith, S. C., Smith, K. A., Smith, B. I., Sodt, A. J., Stewart, N. N., Stumpf, K. R., Sunkin, S. M., Sutram, M., Tam, A., Teemer, C. D., Thaller, C., Thompson, C. L., Varnam, L. R., Visel, A., Whitlock, R. M., Wohnoutka, P. E., Wolkey, C. K., Wong, V. Y., Wood, M., Yaylaoglu, M. B., Young, R. C., Youngstrom, B. L., Yuan, X. F., Zhang, B., Zwingman, T. A., and Jones, A. R. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176.
Lewis, P. M., Dunn, M. P., McMahon, J. A., Logan, M., Martin, J. F., St-Jacques, B., and McMahon, A. P. (2001). Cholesterol modification of sonic hedgehog is required for long-range signaling activity and effective modulation of signaling by Ptc1. Cell 105, 599–612.
Litingtung, Y., and Chiang, C. (2000). Specification of ventral neuron types is mediated by an antagonistic interaction between Shh and Gli3. Nat. Neurosci. 3, 979–985.
Lopez-Bendito, G., and Molnar, Z. (2003). Thalamocortical development: how are we going to get there? Nat. Rev. Neurosci. 4, 276–289.
Magnani, D., Hasenpusch-Theil, K., Jacobs, E. C., Campagnoni, A. T., Price, D. J., and Theil, T. (2010). The Gli3 hypomorphic mutation Pdn causes selective impairment in the growth, patterning, and axon guidance capability of the lateral ganglionic eminence. J. Neurosci. 30, 13883–13894.
Marigo, V., Johnson, R. L., Vortkamp, A., and Tabin, C. J. (1996). Sonic hedgehog differentially regulates expression of GLI and GLI3 during limb development. Dev. Biol. 180, 273–283.
Martinez-de-la-Torre, M., Garda, A. L., Puelles, E., and Puelles, L. (2002). Gbx2 expression in the late embryonic chick dorsal thalamus. Brain Res. Bull. 57, 435–438.
Martinez-Ferre, A., and Martinez, S. (2009). The development of the thalamic motor learning area is regulated by Fgf8 expression. J. Neurosci. 29, 13389–13400.
Matise, M. P., Epstein, D. J., Park, H. L., Platt, K. A., and Joyner, A. L. (1998). Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development 125, 2759–2770.
Matsui, A., Yoshida, A. C., Kubota, M., Ogawa, M., and Shimogori, T. (2011). Mouse in utero electroporation: controlled spatiotemporal gene transfection. J. Vis. Exp. 54, e3024. doi: 10.3791/3024
Maynard, T. M., Jain, M. D., Balmer, C. W., and LaMantia, A. S. (2002). High-resolution mapping of the Gli3 mutation extra-toes reveals a 51.5-kb deletion. Mamm. Genome 13, 58–61.
Methot, N., and Basler, K. (2001). An absolute requirement for cubitus interruptus in hedgehog signaling. Development 128, 733–742.
Miyashita-Lin, E. M., Hevner, R., Wassarman, K. M., Martinez, S., and Rubenstein, J. L. (1999). Early neocortical regionalization in the absence of thalamic innervation. Science 285, 906–909.
Mo, R., Freer, A. M., Zinyk, D. L., Crackower, M. A., Michaud, J., Heng, H. H., Chik, K. W., Shi, X. M., Tsui, L. C., Cheng, S. H., Joyner, A. L., and Hui, C. (1997). Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development 124, 113–123.
Motoyama, J., Liu, J., Mo, R., Ding, Q., Post, M., and Hui, C. C. (1998). Essential function of Gli2 and Gli3 in the formation of lung, trachea and oesophagus. Nat. Genet. 20, 54–57.
Motoyama, J., Milenkovic, L., Iwama, M., Shikata, Y., Scott, M. P., and Hui, C. C. (2003). Differential requirement for Gli2 and Gli3 in ventral neural cell fate specification. Dev. Biol. 259, 150–161.
Mullor, J. L., Dahmane, N., Sun, T., and Ruiz i Altaba, A. (2001). Wnt signals are targets and mediators of Gli function. Curr. Biol. 11, 769–773.
Nakagawa, Y., and O’Leary, D. D. (2001). Combinatorial expression patterns of LIM-homeodomain and other regulatory genes parcellate developing thalamus. J. Neurosci. 21, 2711–2725.
Nguyen, V., Chokas, A. L., Stecca, B., and Ruiz i Altaba, A. (2005). Cooperative requirement of the Gli proteins in neurogenesis. Development 132, 3267–3279.
Niwa, H., Yamamura, K., and Miyazaki, J. (1991). Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108, 193–199.
Park, H. L., Bai, C., Platt, K. A., Matise, M. P., Beeghly, A., Hui, C. C., Nakashima, M., and Joyner, A. L. (2000). Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development 127, 1593–1605.
Pavletich, N. P., and Pabo, C. O. (1993). Crystal structure of a five-finger GLI-DNA complex: new perspectives on zinc fingers. Science 261, 1701–1707.
Persson, M., Stamataki, D., te Welscher, P., Andersson, E., Bose, J., Ruther, U., Ericson, J., and Briscoe, J. (2002). Dorsal–ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev. 16, 2865–2878.
Puelles, L., Amat, J. A., and Martinez-de-la-Torre, M. (1987). Segment-related, mosaic neurogenetic pattern in the forebrain and mesencephalon of early chick embryos: I. Topography of AChE-positive neuroblasts up to stage HH18. J. Comp. Neurol. 266, 247–268.
Puelles, L., and Rubenstein, J. L. (1993). Expression patterns of homeobox and other putative regulatory genes in the embryonic mouse forebrain suggest a neuromeric organization. Trends Neurosci. 16, 472–479.
Quinlan, R., Graf, M., Mason, I., Lumsden, A., and Kiecker, C. (2009). Complex and dynamic patterns of Wnt pathway gene expression in the developing chick forebrain. Neural Dev. 4, 35.
Quinn, J. C., Molinek, M., Mason, J. O., and Price, D. J. (2009). Gli3 is required autonomously for dorsal telencephalic cells to adopt appropriate fates during embryonic forebrain development. Dev. Biol. 327, 204–215.
Rallu, M., Machold, R., Gaiano, N., Corbin, J. G., McMahon, A. P., and Fishell, G. (2002). Dorsoventral patterning is established in the telencephalon of mutants lacking both Gli3 and hedgehog signaling. Development 129, 4963–4974.
Rash, B. G., and Grove, E. A. (2007). Patterning the dorsal telencephalon: a role for sonic hedgehog? J. Neurosci. 27, 11595–11603.
Rash, B. G., and Grove, E. A. (2011). Shh and Gli3 regulate formation of the telencephalic–diencephalic junction and suppress an isthmus-like signaling source in the forebrain. Dev. Biol. 359, 242–250.
Rose, J. E. (1942). The ontogenetic development of the rabbit’s diencephalon. J. Comp. Neurol. 77, 61–129.
Ruiz i Altaba, A. (1998). Combinatorial Gli gene function in floor plate and neuronal inductions by Sonic hedgehog. Development 125, 2203–2212.
Ruiz i Altaba, A., Mas, C., and Stecca, B. (2007). The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends Cell Biol. 17, 438–447.
Ruiz i Altaba, A., Nguyen, V., and Palma, V. (2003). The emergent design of the neural tube: prepattern, SHH morphogen and GLI code. Curr. Opin. Genet. Dev. 13, 513–521.
Saito, T. (2006). In vivo electroporation in the embryonic mouse central nervous system. Nat. Protoc. 1, 1552–1558.
Saito, T., and Nakatsuji, N. (2001). Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev. Biol. 240, 237–246.
Sasaki, H., Nishizaki, Y., Hui, C., Nakafuku, M., and Kondoh, H. (1999). Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 126, 3915–3924.
Scholpp, S., Delogu, A., Gilthorpe, J., Peukert, D., Schindler, S., and Lumsden, A. (2009). Her6 regulates the neurogenetic gradient and neuronal identity in the thalamus. Proc. Natl. Acad. Sci. U.S.A. 106, 19895–19900.
Scholpp, S., Foucher, I., Staudt, N., Peukert, D., Lumsden, A., and Houart, C. (2007). Otx1l, Otx2 and Irx1b establish and position the ZLI in the diencephalon. Development 134, 3167–3176.
Scholpp, S., and Lumsden, A. (2010). Building a bridal chamber: development of the thalamus. Trends Neurosci. 33, 373–380.
Scholpp, S., Wolf, O., Brand, M., and Lumsden, A. (2006). Hedgehog signalling from the zona limitans intrathalamica orchestrates patterning of the zebrafish diencephalon. Development 133, 855–864.
Shimamura, K., Hartigan, D. J., Martinez, S., Puelles, L., and Rubenstein, J. L. (1995). Longitudinal organization of the anterior neural plate and neural tube. Development 121, 3923–3933.
Shin, S. H., Kogerman, P., Lindstrom, E., Toftgard, R., and Biesecker, L. G. (1999). GLI3 mutations in human disorders mimic Drosophila cubitus interruptus protein functions and localization. Proc. Natl. Acad. Sci. U.S.A. 96, 2880–2884.
Shoji, H., Ito, T., Wakamatsu, Y., Hayasaka, N., Ohsaki, K., Oyanagi, M., Kominami, R., Kondoh, H., and Takahashi, N. (1996). Regionalized expression of the Dbx family homeobox genes in the embryonic CNS of the mouse. Mech. Dev. 56, 25–39.
Stamataki, D., Ulloa, F., Tsoni, S. V., Mynett, A., and Briscoe, J. (2005). A gradient of Gli activity mediates graded Sonic hedgehog signaling in the neural tube. Genes Dev. 19, 626–641.
Stecca, B., and Ruiz, I. A. A. (2010). Context-dependent regulation of the GLI code in cancer by HEDGEHOG and non-HEDGEHOG signals. J. Mol. Cell Biol. 2, 84–95.
Stoykova, A., Fritsch, R., Walther, C., and Gruss, P. (1996). Forebrain patterning defects in Small eye mutant mice. Development 122, 3453–3465.
Stoykova, A., and Gruss, P. (1994). Roles of Pax-genes in developing and adult brain as suggested by expression patterns. J. Neurosci. 14, 1395–1412.
Suzuki-Hirano, A., Ogawa, M., Kataoka, A., Yoshida, A. C., Itoh, D., Ueno, M., Blackshaw, S., and Shimogori, T. (2011). Dynamic spatiotemporal gene expression in embryonic mouse thalamus. J. Comp. Neurol. 519, 528–543.
Szabo, N. E., Zhao, T., Cankaya, M., Theil, T., Zhou, X., and Alvarez-Bolado, G. (2009a). Role of neuroepithelial Sonic hedgehog in hypothalamic patterning. J. Neurosci. 29, 6989–7002.
Szabo, N. E., Zhao, T., Zhou, X., and Alvarez-Bolado, G. (2009b). The role of Sonic hedgehog of neural origin in thalamic differentiation in the mouse. J. Neurosci. 29, 2453–2466.
Theil, T. (2005). Gli3 is required for the specification and differentiation of preplate neurons. Dev. Biol. 286, 559–571.
Theil, T., Alvarez-Bolado, G., Walter, A., and Ruther, U. (1999). Gli3 is required for Emx gene expression during dorsal telencephalon development. Development 126, 3561–3571.
Tyurina, O. V., Guner, B., Popova, E., Feng, J., Schier, A. F., Kohtz, J. D., and Karlstrom, R. O. (2005). Zebrafish Gli3 functions as both an activator and a repressor in hedgehog signaling. Dev. Biol. 277, 537–556.
Vieira, C., Garda, A. L., Shimamura, K., and Martinez, S. (2005). Thalamic development induced by Shh in the chick embryo. Dev. Biol. 284, 351–363.
Vieira, C., and Martinez, S. (2006). Sonic hedgehog from the basal plate and the zona limitans intrathalamica exhibits differential activity on diencephalic molecular regionalization and nuclear structure. Neuroscience 143, 129–140.
Vue, T. Y., Bluske, K., Alishahi, A., Yang, L. L., Koyano-Nakagawa, N., Novitch, B., and Nakagawa, Y. (2009). Sonic hedgehog signaling controls thalamic progenitor identity and nuclei specification in mice. J. Neurosci. 29, 4484–4497.
Wang, B., Fallon, J. F., and Beachy, P. A. (2000). Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100, 423–434.
Wang, H., Ge, G., Uchida, Y., Luu, B., and Ahn, S. (2011). Gli3 is required for maintenance and fate specification of cortical progenitors. J. Neurosci. 31, 6440–6448.
Weber, K., Mock, U., Petrowitz, B., Bartsch, U., and Fehse, B. (2010). Lentiviral gene ontology (LeGO) vectors equipped with novel drug-selectable fluorescent proteins: new building blocks for cell marking and multi-gene analysis. Gene Ther. 17, 511–520.
Wijgerde, M., McMahon, J. A., Rule, M., and McMahon, A. P. (2002). A direct requirement for hedgehog signaling for normal specification of all ventral progenitor domains in the presumptive mammalian spinal cord. Genes Dev. 16, 2849–2864.
Willaredt, M. A., Hasenpusch-Theil, K., Gardner, H. A., Kitanovic, I., Hirschfeld-Warneken, V. C., Gojak, C. P., Gorgas, K., Bradford, C. L., Spatz, J., Wolfl, S., Theil, T., and Tucker, K. L. (2008). A crucial role for primary cilia in cortical morphogenesis. J. Neurosci. 28, 12887–12900.
Wilson, S. L., Wilson, J. P., Wang, C., Wang, B., and McConnell, S. K. (2011). Primary cilia and Gli3 activity regulate cerebral cortical size. Dev. Neurobiol. doi: 10.1002/dneu.20985
Winsky, L., Montpied, P., Arai, R., Martin, B. M., and Jacobowitz, D. M. (1992). Calretinin distribution in the thalamus of the rat: immunohistochemical and in situ hybridization histochemical analyses. Neuroscience 50, 181–196.
Wolf, H. K., Buslei, R., Schmidt-Kastner, R., Schmidt-Kastner, P. K., Pietsch, T., Wiestler, O. D., and Blumcke, I. (1996). NeuN: a useful neuronal marker for diagnostic histopathology. J. Histochem. Cytochem. 44, 1167–1171.
Xie, J., Murone, M., Luoh, S. M., Ryan, A., Gu, Q., Zhang, C., Bonifas, J. M., Lam, C. W., Hynes, M., Goddard, A., Rosenthal, A., Epstein, E. H. J., and de Sauvage, F. J. (1998). Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 391, 90–92.
Yu, T., Fotaki, V., Mason, J. O., and Price, D. J. (2009a). Analysis of early ventral telencephalic defects in mice lacking functional Gli3 protein. J. Comp. Neurol. 512, 613–627.
Yu, W., Wang, Y., McDonnell, K., Stephen, D., and Bai, C. B. (2009b). Patterning of ventral telencephalon requires positive function of Gli transcription factors. Dev. Biol. 334, 264–275.
Yuge, K., Kataoka, A., Yoshida, A. C., Itoh, D., Aggarwal, M., Mori, S., Blackshaw, S., and Shimogori, T. (2011). Region-specific gene expression in early postnatal mouse thalamus. J. Comp. Neurol. 519, 544–561.
Zhao, T., Szabo, N., Ma, J., Luo, L., Zhou, X., and Alvarez-Bolado, G. (2008). Genetic mapping of Foxb1-cell lineage shows migration from caudal diencephalon to telencephalon and lateral hypothalamus. Eur. J. Neurosci. 28, 1941–1955.
Zhao, T., Zhou, X., Szabo, N., Leitges, M., and Alvarez-Bolado, G. (2007). Foxb1-driven Cre expression in somites and the neuroepithelium of diencephalon, brainstem, and spinal cord. Genesis 45, 781–787.
Keywords: differentiation, double mutant, Gbx2, Gli2, Gli3, mouse, Sonic hedgehog, thalamus
Citation: Haddad-Tóvolli R, Heide M, Zhou X, Blaess S and Alvarez-Bolado G (2012) Mouse thalamic differentiation: Gli-dependent pattern and Gli-independent prepattern. Front. Neurosci. 6:27. doi: 10.3389/fnins.2012.00027
Received: 24 November 2011; Accepted: 08 February 2012;
Published online: 22 February 2012.
Edited by:
Steffen Scholpp, Karlsruhe Institute of Technology, GermanyReviewed by:
Tomomi Shimogori, RIKEN Brain Science Institute, JapanDavid J. Price, University of Edinburgh, UK
Copyright: © 2012 Haddad-Tóvolli, Heide, Zhou, Blaess and Alvarez-Bolado. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Gonzalo Alvarez-Bolado, Department of Neuroanatomy, Institute of Anatomy and Cell Biology, University of Heidelberg, Im Neuenheimer Feld 307, 69120 Heidelberg, Germany. e-mail:YWx2YXJlekBhbmEudW5pLWhlaWRlbGJlcmcuZGU=