 Sandeep Kumar1
Sandeep Kumar1 Qinglu Ren1 Jonathon H. Beckley1 Todd K. O’Buckley1 Eduardo D. Gigante2 Jessica L. Santerre2
Qinglu Ren1 Jonathon H. Beckley1 Todd K. O’Buckley1 Eduardo D. Gigante2 Jessica L. Santerre2 David F. Werner1,2
David F. Werner1,2 A. Leslie Morrow1,3,4*
A. Leslie Morrow1,3,4*- 1 Bowles Center for Alcohol Studies, University of North Carolina School of Medicine, Chapel Hill, NC, USA
- 2 Department of Psychology, Center for Development and Behavioral Neuroscience, Binghamton University – State University of New York, Binghamton, NY, USA
- 3 Department of Psychiatry, University of North Carolina School of Medicine, Chapel Hill, NC, USA
- 4 Department of Pharmacology, University of North Carolina School of Medicine, Chapel Hill, NC, USA
Protein kinases are implicated in neuronal cell functions such as modulation of ion channel function, trafficking, and synaptic excitability. Both protein kinase C (PKC) and A (PKA) are involved in regulation of γ-aminobutyric acid type A (GABAA) receptors through phosphorylation. However, the role of PKA in regulating GABAA receptors (GABAA-R) following acute ethanol exposure is not known. The present study investigated the role of PKA in the effects of ethanol on GABAA-R α1 subunit expression in rat cerebral cortical P2 synaptosomal fractions. Additionally, GABA-related behaviors were examined. Rats were administered ethanol (2.0–3.5 g/kg) or saline and PKC, PKA, and GABAA-R α1 subunit levels were measured by western blot analysis. Ethanol (3.5 g/kg) transiently increased GABAA-R α1 subunit expression and PKA RIIβ subunit expression at similar time points whereas PKA RIIα was increased at later time points. In contrast, PKC isoform expression remained unchanged. Notably, lower ethanol doses (2.0 g/kg) had no effect on GABAA-R α1 subunit levels, although PKA type II regulatory subunits RIIα and RIIβ were increased at 10 and 60 min when PKC isozymes are also known to be elevated. To determine if PKA activation was responsible for the ethanol-induced elevation of GABAA-R α1 subunits, the PKA antagonist H89 was administered to rats prior to ethanol exposure. H89 administration prevented ethanol-induced increases in GABAA-R α1 subunit expression. Moreover, increasing PKA activity intracerebroventricularly with Sp-cAMP prior to a hypnotic dose of ethanol increased ethanol-induced loss of righting reflex (LORR) duration. This effect appears to be mediated in part by GABAA-R as increasing PKA activity also increased the duration of muscimol-induced LORR. Overall, these data suggest that PKA mediates ethanol-induced GABAA-R expression and contributes to behavioral effects of ethanol involving GABAA-R.
Introduction
Alcohol (ethanol) exposure results in wide ranging neurobehavioral effects including intoxication, hypnosis, tolerance, and dependence, but its mechanism(s) of action are not fully understood. Nonetheless, much evidence clearly implicates γ-aminobutyric acid type A (GABAA) receptors as having a major role in the action(s) of ethanol (Kumar et al., 2009). GABAA receptors (GABAA-R) are heteropentameric ligand-gated ion channels from a family consisting of 19 different subunits; however, the majority of receptors are composed of 2α, 2β and either a γ or δ subunit (Olsen and Sieghart, 2009). The GABAA-R α1 subunit is the most abundant α subunit in the adult brain as it is a component of about 50% of all GABAA-R and can be found expressed in most major brain regions (Fritschy and Mohler, 1995; Kralic et al., 2002).
The regulation of GABAA-R likely contributes to the responses to ethanol exposure. Multiple studies have shown that chronic ethanol exposure regulates GABAA-R expression and function (see: Kumar et al., 2009). In many cases, GABAA-R α1 subunit expression is decreased following chronic ethanol exposure (e.g., Devaud et al., 1997). More recently, studies have demonstrated that acute ethanol exposure also yields similar effects. For instance, a single high dose ethanol exposure decreased α1 subunit expression (Liang et al., 2007). Additionally, in vitro studies indicate that GABAA-R α1 subunit is decreased in as little as 4 h following ethanol exposure in cultured cortical neurons (Kumar et al., 2010).
Protein kinases have been implicated in regulating GABAA-R α1 subunit homeostasis, most likely through phosphorylation. GABAA-R subunits contain consensus sites for both protein kinase A (PKA) and protein kinase C (PKC; Moss et al., 1992a; Brandon et al., 2000) and much attention has focused on PKC regulation of GABAA-R. Ethanol has been routinely demonstrated to increase PKC activity (Messing et al., 1991), and work by our lab has shown that moderate doses of ethanol (2.0 g/kg) differentially regulate PKCβ, γ, and ε expression and translocation to the P2 synaptosomal fraction in cerebral cortical tissue (Kumar et al., 2006), indicative of increased PKC activity following ethanol exposure. In particular, PKCγ co-localization with α1-containing GABAA-R is quickly increased following an acute ethanol exposure in vitro, and is necessary for internalization of α1 subunits in primary cortical neuronal cultures (Kumar et al., 2010). Moreover, PKCγ is also involved in the up-regulation of the surface expression of α4 subunit-containing GABAA-Rs following a short term ethanol exposure (Werner et al., 2011).
In addition to activating PKC, ethanol is known to increase intracellular cyclic adenosine monophosphate (cAMP) via adenylyl cyclase, thereby activating PKA (Diamond and Gordon, 1997). PKA is a tetramer composed of a homodimer of regulatory subunits and two catalytic subunits. Four regulatory subunits denoted as RIα, RIβ, RIIα, RIIβ and two catalytic subunits Cα and Cβ exist (McKnight, 1991). PKA activity appears to play a prominent role in ethanol-related behavior, as noted by studies using genetically modified animals and PKA modulators. For instance, PKA RIIβ knockout mice have decreased ethanol-induced loss of righting reflex (LORR), but increased ethanol consumption (Thiele et al., 2000). Additionally, mutant Drosophila with hypomorphic PKA RII also have reduced sensitivity to ethanol’s hypnotic effects (Park et al., 2000). Also, inhibiting PKA decreases the sedative–hypnotic and motor ataxic effects of ethanol (Lai et al., 2007).
However, relatively little is known about the role of PKA in regulating GABAA-Rs; although limited studies have hinted that PKA activity can regulate GABAA-R expression (Ives et al., 2002; Brandon et al., 2003). Functionally, the effects of PKA on GABAA-R responses are not straightforward and many attempts at understanding these effects have yielded mixed results. PKA activation may either increase or decrease GABAA-R responses (e.g., Leidenheimer et al., 1991; Kano and Konnerth, 1992). Such discrepancies are thought to be dependent on the brain region, cell type and exposure time of PKA modulators. It is possible that PKA’s effects on GABA-related behaviors may be the net result of these effects. Interestingly, the effects of PKA on GABAA-R-related behavioral responses have not been determined. Variations in GABAA-R functional responses have been suggested to be due to differences in GABAA-R subunit composition (Nusser et al., 1999), and the regulation of GABAA-R by PKA likely contributes to the interpretation of these functional responses. Nonetheless, investigation of GABAA-R regulation by PKA has been limited, and no studies to date have assessed PKA involvement in regulating GABAA-Rs in response to ethanol exposure. Importantly, co-application of a PKA activator and ethanol results in increased GABAA-R potentiation over the effects of application of a PKA activator alone (Freund and Palmer, 1997).
As ethanol alters PKA and PKC activity, and both kinase families regulate GABAA-R expression and function, it is quite likely that ethanol-induced regulation of GABAA-R expression is the net result of both PKA and PKC effects. In the present study, we report that GABAA-R α1 subunit expression is altered following acute ethanol exposure in a dose-dependent manner in vivo. We further examined the contribution of PKA to ethanol-induced changes in GABAA-R α1 subunit expression as well as behavioral responses induced by ethanol and direct activation of GABAA-Rs.
Materials and Methods
Animals
Experiments were conducted in accordance with the National Institute of Health Guidelines under Institutional Animal Care and Use Committee-approved protocols at the University of North Carolina at Chapel Hill and at the State University of New York – Binghamton. Adult male Sprague-Dawley rats (190–220 g, approximate age 10–12 weeks) were purchased from Harlan (Indianapolis, IN, USA) or Taconic (Germantown, NY, USA). Animals from Harlan were used for experiments in Figures 1, 2, 4, and 5. Animals from Taconic were used in experiments in Figures 3 and 6. Rats were group-housed for most experiments, with the exception of animals that underwent intracerebroventricular (i.c.v.) surgery. Rats were maintained on a standard 12 h light–dark schedule with ad libitum access to rat chow and water.
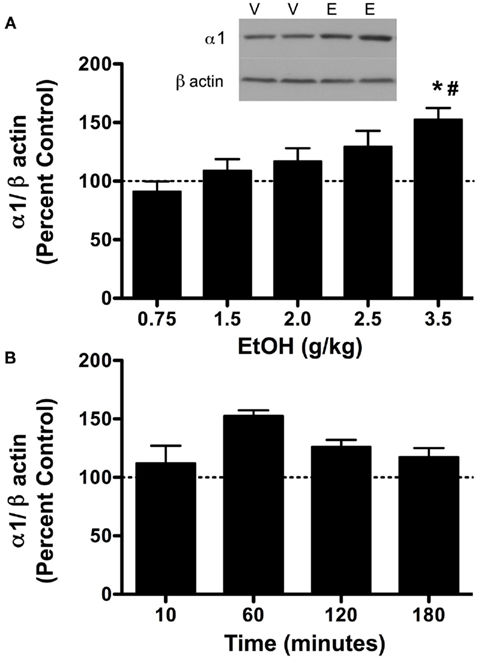
Figure 1. Ethanol (3.5 g/kg) temporally alters GABAA receptor α1 subunit expression. Rats were injected with vehicle (V) or ethanol (EtOH, E) and cerebral cortex was collected at various timepoints. P2 fractions were isolated and analyzed by western blot analysis. (A) GABAA receptor α1 subunit expression was increased by 3.5 g/kg ethanol compared to controls 60 min following ethanol exposure, but not at lower doses. Inset: representative western blot image is shown for 3.5 g/kg ethanol. (B) GABAA receptor α1 subunit expression following 3.5 g/kg ethanol administration. Data represent mean ± SEM. *p < 0.05, compared to controls; #p < 0.01, compared to 0.75 g/kg EtOH. Two-way ANOVA, with Bonferroni post-test, n = 3–6 per group, in duplicate. For clarity, matched control groups are shown by a dotted black line at 100%.
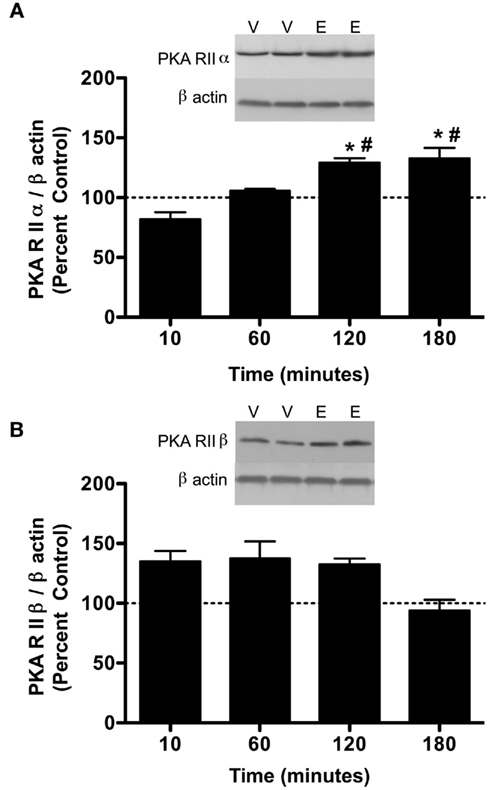
Figure 2. Ethanol (3.5 g/kg) temporally alters PKA subunit expression in the P2 fraction of cerebral cortex. Rats were injected with vehicle (V) or ethanol (E) and cerebral cortex was collected after 10, 60, 120, and 180 min. P2 fraction was analyzed by western blot analysis. (A) PKA RIIα expression was increased at 120 and 180 min following 3.5 g/kg ethanol exposure. (B) PKA RIIβ expression following 3.5 g/kg ethanol exposure. Representative western blot images are shown at 60 min following ethanol exposure. Data are compared to matched controls for each time point. Data represent mean ± SEM. *p < 0.05, compared to controls; #p < 0.01, compared to 10 min. Two-way ANOVA, with Bonferroni post-test, n = 3–4 per group, in duplicate. For clarity, matched control groups are shown by a dotted black line at 100%.
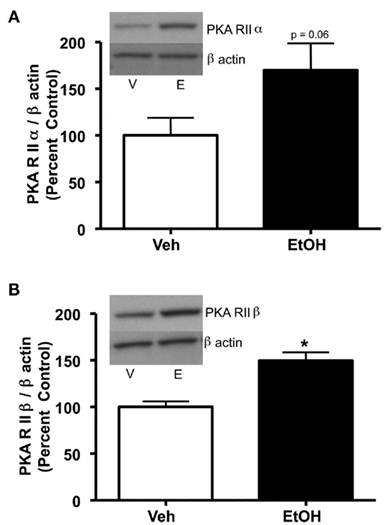
Figure 3. PKA subunit expression is increased in cerebral cortical P2 fractions 24 h after a single 3.5 g/kg ethanol exposure. Rats were injected with vehicle (Veh, V) or ethanol (EtOH, E) and cerebral cortex was collected and analyzed by P2 fraction and western blot analysis. (A) A trend toward increased PKA RIIα was observed following ethanol exposure. (B) PKA RIIβ was increased following ethanol exposure. Data are presented as mean ± SEM. Representative western blots are shown in insets for both. *p < 0.05, Student’s t-test, n = 8 per group.
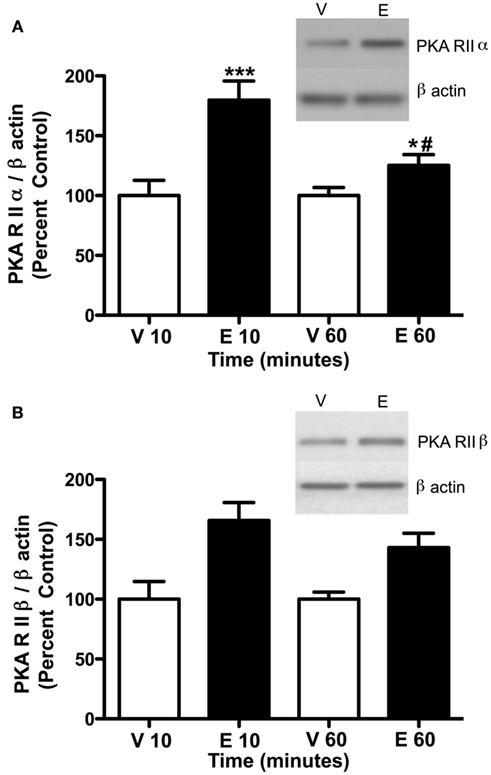
Figure 4. Ethanol (2.0 g/kg) alters PKA subunit expression in the P2 fraction of cerebral cortex. Rats were injected with vehicle (Veh, V) or ethanol (EtOH, E) and cerebral cortex was collected after 10 and 60 min. P2 fraction was analyzed by western blot analysis. (A) PKA RIIα expression was increased at 10 and 60 min following 2.0 g/kg ethanol exposure. (B) PKA RIIβ expression was increased at 10 and 60 min following ethanol exposure. Representative western blot images are shown at 10 min. Data represent mean ± SEM. *p < 0.05, ***p < 0.001, compared to controls; #p < 0.001, compared to 10 min. Two-way ANOVA, with Bonferroni post-test, n = 4–8 per group.
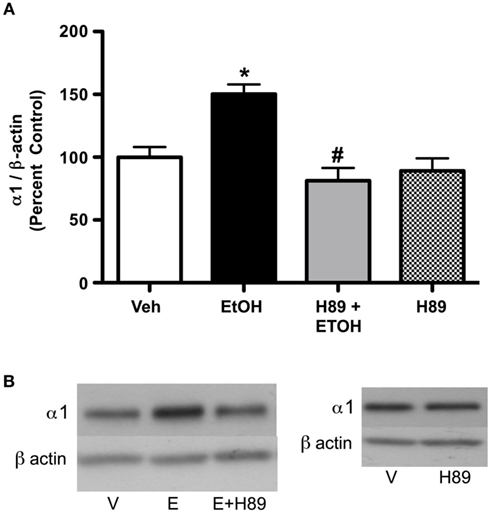
Figure 5. PKA inhibition blocks ethanol-induced (3.5 g/kg) increases in GABAA receptor α1 subunit expression. Rats were injected with H89 (10 mg/kg, s.c.) or vehicle 30 min before ethanol exposure. (A) H89 reversed increases in ethanol-induced GABAA receptor α1 subunit expression. (B) Representative western blots are shown. Data are presented as mean ± SEM. *p < 0.05, compared to vehicle; #p < 0.05, compared to ethanol. Two-way ANOVA, with Bonferroni post-test, n = 4–8 per group.
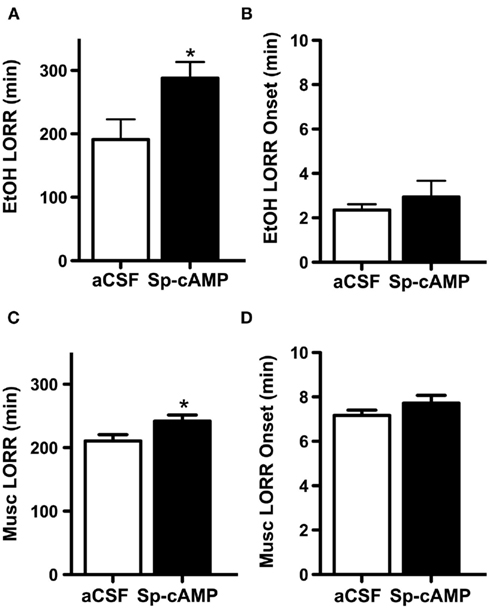
Figure 6. Increasing PKA activity prior to ethanol or muscimol administration increases loss of righting reflex (LORR) duration. Male adult rats were infused i.c.v. with either artificial cerebrospinal fluid (aCSF) or the PKA activator (Sp-cAMP 100 nmol) 15 min prior to an i.p. injection of a hypnotic dose of or ethanol (EtOH, 3.5 g/kg) or muscimol (Musc, 5.0 mg/kg). Times that subjects lost and regained the righting reflex were recorded. (A) Sp-cAMP increased the ethanol-induced LORR duration compared to control rats that received aCSF. (B) aCSF and Sp-cAMP pretreatment did not differ in LORR onset following ethanol exposure. (C) Sp-cAMP increased the muscimol-induced LORR duration. (D) aCSF and Sp-cAMP pretreatment did not differ in LORR onset following muscimol exposure. *p < 0.05 compared to aCSF, Students t-test, n = 8–14 per group.
Drug Exposure
Rats were injected with ethanol (20% v/v in saline, intraperitoneally (i.p.); Pharmco, Brookfield, CT, USA) or saline, and sacrificed after intervals between 10 min and 24 h. Ethanol doses ranged from 0.75 to 3.5 g/kg. This method of ethanol administration was chosen to produce consistent blood ethanol concentrations. H89 (10 mg/kg, subcutaneously (s.c.); Sigma-Aldrich, St. Louis, MO, USA), a PKA inhibitor, was injected 30 min prior to saline or ethanol injection. Sp-adenosine 3′,5′-cyclic monophosphorothioate triethylammonium salt (Sp-cAMP; 100 nmol/rat, i.c.v.; Sigma-Aldrich, St. Louis, MO, USA), a PKA activator, was administered 15 min prior to ethanol or muscimol (5 mg/kg, i.p.; Sigma-Aldrich, St. Louis, MO, USA).
Tissue and Protein Preparations
Rats were immediately sacrificed following ethanol exposure at predetermined time points. The brain was rapidly removed from the skull and the cerebral cortex was dissected out. Tissue was flash frozen and stored at −80°C, until further use. P2 synaptosomal fractions from individual cerebral cortices were prepared by homogenization, low speed centrifugation in 0.32 M sucrose, and centrifugation of the supernatant at 12,000 × g for 20 min. The pellet (P2 fraction) was resuspended in phosphate buffered saline (PBS) with phosphatase inhibitor cocktail (1:100 dilution, proprietary mixture of microcystin LR, cantharidin, and bromotetramisole, Sigma-Aldrich, St. Louis, MO, USA) and stored at −80°C. Protein concentrations were quantified using a bicinchoninic acid method.
Western Blot Analysis
P2 synaptosomal fractions were subjected to sodium-dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using Novex Tris–Glycine gels (8–16%) and transferred to polyvinylidene difluoride (PVDF) membranes (Invitrogen, Carlsbad, CA, USA). Membranes were probed with antibodies for the following proteins: GABAA-R α1 subunit (Novus, Lake Placid, NY, USA); PKCβ, PKCγ, PKCδ, and PKCε (BD Biosciences, San Jose, CA, USA); and PKA RIIα and RIIβ (BD Biosciences). Blots were subsequently exposed to a second primary antibody directed against β-actin to verify equivalent protein loading and transfer. Bands were detected by enhanced chemiluminescence (GE Healthcare, Piscataway, NJ, USA), exposed to X-ray films under non-saturating conditions, and analyzed by densitometric measurements using NIH Image 1.57.
Intracerebroventricular Surgery
Stereotactic surgery was performed to implant guide cannula directed toward the lateral ventricles. Briefly, rats were anesthetized with 3.0% isoflurane and subsequently restrained in a stereotactic frame. Guide cannula (Plastics One, Roanoke, VA, USA) were implanted unilaterally into the lateral cerebral ventricle at coordinates AP -0.8 mm, L+ or -1.2 mm from bregma, and DV -2.5 mm (Paxinos and Watson, 2007). The cannula was secured to the skull using three stainless steel screws and dental cement and the cannula patency was maintained and protected with an internal guide and cap. The skin surrounding the surgical site was sutured to prevent infection. Animals were allowed a 1-week recovery period prior to behavioral testing. Rats were sacrificed 24 h following the completion of testing. Brains were rapidly removed and stored at −80°C until further use. India ink was used to determine i.c.v. cannula placements. Only animals with a positive indication of ink in their ventricles (43/44) were used for subsequent analysis.
Loss of Righting Reflex
To determine the effect of PKA activators on ethanol- or muscimol-induced hypnotic responses, rats were randomly selected to receive either Sp-cAMP (100 nmol/rat) or artificial cerebrospinal fluid (aCSF) 15 min prior to ethanol or muscimol administration. Two microliters of Sp-cAMP or aCSF was infused i.c.v. at a flow rate of 1 μL/min. Injector needles were left in place for an additional minute to ensure proper distribution and prevent backflow of the drug into the cannula. Rats were subsequently administered a hypnotic dose of ethanol (3.5 g/kg, i.p.) or muscimol (5 mg/kg, i.p.) 15 min later. Rats were observed and repeatedly tested until they were unable to right themselves from a supine position. The length of time from ethanol administration until onset of LORR was recorded (LORR Onset). Animals remained in a supine position in v-shaped troughs (90° angle, 12.5 cm × 25.5 cm) until they regained their righting reflex. An animal was deemed to have regained their righting reflex if they were able to right themselves three times in a 60-s period. The duration of LORR was calculated by subtracting the time of onset of LORR from the time at which the animal regained the righting reflex. Rats that did not lose the righting reflex were excluded from the study (n = 9).
Statistical Analysis
For western blots, all comparisons were made within blots. For ethanol dose and time dependent studies, each group was compared to saline controls run in parallel. Analyses were conducted using Student’s t-test, one-way ANOVA with Newman–Keuls post hoc test or two-way ANOVA with Bonferroni post hoc test. For data in Figures 1, 2, and 4, each respective dose or time point had a matched saline control group. Therefore, two-way ANOVAs were used for analysis. LORR data were assessed using Student’s t-test. For all experiments, p < 0.05 was considered significant.
Results
Effects of Acute Ethanol Exposure on GABAA Receptor α1 Subunit Expression
To assess the effects of ethanol exposure on GABAA-R α1 subunit expression, we examined the effects of multiple doses over 1 h of ethanol exposure. Ethanol caused a dose-dependent increase in GABAA-R α1 subunit expression 1 h following ethanol exposure in P2 fractions (Figure 1A). Two-way ANOVA indicated an overall main effect of ethanol dose [F4,33 = 2.71, p < 0.05], treatment [saline vs. ethanol, F1,33 = 9.49, p < 0.01], and an interaction of the two [F4,33 = 2.71, p < 0.05]. Notably, 3.5 g/kg ethanol increased GABAA-R α1 subunit expression (52.6 ± 19%, p < 0.05, compared to controls), but lower ethanol doses were ineffective. To assess the temporal effects of ethanol administration, 3.5 g/kg was administered and rats were sacrificed at various time points. An overall main effect was only observed for treatment [saline vs. ethanol, F1,23 = 9.46, p < 0.01], but not time or an interaction of time × treatment. This suggests that 3.5 g/kg ethanol increased GABAA-R α1 subunit expression irrespective of time (Figure 1B).
Effects of Ethanol Exposure on Protein Kinase Expression
Previous studies have indicated that PKC isoform expression is regulated following 2.0 g/kg ethanol exposure (Kumar et al., 2006) and that PKC plays a role in regulating GABAA-R α1 subunit expression (Kumar et al., 2006, 2010). Therefore, we investigated whether PKC isoform expression was altered following 3.5 g/kg ethanol exposure. With the exception of PKCδ [main effect of treatment, F1,24 = 8.39, p < 0.05], ethanol exposure failed to modulate most PKC isoform expression (Table 1). Because PKC and PKA are known to regulate GABAA-R expression and function, we investigated whether PKA regulatory subunit expression was altered following ethanol exposure. Notably, ethanol exposure (3.5 g/kg) resulted in increases in both PKA RIIα and RIIβ in P2 membranes. For RIIα, overall main effects of time [F3,24 = 5.41, p < 0.01], treatment [saline vs. ethanol, F1,24 = 5.99, p < 0.05], and their interaction [F1,24 = 5.42, 0 < 0.05] were observed. Further analysis revealed that PKA RIIα was increased 120 and 180 min following ethanol exposure (Figure 2A). For RIIβ, an overall effect of treatment [saline vs. ethanol, F1,24 = 15.56, p < 0.001], but no effect of time or interaction was observed, thereby suggesting that 3.5 g/kg ethanol exposure alone increases PKA RIIβ expression irrespective of time. Interestingly, PKA RIIβ expression was elevated 24 h following ethanol exposure while a trend toward increased expression was observed for RIIα (Figures 3A,B). Because of the dose-dependent effects of ethanol on PKC, we determined if changes in PKA expression were also dose-dependent. One hour following a 2.0-g/kg ethanol exposure, an overall main effect of treatment [saline vs. ethanol, F1,20 = 35.65, p < 0.0001], time [F1,20 = 9.65, p < 0.01] and their interaction [F1,20 = 9.65, p < 0.01] were observed for PKA RIIα. Post-test revealed that ethanol increased PKA RIIα at both time points, but the effect of ethanol was lower at 60 min compared to 10 min of ethanol exposure (Figure 4A). For RIIβ, only a main effect of treatment [saline vs. ethanol, F1,20 = 26.97, p < 0.0001] was observed after 2.0 g/kg ethanol, but no main effect of time or interaction of time × treatment. This suggests that 2.0 g/kg ethanol increased PKA RIIβ subunit expression irrespective of time (Figure 4B).
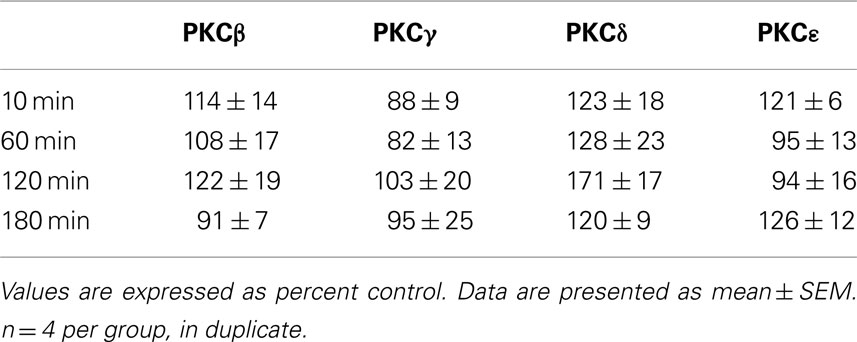
Table 1. PKC isoform expression following 3.5 g/kg ethanol exposure.
Effects of PKA Activity on GABAA Receptor α1 Subunit Expression
Since PKA RIIα and RIIβ expression were altered following 3.5 g/kg ethanol exposure, we investigated whether PKA activation was required for the effect of ethanol on GABAA α1 subunits. Administration of the PKA inhibitor H89 prior to ethanol abolished ethanol-induced increases in GABAA-R α1 subunit expression (Figures 5A,B). Initial analysis indicated a main effect of treatment [saline vs. ethanol, F2,22 = 5.02, p < 0.05], H89 [F1,22 = 17.42, p < 0.001], and an interaction of H89 × treatment [F1,22 = 9.34, p < 0.01]. Further analysis indicated that H89 reversed ethanol’s effects on GABAA-R α1 subunit expression, but H89 alone did not have any effect. In addition, H89 administration alone did not alter PKA RIIβ expression; but a suggestive trend toward a decrease was noted (14.81 ± 7.8%, p = 0.087, not shown).
Effects of PKA Activity on Ethanol- and Muscimol-Induced Hypnotic Responses
Lastly, because PKA activity regulates GABAA-R α1 subunit expression at hypnotic doses, we determined if PKA activity would alter ethanol-induced behavioral responses. We found that rats administered the PKA activator Sp-cAMP prior to ethanol exposure had ∼50% longer duration of LORR than control rats given aCSF (288.1 ± 25.3 and 191.2 ± 31.8 min, respectively, p < 0.05, Figure 6A). No differences were detected in LORR onset (Figure 6B). To further determine if ethanol’s effects were related to PKA activity on GABAA-R, we investigated the effects of PKA activity on muscimol-induced LORR. Sp-cAMP increased the LORR duration by ∼15% compared to aCSF-treated animals (242.1 ± 9.4 and 210.7 ± 9.9 min, respectively, p < 0.05, Figure 6C). Again, no differences were noted in LORR onset (Figure 6D). Overall, these data suggest that PKA activity contributes to ethanol-induced regulation of GABAA-R α1 subunits and that PKA’s effects on GABAA-R contribute to the behavioral effects of ethanol.
Discussion
GABAA receptors, particularly those containing α1 subunits, have been heavily implicated in alcohol’s actions and alcohol dependence In the current study, we investigated in vivo cerebral cortical GABAA-R α1 subunit expression following acute ethanol exposure and found that high doses of ethanol (3.5 g/kg) increased GABAA-R α1 subunit expression in the P2 membrane fraction, but lower doses were ineffective. The effect appears to be mediated by PKA. PKA RIIα and RIIβ subunit expression were increased following ethanol exposure, and prior administration of a PKA inhibitor prevented ethanol-induced increases in GABAA-R α1 subunit expression. Importantly, activating PKA enhanced the duration of the ethanol-induced LORR. The LORR behavioral effect appears to be mediated, in part, by GABAA-Rs as Sp-cAMP also increased muscimol-induced LORR.
The increase in GABAA-R α1 subunit expression in vivo appears to contradict previous studies that indicate α1 subunits are decreased following ethanol exposure (reviewed in Kumar et al., 2009). However, we show here that the effect of ethanol is dependent on dose and time, and is likely due to underlying effects on PKA and PKC. Our previous results indicate that PKCγ plays a major role in decreasing GABAA-R α1 subunit expression (Kumar et al., 2010). However, PKCγ expression was not altered in the cerebral cortex following this ethanol dose. Previous results in cultured murine cerebellar granule cells indicate that activation of PKA results in a post-transcriptional increase in GABAA-R α1 subunit surface expression (Ives et al., 2002). Therefore, it is quite likely that the transient increases in GABAA-R α1 subunit expression are likely due to PKA activity. The mechanism by which PKA increases GABAA-R α1 subunit expression in P2 synaptosomes is unknown. However, the most likely explanation involves trafficking of α1-containing GABAA-R from intracellular stores or extrasynaptic sites (Thomas et al., 2005; Bogdanov et al., 2006). Transcription/translation-related processes are less likely as longer ethanol exposures result in eventual decreases in α1 (Devaud et al., 1997; Cagetti et al., 2003).
The data presented here coupled with our previous studies suggest that PKC and PKA may have antagonistic effects in regulating GABAA-R α1 subunit expression. Indeed, both PKA (presented here) and PKC (Kumar et al., 2006) are increased in P2 synaptosomal fractions at 2.0 g/kg ethanol where GABAA-R α1 subunit expression is unaltered. Further experiments will need to be conducted to further determine this antagonistic role of PKA and PKC on GABAA-R subunit expression. Other studies have suggested that the phosphorylation state of GABAA-R is dependent on both PKA and PKC activity. For instance, Brandon et al. (2000) indicated that PKA activators could only phosphorylate GABAA-R β3 subunits in the presence of PKC inhibitors. It should be noted that antagonistic effects of PKA and PKC have been proposed for other receptor systems. Vaello et al. (1994) have reported that glycine receptors can be phosphorylated in vivo in response to activation of either PKC or PKA with opposite functional consequences. And, more recently, PKC and PKA have been shown to have differential involvement in ghrelin-induced growth hormone and gonadotrophin release (Grey and Chang, 2011). Therefore, our studies lend further support to broader implications that PKC and PKA possibly have opposing roles in the central nervous system. Clearly, more studies need to be conducted to further evaluate this generalization. The role of phosphatase activity also cannot be excluded, but we have previously shown that ethanol exposure does not alter PP1 phosphatase expression in vivo (Kumar et al., 2006) or in vitro (Kumar et al., 2010).
It is unknown if the expression of other GABAA-R subunits is altered in response to fluctuations in PKA activity. However, Ives et al. (2002) reported that β2 and β3 subunit surface expression was increased and decreased, respectively, while α6 subunits and Ro15-4513 (a GABAA-R inverse agonist) binding were unchanged. Given that α1-containing GABAA-Rs are increased following ethanol treatment, it is likely that other GABAA-R subtypes are also affected. This possibility should be investigated further.
Behaviorally, rats that were pretreated with the PKA activator Sp-cAMP exhibited increased sensitivity to ethanol’s sedative–hypnotic effects. This effect is in agreement with other studies. For instance, rats pretreated with a PKA inhibitor display reduced sensitivity to ethanol’s motor ataxic and sedative–hypnotic effects (Lai et al., 2007) as well as ethanol withdrawal-related anxiety (Pandey et al., 2003). Multiple knockout studies also indicate that decreases in the cAMP signaling pathway reduce sensitivity to ethanol. Aside from PKA RIIβ knockout mice (Thiele et al., 2000), ethanol behavioral responses are also reduced in knockouts of adenosine A2a (El Yacoubi et al., 2001; Naassila et al., 2002) and adenylyl cyclase 5 (Kim et al., 2010). However, it should be noted that not all studies are in agreement with this effect (Wand et al., 2001; Yang et al., 2003; Maas et al., 2005). Nonetheless, it is clear that PKA activity contributes to ethanol-induced behavior.
We further investigated whether PKA’s ethanol-related effects were mediated through GABAA-R, and observed that Sp-cAMP also increased muscimol-induced LORR. However, the magnitude of increase in muscimol-induced LORR by Sp-cAMP was less than Sp-cAMP’s effects on ethanol-induced LORR. It is likely that the ethanol-related effects represent a combination of all GABAA-R subtypes and distribution, presynaptic GABA release, and PKA effects at other ion channels. GABAA-R may only partly mediate ethanol hypnosis. Indeed, PKA is known to phosphorylate and regulate NMDA receptors (Ferrani-Kile et al., 2003; Lau et al., 2004). A second possibility for the difference in magnitude may be the overall net effect of GABAA-R inhibition. Multiple electrophysiological studies have noted that PKA activity has differential effects on GABAA-R function. For instance, increasing PKA activity has increased GABAA-R responses in Purkinje neurons (Kano and Konnerth, 1992; Freund and Palmer, 1997), cerebellar interneurons (Nusser et al., 1999), hippocampal dentate granule cells (Kapur and Macdonald, 1996), CA1 neurons (Shew et al., 2000), hypoglossal motoneurons (Saywell and Feldman, 2004) as well as recombinant receptors (Angelotti et al., 1993). Conversely, PKA has been shown to reduce GABAA-R function in microsacs (Leidenheimer et al., 1991), cortical neurons (Tehrani et al., 1989), dorsal root ganglion neurons (White et al., 1992), recombinant receptors as well as primary neuronal cultures (Moss et al., 1992b), cerebellar granule cells (Robello et al., 1993), and hippocampal CA1 neurons (Poisbeau et al., 1999). It should be pointed out that the latter group observed a decrease in mIPSC amplitude, but not an increase in decay; therefore it is possible that even if there is a reduction in receptor numbers, the receptors that are remaining are sensitive to PKA’s potentiating effects. It is also quite possible that PKA’s effects on GABAA-R inhibition depends on GABAA-R subunit composition. Recently Tang et al. (2010) demonstrated that increasing PKA activity increased spontaneous tonic currents. Importantly, it was discovered that this effect was dependent on the concentration of GABA. Nonetheless, further studies need to be conducted to determine PKA activity on specific GABAA-R subtypes. Lastly, the difference in ethanol and muscimol-stimulated effects may be related to ethanol or PKA effects on presynaptic GABA release. Ethanol is well documented to increase GABA release (e.g., Criswell et al., 2008), but recent work has indicated that blocking adenylyl cyclase or PKA activity prevents ethanol from increasing GABA release (Kelm et al., 2008).
While PKA RIIα and RIIβ are up-regulated in response to ethanol exposure, it remains unknown which of these subunits are necessary for GABAA-R regulation and ethanol-related behavior. Given that PKA RIIα and RIIβ are up-regulated following multiple ethanol doses, it is possible that both subunits may contribute to ethanol-related behavior. RIIα and RIIβ were investigated as they may be localized at cell membranes whereas RIα and RIβ tend to be found in the cytoplasm (discussed in Dohrman et al., 1996). Because we observed that only PKA RIIβ was increased at the same time we observed increases in GABAA-R α1 subunit expression, it is quite possible that PKA RIIβ might be mediating this effect. However, additional studies, potentially using PKA RIIβ knockout mice, may be necessary to further define this effect. Alternatively, it may also be important to further determine whether A kinase associated proteins (AKAPs), such as AKAP150, that co-localize with PKA RIIα and RIIβ are responsible for placing specific PKA (Glantz et al., 1992).
Interestingly, both PKA RIIα and RIIβ displayed changes in expression that were dependent upon ethanol dose. At moderate doses, both PKA RIIα and RIIβ were up-regulated. However, at higher ethanol doses, PKA RIIβ exhibited increased expression immediately following ethanol exposure whereas PKA RIIα had a latent increase in expression in P2 synaptosomes. However, both were up-regulated 24 h following high dose ethanol exposure. Although the reasons for the initial and latent increases in PKA RII expression are not completely understood, it is possible that both may be independent effects in response to ethanol exposure. Previous work has suggested that ethanol results in translocation of PKA in two separate phases. Ethanol has been shown to alter translocation at time points earlier than 30 min that returned to baseline after 60 min in vitro (Dohrman et al., 2002). However, a second translocation phase was noted after 12 h that did not require adenosine A2 receptor subtypes or cAMP, and is thought to be due to transcriptional and/or translational processes. It remains unknown whether increases in PKA RII in our results are simply due to activity following ethanol exposure or due to increased production of PKA through transcription and translation-related effects.
In summary, we demonstrate that protein kinases are dose dependently altered in response to ethanol exposure and exhibit temporal differences in expression. Importantly, the pattern of PKA expression likely contributes to the differential regulation of GABAA-R subtypes as well as ethanol and GABAA-R-mediated behavior.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Institute on Alcohol Abuse and Alcoholism, grants 015409 (Sandeep Kumar), 011605 (A. Leslie Morrow), 019367 (LPS – faculty recruitment of David F. Werner).
Abbreviations
aCSF, artificial cerebrospinal fluid; GABA, γ-aminobutyric acid; PKA protein kinase A; PKC, protein kinase C.
References
Angelotti, T. P., Uhler, M. D., and Macdonald, R. L. (1993). Enhancement of recombinant gamma-aminobutyric acid type A receptor currents by chronic activation of cAMP-dependent protein kinase. Mol. Pharmacol. 44, 1202–1210.
Bogdanov, Y., Michels, G., Armstrong-Gold, C., Haydon, P. G., Lindstrom, J., Pangalos, M., and Moss, S. J. (2006). Synaptic GABAA receptors are directly recruited from their extrasynaptic counterparts. EMBO J. 25, 4381–4389.
Brandon, N. J., Delmas, P., Kittler, J. T., McDonald, B. J., Sieghart, W., Brown, D. A., Smart, T. G., and Moss, S. J. (2000). GABAA receptor phosphorylation and functional modulation in cortical neurons by a protein kinase C-dependent pathway. J. Biol. Chem. 275, 38856–38862.
Brandon, N. J., Jovanovic, J. N., Colledge, M., Kittler, J. T., Brandon, J. M., Scott, J. D., and Moss, S. J. (2003). A-kinase anchoring protein 79/150 facilitates the phosphorylation of GABA(A) receptors by cAMP-dependent protein kinase via selective interaction with receptor beta subunits. Mol. Cell. Neurosci. 22, 87–97.
Cagetti, E., Liang, J., Spigelman, I., and Olsen, R. W. (2003). Withdrawal from chronic intermittent ethanol treatment changes subunit composition, reduces synaptic function, and decreases behavioral responses to positive allosteric modulators of GABAA receptors. Mol. Pharmacol. 63, 53–64.
Criswell, H. E., Ming, Z., Kelm, M. K., and Breese, G. R. (2008). Brain regional differences in the effect of ethanol on GABA release from presynaptic terminals. J. Pharmacol. Exp. Ther. 326, 596–603.
Devaud, L. L., Fritschy, J. M., Sieghart, W., and Morrow, A. L. (1997). Bidirectional alterations of GABA(A) receptor subunit peptide levels in rat cortex during chronic ethanol consumption and withdrawal. J. Neurochem. 69, 126–130.
Diamond, I., and Gordon, A. S. (1997). Cellular and molecular neuroscience of alcoholism. Physiol. Rev. 77, 1–20.
Dohrman, D. P., Chen, H. M., Gordon, A. S., and Diamond, I. (2002). Ethanol-induced translocation of protein kinase A occurs in two phases: control by different molecular mechanisms. Alcohol. Clin. Exp. Res. 26, 407–415.
Dohrman, D. P., Diamond, I., and Gordon, A. S. (1996). Ethanol causes translocation of cAMP-dependent protein kinase catalytic subunit to the nucleus. Proc. Natl. Acad. Sci. U.S.A. 93, 10217–10221.
El Yacoubi, M., Ledent, C., Parmentier, M., Daoust, M., Costentin, J., and Vaugeois, J. (2001). Absence of the adenosine A(2A) receptor or its chronic blockade decrease ethanol withdrawal-induced seizures in mice. Neuropharmacology 40, 424–432.
Ferrani-Kile, K., Randall, P. K., and Leslie, S. W. (2003). Acute ethanol affects phosphorylation state of the NMDA receptor complex: implication of tyrosine phosphatases and protein kinase A. Brain Res. Mol. Brain Res. 115, 78–86.
Freund, R. K., and Palmer, M. R. (1997). Beta adrenergic sensitization of gamma-aminobutyric acid receptors to ethanol involves a cyclic AMP/protein kinase A second-messenger mechanism. J. Pharmacol. Exp. Ther. 280, 1192–1200.
Fritschy, J. M., and Mohler, H. (1995). GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J. Comp. Neurol. 359, 154–194.
Glantz, S. B., Amat, J. A., and Rubin, C. S. (1992). cAMP signaling in neurons: patterns of neuronal expression and intracellular localization for a novel protein, AKAP 150, that anchors the regulatory subunit of cAMP-dependent protein kinase II beta. Mol. Biol. Cell 3, 1215–1228.
Grey, C. L., and Chang, J. P. (2011). Differential Involvement of PKC and PKA in ghrelin-induced growth hormone and gonadotrophin release from goldfish (Carassius auratus) pituitary cells. J. Neuroendocrinol. 12, 1273–1287.
Ives, J. H., Drewery, D. L., and Thompson, C. L. (2002). Differential cell surface expression of GABAA receptor alpha1, alpha6, beta2 and beta3 subunits in cultured mouse cerebellar granule cells influence of cAMP-activated signalling. J. Neurochem. 80, 317–327.
Kano, M., and Konnerth, A. (1992). Potentiation of GABA-mediated currents by cAMP-dependent protein kinase. Neuroreport 3, 563–566.
Kapur, J., and Macdonald, R. L. (1996). Cyclic AMP-dependent protein kinase enhances hippocampal dentate granule cell GABAA receptor currents. J. Neurophysiol. 76, 2626–2634.
Kelm, M. K., Criswell, H. E., and Breese, G. R. (2008). The role of protein kinase A in the ethanol-induced increase in spontaneous GABA release onto cerebellar Purkinje neurons. J. Neurophysiol. 100, 3417–3428.
Kim, K. S., Kim, H., Baek, I. S., Lee, K. W., and Han, P. L. (2010). Mice lacking adenylyl cyclase type 5 (AC5) show increased ethanol consumption and reduced ethanol sensitivity. Psychopharmacology (Berl.) 215, 391–398.
Kralic, J. E., O’Buckley, T. K., Khisti, R. T., Hodge, C. W., Homanics, G. E., and Morrow, A. L. (2002). GABA(A) receptor alpha-1 subunit deletion alters receptor subtype assembly, pharmacological and behavioral responses to benzodiazepines and zolpidem. Neuropharmacology 43, 685–694.
Kumar, S., Lane, B. M., and Morrow, A. L. (2006). Differential effects of systemic ethanol administration on protein kinase Cepsilon, gamma, and beta isoform expression, membrane translocation, and target phosphorylation: reversal by chronic ethanol exposure. J. Pharmacol. Exp. Ther. 319, 1366–1375.
Kumar, S., Porcu, P., Werner, D. F., Matthews, D. B., Diaz-Granados, J. L., Helfand, R. S., and Morrow, A. L. (2009). The role of GABA(A) receptors in the acute and chronic effects of ethanol: a decade of progress. Psychopharmacology (Berl.) 205, 529–564.
Kumar, S., Suryanarayanan, A., Boyd, K. N., Comerford, C. E., Lai, M. A., Ren, Q., and Morrow, A. L. (2010). Ethanol reduces GABAA alpha1 subunit receptor surface expression by a protein kinase Cgamma-dependent mechanism in cultured cerebral cortical neurons. Mol. Pharmacol. 77, 793–803.
Lai, C. C., Kuo, T. I., and Lin, H. H. (2007). The role of protein kinase A in acute ethanol-induced neurobehavioral actions in rats. Anesth. Analg. 105, 89–96.
Lau, G. C., Saha, S., Faris, R., and Russek, S. J. (2004). Up-regulation of NMDAR1 subunit gene expression in cortical neurons via a PKA-dependent pathway. J. Neurochem. 88, 564–575.
Leidenheimer, N. J., Machu, T. K., Endo, S., Olsen, R. W., Harris, R. A., and Browning, M. D. (1991). Cyclic AMP-dependent protein kinase decreases gamma-aminobutyric acidA receptor-mediated 36Cl- uptake by brain microsacs. J. Neurochem. 57, 722–725.
Liang, J., Suryanarayanan, A., Abriam, A., Snyder, B., Olsen, R. W., and Spigelman, I. (2007). Mechanisms of reversible GABAA receptor plasticity after ethanol intoxication. J. Neurosci. 27, 12367–12377.
Maas, J. W. Jr., Vogt, S. K., Chan, G. C., Pineda, V. V., Storm, D. R., and Muglia, L. J. (2005). Calcium-stimulated adenylyl cyclases are critical modulators of neuronal ethanol sensitivity. J. Neurosci. 25, 4118–4126.
Messing, R. O., Petersen, P. J., and Henrich, C. J. (1991). Chronic ethanol exposure increases levels of protein kinase C delta and epsilon and protein kinase C-mediated phosphorylation in cultured neural cells. J. Biol. Chem. 266, 23428–23432.
Moss, S. J., Doherty, C. A., and Huganir, R. L. (1992a). Identification of the cAMP-dependent protein kinase and protein kinase C phosphorylation sites within the major intracellular domains of the beta 1, gamma 2S, and gamma 2L subunits of the gamma-aminobutyric acid type A receptor. J. Biol. Chem. 267, 14470–14476.
Moss, S. J., Smart, T. G., Blackstone, C. D., and Huganir, R. L. (1992b). Functional modulation of GABAA receptors by cAMP-dependent protein phosphorylation. Science 257, 661–665.
Naassila, M., Ledent, C., and Daoust, M. (2002). Low ethanol sensitivity and increased ethanol consumption in mice lacking adenosine A2A receptors. J. Neurosci. 22, 10487–10493.
Nusser, Z., Sieghart, W., and Mody, I. (1999). Differential regulation of synaptic GABAA receptors by cAMP-dependent protein kinase in mouse cerebellar and olfactory bulb neurones. J. Physiol. 521(Pt 2), 421–435.
Olsen, R. W., and Sieghart, W. (2009). GABA A receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology 56, 141–148.
Pandey, S. C., Roy, A., and Zhang, H. (2003). The decreased phosphorylation of cyclic adenosine monophosphate (cAMP) response element binding (CREB) protein in the central amygdala acts as a molecular substrate for anxiety related to ethanol withdrawal in rats. Alcohol. Clin. Exp. Res. 27, 396–409.
Park, S. K., Sedore, S. A., Cronmiller, C., and Hirsh, J. (2000). Type II cAMP-dependent protein kinase-deficient Drosophila are viable but show developmental, circadian, and drug response phenotypes. J. Biol. Chem. 275, 20588–20596.
Paxinos, G., and Watson, C. H. (2007). The Rat Brain in Stereotaxic Coordinates, 6th Edn. London: Elsevier.
Poisbeau, P., Cheney, M. C., Browning, M. D., and Mody, I. (1999). Modulation of synaptic GABAA receptor function by PKA and PKC in adult hippocampal neurons. J. Neurosci. 19, 674–683.
Robello, M., Amico, C., and Cupello, A. (1993). Regulation of GABAA receptor in cerebellar granule cells in culture: differential involvement of kinase activities. Neuroscience 53, 131–138.
Saywell, S. A., and Feldman, J. L. (2004). Dynamic interactions of excitatory and inhibitory inputs in hypoglossal motoneurones: respiratory phasing and modulation by PKA. J. Physiol. 554, 879–889.
Shew, T., Yip, S., and Sastry, B. R. (2000). Mechanisms involved in tetanus-induced potentiation of fast IPSCs in rat hippocampal CA1 neurons. J. Neurophysiol. 83, 3388–3401.
Tang, X., Hernandez, C. C., and Macdonald, R. L. (2010). Modulation of spontaneous and GABA-evoked tonic alpha4beta3delta and alpha4beta3gamma2L GABAA receptor currents by protein kinase A. J. Neurophysiol. 103, 1007–1019.
Tehrani, M. H., Hablitz, J. J., and Barnes, E. M. Jr. (1989). cAMP increases the rate of GABAA receptor desensitization in chick cortical neurons. Synapse 4, 126–131.
Thiele, T. E., Willis, B., Stadler, J., Reynolds, J. G., Bernstein, I. L., and McKnight, G. S. (2000). High ethanol consumption and low sensitivity to ethanol-induced sedation in protein kinase A-mutant mice. J. Neurosci. 20, RC75.
Thomas, P., Mortensen, M., Hosie, A. M., and Smart, T. G. (2005). Dynamic mobility of functional GABAA receptors at inhibitory synapses. Nat. Neurosci. 8, 889–897.
Vaello, M. L., Ruiz-Gomez, A., Lerma, J., and Mayor, F. Jr. (1994). Modulation of inhibitory glycine receptors by phosphorylation by protein kinase C and cAMP-dependent protein kinase. J. Biol. Chem. 269, 2002–2008.
Wand, G., Levine, M., Zweifel, L., Schwindinger, W., and Abel, T. (2001). The cAMP-protein kinase A signal transduction pathway modulates ethanol consumption and sedative effects of ethanol. J. Neurosci. 21, 5297–5303.
Werner, D. F., Kumar, S., Criswell, H. E., Suryanarayanan, A., Alex Fetzer, J., Comerford, C. E., and Leslie Morrow, A. (2011). PKCgamma is required for ethanol-induced increases in GABA(A) receptor alpha4 subunit expression in cultured cerebral cortical neurons. J. Neurochem. 116, 554–563.
White, G., Li, C., and Ishac, E. (1992). 1,9-Dideoxyforskolin does not mimic all cAMP and protein kinase A independent effects of forskolin on GABA activated ion currents in adult rat sensory neurons. Brain Res. 586, 157–161.
Keywords: GABAA receptors, ethanol, PKA, PKC, loss of righting reflex
Citation: Kumar S, Ren Q, Beckley JH, O’Buckley TK, Gigante ED, Santerre JL, Werner DF and Morrow AL (2012) Ethanol activation of protein kinase A regulates GABAA receptor subunit expression in the cerebral cortex and contributes to ethanol-induced hypnosis. Front. Neurosci. 6:44. doi: 10.3389/fnins.2012.00044
Received: 25 October 2011; Accepted: 19 March 2012;
Published online: 09 April 2012.
Edited by:
Meredith A. Fox, National Institute of Mental Health, USAReviewed by:
Glenn W. Stevenson, University of New England, USAIgor Spigelman, University of California Los Angeles, USA
Copyright: © 2012 Kumar, Ren, Beckley, O’Buckley, Gigante, Santerre, Werner and Morrow. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: A. Leslie Morrow, Bowles Center for Alcohol Studies, University of North Carolina School of Medicine, 3027 Thurston-Bowles Building, CB # 7178, Chapel Hill, NC 27599, USA. e-mail:bW9ycm93QG1lZC51bmMuZWR1