 Marie-Solenne Felix†
Marie-Solenne Felix† Natalia Popa
Natalia Popa Mehdi Djelloul
Mehdi Djelloul José Boucraut
José Boucraut Patrick Gauthier†
Patrick Gauthier† Sylvian Bauer†
Sylvian Bauer† Valery A. Matarazzo†*
Valery A. Matarazzo†*- Centre de Recherche en Neurobiologie et Neurophysiologie de Marseille, Aix-Marseille Université, CNRS-UMR 6231, Marseille, France
Spinal cord injury (SCI) triggers a complex cellular response at the injury site, leading to the formation of a dense scar tissue. Despite this local tissue remodeling, the consequences of SCI at the cellular level in distant rostral sites (i.e., brain), remain unknown. In this study, we asked whether cervical SCI could alter cell dynamics in neurogenic areas of the adult rat forebrain. To this aim, we quantified BrdU incorporation and determined the phenotypes of newly generated cells (neurons, astrocytes, or microglia) during the subchronic and chronic phases of injury. We find that subchronic SCI leads to a reduction of BrdU incorporation and neurogenesis in the olfactory bulb and in the hippocampal dentate gyrus. By contrast, subchronic SCI triggers an increased BrdU incorporation in the dorsal vagal complex of the hindbrain, where most of the newly generated cells are identified as microglia. In chronic condition 90 days after SCI, BrdU incorporation returns to control levels in all regions examined, except in the hippocampus, where SCI produces a long-term reduction of neurogenesis, indicating that this structure is particularly sensitive to SCI. Finally, we observe that SCI triggers an acute inflammatory response in all brain regions examined, as well as a hippocampal-specific decline in BDNF levels. This study provides the first demonstration that forebrain neurogenesis is vulnerable to a distal SCI.
Introduction
Central nervous insults, such as spinal cord injury (SCI) or traumatic brain injury (TBI), are among the leading causes of mortality and morbidity worldwide. They produce profound neurological dysfunction, due to the progressive destruction of local and distal neuronal networks, resulting from injury-induced tissue damage and subsequent local, cellular, and biochemical reactions. Interestingly, significant spontaneous neuroplasticity occurs over the weeks and months following SCI or TBI, leading to some functional recovery (Raineteau, 2008; Darian-Smith, 2009; Kernie and Parent, 2010; Richardson et al., 2010). The underlying cellular mechanisms remain unclear, but may comprise dendritic remodeling, axonal sprouting, as well as local neuronal circuit reorganization (Darian-Smith, 2009). Another feature that could contribute to this post-injury neuroplasticity is injury-induced neurogenesis (Kazanis, 2009).
Adult neurogenesis in physiological conditions is sustained by neural stem and progenitor cells that persist in two main regions of the mammalian brain, the subventricular zone (SVZ) bordering the lateral ventricle (Alvarez-Buylla and Garcia-Verdugo, 2002), and the subgranular zone (SGZ) of the hippocampal dentate gyrus (Seaberg and van der Kooy, 2002). Several studies have shown that the proliferation of neural progenitors in the adult forebrain is stimulated after various types of brain injury, including stroke, seizure, or TBI. Following TBI, cell proliferation and neurogenesis increase in the hippocampal dentate gyrus (Kernie et al., 2001; Rola et al., 2006; Sun et al., 2007; Urrea et al., 2007; Yu et al., 2008), and a recent study using a mouse model of TBI showed that newly generated neurons in this region persist over time and might participate in the cognitive recovery (Blaiss et al., 2011).
In addition to these two principal brain niches, other areas enriched in markers of morpho-functional neuroplasticity contain adult neural stem cells, albeit to considerably lower levels compared to SVZ and SGZ regions. One such area is the dorsal vagal complex (DVC) of the hindbrain (Bauer et al., 2005; Charrier et al., 2006). Like for the SVZ and SGZ, neuronal markers of plasticity such as PSA-NCAM and BDNF are continuously expressed in the adult DVC (Conner et al., 1997; Bouzioukh et al., 2001). Interestingly, the DVC is responsive to injury since vagotomy produces an increase of microgliosis and astrogliosis in this hindbrain area (Bauer et al., 2005).
The situation becomes more complex for the adult mammalian spinal cord, which is non-neurogenic in physiological conditions. Although it retains multipotent neural stem cells that could generate functional neurons in vitro (Dromard et al., 2008; Meletis et al., 2008), their potential is mainly restricted to the glial lineage in vivo (Horner et al., 2000; Wrathall and Lytle, 2008). Injury to the spinal cord is well-known to induce the formation of reactive astrocytes and the infiltration of immune cells in the vicinity of the lesion site (Horky et al., 2006; Yang et al., 2006; Rolls et al., 2009; Beck et al., 2010), but whether SCI also induces the production of new neurons in vivo remains controversial. Studies have shown that spinal neurogenesis occurs to a limited extent after SCI (Chi et al., 2006; Ke et al., 2006; Shechter et al., 2007), but that it could be stimulated by experimental intervention (Ohori et al., 2006). Overall, available data suggest that the extent of neurogenesis at the spinal level after SCI depends on the localization and severity of the injury (Ohori et al., 2006; Vessal et al., 2007).
Another question concerns the distal consequences of SCI in the forebrain. Indeed, beyond the proximal consequences of SCI at the spinal level, SCI leads to topographic and neuronal reorganizations in the cerebral cortex as well as cortical atrophy indicating that the brain responds to a distant SCI (Endo et al., 2007; Kaas et al., 2008; Nishimura and Isa, 2009; Freund et al., 2011). Furthermore, a recent study showed that cervical dorsal rhizotomy, a model known to induce cortical reorganizations similar to those observed after SCI, also induces reactive neurogenesis in the primary sensorimotor cortex of adult monkeys (Vessal and Darian-Smith, 2010). However, whether neurogenesis in the adult brain is altered after a direct, central injury to the spinal cord has never been investigated.
This study sought to determine if the modulation of neurogenesis in the adult brain could constitute a general mechanism in the neuroplasticity response triggered by SCI. The specific aim was to investigate whether SCI could alter neurogenesis in known neurogenic niches of the adult rat forebrain, hindbrain, as well as at the spinal level, in both subchronic and chronic conditions [15 and 90 days post-injury (dpi), respectively]. We used a partial C2 cervical SCI model because it is reported to enhance post-lesional neuroplasticity (Bareyre et al., 2004; Vinit and Kastner, 2009), and as it severs descending bulbospinal pathways originating from the medulla oblongata (Krassioukov, 2009), we also investigated whether SCI could alter cell dynamics in the hindbrain region of the DVC. Finally, we further examined the possibility that SCI could produce inflammation and variation of BDNF levels in these brain regions. Indeed, these two factors are well-known to regulate adult brain neurogenesis (Lee et al., 2002; Scharfman et al., 2005; Das and Basu, 2008; Li et al., 2008; Whitney et al., 2009). Unexpectedly, our results reveal for the first time that neurogenic regions of the adult brain are vulnerable to SCI despite their distance from the lesion site.
Materials and Methods
Animal Care
Rats were handled and cared in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996) and the European Communities Council Directive of September 22, 2010 (2010/63/EU, 74). Experimental protocols were approved by the institutional Ethical Committee guidelines for animal research with the accreditation no. C13-055-6 from the French Ministry of Agriculture. V. Matarazzo permits number # 13.305. All efforts were made to minimize the number of animals used and their suffering.
Experimental Groups and Surgical Procedures
A total of 51 adult female Sprague-Dawley rats were used for the study (285 ± 3 g). Thirty-seven of them were subjected to SCI and housed for a post-lesional period of 2 (n = 6), 7 (n = 9), 15 (n = 11), and 90 (n = 11) dpi. Fourteen of them were not subjected to SCI and thus categorized as controls.
The animals were submitted to a partial cervical spinal cord hemisection (Anderson et al., 2005; Vinit et al., 2006), which was performed under anesthesia using a mixture of ketamine (80 mg/kg, Imalgène 1000, Virbac, i.p.) – xylazine (20 mg/kg, Bayer, i.p.). Briefly, the rats were placed in a surgery system and their body temperature was maintained between 36.5 and 37.5°C. Skin and muscles covering the first cervical vertebrae were retracted; a laminectomy of the dorsal C2 vertebra was performed and the dura mater was cut and put aside. The left side of the spinal cord was transected laterally at the C2 segmental level, just caudally to the C2 dorsal roots, with microscalpel. After injury, muscles and skin were sutured, and the animal received morphine (0.1 mg/mL, Lavoisier, France), terramycin (8%, Pfizer, France), and glucose (5%, Lavoisier, France) during the first 24 h. Rats were housed in individual cage with food and water ad libitum.
Proliferation Assay and BrdU Pulse-Chase
Twenty-one rats were used for the immunochemistry study. Thirteen of them were subjected to SCI and housed for a post-lesional period of 7 (n = 3), 15 (n = 5), and 90 (n = 5) dpi. Eight of them were not subjected to SCI and thus categorized as controls.
Cumulative in vivo labeling of cell proliferation was performed by means of repetitive i.p. injections of Bromodeoxyuridine (BrdU, 50 mg/kg i.p., Sigma), conforming to protocols commonly used for neurogenesis assessment in the forebrain (Kempermann et al., 1998; Eisch et al., 2000). The 7-dpi animals and the associated control animals received a single injection of BrdU, the day before the sacrifice. The 15-, 90-dpi, and the associated control animals received one injection per day of BrdU for four consecutive days, the first one starting 2 weeks before the sacrifice.
Immunohistochemistry
All animals were sacrificed under deep anesthesia with pentobarbital (100 mg/kg, Ceva, i.p.), perfused transcardially with 300 mL of 0.1 M ice-cold phosphate buffer (pH 7.4) and followed by the same volume of 4% phosphate-buffered paraformaldehyde (pH 7.4). Tissues were immediately dissected, post-fixed for 2 h at 4°C in the same fixative buffer and cryoprotected 24 h at 4°C in 30% sucrose (Sigma). Tissues were then snap-frozen at −40°C for 30 s in isopentane solution and stored at −80°C.
The olfactory bulb (OB), the SVZ, the SGZ, the medulla (containing the DVC), and the C2 cervical spinal cord were cut coronally into 30 μm thick sections using a cryostat (Leica). Sections were mounted on glass microscope slides coated with 0.05% poly-L-lysine (Sigma).
For BrdU labeling, sections were rinsed twice in PBS during 10 min, then incubated in formamide 50% + SSC2X at 65°C during 30 min. Sections were washed in SSC2X during 10 min then in PBS during 10 min with shaking. Sections were incubated in HCl 2N + 0.2% triton X-100 at 37°C during 30 min. Then, sections were washed twice with 0.1 M borate (pH 8.5) during 5 min and twice with PBS during 10 min. Sections were then incubated overnight at 4°C with a rat anti-BrdU (1/50; AbD Serotec) diluted in a solution containing 0.1 M PBS pH 7.4; 0.2% Triton X-100; and 10% normal horse serum (NHS). Sections were then washed twice with PBS, and incubated for 2 h at room temperature with an Alexa Fluor® 488 goat anti-rat (1/400; Invitrogen) which was previously diluted in a solution containing 0.1 M PBS pH 7.4 and 5% NHS.
Neuronal and glial cell immunostainings were performed following BrdU labeling, while microglial cell immunostainings preceded BrdU labeling. Immature neurons were detected using either a mouse anti-Hu (1/1000, Invitrogen) or a goat anti-DCX (1/500, sc-8066, Santa Cruz Biotechnology) antibody. Glial and microglial cells were respectively immunostained by a rabbit anti-GFAP antibody (1/1000, Sigma) and a mouse anti-CD11 clone OX-42 antibody (1/50, Chemicon). Sections were permeabilized with PBS containing 0.2% Triton X-100, immunoblocked with PBS containing 0.2% Triton X-100 and 10% NHS for 45 min, and incubated overnight at 4°C in the same solution with primary antibodies above-cited. Sections were then washed with PBS and incubated for 2 h with secondary antibodies in PBS containing 5% NHS at room temperature. The following secondary antibodies were used: Alexa Fluor® 568 highly cross-adsorbed goat anti-mouse, Alexa Fluor® 568 donkey anti-goat, and Alexa Fluor® 594 goat anti-rabbit (1/400, Invitrogen). For nuclear staining, sections were washed with PBS, and then incubated 15 min with DAPI (0.5 ng/mL, Invitrogen). After a last wash in PBS, slides were rinsed in distilled water and medium mounted with Mowiol® (Calbiochem).
Colabeling of activated-caspase-3 with either Hu C/D or DCX were performed as described above with the exception that sections were blocked with 4% normal donkey serum (NDS), incubated overnight at 4°C with either a mouse anti-Hu C/D or goat anti-DCX and then incubated again overnight at 4°C with a rabbit anti-activated-caspase-3 (1/1000, Cell Signaling) in 0.1 M PBS containing 0.2% Triton X-100; 4% NDS. Secondary antibodies were Alexa Fluor® 568 goat anti-mouse, Alexa Fluor® 568 donkey anti-goat, and Alexa Fluor® 488 donkey anti-rabbit (1/400, Invitrogen).
Cresyl violet staining was achieved to illustrate the SCI. Sections were rinsed in distilled water for 5 min and incubated 3 min in a cresyl violet bath. Sections were then dehydrated through a sequence of ethanol baths (70, 95, and 100%). Sections were finally cleaned in Xylene during 2 min and medium mounted with coverslip using Permount® (Fisher Scientific, Fair Lawn, NJ, USA).
Image Acquisition
Low magnified images were acquired with an Olympus BX50 fluorescent microscope equipped with an Olympus DP50 digital camera and 4× lens.
Confocal images were captured with a Leica TCS SP2 scanning confocal microscope (Leica) equipped with a 40× lens and with Mira-Verdi multiphoton (780 nm), argon (488 nm), and helium (543 nm) lasers and monitored with LCS 2.61 image acquisition software. For all animals in each treatment group, Z-stacks composed of 10 confocal images (1.5 μm focal spacing) were acquired in the external plexiform layer of the OB, the SVZ, the SGZ, the nucleus tractus solitarius (NTS), and at the C2 cervical level of the spinal cord. For the spinal cord, images were performed in the ipsilateral (injured side).
Quantification of BrdU-Positive Cells
We selected for comparisons corresponding coronal sections (30 μm) exhibiting the same cytoarchitectonic features, determined using the rat atlas (Paxinos and Watson, 1986), in all of the injured and non-injured animals. This allowed BrdU+ nuclei to be counted in coronal sections at comparable rostrocaudal positions. BrdU+ nuclei were manually counted under a 40× lens with the profile counts method particularly useful for small object in thick sections (Guillery, 2002). Because profile counts could produce overcounting depending on the thickness and the height of the counted object, we calculated a correction factor of 0.80 using the Abercrombie’s formula (height of BrdU nuclei: 0.75 μm; section thickness: 30 μm; Guillery, 2002). This value was high enough and we thus considered that applying a correction factor on the profile counts was not trivial (Guillery, 2002). Brain and spinal cord structures were sampled either by selecting a predetermined area (OB) or by analyzing entire structures on each section (SVZ, SGZ, DVC, SC). Cell quantification was performed separately from the contralateral and ipsilateral sides for each structure and each animal. However, since no statistical difference was observed between both sides, with exception to the spinal cord, data from ipsilateral and contralateral sides were systematically combined. Hence, the numbers obtained in this study are absolute numbers per 30 μm thick section and are independent of the volume of the structures.
Olfactory bulb
Every 2d section was selected from interaural 16.56–15.60 mm. As depicted in Figure 2, a predetermined area (400 μm × 400 μm) in the external plexiform layer of the OB was analyzed on each section. All BrdU+ nuclei in this selected area were counted and represented as BrdU+ cells/mm2.
Subventricular zone
Every fourth section was selected from interaural 11.28–9.72 mm and analyzed entirely for BrdU+ nuclei bordering the lateral ventricle wall. Cell numbers are represented as BrdU+ cells/section (i.e., positive cells per 30 μm thick SVZ section).
Subgranular zone
Every fourth section was selected from interaural 6.20–4.56 mm and analyzed entirely for BrdU+ nuclei. The granular cell layer was combined with the SGZ, but the hilus and the molecular cell layer were excluded from counting. Cell numbers are represented as BrdU+ cells/section (i.e., positive cells per 30 μm thick dentate gyrus section).
Dorsal vagal complex
Every fourth section from interaural −4.32 to −5.28 mm (region that includes the area postrema, AP) was selected. The AP, the NTS, and the dorsal motor nucleus of the vagus nerve, were entirely analyzed for BrdU+ nuclei. Cell numbers are represented as BrdU+ cells/section (i.e., positive cells per 30 μm thick DVC section).
SC
Every fourth section was selected from 0.5 mm rostral to 0.5 mm caudal of the epicenter of the C2 spinal cord lesion. BrdU+ nuclei were entirely analyzed in the white and gray matter but ipsilateral (injured) and contralateral (uninjured) sides were dissociated. Cell numbers are represented as BrdU+ cells/section (i.e., positive cells per 30 μm thick hemi-spinal cord section).
Statistical analyses were performed with ANOVA one way analysis of variance followed by a Tukey’s post-test using GraphPad Prism version 4.00, GraphPad Software, San Diego, CA, USA, “www.graphpad.com.”
RT-qPCR
From a total of 15 rats, 12 of them were subjected to SCI and housed for a post-lesional period of 2 (n = 3), 7 (n = 3), 15 (n = 3), and 90 (n = 3) dpi. The last three rats were not subjected to SCI and thus categorized as controls (referred as 0 dpi in the graph). Total RNA was separately extracted from C2 spinal cord, DVC, hippocampus, and SVZ tissues using the RNeasy Plus Universal Mini Kit according to the manufacturer’s protocol (Qiagen, France). cDNAs were obtained by reverse transcription using M-MLV reverse transcriptase (Invitrogen, France) starting with 2 μg of total RNA. The PCR reaction was performed using the 7500 Fast Real-Time PCR System (Applied Biosystems). An equivalent of 20 ng initial RNA was subjected to PCR amplification using 0.1 μM of each primer and Power SYBR Green PCR master mix (Applied Biosystems, France). PCR reaction was as follows: 95°C for 10 min once, then 95°C for 15 s, 58°C for 30 s, 72°C for 30 s; 40 cycles. The following forward and reverse primers were used: tumor necrosis-factor alpha 1 (TNFα1; TACTGAACTTCGGGGTGATTGGTCC and CAGCCTTGTCCCTTGAAGAGAACC); CCR2 (CCACCACACCGTATGACTATGAT and ACAGCATGGACAATAGCCAAATA); CCR5 (TGTTTCGCTGTAGGAATGAGAAG and GTTTGACAATGTGTTTTCGGAAG); GAPDH (ATCACCATCTTCCAGGAGCG and TGCATTGCTGACAATCTTGAGG). Samples were subjected to a melting curve analysis to confirm the amplification specificity. Gene expression analysis was performed using the 2−ΔΔCT method (Livak and Schmittgen, 2001) where GAPDH was the reference gene and TNFα1, CCR2, CCR5 were the target genes. Amplification efficiencies were verified between the reference and target genes. Statistical analyses were performed with ANOVA one way analysis of variance followed by a Dunnett’s post-test.
BDNF Immunoassay
From a total of 15 rats, 12 of them were subjected to SCI and housed for a post-lesional period of 2 (n = 3), 7 (n = 3), 15 (n = 3), and 90 (n = 3) dpi. The last three rats were not subjected to SCI and thus categorized as controls (referred as 0 dpi in the graph). Tissues from C2 spinal cord, DVC, hippocampus, and SVZ were extracted and homogenized in RIPA lysis buffer (25 mM Tris–HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) Pierce Biotechnology, Rockford, IL, USA) supplemented with protease and phosphatase inhibitors cocktail (Sigma-Aldrich, France). Samples were treated using 1 mL of RIPA buffer per mg of tissue and incubated 45 min at 4°C. Samples were then centrifuged at 15,000 g, 30 min at 4°C 15,000 g. The supernatants were collected and stored frozen until use. Total protein concentration of each sample was determined by BCA protein assay kit (Pierce Biotechnology, Rockford, IL, USA) and adjusted to 80 μg/mL. Tissular concentrations of BDNF protein were measured in duplicate with a conventional two-site ELISA. BDNF Emax immunoassay system (Promega, France) was performed according to the manufacturer’s instruction. Tissular BDNF concentrations were determined as picograms per milliliters. Statistical analyses were performed with ANOVA one way analysis of variance followed by a Dunnett’s post-test.
Results
The aim of our study was to investigate whether SCI modulates neurogenesis in the adult rat brain (Figure 1A). We performed a partial hemisection at C2 level which interrupts both descending motor and lateral ascending sensory tracts within the lateral funiculi of the spinal cord (Anderson et al., 2005; Vinit et al., 2006; Figure 1B). The effects of SCI were investigated in three supra-spinal brain areas where ongoing adult neurogenesis has been described: the SVZ/OB system (Doetsch and Alvarez-Buylla, 1996), the SGZ (Altman and Das, 1965), and the DVC (Bauer et al., 2005). Pulse/chase BrdU treatment consisted in one daily i.p. injection of BrdU for 4 days, followed by 10 days washout (see Figure 1C for a graphical representation of the experimental paradigm). This protocol was selected in order to maximize the detection of BrdU+ cells that would have undergone neuronal differentiation and would express immature neuronal markers, both in the OB and the SGZ (Brown et al., 2003). To address the possibility that SCI-induced modulation of neurogenesis could vary as a function of time after the lesion, we studied two time points, a subchronic condition 15 dpi and a chronic condition 90 dpi.
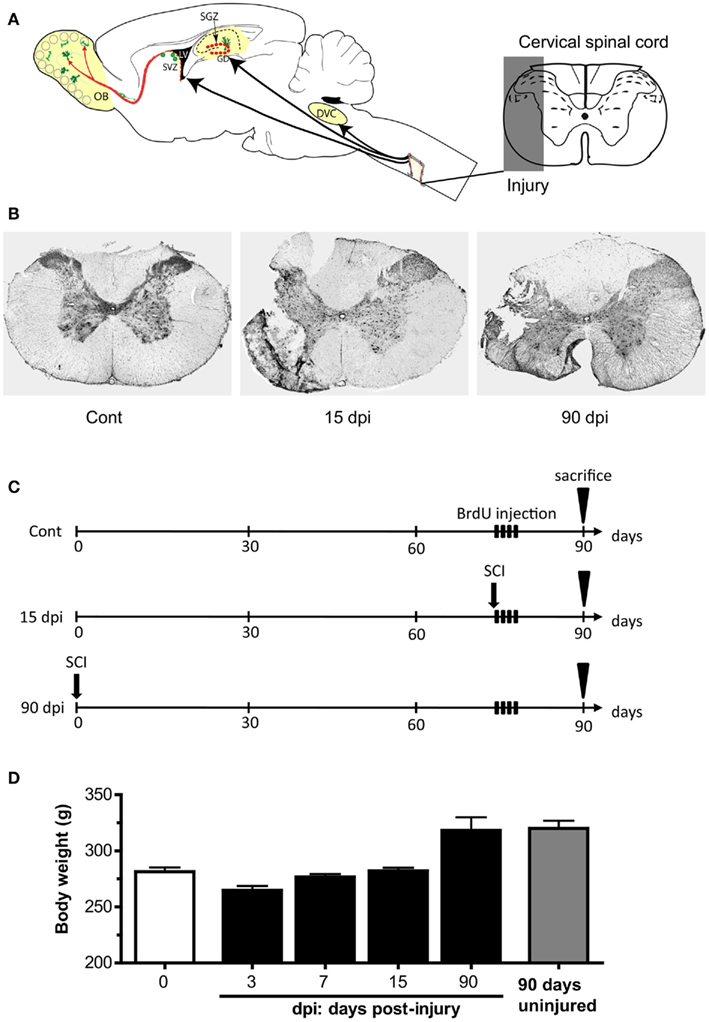
Figure 1. Experimental paradigm. (A) Schematic representation of the spinal cord injury (cervical C2 partial hemisection) and the brain areas examined: the subventricular zone (SVZ) of the lateral ventricle and the olfactory bulb (OB), the subgranular zone (SGZ) of the hippocampus, the dorsal vagal complex (DVC) in the medulla, and spinal cord (SC). (B) Histochemistry for violet cresyl at the site of injury (cervical C2 level) in control, 15 days post-injury (dpi), and 90 dpi conditions. (C) BrdU pulse-chase protocol: for the control, 15, and 90 dpi conditions, animals received one daily injection of BrdU during four consecutive days, the first one starting 14 days before sacrifice. Cont, control non-injured animals; dpi, days post-injury. (D) Variation in body weight before and after the lesion, up to 90 days.
Using the same animal groups, BrdU labeling was also analyzed in the spinal cord, both in the lesion side where previous studies have shown that early proliferating cells accumulate within the lesion site (Frisen et al., 1995), and in the contralateral, uninjured side.
Behavioral Observations and General State of the Animal
Rats were observed daily after the surgery, and showed a rapid recovery with 100% survival rate. After initial weight loss due to the lesion, all rats presented a rapid spontaneous weight recovery with normal overall behavior. Indeed, maximal weight loss was observed 3 dpi and animals spontaneously recovered their weight by 7 dpi (Figure 1D). Although we did not systematically assess performances of the injured rats, behavior in the home-cage and daily handling indicated an initial impairment characterized by a temporary hemiparesis of the ipsilateral forepaw, which regained normal functionality within 3–8 days. These changes did not affect the contralateral forepaw and apparently poorly affected the hind legs since locomotion and body supportive function seemed not affected, as indicated by apparent normal drinking and food pellet reaching (using both forepaws), which required the rats to get upon hind legs to reach for food and water. Furthermore, no signs of pain, prostration and immobility, no lower tolerance to handling, no pupillary dilatation, no avoidance, or abnormal behavior were observed in injured animals. For these reasons there was no need for specific nursing of the animals.
Neurogenesis in the OB is Transiently Decreased during Subchronic SCI
Newly generated neurons in the main OB are generated from neural stem and progenitor cells residing in the SVZ, from where they migrate for about 3–6 days before reaching the OB (Lois and Alvarez-Buylla, 1994). To determine the impact of SCI on OB neurogenesis, we performed dual immunohistochemical staining for BrdU and Doublecortin (DCX) to label newly generated, immature neurons that have migrated out of the SVZ into the main OB. We thus quantified BrdU+ nuclei as well as cells colabeled with BrdU and DCX in the granular cell layer of the OB. We found that the total number of BrdU+ nuclei was significantly decreased 15 dpi compared to control, non-lesioned animals: BrdU+ nuclei in the OB of lesioned animals amounted only 36% of those detected in control animals, hence revealing a 74% reduction in BrdU labeling 15 dpi (Figures 2A–C,G). Similarly, the number of BrdU+–DCX+ cells showed a significant 76% decrease in the OB of lesioned animals 15 dpi (Figures 2D–F,H). However, no difference was observed between the left and right OB in injured animals. These alterations were only transient since both the number of BrdU+ nuclei and the number of BrdU+–DCX+ cells returned to control values in the OB of animals analyzed 90 dpi (Figures 2C,F–H). Despite these profound variations in the number of newly generated OB neurons, the rate of neuronal differentiation, defined as the proportion of BrdU+–DCX+ cells among the total number of BrdU+ nuclei, was not altered by SCI (Figure 2I). Thus, although SCI strongly reduces the number of newly generated neurons that reach the main OB 15 days after the lesion, it does not alter the ability of newly generated cells to differentiate along the neuronal lineage at this time-point, nor in the chronic condition 90 days after the lesion. No modification of astrogliosis or microgliosis was observed in the OB after SCI, at least during these periods (data not shown).
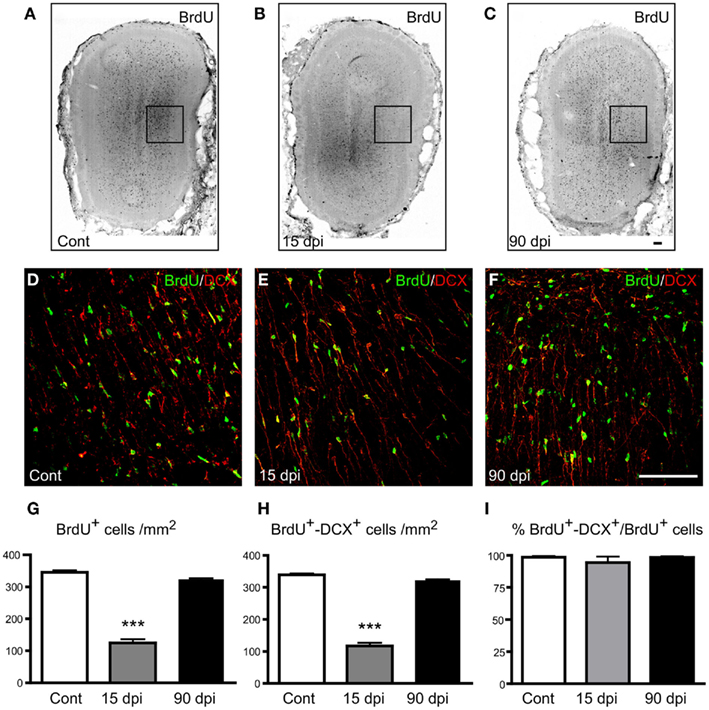
Figure 2. Distribution and quantification of immature neurons in the OB after SCI. (A–C) Immunohistochemistry for BrdU in the OB in control (A), 15 (B), and 90 dpi (C) conditions. (D–F) Immunohistochemistry for BrdU (green) and Doublecortin (DCX; red) in the external plexiform layer of the main OB in control (D), 15 (E), and 90 dpi (F) conditions. Images (D–F) correspond to high magnifications of the boxed areas represented in (A–C). (G–I) Quantification of the number of BrdU+ nuclei (G), number of BrdU+–DCX+ cells per mm2(H), and percentage of BrdU+–DCX+ cells among the total BrdU+ nuclei (I). Cont, control non-injured animals; dpi, days post-injury. All data are presented as mean ± SEM. ***p < 0.001. Scale bar: 100 μm.
Neurogenesis in the SGZ of the Dentate Gyrus is Decreased during Subchronic and Chronic SCI
Having shown that a spinal cord distant lesion produces a transient reduction in the number of newly generated neurons in the adult OB, we next asked whether adult neurogenesis in the dentate gyrus could also be altered by SCI, using the same animals and a similar approach (Figures 3A–C). As observed in the OB, the number of BrdU+ nuclei in the dentate gyrus showed a significant 35% reduction 15 dpi (Figure 3D), whereas the number of BrdU+–DCX+ cells was reduced by 40% compared to control, non-injured animals (Figure 3E). However, in contrast to the transient reduction observed in the OB, the number of BrdU+ nuclei or of BrdU+–DCX+ cells did not return to baseline levels in the dentate gyrus of lesioned animals at 90 dpi. At this time-point, the reduction was sustained and reached values similar to those observed at 15 dpi (Figure 3E). Similar results were observed by quantifying cell profiles that were positive for DCX but BrdU-negative (referred to BrdU−–DCX+ cells in Figure 3G), likely corresponding to immature neurons generated between our last BrdU injection and animal sacrifice. No difference was observed between the left and right dentate gyri in injured animals. Thus, a distant lesion to the spinal cord, in addition to producing a transient reduction in the number of newly generated neurons in the main OB, also produces a long-term reduction in the number of newly generated neurons in the dentate gyrus. No modification of astrogliosis or microgliosis was observed in the dentate gyrus after SCI, at least during these periods (data not shown).
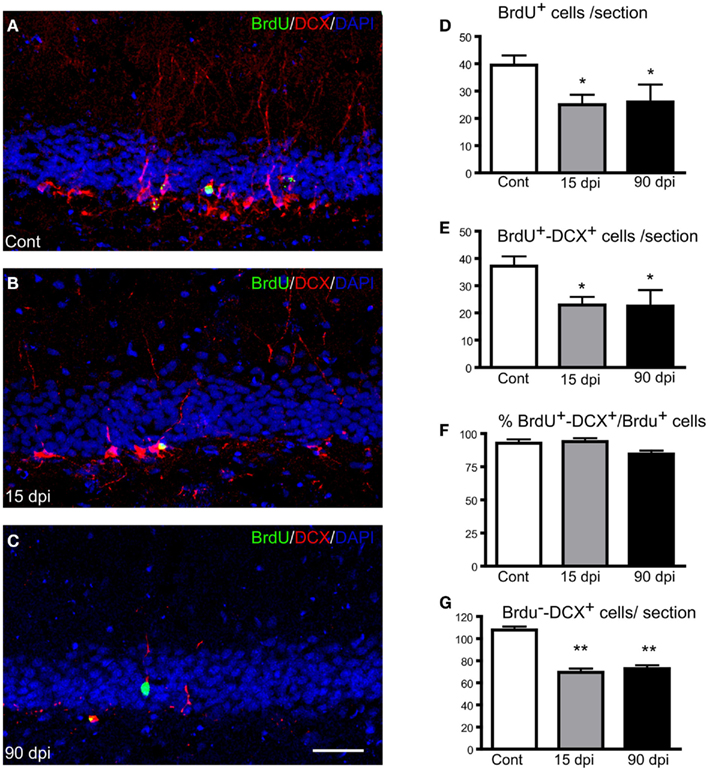
Figure 3. Distribution and quantification of immature neurons in the adult dentate gyrus after SCI. (A–C) Immunohistochemistry for BrdU (green) and Doublecortin (DCX; red) along with nuclear staining (DAPI; blue) in the hippocampal dentate gyrus in control (A), 15 (B), and 90 dpi (C) conditions. Scale bar: 100 μm. (D–G) Quantification of the number of BrdU+ nuclei per dentate gyrus in a 30-μm section (D), number of BrdU+–DCX+ cells per dentate gyrus in a 30-μm section (E), percentage of BrdU+–DCX+ cells among the total BrdU+ nuclei (F), and number of singly labeled DCX+ cells per dentate gyrus in a 30-μm section (G). Cont, control non-injured animals; dpi, days post-injury. Data are presented as mean ± SEM. **p < 0.01, *p < 0.05.
As in the OB, the rate of neuronal differentiation in the dentate gyrus, defined as the proportion of BrdU+–DCX+ cells among the total number of BrdU+ nuclei, was not altered by SCI (Figure 3F), although this ratio appeared slightly reduced at 90 dpi compared to control (84.5 ± 2.6 versus 92.7 ± 1.8) without reaching significance. Thus, SCI does not alter the ability of newly generated cells to differentiate along the neuronal lineage in the hippocampal dentate gyrus either.
SCI-Mediated Decline in Forebrain Neurogenesis Results from Decreased Cell Proliferation with No Increased Cell Death
The diminished number of forebrain newly generated neurons observed after SCI could be explained by a reduced cell proliferation of progenitors and/or an increased cell death of newly formed cells. To gain further insights into these mechanisms, we generated animal groups that were sacrificed 24 h after a single BrdU injection. This allowed us to obtain an index of cell proliferation by counting the number of BrdU+ nuclei in the SVZ and SGZ of these animals (Figures 4A–F). These analyses revealed that SCI produced a significant reduction in the number of BrdU+ nuclei, amounted 39% in the SVZ, (Cont: 196.5 ± 1.4; 7 dpi: 120.1 ± 2.8; p < 0.001), and 58% in the SGZ (Cont: 4.7 ± 1.6; 7 dpi: 2.0 ± 0.6; p < 0.01; Figures 4C–F). In a second set of experiments, we quantified cell death of immature neurons in forebrain neurogenic regions by using immunostaining for active caspase-3 and the neuronal markers DCX or Hu C/D (Figures 4G–N). Cells expressing active caspase-3, alone or in association with neuronal markers, were very scarce in all conditions tested (control, 7, 15, 90 dpi), and we observed no significant difference between control and injured animals (Table 1). Collectively, these results demonstrate that the reduced number of newly generated neurons in the forebrain accounts for a reduction in cell proliferation rather than an increase of cell death. Whether additional factors such as an altered migration of SVZ newly generated neurons en route to the OB are also at play for these neurogenic niches remains to be determined.
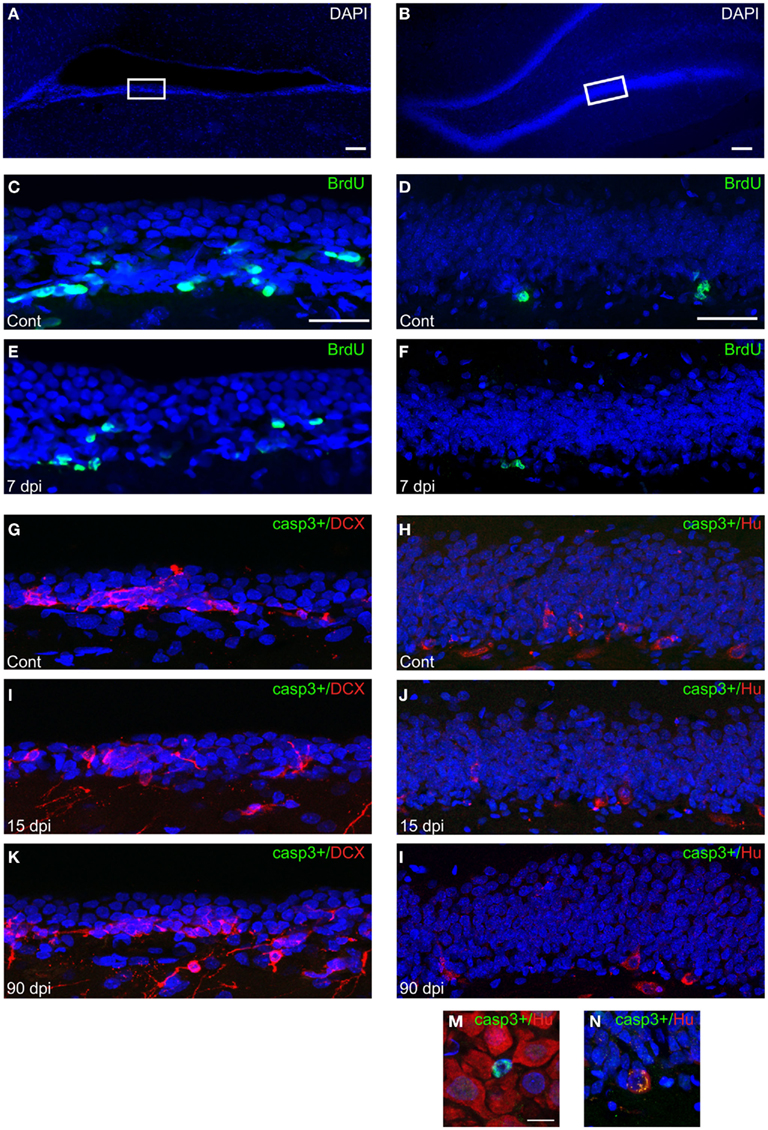
Figure 4. Index of cell proliferation and cell death in the forebrain neurogenic niches after SCI. (A–B) Low magnification pictures of DAPI (blue) staining in the SVZ (A) and SGZ (B). Scale bars: 100 μm. White boxes represent regions where high magnification pictures were captured (C–L). (C–F) Immunohistochemistry for BrdU (green) in the SVZ (C–E) and SGZ (D–F) along with nuclear staining (DAPI, blue) 24 h after a single BrdU i.p. injection. Scale bar: 50 μm. (G–L) Immunohistochemistry for active caspase-3 and DCX in the SVZ (G,I,K) or active caspase-3 and Hu C/D in the SGZ (H,J,L). Cont, control non-injured animals; dpi, days post-injury. Casp3+: active caspase-3. Scale bar: 50 μm. (M,N) High magnification pictures showing positive active caspase-3 – negative Hu C/D (M) or positive Hu C/D (N) cells in the hippocampus. Scale bar: 10 μm.
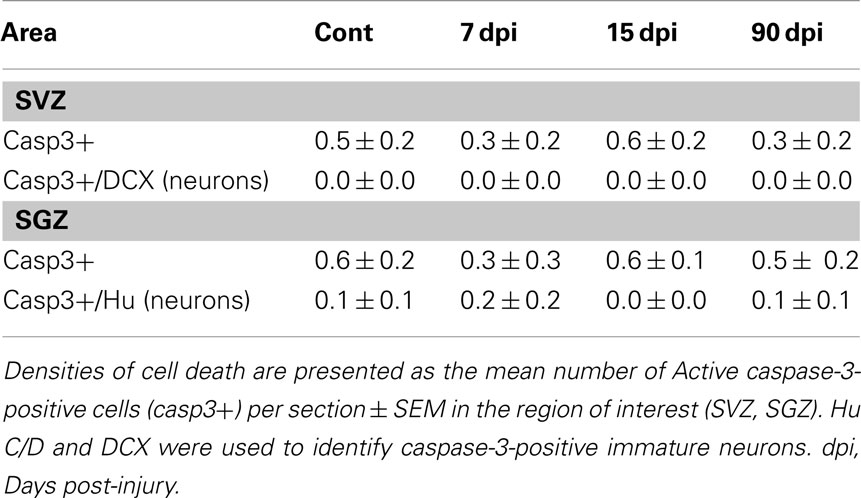
Table 1. Density of cell death in the forebrain neurogenic niches.
Microgliosis in the DVC of the Medulla Oblongata is Transiently Increased during Subchronic SCI
We further asked whether the alteration of adult neural stem cell niches triggered by SCI could be extended to the DVC of the medulla oblongata, a hindbrain region comprising the AP, the NTS, and the dorsal motor nucleus of the vagus nerve (DMX), where adult neural stem cells has recently been described (Bauer et al., 2005). The DVC could be responsive to SCI since it is a major integrative center for the autonomic nervous functions that are severed by high cervical SCI (Krassioukov, 2009).
We found that BrdU+ nuclei were scattered throughout the DVC, with the highest degree of labeling present within the NTS in control animals as well as after SCI, the lesion being without effect on the gross anatomical distribution of BrdU+ nuclei (Figure 5; Table 2). However, in contrast to the decreased BrdU labeling found in the forebrain, the total number of BrdU+ nuclei was significantly increased in all subregions of the DVC 15 dpi compared to control, non-lesioned animals (Table 2). When considering the DVC as a whole, SCI resulted in an 80 ± 9% increase in total BrdU+ nuclei.
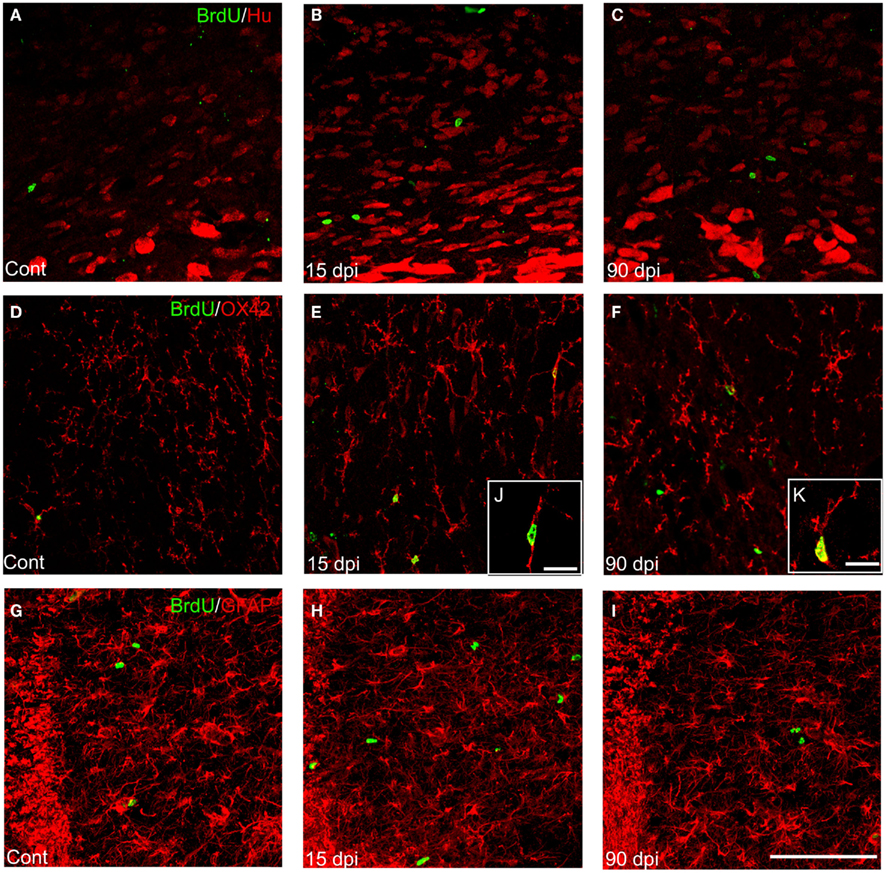
Figure 5. Distribution and quantification of newly generated neurons, astrocytes, and microglia in the DVC after SCI. (A–C) Immunohistochemistry for BrdU+ nuclei (green) and Hu+ cells (red) in the nucleus tractus solitarius (NTS) of the DVC in control (A), 15 (B), and 90 dpi (C) conditions. (D–F) Immunohistochemistry for BrdU+ nuclei (green) and OX-42+ cells (red) in the NTS of the DVC in control (D), 15 (E), and 90 dpi (F) conditions. (G–I) Immunohistochemistry for BrdU+ nuclei (green) and GFAP+ cells (red) in the NTS of the DVC in control (G), 15 (H), and 90 dpi (I) conditions. Scale bar: 100 μm. (J–K) High magnification pictures of BrdU+–OX-42+ cells at 15 (J) and 90 dpi (K) conditions. Scale bars: 15 μm. Cont, control non-injured animals; dpi, days post-injury.
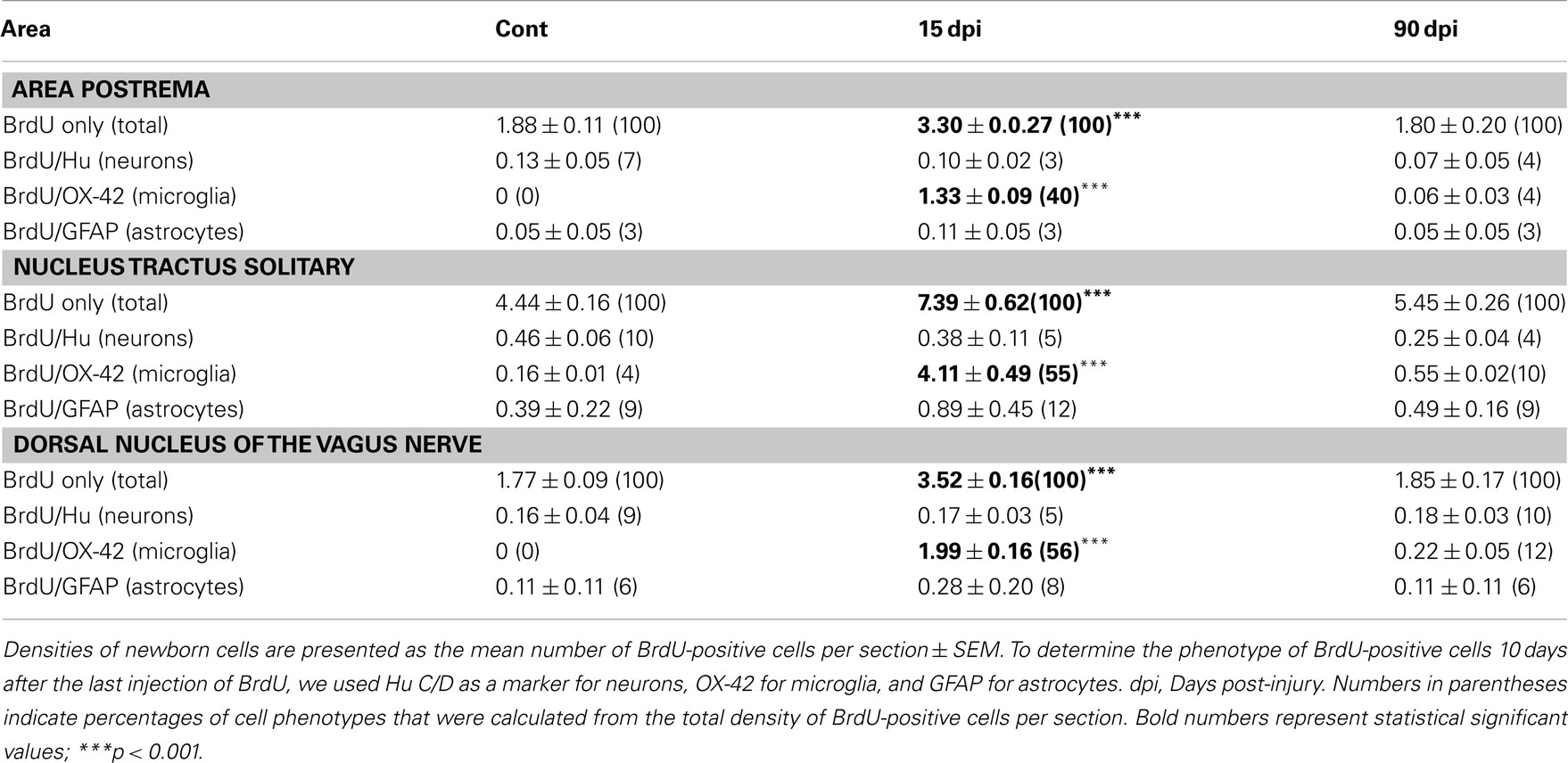
Table 2. Density and cell fate of newborn cells in the dorsal vagal complex.
We next examined the phenotypes of these BrdU+ cells by using Hu C/D, a marker of both immature and mature neurons (Barami et al., 1995), as well as GFAP, a marker of astrocytes. We found no differences in the numbers of BrdU+ cells colabeled with either marker between injured animals at 15 dpi and non-injured controls. However, dual staining for BrdU and OX-42, a marker of microglia/macrophages, revealed that the majority of BrdU+ cells observed within the DVC at 15 dpi also expressed this marker (Figure 5; Table 2). Increases in numbers of BrdU+ nuclei and BrdU+–OX-42+ were transient after SCI, since they both returned to control values by 90 dpi (Table 2). Therefore, cervical SCI triggers a transient and massive increase in the generation and/or proliferation of microglia/macrophages within the DVC at 15 dpi, with no apparent effect on neurogenesis or astrogliogenesis (Table 2).
Astrogliosis and Microgliosis Are Enhanced in the Cervical Spinal Cord Lesion Site during Subchronic SCI
Previous studies have shown that proliferating cells accumulate in the perilesion region over time following spinal cord contusion injury (Tripathi and McTigue, 2007). In order to compare, in the same animal, the proportion of these cells that could differentiate into new neurons (DCX+), astrocytes (GFAP+), or microglia/macrophages (OX-42+), and to confront these results with those obtained in the supra-spinal regions, we quantified the number of BrdU+ cells expressing these specific markers among the total BrdU+ nuclei observed in the cervical spinal cord. Ipsilateral injured and contralateral non-injured sides were analyzed separately (Figures 1 and 6; Table 3).
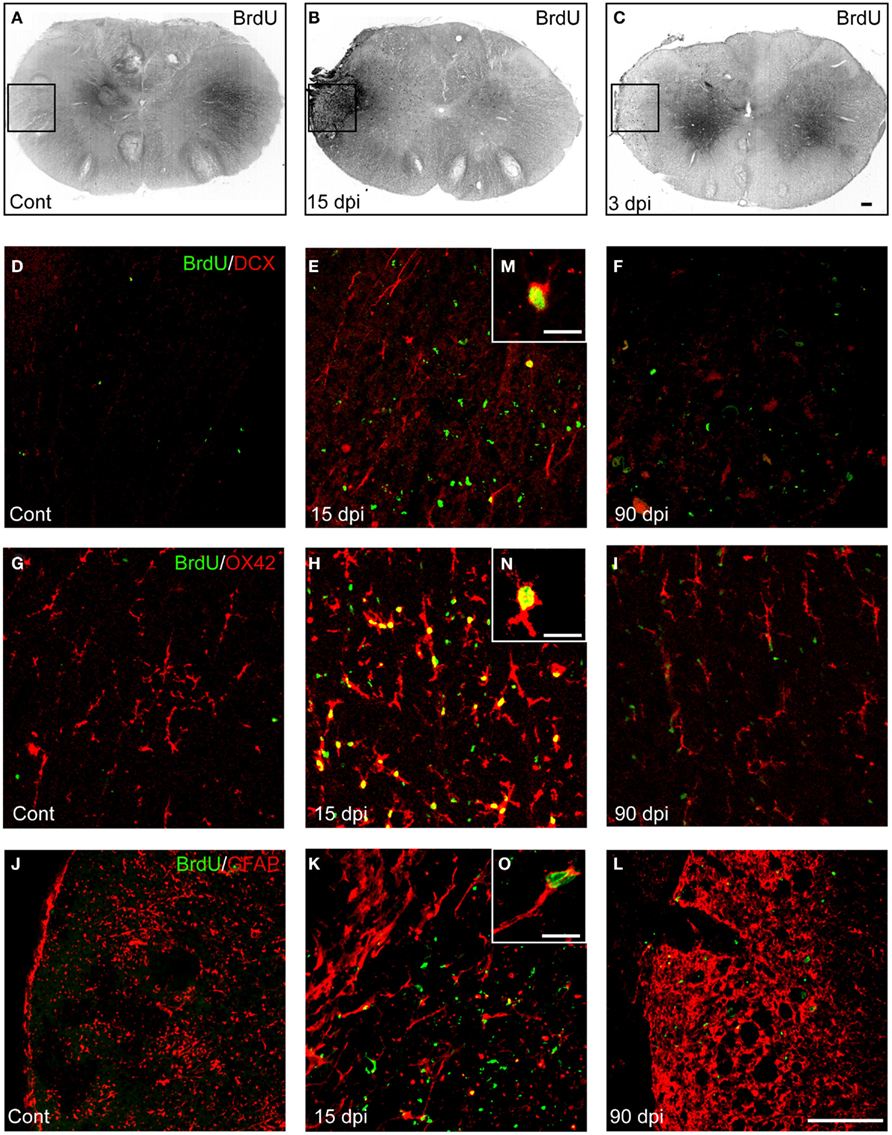
Figure 6. Distribution and quantification of newly generated neurons, astrocytes, and microglia in the spinal cord after SCI. (A–C) Immunohistochemistry for BrdU+ nuclei in the spinal cord in control (A), 15 (B), and 90 dpi (C) conditions. (D–F) Immunohistochemistry for BrdU+ nuclei (green) and Doublecortin (DCX; red) cells at the site of injury in the spinal cord in control (D), 15 (E), and 90 dpi (F) conditions. (G–I) Immunohistochemistry for BrdU+ nuclei (green) and OX-42+ cells (red) at the site of injury in the spinal cord in control (G), 15 (H), and 90 dpi (I) conditions. (J–L) Immunohistochemistry for BrdU+ nuclei (green) and GFAP+ cells (red) at the site of injury in the spinal cord in control (J), 15 (K), and 90 dpi (L) conditions. Scale bar (A–L): 100 μm. Images (D–L) correspond to high magnification of the boxed areas represented in (A–C). (M–O) High magnification pictures of BrdU+–DCX+(M), BrdU+–OX-42+ (N), and BrdU+–GFAP+ (O) cells. Scale bars: 10 μm. Cont, control non-injured animals; dpi, days post-injury.
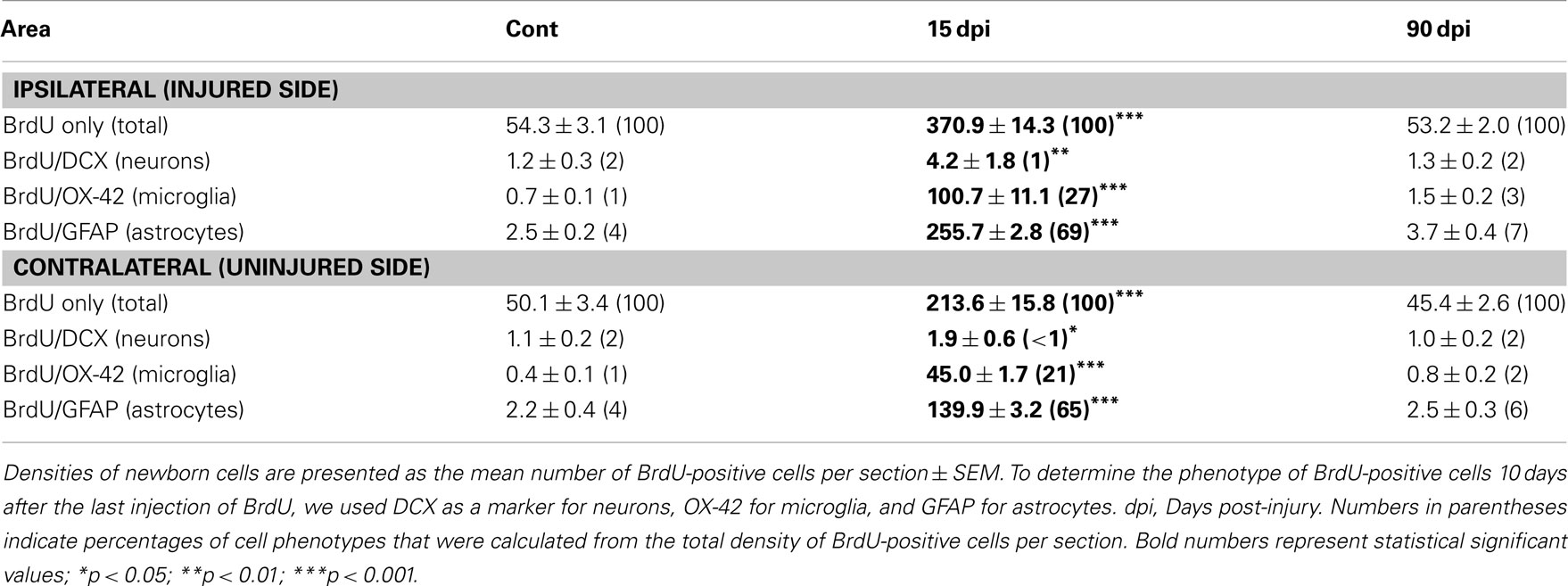
Table 3. Density and cell fate of new born cells in the cervical spinal cord.
Low magnification BrdU immunostaining revealed a considerable increase in BrdU+ cells at 15 dpi in both sides of the injured spinal cord compared to control, non-injured animals (Figures 6A–C), although this increase was greater in the injured side (6.8-fold) compared to the contralateral side (4.3-fold; Table 3). As previously reported (Vessal et al., 2007; Meletis et al., 2008), this increase was transient since the total number of BrdU+ nuclei returned to control values at 90 dpi (Table 3). Phenotype analyses of BrdU+ cells revealed that the production of newly generated immature neurons (BrdU+–DCX+ cells) was absent in the intact spinal cord and rare in the injured one at 15 dpi (Figures 6D–F,M; Table 3). However, the number of proliferating microglial cells (BrdU+–OX-42+) was significantly increased compared to non-injured animals, preferentially in the injured side (144-fold) compared to the contralateral uninjured side (113-fold; Figures 6G–I,N; Table 3). As expected, and consistent with extant literature (White et al., 2010), a large increase was also obtained for BrdU+–GFAP+ cells, again with a preferential increase in the injured side (102-fold) compared to the contralateral uninjured side (64-fold; Figures 6J–L,O; Table 3). Considering the proportion of newly generated neurons, microglia, and astrocytes among the total number of BrdU+ nuclei, we observed that at 15 dpi, newly generated neurons represented only 1% of the BrdU+ nuclei (Table 3). In contrast, microglia, and astrocytes comprised the majority of BrdU-labeled cells, with 27 and 69% of BrdU-containing cells coexpressing respectively the OX-42 microglia/macrophage and GFAP astrocyte markers (Table 3).
SCI Produces an Acute Inflammatory Response in Spinal but also in Brain Regions
It is well-known that, during the acute phase, SCI initiates a robust inflammatory response characterized by the local synthesis of cytokines and chemokines, through a coordinated infiltration of immune cells at the damaged site (Bartholdi and Schwab, 1997; Beck et al., 2010). Furthermore, inflammation has been shown to be detrimental for adult neurogenesis (Monje et al., 2003). Thus, we hypothesized that SCI triggered-inflammation could propagate to distant areas of the brain, which could explain the reduction of neurogenesis observed in the SVZ/OB system and in the hippocampal dentate gyrus, as well as the activation of microglia within the DVC. To analyze brain inflammation after SCI, we ran comparative qPCR experiments aimed at exploring the kinetics of SCI-induced transcriptional activities of candidate cytokines and chemokines in the various brain regions of interest (Figure 7). The chemokine (C–C motif) receptors CCR2 and CCR5 were respectively selected as astrocyte and microglia inflammatory markers (Asensio and Campbell, 1999), while TNFα1 was chosen as an indicator of the cytokine response after SCI (Brewer and Nolan, 2007).
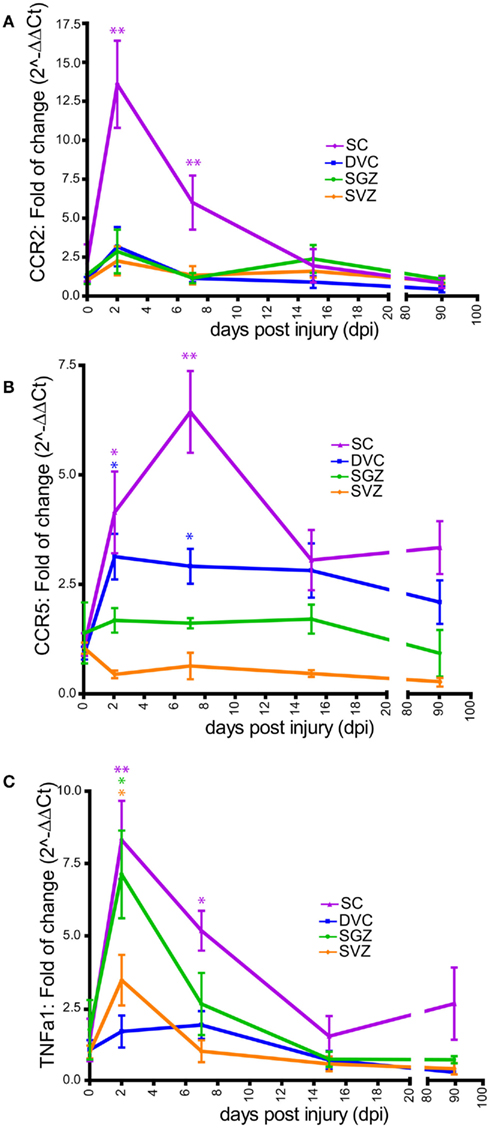
Figure 7. Time course of chemokines and cytokines mRNA expression after SCI. CCR2 (A), CCR5 (B), and TNFα1 (C) mRNAs expression were performed in non-injured rat (referred in the X-axis as 0 dpi) and four SCI rat groups (referred in X-axis as 2, 7, 15, and 90 days after SCI). For all animal groups, measurement of these mRNAs were conducted in the DVC (blue), the SGZ (green), the SVZ (orange), and the spinal cord (SC; violet). Data are presented as mean ± SEM. *p < 0.05; **p < 0.01.
We found that within the cervical spinal cord, CCR2 mRNA expression increased during the acute phase of injury, peaking at 48 h after SCI (13.5 ± 2.7-fold), and remained significantly high up to 15 dpi (Figure 7A). However, within the DVC, SGZ, or SVZ, changes in CCR2 mRNA levels were found to be non-significant at all time points examined. These results appeared consistent with the astrogliosis observed selectively in the injured spinal cord but not in other brain areas (Figure 6; Table 3). For CCR5 mRNA expression, we found a significant increased both in the spinal cord and in the DVC of injured animals during the acute phases of injury, with the highest expression levels observed in the DVC at 2 dpi (3.1 ± 0.5-fold) and in the spinal cord at 7 dpi (6.4 ± 0.9-fold). However, CCR5 mRNA expression remained unchanged in the SVZ and SGZ (Figure 7B). Overall, this pattern of expression appeared consistent with the microgliosis observed selectively in the spinal cord and in the DVC during the acute and subchronic phases of injury, but not in other brain regions examined (Figures 6 and 7; Tables 2 and 3). Finally, we found that TNFα1 mRNA expression increased significantly in the spinal cord, the SGZ, and SVZ at 48 h after SCI (8.3 ± 1.3; 7.1 ± 1.5; 3.5 ± 0.8-fold, respectively), while no significant changes in expression were found within the DVC over the time course of the analysis. TNFα1 mRNA expression steadily diminished to baseline level in the SVZ and SGZ, but remained significantly high in the spinal cord at 7 dpi (5.2 ± 0.6-fold; Figure 7C). Thus, the expression of the cytokine TNFα1 is activated in the forebrain in response to SCI, and could participate in the reduction of adult neurogenesis observed after SCI.
SCI Produces Local Variation of BDNF Protein Levels in Brain and Spinal Cord Regions
As BDNF is known to regulate adult neurogenic niches at least in the SGZ (Lee et al., 2002; Scharfman et al., 2005; Li et al., 2008), we hypothesized that SCI-induced reduction of forebrain neurogenesis could be associated with an alteration of BDNF protein levels. This assumption is supported by studies reporting a reduction of BDNF mRNA in the hippocampus after an acute SCI (Fumagalli et al., 2009), and an increased BDNF receptor TrkB mRNA in the spinal cord until 6 weeks after SCI (Frisen et al., 1992). Using BDNF immunoassays, we found no significant variation of BDNF level within the SVZ over the time course of the analysis (Figure 8), indicating that the transient decrease in OB neurogenesis is not related to variation of BDNF levels. In contrast, within the SGZ, BDNF levels transiently increased during the acute phase, reaching highest values at 2 dpi (non-injured: 51.7 ± 2.0 pg/mL; 2 dpi: 76.6 ± 7.3 pg/mL; p < 0.01). However, this early peak was followed by a significant reduction of BDNF throughout the subchronic and chronic phases of SCI (15 dpi: 26.6 ± 3.4 pg/mL, p < 0.01 and 90 dpi: 36.4 ± 2.7 pg/mL, p < 0.05). Thus, SCI-induced decrease of hippocampal neurogenesis is correlated with the reduction of BDNF protein levels. Reductions of BDNF levels were only found in the hippocampus, as they increased after SCI in the spinal cord and the DVC. Indeed, within the cervical spinal cord, BDNF levels significantly increased during the acute phase, peaking at 7 dpi (non-injured: 14.4 ± 1.4 pg/mL; 7 dpi: 39.5 ± 4.1 pg/mL; p < 0.001), then returning to control values by 90 dpi (19.4 ± 0.7 pg/mL; Figure 8). Within the DVC, BDNF levels were found to be stable until 7 dpi, but then increased as a function of time after injury, achieving significance at 90 dpi (non-injured: 21.3 ± 2.6 pg/mL; 90 dpi: 38.3 ± 3.2 pg/mL; p < 0.05).
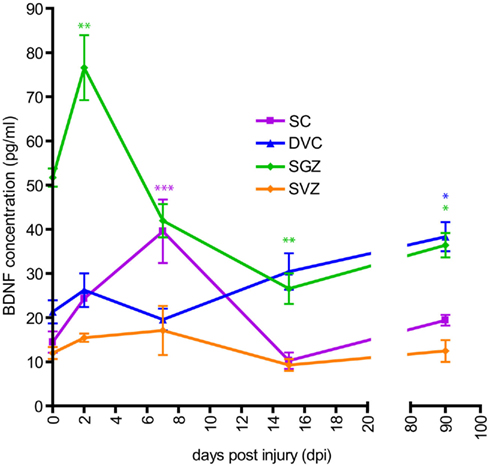
Figure 8. Time course of BDNF protein level after SCI. BDNF immunoassay (ELISA) was performed in non-injured rat (referred in the X-axis as 0 dpi) and four SCI rat groups (referred in X-axis as 2, 7, 15, and 90 days after SCI). For all animal groups, measurement of BDNF protein levels were conducted in the DVC (blue), the SGZ (green), the SVZ (orange), and the spinal cord (SC; violet). Data are presented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
Since the discovery that neural stem and progenitor cells persist into the adult mammalian brain, including in humans, the great hope for regenerative medicine is to induce intrinsic brain repair strategies using this potential. Therefore, injury-induced neurogenesis in the central nervous system has been mostly investigated near the lesion site, to search if newly generated neurons could replace the dying ones within the damaged tissue. Areas more distal to the lesion have yet to receive in-depth attention, although the intrinsic nature of the brain (i.e., a closed system facilitating the dispersion or diffusion of circulating, extracellular agents, and a highly connected organization allowing the transfer of information along neuronal networks) makes it conceivable that the consequences of a focal injury could alter uninjured nervous regions.
Here, we investigated whether a partial cervical spinal cord hemisection could modulate forebrain neurogenesis in the adult rat brain. We chose this partial injury to prevent permanent loss of locomotor activities, since physical activity is a known modulator of adult neurogenesis (van Praag et al., 1999). In addition, this model is reported to enhance post-lesional neuroplasticity and spontaneous motor recovery (Bareyre et al., 2004; Vinit and Kastner, 2009). If our study confirms the work from a plethora of publications describing massive gliosis at the site of injury, which leads to glial scar formation (Yang et al., 2006; Meletis et al., 2008), we reveal for the first time that cell dynamics in adult brain neurogenic niches is altered by a distant lesion (2–3 cm) performed at the cervical spinal cord level. Indeed, we show that SCI decreases neurogenesis in the SVZ/OB system and in the SGZ in subchronic condition. We further show that, whereas OB neurogenesis returns to control levels by 90 dpi, SGZ neurogenesis does not recover and stays 40% lower than controls in chronic condition, suggesting that the SGZ is more sensitive to the deleterious effect of SCI than the OB. Alternatively, the recovery of neurogenesis in the OB 90 days after SCI might reflect the strong plasticity of the SVZ, known to regenerate after ablation of rapidly dividing cells (Doetsch et al., 1999). At the cellular level, we provide evidence suggesting that these alterations are not due to an increase of cell death but rather to a decrease in cell proliferation rate. In the brainstem, we report that microglia is transiently activated in the DVC during the subchronic phase after SCI. Similar microglial activation was reported in this region after unilateral cervical vagotomy (Bauer et al., 2005), although this reaction was clearly ipsilateral to the lesion, a phenomenon we did not observe after partial spinal cord hemisection.
Our study thus demonstrates that, in contrast to injury-induced neurogenesis produced by TBI, SCI does not favor neurogenesis, neither locally (i.e., in the spinal cord) nor distally (i.e., in the brain). On the other hand, cervical dorsal rhizotomy was recently shown to induce reactive neurogenesis within the cervical spinal cord, as well as in the primary sensorimotor cortex of adult monkeys (Vessal and Darian-Smith, 2010). In our study, we observed a minor induction of neurogenesis in the spinal cord (only 1% of the BrdU+ cells) but not in the sensory cortex (data not shown) by comparing control versus injured animals. Many factors might explain these discrepancies, including differences in animal species (monkeys versus rats), and technical aspects of BrdU protocol (administration route, BrdU pulse-chase time period, BrdU detection). However, one major difference may reside in the type of injury (central versus peripheral nervous injury), which might affect the severity of CNS injury. Indeed, Vessal and Darian-Smith (2010) performed rhizotomies by cutting cervical dorsal rootlets through three full cervical segments. Rhizotomy limits direct damage to the spinal cord and thus CNS inflammation, while SCI at C2 level (even partial) produces an important inflammatory response in the CNS with glial scar formation. Differences in brain inflammation might thus play a key role, as inflammation is well-known to downregulate neurogenesis in the adult brain (Das and Basu, 2008; Whitney et al., 2009), whereas anti-inflammatory treatments prevent this effect (Ekdahl et al., 2003; Monje et al., 2003). In support to this neurogenesis–inflammation hypothesis, Vessal et al. (2007) have reported that whilst rhizotomy promotes spinal cord neurogenesis, no such increase is detected after a dorsal SCI. Thus, variation of adult neurogenesis after injury to the CNS is certainly dependent on the degree of inflammatory response (Das and Basu, 2008; Whitney et al., 2009), even if lesion and inflammation occur in remote CNS regions.
Here, we further explored this hypothesis, and show that SCI, performed at the cervical level, produces evidence of increased inflammation in distant forebrain areas (and in other areas) examined, which correlates well with the alterations observed at the anatomical level. Indeed, we found that SCI triggers an increased mRNA expression of the CCR5 chemokines receptor in the DVC in acute and subchronic conditions, and of TNFα1 in the SVZ and in the SGZ in acute condition. These results thus demonstrate an inflammatory reaction in distant brain areas, and are consistent with the increased microgliosis observed in the DVC (paralleled by increased CCR5 expression), and with the decreased neurogenesis observed in the SVZ and the SGZ (paralleled by increased TNFα1 expression). Thus our overall findings suggest that the short-term decrease in forebrain neurogenesis could be caused by brain inflammation.
Adult neurogenesis is a highly regulated process that is modulated by both extrinsic and intrinsic factors (Lledo et al., 2006). Given the consequences of SCI on adult neurogenesis and the long distance between the site of injury and neurogenic niches, other diffusible factors such as growth factors or neurotrophins could mediate these effects. In particular, it is known that endogenous BDNF or its receptor, TrkB, are required to maintain basal neurogenesis in the SGZ (Lee et al., 2002; Li et al., 2008), and that exogenous BDNF stimulates adult rat SGZ neurogenesis (Scharfman et al., 2005). In line with these data, our results suggest that the reduction of BDNF protein levels might be responsible for the decreased SGZ neurogenesis. However, it is important to note that BDNF is expressed at high level in dentate gyrus neurons (Conner et al., 1997; Yan et al., 1997). Therefore, the decline in BDNF protein levels could be instead the consequence of the reduced hippocampal neurogenesis. Importantly, these two mechanisms are not mutually exclusive and could act on each other.
In contrast, we do not find such correlation between neurogenesis and BDNF protein levels in the SVZ after SCI, which is consistent with a recent study demonstrating that BDNF signaling does not affect SVZ neurogenesis in the adult rat (Galvao et al., 2008).
Studies have already reported that SCI induces cortical and subcortical map remodeling, indicating that the mammalian brain is responsive to SCI (Bareyre et al., 2004; Endo et al., 2007; Kaas et al., 2008). Whether such neuroplasticity changes are beneficial or detrimental remains the object of debate given they are balanced with growing evidence derived from human spinal cord injured patients which demonstrate cortical atrophy, deficit of motor brain functions, and abnormalities in emotion-related brain activities (Cramer et al., 2005; Nicotra et al., 2006; Freund et al., 2011). Our work provides the first demonstration that forebrain neurogenesis is vulnerable to SCI and supports the current idea that SCI has a distal negative impact in some part of the brain. Deciphering the mechanisms that underlie this vulnerability would certainly offer strategies aimed at protecting the brain subsequent to SCI.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Marie-Pierre Blanchard for confocal microscopy technical assistance and the Imaging and microscopy center core facility from the Jean-Roche Institute, Marseille, France. We thank Professor François Féron (Aix-Marseille University, France) and Dr. Pascale Durbec for constructive comments on this work. We are grateful to Professor Linda S. Pagani (Montreal University, Canada) for corrections. M. S. Félix was supported by a Ph.D. fellowship from the French Minister for Research and Technology. This work was supported by grants from the Institut de Recherche sur la Moelle épinière et l’Encéphale (IRME #2009–2011, Paris, France) and Centre National de Recherche Scientifique (CNRS #2009-2011, France).
References
Altman, J., and Das, G. D. (1965). Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J. Comp. Neurol. 124, 319–335.
Alvarez-Buylla, A., and Garcia-Verdugo, J. M. (2002). Neurogenesis in adult subventricular zone. J. Neurosci. 22, 629–634.
Anderson, K. D., Gunawan, A., and Steward, O. (2005). Quantitative assessment of forelimb motor function after cervical spinal cord injury in rats: relationship to the corticospinal tract. Exp. Neurol. 194, 161–174.
Asensio, V. C., and Campbell, I. L. (1999). Chemokines in the CNS: plurifunctional mediators in diverse states. Trends Neurosci. 22, 504–512.
Barami, K., Iversen, K., Furneaux, H., and Goldman, S. A. (1995). Hu protein as an early marker of neuronal phenotypic differentiation by subependymal zone cells of the adult songbird forebrain. J. Neurobiol. 28, 82–101.
Bareyre, F. M., Kerschensteiner, M., Raineteau, O., Mettenleiter, T. C., Weinmann, O., and Schwab, M. E. (2004). The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat. Neurosci. 7, 269–277.
Bartholdi, D., and Schwab, M. E. (1997). Expression of pro-inflammatory cytokine and chemokine mRNA upon experimental spinal cord injury in mouse: an in situ hybridization study. Eur. J. Neurosci. 9, 1422–1438.
Bauer, S., Hay, M., Amilhon, B., Jean, A., and Moyse, E. (2005). In vivo neurogenesis in the dorsal vagal complex of the adult rat brainstem. Neuroscience 130, 75–90.
Beck, K. D., Nguyen, H. X., Galvan, M. D., Salazar, D. L., Woodruff, T. M., and Anderson, A. J. (2010). Quantitative analysis of cellular inflammation after traumatic spinal cord injury: evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain 133, 433–447.
Blaiss, C. A., Yu, T. S., Zhang, G., Chen, J., Dimchev, G., Parada, L. F., Powell, C. M., and Kernie, S. G. (2011). Temporally specified genetic ablation of neurogenesis impairs cognitive recovery after traumatic brain injury. J. Neurosci. 31, 4906–4916.
Bouzioukh, F., Tell, F., Jean, A., and Rougon, G. (2001). NMDA receptor and nitric oxide synthase activation regulate polysialylated neural cell adhesion molecule expression in adult brainstem synapses. J. Neurosci. 21, 4721–4730.
Brewer, K. L., and Nolan, T. A. (2007). Spinal and supraspinal changes in tumor necrosis factor-alpha expression following excitotoxic spinal cord injury. J. Mol. Neurosci. 31, 13–21.
Brown, J. P., Couillard-Despres, S., Cooper-Kuhn, C. M., Winkler, J., Aigner, L., and Kuhn, H. G. (2003). Transient expression of doublecortin during adult neurogenesis. J. Comp. Neurol. 467, 1–10.
Charrier, C., Coronas, V., Fombonne, J., Roger, M., Jean, A., Krantic, S., and Moyse, E. (2006). Characterization of neural stem cells in the dorsal vagal complex of adult rat by in vivo proliferation labeling and in vitro neurosphere assay. Neuroscience 138, 5–16.
Chi, L., Ke, Y., Luo, C., Li, B., Gozal, D., Kalyanaraman, B., and Liu, R. (2006). Motor neuron degeneration promotes neural progenitor cell proliferation, migration, and neurogenesis in the spinal cords of amyotrophic lateral sclerosis mice. Stem Cells 24, 34–43.
Conner, J. M., Lauterborn, J. C., Yan, Q., Gall, C. M., and Varon, S. (1997). Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J. Neurosci. 17, 2295–2313.
Cramer, S. C., Lastra, L., Lacourse, M. G., and Cohen, M. J. (2005). Brain motor system function after chronic, complete spinal cord injury. Brain 128, 2941–2950.
Darian-Smith, C. (2009). Synaptic plasticity, neurogenesis, and functional recovery after spinal cord injury. Neuroscientist 15, 149–165.
Das, S., and Basu, A. (2008). Inflammation: a new candidate in modulating adult neurogenesis. J. Neurosci. Res. 86, 1199–1208.
Doetsch, F., and Alvarez-Buylla, A. (1996). Network of tangential pathways for neuronal migration in adult mammalian brain. Proc. Natl. Acad. Sci. U.S.A. 93, 14895–14900.
Doetsch, F., Caille, I., Lim, D. A., Garcia-Verdugo, J. M., and Alvarez-Buylla, A. (1999). Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97, 703–716.
Dromard, C., Guillon, H., Rigau, V., Ripoll, C., Sabourin, J. C., Perrin, F. E., Scamps, F., Bozza, S., Sabatier, P., Lonjon, N., Duffau, H., Vachiery-Lahaye, F., Prieto, M., Tran Van Ba, C., Deleyrolle, L., Boularan, A., Langley, K., Gaviria, M., Privat, A., Hugnot, J. P., and Bauchet, L. (2008). Adult human spinal cord harbors neural precursor cells that generate neurons and glial cells in vitro. J. Neurosci. Res. 86, 1916–1926.
Eisch, A. J., Barrot, M., Schad, C. A., Self, D. W., and Nestler, E. J. (2000). Opiates inhibit neurogenesis in the adult rat hippocampus. Proc. Natl. Acad. Sci. U.S.A. 97, 7579–7584.
Ekdahl, C. T., Claasen, J. H., Bonde, S., Kokaia, Z., and Lindvall, O. (2003). Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. U.S.A. 100, 13632–13637.
Endo, T., Spenger, C., Tominaga, T., Brene, S., and Olson, L. (2007). Cortical sensory map rearrangement after spinal cord injury: fMRI responses linked to Nogo signalling. Brain 130, 2951–2961.
Freund, P., Weiskopf, N., Ward, N. S., Hutton, C., Gall, A., Ciccarelli, O., Craggs, M., Friston, K., and Thompson, A. J. (2011). Disability, atrophy and cortical reorganization following spinal cord injury. Brain. 134, 1610–1622.
Frisen, J., Johansson, C. B., Torok, C., Risling, M., and Lendahl, U. (1995). Rapid, widespread, and longlasting induction of nestin contributes to the generation of glial scar tissue after CNS injury. J. Cell Biol. 131, 453–464.
Frisen, J., Verge, V. M., Cullheim, S., Persson, H., Fried, K., Middlemas, D. S., Hunter, T., Hokfelt, T., and Risling, M. (1992). Increased levels of trkB mRNA and trkB protein-like immunoreactivity in the injured rat and cat spinal cord. Proc. Natl. Acad. Sci. U.S.A. 89, 11282–11286.
Fumagalli, F., Madaschi, L., Caffino, L., Marfia, G., Di Giulio, A. M., Racagni, G., and Gorio, A. (2009). Acute spinal cord injury reduces brain derived neurotrohic factor expression in rat hippocampus. Neuroscience 159, 936–939.
Galvao, R. P., Garcia-Verdugo, J. M., and Alvarez-Buylla, A. (2008). Brain-derived neurotrophic factor signaling does not stimulate subventricular zone neurogenesis in adult mice and rats. J. Neurosci. 28, 13368–13383.
Horky, L. L., Galimi, F., Gage, F. H., and Horner, P. J. (2006). Fate of endogenous stem/progenitor cells following spinal cord injury. J. Comp. Neurol. 498, 525–538.
Horner, P. J., Power, A. E., Kempermann, G., Kuhn, H. G., Palmer, T. D., Winkler, J., Thal, L. J., and Gage, F. H. (2000). Proliferation and differentiation of progenitor cells throughout the intact adult rat spinal cord. J. Neurosci. 20, 2218–2228.
Kaas, J. H., Qi, H. X., Burish, M. J., Gharbawie, O. A., Onifer, S. M., and Massey, J. M. (2008). Cortical and subcortical plasticity in the brains of humans, primates, and rats after damage to sensory afferents in the dorsal columns of the spinal cord. Exp. Neurol. 209, 407–416.
Kazanis, I. (2009). The subependymal zone neurogenic niche: a beating heart in the centre of the brain: how plastic is adult neurogenesis? Opportunities for therapy and questions to be addressed. Brain 132, 2909–2921.
Ke, Y., Chi, L., Xu, R., Luo, C., Gozal, D., and Liu, R. (2006). Early response of endogenous adult neural progenitor cells to acute spinal cord injury in mice. Stem Cells 24, 1011–1019.
Kempermann, G., Kuhn, H. G., and Gage, F. H. (1998). Experience-induced neurogenesis in the senescent dentate gyrus. J. Neurosci. 18, 3206–3212.
Kernie, S. G., Erwin, T. M., and Parada, L. F. (2001). Brain remodeling due to neuronal and astrocytic proliferation after controlled cortical injury in mice. J. Neurosci. Res. 66, 317–326.
Kernie, S. G., and Parent, J. M. (2010). Forebrain neurogenesis after focal ischemic and traumatic brain injury. Neurobiol. Dis. 37, 267–274.
Krassioukov, A. (2009). Autonomic function following cervical spinal cord injury. Respir. Physiol. Neurobiol. 169, 157–164.
Lee, J., Duan, W., and Mattson, M. P. (2002). Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J. Neurochem. 82, 1367–1375.
Li, Y., Luikart, B. W., Birnbaum, S., Chen, J., Kwon, C. H., Kernie, S. G., Bassel-Duby, R., and Parada, L. F. (2008). TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 59, 399–412.
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408.
Lledo, P. M., Alonso, M., and Grubb, M. S. (2006). Adult neurogenesis and functional plasticity in neuronal circuits. Nat. Rev. Neurosci. 7, 179–193.
Lois, C., and Alvarez-Buylla, A. (1994). Long-distance neuronal migration in the adult mammalian brain. Science 264, 1145–1148.
Meletis, K., Barnabe-Heider, F., Carlen, M., Evergren, E., Tomilin, N., Shupliakov, O., and Frisen, J. (2008). Spinal cord injury reveals multilineage differentiation of ependymal cells. PLoS Biol. 6, e182. doi:10.1371/journal.pbio.0060182
Monje, M. L., Toda, H., and Palmer, T. D. (2003). Inflammatory blockade restores adult hippocampal neurogenesis. Science 302, 1760–1765.
National Research Council. (1996). The Guide for the Care and Use of Laboratory Animals. Washington: The National Academies Press.
Nicotra, A., Critchley, H. D., Mathias, C. J., and Dolan, R. J. (2006). Emotional and autonomic consequences of spinal cord injury explored using functional brain imaging. Brain 129, 718–728.
Nishimura, Y., and Isa, T. (2009). Compensatory changes at the cerebral cortical level after spinal cord injury. Neuroscientist 15, 436–444.
Ohori, Y., Yamamoto, S., Nagao, M., Sugimori, M., Yamamoto, N., Nakamura, K., and Nakafuku, M. (2006). Growth factor treatment and genetic manipulation stimulate neurogenesis and oligodendrogenesis by endogenous neural progenitors in the injured adult spinal cord. J. Neurosci. 26, 11948–11960.
Paxinos, G., and Watson, C. (1986). The Rat Brain in Stereotaxic Coordinates, 5th Edn. London: Academic Press.
Richardson, R. M., Singh, A., Sun, D., Fillmore, H. L., Dietrich, D. W. III, and Bullock, M. R. (2010). Stem cell biology in traumatic brain injury: effects of injury and strategies for repair. J. Neurosurg. 112, 1125–1138.
Rola, R., Mizumatsu, S., Otsuka, S., Morhardt, D. R., Noble-Haeusslein, L. J., Fishman, K., Potts, M. B., and Fike, J. R. (2006). Alterations in hippocampal neurogenesis following traumatic brain injury in mice. Exp. Neurol. 202, 189–199.
Rolls, A., Shechter, R., and Schwartz, M. (2009). The bright side of the glial scar in CNS repair. Nat. Rev. Neurosci. 10, 235–241.
Scharfman, H., Goodman, J., Macleod, A., Phani, S., Antonelli, C., and Croll, S. (2005). Increased neurogenesis and the ectopic granule cells after intrahippocampal BDNF infusion in adult rats. Exp. Neurol. 192, 348–356.
Seaberg, R. M., and van der Kooy, D. (2002). Adult rodent neurogenic regions: the ventricular subependyma contains neural stem cells, but the dentate gyrus contains restricted progenitors. J. Neurosci. 22, 1784–1793.
Shechter, R., Ziv, Y., and Schwartz, M. (2007). New GABAergic interneurons supported by myelin-specific T cells are formed in intact adult spinal cord. Stem Cells 25, 2277–2282.
Sun, D., McGinn, M. J., Zhou, Z., Harvey, H. B., Bullock, M. R., and Colello, R. J. (2007). Anatomical integration of newly generated dentate granule neurons following traumatic brain injury in adult rats and its association to cognitive recovery. Exp. Neurol. 204, 264–272.
Tripathi, R., and McTigue, D. M. (2007). Prominent oligodendrocyte genesis along the border of spinal contusion lesions. Glia 55, 698–711.
Urrea, C., Castellanos, D. A., Sagen, J., Tsoulfas, P., Bramlett, H. M., and Dietrich, W. D. (2007). Widespread cellular proliferation and focal neurogenesis after traumatic brain injury in the rat. Restor. Neurol. Neurosci. 25, 65–76.
van Praag, H., Kempermann, G., and Gage, F. H. (1999). Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat. Neurosci. 2, 266–270.
Vessal, M., Aycock, A., Garton, M. T., Ciferri, M., and Darian-Smith, C. (2007). Adult neurogenesis in primate and rodent spinal cord: comparing a cervical dorsal rhizotomy with a dorsal column transection. Eur. J. Neurosci. 26, 2777–2794.
Vessal, M., and Darian-Smith, C. (2010). Adult neurogenesis occurs in primate sensorimotor cortex following cervical dorsal rhizotomy. J. Neurosci. 30, 8613–8623.
Vinit, S., Gauthier, P., Stamegna, J. C., and Kastner, A. (2006). High cervical lateral spinal cord injury results in long-term ipsilateral hemidiaphragm paralysis. J. Neurotrauma 23, 1137–1146.
Vinit, S., and Kastner, A. (2009). Descending bulbospinal pathways and recovery of respiratory motor function following spinal cord injury. Respir. Physiol. Neurobiol. 169, 115–122.
White, R. E., McTigue, D. M., and Jakeman, L. B. (2010). Regional heterogeneity in astrocyte responses following contusive spinal cord injury in mice. J. Comp. Neurol. 518, 1370–1390.
Whitney, N. P., Eidem, T. M., Peng, H., Huang, Y., and Zheng, J. C. (2009). Inflammation mediates varying effects in neurogenesis: relevance to the pathogenesis of brain injury and neurodegenerative disorders. J. Neurochem. 108, 1343–1359.
Wrathall, J. R., and Lytle, J. M. (2008). Stem cells in spinal cord injury. Dis. Markers 24, 239–250.
Yan, Q., Rosenfeld, R. D., Matheson, C. R., Hawkins, N., Lopez, O. T., Bennett, L., and Welcher, A. A. (1997). Expression of brain-derived neurotrophic factor protein in the adult rat central nervous system. Neuroscience 78, 431–448.
Yang, H., Lu, P., McKay, H. M., Bernot, T., Keirstead, H., Steward, O., Gage, F. H., Edgerton, V. R., and Tuszynski, M. H. (2006). Endogenous neurogenesis replaces oligodendrocytes and astrocytes after primate spinal cord injury. J. Neurosci. 26, 2157–2166.
Keywords: adult neurogenesis, cervical trauma, brain derived neurotrophic factor, inflammation, plasticity, hippocampus, subventricular zone, subgranular zone
Citation: Felix M-S, Popa N, Djelloul M, Boucraut J, Gauthier P, Bauer S and Matarazzo VA (2012) Alteration of forebrain neurogenesis after cervical spinal cord injury in the adult rat. Front. Neurosci. 6:45. doi: 10.3389/fnins.2012.00045
Received: 17 November 2011; Accepted: 19 March 2012;
Published online: 09 April 2012.
Edited by:
James E. Goldman, Columbia University, USAReviewed by:
Stephen C. Noctor, University of California Davis, USAFrancesca Ciccolini, University of Heidelberg, Germany
Jozsef Z. Kiss, University of Geneva, Switzerland
Copyright: © 2012 Felix, Popa, Djelloul, Boucraut, Gauthier, Bauer and Matarazzo. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Valery A. Matarazzo, Institut de Neurobiologie de la Méditerranée , INSERM U901, Aix-Marseille Université, 163 Route de Luminy, BP13, 13273, Marseille, France. e-mail:dmFsZXJ5Lm1hdGFyYXp6b0BpbnNlcm0uZnI=
†Present Address: Marie-Solenne Felix, Sylvian Bauer and Valery A. Matarazzo, Institut de Neurobiologie de la Méditerranée, INSERM U901, Aix-Marseille Université, 163 Route de Luminy, BP13, 13273 Marseille, France; Patrick Gauthier, Laboratoire de Neurosciences Intégratives et Adaptatives, Marseille, France.