 Orly Lazarov* and Michael P. Demars
Orly Lazarov* and Michael P. Demars
- Department of Anatomy and Cell Biology, College of Medicine, The University of Illinois at Chicago, Chicago, IL, USA
Recent intriguing evidence suggests that metabolites of amyloid precursor protein (APP), mutated in familial forms of Alzheimer’s disease (AD), play critical roles in developmental and postnatal neurogenesis. Of note is soluble APPα (sAPPα) that regulates neural progenitor cell proliferation. The APP family encompasses a group of ubiquitously expressed and evolutionarily conserved, type I transmembrane glycoproteins, whose functions have yet to be fully elucidated. APP can undergo proteolytic cleavage by mutually exclusive pathways. The subtle structural differences between metabolites generated in the different pathways, as well as their equilibrium, may be crucial for neuronal function. The implications of this new body of evidence are significant. Miscleavage of APP would readily impact developmental and postnatal neurogenesis, which might contribute to cognitive deficits characterizing Alzheimer’s disease. This review will discuss the implications of the role of the APP family in neurogenesis for neuronal development, cognitive function, and brain disorders that compromise learning and memory, such as AD.
Introduction
The adult mammalian brain contains pools of neural stem cells (NSC) in two discrete regions: the subventricular zone (SVZ; Lois and Alvarez-Buylla, 1994; Doetsch et al., 1999), lining the lateral ventricle and the subgranular layer (SGL) of the dentate gyrus (DG) in the hippocampal formation (Cameron et al., 1993; Kuhn et al., 1996; Kempermann et al., 1997a). There is evidence to suggest that neurogenesis takes place in the human brain as well (Eriksson et al., 1998). In the SVZ, newly generated neural progenitor cells (NPC) migrate in chains toward the olfactory bulb (Lois and Alvarez-Buylla, 1994; Lois et al., 1996) where they are thought to replace most of the inner granule layer and 50% of the superficial zone (Imayoshi et al., 2008). In the DG, NPC from the SGL migrate only a short distance to the inner one-third of the granule layer where they functionally incorporate and show characteristic electrophysiological properties (van Praag et al., 2002; Zhao et al., 2006; for review see Ming and Song, 2011). The neurogenic niche is thought to be a tightly regulated environment where a combination of factors allows for the constant self-renewal and proliferation of NSC throughout the lifespan of an organism as displayed by the inability of SVZ NSC to proliferate and form neurons when injected into non-neurogenic regions (Gage, 2000; Lim et al., 2000). Individualized cross talk of the NSC and the neurogenic niche permits cell differentiation, migration, and ultimately incorporation into existing functional circuits (Reviewed in Ihrie and Alvarez-Buylla, 2011; Ming and Song, 2011).
The functional role of neurogenesis in the adult hippocampus has yet to be fully elucidated. Because NPC incorporate into the DG of the hippocampus and the olfactory bulb, regions associated with different types of learning and memory, much of the focus of the biological function of neurogenesis has been on a potential role in these processes. The initial evidence for neurogenic involvement in memory formation came from the seasonal changes in songbird neurogenesis. It was shown that the newly born cells responded to songs and that neurogenic increases could be correlated with seasonal and hormonal changes associated with song learning (Nottebohm, 2004). Studies in mammals using different strains of mice (Kempermann et al., 1997a), environmental enrichment (Kempermann et al., 1997b) and running (van Praag et al., 1999) have each shown a correlation between increases in neurogenesis and enhanced performance on a spatial memory task. Conversely, experiments using different aged rats (Bizon and Gallagher, 2003), stress paradigms (Lemaire et al., 2000), irradiation (Madsen et al., 2003; Raber et al., 2004; Rola et al., 2004), and the DNA methylating agent methylazoxy-methanol acetate (MAM; Shors et al., 2001; Shors et al., 2002) have each shown correlations between a decrease in neurogenesis and impairment on hippocampal-dependent memory tasks though studies have provided conflicting results as to which memory paradigms are affected. Genetic manipulations have provided further evidence for the role of neurogenic processes in memory formation. Deletion of either neurotrophin 3 (NT-3; Shimazu et al., 2006) or methyl-CpG binding protein 1 (MBD1), a methylated DNA binding protein (Zhao et al., 2003), produced a decrease in neurogenesis as well as an impairment on the Morris water-maze task. A study using both irradiation and glial fibrillary acidic protein (GFAP)-thymidine kinase (TK) transgenic mice that express a herpes virus TK in progenitor cells allowing for ablation of these cells through ganciclovir administration showed that spatial memory was retained while contextual fear conditioning was impaired. Long-term potentiation in the DG was also impaired in both models of neurogenic ablation (Saxe et al., 2006). Also of note, another study showed the preferential recruitment of new neurons into spatial memory networks following water-maze learning using the expression of c-fos, an immediate early gene, to examine neuronal excitation (Kee et al., 2007). Furthermore, an inducible deletion of TLX from the adult mouse brain drastically reduced neurogenesis and impaired spatial learning (Zhang et al., 2008). In that regard, ablation of hippocampal neurogenesis by inducible Bax expression in NPC results in deficits in Novelty detection, contextual fear conditioning, place learning, and spatial navigation (Dupret et al., 2008). Emerging evidence also suggests that neurogenesis is crucial to pattern separation in the DG, a set of processes in which converts similar experiences into discrete events (Clelland et al., 2009; Sahay et al., 2011).
In the SVZ-olfactory bulb system, the functional significance of neurogenesis has proven more difficult to elucidate. Though NPC from the SVZ replace nearly all of the inner granule layer and 50% of the superficial zone (Imayoshi et al., 2008), ablation of neural cell adhesion molecule (NCAM) in mice does not impair olfactory discrimination learning, and impaired migration through the (RMS) due to a knock-out of Bax does not impair olfactory discrimination or memory (Schellinck et al., 2004; Kim et al., 2007). However, in another study of the NCAM-null model, it was shown that alterations in NPC migration led to a 40% reduction in inner granule interneurons and fine olfactory discrimination deficits but normal olfactory threshold and memory (Gheusi et al., 2000). More recent evidence has emerged that newly generated neurons are vital to paternal and maternal behaviors intimately associated with olfactory cues (Mak and Weiss, 2010; Sakamoto et al., 2011). Short-term olfactory memory improvement associated with olfactory enrichment depends on newborn neurons (Rochefort et al., 2002). In support of that, ablation of new neurons in the olfactory bulb by AraC treatment results in reduced short-term olfactory memory, but did not affect spontaneous odor discrimination and long-term odor-associative memory tasks (Breton-Provencher et al., 2009). In contrast, deletion of new neurons in the olfactory bulb of adult mice by focal irradiation impaired long-term rather than short-term olfactory memory (Lazarini et al., 2009). Using a combination of behavioral training and pharmacological treatment for the manipulation of neurogenesis in the SVZ, Sultan et al. (2010) show that newborn neurons participate in the neural representation of the learned odor, which is used on recall of the task. Moreover, learning selects new neurons to survive in an odor-specific area of the olfactory bulb. These selected neurons are required for long-term olfaction memory but not for task acquisition (Sultan et al., 2010).
In addition, another study suggests that a more complex olfactory associated tasks such as olfactory perceptual learning seem to require ongoing neurogenesis (Moreno et al., 2009), suggesting that newly formed cells may be specially adapted to handle fine or complex olfactory function. It was recently suggested that conflicting results concerning the functional significance of adult neurogenesis in olfaction may stem from differences in the behavioral paradigms used in these studies. Specifically, this study suggests that the use of an operant component, in which a reward is associated with olfactory task, reinforces an active behavior. This behavior represents more faithfully long-term olfaction memory that is neurogenesis-dependent (Mandairon et al., 2011). Regardless, the difficulty in obtaining consistent results concerning the function of new neurons in olfaction memory and in hippocampus-dependent memory, may underlie, at least in part, controversial data described below, concerning the functional significance of specific metabolites in neurogenesis, and in learning and memory.
Complementary information concerning the functional significance of neurogenesis may be obtained from learning and memory disorders, such as Alzheimer’s disease (AD). AD is a progressive neurodegenerative disease characterized by progressive loss of memory, impaired learning, and cognitive deterioration. Individuals affected with the disease experience loss of olfaction-dependent and hippocampal-dependent function. Intriguingly, increasing evidence suggests that in mouse models of familial Alzheimer’s disease (FAD), neurogenesis is impaired early in life, preceding hallmarks of the disease, such as amyloid deposition, tau hyperphosphorylation, and cognitive impairments (Li et al., 2008; Rodriguez et al., 2008; Demars et al., 2010b; Gang et al., 2011; for review see Lazarov and Marr, 2010). Nevertheless, the mechanism underlying impaired neurogenesis in AD is not fully elucidated. Familial, rare forms of the disease (FAD) are caused by mutations in amyloid precursor protein (APP), presenilin-1, and 2 (PS1,2; For review see Selkoe, 2001). Interestingly, it becomes clear that both APP and PS1, the catalytic core of one of the enzymes that cleaves APP, play a role in regulation of neurogenesis during development and postnatally. This review will discuss emerging information of the role of the APP family members in neurogenesis.
The APP Family
The APP family is a group of ubiquitously expressed and evolutionarily conserved, type I transmembrane glycoproteins, whose functions have yet to be fully elucidated (Figure 1). In mammals, this gene family includes APP, amyloid precursor-like proteins 1 & 2 (APLP1 & 2). Apl-1 and Appl are the worm and the fly homolog, respectively. The APLP’s show strong sequence homology to APP, particularly in the C-terminal domain (Wasco et al., 1992, 1993). APP undergoes extensive proteolysis by at least three enzymatic activities termed α-, β-, γ-secretase, leading to the production of a number of intra- and extracellular metabolites. Of note, APP processing resembles Notch receptor processing (Figure 2), for review see Hartmann et al. (2001). In the non-amyloidogenic pathway, APP is cleaved by α-secretase, followed by regulated intra-membrane proteolysis (RIP) by the aspartyl protease γ-secretase. Several enzymes have been shown to have α-secretase activity including members of the “a disintegrin and metalloproteinase” (ADAM) family, ADAM9, ADAM10 (Lammich et al., 1999), and ADAM17 (Buxbaum et al., 1998; Asai et al., 2003; Postina et al., 2004) and β-site APP cleaving enzyme 2 (BACE2; Yan et al., 2001). This cleavage event releases sAPPα to the extracellular space and leaves a membrane-tethered APP C-terminal fragment (APP-CTF; Figure 2; Sisodia et al., 1990). Recent studies suggest that ADAM10 is the major α-secretase of the brain both in development and in the adult (Jorissen et al., 2010). Subsequent RIP by γ secretase releases the APP intracellular domain (AICD) that can translocate to the nucleus and a P3 peptide (Haass and Selkoe, 1993; Yu et al., 2000; Kimberly et al., 2001; Francis et al., 2002). PS are the catalytic core of the aspartyl protease γ-secretase complex (Wolfe et al., 1999; Yamasaki et al., 2006). In the amyloidogenic cleavage pathway, APP is first cleaved by β-secretase to produce sAPPβ and a βAPP-CTF. This cleavage event is mediated by β-site APP cleaving enzyme 1 (BACE1; Vassar et al., 1999). Subsequent cleavage by γ-secretase yields AICD as well as the amyloid β (Aβ) peptide that aggregates to form the plaques that are one of the pathophysiological hallmarks of AD (Reviewed in Selkoe, 2001). The abundance of metabolites confers great complexity to the function of APP but makes the interpretation of studies on APP transgenic and knockout mice a challenging task. Here we will attempt to dissociate the functions of each of the metabolites as they relate to neurogenesis and try to understand the function of each individually to understand how cleavage patterns may contribute to overall APP function.
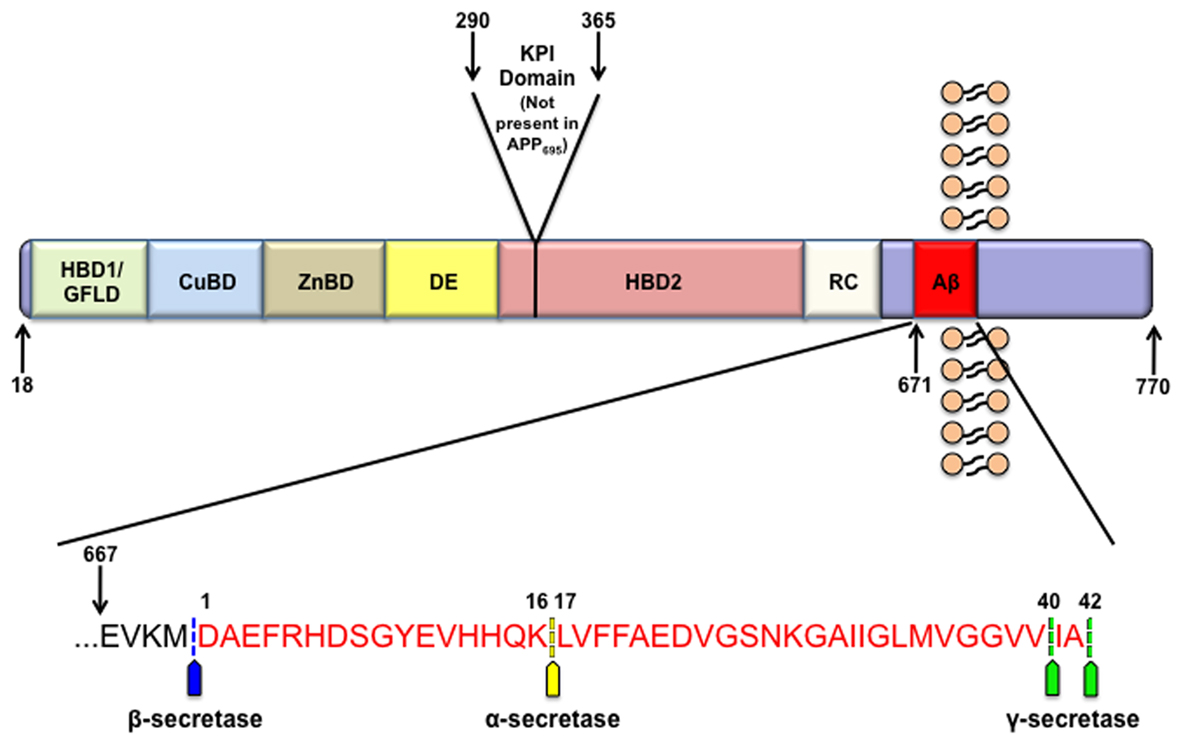
Figure 1. APP structure. APP contains many functional domains as illustrated. The three most abundant isoforms of APP are APP770, APP751, and the predominantly neuronal APP695. From the N-terminal region these domains include a heparin binding and growth factor like domain (HBD1/GFLD), a copper binding domain (CuBD), zinc binding domain (ZnBD), an acidic region (DE), Kunitz-type protease inhibitor domain (KPI; not present in APP695), a second heparin binding domain (HBD2), random coiled region (RC), and the amyloid beta domain (Aβ). The inset displays the human amyloid beta sequence in red along with the sites of APP cleavage by α-, β-, and γ-secretase.
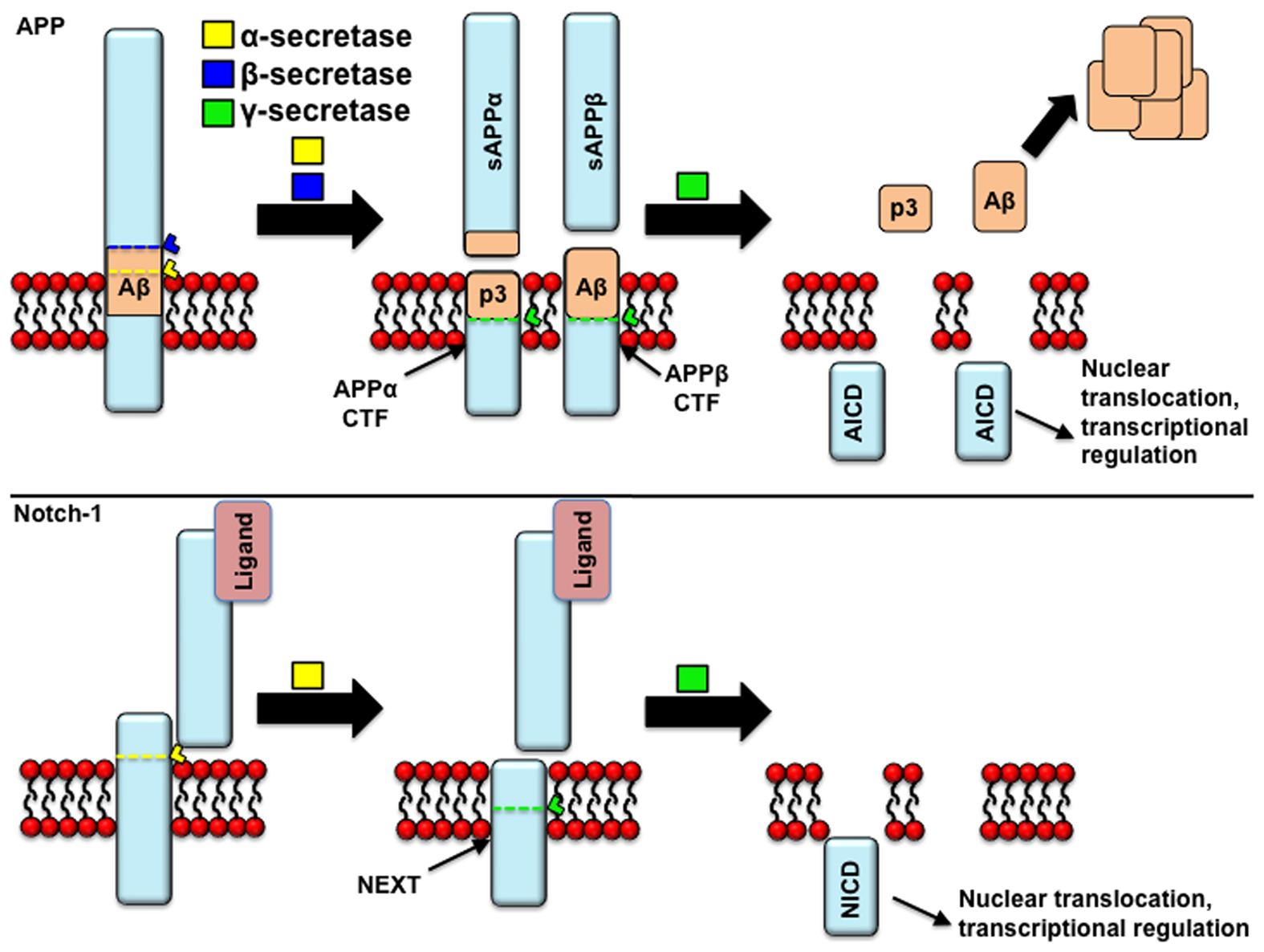
Figure 2. APP processing: similarities to Notch processing. Both APP and Notch receptor may undergo proteolytic cleavage by α-secretase. In the case of APP, this cleavage prevents the formation of beta-amyloid (Aβ) peptides, and induces the release of the p3 fragment and the retention of a membrane-tethered fragment [carboxyl-terminal fragments of APP (APP-CTFs)], that is a substrate of γ-secretase cleavage, yielding APP intracellular domain (AICD) fragments. In the alternative, amyloidogenic pathway, APP is cleaved by β-secretase prior to γ-secretase. In the case of the Notch receptor, the holoprotein is cleaved in the trans Golgi network by a furin-like protease activity in the juxtamembrane extracellular domain, giving rise to two fragments (120 and 180 kDa) that remain associated as a heterodimer. It is thought that Notch reaches the cell membrane in this assembly, where it can undergo activation by ligand binding, following which it gets cleaved by α-secretase to yield the “Notch extracellular truncated” derivative (NEXT). Similarly to the APP-CTFs, only this truncated derivative is then cleaved by γ-secretase. Similarly to NICD, AICD fragments are thought to translocate to the nucleus and activate gene transcription.
APP Metabolites and Neurogenesis
sAPPα: Regulation of Neural Progenitor Cell Proliferation, Survival, and Migration
The first studies to draw a correlation between APP and neurogenic processes showed that embryonic expression of APP peaked during the height of neuronal differentiation and neurite outgrowth (Hung et al., 1992; Salbaum and Ruddle, 1994). Evidence soon emerged that the cleavage of Notch 1, an integral protein involved in the maintenance of NSC pools by enhancing symmetric division (Alexson et al., 2006), and APP undergo strikingly similar proteolytic cleavage patterns by α- and γ-secretase (Figure 2; De Strooper et al., 1999; Lavoie and Selkoe, 2003; van Tetering et al., 2009).
Under non-pathological circumstances, the non-amyloidogenic cleavage pathway of APP dominates through cleavage by α-secretase and release of the soluble APPα (sAPPα) metabolite. sAPPα from the predominantly neuronal APP695 isoform contains a series of domains that include a growth factor domain (D1), a copper binding domain (D2), an acidic region (D3), and a carbohydrate domain (D6; Storey and Cappai, 1999; Reinhard et al., 2005). Importantly, this isoform does not contain either the KPI domain (D4) or the OX-2 domain (D5; Figure 1; Sandbrink et al., 1996). A crystal structure of sAPP suggests that the growth factor like domain is similar to cysteine-rich growth factors (Rossjohn et al., 1999). In light of the appearance of a growth factor like domain, it is not surprising that addition of sAPPα stimulates the proliferation of a number of different cell types. However, the cellular role of sAPPα seems to be broader than that (Table 1). Specifically, sAPPα stimulates the proliferation of fibroblasts (Saitoh et al., 1989), thyroid epithelial cells (Pietrzik et al., 1998), epidermal basal cells (Hoffmann et al., 2000), embryonic stem cells (Ohsawa et al., 1999), and carcinoma cells (Ko et al., 2004). Takayama et al. (2009) suggest that sAPP promotes androgen-dependent proliferation of prostate cancer cells and tumor growth, however, it is yet to be determined whether this effect is solely due to sAPP. Caille et al (2004) showed that rapidly proliferating, EGF-responsive type-C cells of the adult SVZ had saturable binding sites for sAPPα and infusion of sAPPα into the lateral ventricles of adult mice was sufficient to increase NPC numbers (Caille et al., 2004). Recent evidence suggests that inhibiting α-secretase activity reduces NPC proliferation in vitro, and that proliferation can be rescued by the addition of recombinant sAPPα into the medium (Demars et al., 2011). In support of that, a recent study suggests that both sAPPα and sAPPβ increase the proliferation of NPC derived from the SGL and the SVZ of adult rats in vitro, and enhances their glial fate (GFAP immunoreactivity; Baratchi et al., 2011). It is yet to be determined if the observed enhanced GFAP immunoreactivity is due to lack of differentiation or due to more differentiation into astrocytes. In addition, the studies by Demars et al. suggest that sAPPα is not only a potent proliferation factor for adult NPC but for mesenchymal and human placental stem cells as well and that this signaling may be accomplished through the mitogen-activated protein (MAP)-kinase pathway, a pathway integral to NPC proliferation (Li et al., 2001; Jin et al., 2005; Sung et al., 2007; Demars et al., 2011). The observation that sAPPα regulates the proliferation of non-NSC may suggest a potential explanation for the ubiquitous expression of APP and ADAMs. Furthermore, studies on a mouse model exhibiting a knockout of the sortilin related receptor with type-a repeats (SORLA), a sorting receptor that inhibits the processing of APP to form both sAPP and Aβ, showed both increased APP metabolites as well as enhanced proliferation of NPC and neurogenesis in adult mice (Rohe et al., 2008). Interestingly, following cerebral stroke, an increase in ADAM17 proteolytic activity and APP expression leads to increased proliferation in the SVZ (Katakowski et al., 2007). It is yet to be determined whether the sAPP effect on proliferation of NPC and other cell types is a result of an effect on cell survival or proliferation per se.
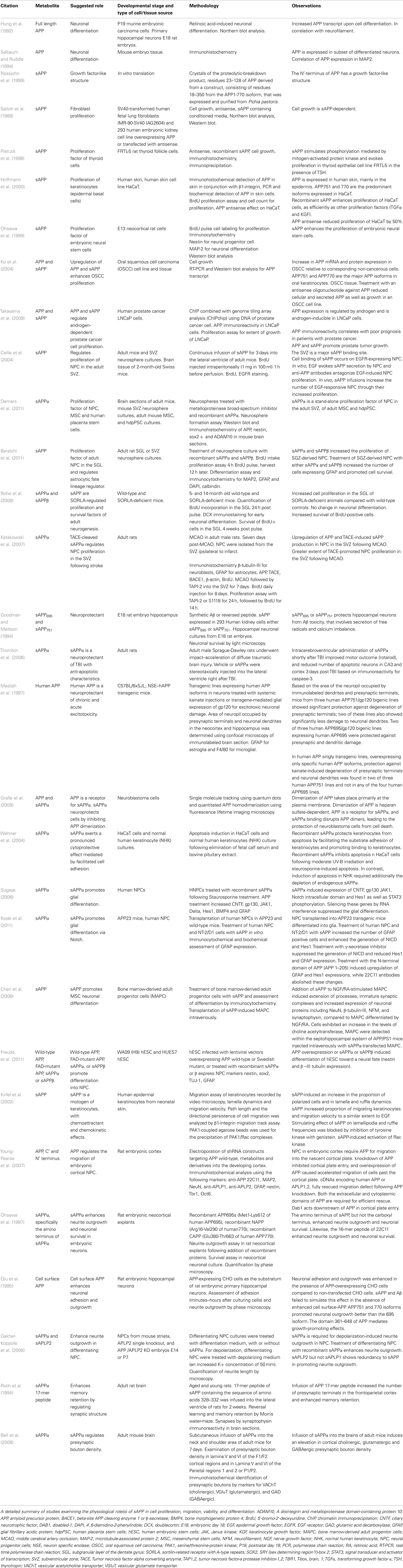
Table 1. Physiological roles of sAPP.
Survival of NPC is an integral, often rate limiting step, in the functional incorporation of newly generated neurons. It is estimated that under normal conditions in the DG only approximately 50% of newly generated neuroblasts survive (Cameron and McKay, 2001). Many neurodegenerative conditions including AD (Verret et al., 2007; for review Lazarov and Marr, 2010), Parkinson’s disease (Hoglinger et al., 2004), and Huntington’s disease (Phillips et al., 2005; Lazic et al., 2006) present with detriments in neurogenesis that may be, at least in part, attributable to alterations in the niche microenvironment. Several studies suggest that sAPPα has a neuroprotective function in association with Aβ toxicity in neurons (Goodman and Mattson, 1994), following traumatic brain injury (Thornton et al., 2006) and excitotoxic infarcts (Masliah et al., 1997). Recent evidence suggests that sAPP may promote cell survival through the disruption of APP dimerization (Gralle et al., 2009). sAPPα also shows a cytoprotective role in human keratinocytes cultured in the absence of fetal calf serum and bovine pituitary extract (Wehner et al., 2004). Alteration in the subcellular localization of APPL in a null-mutant of yata induces progressive eye vacuolization, brain volume reduction, and lifespan shortening (Sone et al., 2009) suggesting that survival of neurons is impaired. While the direct function of sAPP with relation to survival of newly generated neurons has not been fully determined, it is interesting to speculate that sAPP could regulate the physiological survival of NPC and newly generated neurons as well as the survival of NPC under pathological conditions such as those present in neurodegenerative disease.
As the developmental expression of APP corresponds to the timing of neuronal differentiation, it was postulated that APP or APP metabolites might play a role in the neuronal differentiation of NPC. However, transgenic expression of human wild-type APP in the hippocampus of mice disrupted the neuronal differentiation associated with environmental enrichment (Naumann et al., 2010). Nevertheless, the overexpression of hAPP is under the neuronal-specific PDGF-β promoter, and it is not clear whether it is expressed in NPCs as well. Treatment of human NPC with APP also caused a shift toward glial differentiation suggested to be driven by an induction in Notch 1 signaling (Sugaya, 2008; Kwak et al., 2011). In adult bone marrow adult progenitor cells and human embryonic stem cells, sAPPα induced neuronal phenotype in (Chen et al., 2006; Freude et al., 2011). Taken together these results highlight the importance of APP metabolites in cell lineage commitment, and the importance of a direct investigation of the role of specific APP metabolites in the proliferation and differentiation of NPC. It remains a likely possibility that APP metabolites affect the fate of NPC differentially. A special attention should be paid to the neurogenic microenvironment from which these cells are derived, and the proper tools must be implemented in order to resolve these outstanding questions.
Another key factor in the functional integration of newly generated neurons into the proper functional circuits is the migration of neuroblasts, often over long distances in the case of the adult SVZ. Little is known about the effect of sAPP on the migration of neuroblasts, however, administration of nanomolar concentrations of sAPP to epithelial cells, thyrocytes, and keratinocytes in vitro caused a marked increase in not only the proliferation of the cells but also their migration (for review see Schmitz et al., 2002). Thus, sAPP might be a part of a long list of motogens, growth factors (such as EGF), regulating both cell proliferation and migration. In keratinocytes it was shown that this mitogenic property was conferred through a chemo-attraction similar to, but distinct from, EGF (Kirfel et al., 2002). Thus an upregulation of sAPP as seen following stroke could suggest its utility as a chemoattractant guiding NPC to the site of injury in an attempt at recovery. During development, an shRNA mediated knockdown of endogenous APP leads to an increase in NPC that remain in the intermediate zone. Conversely, overexpression of APP in the developing brain leads to migration of progenitors past the cortical plate boundary (Young-Pearse et al., 2007). Further analysis revealed that the C-terminus of APP may interact transiently with the N-terminus of disrupted in schizophrenia 1 (DISC1) altering the subcellular localization of DISC1. Both DISC1 and disabled-1 (Dab1) seem to operate downstream of APP to impact migration (Young-Pearse et al., 2007, 2010). These results suggest that a proper balance of APP expression is necessary for cortical migration during development.
In order to fully mature and functionally incorporate into existing neural networks, a developing neuron must sprout neurites, form dendritic spines and develop synapses. In rat neocortical explant cultures, treatment with sAPP-induced neurite outgrowth which could be blocked by the addition of 22C11 antibodies that recognize the N-terminal domain of APP (Ohsawa et al., 1997). Qiu et al. (1995) showed that APP transfected CHO cells co-cultured with rat primary hippocampal neurons caused an increase in neurite elongation. However, sAPP in the conditioned media was unable to replicate this effect and peptides blocking sAPP failed to disrupt what is an apparent cell surface function of full length APP (Qiu et al., 1995). It was shown that in vitro depolarization of immature neurons derived from either embryonic striatal NPC or early postnatal SVZ NPC, stimulated neurite outgrowth. Inhibition of sAPP secretion through a blockade of α-secretase activity or inactivation of sAPP by antibody administration could abolish depolarization-induced neurite outgrowth. Further, administration of recombinant sAPPα alone was sufficient to promote neurite outgrowth in these cells through stimulation of the MAP-kinase pathway (Gakhar-Koppole et al., 2008). A 17-mer peptide mimicking the trophic domain of sAPP significantly enhanced synaptic density and memory retention when injected into rat brains (Roch et al., 1994). Another study utilizing a moderately overexpressing ADAM10 mouse model showed an increase in cortical synaptogenesis of cholinergic, glutamatergic, and GABAergic synapses at both 8 and 18 months of age. In order to rule out other ADAM10 substrates, the authors infused sAPPα into non-transgenic mice for 1 week and showed similar results suggesting that sAPPα mediates synaptogenesis of these neuronal populations (Bell et al., 2008).
AICD: A Negative Regulator of Neurogenesis
The AICD is formed following cleavage of APP by either α- or β-secretase and subsequent RIP through the activity of the γ-secretase complex (Haass and Selkoe, 1993; Kimberly et al., 2001). It has been shown to contain a YENPTY domain that is a binding site for phosphotyrosine binding domain-containing adapter proteins (Kavanaugh and Williams, 1994). The AICD itself can also undergo modification through phosphorylation events that may underlie docking of some of its many adapter proteins (Tarr et al., 2002). The AICD has been shown to form a transcriptionally active complex with Fe65 and Tip60, a histone acetyltransferase, that translocates to the nucleus and regulates transcription of a number of genes (Cao and Sudhof, 2001; Gao and Pimplikar, 2001; Muller et al., 2008). Much interest has evolved in the relationship between AICD and neurogenesis due to the finding that the cleavage pattern, formation of an ICD, and its translocation to the nucleus mimic the integral neurogenesis protein Notch 1 (Figure 2, for review Ebinu and Yankner, 2002).
Conversely to sAPPα function, AICD has been shown to be a negative regulator of proliferation in NPC. First, evidence suggests that AICD negatively regulates the transcription of the epidermal growth factor receptor (EGFR), a receptor that drives NPC proliferation (Zhang et al., 2007b; Ayuso-Sacido et al., 2010). Ma et al. showed that the glycophosphatidylinositol (GPI)-linked recognition protein, TAG1, extracellularly interacts with APP to increase AICD release. Using a TAG1 null mouse model with aberrantly high proliferation of embryonic NPC, they showed that either administration of recombinant TAG1 or transfection with AICD59 in vitro could ameliorate enhanced neurogenesis. However, transfection of AICD with a mutated Fe65 binding site had no effect (Ma et al., 2008). This suggests that AICD is a negative regulator of NPC proliferation and that the binding to Fe65 is necessary for its action. Additionally, in APPKO mice expressing AICD there is a reduction in proliferation and survival of NPC in the adult DG, but no change in their differentiation. Notably, this deficit in neurogenesis is Aβ-independent, and is suggested to be due, at least in part, to neuroinflammation in light of the fact that ibuprofen or naproxen treatment could reverse these alterations (Ghosal et al., 2010). In trisomic NPC, enhanced AICD binding to the sonic hedgehog (shh) receptor, patched1 (ptch1), promoter was sufficient to decrease proliferation. Shh binding to ptch1 results in the suppression of second receptor smoothened (Smo) and repressed signaling (Goodrich et al., 1999; Taipale et al., 2002; Trazzi et al., 2010). Interestingly, in post-mitotic neurons it has been suggested that the interaction of the intracellular domain of APP with APP binding protein 1 (APP-BP1) results in re-entry to the cell cycle and ultimately apoptosis (Chen et al., 2003).
The targeted cell death of newly formed neurons, as in embryonic development, may be an important factor underlying functional adult neurogenesis. Indeed, the evidence that only 50% of newly generated neurons in the adult hippocampus survive suggests that targeted cell death is a physiological property and some speculate the tight regulation may be important to normal function. The induction of apoptosis seems to be a common theme in studies of AICD function. Unlike sAPP, which serves a cytoprotective role, the AICD has been shown to induce neuronal apoptosis. The first evidence for this came from APP overexpression studies in post-mitotic neurons in vitro. The overexpression of APP was shown to be sufficient to cause apoptosis in these neurons (Yoshikawa et al., 1992). Further, studies on embryonic carcinoma P19 cells overexpressing AICD display neuron specific apoptosis upon differentiation as shown by TUNEL + Tuj-1+ positive cells. This group suggests that gene changes associated with AICD translocation are responsible for the apoptotic events (Nakayama et al., 2008; Ohkawara et al., 2011). Indeed, research from other groups has shown that AICD interaction with Tip60 in H4 cells derived from human neuroglioma cells (Kinoshita et al., 2002) or p53 in either H4 neuroglioma or SH-SY5Y neuroblastoma cell line (Ozaki et al., 2006) can mediate AICD driven apoptotic events. Alternatively, the APP-CTF can be cleaved by caspases to form a 31 amino acid peptide (C31) that signals apoptotic cell death through initiator caspases 8 and 9 in rat neuroblastoma cell line (Bertrand et al., 2001; Lu et al., 2003) and in embryonic mouse cortical cultures (Bertrand et al., 2001). This cleavage appears to be regulated by Aβ and C31 was shown to be upregulated in the brains of AD patients (Bertrand et al., 2001; Lu et al., 2003). Currently, it is not known whether AICD or C31 induced apoptosis occurs under physiological conditions and thus may represent a purely pathological phenomenon in AD brains.
Cytoskeletal dynamics are an important function in the maturation of neurons, including growth cone elongation (Geraldo and Gordon-Weeks, 2009) and dendritic spine formation (Hotulainen and Hoogenraad, 2010). Sabo et al. (2001, 2003) showed that APP and Fe65 binding in dynamic adhesion actin-rich sites could induce enhanced migration in MDCK wound healing assay. Further, they show the localization of APP/Fe65 complexes in synapses and growth cones. More recently, utilizing an inducible expression system for either AICD, Fe65 or AICD, and Fe65 in a human neuroblastoma cell line, Tet21, it was shown that AICD expression activated genes associated with actin remodeling including transgelin and tropomyosin 1 and co-expression of AICD and Fe65 induced enhanced cytoskeletal dynamics (Muller et al., 2007).
Aβ: A Puzzling Metabolite
The Aβ peptide is produced from the sequential cleavage of APP by β- and γ-secretase and is most well known for its aggregation to form amyloid plaques in AD. Studies on the effect of Aβ with respect to NPC proliferation have generally met with conflicting results. Haughey et al. (2002a) showed that treatment with Aβ in human cortical NPC culture in vitro and following intraventricular infusion of Aβ in vivo, NPC proliferation was drastically reduced (Haughey et al., 2002a; He and Shen, 2009). He and Shen (2009) observed that Aβ could reduce β-catenin signaling and propose this as a mechanism for Aβ action. These findings were in agreement with the observation that Aβ restricted the formation of colonies from human NPC (Mazur-Kolecka et al., 2006). However, other studies have shown that Aβ could stimulate the proliferation of adult murine SVZ derived NPC (Lopez-Toledano and Shelanski, 2004) and that this action may be through the p75 neurotrophin receptor (Sotthibundhu et al., 2009). This contradiction may be due to the use of different isoforms of Aβ or the use of NPC of different species or culture system, and potentially different aggregation states of Aβ. Thus the physiological role of Aβ with respect to proliferation of NPC remains unclear.
Studies have also shown that Aβ may play a role in the fate determination of NPC. One such study determined that treatment of SVZ derived NPC with oligomeric Aβ42 induced both proliferation and pushed differentiation toward a neuronal fate in vitro, as determined by both Western blotting for NCAM and immunohistological analyses. In addition, the authors showed that oligomeric Aβ42 enhanced the migration of immature neurons, in vitro. Increasing concentrations, however, proved to be neurotoxic (Heo et al., 2007). Chen and Dong (2009) showed that Aβ40 was sufficient to promote neuronal differentiation in embryonic cortical NPC culture, while Aβ42 induced astrocytic differentiation.
While studies that attempt to uncover the physiological significance of the p3 peptide derived from γ-secretase cleavage in the non-amyloidogenic pathway are virtually non-existent, the above discussion of the effects of APP metabolites in relation to neurogenesis suggest great complexity in potential signaling. The complexity of cleavage and the number of metabolites produced confer great convolution in the study of APP function. By attempting to understand the physiological role of each individual metabolite, we are able to better understand the signaling pathways associated. However, most of these studies have been done in vitro and in many cases do not represent the interplay of cleavage events that potentially takes place in vivo. While it is advisable to understand the role of each metabolite individually, understanding the interplay of these metabolites may be the key to understanding the true role of APP in neurogenic processes (Figure 3). Parsing out the individual roles of APP metabolites with respect to neurogenesis in transgenic models has proven complex as well. Not only is the metabolism of APP intricate but the potential functional redundancy of APP homologs APLP1 & 2 has only enhanced the complexity of understanding APP function. To attempt to understand how we may approach new models for the study of APP metabolites and APP in neurogenesis we will review the models currently available for this purpose and highlight the deficiencies of each (Table 2).
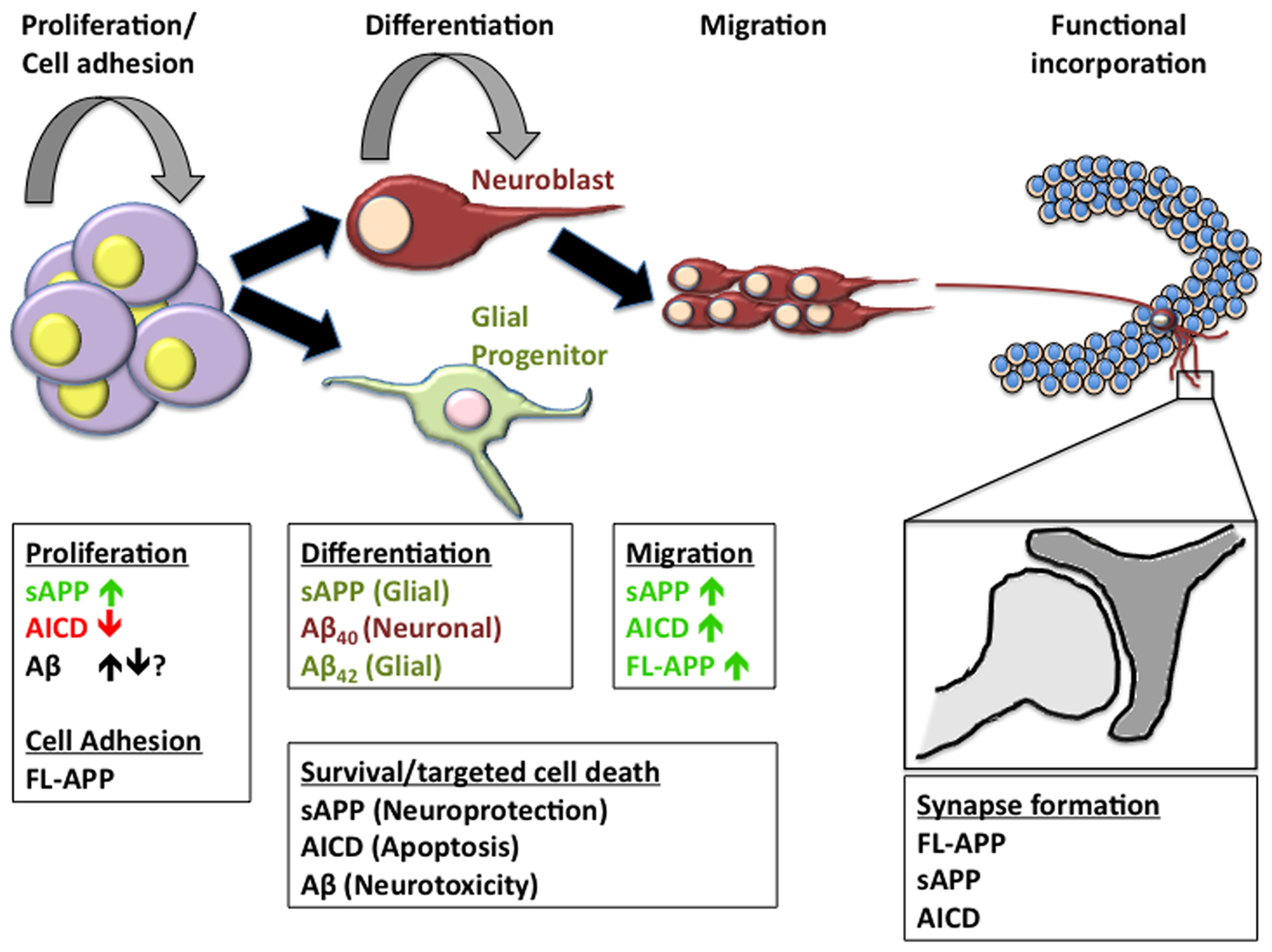
Figure 3. APP metabolites in neurogenesis. The formation of new neurons or astroglia in the adult brain is a multifaceted process including the necessity of a niche that supports continuous proliferation of neural progenitor cells, the determination of neural lineage, migration of immature cells often across large distances, and the functional incorporation into existing neural networks. Many of the metabolites of APP have been implicated in one or more of these processes. In order to fully understand the impact of APP metabolism on neurogenic processes, we must first unravel the individual functions of each of the metabolites in order to better realize the implications of alterations in the cleavage pattern of APP.
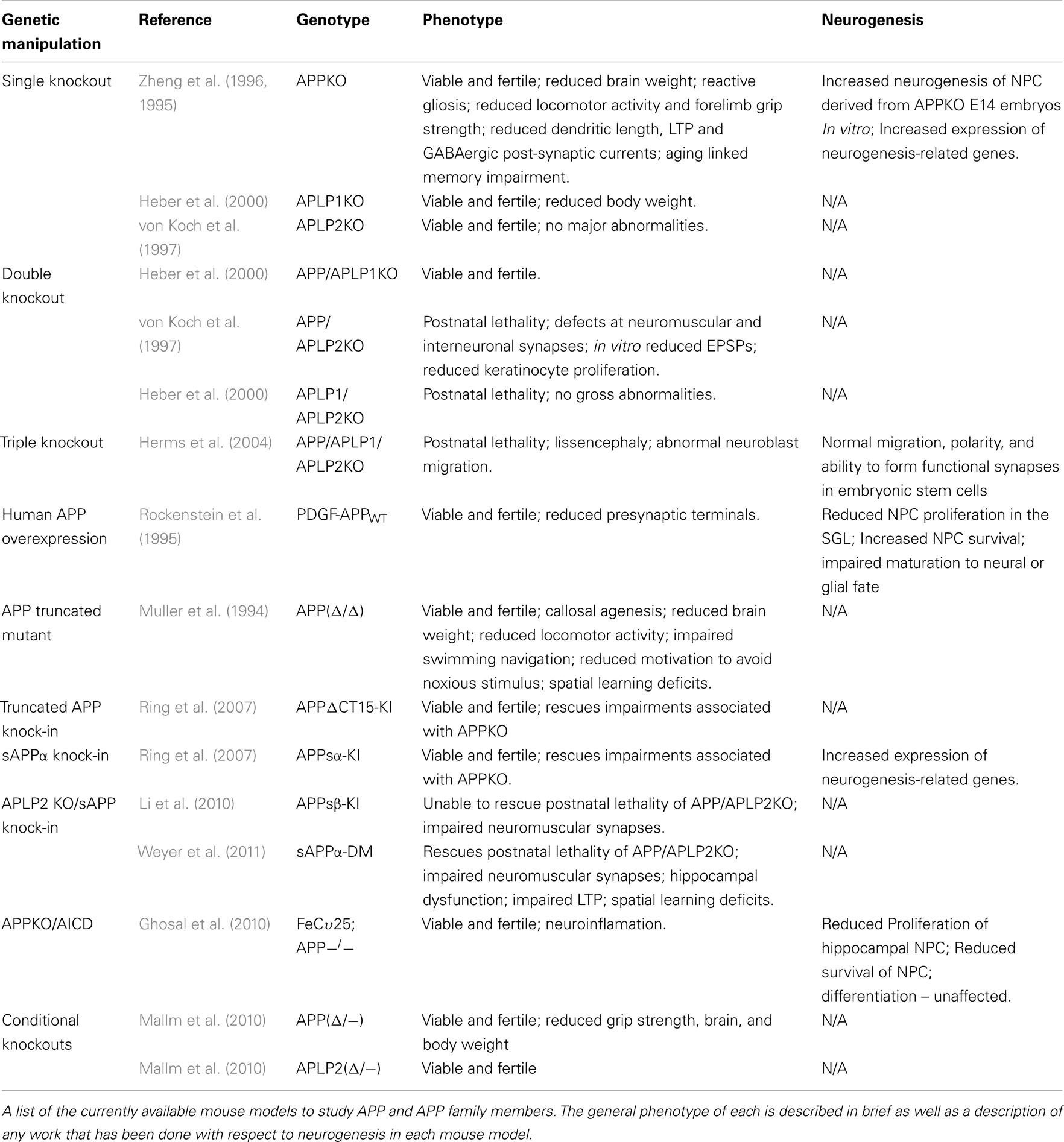
Table 2. APP animal models and neurogenesis.
Alzheimer’s Disease Animal Models: Adequate for the Examination of Neurogenesis?
The first transgenic models to delve into the question of APP function in neurogenesis were FAD mutants that possess either mutations in APP or PS1, most times causing a shift in cleavage patterns toward the amyloidogenic pathway, up-regulating β- or γ-secretase cleavage and producing higher levels of Aβ (for review Selkoe, 2001). The effect of these mutations on neurogenic processes has been reviewed extensively (Lazarov and Marr, 2010). Briefly, APP mutants have been shown to have both decreased (Haughey et al., 2002a,b; Donovan et al., 2006) and increased (Lopez-Toledano and Shelanski, 2007; Kolecki et al., 2008) proliferation of NPC derived from the SGL or the SVZ of adult transgenic mice. In mice exhibiting mutations in both APP and PS1, studies have for the most part shown impaired proliferation (Taniuchi et al., 2007; Zhang et al., 2007a; Ermini et al., 2008). These studies have all shown impairments in mice after amyloid deposition and plaque formation. As we first described in the APPSwe/PS1ΔE9 mouse, impairments in proliferation, and neurogenesis occur before memory impairment or pathological alterations in these mice (Demars et al., 2010a). Furthermore, when NPC are cultured from the SVZ of 2-month-old APPSwe/PS1ΔE9 mice, they show impairments in proliferation in vitro suggesting that mutations may cause intrinsic impairments in NPC proliferation irrespective of environmental or niche cues (Demars et al., 2010a).
In order to determine a physiological role for APP, the first logical step was to create an APP-null mouse model and to infer from any impairment what the function of APP may be. Surprisingly, the APP knockout mouse was viable and fertile, giving the first clue that APP homologs may possess functional redundancy with APP. The mice weighed 15–20% less than their wild-type littermates and showed impaired locomotor activity and decreased forelimb grip strength. In the CNS, the only gross alteration was a reactive gliosis in some mice (Zheng et al., 1995, 1996). Further investigation revealed that APP-null mice did exhibit reduced brain weight, reduced size of forebrain commissures, and an increased frequency of neuroanatomical abnormalities such as callosal agenisis but these effects were mouse background specific (Magara et al., 1999). APP-null mice also showed aging linked impairments in dendritic length and synaptic plasticity along with both decreased long-term potentiation and impaired GABAergic post-synaptic currents. These mice also display aging linked impairments learning and memory (Dawson et al., 1999; Seabrook et al., 1999). Another model of APP deficiency that utilizes a truncated mutant lacking amino acids 20–75 and expressed at 5% of normal levels (APPΔ/Δ) also shows abnormalities such as callosal agenesis, reduced brain weight, and decreased locomotor activity (Muller et al., 1994; Magara et al., 1999). The APPΔ/Δ mice also showed cognitive impairments associated with swimming navigation during the Morris water-maze task and an apparent decline in motivation to avoid a noxious stimuli (Muller et al., 1994). Interestingly, mice tested at early postnatal stages (pd 3–10) did not show impairments while major impairments only became evident at later postnatal stages (pd 11–19) and while reduced grip strength persisted into adulthood, deficits in spatial learning, contrary to phenotypic expectation, could not be shown (Tremml et al., 1998). Further investigation revealed that handling during these early periods resulted in rescued impairment in spatial learning as tested by the Morris water-maze but failed to rescue impaired activity in an open field test (Tremml et al., 2002). This result suggests that impairments caused by APP deficiency do not completely disrupt plasticity or the ability of environmental enrichment to enhance cognitive function. In fact, studies on NPC derived from E14 APP-null mice showed aberrantly increased neurogenesis (Ma et al., 2008). While the direct affect of APP disruption on neurogenesis in vivo in the adult has not been reported, studies on a PDGF-APPWT mouse line overexpressing human APP have shown that the overexpression results in decreased proliferation of NPC in the adult hippocampus, an increased survival rate of these NPC and impaired maturation of NPC to either neural or glial fate (Naumann et al., 2010). However, these animals have also been shown to display high levels of Aβ42, synaptic dysfunction, neuronal degeneration, and alterations in isoform expression patterns (Rockenstein et al., 1995; Mucke et al., 2000; Simon et al., 2009). Deletion of APP homologs, APLP1 and APLP2, has also been attempted. Like APP-null mice, both APLP1 and APLP2 knockout mice are viable and fertile (von Koch et al., 1997; Heber et al., 2000). APLP2 knockout mice present with normal size and weight, no gross tissue abnormalities, normal grip strength, normal posture, and no apparent cognitive deficits (von Koch et al., 1997). This is in contrast to APP-null mice which exhibit reduced weight, forelimb grip strength, and aging linked cognitive deficits. Mice exhibiting a knockout of APLP1 show approximately 10% reduction in body weight but are absent of most other abnormalities associated with APP-null mice including grip strength, locomotor activity, callosal agenesis, and spatial learning (Heber et al., 2000). The generation of mouse lines designed to either ablate or overexpress APP and APP homologs present a number of problems with respect to the study of APP function. First, the APP family has been shown to confer much of its activity through its extensive proteolytic cleavage and release of various metabolites. Many of these metabolites have been shown to have potential synergistic and/or antagonistic activities on a number of different cellular processes. The regulation of APP function may not be solely driven by the expression of APP, but through the activity of proteases and formation of metabolites. Simply ablating APP or APP homologs may be valuable in discerning the dominant pathway but does not necessarily segregate between physiological signaling pathways of APP metabolites. Second, a number of processes have been shown to affect neurogenesis including neuronal excitability, learning, inflammation, physical activity, enriched environment, and stress among others (Reviewed in Ming and Song, 2005). None of the knockout models described above infer spatial or temporal specificity. As such, all systems, cell types and regions are void of APP or APP homologs. Lack of cell type specificity could lead to alterations in mature neuronal or glial function that could alter neurogenic processes. Impaired LTP (Seabrook et al., 1999), decreased locomotor activity or reactive gliosis (Zheng et al., 1995) as seen in APP-null mice could compromise the study of neurogenesis in these mice. Lack of temporal regulation could also compromise the study of APP function by allowing for compensatory alterations during development that may mask some of the functions of the protein. Finally, functional redundancy between the homologs could hinder the study of single knockouts by masking the role of a single homolog. For example, APP-null mice express both APLP1 and APLP2 and were shown to be viable and fertile despite the belief that APP plays a major role in development.
To overcome the issue of redundancy APP/APLP1 knockout, APP/APLP2 knockout, APLP1/APLP2 knockout, and APP/APLP1/APLP2 triple knockout mice were generated (Heber et al., 2000; Herms et al., 2004). APP/APLP2 and APLP1/APLP2 knockout mice proved to be postnatally lethal, while APP/APLP1 knockout mice were viable and fertile (von Koch et al., 1997; Heber et al., 2000). These results further suggest a functional redundancy between various APP homologs and imply that various homologs may be involved in differential functions. At first there seemed to be few gross abnormalities in double knockouts, even in mice that were postnatally lethal. However, further investigation has revealed that APP/APLP2 double knockout mice have defects at neuromuscular synaptic junctions including increased nerve terminal sprouting, reduced presynaptic vesicles, deficits in neurotransmitter release, and a higher incidence of synaptic failure (Wang et al., 2005). This phenomenon was also seen in interneuronal synapses at the submandibular ganglion (Yang et al., 2005). In neurons derived from APP/APLP2 knockout embryonic stem cells, a decrease in vGLUT2 was correlated with a reduction in field excitatory post-synaptic potentials signaling further impairments in neuronal function (Schrenk-Siemens et al., 2008). Keratinocyes derived from APP/APLP2 double knockout mice also show impaired proliferation in vivo and in vitro that can be rescued by exogenous addition of sAPPα (Siemes et al., 2006). APP and APLP2 have been implicated in lipoprotein and cholesterol metabolism in the brain as well through modulation of the expression of LRP1, a major lipoprotein receptor. APP/APLP2 knockout mice show increased expression of LRP1 that is directly attributable to the lack of AICD as AICD was shown to bind to the LRP1 promoter and repress transcription (Liu et al., 2007). Insulin and glucose homeostasis was shown to be impaired in APP/APLP2 double knockout even further than that of single knockouts, which grow to hypoglycemic adults (Needham et al., 2008). The APP/APLP1/APLP2 triple knockout appears to have the most severe phenotype with cranial abnormalities and cortical dysplasia resembling type 2 lissencephaly where neuroblasts have migrated through the basal lamina and pial membrane (Herms et al., 2004). This phenotype would suggest a critical role for the APP family in migration and adhesion. However, embryonic stem cells derived from triple knockouts were reported to show normal migration, polarity, and to form functional synapses (Bergmans et al., 2010). While these models addressed the downfall of previous models in so much as they accounted for functional redundancy, they nonetheless failed to address which metabolite could be ascribed to each phenotypic change in these mice. Ring et al. (2007) addressed this question by developing a pair of mutant mice, one a sAPPα knock-in and the other an APP knock-in with a truncated C-terminus lacking the YENPTY motif, to understand if sAPPα could rescue impairments associated with the APP-null mouse. Indeed, both mutants either attenuated or completely reversed phenotypic alterations in APP-null mice including brain and body weight, locomotor deficits, grip strength impairment, exploratory behavior, long-term potentiation, and spatial learning deficits (Ring et al., 2007). Studies using similar knock-in models crossed with an APLP2 knockout mutant showed mixed results with the addition of sAPP recovering postnatal lethality in one of the models but unable to ameliorate postnatal lethality in the other (Li et al., 2010; Weyer et al., 2011). Both studies showed that impairments in neuromuscular synapses persisted in sAPPα knock-in double mutants. Further, Weyer et al. (2011) went on to show that while sAPPα knock-in double mutants had normal CNS morphology, they displayed hippocampal dysfunction, spatial learning deficits, and impaired long-term potentiation (Weyer et al., 2011). Analysis of differential mRNA expression in the prefrontal cortex of adult wild-type, APP-null, APLP2 knockout, and sAPPα knock-in mice showed that APP-null and sAPPα knock-in mice had very similar profiles suggesting a potentially dominant role of AICD in transcriptional regulation (Aydin et al., 2011). Recently, the development of APP and APLP2 conditional knockout mice has been accomplished with the hope that they may be crossed with viable single knockouts to allow for postnatal and adult study of the effect of these ablations. The APP conditional knockout mice showed similar deficits in brain weight, body weight, and grip strength to the classic APP-null mouse model (Mallm et al., 2010).
In order to develop better mouse models designed to study the physiological role of APP family members with respect to neurogenic processes, three crucial factors must be considered. First, it has become apparent that APP family members exhibit some degree of functional redundancy and thus models such as the double and triple knockouts of the APP family of proteins potentially provide critical phenotypic evidence that is masked in single knockouts. This may be essential to understanding the individual roles of APP homologs and ultimately understanding the function of the APP family. Simply examining the nature of knockouts of holo-APP family members, however, does not take into account the proteolytic processing and potential for metabolites to exert synergistic or antagonistic roles on certain neurogenic processes. Models such as the APP/APLP2 knockout sAPPα knock-in or the APP-null AICD mice allow for examination of the expression of a single metabolite and may allow for better understanding of the role of each in neurogenic function. Further, study of NPC derived from knockout models may allow for the exogenous addition of soluble factors or expression of metabolites such as AICD, permitting their study in vitro. Finally, none of the APP mouse models to date have provided for temporal and spatial regulation of ablation or expression. Utilizing technology such as inducible knockout systems, promoter selection for NPC specificity and lentiviral expression for reintroduction of single APP metabolites may provide a more precise system in which to study the direct effect of each metabolite on neurogenic processes. As technology develops, inducible systems will provide the greatest control over temporal and spatial transgenic expression and have the ability to greatly enhance our understanding of the APP family and their metabolites.
Conclusion Remarks
The increasing knowledge of the role of APP metabolites in developmental and postnatal neurogenesis opens up new research directions that may lead to greater understanding of cognitive pathways in health and brain disorders. The APP is a family of ubiquitously expressed homologs, some of which exhibit functional redundancy. APP yields a complex set of proteolytic metabolites. New animal models are required in order to dissect out the functional significance of each APP metabolite and their role in neurogenesis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was supported by the NIA AG033570 (Orly Lazarov), Alzheimer’s Association Young Investigator Award (Orly Lazarov), the Illinois Department of Public Health ADRF award (Orly Lazarov), and the Brain Research Foundation (Orly Lazarov).
Abbreviations
Aβ, beta amyloid; AD, Alzheimer’s disease; ADAM, a disintegrin and metalloproteinase; AICD, amyloid precursor protein intracellular domain; APP, amyloid precursor protein; APP-BP1, APP binding protein 1; APP-CTFs, amyloid precursor protein carboxyl-terminal fragments; APLP1, amyloid precursor-like proteins 1; APLP2, amyloid precursor-like proteins 2; BACE1, β-site APP cleaving enzyme 1; BACE2, β-site APP cleaving enzyme 2; BMP4, bone morphogenetic protein 4; BrdU, 5′-Bromo-2′-deoxyuridine; C31, caspase-cleaved APP-CTF 31 amino acid peptide; ChIP, chromatin immunoprecipitation; CNTF, ciliary neurotrophic factor; CuBD, copper binding domain; DAB1, disabled-1; DAPI, 4′,6-diamidino-2-phenylindole; DCX, doublecortin; DG, dentate gyrus; DISC1, disrupted in schizophrenia 1; E18, embryonic day 18; EGFR, epidermal growth factor receptor; FAD, familial Alzheimer’s disease; GAD, glutamic acid decarboxylase; GFAP, glial fibrillary acidic protein; GPI, glycophosphatidylinositol; HBD1/GFLD, heparin binding and growth factor like domain; hdpPSC, human placenta stem cells; hESC, human embryonic stem cells; JAK, Janus kinase; KPI, Kunitz-type protease inhibitor domain; MAM, methylazoxy-methanol acetate; MAP, mitogen-activated protein; MAP2, microtubule-associated protein 2; MAPC, bone marrow-derived adult progenitor cells; MBD1, methyl-CpG binding protein 1; MCAO, middle cerebral artery occlusion; NCAM, neural cell adhesion molecule; MSC, mesenchymal stem cells; NEXT, Notch extracellular truncated; NFM, neurofilament; NGF, nerve growth factor; NHK, normal human keratinocyte; NICD, Notch intracellular domain; NSC, neural stem cells; NSE, neuron specific enolase; NPC, neural progenitor cells; NT-3, neurotrophin 3; OSCC, oral squamous cell carcinoma; P19, postnatal day 19; PAK1, serine/threonine-protein kinase; PCR, Polymerase chain reaction; PTCH1, patched1; PS1, presenilin-1; PS2, presenilin 2; RA, retinoic acid; RC, random coiled region; RIP, regulated intra-membrane proteolysis; RMS, rostral migratory stream; RT-PCR, real time polymerase chain reaction; sAPPα, soluble amyloid precursor protein alpha; SGL, subgranular layer of the dentate gyrus; SHH, sonic hedgehog; SMO, smoothened; SORLA, sortilin related receptor with type-a repeats; SOX2, SRY (sex determining region Y)-box 2; STAT3, signal transducer and activator of transcription; SVZ, subventricular zone; TACE, tumor necrosis factor alpha converting enzyme; TAP1,2, tumor necrosis factor-a protease inhibitor-1,2; TBR1, T-box, brain, 1; TGFα, transforming growth factor α; TK, thymidine kinase; TSH, thyrotropin; VAChT, vesicular acetylcholine transporter; VGluT, vesicular glutamate transporter; ZnBD, zinc binding domain.
References
Alexson, T. O., Hitoshi, S., Coles, B. L., Bernstein, A., and Van Der Kooy, D. (2006). Notch signaling is required to maintain all neural stem cell populations – irrespective of spatial or temporal niche. Dev. Neurosci. 28, 34–48.
Asai, M., Hattori, C., Szabo, B., Sasagawa, N., Maruyama, K., Tanuma, S., and Ishiura, S. (2003). Putative function of ADAM9, ADAM10, and ADAM17 as APP alpha-secretase. Biochem. Biophys. Res. Commun. 301, 231–235.
Aydin, D., Filippov, M. A., Tschape, J. A., Gretz, N., Prinz, M., Eils, R., Brors, B., and Muller, U. C. (2011). Comparative transcriptome profiling of amyloid precursor protein family members in the adult cortex. BMC Genomics 12, 160. doi:10.1186/1471-2164-12-160
Ayuso-Sacido, A., Moliterno, J. A., Kratovac, S., Kapoor, G. S., O’Rourke, D. M., Holland, E. C., Garcia-Verdugo, J. M., Roy, N. S., and Boockvar, J. A. (2010). Activated EGFR signaling increases proliferation, survival, and migration and blocks neuronal differentiation in post-natal neural stem cells. J. Neurooncol. 97, 323–337.
Baratchi, S., Evans, J., Tate, W. P., Abraham, W. C., and Connor, B. (2011). Secreted amyloid precursor proteins promote proliferation and glial differentiation of adult hippocampal neural progenitor cells. Hippocampus. doi: 10.1002/hipo.20988. [Epub ahead of print].
Bell, K. F., Zheng, L., Fahrenholz, F., and Cuello, A. C. (2008). ADAM-10 over-expression increases cortical synaptogenesis. Neurobiol. Aging 29, 554–565.
Bergmans, B. A., Shariati, S. A., Habets, R. L., Verstreken, P., Schoonjans, L., Muller, U., Dotti, C. G., and De Strooper, B. (2010). Neurons generated from APP/APLP1/APLP2 triple knockout embryonic stem cells behave normally in vitro and in vivo: lack of evidence for a cell autonomous role of the amyloid precursor protein in neuronal differentiation. Stem Cells 28, 399–406.
Bertrand, E., Brouillet, E., Caille, I., Bouillot, C., Cole, G. M., Prochiantz, A., and Allinquant, B. (2001). A short cytoplasmic domain of the amyloid precursor protein induces apoptosis in vitro and in vivo. Mol. Cell. Neurosci. 18, 503–511.
Bizon, J. L., and Gallagher, M. (2003). Production of new cells in the rat dentate gyrus over the lifespan: relation to cognitive decline. Eur. J. Neurosci. 18, 215–219.
Breton-Provencher, V., Lemasson, M., Peralta, M. R. III, and Saghatelyan, A. (2009). Interneurons produced in adulthood are required for the normal functioning of the olfactory bulb network and for the execution of selected olfactory behaviors. J. Neurosci. 29, 15245–15257.
Buxbaum, J. D., Liu, K. N., Luo, Y., Slack, J. L., Stocking, K. L., Peschon, J. J., Johnson, R. S., Castner, B. J., Cerretti, D. P., and Black, R. A. (1998). Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J. Biol. Chem. 273, 27765–27767.
Caille, I., Allinquant, B., Dupont, E., Bouillot, C., Langer, A., Muller, U., and Prochiantz, A. (2004). Soluble form of amyloid precursor protein regulates proliferation of progenitors in the adult subventricular zone. Development 131, 2173–2181.
Cameron, H. A., and McKay, R. D. (2001). Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J. Comp. Neurol. 435, 406–417.
Cameron, H. A., Woolley, C. S., Mcewen, B. S., and Gould, E. (1993). Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. Neuroscience 56, 337–344.
Cao, X., and Sudhof, T. C. (2001). A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293, 115–120.
Chen, C. W., Boiteau, R. M., Lai, W. F., Barger, S. W., and Cataldo, A. M. (2006). sAPPalpha enhances the transdifferentiation of adult bone marrow progenitor cells to neuronal phenotypes. Curr. Alzheimer Res. 3, 63–70.
Chen, Y., and Dong, C. (2009). Abeta40 promotes neuronal cell fate in neural progenitor cells. Cell Death Differ. 16, 386–394.
Chen, Y., Liu, W., Mcphie, D. L., Hassinger, L., and Neve, R. L. (2003). APP-BP1 mediates APP-induced apoptosis and DNA synthesis and is increased in Alzheimer’s disease brain. J. Cell Biol. 163, 27–33.
Clelland, C. D., Choi, M., Romberg, C., Clemenson, G. D. Jr., Fragniere, A., Tyers, P., Jessberger, S., Saksida, L. M., Barker, R. A., Gage, F. H., and Bussey, T. J. (2009). A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science 325, 210–213.
Dawson, G. R., Seabrook, G. R., Zheng, H., Smith, D. W., Graham, S., O’Dowd, G., Bowery, B. J., Boyce, S., Trumbauer, M. E., Chen, H. Y., Van Der Ploeg, L. H., and Sirinathsinghji, D. J. (1999). Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein. Neuroscience 90, 1–13.
De Strooper, B., Annaert, W., Cupers, P., Saftig, P., Craessaerts, K., Mumm, J. S., Schroeter, E. H., Schrijvers, V., Wolfe, M. S., Ray, W. J., Goate, A., and Kopan, R. (1999). A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522.
Demars, M., Hu, Y. S., Gadadhar, A., and Lazarov, O. (2010a). Impaired neurogenesis is an early event in the etiology of familial Alzheimer’s disease in transgenic mice. J. Neurosci. Res. 88, 2103–2117.
Demars, M., Hu, Y. S., Gadadhar, A., and Lazarov, O. (2010b). Impaired neurogenesis is an early event in the etiology of familial Alzheimer’s disease in transgenic mice. J. Neurosci. Res. 88, 2103–2117.
Demars, M. P., Bartholomew, A., Strakova, Z., and Lazarov, O. (2011). Soluble amyloid precursor protein: a novel proliferation factor of adult progenitor cells of ectodermal and mesodermal origin. Stem Cell Res. Ther. 2, 36.
Doetsch, F., Caille, I., Lim, D. A., Garcia-Verdugo, J. M., and Alvarez-Buylla, A. (1999). Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97, 703–716.
Donovan, M. H., Yazdani, U., Norris, R. D., Games, D., German, D. C., and Eisch, A. J. (2006). Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer’s disease. J. Comp. Neurol. 495, 70–83.
Dupret, D., Revest, J. M., Koehl, M., Ichas, F., De Giorgi, F., Costet, P., Abrous, D. N., and Piazza, P. V. (2008). Spatial relational memory requires hippocampal adult neurogenesis. PLoS ONE 3, e1959. doi:10.1371/journal.pone. 0001959
Ebinu, J. O., and Yankner, B. A. (2002). A RIP tide in neuronal signal transduction. Neuron 34, 499–502.
Eriksson, P. S., Perfilieva, E., Bjork-Eriksson, T., Alborn, A. M., Nordborg, C., Peterson, D. A., and Gage, F. H. (1998). Neurogenesis in the adult human hippocampus. Nat. Med. 4, 1313–1317.
Ermini, F. V., Grathwohl, S., Radde, R., Yamaguchi, M., Staufenbiel, M., Palmer, T. D., and Jucker, M. (2008). Neurogenesis and alterations of neural stem cells in mouse models of cerebral amyloidosis. Am. J. Pathol. 172, 1520–1528.
Francis, R., Mcgrath, G., Zhang, J., Ruddy, D. A., Sym, M., Apfeld, J., Nicoll, M., Maxwell, M., Hai, B., Ellis, M. C., Parks, A. L., Xu, W., Li, J., Gurney, M., Myers, R. L., Himes, C. S., Hiebsch, R., Ruble, C., Nye, J. S., and Curtis, D. (2002). aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev. Cell 3, 85–97.
Freude, K. K., Penjwini, M., Davis, J. L., Laferla, F. M., and Blurton-Jones, M. (2011). Soluble amyloid precursor protein induces rapid neural differentiation of human embryonic stem cells. J. Biol. Chem. 286, 24264–24274.
Gakhar-Koppole, N., Hundeshagen, P., Mandl, C., Weyer, S. W., Allinquant, B., Muller, U., and Ciccolini, F. (2008). Activity requires soluble amyloid precursor protein alpha to promote neurite outgrowth in neural stem cell-derived neurons via activation of the MAPK pathway. Eur. J. Neurosci. 28, 871–882.
Gang, B., Yue, C., Han, N., Xue, H., Li, B., Sun, L., Li, X., and Zhao, Q. (2011). Limited hippocampal neurogenesis in SAMP8 mouse model of Alzheimer’s disease. Brain Res. 1389, 183–193.
Gao, Y., and Pimplikar, S. W. (2001). The gamma-secretase-cleaved C-terminal fragment of amyloid precursor protein mediates signaling to the nucleus. Proc. Natl. Acad. Sci. U.S.A. 98, 14979–14984.
Geraldo, S., and Gordon-Weeks, P. R. (2009). Cytoskeletal dynamics in growth-cone steering. J. Cell. Sci. 122, 3595–3604.
Gheusi, G., Cremer, H., Mclean, H., Chazal, G., Vincent, J. D., and Lledo, P. M. (2000). Importance of newly generated neurons in the adult olfactory bulb for odor discrimination. Proc. Natl. Acad. Sci. U.S.A. 97, 1823–1828.
Ghosal, K., Stathopoulos, A., and Pimplikar, S. W. (2010). APP intracellular domain impairs adult neurogenesis in transgenic mice by inducing neuroinflammation. PLoS ONE 5, e11866. doi:10.1371/journal.pone. 0011866
Goodman, Y., and Mattson, M. P. (1994). Secreted forms of beta-amyloid precursor protein protect hippocampal neurons against amyloid beta-peptide-induced oxidative injury. Exp. Neurol. 128, 1–12.
Goodrich, L. V., Jung, D., Higgins, K. M., and Scott, M. P. (1999). Overexpression of ptc1 inhibits induction of Shh target genes and prevents normal patterning in the neural tube. Dev. Biol. 211, 323–334.
Gralle, M., Botelho, M. G., and Wouters, F. S. (2009). Neuroprotective secreted amyloid precursor protein acts by disrupting amyloid precursor protein dimers. J. Biol. Chem. 284, 15016–15025.
Haass, C., and Selkoe, D. J. (1993). Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell 75, 1039–1042.
Hartmann, D., Tournoy, J., Saftig, P., Annaert, W., and De Strooper, B. (2001). Implication of APP secretases in notch signaling. J. Mol. Neurosci. 17, 171–181.
Haughey, N. J., Liu, D., Nath, A., Borchard, A. C., and Mattson, M. P. (2002a). Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid beta-peptide: implications for the pathogenesis of Alzheimer’s disease. Neuromolecular Med. 1, 125–135.
Haughey, N. J., Nath, A., Chan, S. L., Borchard, A. C., Rao, M. S., and Mattson, M. P. (2002b). Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer’s disease. J. Neurochem. 83, 1509–1524.
He, P., and Shen, Y. (2009). Interruption of beta-catenin signaling reduces neurogenesis in Alzheimer’s disease. J. Neurosci. 29, 6545–6557.
Heber, S., Herms, J., Gajic, V., Hainfellner, J., Aguzzi, A., Rulicke, T., Von Kretzschmar, H., Von Koch, C., Sisodia, S., Tremml, P., Lipp, H. P., Wolfer, D. P., and Muller, U. (2000). Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J. Neurosci. 20, 7951–7963.
Heo, C., Chang, K. A., Choi, H. S., Kim, H. S., Kim, S., Liew, H., Kim, J. A., Yu, E., Ma, J., and Suh, Y. H. (2007). Effects of the monomeric, oligomeric, and fibrillar Abeta42 peptides on the proliferation and differentiation of adult neural stem cells from subventricular zone. J. Neurochem. 102, 493–500.
Herms, J., Anliker, B., Heber, S., Ring, S., Fuhrmann, M., Kretzschmar, H., Sisodia, S., and Muller, U. (2004). Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 23, 4106–4115.
Hoffmann, J., Twiesselmann, C., Kummer, M. P., Romagnoli, P., and Herzog, V. (2000). A possible role for the Alzheimer amyloid precursor protein in the regulation of epidermal basal cell proliferation. Eur. J. Cell Biol. 79, 905–914.
Hoglinger, G. U., Rizk, P., Muriel, M. P., Duyckaerts, C., Oertel, W. H., Caille, I., and Hirsch, E. C. (2004). Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nat. Neurosci. 7, 726–735.
Hotulainen, P., and Hoogenraad, C. C. (2010). Actin in dendritic spines: connecting dynamics to function. J. Cell Biol. 189, 619–629.
Hung, A. Y., Koo, E. H., Haass, C., and Selkoe, D. J. (1992). Increased expression of beta-amyloid precursor protein during neuronal differentiation is not accompanied by secretory cleavage. Proc. Natl. Acad. Sci. U.S.A. 89, 9439–9443.
Ihrie, R. A., and Alvarez-Buylla, A. (2011). Lake-front property: a unique germinal niche by the lateral ventricles of the adult brain. Neuron 70, 674–686.
Imayoshi, I., Sakamoto, M., Ohtsuka, T., Takao, K., Miyakawa, T., Yamaguchi, M., Mori, K., Ikeda, T., Itohara, S., and Kageyama, R. (2008). Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat. Neurosci. 11, 1153–1161.
Jin, K., Mao, X. O., Del Rio Guerra, G., Jin, L., and Greenberg, D. A. (2005). Heparin-binding epidermal growth factor-like growth factor stimulates cell proliferation in cerebral cortical cultures through phosphatidylinositol 3’-kinase and mitogen-activated protein kinase. J. Neurosci. Res. 81, 497–505.
Jorissen, E., Prox, J., Bernreuther, C., Weber, S., Schwanbeck, R., Serneels, L., Snellinx, A., Craessaerts, K., Thathiah, A., Tesseur, I., Bartsch, U., Weskamp, G., Blobel, C. P., Glatzel, M., De Strooper, B., and Saftig, P. (2010). The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J. Neurosci. 30, 4833–4844.
Katakowski, M., Chen, J., Zhang, Z. G., Santra, M., Wang, Y., and Chopp, M. (2007). Stroke-induced subventricular zone proliferation is promoted by tumor necrosis factor-alpha-converting enzyme protease activity. J. Cereb. Blood Flow Metab. 27, 669–678.
Kavanaugh, W. M., and Williams, L. T. (1994). An alternative to SH2 domains for binding tyrosine-phosphorylated proteins. Science 266, 1862–1865.
Kee, N., Teixeira, C. M., Wang, A. H., and Frankland, P. W. (2007). Preferential incorporation of adult-generated granule cells into spatial memory networks in the dentate gyrus. Nat. Neurosci. 10, 355–362.
Kempermann, G., Kuhn, H. G., and Gage, F. H. (1997a). Genetic influence on neurogenesis in the dentate gyrus of adult mice. Proc. Natl. Acad. Sci. U.S.A. 94, 10409–10414.
Kempermann, G., Kuhn, H. G., and Gage, F. H. (1997b). More hippocampal neurons in adult mice living in an enriched environment. Nature 386, 493–495.
Kim, W. R., Kim, Y., Eun, B., Park, O. H., Kim, H., Kim, K., Park, C. H., Vinsant, S., Oppenheim, R. W., and Sun, W. (2007). Impaired migration in the rostral migratory stream but spared olfactory function after the elimination of programmed cell death in Bax knock-out mice. J. Neurosci. 27, 14392–14403.
Kimberly, W. T., Zheng, J. B., Guenette, S. Y., and Selkoe, D. J. (2001). The intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J. Biol. Chem. 276, 40288–40292.
Kinoshita, A., Whelan, C. M., Berezovska, O., and Hyman, B. T. (2002). The gamma secretase-generated carboxyl-terminal domain of the amyloid precursor protein induces apoptosis via Tip60 in H4 cells. J. Biol. Chem. 277, 28530–28536.
Kirfel, G., Borm, B., Rigort, A., and Herzog, V. (2002). The secretory beta-amyloid precursor protein is a motogen for human epidermal keratinocytes. Eur. J. Cell Biol. 81, 664–676.
Ko, S. Y., Lin, S. C., Chang, K. W., Wong, Y. K., Liu, C. J., Chi, C. W., and Liu, T. Y. (2004). Increased expression of amyloid precursor protein in oral squamous cell carcinoma. Int. J. Cancer 111, 727–732.
Kolecki, R., Lafauci, G., Rubenstein, R., Mazur-Kolecka, B., Kaczmarski, W., and Frackowiak, J. (2008). The effect of amyloidosis-beta and ageing on proliferation of neuronal progenitor cells in APP-transgenic mouse hippocampus and in culture. Acta Neuropathol. 116, 419–424.
Kuhn, H. G., Dickinson-Anson, H., and Gage, F. H. (1996). Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J. Neurosci. 16, 2027–2033.
Kwak, Y. D., Marutle, A., Dantuma, E., Merchant, S., Bushnev, S., and Sugaya, K. (2011). Involvement of notch signaling pathway in amyloid precursor protein induced glial differentiation. Eur. J. Pharmacol. 650, 18–27.
Lammich, S., Kojro, E., Postina, R., Gilbert, S., Pfeiffer, R., Jasionowski, M., Haass, C., and Fahrenholz, F. (1999). Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. U.S.A. 96, 3922–3927.
Lavoie, M. J., and Selkoe, D. J. (2003). The Notch ligands, Jagged and Delta, are sequentially processed by alpha-secretase and presenilin/gamma-secretase and release signaling fragments. J. Biol. Chem. 278, 34427–34437.
Lazarini, F., Mouthon, M. A., Gheusi, G., De Chaumont, F., Olivo-Marin, J. C., Lamarque, S., Abrous, D. N., Boussin, F. D., and Lledo, P. M. (2009). Cellular and behavioral effects of cranial irradiation of the subventricular zone in adult mice. PLoS ONE 4, e7017. doi:10.1371/journal.pone.0007017
Lazarov, O., and Marr, R. A. (2010). Neurogenesis and Alzheimer’s disease: at the crossroads. Exp. Neurol. 223, 267–281.
Lazic, S. E., Grote, H. E., Blakemore, C., Hannan, A. J., Van Dellen, A., Phillips, W., and Barker, R. A. (2006). Neurogenesis in the R6/1 transgenic mouse model of Huntington’s disease: effects of environmental enrichment. Eur. J. Neurosci. 23, 1829–1838.
Lemaire, V., Koehl, M., Le Moal, M., and Abrous, D. N. (2000). Prenatal stress produces learning deficits associated with an inhibition of neurogenesis in the hippocampus. Proc. Natl. Acad. Sci. U.S.A. 97, 11032–11037.
Li, B., Yamamori, H., Tatebayashi, Y., Shafit-Zagardo, B., Tanimukai, H., Chen, S., Iqbal, K., and Grundke-Iqbal, I. (2008). Failure of neuronal maturation in Alzheimer disease dentate gyrus. J. Neuropathol. Exp. Neurol. 67, 78–84.
Li, B. S., Ma, W., Zhang, L., Barker, J. L., Stenger, D. A., and Pant, H. C. (2001). Activation of phosphatidylinositol-3 kinase (PI-3K) and extracellular regulated kinases (Erk1/2) is involved in muscarinic receptor-mediated DNA synthesis in neural progenitor cells. J. Neurosci. 21, 1569–1579.
Li, H., Wang, B., Wang, Z., Guo, Q., Tabuchi, K., Hammer, R. E., Sudhof, T. C., and Zheng, H. (2010). Soluble amyloid precursor protein (APP) regulates transthyretin and Klotho gene expression without rescuing the essential function of APP. Proc. Natl. Acad. Sci. U.S.A. 107, 17362–17367.
Lim, D. A., Tramontin, A. D., Trevejo, J. M., Herrera, D. G., Garcia-Verdugo, J. M., and Alvarez-Buylla, A. (2000). Noggin antagonizes BMP signaling to create a niche for adult neurogenesis. Neuron 28, 713–726.
Liu, Q., Zerbinatti, C. V., Zhang, J., Hoe, H. S., Wang, B., Cole, S. L., Herz, J., Muglia, L., and Bu, G. (2007). Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 56, 66–78.
Lois, C., and Alvarez-Buylla, A. (1994). Long-distance neuronal migration in the adult mammalian brain. Science 264, 1145–1148.
Lois, C., Garcia-Verdugo, J. M., and Alvarez-Buylla, A. (1996). Chain migration of neuronal precursors. Science 271, 978–981.
Lopez-Toledano, M. A., and Shelanski, M. L. (2004). Neurogenic effect of beta-amyloid peptide in the development of neural stem cells. J. Neurosci. 24, 5439–5444.
Lopez-Toledano, M. A., and Shelanski, M. L. (2007). Increased neurogenesis in young transgenic mice overexpressing human APP(Sw, Ind). J. Alzheimers Dis. 12, 229–240.
Lu, D. C., Soriano, S., Bredesen, D. E., and Koo, E. H. (2003). Caspase cleavage of the amyloid precursor protein modulates amyloid beta-protein toxicity. J. Neurochem. 87, 733–741.
Ma, Q. H., Futagawa, T., Yang, W. L., Jiang, X. D., Zeng, L., Takeda, Y., Xu, R. X., Bagnard, D., Schachner, M., Furley, A. J., Karagogeos, D., Watanabe, K., Dawe, G. S., and Xiao, Z. C. (2008). A TAG1-APP signalling pathway through Fe65 negatively modulates neurogenesis. Nat. Cell Biol. 10, 283–294.
Madsen, T. M., Kristjansen, P. E., Bolwig, T. G., and Wortwein, G. (2003). Arrested neuronal proliferation and impaired hippocampal function following fractionated brain irradiation in the adult rat. Neuroscience 119, 635–642.
Magara, F., Muller, U., Li, Z. W., Lipp, H. P., Weissmann, C., Stagljar, M., and Wolfer, D. P. (1999). Genetic background changes the pattern of forebrain commissure defects in transgenic mice underexpressing the beta-amyloid-precursor protein. Proc. Natl. Acad. Sci. U.S.A. 96, 4656–4661.
Mak, G. K., and Weiss, S. (2010). Paternal recognition of adult offspring mediated by newly generated CNS neurons. Nat. Neurosci. 13, 753–758.
Mallm, J. P., Tschape, J. A., Hick, M., Filippov, M. A., and Muller, U. C. (2010). Generation of conditional null alleles for APP and APLP2. Genesis 48, 200–206.
Mandairon, N., Sultan, S., Nouvian, M., Sacquet, J., and Didier, A. (2011). Involvement of newborn neurons in olfactory associative learning? The operant or non-operant component of the task makes all the difference. J. Neurosci. 31, 12455–12460.
Masliah, E., Westland, C. E., Rockenstein, E. M., Abraham, C. R., Mallory, M., Veinberg, I., Sheldon, E., and Mucke, L. (1997). Amyloid precursor proteins protect neurons of transgenic mice against acute and chronic excitotoxic injuries in vivo. Neuroscience 78, 135–146.
Mazur-Kolecka, B., Golabek, A., Nowicki, K., Flory, M., and Frackowiak, J. (2006). Amyloid-beta impairs development of neuronal progenitor cells by oxidative mechanisms. Neurobiol. Aging 27, 1181–1192.
Ming, G. L., and Song, H. (2005). Adult neurogenesis in the mammalian central nervous system. Annu. Rev. Neurosci. 28, 223–250.
Ming, G. L., and Song, H. (2011). Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70, 687–702.
Moreno, M. M., Linster, C., Escanilla, O., Sacquet, J., Didier, A., and Mandairon, N. (2009). Olfactory perceptual learning requires adult neurogenesis. Proc. Natl. Acad. Sci. U.S.A. 106, 17980–17985.
Mucke, L., Yu, G. Q., Mcconlogue, L., Rockenstein, E. M., Abraham, C. R., and Masliah, E. (2000). Astroglial expression of human alpha(1)-antichymotrypsin enhances alzheimer-like pathology in amyloid protein precursor transgenic mice. Am. J. Pathol. 157, 2003–2010.
Muller, T., Concannon, C. G., Ward, M. W., Walsh, C. M., Tirniceriu, A. L., Tribl, F., Kogel, D., Prehn, J. H., and Egensperger, R. (2007). Modulation of gene expression and cytoskeletal dynamics by the amyloid precursor protein intracellular domain (AICD). Mol. Biol. Cell 18, 201–210.
Muller, T., Meyer, H. E., Egensperger, R., and Marcus, K. (2008). The amyloid precursor protein intracellular domain (AICD) as modulator of gene expression, apoptosis, and cytoskeletal dynamics-relevance for Alzheimer’s disease. Prog. Neurobiol. 85, 393–406.
Muller, U., Cristina, N., Li, Z. W., Wolfer, D. P., Lipp, H. P., Rulicke, T., Brandner, S., Aguzzi, A., and Weissmann, C. (1994). Behavioral and anatomical deficits in mice homozygous for a modified beta-amyloid precursor protein gene. Cell 79, 755–765.
Nakayama, K., Ohkawara, T., Hiratochi, M., Koh, C. S., and Nagase, H. (2008). The intracellular domain of amyloid precursor protein induces neuron-specific apoptosis. Neurosci. Lett. 444, 127–131.
Naumann, N., Alpar, A., Ueberham, U., Arendt, T., and Gartner, U. (2010). Transgenic expression of human wild-type amyloid precursor protein decreases neurogenesis in the adult hippocampus. Hippocampus 20, 971–979.
Needham, B. E., Wlodek, M. E., Ciccotosto, G. D., Fam, B. C., Masters, C. L., Proietto, J., Andrikopoulos, S., and Cappai, R. (2008). Identification of the Alzheimer’s disease amyloid precursor protein (APP) and its homologue APLP2 as essential modulators of glucose and insulin homeostasis and growth. J. Pathol. 215, 155–163.
Nottebohm, F. (2004). The road we travelled: discovery, choreography, and significance of brain replaceable neurons. Ann. N. Y. Acad. Sci. 1016, 628–658.
Ohkawara, T., Nagase, H., Koh, C. S., and Nakayama, K. (2011). The amyloid precursor protein intracellular domain alters gene expression and induces neuron-specific apoptosis. Gene 475, 1–9.
Ohsawa, I., Takamura, C., and Kohsaka, S. (1997). The amino-terminal region of amyloid precursor protein is responsible for neurite outgrowth in rat neocortical explant culture. Biochem. Biophys. Res. Commun. 236, 59–65.
Ohsawa, I., Takamura, C., Morimoto, T., Ishiguro, M., and Kohsaka, S. (1999). Amino-terminal region of secreted form of amyloid precursor protein stimulates proliferation of neural stem cells. Eur. J. Neurosci. 11, 1907–1913.
Ozaki, T., Li, Y., Kikuchi, H., Tomita, T., Iwatsubo, T., and Nakagawara, A. (2006). The intracellular domain of the amyloid precursor protein (AICD) enhances the p53-mediated apoptosis. Biochem. Biophys. Res. Commun. 351, 57–63.
Phillips, W., Morton, A. J., and Barker, R. A. (2005). Abnormalities of neurogenesis in the R6/2 mouse model of Huntington’s disease are attributable to the in vivo microenvironment. J. Neurosci. 25, 11564–11576.
Pietrzik, C. U., Hoffmann, J., Stober, K., Chen, C. Y., Bauer, C., Otero, D. A., Roch, J. M., and Herzog, V. (1998). From differentiation to proliferation: the secretory amyloid precursor protein as a local mediator of growth in thyroid epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 95, 1770–1775.
Postina, R., Schroeder, A., Dewachter, I., Bohl, J., Schmitt, U., Kojro, E., Prinzen, C., Endres, K., Hiemke, C., Blessing, M., Flamez, P., Dequenne, A., Godaux, E., Van Leuven, F., and Fahrenholz, F. (2004). A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Invest. 113, 1456–1464.
Qiu, W. Q., Ferreira, A., Miller, C., Koo, E. H., and Selkoe, D. J. (1995). Cell-surface beta-amyloid precursor protein stimulates neurite outgrowth of hippocampal neurons in an isoform-dependent manner. J. Neurosci. 15, 2157–2167.
Raber, J., Rola, R., Lefevour, A., Morhardt, D., Curley, J., Mizumatsu, S., Vandenberg, S. R., and Fike, J. R. (2004). Radiation-induced cognitive impairments are associated with changes in indicators of hippocampal neurogenesis. Radiat. Res. 162, 39–47.
Reinhard, C., Hebert, S. S., and De Strooper, B. (2005). The amyloid-beta precursor protein: integrating structure with biological function. EMBO J. 24, 3996–4006.
Ring, S., Weyer, S. W., Kilian, S. B., Waldron, E., Pietrzik, C. U., Filippov, M. A., Herms, J., Buchholz, C., Eckman, C. B., Korte, M., Wolfer, D. P., and Muller, U. C. (2007). The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J. Neurosci. 27, 7817–7826.
Roch, J. M., Masliah, E., Roch-Levecq, A. C., Sundsmo, M. P., Otero, D. A., Veinbergs, I., and Saitoh, T. (1994). Increase of synaptic density and memory retention by a peptide representing the trophic domain of the amyloid beta/A4 protein precursor. Proc. Natl. Acad. Sci. U.S.A. 91, 7450–7454.
Rochefort, C., Gheusi, G., Vincent, J. D., and Lledo, P. M. (2002). Enriched odor exposure increases the number of newborn neurons in the adult olfactory bulb and improves odor memory. J. Neurosci. 22, 2679–2689.
Rockenstein, E. M., Mcconlogue, L., Tan, H., Power, M., Masliah, E., and Mucke, L. (1995). Levels and alternative splicing of amyloid beta protein precursor (APP) transcripts in brains of APP transgenic mice and humans with Alzheimer’s disease. J. Biol. Chem. 270, 28257–28267.
Rodriguez, J. J., Jones, V. C., Tabuchi, M., Allan, S. M., Knight, E. M., Laferla, F. M., Oddo, S., and Verkhratsky, A. (2008). Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS ONE 3, e2935. doi:10.1371/journal.pone.0002935
Rohe, M., Carlo, A. S., Breyhan, H., Sporbert, A., Militz, D., Schmidt, V., Wozny, C., Harmeier, A., Erdmann, B., Bales, K. R., Wolf, S., Kempermann, G., Paul, S. M., Schmitz, D., Bayer, T. A., Willnow, T. E., and Andersen, O. M. (2008). Sortilin-related receptor with A-type repeats (SORLA) affects the amyloid precursor protein-dependent stimulation of ERK signaling and adult neurogenesis. J. Biol. Chem. 283, 14826–14834.
Rola, R., Raber, J., Rizk, A., Otsuka, S., Vandenberg, S. R., Morhardt, D. R., and Fike, J. R. (2004). Radiation-induced impairment of hippocampal neurogenesis is associated with cognitive deficits in young mice. Exp. Neurol. 188, 316–330.
Rossjohn, J., Cappai, R., Feil, S. C., Henry, A., Mckinstry, W. J., Galatis, D., Hesse, L., Multhaup, G., Beyreuther, K., Masters, C. L., and Parker, M. W. (1999). Crystal structure of the N-terminal, growth factor-like domain of Alzheimer amyloid precursor protein. Nat. Struct. Biol. 6, 327–331.
Sabo, S. L., Ikin, A. F., Buxbaum, J. D., and Greengard, P. (2001). The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J. Cell Biol. 153, 1403–1414.
Sabo, S. L., Ikin, A. F., Buxbaum, J. D., and Greengard, P. (2003). The amyloid precursor protein and its regulatory protein, FE65, in growth cones and synapses in vitro and in vivo. J. Neurosci. 23, 5407–5415.
Sahay, A., Scobie, K. N., Hill, A. S., O’Carroll, C. M., Kheirbek, M. A., Burghardt, N. S., Fenton, A. A., Dranovsky, A., and Hen, R. (2011). Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature 472, 466–470.
Saitoh, T., Sundsmo, M., Roch, J. M., Kimura, N., Cole, G., Schubert, D., Oltersdorf, T., and Schenk, D. B. (1989). Secreted form of amyloid beta protein precursor is involved in the growth regulation of fibroblasts. Cell 58, 615–622.
Sakamoto, M., Imayoshi, I., Ohtsuka, T., Yamaguchi, M., Mori, K., and Kageyama, R. (2011). Continuous neurogenesis in the adult forebrain is required for innate olfactory responses. Proc. Natl. Acad. Sci. U.S.A. 108, 8479–8484.
Salbaum, J. M., and Ruddle, F. H. (1994). Embryonic expression pattern of amyloid protein precursor suggests a role in differentiation of specific subsets of neurons. J. Exp. Zool. 269, 116–127.
Sandbrink, R., Masters, C. L., and Beyreuther, K. (1996). APP gene family. Alternative splicing generates functionally related isoforms. Ann. N. Y. Acad. Sci. 777, 281–287.
Saxe, M. D., Battaglia, F., Wang, J. W., Malleret, G., David, D. J., Monckton, J. E., Garcia, A. D., Sofroniew, M. V., Kandel, E. R., Santarelli, L., Hen, R., and Drew, M. R. (2006). Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc. Natl. Acad. Sci. U.S.A. 103, 17501–17506.
Schellinck, H. M., Arnold, A., and Rafuse, V. F. (2004). Neural cell adhesion molecule (NCAM) null mice do not show a deficit in odour discrimination learning. Behav. Brain Res. 152, 327–334.
Schmitz, A., Tikkanen, R., Kirfel, G., and Herzog, V. (2002). The biological role of the Alzheimer amyloid precursor protein in epithelial cells. Histochem. Cell Biol. 117, 171–180.
Schrenk-Siemens, K., Perez-Alcala, S., Richter, J., Lacroix, E., Rahuel, J., Korte, M., Muller, U., Barde, Y. A., and Bibel, M. (2008). Embryonic stem cell-derived neurons as a cellular system to study gene function: lack of amyloid precursor proteins APP and APLP2 leads to defective synaptic transmission. Stem Cells 26, 2153–2163.
Seabrook, G. R., Smith, D. W., Bowery, B. J., Easter, A., Reynolds, T., Fitzjohn, S. M., Morton, R. A., Zheng, H., Dawson, G. R., Sirinathsinghji, D. J., Davies, C. H., Collingridge, G. L., and Hill, R. G. (1999). Mechanisms contributing to the deficits in hippocampal synaptic plasticity in mice lacking amyloid precursor protein. Neuropharmacology 38, 349–359.
Shimazu, K., Zhao, M., Sakata, K., Akbarian, S., Bates, B., Jaenisch, R., and Lu, B. (2006). NT-3 facilitates hippocampal plasticity and learning and memory by regulating neurogenesis. Learn. Mem. 13, 307–315.
Shors, T. J., Miesegaes, G., Beylin, A., Zhao, M., Rydel, T., and Gould, E. (2001). Neurogenesis in the adult is involved in the formation of trace memories. Nature 410, 372–376.
Shors, T. J., Townsend, D. A., Zhao, M., Kozorovitskiy, Y., and Gould, E. (2002). Neurogenesis may relate to some but not all types of hippocampal-dependent learning. Hippocampus 12, 578–584.
Siemes, C., Quast, T., Kummer, C., Wehner, S., Kirfel, G., Muller, U., and Herzog, V. (2006). Keratinocytes from APP/APLP2-deficient mice are impaired in proliferation, adhesion and migration in vitro. Exp. Cell Res. 312, 1939–1949.
Simon, A. M., Schiapparelli, L., Salazar-Colocho, P., Cuadrado-Tejedor, M., Escribano, L., Lopez De Maturana, R., Del Rio, J., Perez-Mediavilla, A., and Frechilla, D. (2009). Overexpression of wild-type human APP in mice causes cognitive deficits and pathological features unrelated to Abeta levels. Neurobiol. Dis. 33, 369–378.
Sisodia, S. S., Koo, E. H., Beyreuther, K., Unterbeck, A., and Price, D. L. (1990). Evidence that beta-amyloid protein in Alzheimer’s disease is not derived by normal processing. Science 248, 492–495.
Sone, M., Uchida, A., Komatsu, A., Suzuki, E., Ibuki, I., Asada, M., Shiwaku, H., Tamura, T., Hoshino, M., Okazawa, H., and Nabeshima, Y. (2009). Loss of yata, a novel gene regulating the subcellular localization of APPL, induces deterioration of neural tissues and lifespan shortening. PLoS ONE 4, e4466. doi:10.1371/journal.pone.0004466
Sotthibundhu, A., Li, Q. X., Thangnipon, W., and Coulson, E. J. (2009). Abeta(1-42) stimulates adult SVZ neurogenesis through the p75 neurotrophin receptor. Neurobiol. Aging 30, 1975–1985.
Storey, E., and Cappai, R. (1999). The amyloid precursor protein of Alzheimer’s disease and the Abeta peptide. Neuropathol. Appl. Neurobiol. 25, 81–97.
Sugaya, K. (2008). Mechanism of glial differentiation of neural progenitor cells by amyloid precursor protein. Neurodegener. Dis. 5, 170–172.
Sultan, S., Mandairon, N., Kermen, F., Garcia, S., Sacquet, J., and Didier, A. (2010). Learning-dependent neurogenesis in the olfactory bulb determines long-term olfactory memory. FASEB J. 24, 2355–2363.
Sung, S. M., Jung, D. S., Kwon, C. H., Park, J. Y., Kang, S. K., and Kim, Y. K. (2007). Hypoxia/reoxygenation stimulates proliferation through PKC-dependent activation of ERK and Akt in mouse neural progenitor cells. Neurochem. Res. 32, 1932–1939.
Taipale, J., Cooper, M. K., Maiti, T., and Beachy, P. A. (2002). Patched acts catalytically to suppress the activity of Smoothened. Nature 418, 892–897.
Takayama, K., Tsutsumi, S., Suzuki, T., Horie-Inoue, K., Ikeda, K., Kaneshiro, K., Fujimura, T., Kumagai, J., Urano, T., Sakaki, Y., Shirahige, K., Sasano, H., Takahashi, S., Kitamura, T., Ouchi, Y., Aburatani, H., and Inoue, S. (2009). Amyloid precursor protein is a primary androgen target gene that promotes prostate cancer growth. Cancer Res. 69, 137–142.
Taniuchi, N., Niidome, T., Goto, Y., Akaike, A., Kihara, T., and Sugimoto, H. (2007). Decreased proliferation of hippocampal progenitor cells in APPswe/PS1dE9 transgenic mice. Neuroreport 18, 1801–1805.
Tarr, P. E., Roncarati, R., Pelicci, G., Pelicci, P. G., and D’Adamio, L. (2002). Tyrosine phosphorylation of the beta-amyloid precursor protein cytoplasmic tail promotes interaction with Shc. J. Biol. Chem. 277, 16798–16804.
Thornton, E., Vink, R., Blumbergs, P. C., and Van Den Heuvel, C. (2006). Soluble amyloid precursor protein alpha reduces neuronal injury and improves functional outcome following diffuse traumatic brain injury in rats. Brain Res. 1094, 38–46.
Trazzi, S., Steger, M., Mitrugno, V. M., Bartesaghi, R., and Ciani, E. (2010). CB1 cannabinoid receptors increase neuronal precursor proliferation through AKT/glycogen synthase kinase-3beta/beta-catenin signaling. J. Biol. Chem. 285, 10098–10109.
Tremml, P., Lipp, H. P., Muller, U., Ricceri, L., and Wolfer, D. P. (1998). Neurobehavioral development, adult openfield exploration and swimming navigation learning in mice with a modified beta-amyloid precursor protein gene. Behav. Brain Res. 95, 65–76.
Tremml, P., Lipp, H. P., Muller, U., and Wolfer, D. P. (2002). Enriched early experiences of mice underexpressing the beta-amyloid precursor protein restore spatial learning capabilities but not normal openfield behavior of adult animals. Genes Brain Behav. 1, 230–241.
van Praag, H., Kempermann, G., and Gage, F. H. (1999). Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat. Neurosci. 2, 266–270.
van Praag, H., Schinder, A. F., Christie, B. R., Toni, N., Palmer, T. D., and Gage, F. H. (2002). Functional neurogenesis in the adult hippocampus. Nature 415, 1030–1034.
van Tetering, G., Van Diest, P., Verlaan, I., Van Der Wall, E., Kopan, R., and Vooijs, M. (2009). Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J. Biol. Chem. 284, 31018–31027.
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., Teplow, D. B., Ross, S., Amarante, P., Loeloff, R., Luo, Y., Fisher, S., Fuller, J., Edenson, S., Lile, J., Jarosinski, M. A., Biere, A. L., Curran, E., Burgess, T., Louis, J. C., Collins, F., Treanor, J., Rogers, G., and Citron, M. (1999). Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741.
Verret, L., Jankowsky, J. L., Xu, G. M., Borchelt, D. R., and Rampon, C. (2007). Alzheimer’s-type amyloidosis in transgenic mice impairs survival of newborn neurons derived from adult hippocampal neurogenesis. J. Neurosci. 27, 6771–6780.
von Koch, C. S., Zheng, H., Chen, H., Trumbauer, M., Thinakaran, G., Van Der Ploeg, L. H., Price, D. L., and Sisodia, S. S. (1997). Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol. Aging 18, 661–669.
Wang, P., Yang, G., Mosier, D. R., Chang, P., Zaidi, T., Gong, Y. D., Zhao, N. M., Dominguez, B., Lee, K. F., Gan, W. B., and Zheng, H. (2005). Defective neuromuscular synapses in mice lacking amyloid precursor protein (APP) and APP-Like protein 2. J. Neurosci. 25, 1219–1225.
Wasco, W., Bupp, K., Magendantz, M., Gusella, J. F., Tanzi, R. E., and Solomon, F. (1992). Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid beta protein precursor. Proc. Natl. Acad. Sci. U.S.A. 89, 10758–10762.
Wasco, W., Gurubhagavatula, S., Paradis, M. D., Romano, D. M., Sisodia, S. S., Hyman, B. T., Neve, R. L., and Tanzi, R. E. (1993). Isolation and characterization of APLP2 encoding a homologue of the Alzheimer’s associated amyloid beta protein precursor. Nat. Genet. 5, 95–100.
Wehner, S., Siemes, C., Kirfel, G., and Herzog, V. (2004). Cytoprotective function of sAppalpha in human keratinocytes. Eur. J. Cell Biol. 83, 701–708.
Weyer, S. W., Klevanski, M., Delekate, A., Voikar, V., Aydin, D., Hick, M., Filippov, M., Drost, N., Schaller, K. L., Saar, M., Vogt, M. A., Gass, P., Samanta, A., Jaschke, A., Korte, M., Wolfer, D. P., Caldwell, J. H., and Muller, U. C. (2011). APP and APLP2 are essential at PNS and CNS synapses for transmission, spatial learning and LTP. EMBO J. 30, 2266–2280.
Wolfe, M. S., Xia, W., Ostaszewski, B. L., Diehl, T. S., Kimberly, W. T., and Selkoe, D. J. (1999). Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 398, 513–517.
Yamasaki, A., Eimer, S., Okochi, M., Smialowska, A., Kaether, C., Baumeister, R., Haass, C., and Steiner, H. (2006). The GxGD motif of presenilin contributes to catalytic function and substrate identification of gamma-secretase. J. Neurosci. 26, 3821–3828.
Yan, R., Munzner, J. B., Shuck, M. E., and Bienkowski, M. J. (2001). BACE2 functions as an alternative alpha-secretase in cells. J. Biol. Chem. 276, 34019–34027.
Yang, G., Gong, Y. D., Gong, K., Jiang, W. L., Kwon, E., Wang, P., Zheng, H., Zhang, X. F., Gan, W. B., and Zhao, N. M. (2005). Reduced synaptic vesicle density and active zone size in mice lacking amyloid precursor protein (APP) and APP-like protein 2. Neurosci. Lett. 384, 66–71.
Yoshikawa, K., Aizawa, T., and Hayashi, Y. (1992). Degeneration in vitro of post-mitotic neurons overexpressing the Alzheimer amyloid protein precursor. Nature 359, 64–67.
Young-Pearse, T. L., Bai, J., Chang, R., Zheng, J. B., Loturco, J. J., and Selkoe, D. J. (2007). A critical function for beta-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J. Neurosci. 27, 14459–14469.
Young-Pearse, T. L., Suth, S., Luth, E. S., Sawa, A., and Selkoe, D. J. (2010). Biochemical and functional interaction of disrupted-in-schizophrenia 1 and amyloid precursor protein regulates neuronal migration during mammalian cortical development. J. Neurosci. 30, 10431–10440.
Yu, G., Nishimura, M., Arawaka, S., Levitan, D., Zhang, L., Tandon, A., Song, Y. Q., Rogaeva, E., Chen, F., Kawarai, T., Supala, A., Levesque, L., Yu, H., Yang, D. S., Holmes, E., Milman, P., Liang, Y., Zhang, D. M., Xu, D. H., Sato, C., Rogaev, E., Smith, M., Janus, C., Zhang, Y., Aebersold, R., Farrer, L. S., Sorbi, S., Bruni, A., Fraser, P., and St George-Hyslop, P. (2000). Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 407, 48–54.
Zhang, C., Mcneil, E., Dressler, L., and Siman, R. (2007a). Long-lasting impairment in hippocampal neurogenesis associated with amyloid deposition in a knock-in mouse model of familial Alzheimer’s disease. Exp. Neurol. 204, 77–87.
Zhang, Y. W., Wang, R., Liu, Q., Zhang, H., Liao, F. F., and Xu, H. (2007b). Presenilin/gamma-secretase-dependent processing of beta-amyloid precursor protein regulates EGF receptor expression. Proc. Natl. Acad. Sci. U.S.A. 104, 10613–10618.
Zhang, C. L., Zou, Y., He, W., Gage, F. H., and Evans, R. M. (2008). A role for adult TLX-positive neural stem cells in learning and behaviour. Nature 451, 1004–1007.
Zhao, C., Teng, E. M., Summers, R. G. Jr., Ming, G. L., and Gage, F. H. (2006). Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus. J. Neurosci. 26, 3–11.
Zhao, X., Ueba, T., Christie, B. R., Barkho, B., Mcconnell, M. J., Nakashima, K., Lein, E. S., Eadie, B. D., Willhoite, A. R., Muotri, A. R., Summers, R. G., Chun, J., Lee, K. F., and Gage, F. H. (2003). Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proc. Natl. Acad. Sci. U.S.A. 100, 6777–6782.
Zheng, H., Jiang, M., Trumbauer, M. E., Hopkins, R., Sirinathsinghji, D. J., Stevens, K. A., Conner, M. W., Slunt, H. H., Sisodia, S. S., Chen, H. Y., and Van Der Ploeg, L. H. (1996). Mice deficient for the amyloid precursor protein gene. Ann. N. Y. Acad. Sci. 777, 421–426.
Zheng, H., Jiang, M., Trumbauer, M. E., Sirinathsinghji, D. J., Hopkins, R., Smith, D. W., Heavens, R. P., Dawson, G. R., Boyce, S., Conner, M. W., Stevens, K. A., Slunt, H. H., Sisoda, S. S., Chen, H. Y., and Van Der Ploeg, L. H. (1995). beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 81, 525–531.
Keywords: neurogenesis, amyloid precursor protein, learning and memory, aging, Alzheimer’s disease, neuronal plasticity
Citation: Lazarov O and Demars MP (2012) All in the family: how the APPs regulate neurogenesis. Front. Neurosci. 6:81. doi: 10.3389/fnins.2012.00081
Received: 07 March 2012; Accepted: 14 May 2012;
Published online: 04 June 2012.
Edited by:
Laura López-Mascaraque, Instituto Cajal-CSIC, SpainReviewed by:
Alino Martinez-Marcos, Universidad de Castilla, SpainJosé Luis Trejo, Institute Cajal-CSIC, Spain
Copyright: © 2012 Lazarov and Demars. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Orly Lazarov, Department of Anatomy and Cell Biology, College of Medicine, The University of Illinois at Chicago, 808 South Wood Street M/C 512, Chicago, IL 60612, USA. e-mail:b2xhemFyb3ZAdWljLmVkdQ==