

- Max F. Perutz Laboratories, Department of Chromosome Biology, University of Vienna, Vienna, Austria
RNA editing by adenosine deaminases that act on RNA converts adenosines to inosines in coding and non-coding regions of mRNAs. Inosines are interpreted as guanosines and hence, this type of editing can change codons, alter splice patterns, or influence the fate of an RNA. A to I editing is most abundant in the central nervous system (CNS). Here, targets for this type of nucleotide modification frequently encode receptors and channels. In many cases, the editing-induced amino acid exchanges alter the properties of the receptors and channels. Consistently, changes in editing patterns are frequently found associated with diseases of the CNS. In this review we describe the mechanisms of RNA editing and focus on target mRNAs of editing that are functionally relevant to normal and aberrant CNS activity.
RNA Editing
RNA editing is a site specific, post transcriptional modification of RNA. Two types of RNA editing can be distinguished. On the one hand, insertion-deletion type editing inserts or removes single or multiple nucleotides from an RNA molecule (Simpson et al., 2000). This type of editing is mostly found in organelles of various phyla. Deamination type editing, on the other hand, changes the identity of a base by deaminating cytidine to uracil or adenosine to inosine, respectively (Carter, 1995). Deamination type editing has been found in all kingdoms of life. Deamination of cytidines was first discovered in the mRNA encoding apolipoprotein B which is deaminated by Apobec1 a member of the apobec/AID cytidine deaminase family that mostly target cytidines in DNA. Recently, abundant cytidine deamination by Apobec1 was identified in the 3′ UTRs of many mouse mRNAs (Rosenberg et al., 2011). The function of these editing events remains to be determined, however.
Adenosine deamination by adenosine deaminases that act on RNA (ADAR) seemingly only affects metazoan nuclear encoded RNAs. Deamination of adenosines leads to the formation of inosines which are recognized as guanosines by most cellular machineries (Bass and Weintraub, 1988). Editing in coding regions of mRNAs can therefore lead to a codon exchange and the subsequent translation of a protein that differs from the genomically encoded version. Moreover, editing can also introduce or remove splice sites and thereby lead to the formation of novel mRNAs (Rueter et al., 1999). Finally, inosines in RNA can change the base-pairing propensity of an RNA and therefore alter their folding and change their signature for RNA-binding proteins (Nishikura, 1992). Thus, also editing in non-translated regions of an mRNA may have profound impact on the fate of the affected RNA. Besides mRNAs, also primary and precursor miRNAs can be targets for RNA editing by ADARs. Editing of pri- and pre-miRNAs can alter their processing but also their base-pairing potential with target mRNAs. Therefore, editing of miRNAs can indirectly change the abundance and translatability of their target mRNAs.
Editing of mRNAs was originally believed to be a rare event. In recent years, however, editing was found to be widespread in mRNAs of higher eukaryotes (Athanasiadis et al., 2004; Levanon et al., 2004). In all organisms editing by ADARs is most abundant in the nervous system. The profound alterations of the transcriptome and proteome introduced by RNA editing may thus help to solve a long lasting biological paradigm, namely, how biological complexity can be achieved with an almost constant number of genes: editing-induced alterations of splice patterns and coding potential of mRNAs may, together with alternative splicing, contribute to the formation of a complex proteome from a limited number of genes. Consistently, alterations in the editing patterns or loss of editing is accompanied by pathologic conditions and disease (Morabito and Emeson, 2009).
The ADAR Protein Family
ADARs were first discovered in Xenopus laevis as an unwinding activity that destabilizes RNA duplexes upon A to I editing (Bass and Weintraub, 1987; Rebagliati and Melton, 1987). ADARs have been well characterized in many organisms including insects, worms, and vertebrates (Bass, 2002). The first ADAR gene identified was vertebrate ADAR1 harboring three double-stranded RNA-binding domains (dsRBDs) and a conserved deaminase domain with zinc binding motifs. Subsequent screens led to the identification of ADAR2 (Melcher et al., 1996; O’Connell et al., 1997). Recent analyses have shown the presence of ADAR1 and ADAR2 in many species including sea urchin and sea anemones (Jin et al., 2009). The vertebrate genome encodes two additional, ADAR proteins. ADAR3, which presumably arose from ADAR2 by gene duplication, contains all functional domains. However, no function has been ascribed to this isoform (Chen et al., 2000). The fourth ADAR-like gene, termed TENR, is expressed in the male germ line and has one dsRBD. TENR lacks conserved zinc chelating residues in the deaminase domain thus explaining its inactivity (Hough and Bass, 1997). ADARs are related to the tRNA editing family of ADATs which are found in all kingdoms of life (Jin et al., 2009).
Phenotypes of ADAR Deficiency
Different phenotypes are associated with the lack of individual ADAR isoforms. ADAR2 null mice have episodes of epileptic seizures and show subsequent postnatal death. A key substrate of ADAR 2 is the mRNA encoding GluA2. Underedited GluA2 allows increased influx of Ca2+ leading to death of neurons (Brusa et al., 1995). Consistently, ADAR2−/−mice can be rescued by replacing the genomic, unedited GluA2 copy with a “preedited” gene copy (Higuchi et al., 2000). Still, even the rescued ADAR2−/−mice display a range of subtle phenotypes ranging from a decreased acoustic startle response to decreased blood glucose level. The molecular mechanisms underlying these changes are still to be determined (Horsch et al., 2011). ADAR2 overexpressing mice, in contrast, display hyperphagia and obesity (Singh et al., 2007). This phenotype can be reproduced by a catalytically inactive version of ADAR2 that retains its RNA-binding ability. This suggests that RNA binding of ADARs can lead to editing-independent phenotypes (Singh et al., 2007).
Mice lacking ADAR1 die during embryonic development, show defective hematopoiesis, widespread apoptosis, and liver disintegration (Hartner et al., 2004). Molecularly, ADAR1 deficient mice show an increase in interferon signaling with the precise molecular mechanisms leading to death remaining unknown (Hartner et al., 2009).
In Drosophila melanogaster inactivation of the single ADAR gene causes tremors, lack of coordination, mating defects, and neurodegeneration presumably resulting from underediting of important dADAR target genes such as Na+ (para), Ca2+(cac), and glutamate-gated Cl− channels (DrosGluCl-α; Palladino et al., 2000). Caenorhabditis elegans strains with homozygous deletions in either of the two ADARs adr-1 or adr-2 show chemotactic defects, also indicating a role in the editing of neuronally expressed substrates (Tonkin et al., 2002).
Substrates of ADAR
RNA editing by adenosine deaminases can affect coding and non-coding RNA sequences. Substrate RNAs are recognized by the dsRBDs located in ADARs. These domains bind to A-form helices formed by double-stranded RNAs. Thus, editing sites are defined by base-paired regions of 20 or more nucleotides in length. A-form helices display a wide minor groove and a narrow major groove. Sequence specific information of the bases cannot be easily contacted making a sequence specific positioning of ADARs difficult (Ryter and Schultz, 1998). Still, various mechanisms can contribute to substrate and editing specificity. Multiple dsRBDs found in ADARs can coordinately bind to substrates (Stefl et al., 2005). Most double-stranded structures formed by endogenous RNAs are disrupted by bulges. These bulges set natural boundaries for the binding of dsRBDs (Lehmann and Bass, 1999). If two or more dsRBDs need to bind to a double-stranded region of limited length they can help to position each other. Structural analysis of ADAR2 bound to a stem loop substrate shows nicely that some dsRBDs can also bind to terminal loops thus helping to increase substrate specific binding (Stefl et al., 2006). Recently, specific minor groove interactions between dsRBDs and nucleotides have been identified. These interactions can increase sequence specificity dramatically, therefore aiding in selecting specific adenosines within a stretch of double-stranded RNA (Stefl et al., 2010). Finally, also the deaminase domains of ADAR1 and ADAR2 display substrate specificities that preferentially select certain adenosines depending on their local sequence context (Polson and Bass, 1994; Eggington et al., 2011). As the adenosine to be edited typically lies within a double-stranded structure the target adenosine needs to be accessed through a base flipping mechanism (Stephens et al., 2000; Yi-Brunozzi et al., 2001).
The altered base-pairing potential of inosines can lead to an alteration of the RNA secondary structure. Thus, editing in the non-translated regions of mRNAs may alter their localization, stability, and translatability. However, the biological consequences of editing in these targets is still under debate. The consequences of adenosine deamination in coding regions of mRNAs and pri-miRNAs on the other hand are more easy to understand. As inosines are read as guanosines during translation, inosines can alter the coding potential or targeting specificity of mRNAs and miRNAs, respectively (Vesely et al., 2012).
The proteins encoded by edited pre-mRNAs vary widely in their function. However, frequently editing-induced amino acid exchanges affect receptors and ion channels expressed in the brain. Another class of proteins affected by RNA editing play a role in cytoskeletal remodeling which also plays an important role in neuronal outgrowth and plasticity. In the following, representative examples of both classes of proteins and the functional implications of their editing will be described.
Glutamate-Gated Ion Channels
Five subunits of the glutamate receptor (GluA2, GluA3, GluA4, GluK1, and GluK2) are found to undergo ADAR-mediated RNA editing (Bass, 2002). A total of four editing sites that result in amino acid changes have been identified, namely glutamine to arginine (Q/R), arginine to glycine (R/G), isoleucine to valine (I/V), and tyrosine to cysteine (Y/C; see Table 1).
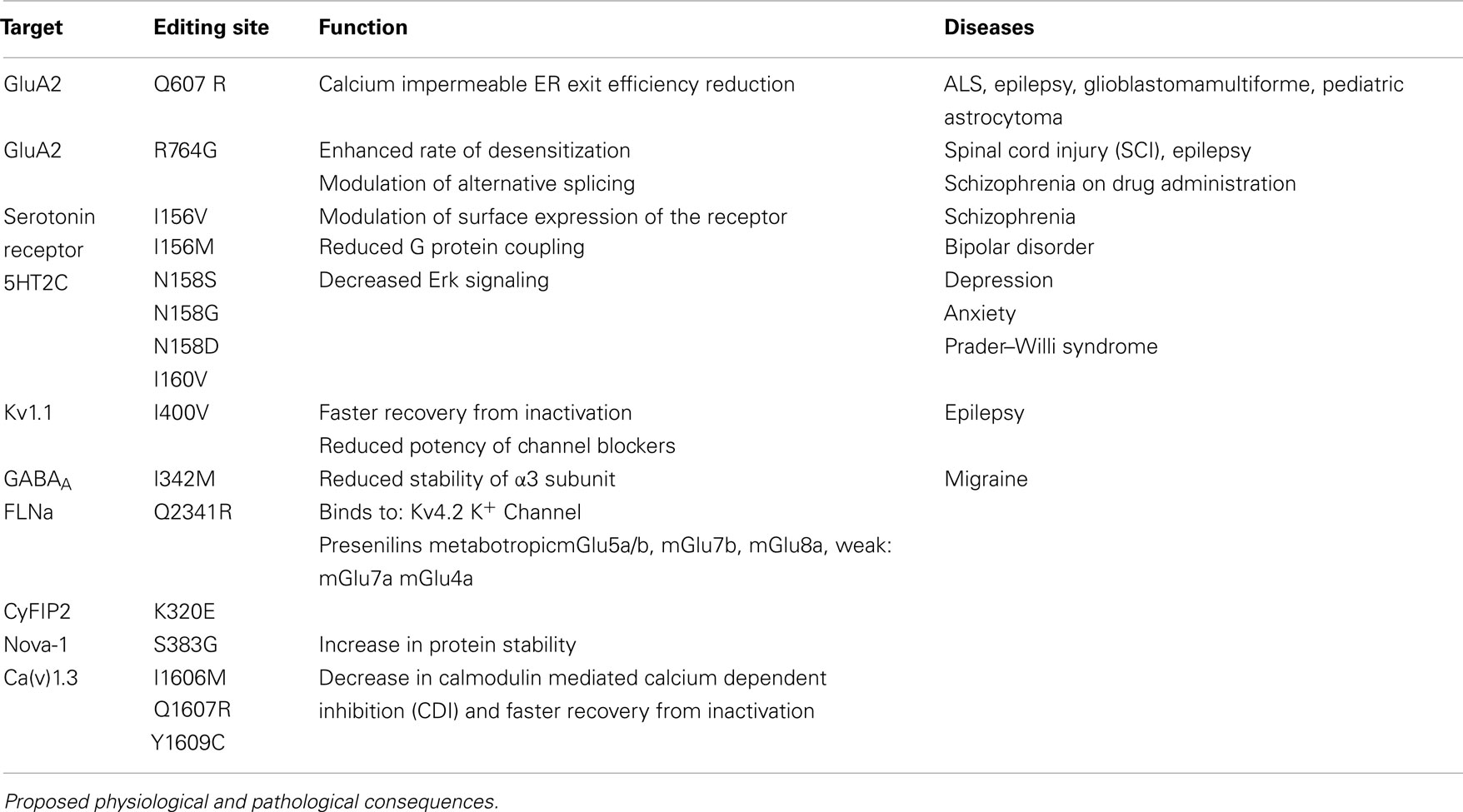
Table 1. Selected editing events in the CNS.
AMPA GluA2 subunit mRNA was the first target discovered. It is edited mainly at two coding sites leading to a glutamine to arginine and arginine to glycine conversion (Sommer et al., 1991; Lomeli et al., 1994; see Figure 2).
Two additional editing sites are found in intron 11 of GluA2 mRNA, called hotspot 1 (or +60 site) and hotspot 2 (or+262/263/264 site), respectively (Higuchi et al., 1993). Editing at the Q/R site reduces Ca2+ permeability (see Figure 1). The edited GluA2R isoforms also show reduced endoplasmic reticulum (ER) exit efficiency, whereas unedited GluA2Q isoforms readily tetramerize and are transported to the synaptic membrane (Greger et al., 2002, 2003). GluA2 in the unedited Q form leads to epileptic seizures and subsequent postnatal death. This toxic effect has been attributed to increased calcium influx (Higuchi et al., 2000). Additionally increased receptor density due to faster ER exit may also contribute to this effect (Greger et al., 2002; see Figure 1).
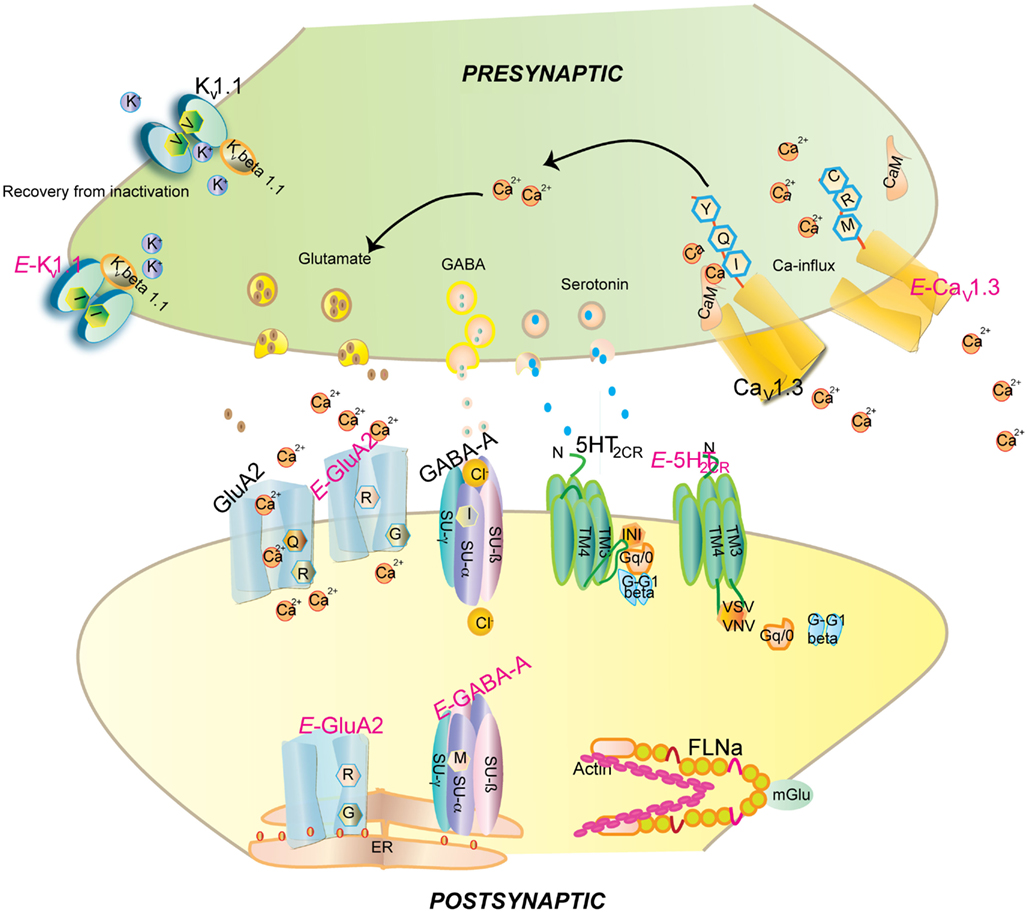
Figure 1. Impact of editing on selected neuronal receptors and proteins. Shown are several receptors and channels in their unedited (black) and edited (E, in pink) versions. Editing at the Q/R site of ionotropic glutamate receptor GluA2 subunit decreases Ca2+permeability and endoplasmic reticulum exit efficiency. Membrane trafficking of the GABAA receptor is reduced by editing of I to M in the alpha3 subunit. Editing of the serotonin 5-HT2c receptor converts the amino acids I-N-I to V-S-V or V-N-V. This reduces G-protein coupling in the receptor. The editing-induced I to V exchange in Kv1.1 (I/V) alters the interaction with Kv β1.1 (see Figure 3 for detail). Editing of the IQ motiv in Cav1.3 to MR abolishes calmodulin binding. Filamin alpha (FLNa) is edited in a region that is known to interact with metabotropic glutamate receptor mGlu7b and some of its relatives.
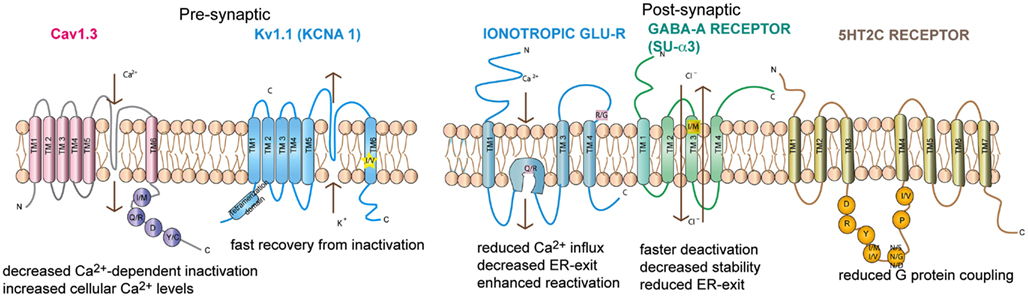
Figure 2. Topography of the neuronal channels containing editing sites Ionotropic glutamate receptor, GABAA receptor subunit α3, Kv1.1, Cav1.3, and 5-HT2c receptors are shown. The relative position of the editing sites within these receptors are highlighted. Consequences of editing are indicated.
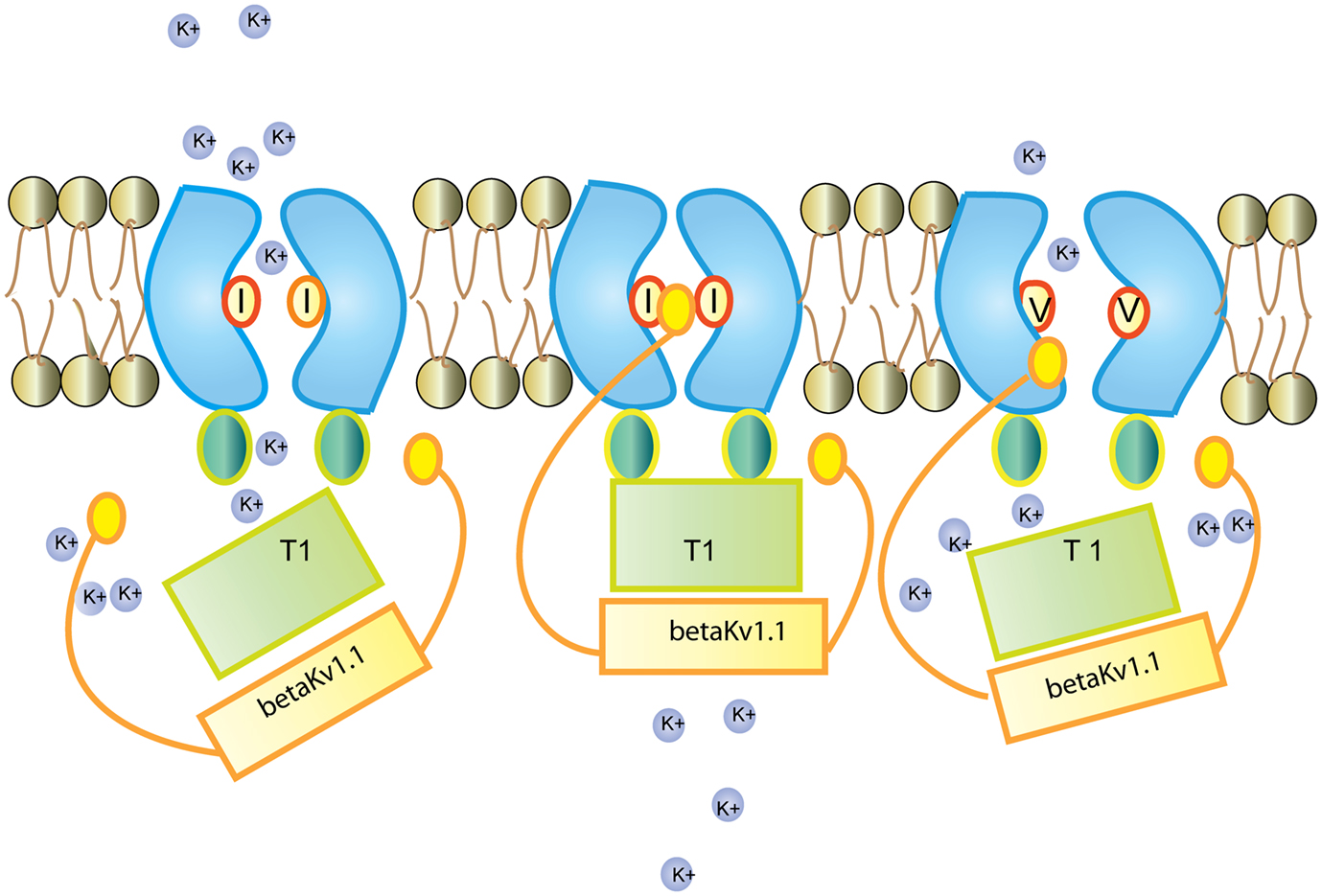
Figure 3. Regulation of the Kv1.1 channel by Kv β1.1 Kv β1.1 has an inactivation gate which interacts with the unedited (I) form of the Kv1.1 receptor. Editing of Kv1.1 changes the isoleucine to valine (V). This reduces the affinity for Kv β1.1 and enhances recovery from inactivation. Figure adapted from (Bezanilla, 2004).
The R/G conversion reduces the assembly of homomeric receptors and slows down receptor maturation in the ER (Greger et al., 2006). Additionally, R/G site editing results in enhanced recovery from desensitization (Lomeli et al., 1994). Editing events in the GluA2 pre-mRNA also affect splicing of nearby introns. Editing at the R/G site of GluA2 takes place two nucleotides upstream of the 5′ splice site in intron13. The Q/R site is located in exon 11, 25 nucleotides upstream of the 5′ splice site of intron 11 (Higuchi et al., 1993). Editing at the Q/R site and the intronic hotspot enhances splicing of the nearby intron, while editing at the R/G site represses splicing at the downstream intron (Schoft et al., 2007). Editing at the R/G site may affect base-pairing of the pre-mRNA with the U1snRNA (Schoft et al., 2007). R/G site editing also influences the alternative splicing of the two downstream exons as editing promotes inclusion of exon15 (flip) over exon14 (flop). GluA2 protein with an edited G and the flip variant undergoes rapid maturation in the ER relative to the flop form. The flip variant also stimulates dendritic growth (Hamad et al., 2011). The flop isoform, in turn, promotes assembly of heteromeric AMPA receptors (Penn and Greger, 2009).
Also kainate receptor subunits GluK1 and -2 are edited at the Q/R position. GluK2 undergoes additional editing at the I/V and Y/C sites, located at positions 621, 567, and 571 respectively, which may lead to higher calcium permeability (Kohler et al., 1993).
GABAA Receptor
GABAA receptors are ligand gated chloride channels consisting of five subunits: 2 α subunits, 2 β subunits, and either a γ or a δ subunit (Hevers and Luddens, 1998). The existence of 6 α, 3 β, 3 γ, and 4 δ subunits allows for the assembly of a wide variety of stoichometries. Position 342 of the α3 subunit is highly edited, resulting in an isoleucine (AUA) to methionine (AUI) codon change (Ohlson et al., 2007; see Figure 2). The editing site is defined by a specific RNA structure marked by bulges at a defined distance from the editing site as well as a specific terminal loop structure (Tian et al., 2011). With age, the two α subunits show opposing expression patterns. While α1 expression increases with age the α3 subunit is predominant at embryonic level (Hutcheon et al., 2004). Moreover, editing is developmentally regulated. The pre-mRNA is mostly found unedited around day e15 but is edited from 80% to 100% at postnatal day 7 (p7; Ohlson et al., 2007; Rula et al., 2008)
The I/M change in GABAA receptor causes a delay in currents and faster deactivation upon stimulation by GABA (Rula et al., 2008). Expression of unedited GABAA receptor in the developing brain is crucial for synapse formation (Ben-Ari et al., 2007). Recently, editing has been proposed to affect the stability of the α3 subunit as the edited version displays low cell surface expression. The M version of the receptor maintains the hydrophobic environment but can influence the interaction between α and γ subunits or ligand interaction (Daniel et al., 2011; see Table 1; Figure 1).
Voltage-Gated Potassium Channels
Neuronal Kv1.1 channels are built of a tetramer of pore forming α subunits along with four regulatory beta subunits and accessory subunits. The channels regulate action potential and modulate neuronal excitability by opening and closing of a potassium selective pore. The human Kv1.1 (KCNA1) gene is intronless and undergoes A to I RNA editing leading to an isoleucine to valine exchange (see Figure 1). The amino acid exchange is located within the sixth transmembrane segment (S6) which lies at the ion conducting pore (Bhalla et al., 2004; see Figure 2). Kv1.1 channels are edited up to 65–80% in medulla, thalamus and spinal cord (Decher et al., 2010). The I–V change is evolutionarily conserved and also occurs in Kv2 (DmShab Shaker) channels in Drosophila melanogaster together with four other editing events (Bhalla et al., 2004; Ryan et al., 2008).
Kv1.1 associates with the redox sensor Kv β1 in the ER (Pan et al., 2008). Kv β harbors an N-terminal inactivation domain that controls inactivation and lag time of Kv1.1. The edited Kv1.1 shows a 20 fold higher recovery from Kv β1 mediated inactivation than the unedited version of the channel (Bezanilla, 2004; Bhalla et al., 2004; see Figure 3).
The Kv channel blocker 4-aminopyridine (4-AP) has been shown to induce epileptic seizures. RNA editing makes the channel insensitive to 4-AP by disrupting the interaction between the pore lining and the channel blocker (see Table 1; Streit et al., 2011). A similar insensitivity was observed against arachidonic acid (Decher et al., 2010).
Squid Kv 1.1A has also been shown to be extensively edited (Rosenthal and Bezanilla, 2002). Here editing not only affects channel gating but also influences the tetramerization of the channel.
Voltage-Gated Calcium channels
Voltage-gated calcium channels (VGCC) are classified into two types: Low voltage activated (LVA) and High voltage activated (HVA) channels (Lacinova, 2005). LVA L-type calcium channels are involved in a broad range of neuronal processes such as neuronal pacemaking, secretion of neurotransmitters, synaptic transmission, mRNA stability, and modulation of other ion channels (Singh et al., 2008). The opening of these channels permits calcium influx. The channels are inactivated by voltage dependent inhibition (VDI) and intracellular calcium dependent inhibition (CDI). The pore forming α1 subunit contains four domains (I–IV), each domain consisting of six transmembrane segments (S1–S6; Catterall et al., 2005; see Figure 2). S5 and S6 of all four domains form the central pore with S6 lining the inner surface of the pore and occluding the pore in the inactive state. S1 to S4 from each domain form the voltage sensing domain and on activation the S4 segment moves outward triggering S6 movement leading to gate opening (Swartz, 2004).
Calmodulin (CaM) binds to the IQ domain located at the C-terminus of the pore forming α1 subunit. The formation of a Ca2+-CaM complex results in CDI (Tadross et al., 2008). Calcium binding to the N- and C-terminal CaM lobes can induce distinct channel regulation (Dick et al., 2008). Recently, editing of the core sequence of the IQ domain of Cav1.3 by ADAR2 has been discovered. The core sequence comprises of the 4 amino acids IQDY. Upon editing different isoforms such as MQDY, IRDY, MQDC, MRDY, MRDC, or IQDC can be generated (see Figures 1 and 4). This editing event is restricted to the central nervous system (CNS; Huang et al., 2012). Additionally, the pattern of editing is developmentally regulated. It is negligible at e14 and prominently increases at p4. RNA editing of the IQ domain shows spatial distribution being highest at the frontal cortex and hippocampus (Huang et al., 2012). The MQ and IR versions show weaker CDI while the MR variant exhibits up to 50% reduction of CDI and faster recovery from inactivation. Reduction in CDI consequently increases the cellular calcium load (see Table 1).
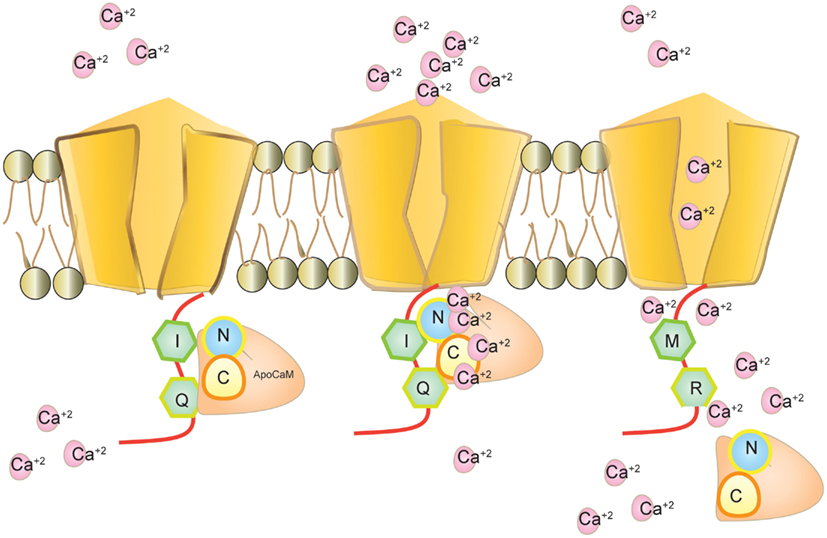
Figure 4. Cav1.3 and calmodulin interaction. Calmodulin binds to Cav1.3 without calcium as apoCaM at the IQ motif. Calmodulin binds to calcium through its N-terminal and C-terminal loop and mediates calcium dependent inhibition (CDI). Editing modifies IQ to MR and inhibits calmodulin binding. Intracellular calcium increases in the absence of CDI.
Serotonin 2C Receptor
The mammalian 5-hydroxytryptamine receptor subtype 2C (5-HT2c) is found widely distributed in the CNS. 5-HT2cR belongs to the G-protein-coupled receptor superfamily that stimulates phospholipase C (PLC) activity (Hoyer et al., 2002). The 5-HT2c receptor interacts with the multi PDZ-domain bearing protein (MPDZ). Both these proteins co-localize on the apical membrane of choroid plexus cells (Drago and Serretti, 2009). The pre-mRNA encoding serotonin receptor 5-HT2c is edited at five sites termed A, B, C, D, and E. Editing can lead to the exchange of three amino acids that are located within the second intracellular loop of the receptor (see Figure 2). This region has been shown to be important for efficient G-protein coupling (Niswender et al., 1998). Editing at the five positions in RNA can, in principle, yield a combination of 32 different mRNAs which encode 24 different protein isoforms (Burns et al., 1997; Niswender et al., 1999). In mouse brain, however, only eight major receptor variants can be detected at significant levels. Also sequencing analysis of human brain samples only detected 12 possible isoforms derived from RNA editing. This suggests that not all possible combinations of editing do exist in vivo (Wang et al., 2000; Wahlstedt et al., 2009). Moreover, the repertoire of possible editing combinations varies throughout development (Wahlstedt et al., 2009). Editing at sites A and B is accomplished by ADAR1, sites C, D, and E, however, are preferentially deaminated by ADAR2 (Hartner et al., 2004).
Upon editing, reduced G-protein coupling is observed in the edited states (Burns et al., 1997; see Figure 1). Editing not only reduces the constitutive activity of the receptor but also diminishes agonist potency and calcium release (Price et al., 2001). At the cellular level, editing changes the surface expression of the receptor. The subcellular distribution of the receptor depends on β arrestin-2 interaction with inverse correlation to the constitutive activity of the receptor. Editing decreases the constitutive activity and enhances surface expression. The fully edited VGV isoform displays complete surface expression. The unedited INI isoforms exhibit endosomal accumulation whereas the edited VSV isoforms with moderate activity display vesicular and cell surface expression (Marion et al., 2004). Editing also modulates the expression of the receptor through splicing. Unedited 5-HT2c transcripts result in a splice version that yields a shorter protein, while edited transcripts form the full length receptor (Flomen et al., 2004). However, the underlying factors resulting in alternative splicing are still not entirely clear. It was shown, for instance, that the human and mouse C/D box snoRNAs HBII-52 and MBII-52, respectively, can inhibit site C editing via base-pairing (Vitali et al., 2005). This base-pairing event also seemingly masks a silencer element important for the regulation of splicing (Kishore and Stamm, 2006).
The serotonin 2c receptors can also activate extracellular signal-regulated kinase (ERK) independent of G-protein coupling. Expression of the unedited INI isoform increases ERK1/2 phosphorylation in transfected HEK293 cells while expression of the edited VGV form decreases phosphorylation. However, this activation is significantly reduced upon β-arrestin depletion (Labasque et al., 2010). Editing also decreases downstream ERK signaling. Consistently, a shift toward the edited isoforms leads to reduced ERK signaling in prefrontal cortex of ADAR2 transgenic mice (Singh et al., 2011). Overexpression of ADAR2 and hyperediting of the 5-HT-2c receptor is also correlated with depressive behavior (Singh et al., 2009).
Mice carrying either a completely unedited “INI” version or a completely edited “VGV” version of the 5-HT-2c receptor have been generated (Kawahara et al., 2008). While the INI mice grow normally but are rather immobile in a forced swim assay, resembling a depressive behavior (Mombereau et al., 2010). It has also been shown that a decrease in the INI form of the receptor can lead to a decrease in ERK1/2 phosphorylation in transgenic ADAR2 mice (Singh et al., 2011). Aberrant ERK1/2 phosphorylation in turn is linked to depression and suicidal behavior as ERK1/2 plays a critical role in synaptic plasticity (Dwivedi et al., 2009).
Mice expressing the fully edited VGV version of the receptor, in contrast, have reduced fat mass, growth retardation, and high energy expenditure most likely due to hyperactivation of the sympathetic nervous system (Kawahara et al., 2008). Mutant mice with fully edited VGV isoforms have also been shown to display symptoms resembling those of the Prader–Willi syndrome (PWS; Morabito et al., 2010).
Thus, data from transgenic mice clearly demonstrate that the editing status of 5-HT2c-R can directly influence behavior underscoring the importance of RNA editing for the etiology of psychiatric disorders.
Actin Organization by Filamins
Two actin cross-linking proteins Filamin A (FLNa) and Filamin B (FLNb) are amongst a group of newly identified mammalian editing targets (Levanon et al., 2005; Nishimoto et al., 2008; Li et al., 2009). The two 280 kDa proteins form homo- and heterodimers and mediate orthogonal branching of actin filaments (Fucini et al., 1997; Sheen et al., 2002; Popowicz et al., 2006). Mammalian filamins are built of 24 immunoglobulin (Ig) like repeats divided into two rod segments. Rod 1 consisting of repeats 1–15 interacts with actin filaments whereas rod 2 is built from repeats 16–23 and interacts with several proteins (Chen et al., 2011). Repeat 24 is required for dimerization. Actin reorganization is essential for cell motility and migration and is an important determinant in dendritic spine and synapse formation (Dillon and Goda, 2005; Popowicz et al., 2006). Depletion of FLNa leads to embryonic lethality with severe cardiovascular and bone development defects (Feng et al., 2006; Hart et al., 2006). Also FLNb deficient mice show defective microvasculature and bone malformation (Zhou et al., 2007). Editing of FLNa or FLNb leads to a conserved glutamine (Q) to arginine (R) codon exchange in repeat 22 (Li et al., 2009) that is developmentally regulated (Wahlstedt et al., 2009). Repeat 22 has been shown to be involved in the interaction with a broad range of proteins (Popowicz et al., 2006; see Table 1).
FLNa interacts with the C-terminus of the metabotrobic glutamate receptor mGlu5a, 5b, 7b, and 8a (see Figure 1). Moreover, low affinity binding was also detected for mGlu4a and mGlu7a. Repeats 21 and 22of FLNa harboring the edited amino acid represent the minimal region critical for this interaction (Enz, 2002). Editing may thus regulate this interaction, the potential consequences of which remain to be determined.
FLNa also interacts with potassium channel Kv4.2 at filipodial roots and shows overlapping expression in cortical and hippocampal neurons. A “PTPP” amino acid motif in Kv4.2 (AA 601–604) is critical for this interaction. Again, FLNa repeats 21–24 are involved in this interaction. Coexpression of filamin in heterologous cells enhances the whole cell current density by ~2.7-fold most likely by properly positioning functional KV4.2 receptors at the cell surface (Petrecca et al., 2000).
Presenilins (PS) belong to a conserved protein family that were the first proteins identified responsible for familial Alzheimer disease (FAD; Nelson et al., 2010). Presenilins harbor eight transmembrane domains. PS1 and PS2 were identified in a yeast two hybrid assay to interact with repeats 21–24 of FLNa. A region between TM6 and TM7 of the presenilins is responsible for this interaction. The same loop harbors 14 different mutations that are associated with FAD. FLNa and PS1 co-localize in astrocytes (Zhang et al., 1998). Moreover, overexpression of PS1 in cultured HEK293 cells redistributes FLN from the cell periphery to the cytoplasm. The FAD-linked mutation PS1M146L induces FLNa expression (Lu et al., 2010). The FLNa PS1interactionis well conserved and could be physically and genetically demonstrated in Drosophila melanogaster (Guo et al., 2000).
FLNa also co-localizes with the neuronal microtubule associated protein Tau. Tau is involved in polymerization and stability of microtubules. Tau protein is abnormally phosphorylated and forms neurofibrillary tangles in the hippocampus in Alzheimer patients. It is believed that Tau induced FLNa depletion leads to actin network destabilization and consequently to synaptic loss (Feuillette et al., 2010).
The functional implication of editing-induced Q2341R amino acid exchange in repeat 22of FLNa is still unknown. However, it may have an effect on a broad range of interactions (Chen et al., 2011). One example is the interaction of FLNa with β-integrin. Repeat 21 cannot interact with β-integrin unless repeat 20 disassociates from it (Lad et al., 2007). Similarly, FLNa editing may change neuronal receptor organization as well as synaptic transmission by altering the interaction profile with binding partners.
Cytoplasmic FMRP Interacting Protein 2 (CyFIP2)
CyFIP2 was identified as an interaction partner of the fragile X mental retardation protein (FMRP) in a yeast two hybrid screen. The region of interaction between CyFIP2 and FMRP overlaps with the FMRP dimerization site (Schenck et al., 2001). CyFIP2 also interacts with FMRP-related proteins FXR1P and FXR2P (Schenck et al., 2001). The CyFIP2 encoding pre-mRNA is primarily edited by ADAR2 introducing a single K320Eamino acid exchange in mouse and human CyFIP2 (Levanon et al., 2005; Nishimoto et al., 2008).
CyFIP2 is a member of the WAVE/SCAR complex and is involved in actin remodeling. It plays a pivotal role in neuronal wiring as it directly interacts with FMRP and Rac-1 (Schenck et al., 2003). Flies have a single Cyfip gene which is 67% identical to human CyFIP1 and CyFIP2. Mutant Cyfip flies display shorter synapses and profound axonal path finding, growth, and branching defects (Schenck et al., 2003, 2004). CyFIP2 is mainly involved in maintaining synaptic plasticity as it is involved in translational regulation impeded in fragile X mental retardation. In vertebrates like Zebrafish that harbor both CyFIP1 and CyFIP2, cyfip2 mutants exhibit dorso-nasal axonal pathfinding defects (Pittman et al., 2010). RNAi of CyFIP2 in murine melanoma cells leads to aberrant lamellipodia proving the functionality of Cyfip2 in actin remodeling and cell motility (Steffen et al., 2004).
Editing at the K/E position of CyFIP2 increases during mouse brain development ranging from 4% at e15 to 75% editing at p21 (Wahlstedt et al., 2009). However, there seems a significant decline in CyFIP2 editing with age in human brain (Nicholas et al., 2010).
The biological significance of Cyfip2 editing is currently not clear. One possibility would be that the migratory behavior of cells is regulated by CyFIP2 by antagonizing Rac-1. However, interactions with FMR1 or the nucleo-cytoplasmic shuttling of Cyfip2 might equally be affected by editing. With the new discovery of modulation of ADAR by FMR1 in flies the possibility of a feed back loop of CyFip2 and ADAR regulation also appears possible as Cyfip antagonizes FMR1 in flies (Schenck et al., 2003).
Hu Proteins
Hu proteins are RNA-binding proteins which play an essential role in neuronal differentiation and plasticity. HuB, HuC, and Hu D are neuron specific whereas HuR is associated with cell stress responses. Each Hu protein has three RNA recognition motifs (RRM1–3). Hu proteins preferentially bind to AU rich RNA elements (ARE) where they can act as RNA stabilizers and regulators of polyadenylation and translation (Mobarak et al., 2000; Zhu et al., 2006; Hinman and Lou, 2008). Recently, five editing sites were discovered in HuD and HuB in a bioinformatic screen of deep-sequencing data (Enstero et al., 2010). The functional implication of editing is unknown. However, it is likely that editing of Hu proteins can alter the landscape of the brain transcriptome (Paz-Yaacov et al., 2010).
Nova-1
Recently, another RNA-binding protein, NOVA-1, was found to be edited (Irimia et al., 2012). NOVA-1 is a key regulator of alternative splicing of RNAs encoding synaptic proteins involved in neuronal activity in the CNS. NOVA-1 binds pre-mRNAs in a sequence dependent manner and diversifies proteins by splicing regulation. Nova-1 null mice die postnatally from motor neuron death due to spinal and brainstem neuron apoptosis (Jensen et al., 2000). The splicing regulation by NOVA-1/2 is well conserved from mammals to insects. Both Nova-1/2 and the Drosophila melanogaster ortholog PASILLA (PS) binds to YCAY enriched regions located upstream of repressed exons and downstream of activated exons (Brooks et al., 2011). RNA editing increases the Nova-1 half life by decreasing its susceptibility to proteasome degradation (Irimia et al., 2012). This stabilization of Nova-1 by RNA editing can create another layer of complexity in diversification of brain specific transcripts.
Dysregulation of A to I editing has been found associated with a number of diseases, ranging from mental disorders to cancers (Paz et al., 2007). The following sections will give an overview on diseases that are strongly influenced by ADAR-mediated editing.
Astrocytoma
This glial cell tumor is classified on the basis of malignancy into four grades (I–IV). Glioblastomamultiforme (GBM) is a grade IV tumor with a survival rate of less than 18 months in children and adults (Stupp et al., 2005). Glial cells respond to external stimuli via neuronal receptors (Bergles et al., 2000; Gallo and Ghiani, 2000). Hypoediting of GluA2 at the Q/R site has been observed in GBM leading to increased Ca2+ influx and activation of the Akt pathway through phosphorylation (Ishiuchi et al., 2007). Also in pediatric astrocytoma the malignancy increases with a decrease in editing. GBM cells show strong migratory activity which is reduced upon ADAR2 expression. Furthermore a decrease in GluK2 editing at the I/V and Y/C sites is observed in different brain regions (Cenci et al., 2008). Since both GluA2 and GluK2 are edited by ADAR2, ADAR2 overexpression strongly inhibits cell proliferation and slows down the cell cycle. Mutation in the ADAR2 deaminase domain does not affect tumor malignancy proving the necessity of editing in tumor progression (Cenci et al., 2008). In this type of tumor ADAR2 is expressed at a normal level, while ADAR1 and ADAR3 are overexpressed leading to the assumption that higher concentrations of ADAR1 and ADAR3 may inhibit the activity of ADAR2 (Cenci et al., 2008).
In pediatric astrocytoma high levels of interferon induced ADAR1 p150 are found. Overexpression of ADAR1 might again interfere with ADAR2 activity (Chen et al., 2000; Cenci et al., 2008).
Amyotrophic Lateral Sclerosis
Amyotrophic Lateral Sclerosis (ALS) is characterized by slow degeneration of upper and lower motor neurons with a consequent loss of voluntary movement (Rothstein, 2009). Different mechanisms are proposed to be the underlying causes of this disease. Decreased editing at the Q/R site leading to increased Ca2+ influx has been observed in mice displaying late onset ALS (Kuner et al., 2005). The editing efficiency at the GluA2 Q/R site also decreases dramatically in ALS patients (Kawahara et al., 2004). Consistent with reduced Q/R site editing, a significant decrease in ADAR2 expression has been observed in spinal motor neurons of ALS patients (Hideyama et al., 2011). However, no decline in editing of Q/R in upper motor neurons was observed.
Additionally, the flip-flop alternative splicing pattern of GluA2, downstream of the R/G editing site is pushed toward flip-bearing transcripts in ALS patients (Kawahara and Kwak, 2005). The flip form of GluA2 promotes assembly of slowly desensitizing AMPA receptors (Tomiyama et al., 2002).
Clearing of glutamate from the synaptic cleft is accomplished through glutamate transporters that prevent repeated firing and excitotoxicity. The astroglial EAAT2 glutamate transport is responsible for clearing glutamate from the cleft. ALS patients show 50% decreased EAAT2 protein levels as editing generates a cryptic polyadenylation site leading to intron 7 retention (Flomen and Makoff, 2011). Depletion of EAAT2 leads to neuronal death in transgenic mice (Rothstein et al., 1996).
Prader–Willi Syndrome
The Prader–Willi locus is genomically imprinted and only expressed from the paternally inherited chromosome, while the maternal copy is transcriptionally silenced (Constancia et al., 2004). Loss of expression or mutation of the paternal 15q11–q13 locus therefore leads to the formation of the Pader–Willi disease phenotype. Patients have growth defects in both sexes due to growth hormone deficiency, and cognition problems (Butler, 2011). Amongst several other transcripts the small C/D box snoRNA MBII-52 is located within the Prader–Willi locus. This snoRNA contains 18 nucleotides that are complementary to the editing site C of the serotonin 5-HT2c receptor. When expressed in nucleoli the 5-HT2c pre-mRNA can even be targeted for 2′-O-methylation (Vitali et al., 2005). Loss of MBII-52 causes an increase in editing. Mice with a deleted PWS imprinted control region show enhanced locomotor activity and aberrant discriminative behavior (Doe et al., 2009). Altered 5-HT2CR editing can also lead to phenotypes that mimic PWS. Mice expressing the fully edited VGV form of the serotonin receptor also exhibit PWS-like phenotypes such as hyperphagia, hypotonia, increased metabolism, and slim stature (Morabito et al., 2010). Molecularly, this isoform exhibits blunted G-protein coupling, reduced constitutive activity and enhanced serotonergic neurotransmission possibly as a consequence of increased surface expression (Kawahara et al., 2008; Morabito et al., 2010).
Transient Forebrain Ischemia
Cerebral ischemia in CA1 pyramidal neurons is caused by reduced oxygen supply, primarily as a consequence of heart attacks or occlusions of arteries. Neuronal damage is caused due to increased Ca2+ influx because of increased GluA2Q expression (Liu et al., 2004). Increase in calcium activates Cdk5 which phosphorylates NMDA receptors (Liu et al., 2004). Phosphorylation, in turn, prolongs opening of NMDA receptors which can activate nitric oxide synthase leading to the formation of toxic peroxynitrite that induces neuronal death (Fiskum et al., 1999; Bossy-Wetzel et al., 2004). During experimental induction of ischemia in rat brain ADAR2 expression is reduced. Consistently, recovery from ischemia can be accomplished through increased ADAR2 expression (Peng et al., 2006).
Downregulation of R/G site editing has been observed during spinal cord injury (SCI). Reduced editing at this site may limit cell death progression by suppressing postsynaptic excitation. Thus, editing might influence recovery after SCI (Barbon et al., 2010). Reduced editing at the R/G site was also observed in rat prefrontal cortex upon treatment with phencyclidene (PCP) that instigates schizophrenia like behavior (Barbon et al., 2007).
Epilepsy
Epilepsy is a common neurological disorder characterized by seizures caused by neuronal hyperexcitability (Bozzi et al., 2012). Decreased editing of the AMPA receptor Q/R site leads to calcium permeable channels. Mice heterozygous for an editing deficient GluA2 allele develop seizures and die at 3 weeks of age while complete absence of GluA2 expression does not provoke seizures (Brusa et al., 1995).
Increased editing at the R/G site of the GluA2 transcripts and also of Kv1.1 have also been linked to seizures (Vollmar et al., 2004). Editing at the R/G site enhances glutamate response of the receptor and modulates neuronal excitability (Lomeli et al., 1994). The editing-induced I/V change in Kv1.1 channels lies in the S6 segment. This is the target site of many drugs blocking the channel (Decher et al., 2010). Interestingly, the Kv channel blocker 4-aminopyridine (4-AP) also induces seizure like events in rats. RNA editing, in turn, reduces the affinity of 4-AP and serves as a compensatory mechanism against epileptic seizures (Streit et al., 2011).
Psychiatric Disorders
Changes in the editing pattern of 5-HT2C pre-mRNA have been linked to different psychiatric disorders such as schizophrenia, depression, and bipolar disorder (Table 1). Editing leads to reduced G-protein activation resulting in decreased basal activity (Niswender et al., 1999). However, the observed correlations do not allow a clear-cut conclusion. Sample sizes are typically small and the investigated samples are rarely well controlled and matched, therefore giving a heterogeneous picture. For instance, overexpression of the edited VSV receptor isoform has been observed in patients suffering from schizophrenia and bipolar disorders (Dracheva et al., 2008). Previously, in two different studies on suicide victims suffering schizophrenia, a significantly under edited B site and a hyper edited A site has been observed (Niswender et al., 2001; Sodhi et al., 2001). Analysis on suicide victims suffering major depression, in contrast, revealed an increase in editing at the C and C′ site accompanied by decreased D site editing. Treatment with fluoxetine, a serotonin selective uptake inhibitor, causes opposing effects on editing of these sites indicating site specific serotonin dependent regulation (Gurevich et al., 2002). Deregulation of A to I editing in schizophrenia and bipolar disorder (type I) patients and underediting of I/V site in GRIK2 resulting in high calcium influx has also been related to over expression of ADAR2 isoforms with diminished catalytic activity (Silberberg and Ohman, 2011; Silberberg et al., 2012). However, increase in ADAR1 expression has also been suggested as an inhibitor of ADAR2 activity (Simmons et al., 2010).
Outlook
Current studies on RNA editing have clearly shown that adenosine deamination is most abundant in the CNS where it plays a major role in the diversification of the transcriptome. Three major processes seem to be primarily affected by A to I editing: first, many receptors and channels are modulated in their primary response and sensitivity to stimuli. Second, in many cases receptor assembly and retention in the ER seems to be affected by RNA editing. Finally, cytoskeletal components required for both outgrowth of neurons but also to the structuring of the cortical cytoskeleton and the anchoring of receptors is affected by RNA editing. It is one of the challenges to understand how these three processes are interconnected possibly being regulated through neuronal activity that may feed back on the process of RNA editing itself.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in the lab of Michael F. Jantsch is supported by the Austrian Science Foundation grant Nr F4313-B09. Aamira Tariq was supported by a grant from the Pakistani Higher Education Commission.
References
Athanasiadis, A., Rich, A., and Maas, S. (2004). Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2, e391. doi:10.1371/journal.pbio.0020391
Barbon, A., Fumagalli, F., Caracciolo, L., Madaschi, L., Lesma, E., Mora, C., Carelli, S., Slotkin, T. A., Racagni, G., Di Giulio, A. M., Gorio, A., and Barlati, S. (2010). Acute spinal cord injury persistently reduces R/G RNA editing of AMPA receptors. J. Neurochem. 114, 397–407.
Barbon, A., Fumagalli, F., La Via, L., Caracciolo, L., Racagni, G., Riva, M. A., and Barlati, S. (2007). Chronic phencyclidine administration reduces the expression and editing of specific glutamate receptors in rat prefrontal cortex. Exp. Neurol. 208, 54–62.
Bass, B. (2002). RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 71, 817–846.
Bass, B. L., and Weintraub, H. (1987). A developmentally regulated activity that unwinds RNA duplexes. Cell 48, 607–613.
Bass, B. L., and Weintraub, H. (1988). An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 55, 1089–1098.
Ben-Ari, Y., Gaiarsa, J. L., Tyzio, R., and Khazipov, R. (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215–1284.
Bergles, D. E., Roberts, J. D., Somogyi, P., and Jahr, C. E. (2000). Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 405, 187–191.
Bezanilla, F. (2004). RNA editing of a human potassium channel modifies its inactivation. Nat. Struct. Mol. Biol. 11, 915–916.
Bhalla, T., Rosenthal, J. J., Holmgren, M., and Reenan, R. (2004). Control of human potassium channel inactivation by editing of a small mRNA hairpin. Nat. Struct. Mol. Biol. 11, 950–956.
Bossy-Wetzel, E., Talantova, M. V., Lee, W. D., Scholzke, M. N., Harrop, A., Mathews, E., Gotz, T., Han, J., Ellisman, M. H., Perkins, G. A., and Lipton, S. A. (2004). Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron 41, 351–365.
Bozzi, Y., Casarosa, S., and Caleo, M. (2012). Epilepsy as a neurodevelopmental disorder. Front. Psychiatry 3:19. doi:10.3389/fpsyt.2012.00019
Brooks, A. N., Yang, L., Duff, M. O., Hansen, K. D., Park, J. W., Dudoit, S., Brenner, S. E., and Graveley, B. R. (2011). Conservation of an RNA regulatory map between Drosophila and mammals. Genome Res. 21, 193–202.
Brusa, R., Zimmermann, F., Koh, D., Feldmeyer, D., Gass, P., Seeburg, P., and Sprengel, R. (1995). Early onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science 270, 1677–1680.
Burns, C., Chu, H., Rueter, S., Hutchinson, L., Canton, H., Sanders-Bush, E., and Emeson, R. (1997). Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature 387, 303–308.
Butler, M. G. (2011). Prader-Willi syndrome: obesity due to genomic imprinting. Curr. Genomics 12, 204–215.
Carter, C. W. Jr. (1995). The nucleoside deaminases for cytidine and adenosine: structure, transition state stabilization, mechanism, and evolution. Biochimie 77, 92–98.
Catterall, W. A., Perez-Reyes, E., Snutch, T. P., and Striessnig, J. (2005). International union of pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 57, 411–425.
Cenci, C., Barzotti, R., Galeano, F., Corbelli, S., Rota, R., Massimi, L., Di Rocco, C., O’Connell, M. A., and Gallo, A. (2008). Down-regulation of RNA editing in pediatric astrocytomas: ADAR2 editing activity inhibits cell migration and proliferation. J. Biol. Chem. 283, 7251–7260.
Chen, C. X., Cho, D. S., Wang, Q., Lai, F., Carter, K. C., and Nishikura, K. (2000). A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 6, 755–767.
Chen, H., Zhu, X., Cong, P., Sheetz, M. P., Nakamura, F., and Yan, J. (2011). Differential mechanical stability of filamin A rod segments. Biophys. J. 101, 1231–1237.
Daniel, C., Wahlstedt, H., Ohlson, J., Bjork, P., and Ohman, M. (2011). Adenosine-to-inosine RNA editing affects trafficking of the gamma-aminobutyric acid type A (GABA(A)) receptor. J. Biol. Chem. 286, 2031–2040.
Decher, N., Streit, A. K., Rapedius, M., Netter, M. F., Marzian, S., Ehling, P., Schlichthorl, G., Craan, T., Renigunta, V., Kohler, A., Dodel, R. C., Navarro-Polanco, R. A., Preisig-Muller, R., Klebe, G., Budde, T., Baukrowitz, T., and Daut, J. (2010). RNA editing modulates the binding of drugs and highly unsaturated fatty acids to the open pore of Kv potassium channels. EMBO J. 29, 2101–2113.
Dick, I. E., Tadross, M. R., Liang, H., Tay, L. H., Yang, W., and Yue, D. T. (2008). A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature 451, 830–834.
Dillon, C., and Goda, Y. (2005). The actin cytoskeleton: integrating form and function at the synapse. Annu. Rev. Neurosci. 28, 25–55.
Doe, C. M., Relkovic, D., Garfield, A. S., Dalley, J. W., Theobald, D. E., Humby, T., Wilkinson, L. S., and Isles, A. R. (2009). Loss of the imprinted snoRNA mbii-52 leads to increased 5htr2c pre-RNA editing and altered 5HT2CR-mediated behaviour. Hum. Mol. Genet. 18, 2140–2148.
Dracheva, S., Patel, N., Woo, D. A., Marcus, S. M., Siever, L. J., and Haroutunian, V. (2008). Increased serotonin 2C receptor mRNA editing: a possible risk factor for suicide. Mol. Psychiatry 13, 1001–1010.
Drago, A., and Serretti, A. (2009). Focus on HTR2C: a possible suggestion for genetic studies of complex disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 601–637.
Dwivedi, Y., Rizavi, H. S., Zhang, H., Roberts, R. C., Conley, R. R., and Pandey, G. N. (2009). Aberrant extracellular signal-regulated kinase (ERK)1/2 signalling in suicide brain: role of ERK kinase 1 (MEK1). Int. J. Neuropsychopharmacol. 12, 1337–1354.
Eggington, J. M., Greene, T., and Bass, B. L. (2011). Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun. 2, 319.
Enstero, M., Akerborg, O., Lundin, D., Wang, B., Furey, T. S., Ohman, M., and Lagergren, J. (2010). A computational screen for site selective A-to-I editing detects novel sites in neuron specific Hu proteins. BMC Bioinformatics 11, 6. doi:10.1186/1471-2105-11-6
Enz, R. (2002). The actin-binding protein Filamin-A interacts with the metabotropic glutamate receptor type 7. FEBS Lett. 514, 184–188.
Feng, Y., Chen, M. H., Moskowitz, I. P., Mendonza, A. M., Vidali, L., Nakamura, F., Kwiatkowski, D. J., and Walsh, C. A. (2006). Filamin A (FLNA) is required for cell-cell contact in vascular development and cardiac morphogenesis. Proc. Natl. Acad. Sci. U.S.A. 103, 19836–19841.
Feuillette, S., Deramecourt, V., Laquerriere, A., Duyckaerts, C., Delisle, M. B., Maurage, C. A., Blum, D., Buee, L., Frebourg, T., Campion, D., and Lecourtois, M. (2010). Filamin-A and myosin VI colocalize with fibrillary tau protein in Alzheimer’s disease and FTDP-17 brains. Brain Res. 1345, 182–189.
Fiskum, G., Murphy, A. N., and Beal, M. F. (1999). Mitochondria in neurodegeneration: acute ischemia and chronic neurodegenerative diseases. J. Cereb. Blood Flow Metab. 19, 351–369.
Flomen, R., Knight, J., Sham, P., Kerwin, R., and Makoff, A. (2004). Evidence that RNA editing modulates splice site selection in the 5-HT2C receptor gene. Nucleic Acids Res. 32, 2113–2122.
Flomen, R., and Makoff, A. (2011). Increased RNA editing in EAAT2 pre-mRNA from amyotrophic lateral sclerosis patients: involvement of a cryptic polyadenylation site. Neurosci. Lett. 497, 139–143.
Fucini, P., Renner, C., Herberhold, C., Noegel, A. A., and Holak, T. A. (1997). The repeating segments of the F-actin cross-linking gelation factor (ABP-120) have an immunoglobulin-like fold. Nat. Struct. Biol. 4, 223–230.
Gallo, V., and Ghiani, C. A. (2000). Glutamate receptors in glia: new cells, new inputs and new functions. Trends Pharmacol. Sci. 21, 252–258.
Greger, I. H., Akamine, P., Khatri, L., and Ziff, E. B. (2006). Developmentally regulated, combinatorial RNA processing modulates AMPA receptor biogenesis. Neuron 51, 85–97.
Greger, I. H., Khatri, L., Kong, X., and Ziff, E. B. (2003). AMPA receptor tetramerization is mediated by Q/R editing. Neuron 40, 763–774.
Greger, I. H., Khatri, L., and Ziff, E. B. (2002). RNA editing at arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron 34, 759–772.
Guo, Y., Zhang, S. X., Sokol, N., Cooley, L., and Boulianne, G. L. (2000). Physical and genetic interaction of filamin with presenilin in Drosophila. J. Cell Sci. 113(Pt 19), 3499–3508.
Gurevich, I., Tamir, H., Arango, V., Dwork, A. J., Mann, J. J., and Schmauss, C. (2002). Altered editing of serotonin 2C receptor pre-mRNA in the prefrontal cortex of depressed suicide victims. Neuron 34, 349–356.
Hamad, M. I., Ma-Hogemeier, Z. L., Riedel, C., Conrads, C., Veitinger, T., Habijan, T., Schulz, J. N., Krause, M., Wirth, M. J., Hollmann, M., and Wahle, P. (2011). Cell class-specific regulation of neocortical dendrite and spine growth by AMPA receptor splice and editing variants. Development 138, 4301–4313.
Hart, A. W., Morgan, J. E., Schneider, J., West, K., Mckie, L., Bhattacharya, S., Jackson, I. J., and Cross, S. H. (2006). Cardiac malformations and midline skeletal defects in mice lacking filamin A. Hum. Mol. Genet. 15, 2457–2467.
Hartner, J. C., Schmittwolf, C., Kispert, A., Muller, A. M., Higuchi, M., and Seeburg, P. H. (2004). Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J. Biol. Chem. 279, 4894–4902.
Hartner, J. C., Walkley, C. R., Lu, J., and Orkin, S. H. (2009). ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat. Immunol. 10, 109–115.
Hevers, W., and Luddens, H. (1998). The diversity of GABAA receptors. Pharmacological and electrophysiological properties of GABAA channel subtypes. Mol. Neurobiol. 18, 35–86.
Hideyama, T., Yamashita, T., Aizawa, H., Tsuji, S., Kakita, A., Takahashi, H., and Kwak, S. (2011). Profound downregulation of the RNA editing enzyme ADAR2 in ALS spinal motor neurons. Neurobiol. Dis. 45, 1121–1128.
Higuchi, M., Maas, S., Single, F., Hartner, J., Rozov, A., Burnashev, N., Feldmeyer, D., Sprengel, R., and Seeburg, P. (2000). Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406, 78–81.
Higuchi, M., Single, F. N., Kohler, M., Sommer, B., Sprengel, R., and Seeburg, P. H. (1993). RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell 75, 1361–1370.
Hinman, M. N., and Lou, H. (2008). Diverse molecular functions of Hu proteins. Cell. Mol. Life Sci. 65, 3168–3181.
Horsch, M., Seeburg, P. H., Adler, T., Aguilar-Pimentel, J. A., Becker, L., Calzada-Wack, J., Garrett, L., Gotz, A., Hans, W., Higuchi, M., Holter, S. M., Naton, B., Prehn, C., Puk, O., Racz, I., Rathkolb, B., Rozman, J., Schrewe, A., Adamski, J., Busch, D. H., Esposito, I., Graw, J., Ivandic, B., Klingenspor, M., Klopstock, T., Mempel, M., Ollert, M., Schulz, H., Wolf, E., Wurst, W., Zimmer, A., Gailus-Durner, V., Fuchs, H., De Angelis, M. H., and Beckers, J. (2011). Requirement of the RNA-editing enzyme ADAR2 for normal physiology in mice. J. Biol. Chem. 286, 18614–18622.
Hough, R. F., and Bass, B. L. (1997). Analysis of Xenopus dsRNA adenosine deaminase cDNAs reveals similarities to DNA methyltransferases. RNA 3, 356–370.
Hoyer, D., Hannon, J. P., and Martin, G. R. (2002). Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol. Biochem. Behav. 71, 533–554.
Huang, H., Tan, B. Z., Shen, Y., Tao, J., Jiang, F., Sung, Y. Y., Ng, C. K., Raida, M., Kohr, G., Higuchi, M., Fatemi-Shariatpanahi, H., Harden, B., Yue, D. T., and Soong, T. W. (2012). RNA editing of the IQ domain in Ca(v)1.3 channels modulates their Ca(2)-dependent inactivation. Neuron 73, 304–316.
Hutcheon, B., Fritschy, J. M., and Poulter, M. O. (2004). Organization of GABA receptor alpha-subunit clustering in the developing rat neocortex and hippocampus. Eur. J. Neurosci. 19, 2475–2487.
Irimia, M., Denuc, A., Ferran, J. L., Pernaute, B., Puelles, L., Roy, S. W., Garcia-Fernandez, J., and Marfany, G. (2012). Evolutionarily conserved A-to-I editing increases protein stability of the alternative splicing factor Nova1. RNA Biol. 9, 12–21.
Ishiuchi, S., Yoshida, Y., Sugawara, K., Aihara, M., Ohtani, T., Watanabe, T., Saito, N., Tsuzuki, K., Okado, H., Miwa, A., Nakazato, Y., and Ozawa, S. (2007). Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J. Neurosci. 27, 7987–8001.
Jensen, K. B., Dredge, B. K., Stefani, G., Zhong, R., Buckanovich, R. J., Okano, H. J., Yang, Y. Y., and Darnell, R. B. (2000). Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron 25, 359–371.
Jin, Y., Zhang, W., and Li, Q. (2009). Origins and evolution of ADAR-mediated RNA editing. IUBMB Life 61, 572–578.
Kawahara, Y., Grimberg, A., Teegarden, S., Mombereau, C., Liu, S., Bale, T. L., Blendy, J. A., and Nishikura, K. (2008). Dysregulated editing of serotonin 2C receptor mRNAs results in energy dissipation and loss of fat mass. J. Neurosci. 28, 12834–12844.
Kawahara, Y., Ito, K., Sun, H., Aizawa, H., Kanazawa, I., and Kwak, S. (2004). Glutamate receptors: RNA editing and death of motor neurons. Nature 427, 801.
Kawahara, Y., and Kwak, S. (2005). Excitotoxicity and ALS: what is unique about the AMPA receptors expressed on spinal motor neurons? Amyotroph. Lateral Scler. Other Motor Neuron Disord. 6, 131–144.
Kishore, S., and Stamm, S. (2006). The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science 311, 230–232.
Kohler, M., Burnashev, N., Sakmann, B., and Seeburg, P. H. (1993). Determinants of Ca2+ permeability in both TM1 and TM2 of high affinity kainate receptor channels: diversity by RNA editing. Neuron 10, 491–500.
Kuner, R., Groom, A. J., Bresink, I., Kornau, H. C., Stefovska, V., Muller, G., Hartmann, B., Tschauner, K., Waibel, S., Ludolph, A. C., Ikonomidou, C., Seeburg, P. H., and Turski, L. (2005). Late-onset motoneuron disease caused by a functionally modified AMPA receptor subunit. Proc. Natl. Acad. Sci. U.S.A. 102, 5826–5831.
Labasque, M., Meffre, J., Carrat, G., Becamel, C., Bockaert, J., and Marin, P. (2010). Constitutive activity of serotonin 2C receptors at G protein-independent signaling: modulation by RNA editing and antidepressants. Mol. Pharmacol. 78, 818–826.
Lad, Y., Kiema, T., Jiang, P., Pentikainen, O. T., Coles, C. H., Campbell, I. D., Calderwood, D. A., and Ylanne, J. (2007). Structure of three tandem filamin domains reveals auto-inhibition of ligand binding. EMBO J. 26, 3993–4004.
Lehmann, K. A., and Bass, B. L. (1999). The importance of internal loops within RNA substrates of ADAR1. J. Mol. Biol. 291, 1–13.
Levanon, E. Y., Eisenberg, E., Yelin, R., Nemzer, S., Hallegger, M., Shemesh, R., Fligelman, Z. Y., Shoshan, A., Pollock, S. R., Sztybel, D., Olshansky, M., Rechavi, G., and Jantsch, M. F. (2004). Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 22, 1001–1005.
Levanon, E. Y., Hallegger, M., Kinar, Y., Shemesh, R., Djinovic-Carugo, K., Rechavi, G., Jantsch, M. F., and Eisenberg, E. (2005). Evolutionarily conserved human targets of adenosine to inosine RNA editing. Nucleic Acids Res. 33, 1162–1168.
Li, J. B., Levanon, E. Y., Yoon, J. K., Aach, J., Xie, B., Leproust, E., Zhang, K., Gao, Y., and Church, G. M. (2009). Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 324, 1210–1213.
Liu, S., Lau, L., Wei, J., Zhu, D., Zou, S., Sun, H. S., Fu, Y., Liu, F., and Lu, Y. (2004). Expression of Ca(2+)-permeable AMPA receptor channels primes cell death in transient forebrain ischemia. Neuron 43, 43–55.
Lomeli, H., Mosbacher, J., Melcher, T., Hoger, T., Geiger, J. R., Kuner, T., Monyer, H., Higuchi, M., Bach, A., and Seeburg, P. H. (1994). Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science 266, 1709–1713.
Lu, Q., Ding, K., Frosch, M. P., Jones, S., Wolfe, M., Xia, W., and Lanford, G. W. (2010). Alzheimer’s disease-linked presenilin mutation (PS1M146L) induces filamin expression and gamma-secretase independent redistribution. J. Alzheimers Dis. 22, 235–245.
Marion, S., Weiner, D. M., and Caron, M. G. (2004). RNA editing induces variation in desensitization and trafficking of 5-hydroxytryptamine 2c receptor isoforms. J. Biol. Chem. 279, 2945–2954.
Melcher, T., Maas, S., Herb, A., Sprengel, R., Seeburg, P., and Higuchi, M. (1996). A mammalian RNA editing enzyme. Nature 379, 460–464.
Mobarak, C. D., Anderson, K. D., Morin, M., Beckel-Mitchener, A., Rogers, S. L., Furneaux, H., King, P., and Perrone-Bizzozero, N. I. (2000). The RNA-binding protein HuD is required for GAP-43 mRNA stability, GAP-43 gene expression, and PKC-dependent neurite outgrowth in PC12 cells. Mol. Biol. Cell 11, 3191–3203.
Mombereau, C., Kawahara, Y., Gundersen, B. B., Nishikura, K., and Blendy, J. A. (2010). Functional relevance of serotonin 2C receptor mRNA editing in antidepressant- and anxiety-like behaviors. Neuropharmacology 59, 468–473.
Morabito, M. V., Abbas, A. I., Hood, J. L., Kesterson, R. A., Jacobs, M. M., Kump, D. S., Hachey, D. L., Roth, B. L., and Emeson, R. B. (2010). Mice with altered serotonin 2C receptor RNA editing display characteristics of Prader-Willi syndrome. Neurobiol. Dis. 39, 169–180.
Morabito, M. V., and Emeson, R. B. (2009). RNA editing as a therapeutic target for CNS disorders. Neuropsychopharmacology 34, 246.
Nelson, O., Supnet, C., Liu, H., and Bezprozvanny, I. (2010). Familial Alzheimer’s disease mutations in presenilins: effects on endoplasmic reticulum calcium homeostasis and correlation with clinical phenotypes. J. Alzheimers Dis. 21, 781–793.
Nicholas, A., De Magalhaes, J. P., Kraytsberg, Y., Richfield, E. K., Levanon, E. Y., and Khrapko, K. (2010). Age-related gene-specific changes of A-to-I mRNA editing in the human brain. Mech. Ageing Dev. 131, 445–447.
Nishikura, K. (1992). Modulation of double-stranded RNAs in vivo by RNA duplex unwindase. Ann. N. Y. Acad. Sci. 660, 240–250.
Nishimoto, Y., Yamashita, T., Hideyama, T., Tsuji, S., Suzuki, N., and Kwak, S. (2008). Determination of editors at the novel A-to-I editing positions. Neurosci. Res. 61, 201–206.
Niswender, C., Herrick-Davis, K., Dilley, G., Meltzer, H., Overholser, J., Stockmeier, C., Emeson, R., and Sanders-Bush, E. (2001). RNA editing of the human serotonin 5-HT2C receptor: alterations in suicide and implications for serotonergic pharmacotherapy. Neuropsychopharmacology 24, 478–491.
Niswender, C. M., Copeland, S. C., Herrick-Davis, K., Emeson, R. B., and Sanders-Bush, E. (1999). RNA editing of the human serotonin 5-hydroxytryptamine 2C receptor silences constitutive activity. J. Biol. Chem. 274, 9472–9478.
Niswender, C. M., Sanders-Bush, E., and Emeson, R. B. (1998). Identification and characterization of RNA editing events within the 5- HT2C receptor. Ann. N. Y. Acad. Sci. 861, 38–48.
O’Connell, M. A., Gerber, A., and Keller, W. (1997). Purification of human double-stranded RNA-specific editase 1 (hRED1) involved in editing of brain glutamate receptor B pre-mRNA. J. Biol. Chem. 272, 473–478.
Ohlson, J., Pedersen, J. S., Haussler, D., and Ohman, M. (2007). Editing modifies the GABA(A) receptor subunit alpha3. RNA 13, 698–703.
Palladino, M. J., Keegan, L. P., O’Connell, M. A., and Reenan, R. A. (2000). A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell 102, 437–449.
Pan, Y., Weng, J., Cao, Y., Bhosle, R. C., and Zhou, M. (2008). Functional coupling between the Kv1.1 channel and aldoketoreductase Kvbeta1. J. Biol. Chem. 283, 8634–8642.
Paz, N., Levanon, E., Amariglio, N., Heimberger, A., Ram, Z., Constantini, S., Barbash, Z., Adamsky, K., Safran, M., Hirschberg, A., Krupsky, M., Ben-Dov, I., Cazacu, S., Mikkelsen, T., Brodie, C., Eisenberg, E., and Rechavi, G. (2007). Altered adenosine-to-inosine RNA editing in human cancer. Genome Res. 17, 1586–1595.
Paz-Yaacov, N., Levanon, E. Y., Nevo, E., Kinar, Y., Harmelin, A., Jacob-Hirsch, J., Amariglio, N., Eisenberg, E., and Rechavi, G. (2010). Adenosine-to-inosine RNA editing shapes transcriptome diversity in primates. Proc. Natl. Acad. Sci. U.S.A. 107, 12174–12179.
Peng, P. L., Zhong, X., Tu, W., Soundarapandian, M. M., Molner, P., Zhu, D., Lau, L., Liu, S., Liu, F., and Lu, Y. (2006). ADAR2-dependent RNA editing of AMPA receptor subunit GluR2 determines vulnerability of neurons in forebrain ischemia. Neuron 49, 719–733.
Penn, A. C., and Greger, I. H. (2009). Sculpting AMPA receptor formation and function by alternative RNA processing. RNA Biol. 6, 517–521.
Petrecca, K., Miller, D. M., and Shrier, A. (2000). Localization and enhanced current density of the Kv4.2 potassium channel by interaction with the actin-binding protein filamin. J. Neurosci. 20, 8736–8744.
Pittman, A. J., Gaynes, J. A., and Chien, C. B. (2010). Nev (cyfip2) is required for retinal lamination and axon guidance in the zebrafish retinotectal system. Dev. Biol. 344, 784–794.
Polson, A., and Bass, B. (1994). Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO J. 13, 5701–5711.
Popowicz, G. M., Schleicher, M., Noegel, A. A., and Holak, T. A. (2006). Filamins: promiscuous organizers of the cytoskeleton. Trends Biochem. Sci. 31, 411–419.
Price, R. D., Weiner, D. M., Chang, M. S., and Sanders-Bush, E. (2001). RNA editing of the human serotonin 5-HT2C receptor alters receptor-mediated activation of G13 protein. J. Biol. Chem. 276, 44663–44668.
Rebagliati, M. R., and Melton, D. A. (1987). Antisense RNA injections in fertilized frog eggs reveal an RNA duplex unwinding activity. Cell 48, 599–605.
Rosenberg, B. R., Hamilton, C. E., Mwangi, M. M., Dewell, S., and Papavasiliou, F. N. (2011). Transcriptome-wide sequencing reveals numerous APOBEC1 mRNA-editing targets in transcript 3′ UTRs. Nat. Struct. Mol. Biol. 18, 230–236.
Rosenthal, J. J., and Bezanilla, F. (2002). Extensive editing of mRNAs for the squid delayed rectifier K+ channel regulates subunit tetramerization. Neuron 34, 743–757.
Rothstein, J. D. (2009). Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann. Neurol. 65(Suppl. 1), S3–S9.
Rothstein, J. D., Dykes-Hoberg, M., Pardo, C. A., Bristol, L. A., Jin, L., Kuncl, R. W., Kanai, Y., Hediger, M. A., Wang, Y., Schielke, J. P., and Welty, D. F. (1996). Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16, 675–686.
Rueter, S. M., Dawson, T. R., and Emeson, R. B. (1999). Regulation of alternative splicing by RNA editing. Nature 399, 75–80.
Rula, E. Y., Lagrange, A. H., Jacobs, M. M., Hu, N., Macdonald, R. L., and Emeson, R. B. (2008). Developmental modulation of GABA(A) receptor function by RNA editing. J. Neurosci. 28, 6196–6201.
Ryan, M. Y., Maloney, R., Reenan, R., and Horn, R. (2008). Characterization of five RNA editing sites in Shab potassium channels. Channels 2, 202–209.
Ryter, J. M., and Schultz, S. C. (1998). Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J. 17, 7505–7513.
Schenck, A., Bardoni, B., Langmann, C., Harden, N., Mandel, J. L., and Giangrande, A. (2003). CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron 38, 887–898.
Schenck, A., Bardoni, B., Moro, A., Bagni, C., and Mandel, J. L. (2001). A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc. Natl. Acad. Sci. U.S.A. 98, 8844–8849.
Schenck, A., Qurashi, A., Carrera, P., Bardoni, B., Diebold, C., Schejter, E., Mandel, J. L., and Giangrande, A. (2004). WAVE/SCAR, a multifunctional complex coordinating different aspects of neuronal connectivity. Dev. Biol. 274, 260–270.
Schoft, V. K., Schopoff, S., and Jantsch, M. F. (2007). Regulation of glutamate receptor B pre-mRNA splicing by RNA editing. Nucleic Acids Res. 35, 3723–3732.
Sheen, V. L., Feng, Y., Graham, D., Takafuta, T., Shapiro, S. S., and Walsh, C. A. (2002). Filamin A and Filamin B are co-expressed within neurons during periods of neuronal migration and can physically interact. Hum. Mol. Genet. 11, 2845–2854.
Silberberg, G., Lundin, D., Navon, R., and Ohman, M. (2012). Deregulation of the A-to-I RNA editing mechanism in psychiatric disorders. Hum. Mol. Genet. 21, 311–321.
Silberberg, G., and Ohman, M. (2011). The edited transcriptome: novel high throughput approaches to detect nucleotide deamination. Curr. Opin. Genet. Dev. 21, 401–406.
Simmons, M., Meador-Woodruff, J. H., and Sodhi, M. S. (2010). Increased cortical expression of an RNA editing enzyme occurs in major depressive suicide victims. Neuroreport 21, 993–997.
Simpson, L., Thiemann, O. H., Savill, N. J., Alfonzo, J. D., and Maslov, D. A. (2000). Evolution of RNA editing in trypanosome mitochondria. Proc. Natl. Acad. Sci. U.S.A. 97, 6986–6993.
Singh, A., Gebhart, M., Fritsch, R., Sinnegger-Brauns, M. J., Poggiani, C., Hoda, J. C., Engel, J., Romanin, C., Striessnig, J., and Koschak, A. (2008). Modulation of voltage- and Ca2+-dependent gating of CaV1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J. Biol. Chem. 283, 20733–20744.
Singh, M., Kesterson, R. A., Jacobs, M. M., Joers, J. M., Gore, J. C., and Emeson, R. B. (2007). Hyperphagia-mediated obesity in transgenic mice misexpressing the RNA-editing enzyme ADAR2. J. Biol. Chem. 282, 22448–22459.
Singh, M., Singh, M. M., Na, E., Agassandian, K., Zimmerman, M. B., and Johnson, A. K. (2011). Altered ADAR 2 equilibrium and 5HT(2C) R editing in the prefrontal cortex of ADAR 2 transgenic mice. Genes Brain Behav. 10, 637–647.
Singh, M., Zimmerman, M. B., Beltz, T. G., and Johnson, A. K. (2009). Affect-related behaviors in mice misexpressing the RNA editing enzyme ADAR2. Physiol. Behav. 97, 446–454.
Sodhi, M. S., Burnet, P. W., Makoff, A. J., Kerwin, R. W., and Harrison, P. J. (2001). RNA editing of the 5-HT(2C) receptor is reduced in schizophrenia. Mol. Psychiatry 6, 373–379.
Sommer, B., Kohler, M., Sprengel, R., and Seeburg, P. H. (1991). RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67, 11–19.
Steffen, A., Rottner, K., Ehinger, J., Innocenti, M., Scita, G., Wehland, J., and Stradal, T. E. (2004). Sra-1 and Nap1 link Rac to actin assembly driving lamellipodia formation. EMBO J. 23, 749–759.
Stefl, R., Oberstrass, F. C., Hood, J. L., Jourdan, M., Zimmermann, M., Skrisovska, L., Maris, C., Peng, L., Hofr, C., Emeson, R. B., and Allain, F. H. (2010). The solution structure of the ADAR2 dsRBM-RNA complex reveals a sequence-specific readout of the minor groove. Cell 143, 225–237.
Stefl, R., Skrisovska, L., Xu, M., Emeson, R. B., and Allain, F. H. (2005). Resonance assignments of the double-stranded RNA-binding domains of adenosine deaminase acting on RNA 2 (ADAR2). J. Biomol. NMR 31, 71–72.
Stefl, R., Xu, M., Skrisovska, L., Emeson, R. B., and Allain, F. H. (2006). Structure and specific RNA binding of ADAR2 double-stranded RNA binding motifs. Structure 14, 345–355.
Stephens, O. M., Yi-Brunozzi, H. Y., and Beal, P. A. (2000). Analysis of the RNA-editing reaction of ADAR2 with structural and fluorescent analogues of the GluR-B R/G editing site. Biochemistry 39, 12243–12251.
Streit, A. K., Derst, C., Wegner, S., Heinemann, U., Zahn, R. K., and Decher, N. (2011). RNA editing of Kv1.1 channels may account for reduced ictogenic potential of 4-aminopyridine in chronic epileptic rats. Epilepsia 52, 645–648.
Stupp, R., Van Den Bent, M. J., and Hegi, M. E. (2005). Optimal role of temozolomide in the treatment of malignant gliomas. Curr. Neurol. Neurosci. Rep. 5, 198–206.
Swartz, K. J. (2004). Towards a structural view of gating in potassium channels. Nat. Rev. Neurosci. 5, 905–916.
Tadross, M. R., Dick, I. E., and Yue, D. T. (2008). Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel. Cell 133, 1228–1240.
Tian, N., Yang, Y., Sachsenmaier, N., Muggenhumer, D., Bi, J., Waldsich, C., Jantsch, M. F., and Jin, Y. (2011). A structural determinant required for RNA editing. Nucleic Acids Res. 39, 5669–5681.
Tomiyama, M., Rodriguez-Puertas, R., Cortes, R., Pazos, A., Palacios, J. M., and Mengod, G. (2002). Flip and flop splice variants of AMPA receptor subunits in the spinal cord of amyotrophic lateral sclerosis. Synapse 45, 245–249.
Tonkin, L. A., Saccomanno, L., Morse, D. P., Brodigan, T., Krause, M., and Bass, B. L. (2002). RNA editing by ADARs is important for normal behavior in Caenorhabditis elegans. EMBO J. 21, 6025–6035.
Vesely, C., Tauber, S., Sedlazeck, F. J., Von Haeseler, A., and Jantsch, M. F. (2012). Adenosine deaminases that act on RNA induce reproducible changes in abundance and sequence of embryonic miRNAs. Genome Res.doi: 10.1101/gr.133025.111
Vitali, P., Basyuk, E., Le Meur, E., Bertrand, E., Muscatelli, F., Cavaille, J., and Huttenhofer, A. (2005). ADAR2-mediated editing of RNA substrates in the nucleolus is inhibited by C/D small nucleolar RNAs. J. Cell Biol. 169, 745–753.
Vollmar, W., Gloger, J., Berger, E., Kortenbruck, G., Kohling, R., Speckmann, E. J., and Musshoff, U. (2004). RNA editing (R/G site) and flip-flop splicing of the AMPA receptor subunit GluR2 in nervous tissue of epilepsy patients. Neurobiol. Dis. 15, 371–379.
Wahlstedt, H., Daniel, C., Enstero, M., and Ohman, M. (2009). Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res. 19, 978–986.
Wang, Q., O’Brien, P. J., Chen, C. X., Cho, D. S., Murray, J. M., and Nishikura, K. (2000). Altered G protein-coupling functions of RNA editing isoform and splicing variant serotonin2C receptors. J. Neurochem. 74, 1290–1300.
Yi-Brunozzi, H. Y., Stephens, O. M., and Beal, P. A. (2001). Conformational changes that occur during an RNA-editing adenosine deamination reaction. J. Biol. Chem. 276, 37827–37833.
Zhang, W., Han, S. W., Mckeel, D. W., Goate, A., and Wu, J. Y. (1998). Interaction of presenilins with the filamin family of actin-binding proteins. J. Neurosci. 18, 914–922.
Zhou, X., Tian, F., Sandzen, J., Cao, R., Flaberg, E., Szekely, L., Cao, Y., Ohlsson, C., Bergo, M. O., Boren, J., and Akyurek, L. M. (2007). Filamin B deficiency in mice results in skeletal malformations and impaired microvascular development. Proc. Natl. Acad. Sci. U.S.A. 104, 3919–3924.
Keywords: calcium channel, glutamate receptor, inosine, potassium channel, RNA modification, RNA editing, serotonin receptor
Citation: Tariq A and Jantsch MF (2012) Transcript diversification in the nervous system: A to I RNA editing in CNS function and disease development. Front. Neurosci. 6:99. doi: 10.3389/fnins.2012.00099
Received: 01 May 2012; Accepted: 14 June 2012;
Published online: 09 July 2012.
Edited by:
Michael A. Kiebler, Medical University of Vienna, AustriaReviewed by:
Ingo H. Greger, MRC Laboratory of Molecular Biology, UKMarie Öhman, Stockholm University, Sweden
Mary Anne O’Connell, Medical Research Council, UK
Copyright: © 2012 Tariq and Jantsch. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Michael F. Jantsch, Max F. Perutz Laboratories, Department of Chromosome Biology, University of Vienna, Dr. Bohr Gasse 1, A-1030 Vienna, Austria. e-mail:bWljaGFlbC5qYW50c2NoQHVuaXZpZS5hYy5hdA==