

- FAS Center for Systems Biology, Harvard University, Cambridge, MA, USA
Alternative pre-mRNA splicing has the potential to greatly diversify the repertoire of transcripts in multicellular organisms. Increasing evidence suggests that this expansive layer of gene regulation plays a particularly important role in the development and function of the nervous system, one of the most complex organ systems found in nature. In this review, we highlight recent studies that continue to emphasize the influence and contribution of alternative splicing regulation to various aspects of neuronal development in addition to its role in the mature nervous system.
Introduction
The nervous system is a uniquely complex structure, composed of diverse classes of neuronal cells found in close proximity to one another. These many classes of neurons must form precise synaptic connections with other neurons that can often be separated by large distances, establishing the circuitry governing the ability to sense, interpret, and appropriately respond to stimuli. It is of great interest to understand how the developmental steps of neurogenesis, migration, pathfinding, synapse formation, and maintenance are controlled with such precision. Defects in any of these processes in humans lead to numerous cognitive and motor disabilities (Mitchell, 2011). It is also of importance to better grasp the mechanisms governing the modulation of synaptic strength and plasticity in the mature nervous system, which plays a key role in sensory adaptation, learning and memory, and other behaviors.
Diverse spatio-temporal gene regulatory mechanisms have proven vital for the control of patterning in the nervous system, including regulation of mRNA synthesis by transcription factors (West and Greenberg, 2011), the dynamic alteration of chromatin states through modifying enzymes (Ooi and Wood, 2008; Yoo et al., 2009), turnover or translational repression by microRNAs (Meza-Sosa et al., 2012), RNA decay pathways, and regulation by post-translational modifications (Fukushima et al., 2009). Alternative splicing, the process in which multiple mRNA isoforms can be generated through differential splice site selection in precursor transcripts, is an additional important mechanism of gene regulation, contributing to transcriptomic and proteomic diversity in metazoans (Nilsen and Graveley, 2010). Greater than 95% of human genes undergo alternative splicing (Pan et al., 2008; Wang et al., 2008), and disruption of splicing contributes to a number of genetic diseases (Chakarova et al., 2002; Briese et al., 2005; Winkler et al., 2005; Mordes et al., 2006; Wang and Cooper, 2007). The nervous system exhibits particularly high levels of alternative splicing (Yeo et al., 2004; Grosso et al., 2008). Indeed, a recent large scale study of human tissues found that the cerebellum exhibited the highest degree of alternative splicing among 11 tested tissue samples, containing 50% more differentially expressed alternative exons than the next highest tissue (the testes; de la Grange et al., 2010). These results suggest that regulated splicing can serve as a potential mechanism for generating the high levels of molecular and cellular diversity observed in the nervous system (Lipscombe, 2005; Li et al., 2007).
The fidelity and efficiency of splicing depends on the action of five small nuclear RNAs (snRNAs) functioning as components of ribonucleoprotein particles called snRNPs, in conjunction with up to hundreds of additional auxiliary proteins (Wahl et al., 2009). This elaborate and highly dynamic complex known as the spliceosome regulates splicing with single nucleotide precision (Will and Luhrmann, 2011). Decades of research have begun to elucidate the “splicing code,” the complete set of cis-acting RNA features (for example, sequence motifs, exon and intron length, secondary structure) and trans-acting splicing factors that dictate where and in what context differential splicing will occur in transcripts (Wang and Burge, 2008; Barash et al., 2010). The combination of detailed biochemical experiments with more recent genome-wide approaches and computational analyses have revealed diverse mechanisms by which alternative splicing can occur, and have been described in greater detail in several recent excellent reviews (Blencowe, 2006; Chen and Manley, 2009; Licatalosi and Darnell, 2010; Han et al., 2011; McManus and Graveley, 2011; Irimia and Blencowe, 2012).
In this focused review, we examine the role of alternative splicing during neuronal development and in response to neuronal activity. Although a large number of alternative isoforms derived from important neuronal genes have been reported in the literature, special emphasis is given here to recent findings illuminating the role of specific trans-acting splicing factors and select target splicing events they regulate in the biogenesis and function of neurons.
Alternative Splicing in Neuronal Development and Maintenance
Alternative splicing plays an important role in generating diversity and specificity in the developing and mature nervous system. In this section we describe several recent examples demonstrating the role of alternative splicing in neuronal differentiation, migration and pathfinding, and synapse formation and function (Figure 1).
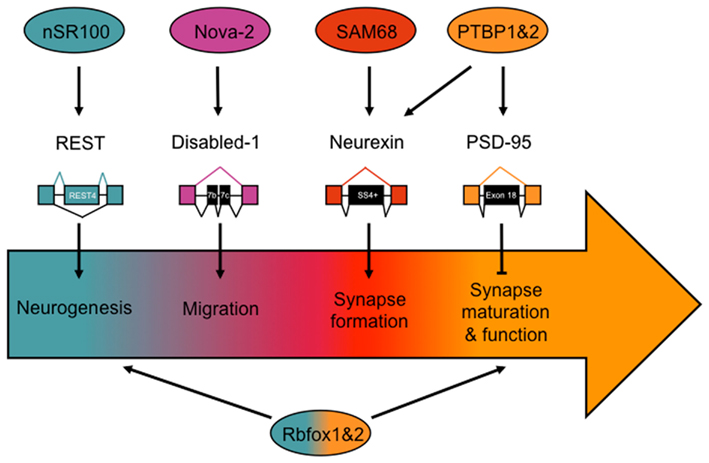
Figure 1. Notable alternative splicing events important for nervous system development, and the factors implicated in regulating their splicing patterns. Developmental stages are represented by the arrow, beginning with neurogenesis, and ending with synapse maturation and function. Splicing factors and target transcripts are correspondingly color coded to the developmental stage in which they are known to be important.
The RS Domain-Containing Splicing Factor nSR100/SRRM4 Controls Neurogenesis
The neural-specific SR-related protein of 100 kDa (nSR100/SRRM4) was identified in a computational and expression based survey of genes encoding RS domain-containing proteins (Calarco et al., 2009). In a similar manner as other members of the SR and SR-related family of proteins, nSR100 was found to regulate alternative splicing decisions. Microarray profiling experiments in mouse neuroblastoma cells and tissues revealed that depletion of nSR100 results in increased skipping of alternatively spliced exons normally included in the brain, suggesting that it mainly acts to promote the inclusion of alternative exons. A significant fraction of genes containing these regulated exons are known to be important for regulating neuronal differentiation, raising the possibility that specific splice variants modulated by nSR100 could contribute to this process. Consistent with this notion, nSR100 was found to play a critical role in neuronal differentiation and neurite extension in vitro as well as nervous system and sensory organ development in zebrafish embryos in vivo (Calarco et al., 2009). The specific mechanism by which nSR100 regulates alternative splicing in the nervous system remains to be elucidated. However, nSR100 was shown to be required for proper inclusion of a target neural-specific exon using in vitro splicing extracts, indicating that it plays a direct biochemical role in promoting exon inclusion (Calarco et al., 2009). It was also found that the introns flanking alternative exons regulated by nSR100 are enriched in pyrimidine rich motifs (Calarco et al., 2009). The majority of these motifs are likely recognized by the polypyrimidine tract binding protein PTBP1 and its tissue-specific paralog PTBP2 (also called neural- or brain-enriched n/brPTB). Consistent with a link between these regulators in modulating neural-specific alternative splicing, many nSR100-dependent alternative splicing events are also regulated by PTBP1 and PTBP2.
More recently, it was discovered that nSR100 indirectly controls the steady-state abundance of a network of transcripts in neuronal cells distinct from the population of mRNAs that it regulates at the level of splicing. Depletion of nSR100 in mouse neuroblastoma cells led to decreased levels of hundreds of transcripts, and a subset of these changes were shown to be dependent on repressor element 1 silencing transcription factor (REST, also known as NRSF), a transcriptional repressor of genes involved in neurogenesis (Raj et al., 2011). In neuronal cells, REST transcripts include an additional exon that results in the introduction of a stop codon and production of a truncated protein lacking domains required for its repressive activity. Raj et al. (2011) found that nSR100 plays a critical role in promoting the inclusion of this alternative exon, suggesting that the expression of nSR100 in neurons contributes to the reduced activity of REST upon differentiation to the neural lineage. Importantly however, REST was also found to directly repress nSR100 transcription in non-neuronal cells and thus indirectly inhibit neural-specific alternative splicing. This negative feedback loop between two gene regulatory levels was found to be important for developmental outcomes in the nervous system, as inhibiting nSR100 expression in mouse brain disrupted cortical neurogenesis, preventing neuronal precursor cells from committing to a neuronal fate (Raj et al., 2011). These results are in agreement with previous studies showing that loss of REST de-represses neuronal transcripts in non-neuronal tissues, while REST overexpression inhibits the expression of transcripts in neuronal tissues, which in one study was shown to result in axon pathfinding errors in chick embryos (Chen et al., 1998; Paquette et al., 2000).
The Splicing Factor Nova-2 Controls Neuronal Migration
The Nova RNA binding proteins were among the first tissue-specific regulators of alternative splicing to be identified. Initial studies in knockout mice indicated that Nova-1 plays a critical role in the maintenance of brainstem and spinal cord neurons, likely through the regulation of alternative splicing in these cells (Jensen et al., 2000). Subsequent studies utilizing splicing-sensitive microarray profiling in mice lacking Nova proteins identified a network of brain-specific splicing events coordinated by these factors (Ule et al., 2005). Importantly, transcripts with Nova-regulated exons encoded proteins that were significantly enriched in functions associated with the synapse (Ule et al., 2005). Integrating this alternative splicing regulatory network with genome-wide cross-linking and immunoprecipitation (CLIP; Licatalosi et al., 2008; Zhang et al., 2010) studies identifying in vivo Nova binding sites in the transcriptome has provided two key advances. First, these datasets have enabled the formulation of “RNA maps” correlating cognate Nova YCAY cis-element locations in pre-mRNA transcripts with effects on splicing regulation, leading to mechanistic insights into how the Nova proteins modulate alternative splicing (Ule et al., 2006). Second, these networks identify target isoforms and pathways that likely contribute to aspects of neuronal physiology. In agreement with this latter point, subsequent phenotypic exploration of the role of target isoforms in Nova knockout mice has identified a role for Nova in modulating slow synaptic inhibition and in neuromuscular junction formation (Huang et al., 2005; Ule et al., 2005; Ruggiu et al., 2009).
Recently, Nova-2 was demonstrated to be important for the migration of late-born cortical and Purkinje neurons in mice (Yano et al., 2010). These migration defects were due largely to the aberrant splicing of transcripts from a single gene, the Reelin signaling adaptor Disabled-1. Reelin signaling is an important pathway regulating neuronal migration in the cortex, cerebellum, and hippocampus. Binding of the Reelin ligand to its receptors ApoER2 and Vldlr leads to phosphorylation of Disabled-1, which recruits various adaptor proteins that mediate cytoskeletal rearrangements and appropriate neuronal migration and positioning (Bar et al., 2000; Ayala et al., 2007). Disabled-1 is thus a critical effector protein in the Reelin signaling pathway.
Nova-2 suppresses the inclusion of Disabled-1 exons 7b and 7c (7bc+), which encode an additional 33 amino acid peptide of unknown function, the inclusion of which could produce a protein isoform with dominant negative activity. Expression of Disabled-1 (7bc+) in the E14.5 mouse cortex was sufficient to cause migration defects similar to the Nova-2 knockout, while Disabled-1 (7bc−) substantially rescued the Nova-2 knockout migration defects (Yano et al., 2010). Thus, Nova-2 controls the sensitivity of neurons to the Reelin signaling pathway, presumably by affecting the balance of Disabled-1 (7bc−), which is activated by the Reelin signaling pathway, and Disabled-1 (7bc+), which, through a currently unknown mechanism, impairs the Reelin signaling pathway. The alternative splicing of Disabled-1 is developmentally regulated, with Nova2-dependent suppression of Disabled-1 (7bc+) highest during the critical window of migration for late-born neurons (E14.5–E16.5), suggesting Nova2-mediated alternative splicing of Disabled-1 as a mechanism to control neuronal sensitivity to Reelin signaling throughout development (Yano et al., 2010).
PTBP1 and PTBP2 Regulate Synapse Formation and Maintenance
PTBP1 and PTBP2 display mutually exclusive patterns of expression in the developing brain, with PTBP1 found in glial and non-neuronal cells, and PTBP2 in neurons (Boutz et al., 2007). This non-overlapping pattern of expression is established by an elegant cross-regulatory network where PTBP1 normally suppresses the inclusion of an exon in PTBP2 transcripts, leading to a non-functional isoform degraded by the nonsense-mediated mRNA decay (NMD) pathway (Boutz et al., 2007; Spellman et al., 2007). In neurons however, PTBP1 is silenced by miR-124, a neuron-specific microRNA, leading to the de-repression of PTBP2 (Makeyev et al., 2007). The consequences of modulating the relative levels of PTBP1 and PTBP2 in neuronal cells have been initially revealed through splicing-sensitive microarray profiling of mouse neuroblastoma cells depleted of these factors (Boutz et al., 2007). Analogous to the Nova-regulated alternative splicing network, PTBP1- and PTBP2-dependent alternative splicing events are frequently found in transcripts expressed from genes with known roles in neuronal differentiation and physiology.
A role for PTBP1 and PTBP2 in regulating the expression of PSD-95, an important scaffolding protein essential for synaptic maturation and plasticity of excitatory neurons, has recently been identified. Overexpression of PTBP1 and PTBP2 in cultured hippocampal neurons was shown to repress synaptic activity, dendritic spine formation, and reduce levels of PSD-95 transcripts (Zheng et al., 2012). This reduced mRNA abundance is caused by PTBP1 and PTBP2 binding to a pyrimidine rich cis-element upstream of PSD-95 exon 18, leading to increased exon skipping and the production of a transcript containing a premature termination codon that is targeted for degradation by the NMD pathway. Importantly, the increased expression of PSD-95 in developing neurons in the cortex was found to correlate with three distinct phases of PTBP1 and PTBP2 expression. At the neural progenitor stage, when PTBP1 levels are high, PSD-95 expression is at its lowest. In embryonic neurons, the weaker repressor PTBP2 is more highly expressed while PTBP1 expression is lost, leading to intermediate levels of PSD-95. Finally, in post-natal cortical neurons, PTBP2 is no longer expressed, allowing PSD-95 abundance to reach its highest levels (Zheng et al., 2012). These results indicate that the sequential changes in relative expression of PTBP1 and PTBP2 can allow for distinct splicing regulatory programs to be established at different stages in neuronal maturation.
Alternative Splicing of Neurexins and Neuroligins in Synapse Formation and Maintenance
A number of studies in recent years have demonstrated the importance of alternative splicing of neurexins and their binding partners neuroligins in establishing and/or maintaining synapses (Boucard et al., 2005; Chih et al., 2006; Graf et al., 2006). Neurexins and neuroligins function as adhesion proteins across the synaptic cleft, and increasing evidence suggest that these factors are central organizing proteins at both glutamatergic and GABAergic synapses in the brain (Craig and Kang, 2007). The Neurexin gene loci are highly complex, with the capacity of generating thousands of potential transcript variants pre-synaptically in mammals through the use of alternative promoters and alternative splicing (Boucard et al., 2005; Chih et al., 2006). The post-synaptic neuroligins also undergo alternative splicing, but to a lesser degree. Several key variants from each of these factors have been functionally characterized in cell culture, leading to the proposal of a trans-synaptic adhesive splicing “code” in which particular neurexin isoforms have specific affinity to particular neuroligin isoforms, and the isoforms utilized in neurons affect the functional properties of the synapse (Boucard et al., 2005; Chih et al., 2006; Graf et al., 2006). For instance, the addition of an alternative exon (B+) to neuroligin 1 decreased its ability to recruit GABAergic synaptic components but increased its glutamatergic synaptic recruitment. This change in activity was due to the reduced binding of neuroligin 1 (B+) isoforms to neurexin variants with splice site #4 selected (SS4+; Chih et al., 2006). Neuroligin (B+) bound neurexin (SS4−) strongly but exhibited only weak binding with neurexin (SS4+), while neuroligin (B−) had strong interaction with both neurexin (SS4+) and (SS4−) isoforms. These results point toward a role of neurexin and neuroligin alternative splicing in shaping the strength and class of synapses (Chih et al., 2006).
Several factors involved in the splicing of neurexin transcripts have been identified. The first was PTBP2, which was demonstrated to suppress selection of SS4 in neurexin-2α (Resnick et al., 2008). More recently it has been demonstrated that the KH domain RNA binding protein SAM68 regulates selection of the SS4 in neurexin 1 and neurexin 3, and that it does so in a neuronal activity-dependent fashion (further discussed below; Iijima et al., 2011).
Rbfox-1/A2BP1 and Rbfox-2/Rbm9 Play a Role in Neuronal Development and Function
Members of the Rbfox family of RNA binding proteins display enriched or highly specific expression patterns in the neuromuscular system, and regulate alternative splicing decisions through interactions with the highly conserved cis-element (U)GCAUG (Underwood et al., 2005; Zhang et al., 2008; Zhou and Lou, 2008; Sun et al., 2012). Focused biochemical studies and several genome-wide analyses have demonstrated that the Rbfox proteins can function as activators or repressors of splicing, depending on the location of (U)GCAUG elements in target pre-mRNA transcripts (Jin et al., 2003; Zhang et al., 2008; Zhou and Lou, 2008; Sun et al., 2012). Together, these studies have begun to shed light on the relevant networks of transcripts modulated by these factors, although the role of the Rbfox proteins in nervous system development and function in vivo has remained somewhat unclear.
Two recent studies from the Black laboratory using Rbfox knockout mice have provided further insight toward the functional importance of these proteins in the nervous system (Gehman et al., 2011, 2012). Deletion of Rbfox1 specifically in the nervous system of transgenic mice did not seem to have any effects on neuronal development or morphology in the brain. However, loss of Rbfox1 did lead to spontaneous seizures, increased sensitivity to induced seizures, and increased excitability in neurons of the dentate gyrus. Integration of splicing-sensitive microarray profiling and CLIP-Seq datasets identified alternative splicing events differentially regulated in the brains of Rbfox1−/− mice, several of which were linked to genes known to be associated with epilepsy and others with roles in synaptic function (Gehman et al., 2011).
In contrast to loss of Rbfox1 in the nervous system, deletion of the gene encoding Rbfox2 in the nervous system led to pronounced defects in cerebellar development. Rbfox2−/− animals have much smaller cerebella than wild-type littermates, defects in Purkinje cell migration and dendritic arborization, and reduction in the migration and number of granule cells (Gehman et al., 2012). Again, splicing-sensitive microarray profiling experiments were performed, revealing alternative splicing events displaying significant changes upon loss of Rbfox2. Genes with affected exons were associated with neuronal development and function, and a subset of Rbfox2-dependent alternative splicing events were also regulated by Rbfox1, suggesting partial redundancy between the two factors. In agreement with these data, double knockout mice displayed far more severe phenotypes than those observed in either single knockout mutant (Gehman et al., 2012). Finally, in an attempt to separate a possible role for both Rbfox proteins in the mature nervous system from their collective role in development, transgenic animals were generated that deleted these two factors specifically in Purkinje cells. Intriguingly, these double knockout mice possess no gross morphological or developmental abnormalities, but display impaired motor skills and significant reductions in spontaneous firing frequency of Purkinje cells, demonstrating that the Rbfox proteins also play an important role in mature neural circuitry in addition to their contribution to development (Gehman et al., 2012).
Neuronal Activity-Dependent Alternative Splicing Regulation
Depolarization Causes Changes in Splicing of Neuronal Transcripts
Calcium signaling has long been recognized to play an important role in various cellular processes such as muscle contraction and gene transcription, and in neurons it is critical for modulating neuronal activity and for learning and memory (West et al., 2001). Increasing evidence suggests that depolarization-induced calcium influx can also regulate alternative splicing in neurons. One of the early studies showing splicing differences in response to neuronal depolarization utilized the cholinergic agonist pilocarpine administered to the brains of rats. Chronic induction of depolarization with pilocarpine caused altered alternative splicing patterns in a number of key neuronal transcripts in the rat hippocampus and cortex, including tra2-beta, clathrin light chain B, NMDAR1, and c-src (Daoud et al., 1999).
A number of further studies using chemical treatments to induce or inhibit calcium signaling in neurons have revealed additional calcium-dependent alternative splicing events. For example, Ania-6, an RNA polymerase II-associated cyclin, exhibited increased inclusion of intron 6 upon glutamate stimulation, but decreased inclusion when stimulated by depolarizing concentrations of KCl. Increased intron inclusion leads to altered protein localization such that the longer isoform is found in nuclear speckles and is associated with hyperphosphorylated RNA Pol II (Berke et al., 2001; Sgambato et al., 2003). In a separate study, mature transcripts encoding SNAP25, a membrane-bound component of the SNARE complex essential for synaptic vesicle fusion, were found to include one of two mutually exclusive alternative exons (5a or 5b). Chronic depolarization of PC12 cells or of cerebellar granule cells by exposure to elevated extracellular K+ resulted in altered splicing in which the abundance of the 5b isoform is increased (Hepp et al., 2001).
Modulation of Neuronal Activity during Circadian Rhythms
Another potentially interesting physiological process that influences neuronal activity over sufficiently long timescales to involve new gene synthesis and RNA processing lies in the regulation of circadian rhythms. The connection between regulated splicing and circadian rhythms in the nervous system has been supported by a recent study using RNA-Seq in the Drosophila brain, which identified numerous splicing events regulated in response to circadian time or period, including splicing of key circadian genes (Hughes et al., 2012). Furthermore, Sanchez et al. (2010) identified the arginine methyl transferase PRMT5, which methylates arginine residues in the spliceosomal Sm proteins, in a screen for novel genes affecting circadian clock regulation in Arabidopsis. Mutations in PRMT5 were found to affect both transcription and alternative splicing of many transcripts, including several components of the circadian clock (Sanchez et al., 2010). The authors further demonstrated that a mutation in the Drosophila prmt5 ortholog causes aberrant circadian-dependent behavior as well as altered mRNA splicing patterns. In both organisms, loss of PRMT5 led predominantly to increased intron retention. Taken together, these findings suggest that PRMT5 directly methylates splicing factors, though an alternative model in which PRMT5 leads to epigenetic changes cannot be ruled out (Sanchez et al., 2010). Continued exploration of the mechanisms controlling circadian regulation of alternative splicing will undoubtedly reveal novel insights.
Mechanisms of Activity-Dependent Alternative Splicing
Although key neuronal transcripts undergoing depolarization-dependent alternative splicing have been discovered, our understanding of the mechanisms controlling this phenomenon is still in its infancy. In the sections that follow, we will highlight current progress in elucidating the role of cis-elements, chromatin states, and RNA binding protein modification as regulators of activity-dependent splicing (see Figure 2 for an outline of examples).
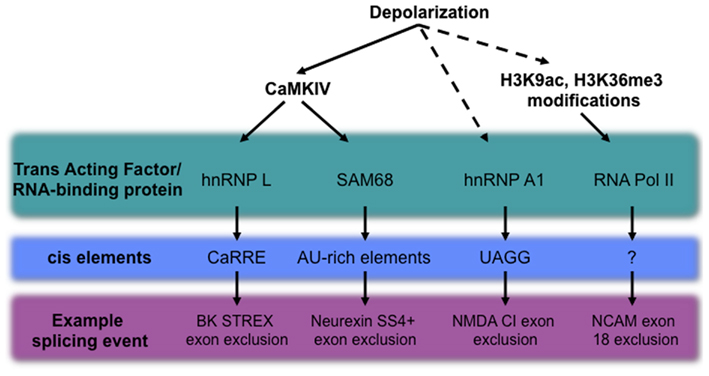
Figure 2. Mechanisms of depolarization-dependent alternative splicing, including known trans-acting factors, the cis-elements with which they interact, and a representative alternative splicing event regulated by each specific pathway. Established mechanisms are shown as solid arrows, while activities with unknown intermediates are shown with dashed lines.
mRNA Elements Necessary for Activity-Dependent Splicing
Several cis-elements regulating alternative splicing changes in response to neuronal depolarization have been identified. The BK (Big Potassium) channel encoded by the Slo gene in mammals is important for determining calcium and voltage sensitivity in neurons. The STREX exon of Slo, which contributes to enhanced neuronal sensitivity to Calcium when included in transcripts (Saito et al., 1997; Xie and Black, 2001), undergoes increased exon exclusion in response to KCl-mediated depolarization in cultured cells (Xie and Black, 2001). This depolarization-dependent alternative splicing required the Ca2+/calmodulin-dependent protein kinase CaMKIV. The cis-elements that conferred responsiveness to CAMKIV signaling were identified, and came to be known as CaRREs (CaMKIV-responsive RNA elements; Xie and Black, 2001). Since then, a number of additional depolarization-dependent alternatively spliced transcripts have been shown to contain CaRREs and to be responsive to CaMKIV (Xie et al., 2005; Lee et al., 2007). Candidate RNA binding proteins that act downstream of CaMKIV signaling and bind to the CaRREs had remained elusive until recent years, when the heterogeneous ribonucleoprotein hnRNP L was identified to interact with CaRRE1 at the upstream 3′ splice site of the STREX exon (Yu et al., 2009). Knockdown of hnRNP L led to increased inclusion of the STREX exon prior to depolarization. However, upon treatment with KCl, cells with reduced hnRNP L levels had smaller STREX exon inclusion changes relative to control cells, but still showed some response to depolarization (Yu et al., 2009). These results suggest that other factors in addition to hnRNP L play a role in the observed depolarization-mediated effects. Indeed, a more recent study has now demonstrated that hnRNP L-like (hnRNP LL) acts redundantly with hnRNP L for the complete modulation of the STREX exon in response to depolarization (Liu et al., 2012).
An additional cis-element involved in activity-dependent splicing was uncovered in experiments demonstrating that K+-induced alternative splicing of the CI cassette exon of the NMDA R1 receptor relied on the presence of two exonic UAGG silencing motifs. These motifs were previously identified as part of a multicomponent regulatory code involving 5′ splice site proximal GGGG elements in coordinating tissue-specific splicing regulation of the CI exon (Han et al., 2005). Introduction of the UAGG silencing motif into a constitutively spliced exon in an unrelated gene led to an increase in exon skipping, and importantly, further increased skipping in response to extracellular K+-induced depolarization (An and Grabowski, 2007). Biochemical studies demonstrated that the association of the heterogeneous nuclear ribonucleoprotein hnRNP A1 with these UAGG motifs was increased in response to cellular excitation (An and Grabowski, 2007). Although it is currently unclear how this increased association is induced, these results suggest that a signaling cascade must exist that connects responses to depolarization at the plasma membrane and in the cytoplasm with subsequent effects on activity in the nucleus. Interestingly, hnRNP A1 has been demonstrated to alter its shuttling state between the nucleus and cytoplasm in response to osmotic shock (Allemand et al., 2005). It will be interesting to determine whether depolarization leads to increased shuttling of hnRNP A1 to the nucleus, which would offer a possible explanation for the elevated association with UAGG elements observed.
Modification of Chromatin
Nucleosome positioning and chromatin modification have been recognized as important factors in memory formation and consolidation (Guan et al., 2002; Levenson et al., 2004). More recently, chromatin state and epigenetic marks, including post-translational histone-tail modifications and DNA methylation, have been found to play an important role in alternative splicing regulation (Hnilicova and Stanek, 2011; Luco and Misteli, 2011). Genome-wide analyses have indicated that nucleosomes have increased occupancy in exons compared to flanking intronic regions, and that local enrichment of certain histone modifications can facilitate alternative exon inclusion (see below; Nahkuri et al., 2009; Schwartz et al., 2009; Tilgner et al., 2009; Huff et al., 2010). Conversely, splicing has also been found to be important for the establishment of histone H3 lysine 36 methylation in intron-containing genes, suggesting a bi-directional communication (de Almeida et al., 2011; Kim et al., 2011). Additionally, and not mutually exclusive with the influence of chromatin state, the rate of RNA polymerase II (pol II) elongation during nascent transcript synthesis has also been found to affect alternative splicing (de la Mata et al., 2003). Two models have been proposed to possibly account for how pol II elongation can regulate alternative splicing. First, the recruitment model suggests that splicing factors can directly associate with pol II, likely via the C-terminal domain (CTD) of its largest subunit. These pol II-recruited splicing factors would then be available to recognize cognate cis-elements found in nascent pre-mRNA transcripts. Second, the kinetic model posits that chromatin structure influences the local rate of pol II transcription elongation, potentially exposing normally weak splice sites for extended periods of time, thereby allowing them to be more efficiently recognized by the spliceosome (Kornblihtt, 2007; Munoz et al., 2010).
Several recent studies have reinforced the hypothesis that local enrichment of distinct histone marks and DNA methylation status at alternative exons and flanking sequences can modulate pol II elongation rate and alternative splicing. First, Luco et al. (2010) have demonstrated using chromatin-immunoprecipitation assays that H3-K36me3 marks are enriched around a set of alternative splicing events regulated by PTBP1. The histone-tail binding protein MRG15, which specifically recognizes H3-K36me3, was also found to be in a physical complex with PTBP1, thus providing a link between histone modifications and the potential recruitment of splicing factors (Luco et al., 2010). In a separate study, Shukla et al. (2011) discovered that the DNA-binding protein CCCTC-binding factor (CTCF) can mediate local pausing of pol II and inclusion of weak alternative exons. These authors further revealed that the action of CTCF is inhibited by DNA methylation at these regulated exons, suggesting a mechanistic link between elongation rate, methylation, and CTCF binding in modulating alternative splicing. Finally, Close et al. (2012) have identified a novel polymerase-associated complex called DBIRD that was found to promote exon skipping. Depletion of components of the DBIRD complex was found to predominantly increase pol II occupancy surrounding regulated exons, leading to more inclusion. In a search for cis-elements associated with these DBIRD-sensitive exons, the authors identified enrichment of (A + T) rich sequences, which have been shown in previous studies to act as pol II elongation pause sites in vitro (Close et al., 2012).
An intriguing study has also implicated neuronal depolarization in the control of alternative splicing by affecting RNA Pol II transcription kinetics. Exon 18 of Neural Cell Adhesion Molecule (NCAM) transcripts undergoes developmentally regulated alternative splicing in which the exon-excluded isoform (NCAM140) is abundant in neuronal precursors while the exon-included isoform (NCAM180) is increasingly expressed throughout the process of neuronal differentiation (Pollerberg et al., 1985, 1986; Cunningham et al., 1987). Schor et al. (2009) demonstrated that NCAM exon 18 skipping increased in response to neuronal depolarization with KCl. This depolarization-mediated increase in exon skipping was not dependent on CaMKIV (Schor et al., 2009), but instead involved histone modification changes specifically in the vicinity of exon 18. Depolarization led to an increase in histone H3-K9 acetylation and H3-K36 tri-methylation exclusively in the region between exons 17 and 19, as well as a local increase in chromatin relaxation and accessibility. Furthermore, exon 18 inclusion could be artificially recapitulated by either using a mutant “slow” pol II or by applying the drug trichostatin which inhibits histone deacetylation. These results are consistent with a kinetic coupling model in which depolarization leads to specific local histone modifications in the region of the alternative exon causing local chromatin relaxation, in turn increasing the speed of pol II transit through the exon and facilitating increased exon 18 exclusion (Schor et al., 2009).
Regulation of RNA Binding Splicing Factors
The activity or levels of RNA binding proteins that regulate alternative splicing can be modified in response to neuronal activity. For instance, phosphorylation of hnRNP L on serine 513 by CaMKIV was found to play a crucial role in the differential regulation of STREX exon inclusion upon treatment of cells with KCl (Liu et al., 2012), providing a molecular link between signaling downstream of the stimulus and subsequent effects on alternative splicing. Two recent studies have implicated the RNA binding protein Rbfox-1/A2BP1 as an important splicing factor mediating activity-dependent alternative splicing (Lee et al., 2009; Amir-Zilberstein et al., 2012). First, transcripts encoding Rbfox-1 itself were identified as a target of depolarization-dependent splicing in mouse cells, where exon 19 was found to be excluded in response to depolarization. Exclusion of exon 19 led to the accumulation of a Rbfox-1 protein isoform targeted to the nucleus, where it re-activated the inclusion of target exons initially displaying more skipping upon depolarization (Lee et al., 2009). Thus, Rbfox-1 lacking exon 19 counteracted the effects of depolarization-dependent exon exclusion, suggesting a novel feedback-based mechanism for adapting to chronic neuronal depolarization. A second study implicated Rbfox-1 as an important downstream target of the hypothalamic transcription factor Orthopedia (Otp) in response to stress (Amir-Zilberstein et al., 2012). Rbfox-1 was demonstrated to be a transcriptional target of Otp, and Rbfox-1 transcript levels were upregulated in response to stress in mice. Rbfox-1 in turn was responsible for an increase in exon 14 inclusion in the pituitary adenylate cyclase-activating peptide (PACAP) receptor PAC1. Inclusion of PAC1 exon 14 led to a decrease in the levels of stress-induced corticotropin-releasing hormone (CRH). These results suggest that Rbfox-1-mediated inclusion of PAC1 exon 14 creates an isoform that helps terminate stress-induced transcription of CRH. In agreement with this model, zebrafish injected with morpholinos inhibiting PAC1 exon 14 inclusion exhibited abnormal “anxiety like” behavior and sustained expression of CRH transcription (Amir-Zilberstein et al., 2012).
Another recent study has provided insight into the mechanism controlling depolarization-dependent splicing of the neurexin SS4+ alternative isoform. As discussed above, alternative splicing of SS4 can modulate the affinity of neurexins for their post-synaptic ligands. Iijima et al. (2011) have now shown that SS4 selection can be suppressed in response to induced depolarization by various methods in cultured mouse neurons. This suppression of exon inclusion is dependent on CaMKIV and results in altered trans-synaptic signaling in response to depolarization. The STAR family RNA binding protein SAM68 was shown to be required for depolarization-dependent splicing of SS4, and to bind directly to AU-rich response elements in the neurexin pre-mRNA. Although SAM68 protein levels and localization were not affected by depolarization, a serine residue within a consensus CaMKIV recognition motif was found to be more highly phosphorylated following depolarization. These results suggest a model where neuronal depolarization affects CaMKIV due to increased intracellular calcium, leading to the phosphorylation of SAM68, which then alters neurexin splicing. Interestingly, the neurexin pre-mRNA does not contain recognizable CaRRE sequences, and a number of transcripts containing CaRRE sequences were not affected by loss of SAM68 (Iijima et al., 2011). Thus, it appears that CamKIV-dependent alternative splicing regulation depends on multiple downstream RNA binding proteins binding to distinct cis-elements.
Perspectives and Future Directions
Neuronal depolarization can affect the splicing of many transcripts in the nervous system, but the mechanisms by which it does so still remain largely unknown. A major future challenge will be to identify the signaling cascades in addition to the CAMKIV pathway linking cellular excitation to alternative splicing via changes in the activity of splicing factors, chromatin state, and perhaps additional mechanisms. Moreover, experiments thus far revealing a role of depolarization in nervous system alternative splicing have relied on in vitro cell culture models requiring chronic depolarization for many hours to modulate the firing activity of neurons. It will now be important to understand how chronic neuronal depolarization affects alternative splicing regulation in vivo, and what consequences the affected isoforms have on neuronal physiology. Many depolarization-responsive alternative splicing events that have been identified are found in transcripts encoding channel proteins, neurotransmitter receptors, and other modulators of synaptic strength (Xie and Black, 2001; Lee et al., 2007; Iijima et al., 2011). Phenomena such as synaptic gain control and homeostasis, where synapses can alter their sensitivity in response to chronic hyper- or hypo-stimulation (Burrone and Murthy, 2003), are thought to occur over the course of hours. This time-frame overlaps well with the temporal dynamics of depolarization-induced alternative splicing changes observed in vitro. While it may be a technically challenging feat, genome-wide analyses of splicing changes in organisms maintained under differing stimuli or behavioral paradigms inducing such synaptic gain control or homeostatic maintenance would provide further insight into the mechanisms and relevance of neuronal activity in regulating alternative splicing in vivo.
The fact that the activity of master regulators of gene expression such as transcription factors can be modified by alternative splicing has blurred the lines of how differentiation programs in cell lineages are established (Gabut et al., 2011; Raj et al., 2011). In a broader sense, these results raise an interesting implication, namely, that regulation of alternative splicing events by RNA binding proteins can play a causal rather than simply consequential role in developmental transitions and activity states of neurons. Future experiments establishing the contribution of splicing and other RNA binding regulators to the identity and fate of neuronal lineages represents an important goal in basic research but also in biomedical applications such as regenerative medicine. While a handful of neuronal-specific splicing regulators have been discovered, it is unlikely that the current repertoire of known factors is sufficient to account for the remarkable degree of splicing complexity observed in the nervous system. Hundreds of RNA binding proteins have been identified in metazoan genomes, and many of them remain uncharacterized. As such, identification and characterization of novel regulators of splicing will be important. Large scale RNAi and/or cDNA overexpression studies in cell culture models will be useful for identifying factors whose inhibition or overexpression affect splicing in the nervous system. Importantly, invertebrate model organisms provide a valuable platform for performing forward and reverse genetic screens to identify previously uncharacterized factors affecting splicing. In addition to identifying RNA binding proteins, such unbiased screens may also uncover novel classes of genes not previously known to regulate alternative splicing, such as non-coding RNAs and chromatin regulators (Luco et al., 2010; Tripathi et al., 2010). Furthermore, it will be important to understand splicing regulation in the nervous system in a more spatially resolved manner, for example, by identifying brain sub-region and neuronal-subtype specific alternative splicing events, the factors that control these events, and the effects they have on specification and function of individual classes of neurons.
Although progress has been made toward understanding splicing changes in the development of the nervous system, much less is known about the interplay between alternative splicing regulation and aging in the brain. It has been known for some time that aberrant splicing of the LMNA gene leads to an accelerated aging phenotype found in individuals with Hutchinson-Gilford Progeria Syndrome (HGPS; Todorova et al., 2003). Recent studies using patient cell lines and HGPS mouse models have identified candidate regulators involved in the cryptic splicing of LMNA (Lopez-Mejia et al., 2011). It is tempting to speculate that analysis of the aging brain will also implicate splicing factors in both aging-related splicing changes and the gradual deterioration of the nervous system. Indeed, a recent study has identified dynamic alterations in splicing during normal brain aging consistent with an increase in PTB-dependent splicing, as well as splicing changes in diseased brain consistent with decreased NOVA-dependent splicing (Tollervey et al., 2011). In the future, it will be important to more directly understand the mechanisms and the consequences of splicing in the aging brain.
Finally, mutations in several RNA binding proteins expressed in the nervous system have been associated with neurodevelopmental disorders (Grabowski and Black, 2001; Wang and Cooper, 2007; Morikawa and Manabe, 2010). Advances in genome-wide approaches to globally monitor transcripts bound by these proteins and their effects on aspects of mRNA metabolism, including alternative splicing, are beginning to shed light on underlying mechanisms of action and provide a more detailed understanding of disease etiology. Deeper investigation of the transcript networks regulated by these RNA binding proteins will hopefully provide promising new insight into the development of treatments for some of these disorders.
As demonstrated by some of the examples described above, alternative splicing has the potential to generate multiple protein isoforms, but can also modulate other properties of mRNA transcripts, including their stability. Two recent studies have suggested that tissue-specific alternative exons can frequently encode structurally disordered regions in proteins and have the potential to influence post-translational modification and protein–protein interaction interfaces (Buljan et al., 2012; Ellis et al., 2012). These observations collectively indicate that it remains an important goal to develop additional techniques and approaches that will facilitate the characterization of the functional consequences of alternative splicing events in biological pathways.
We are embarking on an exciting time where techniques for large scale analysis of nervous system transcriptome dynamics in distinct cellular subtypes, multiple developmental states, and in response to environmental cues, are constantly improving. These approaches are already revealing a previously unappreciated role for post-transcriptional gene regulatory mechanisms in almost all aspects of nervous system physiology. Continued research taking advantage of these techniques, coupled with emerging computational approaches and more traditional biochemical and molecular genetic assays, will produce a more comprehensive understanding of alternative splicing regulation. These integrative analyses should also shed further light on the interplay between alternative splicing and other layers of gene regulation in generating the constellation of neuronal subtypes and their diverse functional properties.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Ben Blencowe, Mathieu Gabut, and Arneet Saltzman for helpful feedback and comments on the manuscript. Research from our group is supported by Harvard University and the NIH (grant 1DP5OD009153). We would also like to apologize to colleagues whose work we were unable to reference due to space limitations.
References
Allemand, E., Guil, S., Myers, M., Moscat, J., Caceres, J. F., and Krainer, A. R. (2005). Regulation of heterogenous nuclear ribonucleoprotein A1 transport by phosphorylation in cells stressed by osmotic shock. Proc. Natl. Acad. Sci. U.S.A. 102, 3605–3610.
Amir-Zilberstein, L., Blechman, J., Sztainberg, Y., Norton, W. N. J., Reuveny, A., Borodovsky, N., Tahor, M., Bonkowsky, J. L., Bally-Cuif, L., Chen, A., and Levkowitz, G. (2012). Homeodomain protein otp and activity-dependent splicing modulate neuronal adaptation to stress. Neuron 73, 279–291.
An, P., and Grabowski, P. J. (2007). Exon silencing by UAGG motifs in response to neuronal excitation. PLoS Biol. 5, e36. doi:10.1371/journal.pbio.0050036
Ayala, R., Shu, T. Z., and Tsai, L. H. (2007). Trekking across the brain: the journey of neuronal migration. Cell 128, 29–43.
Bar, I., de Rouvroit, C. L., and Goffinet, A. M. (2000). The Reelin signaling pathway in mouse cortical development. Eur. J. Morphol. 38, 321–325.
Barash, Y., Calarco, J. A., Gao, W. J., Pan, Q., Wang, X. C., Shai, O., Blencowe, B. J., and Frey, B. J. (2010). Deciphering the splicing code. Nature 465, 53–59.
Berke, J. D., Sgambato, V., Zhu, P. P., Lavoie, B., Vincent, M., Krause, M., and Hyman, S. E. (2001). Dopamine and glutamate induce distinct striatal splice forms of Ania-6, an RNA polymerase II-associated cyclin. Neuron 32, 277–287.
Boucard, A. A., Chubykin, A. A., Comoletti, D., Taylor, P., and Sudhof, T. C. (2005). A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron 48, 229–236.
Boutz, P. L., Stoilov, P., Li, Q., Lin, C. H., Chawla, G., Ostrow, K., Shiue, L., Ares, M., and Black, D. L. (2007). A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 21, 1636–1652.
Briese, M., Esmaeili, B., and Sattelle, D. B. (2005). Is spinal muscular atrophy the result of defects in motor neuron processes? Bioessays 27, 946–957.
Buljan, M., Chalancon, G., Eustermann, S., Wagner, G. P., Fuxreiter, M., Bateman, A., and Babu, M. M. (2012). Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol. Cell 46, 871–883.
Burrone, J., and Murthy, V. N. (2003). Synaptic gain control and homeostasis. Curr. Opin. Neurobiol. 13, 560–567.
Calarco, J. A., Superina, S., O’Hanlon, D., Gabut, M., Raj, B., Pan, Q., Skalska, U., Clarke, L., Gelinas, D., van der Kooy, D., Zhen, M., Ciruna, B., and Blencowe, B. J. (2009). Regulation of vertebrate nervous system alternative splicing and development by an SR-related protein. Cell 138, 898–910.
Chakarova, C. F., Hims, M. M., Bolz, H., Abu-Safieh, L., Patel, R. J., Papaioannou, M. G., Inglehearn, C. F., Keen, T. J., Willis, C., Moore, A. T., Rosenberg, T., Webster, A. R., Bird, A. C., Gal, A., Hunt, D., Vithana, E. N., and Bhattacharya, S. S. (2002). Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum. Mol. Genet. 11, 87–92.
Chen, M., and Manley, J. L. (2009). Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 10, 741–754.
Chen, Z. F., Paquette, A. J., and Anderson, D. J. (1998). NRSF/REST is required in vivo for repression of multiple neuronal target genes during embryogenesis. Nat. Genet. 20, 136–142.
Chih, B., Gollan, L., and Scheiffele, P. (2006). Alternative splicing controls selective trans-synaptic interactions of the neuroligin-neurexin complex. Neuron 51, 171–178.
Close, P., East, P., Dirac-Svejstrup, A. B., Hartmann, H., Heron, M., Maslen, S., Chariot, A., Soding, J., Skehel, M., and Svejstrup, J. Q. (2012). DBIRD complex integrates alternative mRNA splicing with RNA polymerase II transcript elongation. Nature 484, 386–389.
Craig, A. M., and Kang, Y. (2007). Neurexin-neuroligin signaling in synapse development. Curr. Opin. Neurobiol. 17, 43–52.
Cunningham, B. A., Hemperly, J. J., Murray, B. A., Prediger, E. A., Brackenbury, R., and Edelman, G. M. (1987). Neural cell adhesion molecule: structure, immunoglobulin-like domains, cell surface modulation, and alternative RNA splicing. Science 236, 799–806.
Daoud, R., Da Penha Berzaghi, M., Siedler, F., Hubener, M., and Stamm, S. (1999). Activity-dependent regulation of alternative splicing patterns in the rat brain. Eur. J. Neurosci. 11, 788–802.
de Almeida, S. F., Grosso, A. R., Koch, F., Fenouil, R., Carvalho, S., Andrade, J., Levezinho, H., Gut, M., Eick, D., Gut, I., Andrau, J. C., Ferrier, P., and Carmo-Fonseca, M. (2011). Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nat. Struct. Mol. Biol. 18, 977–983.
de la Grange, P., Gratadou, L., Delord, M., Dutertre, M., and Auboeuf, D. (2010). Splicing factor and exon profiling across human tissues. Nucleic Acids Res. 38, 2825–2838.
de la Mata, M., Alonso, C. R., Kadener, S., Fededa, J. P., Blaustein, M., Pelisch, F., Cramer, P., Bentley, D., and Kornblihtt, A. R. (2003). A slow RNA polymerase II affects alternative splicing in vivo. Mol. Cell 12, 525–532.
Ellis, J. D., Barrios-Rodiles, M., Colak, R., Irimia, M., Kim, T., Calarco, J. A., Wang, X., Pan, Q., O’Hanlon, D., Kim, P. M., Wrana, J. L., and Blencowe, B. J. (2012). Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol. Cell 46, 884–892.
Fukushima, N., Furuta, D., Hidaka, Y., Moriyama, R., and Tsujiuchi, T. (2009). Post-translational modifications of tubulin in the nervous system. J. Neurochem. 109, 683–693.
Gabut, M., Samavarchi-Tehrani, P., Wang, X., Slobodeniuc, V., O’Hanlon, D., Sung, H. K., Alvarez, M., Talukder, S., Pan, Q., Mazzoni, E. O., Nedelec, S., Wichterle, H., Woltjen, K., Hughes, T. R., Zandstra, P. W., Nagy, A., Wrana, J. L., and Blencowe, B. J. (2011). An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell 147, 132–146.
Gehman, L. T., Meera, P., Stoilov, P., Shiue, L., O’Brien, J. E., Meisler, M. H., Ares, M., Otis, T. S., and Black, D. L. (2012). The splicing regulator Rbfox2 is required for both cerebellar development and mature motor function. Genes Dev. 26, 445–460.
Gehman, L. T., Stoilov, P., Maguire, J., Damianov, A., Lin, C. H., Shiue, L., Ares, M., Mody, I., and Black, D. L. (2011). The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat. Genet. 43, 706-U133.
Grabowski, P. J., and Black, D. L. (2001). Alternative RNA splicing in the nervous system. Prog. Neurobiol. 65, 289–308.
Graf, E. R., Kang, Y., Hauner, A. M., and Craig, A. M. (2006). Structure function and splice site analysis of the synaptogenic activity of the neurexin-1 beta LNS domain. J. Neurosci. 26, 4256–4265.
Grosso, A. R., Gomes, A. Q., Barbosa-Morais, N. L., Caldeira, S., Thorne, N. P., Grech, G., von Lindern, M., and Carmo-Fonseca, M. (2008). Tissue-specific splicing factor gene expression signatures. Nucleic Acids Res. 36, 4823–4832.
Guan, Z. H., Giustetto, M., Lomvardas, S., Kim, J. H., Miniaci, M. C., Schwartz, J. H., Thanos, D., and Kandel, E. R. (2002). Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell 111, 483–493.
Han, J. H., Xiong, J., Wang, D., and Fu, X. D. (2011). Pre-mRNA splicing: where and when in the nucleus. Trends Cell Biol. 21, 336–343.
Han, K., Yeo, G., An, P., Burge, C. B., and Grabowski, P. J. (2005). A combinatorial code for splicing silencing: UAGG and GGGG motifs. PLoS Biol. 3, e158. doi:10.1371/journal.pbio.0030158
Hepp, R., Dupont, J. L., Aunis, D., Langley, K., and Grant, N. J. (2001). NGF enhances depolarization effects on SNAP-25 expression: induction of SNAP-25b isoform. Neuroreport 12, 673–677.
Huang, C. S., Shi, S. H., Ule, J., Ruggiu, M., Barker, L. A., Darnell, R. B., Jan, Y. N., and Jan, L. Y. (2005). Common molecular pathways mediate long-term potentiation of synaptic excitation and slow synaptic inhibition. Cell 123, 105–118.
Huff, J. T., Plocik, A. M., Guthrie, C., and Yamamoto, K. R. (2010). Reciprocal intronic and exonic histone modification regions in humans. Nat. Struct. Mol. Biol. 17, 1495–1499.
Hughes, M. E., Grant, G. R., Paquin, C., Qian, J., and Nitabach, M. N. (2012). Deep sequencing the circadian and diurnal transcriptome of Drosophila brain. Genome Res. 22, 1266–1281.
Iijima, T., Wu, K. R., Witte, H., Hanno-Iijima, Y., Glatter, T., Richard, S., and Scheiffele, P. (2011). SAM68 regulates neuronal activity-dependent alternative splicing of neurexin-1. Cell 147, 1601–1614.
Irimia, M., and Blencowe, B. J. (2012). Alternative splicing: decoding an expansive regulatory layer. Curr. Opin. Cell Biol. 24, 323–332.
Jensen, K. B., Dredge, B. K., Stefani, G., Zhong, R., Buckanovich, R. J., Okano, H. J., Yang, Y. Y. L., and Darnell, R. B. (2000). Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron 25, 359–371.
Jin, Y., Suzuki, H., Maegawa, S., Endo, H., Sugano, S., Hashimoto, K., Yasuda, K., and Inoue, K. (2003). A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. EMBO J. 22, 905–912.
Kim, S., Kim, H., Fong, N., Erickson, B., and Bentley, D. L. (2011). Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proc. Natl. Acad. Sci. U.S.A. 108, 13564–13569.
Kornblihtt, A. R. (2007). Coupling transcription and alternative splicing. Adv. Exp. Med. Biol. 623, 175–189.
Lee, J. A., Tang, Z. Z., and Black, D. L. (2009). An inducible change in Fox-1/A2BP1 splicing modulates the alternative splicing of downstream neuronal target exons. Genes Dev. 23, 2284–2293.
Lee, J. A., Xing, Y., Nguyen, D., Xie, J. Y., Lee, C. J., and Black, D. L. (2007). Depolarization and CaM kinase IV modulate NMDA receptor splicing through two essential RNA elements. PLoS Biol. 5, 281–294. doi:10.1371/journal.pbio.0050040
Levenson, J. M., O’Riordan, K. J., Brown, K. D., Trinh, M. A., Molfese, D. L., and Sweatt, J. D. (2004). Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 279, 40545–40559.
Li, Q., Lee, J. A., and Black, D. L. (2007). Neuronal regulation of alternative pre-mRNA splicing. Nat. Rev. Neurosci. 8, 819–831.
Licatalosi, D. D., and Darnell, R. B. (2010). RNA processing and its regulation: global insights into biological networks. Nat. Rev. Genet. 11, 75–87.
Licatalosi, D. D., Mele, A., Fak, J. J., Ule, J., Kayikci, M., Chi, S. W., Clark, T. A., Schweitzer, A. C., Blume, J. E., Wang, X., Darnell, J. C., and Darnell, R. B. (2008). HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456, 464–469.
Lipscombe, D. (2005). Neuronal proteins custom designed by alternative splicing. Curr. Opin. Neurobiol. 15, 358–363.
Liu, G., Razanau, A., Hai, Y., Yu, J., Sohail, M., Lobo, V. G., Chu, J., Kung, S. K., and Xie, J. (2012). A conserved serine of heterogeneous nuclear ribonucleoprotein L (hnRNP L) mediates depolarization-regulated alternative splicing of potassium channels. J. Biol. Chem. 287, 22709–22716.
Lopez-Mejia, I. C., Vautrot, V., De Toledo, M., Behm-Ansmant, I., Bourgeois, C. F., Navarro, C. L., Osorio, F. G., Freije, J. M., Stevenin, J., De Sandre-Giovannoli, A., Lopez-Otin, C., Lévy, N., Branlant, C., and Tazi, J. (2011). A conserved splicing mechanism of the LMNA gene controls premature aging. Hum. Mol. Genet. 20, 4540–4555.
Luco, R. F., and Misteli, T. (2011). More than a splicing code: integrating the role of RNA, chromatin and non-coding RNA in alternative splicing regulation. Curr. Opin. Genet. Dev. 21, 366–372.
Luco, R. F., Pan, Q., Tominaga, K., Blencowe, B. J., Pereira-Smith, O. M., and Misteli, T. (2010). Regulation of alternative splicing by histone modifications. Science 327, 996–1000.
Makeyev, E. V., Zhang, J. W., Carrasco, M. A., and Maniatis, T. (2007). The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell 27, 435–448.
McManus, C. J., and Graveley, B. R. (2011). RNA structure and the mechanisms of alternative splicing. Curr. Opin. Genet. Dev. 21, 373–379.
Meza-Sosa, K. F., Valle-Garcia, D., Pedraza-Alva, G., and Perez-Martinez, L. (2012). Role of microRNAs in central nervous system development and pathology. J. Neurosci. Res. 90, 1–12.
Mitchell, K. J. (2011). The genetics of neurodevelopmental disease. Curr. Opin. Neurobiol. 21, 197–203.
Mordes, D., Luo, X. Y., Kar, A., Kuo, D., Xu, L. L., Fushimi, K., Yu, G. W., Sternberg, P., and Wu, J. Y. (2006). Pre-mRNA splicing and retinitis pigmentosa. Mol. Vis. 12, 1259–1271.
Morikawa, T., and Manabe, T. (2010). Aberrant regulation of alternative pre-mRNA splicing in schizophrenia. Neurochem. Int. 57, 691–704.
Munoz, M. J., de la Mata, M., and Kornblihtt, A. R. (2010). The carboxy terminal domain of RNA polymerase II and alternative splicing. Trends Biochem. Sci. 35, 497–504.
Nahkuri, S., Taft, R. J., and Mattick, J. S. (2009). Nucleosomes are preferentially positioned at exons in somatic and sperm cells. Cell Cycle 8, 3420–3424.
Nilsen, T. W., and Graveley, B. R. (2010). Expansion of the eukaryotic proteome by alternative splicing. Nature 463, 457–463.
Ooi, L., and Wood, I. C. (2008). Regulation of gene expression in the nervous system. Biochem. J. 414, 327–341.
Pan, Q., Shai, O., Lee, L. J., Frey, B. J., and Blencowe, B. J. (2008). Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 40, 1413–1415.
Paquette, A. J., Perez, S. E., and Anderson, D. J. (2000). Constitutive expression of the neuron-restrictive silencer factor (NRSF)/REST in differentiating neurons disrupts neuronal gene expression and causes axon pathfinding errors in vivo. Proc. Natl. Acad. Sci. U.S.A. 97, 12318–12323.
Pollerberg, E. G., Sadoul, R., Goridis, C., and Schachner, M. (1985). Selective expression of the 180-kD component of the neural cell adhesion molecule N-CAM during development. J. Cell Biol. 101, 1921–1929.
Pollerberg, G. E., Schachner, M., and Davoust, J. (1986). Differentiation state-dependent surface mobilities of two forms of the neural cell adhesion molecule. Nature 324, 462–465.
Raj, B., O’Hanlon, D., Vessey, J. P., Pan, Q., Ray, D., Buckley, N. J., Miller, F. D., and Blencowe, B. J. (2011). Cross-regulation between an alternative splicing activator and a transcription repressor controls neurogenesis. Mol. Cell 43, 843–850.
Resnick, M., Segall, A., Rozic-Kotliroff, G., Lupowitz, Z., and Zisapel, N. (2008). Alternative splicing of neurexins: a role for neuronal polypyrimidine tract binding protein. Neurosci. Lett. 439, 235–240.
Ruggiu, M., Herbst, R., Kim, N., Jevsek, M., Fak, J. J., Mann, M. A., Fischbach, G., Burden, S. J., and Darnell, R. B. (2009). Rescuing Z+ agrin splicing in Nova null mice restores synapse formation and unmasks a physiologic defect in motor neuron firing. Proc. Natl. Acad. Sci. U.S.A. 106, 3513–3518.
Saito, M., Nelson, C., Salkoff, L., and Lingle, C. J. (1997). A cysteine-rich domain defined by a novel exon in a slo variant in rat adrenal chromaffin cells and PC12 cells. J. Biol. Chem. 272, 11710–11717.
Sanchez, S. E., Petrillo, E., Beckwith, E. J., Zhang, X., Rugnone, M. L., Hernando, C. E., Cuevas, J. C., Herz, M. A. G., Depetris-Chauvin, A., Simpson, C. G., Brown, J. W., Cerdán, P. D., Borevitz, J. O., Mas, P., Ceriani, M. F., Kornblihtt, A. R., and Yanovsky, M. J. (2010). A methyl transferase links the circadian clock to the regulation of alternative splicing. Nature 468, 112–116.
Schor, I. E., Rascovan, N., Pelisch, F., Allo, M., and Kornblihtt, A. R. (2009). Neuronal cell depolarization induces intragenic chromatin modifications affecting NCAM alternative splicing. Proc. Natl. Acad. Sci. U.S.A. 106, 4325–4330.
Schwartz, S., Meshorer, E., and Ast, G. (2009). Chromatin organization marks exon-intron structure. Nat. Struct. Mol. Biol. 16, 990–995.
Sgambato, V., Minassian, R., Nairn, A. C., and Hyman, S. E. (2003). Regulation of ania-6 splice variants by distinct signaling pathways in striatal neurons. J. Neurochem. 86, 153–164.
Shukla, S., Kavak, E., Gregory, M., Imashimizu, M., Shutinoski, B., Kashlev, M., Oberdoerffer, P., Sandberg, R., and Oberdoerffer, S. (2011). CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479, 74-U99.
Spellman, R., Llorian, M., and Smith, C. W. J. (2007). Crossregulation and functional redundancy between the splicing regulator PTB and its paralogs nPTB and ROD1. Mol. Cell 27, 420–434.
Sun, S., Zhang, Z., Fregoso, O., and Krainer, A. R. (2012). Mechanisms of activation and repression by the alternative splicing factors RBFOX1/2. RNA 18, 274–283.
Tilgner, H., Nikolaou, C., Althammer, S., Sammeth, M., Beato, M., Valcarcel, J., and Guigo, R. (2009). Nucleosome positioning as a determinant of exon recognition. Nat. Struct. Mol. Biol. 16, 996-U124.
Todorova, A., Halliger-Keller, B., Walter, M. C., Dabauvalle, M. C., Lochmuller, H., and Muller, C. R. (2003). A synonymous codon change in the LMNA gene alters mRNA splicing and causes limb girdle muscular dystrophy type 1B. J. Med. Genet. 40, e115.
Tollervey, J. R., Wang, Z., Hortobagyi, T., Witten, J. T., Zarnack, K., Kayikci, M., Clark, T. A., Schweitzer, A. C., Rot, G., Curk, T., Zupan, B., Rogelj, B., Shaw, C. E., and Ule, J. (2011). Analysis of alternative splicing associated with aging and neurodegeneration in the human brain. Genome Res. 21, 1572–1582.
Tripathi, V., Ellis, J. D., Shen, Z., Song, D. Y., Pan, Q., Watt, A. T., Freier, S. M., Bennett, C. F., Sharma, A., Bubulya, P. A., Blencowe, B. J., Prasanth, S. G., and Prasanth, K. V. (2010). The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 39, 925–938.
Ule, J., Stefani, G., Mele, A., Ruggiu, M., Wang, X., Taneri, B., Gaasterland, T., Blencowe, B. J., and Darnell, R. B. (2006). An RNA map predicting Nova-dependent splicing regulation. Nature 444, 580–586.
Ule, J., Ule, A., Spencer, J., Williams, A., Hu, J. S., Cline, M., Wang, H., Clark, T., Fraser, C., Ruggiu, M., Zeeberg, B. R., Kane, D., Weinstein, J. N., Blume, J., and Darnell, R. B. (2005). Nova regulates brain-specific splicing to shape the synapse. Nat. Genet. 37, 844–852.
Underwood, J. G., Boutz, P. L., Dougherty, J. D., Stoilov, P., and Black, D. L. (2005). Homologues of the Caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Mol. Cell. Biol. 25, 10005–10016.
Wahl, M. C., Will, C. L., and Luhrmann, R. (2009). The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701–718.
Wang, E. T., Sandberg, R., Luo, S. J., Khrebtukova, I., Zhang, L., Mayr, C., Kingsmore, S. F., Schroth, G. P., and Burge, C. B. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476.
Wang, G. S., and Cooper, T. A. (2007). Splicing in disease: disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 8, 749–761.
Wang, Z. F., and Burge, C. B. (2008). Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA 14, 802–813.
West, A. E., Chen, W. G., Dalva, M. B., Dolmetsch, R. E., Kornhauser, J. M., Shaywitz, A. J., Takasu, M. A., Tao, X., and Greenberg, M. E. (2001). Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. U.S.A. 98, 11024–11031.
West, A. E., and Greenberg, M. E. (2011). Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb. Perspect. Biol. 3, pii: a005744.
Will, C. L., and Luhrmann, R. (2011). Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 3, pii: a003707.
Winkler, C., Eggert, C., Gradl, D., Meister, G., Giegerich, M., Wedlich, D., Laggerbauer, B., and Fischer, U. (2005). Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes Dev. 19, 2320–2330.
Xie, J. Y., and Black, D. L. (2001). A CaMK IV responsive RNA element mediates depolarization-induced alternative splicing of ion channels. Nature 410, 936–939.
Xie, J. Y., Jan, C., Stoilov, P., Park, J., and Black, D. L. (2005). A consensus CaMK IV-responsive RNA sequence mediates regulation of alternative exons in neurons. RNA 11, 1825–1834.
Yano, M., Hayakawa-Yano, Y., Mele, A., and Darnell, R. B. (2010). Nova2 regulates neuronal migration through an RNA switch in disabled-1 signaling. Neuron 66, 848–858.
Yeo, G., Holste, D., Kreiman, G., and Burge, C. B. (2004). Variation in alternative splicing across human tissues. Genome Biol. 5, R74.
Yoo, A. S., Staahl, B. T., Chen, L., and Crabtree, G. R. (2009). MicroRNA-mediated switching of chromatin-remodelling complexes in neural development. Nature 460, 642-U112.
Yu, J. K., Hai, Y., Liu, G. D., Fang, T. L., Kung, S. K. P., and Xie, J. Y. (2009). The heterogeneous nuclear ribonucleoprotein L is an essential component in the Ca2+/calmodulin-dependent protein kinase IV-regulated alternative splicing through cytidine-adenosine repeats. J. Biol. Chem. 284, 1505–1513.
Zhang, C., Frias, M. A., Mele, A., Ruggiu, M., Eom, T., Marney, C. B., Wang, H., Licatalosi, D. D., Fak, J. J., and Darnell, R. B. (2010). Integrative modeling defines the Nova splicing-regulatory network and its combinatorial controls. Science 329, 439–443.
Zhang, C., Zhang, Z., Castle, J., Sun, S., Johnson, J., Krainer, A. R., and Zhang, M. Q. (2008). Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev. 22, 2550–2563.
Zheng, S., Gray, E. E., Chawla, G., Porse, B. T., O’Dell, T. J., and Black, D. L. (2012). PSD-95 is post-transcriptionally repressed during early neural development by PTBP1 and PTBP2. Nat. Neurosci. 15, 381-U202.
Keywords: alternative splicing, genomics, nervous system, RNA processing, gene regulation
Citation: Norris AD and Calarco JA (2012) Emerging roles of alternative pre-mRNA splicing regulation in neuronal development and function. Front. Neurosci. 6:122. doi: 10.3389/fnins.2012.00122
Received: 29 May 2012; Paper pending published: 16 June 2012;
Accepted: 02 August 2012; Published online: 21 August 2012.
Edited by:
Jernej Ule, MRC Laboratory of Molecular Biology, UKCopyright: © 2012 Norris and Calarco. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: John A. Calarco, FAS Center for Systems Biology, Harvard University, 52 Oxford Street, Room 457.40, Cambridge, MA 02138, USA. e-mail:amNhbGFyY29AZmFzLmhhcnZhcmQuZWR1