 G. D. Anderson1*
G. D. Anderson1* T. C. Peterson2 F. M. Farin3 T. K. Bammler3 R. P. Beyer3 E. D. Kantor1
T. C. Peterson2 F. M. Farin3 T. K. Bammler3 R. P. Beyer3 E. D. Kantor1 M. R. Hoane2
M. R. Hoane2
- 1Department of Pharmacy, University of Washington, Seattle, WA, USA
- 2Department of Psychology, Southern Illinois University, Carbondale, IL, USA
- 3Department of Environmental and Occupational Health Sciences, University of Washington, Seattle, WA, USA
Microarray-based transcriptional profiling was used to determine the effect of nicotinamide on gene expression in an experimental traumatic brain injury (TBI) model. Ingenuity Pathway Analysis (IPA) was used to evaluate the effect on relevant functional categories and canonical pathways. At 24 h, 72 h, and 7 days, respectively, 70, 58, and 76%, of the differentially expressed genes were up-regulated in the vehicle treated compared to the sham animals. At 24 h post-TBI, there were 150 differentially expressed genes in the nicotinamide treated animals compared to vehicle; the majority (82%) down-regulated. IPA analysis identified a significant effect of nicotinamide on the functional categories of cellular movement, cell-to-cell-signaling, antigen presentation and cellular compromise, function, and maintenance and cell death. The canonical pathways identified were signaling pathways primarily involved with the inflammatory process. At 72 h post-cortical contusion injury, there were 119 differentially expressed genes in the nicotinamide treated animals compared to vehicle; the majority (90%) was up-regulated. IPA analysis identified a significant effect of nicotinamide on cell signaling pathways involving neurotransmitters, neuropeptides, growth factors, and ion channels with little to no effect on inflammatory pathways. At 7 days post-TBI, there were only five differentially expressed genes with nicotinamide treatment compared to vehicle. Overall, the effect of nicotinamide on counteracting the effect of TBI resulted in significantly decreased number of genes differentially expressed by TBI. In conclusion, the mechanism of the effect of nicotinamide on secondary injury pathways involves effects on inflammatory response, signaling pathways, and cell death.
Introduction
The Centers for Disease Control and Prevention have stated that traumatic brain injury (TBI) is among the leading causes of acute and chronic disability in the United States and each year 1.7 million Americans endure a TBI, and 52,000 die (Faul et al., 2010). Although more individuals survive TBI than in the past, the survivors endure residual physical, cognitive, emotional, and/or behavioral impairments from the cascade of central pathological responses resulting from TBI.
The field of TBI recognizes two distinct forms or stages of brain injury. The first or primary injury relates to the initial injury caused by direct damage to the brain. It is believed that only injury prevention or reduction will reduce the consequences of the primary injury. The second is indirect and progressive and is referred to as secondary injury.
The etiology of secondary brain injury is multi-factorial, with a host of likely inter-related processes including mitochondrial energy failure, excessive generation of reactive oxygen species, activation of destructive enzymes such as poly (ADP-ribose) polymerase (PARP) and caspase family of proteases, membrane disruption, neuronal death, thrombosis due to intravascular coagulation in small vessels, increased synaptic concentrations of excitatory amino acids, and activation of innate inflammatory responses (Schouten, 2007). To date, no single pharmacological agent has been shown to improve the outcome of TBI. Several reviews have suggested the need for pharmacological treatments that target multiple secondary factors or combination treatment strategies (Faden and Stoica, 2007; Schouten, 2007).
Nicotinamide has been shown to improve neurological outcome and reduce the amount of tissue damage in various animal models TBI (Hoane et al., 2003, 2006a,b,c, 2008a,b; Holland et al., 2008; Goffus et al., 2010; Vonder Haar et al., 2011) and stroke (Ayoub et al., 1999; Sakakibara et al., 2002; Yang et al., 2002a,b,c). Recovery of function of both sensory and motor tasks after TBI have been found with a variety of nicotinamide dosing regiments which include doses ranging from 50 mg/kg to 500 mg/day kg administered up to 24 h post-injury to sustained infusion studies using 150 mg/day for 7 days.
Nicotinamide is the precursor of β-nicotinamide adenine dinucleotide (NAD+) and is important for the synthesis of nicotinamide adenine dinucleotide phosphate (NADP+). NAD+ and NADP+ are coenzymes that are vital to many oxidation-reduction reactions in cell metabolism. NAD+ is a precursor for ATP, and NAD+ increases neuronal ATP concentration (Klaidman et al., 1996). After TBI, nicotinamide may prevent the depletion of NAD+, thus protecting against ATP depletion and increasing neuroprotection (Li et al., 2006). Previous experimental studies have demonstrated that nicotinamide has anti-inflammatory and anti-oxidant effects, prevents apoptosis, and decreases cerebral edema (for reviews, see Yang and Adams, 2004; Li et al., 2006). However, the predominance of the preclinical studies evaluating the proposed mechanism of actions of nicotinamide have evaluated single or multiple 500 mg/kg doses, which result in peak concentrations of ∼300 μg/mL (Hoane et al., 2006c), 10–30 times higher than clinically relevant concentrations. Nicotinamide doses of 1–3 g/day (15–45 mg/kg/day; Knip et al., 2000) have been administered in the experimental treatment or prevention of diabetes type 2 and result in peak nicotinamide serum concentrations of ∼25–70 μg/mL (Horsman et al., 1993).
The objective of this study was to use microarray analysis to determine the effect of clinically relevant doses of nicotinamide on gene expression patterns and profiles following experimental TBI in the rat. By understanding the mechanism of the effect of nicotinamide on the secondary injury response in TBI, we can develop future treatment strategies using a combination of drugs to target the multiple pathways involved in secondary injury.
Materials and Methods
Animals
Male, Sprague-Dawley rats (Harlan, Indianapolis, IN, USA), 4-months of age were used in this study. All animal and surgical procedures were adhered to as described in the NIH Guide for the Care and Use of Laboratory Animals. The Southern Illinois University Institutional Animal Care and Use Committee (IACUC) and the University of Washington’s IACUC reviewed and approved all experimental procedures. Before and after injury, animals were housed in the university-maintained vivarium, with a 12-h light/dark schedule and a controlled environmental temperature of 22°C in standard housing cages with food and water available ad libitum.
Pharmacokinetic Studies
Male Sprague-Dawley rats (309 ± 17 g) with surgically implanted jugular vein catheters were obtained from Harlan Laboratories. Four animals were anesthetized using a mixture of isoflurane (2–4%) and oxygen (0.8 L/min). Alzet osmotic mini-pumps (Alzet model 2ML1, pumping rate of 10 μL/h) were implanted subcutaneously (s.c.) ∼2.0 cm posterior to the scapulae and shifted to the right of midline. The infusion pumps were weighed prior to and after loading the nicotinamide solution. After filling, the pumps were soaked overnight in a water bath at 37°C, and taken out just prior to placing in rat to insure that the pump was operating at the desired flow rate at the time of implantation. Based on the pharmacokinetic results of a previous single dose study (data not shown), a loading dose of 75 mg/kg of nicotinamide (Sigma-Aldrich Co., St. Louis, MO, USA) dissolved in phosphate buffered saline (PBS) was administered by s.c. injection and an infusion rate of 12.0 mg/h kg were used to target a steady state concentration of ∼50 μg/mL. Blood samples were collected in microtubes immediately prior to loading dose, and at 1, 4, 24, 48, and 72 h, and stored at −80°C until assayed using a previously published assay (Goffus et al., 2010).
Experimental Injury Model
The cortical contusion injury (CCI) model utilized in the present study was based on previous studies and was intended to produce a moderately severe injury (Quigley et al., 2009; Goffus et al., 2010; Anderson et al., 2011). Animals, ∼3 months of age with a weight of 335 ± 28 g were anesthetized using a mixture of isoflurane (2–4%) and oxygen (0.8 L/min). When the animal became unresponsive (no ocular or pedal reflexes) the head was shaved and scrubbed with 70% alcohol followed by betadine and placed into a stereotaxic device. A midline incision was made in the skin as well as through the underlying fascia. A circular craniotomy (5.0 mm) was centered 2.4 mm posterior to and 2.4 mm lateral (left) to bregma. The contusion injury was created with the Benchmark™ stereotaxic impactor with a 4.0-mm diameter impactor tip (St. Louis, MO, USA)1. A moderate injury was induced with an impact speed of 3.0 m/s and an impact depth of 2.5 mm (Swan et al., 2011). The impact tip maintained contact with the brain tissue for 0.5 s before retraction. Normal body temperature (37°C) during surgery and recovery was maintained with a warm water recycling bed and pump system (EZ Anesthesia, Palmer, PA, USA). Four hours post-CCI, osmotic mini-pumps were implanted as described above. Rats receiving sham surgeries underwent identical surgical preparation as the injured animals but did not receive craniotomies or injuries, and were then sutured and allowed to recover.
Based on the pharmacokinetic studies, each animal received a 75-mg/kg s.c. loading dose of nicotinamide or vehicle (1.0 mL/kg PBS) at time of implantation of the infusion pump 4 h after the CCI injury. The nicotinamide infusion rate was 12 mg/h kg or vehicle (0.9% PBS) for 72 h post-injury as described above. Animals were randomly assigned to one of three groups: Group 1: Intact sham (n = 5), Group 2. CCI injured nicotinamide (n = 15), and Group 3. CCI injured vehicle (n = 15).
Tissue Harvest
Five intact sham animals and five animals in each treatment group at specified time point’s post-CCI (24 h, 72 h, and 7 days) were overdosed with a mixture of CO2 (80%) and O2 (20%). The rats were then decapitated; a cardiac blood sample collected and brains were rapidly extracted. For the animals in the 7-day group, the infusion pump was explanted under anesthesia at 72 h post-TBI and a tail snip blood sample was obtained. To maintain quality control and to assure that all of the brains were injured, each brain was assigned a rating score (1 = no visual sign of trauma; 2 = bruised and swollen cortex; 3 = no remaining cortex; or extensive damage). Only brains with a score of 2 were used in the subsequent analyses. An example of representative dorsal images of the injury in each group prior to the harvesting of the tissue for analysis is shown in Figure 1.
Figure 1. Histology plate. Shown are representative dorsal images of the injury in each group prior to the harvesting of the tissue for analysis. Image bar is 2.0 mm.
The brain was then cut into a 4.0-mm coronal slab containing the injury site in a brain matrix (Braintree Scientific, Inc., Braintree, MA, USA) and was placed onto an RNAase free cold plate and a 5.0-mm biopsy bunch was used to collect the injury site and surrounding cortical tissue (Hoane et al., 2006a; Anderson et al., 2011). The tissue punch included all injured cortical tissue and a small strip of pericontusional tissue, with the ventral extent of the punch extending to the corpus callosum. Tissue punches were placed into microcentrifuge tubes, snap frozen, and then stored at −80°C. All samples were shipped by overnight carrier to the University of Washington on dry ice.
Processing of Samples for Microarray Analysis
Integrity of RNA samples was assessed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA) which is the method of choice and the recognized standard in the field. RNA integrity was judged by observing distinct and sharp 18 and 28 s ribosomal RNA peaks that were baseline separated. RNA quantity was determined by measuring OD260 with a Thermo Scientific NanoDrop™ 1000 Spectrophotometer (Thermo Fisher Scientific, Inc., Wilmington, DE, USA). The NanoDrop instrument was also used to determine purity of RNA samples by measuring OD260/280 and OD260/230 ratios. Only samples passing this stringent quality control were processed further. Processing of the RNA samples was carried out according to the Affymetrix GeneChip Whole Transcript Sense Target labeling protocol. Briefly, double-stranded cDNA was synthesized with random hexamers tagged with a T7 promoter sequence. The double-stranded cDNA was subsequently used as a template and amplified by T7 RNA polymerase producing many copies of antisense cRNA. In the second cycle of cDNA synthesis, random hexamers were used to prune reverse transcription of the cRNA from the first cycle to produce single-stranded DNA in the sense orientation. In order to reproducibly fragment the single-stranded DNA and improve the robustness of the assay, dUTP was incorporated in the DNA during the second cycle, first-strand reverse transcription reaction. This single-stranded DNA sample was then treated with a combination of uracil DNA glycosylase (UDG) and apurinic/apyrimidinic endonuclease 1 (APE 1) that specifically recognizes the unnatural dUTP residues and breaks the DNA strand. DNA was labeled by terminal deoxynucleotidyltransferase (TdT) with the Affymetrix® proprietary DNA Labeling Reagent that is covalently linked to biotin. The biotin labeled DNA fragments were hybridized to Affymetrix GeneChip Rat Gene 1.0 ST arrays, washed, and stained with fluorescent anti streptavidin biotinylated antibody. Following an additional wash step, the arrays were scanned with an Affymetrix GeneChip® 3000 scanner. Image generation and feature extraction was performed using Affymetrix GeneChip Command Console Software.
Analysis of the Microarray Data
Raw microarray data was processed and analyzed with the Bioconductor aroma package (Bengtsson et al., 2008) and normalized using the RMA method from the Bioconductor Affy package. The quantile normalization step of the RMA normalization was performed at the probeset level. Using the normalized data, genes with significant evidence for differential expression were identified with the limma package (Smyth, 2004) in Bioconductor. The limma methodology calculates a p-value for each gene using a modified t-test in conjunction with an empirical Bayesian method to moderate the standard errors of the estimated log-fold changes. This method of detecting differentially expressed genes draws strength across genes for more robust and accurate detection of differentially expressed genes. Such an adjustment has repeatedly been shown to avoid an excess of false positives when identifying differentially expressed genes (Allison et al., 2006). Using the p-values from limma, we used the Bioconductor package p.adjust (Benjamini and Hochberg, 1995) to estimate the false discovery rate associated with our list of differentially expressed genes. This methodology allows us to address the multiple testing problem without resorting to an excessively conservative approach that controls the familywise error, such as a Bonferroni correction. Venn diagrams were generated with the Bioconductor limma package.
Ingenuity Pathway Analysis (IPA; Build 131235; Version 11904312; Database Status 02.20.2012)2 was used to analyze differentially expressed genes (>1.5-fold up or down-regulated, p < 0.05) using the Core Analysis feature. IPA is a commercial tool that is based on a proprietary database (see text footnote 2) to facilitate the identification of biological themes, in microarray gene expression data. IPA uses a Right-tailed Fisher’s exact test to calculate a p-value determining the probability that each biological function, canonical pathway, or transcriptional network assigned to the data set is due to chance alone.
Validation of Data Obtained with Microarrays Using Fluorogenic 5′-Nuclease-Based Assay and Quantitative RT-PCR
Quantitative TaqMan based RT-PCR (qPCR) analysis has a greater dynamic range for changes in gene expression levels compared to microarray-based analysis. Therefore, we used quantitative-PCR (qPCR) to validate expression changes of 15 genes of interest that had been identified by microarray analysis with significant changes by either TBI or nicotinamide treatment. Briefly, reverse transcription was performed according to the manufacturer’s established protocol using total RNA and the SuperScript® III First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA). For gene expression measurements, 2 μL of cDNA were included in a PCR reaction (12 μL final volume) that also consisted of the ABI inventoried TaqMan® Gene Expression Assays mix and the TaqMan Gene Expression Master Mix according to the manufacturer’s protocol (Applied Biosystems, Inc., Foster City, CA, USA). Amplification and detection of PCR amplicons were performed with the ABI PRISM 7900 system (Applied Biosystems, Inc., Foster City, CA, USA) with the following PCR reaction profile: 1 cycle of 95°C for 10 min, 40 cycles of 95°C for 30 s, and 60°C for 1 min. β-Actin amplification plots derived from serial dilutions of an established reference sample were used to create a linear regression formula in order to calculate expression levels. β-Actin gene expression levels were utilized as an internal control to normalize the data.
Results
Nicotinamide Concentrations
In the healthy animals, 75 mg/kg s.c. bolus plus an infusion rate of 12.0 mg/h kg of nicotinamide resulted in peak concentrations of 68 ± 8 μg/mL at 1 h with steady state concentrations of 41 ± 7, 36 ± 8, and 33 ± 9 μg/mL at 24, 48, and 72 h post-infusion, respectively. In the CCI injured animals, nicotinamide concentrations were 45 ± 12 and 55 ± 4 μg/mL at 24 and 72 h, respectively at time of sacrifice. The nicotinamide concentrations was 50 ± 13 μg/mL obtained by tail snip at 72 h at the time of removal of the osmotic pump in the animals sacrificed at 7 days. Nicotinamide concentrations were below the detectable limit at the time of sacrifice at 7 days post-CCI.
Gene Expression Study
The microarray data passed all the standard and advanced quality control metrics. The number of differentially expressed genes (>1.5-fold change, p < 0.05) at 24 h, 72 h, and 7 days is presented in Table 1. The vehicle to sham (no injury) comparison reflects the effect of the TBI without treatment relative to sham controls. At 24 h, 72 h, and 7 days, respectively, 70, 58, and 76%, of the differentially expressed genes were up-regulated in the vehicle treated animals compared to the sham animals (Table 1). The nicotinamide to sham comparison reflects the effect of TBI with treatment compared to sham controls. The Venn diagrams show the number of genes differentially expressed more than 1.5-fold (p < 0.05) in the nicotinamide compared to sham and vehicle compared to sham contrasts at the 24-h, 72 h, and 7 day time points (Figure 2). At 24 h, vehicle treatment resulted in increased expression of 808 genes (>1.5-fold, p < 0.05) compared to non-injured sham animals, whereas only 621 genes in the nicotinamide treated animals showed elevated expression. Similarly, at 24 h post-TBI, mRNA levels of 351 genes were decreased more than 1.5-fold (p < 0.05) compared to non-injured sham animals, whereas only 106 genes exhibited decreased expression in the nicotinamide treated animals. At 72 h, vehicle treatment resulted in increased expression of 1739 genes (>1.5-fold, p < 0.05) compared to non-injured sham animals, whereas only 1332 genes in the nicotinamide treated animals showed elevated expression. In the same way, 72 h post-TBI, mRNA levels of 1264 genes were decreased more than 1.5-fold (p < 0.05) compared to non-injured sham animals, whereas only 479 genes exhibited decreased expression in the nicotinamide treated animals. By 7 day post-TBI, vehicle treatment resulted in increased expression of 1108 genes (>1.5-fold, p < 0.05) compared to non-injured sham animals, whereas only 886 genes in the nicotinamide treated animals showed elevated expression. Likewise, mRNA levels of 351 genes were decreased more than 1.5-fold (p < 0.05) compared to non-injured sham animals, whereas only 247 genes exhibited decreased expression in the nicotinamide treated animals. Overall, the Venn diagrams demonstrate the effect of nicotinamide on counteracting the effect of TBI which results in significantly decreased number of genes differentially expressed by TBI.
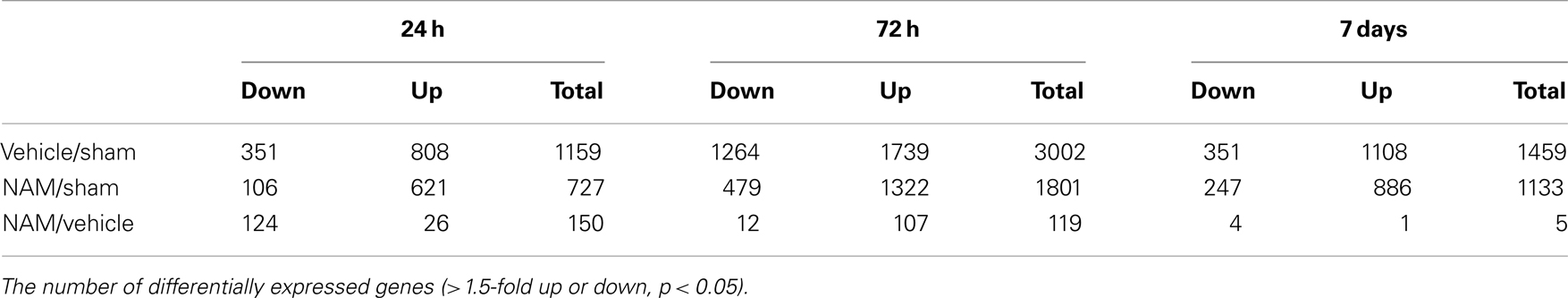
Table 1. Brain gene expression data.
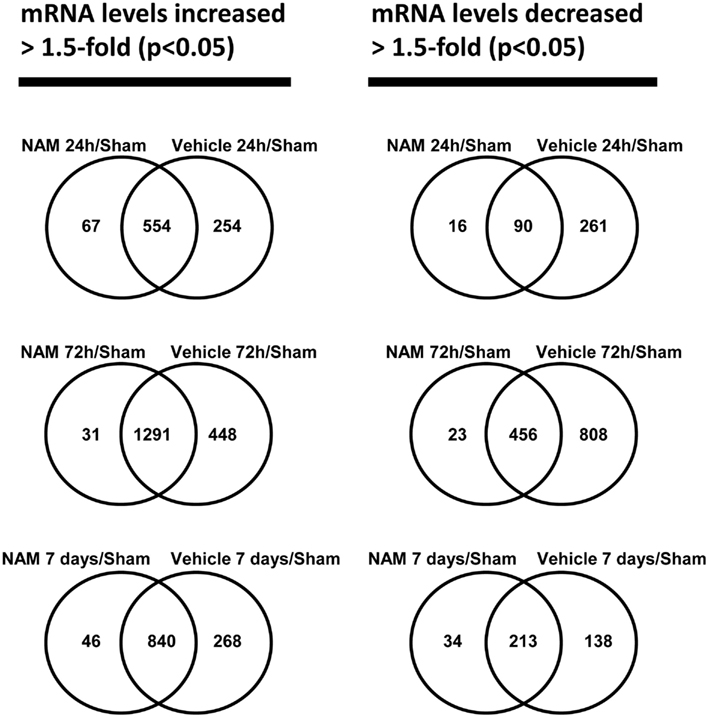
Figure 2. The Venn diagrams show the number of genes whose expression was up or down-regulated more than 1.5-fold (p < 0.05) in the NAM/Sham and Vehicle/Sham contrasts at the 24 h, 72 h, and 7-day time points. Venn diagrams were generated with the Bioconductor limma package
The nicotinamide (CCI animals that received nicotinamide) to vehicle (CCI animals that received vehicle) comparison evaluates the effect of nicotinamide treatment by separating out the effect of the TBI on gene expression. At 24 h post-TBI, the majority (82%) of the 150 differentially expressed genes in the nicotinamide treated animals compared to vehicle were down-regulated (Table 1). At 72 h post-TBI, there were 119 differentially expressed genes in the nicotinamide treated animals compared to vehicle with the majority (90%) being up-regulated. At 7 days post-TBI (4 days post-nicotinamide treatment), there were only five differentially expressed genes in the nicotinamide compared to vehicle treated animals.
Ingenuity Pathway Analysis was used to facilitate the identification of biological themes in the microarray data. IPA shifts the emphasis from the evaluation of single genes to an evaluation of molecular pathways, networks, and biological functions. Functional categories were identified by molecular and cellular functions. Canonical pathways include signaling and metabolic pathways. IPA was used to generate Figures 3–5; Figures 3 and 4 show bar-graphs for the top 10 functional categories and canonical pathways for the NAM/Vehicle comparison ranked according to statistical significance (p < 0.05). Figure 5 depicts the second most significant functional category, i.e., the Cell-to-Cell-Signaling and Interaction network (p < 0.05) in detail; IPA identified 24 and 27 significant (p < 0.05) functional categories, and 120 and 81 significant (p < 0.05) canonical pathways at 24 and 72 h post-TBI for the NAM/vehicle contrasts, respectively. The differentially expressed genes associated with each of the top 10 functional categories and canonical pathways are listed in Tables 2 and 3, whereas their degree of differential expression for the NAM/Vehicle and Vehicle/Sham contrasts at 24 and 72 h are provided in Tables 4 and 5 respectively.
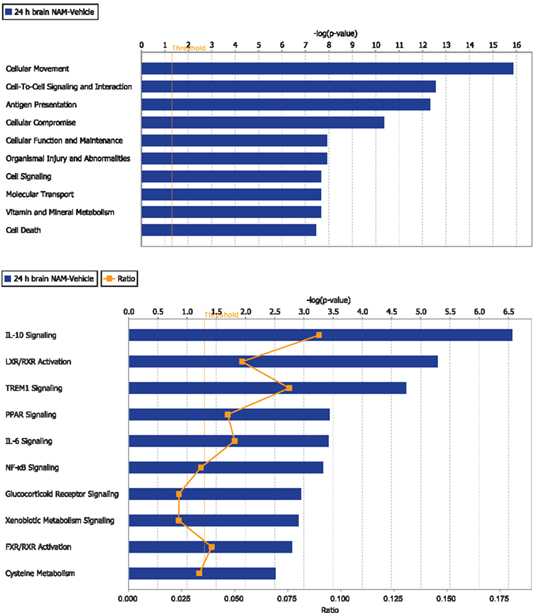
Figure 3. The top 10 functional categories and canonical pathways involving significantly regulated genes in the nicotinamide treated compared to vehicle treated CCI injured animals for the 24-h time point. The length of the bar is the negative log of the p-value and is significant if it extends to the right of the threshold line −log(p < 0.05). The ratio number, is defined as the number of significant molecules in the given pathways divided by the total number of molecules that are included in the pathway.
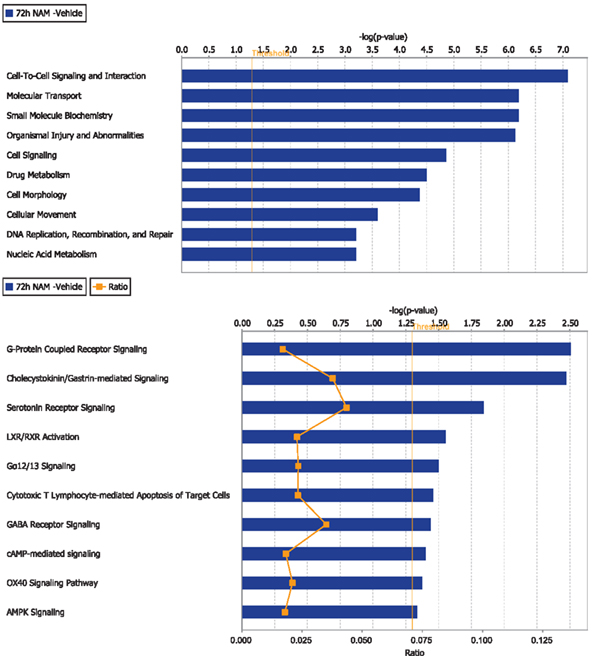
Figure 4. The top 10 functional categories and canonical pathways involving significantly regulated genes in the nicotinamide treated compared to vehicle treated CCI injured animals for the 72-h time point. The length of the bar is the negative log of the p-value and is significant if it extends to the right of the threshold line [−log(p < 0.05)]. The ratio number, is defined as the number of significant molecules in the given pathways divided by the total number of molecules that are included in the pathway.
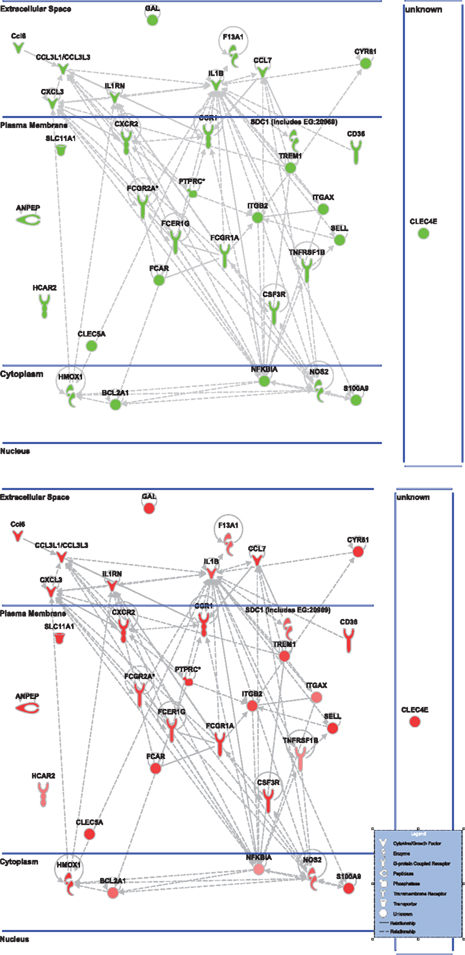
Figure 5. Cell-to-cell signaling network generated by ingenuity pathway analysis of differentially expressed genes: Molecules are represented as nodes, and the biological relationship between two nodes is represented as an edge (line). All edges are supported by at least one reference from the literature, from a textbook, or from canonical information stored in the Ingenuity Knowledge Base. The intensity of the node color indicates the degree of up- (red) or down- (green) regulation. Nodes are displayed using various shapes that represent the functional class of the gene product. Edges are displayed with various labels that describe the nature of the relationship between the nodes. Top: the nicotinamide treated injured animals compared to vehicle treated injured animal. Bottom: the vehicle treated injured animals compared to sham controls highlighting only the genes affected by nicotinamide treatment shown in the top figure.

Table 2. Regulated genes in the functional categories and canonical pathways at 24 h post-TBI.

Table 3. Regulated genes in the functional categories and canonical pathways at 72 h post-TBI.
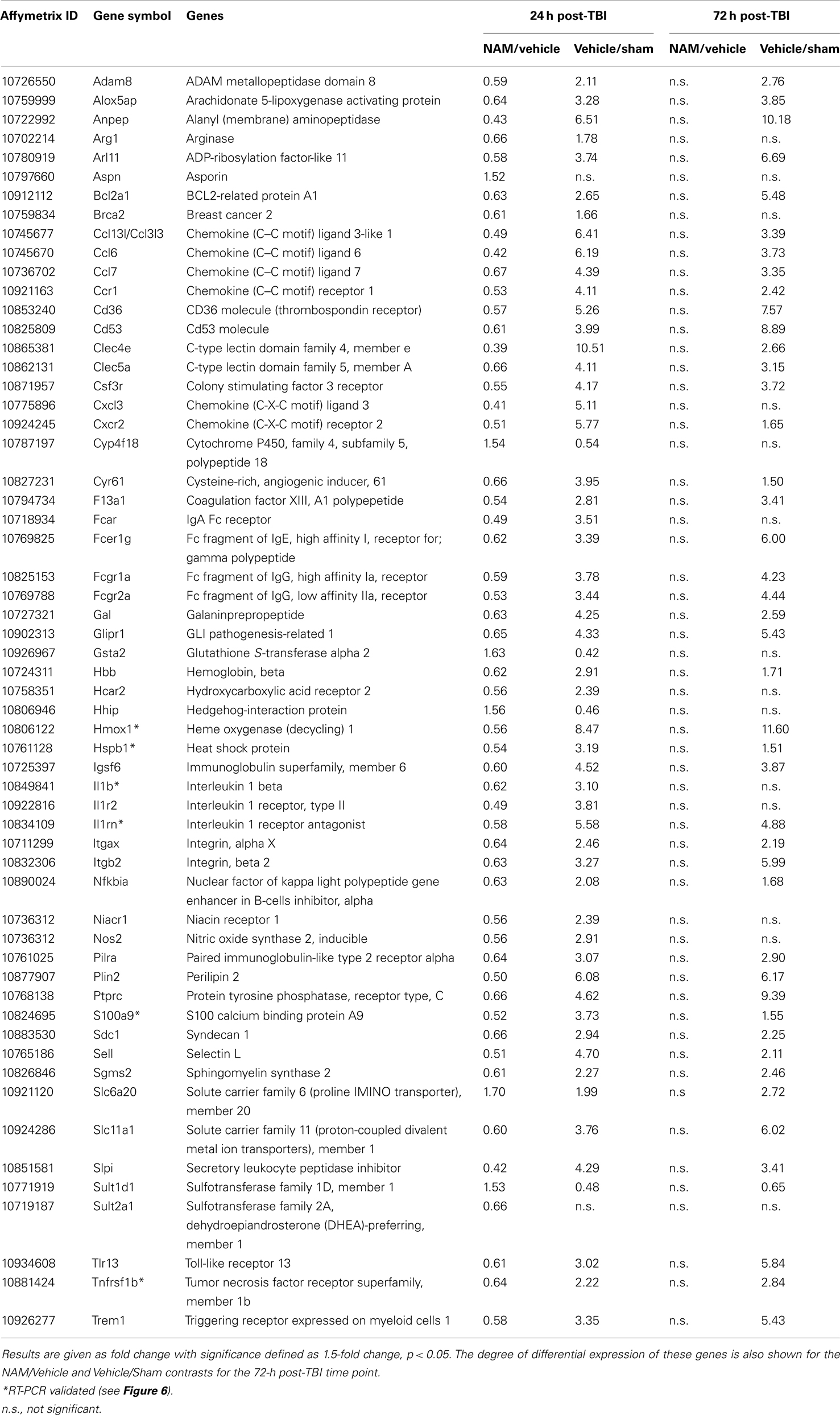
Table 4. The effect of NAM treatment (NAM/vehicle) and TBI (vehicle/sham) on differentially expressed genes associated with significant functional categories and canonical pathways identified by IPA at 24 h post-TBI.
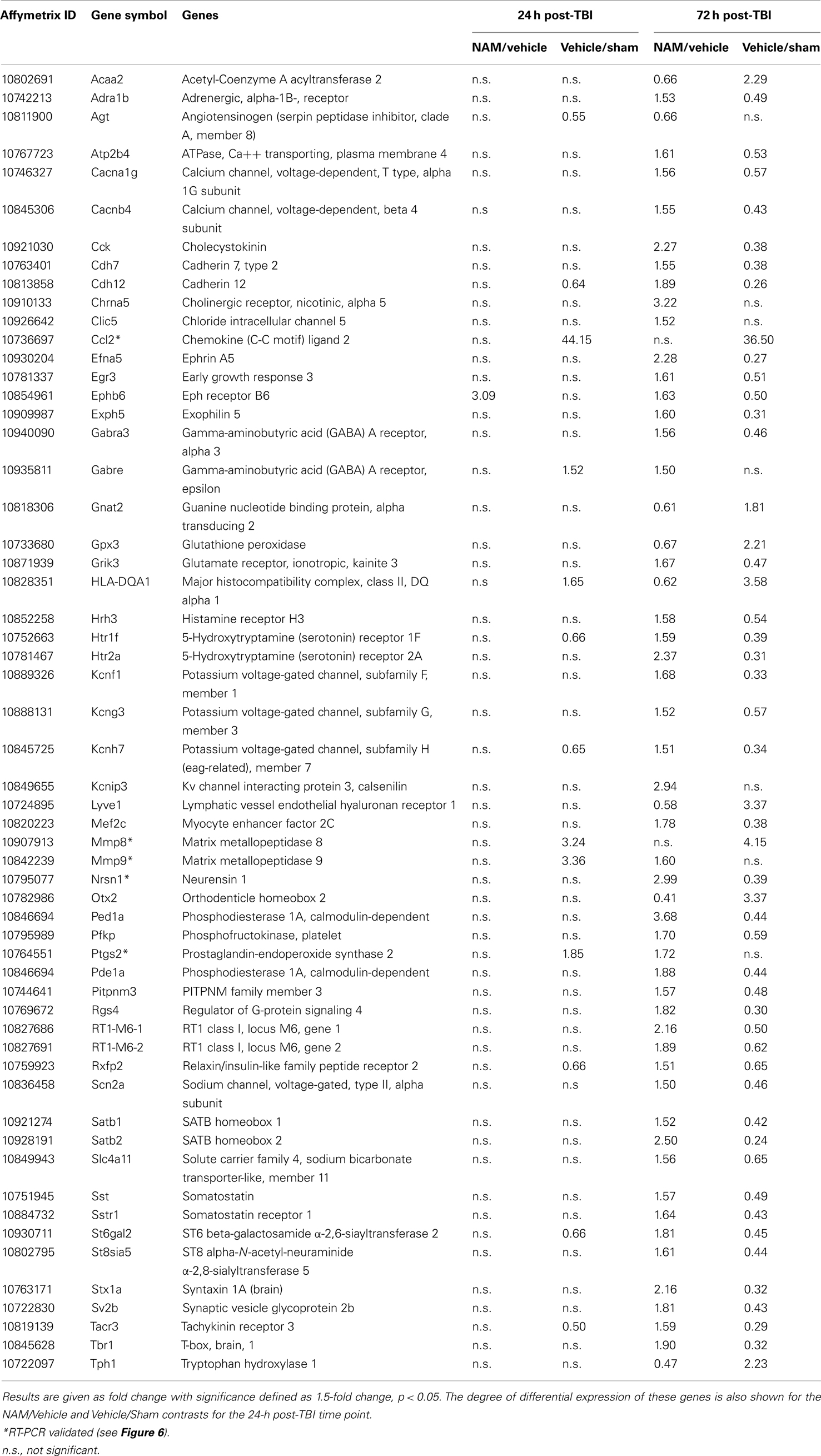
Table 5. The effect of NAM treatment (NAM/vehicle) and TBI (vehicle/sham) on differentially expressed genes associated with significant functional categories and canonical pathways identified by IPA at 24 h post-TBI.
At 24 h post-TBI, 58 unique genes were over-represented in the IPA identified significant functional categories and canonical pathways in the NAM/vehicle contrast with many genes included in multiple categories (Table 4). The majority of the regulated genes was involved in cell movement, cell signaling, cell death, and the inflammatory/immune system and included several cytokines, cytokine receptors, and chemokines as well as other molecules involved in the inflammatory/immune response. The expression of the majority of the regulated genes down-regulated by nicotinamide, were up-regulated by TBI as shown in Table 4. At 72 h, there was still significantly increased expression in the vehicle treated injured compared to non-injured sham animals in the majority of the genes affected by nicotinamide at 24 h (Table 4). However, there was not a continued effect of nicotinamide treatment on the expression of the genes at 72 h although nicotinamide concentrations were maintained until time of sacrifice.
By 72 h post-TBI, 57 unique genes were significantly over-represented in the IPA identified significant pathways and categories in the NAM/vehicle contrast (Table 5). IPA analysis identified a significant effect of nicotinamide on cell-to-cell-signaling and interactions, molecular transport, small molecular biochemistry, cell, and drug metabolism. All of the top 10 canonical pathways identified were signaling pathways. In contrast to the effects at 24 h, the majority of the genes differentially expressed were up-regulated by nicotinamide in the NAM/vehicle contrast, were down-regulated by TBI. The genes differentially expressed with nicotinamide at 72 h were not affected by nicotinamide treatment at the earlier 24 h time point (Table 5).
Validation of Microarray Data
Validation of microarray data
To validate gene expression changes, 15 genes were selected from specific pathways of interest. Figure 6 shows the gene expression results generated via microarray and qPCR (normalized to β-actin) for the nicotinamide versus vehicle group contrasts; Figures 6A,B show the data for the 24 and 72-h time points respectively. The qPCR findings were highly correlated with the microarray data (Slope = 1.187, Pearson’s R = 0.54).
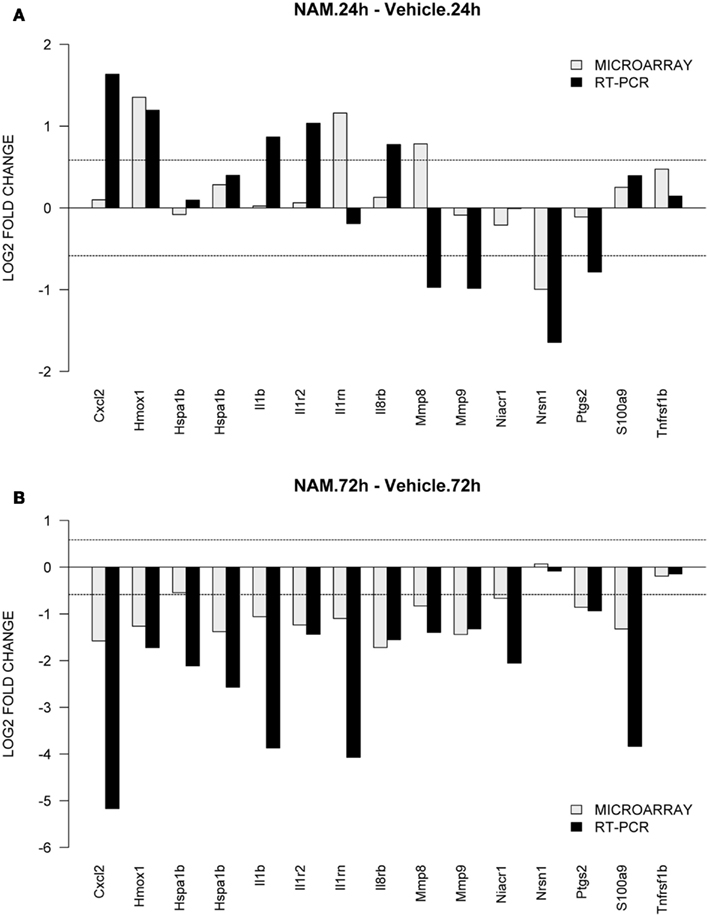
Figure 6. TaqMan based RT-PCR validation of the microarray data for the selected genes: Cxc12, chemokine (C-X-C motif) ligand 2; Hmox1, heme oxygenase 1; Hspa1b, heat shock protein 1b; Il1b, interleukin 1beta; Il1rn, interleukin 1 receptor antagonist; Il8rb, interleukin 8 receptor beta; Mmp8, matrix metallopeptidase 8; Mmp9, matrix metallopeptidase 9; Niacr1, niacin receptor 1; Nrsn1, neurensin 1; Ptgs2, prostaglandin-endoperoxide synthase 2; S100a9, S100 calcium binding protein A9; Tnfrsf1b, tumor necrosis factor receptor superfamily, member 1b. The RT-PCR data was normalized to the housekeeping gene β-actin. In order to compare the microarray data to the RT-PCR data, the microarray data for each gene was divided by the beta-actin data as it was measured by the microarray analysis. The gray bars show the microarray data and the black bars display the RT-PCR data for the contrast. (A,B) Show the data from the 24 and 72-h time point for the contrasts nicotinamide treated injured animals compared to the vehicle treated injured animals.
Discussion
In a study comparing a nicotinamide infusion to two doses of progesterone on functional recovery in the CCI model, the neuroprotective effect of nicotinamide, was equally effective asa low dose progesterone (10 mg/kg i.p. every 12 h for 72 h; Peterson et al., 2012). The functional recovery study used the same experimental conditions (rat strain, injury model, nicotinamide dosing regimen, and nicotinamide dose) as this study used. In fact, the samples that were used in this study were generated by the same laboratory. Therefore, the phenotypic data described by Peterson and colleagues can be directly compared to the transcriptional changes described in this study.
Both the nicotinamide infusion and the low dose progesterone were significantly more effective than the higher dose progesterone (20 mg/kg i.p. every 12 h for 72 h; Peterson et al., 2012). Functional recovery was assessed with two spatial memory tasks in the Morris water maze, the acquisition of a reference memory task and a reversal learning task. Neuropathological assessments were conducted in the cortex and hippocampus. Both low dose progesterone and nicotinamide improved reference memory acquisition and reversal learning in the Morris water maze and reduced tissue loss in the injured cortex and ipsilateral hippocampus compared to vehicle treatment. As determined in this study, the mechanism of the effect of nicotinamide on secondary injury pathways involves effects on inflammatory response, signaling pathways, and cell death.
Inflammatory Response
At 24 h, nicotinamide down-regulated the expression of 23 genes involved in immune or inflammatory processes. All 23 were significantly up-regulated in the TBI animals compared to sham controls and included cytokines, cytokine receptors, and chemokines as well as others involved in the inflammatory/immune response. Nicotinamide treatment down-regulated the expression of Il-1β, Il-receptor type II (Ilr2), Il-1 receptor antagonist (Il1rn), Il-8 receptor β (Il-8rβ) tumor necrosis factor receptor superfamily, member 1β (Tnfrsf1β), and other genes involved in the inflammatory response (Tables 2 and 4). Nicotinamide treatment significantly altered the expression of genes involved in Il-10, Trem1, Il-6, Nf-κB. LXR/RXR, PPAR, NF-κB, and glucocorticoid signaling pathways at 24 h post-TBI (see Table 2 for specific genes involved in these pathways). Il-10 is the principal anti-inflammatory cytokine and inhibits the expression of the pro-inflammatory mediators, TNFα, Il-6, Il-1, Mmp9, and the generation of Nos2 (Murray and Smale, 2012). Interesting, neither TBI nor nicotinamide treatment changed expression of the Il-6 or Il-10 genes themselves at the time points evaluated. The expression of Trem1 was increased by TBI and decreased by nicotinamide treatment (Table 4). Trem1 increases the secretion of pro-inflammatory cytokines and chemokines (Colonna, 2003). Nicotinamide decreased the expression of c-type lectin domain family 4, member e (Clec4e), c-type lectin domain family 4, member d (Clec4d), and S100 calcium binding protein A9 (S100a9). In the CCI injured animals compared to sham control, TBI increased the expression of Clec4d and Clec4e, 6- and 10-fold respectively. Clec4d and Clec4e are induced by inflammatory stimuli and expression leads to cytokine and chemokine production (Yamasaki et al., 2008; Wong et al., 2011). S100A9 was increased 15-fold by TBI and regulates vascular inflammation by promoting leukocyte recruitment (Bourke et al., 2003; Croce et al., 2009).
Chemokines are regulatory peptides whose major role is to guide the migration and activation of leukocyte cells into inflammatory sites and are considered pro-inflammatory. Chemokines act with chemokine receptors, G-protein-linked transmembrane receptors found on surface of target cells. Chemokine ligand 2 (Ccl2) and its receptor chemokine receptor 2 (Ccr2) are both increased in traumatic and ischemic injuries and have been implicated in their neuropathologies (Semple et al., 2010c). TBI significantly increased the gene expression of several chemokines and chemokine receptors including a 44-fold increase in Ccl2 (Table 5) and 3-fold increased expression in its receptor Ccr2. In a knock-out mouse model of closed head injury, Ccl2−/− mice had smaller cortical lesions and decreased neuronal loss (Semple et al., 2010a,b). Treatment with nicotinamide significantly down-regulated the gene expression of Ccl3, Ccl6, Ccl7, Cxc12, and colony stimulating factor 3 receptor (Csf3r). Nicotinamide did not decrease the expression of either Ccl2 or Ccr2.
LXR, PPAR, and FXR are nuclear receptors which when activated, translocate into the nucleus, form a dimer, or heterodimer with RXR to up or down regulate gene expression. Nicotinamide decrease the gene expression of only one nuclear receptor, Nfkbia (Table 4). Nfkbia, is a member of the NF-κ-B inhibitor family, which are involved in the expression of the pro-inflammatory cytokines, immune response, and anti-apoptotic genes (Tak and Firestein, 2001). Polymorphisms of the Nfkbia gene are associated with susceptibility to autoimmune and inflammatory diseases (Zhang et al., 2011).
At 72 h, post-TBI, nicotinamide increased the expression of prostaglandin-endoperoxide synthase 2 (Ptgs2), also known as COX-2 (Table 5). Ptgs2 can be either pro-inflammatory or anti-inflammatory (Choi et al., 2009). Ptgs2 is localized in neurons and is mainly induced in response to inflammatory stimuli. Animal studies have shown that during the early phase of inflammation, Ptgs2 is pro-inflammatory, but during the later phases of inflammation dominated by mononuclear cells, Ptgs2 appears to have anti-inflammatory effects by generating an alternate set of anti-inflammatory prostaglandins (Gilroy et al., 1999). Nicotinamide treatment resulted in increased gene expression of the RT1 and RTII class proteins, the major histocompatibility complex (MHC) of the rat (Gunther and Walter, 2001). In addition to their function in adaptive immunity, MHC class 1 molecules have been shown to be important in brain development, neuronal differentiation, and synaptic plasticity (Boulanger and Shatz, 2004).
Signaling Pathways
At 72 h, nicotinamide increased the expression of several hormones and receptors including the expression of two subunits of the GABAA receptor, GABAA receptor epsilon and GABAA receptor, α3 (Table 5). The GABAA receptor is a ligand-gated chloride channel, which binds GABA, the major inhibitory neurotransmitter in the brain. Nicotinamide also increased the gene expression of the glutamate receptor, ionotropic, kainate 3 (Grik3), serotonin receptors 1F and 2A, adrenergic α-1B, receptor, histamine receptor H3, relaxin/insulin-like family peptic receptor 2, and the somatostatin receptor 1 (Table 5). In the brain, the histamine receptor H3, is an auto-receptor, inhibiting the release of histamine (Schwartz, 2011). Nicotinamide increased the expression of somatostatin and its receptor. Somatostatin is a growth hormone-inhibition hormone, responsible for inhibiting the release of numerous secondary hormones. Somatostatin has been shown to have anticonvulsant activity in models of status epilepticus (Vezzani et al., 1991; Tallent and Qiu, 2008).
Nicotinamide increased the gene expression of several potassium and sodium voltage-gated ion channels, as well as, calcium voltage-gated ion channels. Specifically, up-regulation of potassium channels has been associated with diminished neural excitability. Potassium channel modulators have been proposed as neuroprotective drugs (Leung, 2010). The expression of synaptic vesicle glycoprotein 2b (Sv2b) was decreased with TBI and increased by nicotinamide. Synaptic vesicle proteins have been implicated with the etiology of epilepsy (Janz et al., 1999; Feng et al., 2009). SV2A has been identified as a target of the antiepileptic drug, levetiracetam (De Smedt et al., 2007), and has recently been found in patients with temporal lobe epilepsy (Janz et al., 1999; Feng et al., 2009). SV2B has been shown to regulate presynaptic Ca2+ signaling and has also been suggested as a target for treatment of epilepsy (Wan et al., 2010).
Apoptosis
The functional category “cell death” was among the top 10 categories with genes significantly over-represented at the 24-h time point (Nam/vehicle; Figure 3). Nicotinamide is a low potency endogenous inhibitor of PARP (Cantoni et al., 1987; Virag and Szabo, 2002). PARP is an enzyme found in the nuclei of cells and functions to sense DNA damage, binds to the damaged DNA, catalyzes the cleavage of NAD+ into nicotinamide and ADP-ribose, and then uses the ADP-ribose to synthesize polymers which attach to nuclear acceptor proteins (Virag and Szabo, 2002). In addition, to the effect of PARP on DNA repair and apoptosis, PARP enhances NF-(B-mediated transcription, and regulates the formation of inflammatory mediators of inducible nitric oxide synthase (Nos2). Nitric oxide is a major signaling molecule in the nervous, immune, and circulatory systems with its action restricted to local effects due to its instability. Nos2 was up-regulated following TBI compared to sham controls at 24 h after injury and nicotinamide decreased the gene expression (Table 4). In the present study there were no effects of TBI or nicotinamide treatment on the expression of PARP1, the major isoform present in the nucleus.
Conclusion
The proposed mechanism of the beneficial effect of nicotinamide (anti-inflammatory, anti-oxidant, and preventing apoptosis) has been based on experimental studies with 10–30 times higher concentrations than used in this study (Yang and Adams, 2004; Li et al., 2006). In this study, we demonstrated that at lower concentrations, the primary effect of nicotinamide is on inflammatory pathways at 24 h post-TBI. By 72 h post-TBI, the primary effect of nicotinamide is on cell signaling pathways involving neurotransmitters, neuropeptides, growth factors, and ion channels with little to no effect on inflammatory pathways.
Another potential promising treatment for TBI is progesterone which has been studied in preclinical models of TBI for several decades (Stein and Wright, 2010) and there are currently two large multi-center clinical TBI studies on-going. In a study evaluating the effect of the progesterone dose on gene expression in the same CCI injury model (Anderson et al., 2011), there was a significant effect of progesterone on inflammatory responses, apoptosis, regulating the DNA damage response, cell proliferation, and blood vessel remodeling following TBI. Low dose progesterone treatment significantly altered the expression of 14 and 40 genes at 24 and 72 h, respectively with approximately equal number up- or down-regulated. At 7 days, low dose progesterone up-regulated 383 of 551 (70%) genes compared to vehicle compared to the lack of effect of nicotinamide on gene expression at 7 days. In general, nicotinamide treatment counteracted the changes in genes differentially expressed due to TBI, while progesterone treatment primarily altered expression of genes that were not affected directly by TBI itself.
In conclusion, the mechanism of the effect of nicotinamide on secondary injury pathways involves effects on inflammatory response, signaling pathways, and cell death. The unique effect of nicotinamide compared to progesterone on gene expression suggests that a combination of nicotinamide and low dose progesterone may be an effective combination of treatment for TBI.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by a grant from the National Institutes of Health/National Institute of Child, Health and Development (R01 HD061944-01). This work was also supported in part by the UW NIEHS sponsored Center for Ecogeneticsand Environmental Health (P30ES07033).
Footnotes
References
Allison, D. B., Cui, X., Page, G. P., and Sabripour, M. (2006). Microarray data analysis: from disarray to consolidation and consensus. Nat. Rev. Genet. 7, 55–65.
Anderson, G. D., Farin, F. M., Bammler, T. K., Beyer, R. P., Swan, A. A., Wilkerson, H. W., et al. (2011). The effect of progesterone dose on gene expression after traumatic brain injury. J. Neurotrauma 28, 1827–1843.
Ayoub, I. A., Lee, E. J., Ogilvy, C. S., Beal, M. F., and Maynard, K. I. (1999). Nicotinamide reduces infarction up to two hours after the onset of permanent focal cerebral ischemia in Wistar rats. Neurosci. Lett. 259, 21–24.
Bengtsson, H., Simpson, K., Bullard, J., and Hansen, K. (2008). Aroma.Affymetrix: A Generic Framework in R for Analyzing Small to Very Large Affymetrix Data sets in Bounded Memory. Berkeley: University of California.
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300.
Boulanger, L. M., and Shatz, C. J. (2004). Immune signalling in neural development, synaptic plasticity and disease. Nat. Rev. 5, 521–531.
Bourke, E., Cassetti, A., Villa, A., Fadlon, E., Colotta, F., and Mantovani, A. (2003). IL-1 beta scavenging by the type II IL-1 decoy receptor in human neutrophils. J. Immunol. 170, 5999–6005.
Cantoni, O., Sestili, P., Spadoni, G., Balsamini, C., Cucchiarini, L., and Cattabeni, F. (1987). Analogues of benzamide containing a sulfur atom as poly(ADP-ribose) transferase inhibitors. Biochem. Int. 15, 329–337.
Choi, S. H., Aid, S., and Bosetti, F. (2009). The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: implications for translational research. Trends Pharmacol. Sci. 30, 174–181.
Croce, K., Gao, H., Wang, Y., Mooroka, T., Sakuma, M., Shi, C., et al. (2009). Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation 120, 427–436.
De Smedt, T., Raedt, R., Vonck, K., and Boon, P. (2007). Levetiracetam: the profile of a novel anticonvulsant drug-part I: preclinical data. CNS Drug Rev. 13, 3–56.
Faden, A. I., and Stoica, B. (2007). Neuroprotection: challenges and opportunities. Arch. Neurol. 64, 794–800.
Faul, M., Xu, L., Wald, M., and Coronado, V. (2010). Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths. Atlanta, GA: Centers for Disease Control and Prevention, National Center for Injury Prevention and Control.
Feng, G., Xiao, F., Lu, Y., Huang, Z., Yuan, J., Xiao, Z., et al. (2009). Down-regulation synaptic vesicle protein 2A in the anterior temporal neocortex of patients with intractable epilepsy. J. Mol. Neurosci. 39, 354–359.
Gilroy, D. W., Colville-Nash, P. R., Willis, D., Chivers, J., Paul-Clark, M. J., and Willoughby, D. A. (1999). Inducible cyclooxygenase may have anti-inflammatory properties. Nat. Med. 5, 698–701.
Goffus, A. M., Anderson, G. D., and Hoane, M. (2010). Sustained delivery of nicotinamide limits cortical injury and improves functional recovery following traumatic brain injury. Oxid. Med. Cell. Longev. 3, 145–152.
Gunther, E., and Walter, L. (2001). The major histocompatibility complex of the rat (Rattus norvegicus). Immunogenetics 53, 520–542.
Hoane, M. R., Akstulewicz, S. L., and Toppen, J. (2003). Treatment with vitamin B3 improves functional recovery and reduces GFAP expression following traumatic brain injury in rats. J. Neurotrauma 20, 1189–1199.
Hoane, M. R., Gilbert, D. R., Holland, M. A., and Pierce, J. L. (2006a). Nicotinamide reduces acute cortical neuronal death and edema in the traumatically injured brain. Neurosci. Lett. 408, 35–39.
Hoane, M. R., Kaplan, S. A., and Ellis, A. L. (2006b). The effects of nicotinamide on apoptosis and blood-brain barrier breakdown following traumatic brain injury. Brain Res. 1125, 185–193.
Hoane, M. R., Tan, A. A., Pierce, J. L., Anderson, G. D., and Smith, D. C. (2006c). Nicotinamide treatment reduces behavioral impairments and provides cortical protection after fluid percussion injury in the rat. J. Neurotrauma 23, 1535–1548.
Hoane, M. R., Pierce, J. L., Holland, M. A., and Anderson, G. D. (2008a). Nicotinamide treatment induces behavioral recovery when administered up to 4 hours following cortical contusion injury in the rat. Neuroscience 154, 861–868.
Hoane, M. R., Pierce, J. L., Kauffman, N. A., and Beare, J. (2008b). Variation in chronic nicotinamide treatment after traumatic brain injury can alter components of functional recovery independent of histological damage. Oxid. Med. Cell. Longev. 1, 46–53.
Holland, M. A., Tan, A. A., Smith, D. C., and Hoane, M. R. (2008). Nicotinamide treatment provides acute neuroprotection and GFAP regulation following fluid percussion injury. J. Neurotrauma 25, 140–152.
Horsman, M. R., Hoyer, M., Honess, D. J., Dennis, I. F., and Overgaard, J. (1993). Nicotinamide pharmacokinetics in humans and mice: a comparative assessment and the implications for radiotherapy. Radiother. Oncol. 27, 131–139.
Janz, R., Goda, Y., Geppert, M., Missler, M., and Sudhof, T. C. (1999). SV2A and SV2B function as redundant Ca2+ regulators in neurotransmitter release. Neuron 24, 1003–1016.
Klaidman, L. K., Mukherjee, S. K., Hutchin, T. P., and Adams, J. D. (1996). Nicotinamide as a precursor for NAD+ prevents apoptosis in the mouse brain induced by tertiary-butylhydroperoxide. Neurosci. Lett. 206, 5–8.
Knip, M., Douek, I. F., Moore, W. P., Gillmor, H. A., McLean, A. E., Bingley, P. J., et al. (2000). Safety of high-dose nicotinamide: a review. Diabetologia 43, 1337–1345.
Leung, Y. M. (2010). Voltage-gated K+ channel modulators as neuroprotective agents. Life Sci. 86, 775–780.
Li, F., Chong, Z. Z., and Maiese, K. (2006). Cell Life versus cell longevity: the mysteries surrounding the NAD+ precursor nicotinamide. Curr. Med. Chem. 13, 883–895.
Murray, P. J., and Smale, S. T. (2012). Restraint of inflammatory signaling by interdependent strata of negative regulatory pathways. Nat. Immunol. 13, 916–924.
Peterson, T., Ward, J., Logue, M., Anderson, G. D., and Hoane, M. R. (2012). An evaluation of the neuroprotective effects of progesterone and nicotinamide on functional recovery following cortical contusion injury in the rat. J. Neurotrauma 29, 2823–2830.
Quigley, A., Tan, A. A., and Hoane, M. R. (2009). The effects of hypertonic saline and nicotinamide on sensorimotor and cognitive function following cortical contusion injury in the rat. Brain Res. 1304, 138–148,
Sakakibara, Y., Mitha, A. P., Ayoub, I. A., Ogilvy, C. S., and Maynard, K. I. (2002). Delayed treatment with nicotinamide (vitamin B3) reduces the infarct volume following focal cerebral ischemia in spontaneously hypertensive rats, diabetic and non-diabetic Fischer 344 rats. Brain Res. 931, 68–73.
Schouten, J. W. (2007). Neuroprotection in traumatic brain injury: a complex struggle against the biology of nature. Curr. Opin. Crit. Care 13, 134–142.
Schwartz, J. C. (2011). The histamine H3 receptor: from discovery to clinical trials with pitolisant. Br. J. Pharmacol. 163, 713–721.
Semple, B. D., Bye, N., Rancan, M., Ziebell, J. M., and Morganti-Kossmann, M. C. (2010a). Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2-/- mice. J. Cereb. Blood Flow Metab. 30, 769–782.
Semple, B. D., Bye, N., Ziebell, J. M., and Morganti-Kossmann, M. C. (2010b). Deficiency of the chemokine receptor CXCR2 attenuates neutrophil infiltration and cortical damage following closed head injury. Neurobiol. Dis. 40, 394–403.
Semple, B. D., Kossmann, T., and Morganti-Kossmann, M. C. (2010c). Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J. Cereb. Blood Flow Metab. 30, 459–473.
Smyth, G. K. (2004). Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article 3.
Stein, D. G., and Wright, D. W. (2010). Progesterone in the clinical treatment of acute traumatic brain injury. Expert Opin. Investig. Drugs 19, 847–857.
Swan, A. A., Chandrashekar, R., Beare, J., and Hoane, M. R. (2011). Preclinical efficacy testing in middle-aged rats: nicotinamide, a novel neuroprotectant, demonstrates diminished preclinical efficacy after controlled cortical impact. J. Neurotrauma 28, 431–440.
Tak, P. P., and Firestein, G. S. (2001). NF-kappaB: a key role in inflammatory diseases. J. Clin. Invest. 107, 7–11.
Tallent, M. K., and Qiu, C. (2008). Somatostatin: an endogenous antiepileptic. Mol. Cell. Endocrinol. 286, 96–103.
Vezzani, A., Serafini, R., Stasi, M. A., Vigano, G., Rizzi, M., and Samanin, R. (1991). A peptidase-resistant cyclic octapeptide analogue of somatostatin (SMS 201-995) modulates seizures induced by quinolinic and kainic acids differently in the rat hippocampus. Neuropharmacology 30, 345–352.
Virag, L., and Szabo, C. (2002). The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev. 54, 375–429.
Vonder Haar, C., Anderson, G. D., and Hoane, M. R. (2011). Continuous nicotinamide administration improves behavioral recovery and reduces lesion size following bilateral frontal controlled cortical impact injury. Behav. Brain Res. 224, 311–317.
Wan, Q. F., Zhou, Z. Y., Thakur, P., Vila, A., Sherry, D. M., Janz, R., et al. (2010). SV2 acts via presynaptic calcium to regulate neurotransmitter release. Neuron 66, 884–895.
Wong, K. L., Tai, J. J., Wong, W. C., Han, H., Sem, X., Yeap, W. H., et al. (2011). Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 118, e16–e31.
Yamasaki, S., Ishikawa, E., Sakuma, M., Hara, H., Ogata, K., and Saito, T. (2008). Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat. Immunol. 9, 1179–1188.
Yang, J., and Adams, J. D. (2004). Nicotinamide and its pharmacological properties for clinical therapy. Drug Design Rev. 1, 43–52.
Yang, J., Klaidman, L. K., and Adams, J. D. (2002a). Medicinal chemistry of nicotinamide in the treatment of ischemia and reperfusion. Mini Rev. Med. Chem. 2, 125–134.
Yang, J., Klaidman, L. K., Chang, M. L., Kem, S., Sugawara, T., Chan, P., et al. (2002b). Nicotinamide therapy protects against both necrosis and apoptosis in a stroke model. Pharmacol. Biochem. Behav. 73, 901–910.
Yang, J., Klaidman, L. K., Nalbandian, A., Oliver, J., Chang, M. L., Chan, P. H., et al. (2002c). The effects of nicotinamide on energy metabolism following transient focal cerebral ischemia in Wistar rats. Neurosci. Lett. 333, 91–94.
Keywords: nicotinamide, gene expression, cortical contusion model, traumatic brain injury
Citation: Anderson GD, Peterson TC, Farin FM, Bammler TK, Beyer RP, Kantor ED and Hoane MR (2013) The effect of nicotinamide on gene expression in a traumatic brain injury model. Front. Neurosci. 7:21. doi: 10.3389/fnins.2013.00021
Received: 15 November 2012; Accepted: 06 February 2013;
Published online: 26 February 2013.
Edited by:
Nick Andrews, Harvard Medical School, USAReviewed by:
Muzamil Ahmad, Indian Institute of Integrative Medicine, IndiaFiras H. Kobeissy, University of Florida, USA
Copyright: © 2013 Anderson, Peterson, Farin, Bammler, Beyer, Kantor and Hoane. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: G. D. Anderson, Department of Pharmacy, University of Washington, Box 357630, Health Science Complex H-361A, Seattle, WA 98195, USA. e-mail:Z2FpbGFAd2FzaGluZ3Rvbi5lZHU=