

- Department of Psychiatry, University of California, San Diego, San Diego, CA, USA
Concerns regarding a drought in psychopharmacology have risen from many quarters. From one perspective, the wellspring of bedrock medications for anxiety disorders, depression, and schizophrenia was serendipitously discovered over 30 year ago, the swell of pharmaceutical investment in drug discovery has receded, and the pipeline's flow of medications with unique mechanisms of action (i.e., glutamatergic agents, CRF antagonists) has slowed to a trickle. Might oxytocin (OT)-based therapeutics be an oasis? Though a large basic science literature and a slowly increasing number of studies in human diseases support this hope, the bulk of extant OT studies in humans are single-dose studies on normals, and do not directly relate to improvements in human brain-based diseases. Instead, these studies have left us with a field pregnant with therapeutic possibilities, but barren of definitive treatments. In this clinically oriented review, we discuss the extant OT literature with an eye toward helping OT deliver on its promise as a therapeutic agent. To this end, we identify 10 key questions that we believe future OT research should address. From this overview, several conclusions are clear: (1) the OT system represents an extremely promising target for novel CNS drug development; (2) there is a pressing need for rigorous, randomized controlled clinical trials targeting actual patients; and (3) in order to inform the design and execution of these vital trials, we need further translational studies addressing the questions posed in this review. Looking forward, we extend a cautious hope that the next decade of OT research will birth OT-targeted treatments that can truly deliver on this system's therapeutic potential.
Oxytocin: Tool or Treatment?
Over the last several decades, the nonapeptide oxytocin (OT) has been cast in two roles on the stage of human neuroscience. First and most dramatic has been its remarkable and ever-expanding role as a powerful mediator of myriad aspects of our uniquely social brains (MacDonald and MacDonald, 2010). Beginning with its evolutionary origin 10,000 years ago—when the progenitor nonapeptide vasotocin was orchestrating decision-making in marine animals (Grimmelikhuijzen and Hauser, 2012)—a series of vital advances have ratcheted forward our understanding of this vital central system. These include its initial discovery as a uterotonic component of pituitary extract over a century ago (Dale, 1906); the concept (novel at the time) of neurosecretion, the “glandular activity” of hormone-secreting neurons (Scharrer and Scharrer, 1945); the vital technique of immunoflourescent visualization of OT-producing neurons (Swaab et al., 1975) which allowed the subsequent histological characterization of the human central OT system (Loup et al., 1991); the more recent sequencing and synthesis of the peptide (Du Vigneaud, 1956), and the gene for the receptor (Kimura et al., 1992); and finally, increasingly sophisticated translational research using techniques like gene knockout and optogenetic manipulation of specific central circuits (Stoop, 2012). Each of these progressive steps has allowed us to ask and answer increasingly specific questions about the nature of the central OT system and its contribution to the bonded, social nature we share with fellow mammals. The culmination of several decades of sophisticated translational neuroscience research has been a decade-long groundswell of human studies which have ensconced OT and its sister nanopeptide vasopressin with testosterone, estrogen, and cortisol in the pantheon of centrally active hormones critical to understanding human behavior.
OT has also been cast for a second, as-yet unfulfilled role as a therapeutic tool to ameliorate suffering from brain-based disease. Promise notwithstanding, its performance here has been a bit more pedestrian. Although it has been safely used for decades in obstetrics to induce and augment labor, the suggestion that OT may have therapeutic value in the treatment of a host of brain-based conditions (addiction, anxiety, autism, mood disorders, and schizophrenia) has not for the most part been bolstered by clinical investigations with meaningful therapeutic endpoints. More precisely, despite an enormous amount of anticipation that OT's effects in preclinical studies can be translated into OT-based treatments for psychiatric disorders, few studies have actually delivered OT as a bona-fide therapeutic agent, using chronic daily dosing, targeting core symptoms of specific disease states, and assessing safety, tolerability, and clinical outcomes. From a clinical perspective, the current portfolio of OT research is flush with more therapeutic hints than help, and exogenous OT has—thus far—acted more decisively in its role as a pharmacological probe than a therapeutic palliative.
These contrasting roles come into apposition at a time in the history of psychiatric therapeutics which some have called a “crisis” (Fibiger, 2012). To whit, despite significant advances in our understanding of the brain bases of human psychiatric disease, our abilities to reduce suffering and restore function in psychiatric disease remains woefully inadequate (Holma et al., 2008; Lieberman and Stroup, 2011; Volkow and Skolnick, 2012). Many or most of the foundational therapeutic medications in our psychiatric armamentarium (i.e., antidepressants, antipsychotics) were discovered serendipitously decades ago. Several promising new therapeutic classes of drugs for CNS conditions have failed to pass late-stage drug development. Stymied by these repeated, costly product failures, many pharmaceutical companies have abjured investing in this therapeutic arena. These pharmaceutical realities stand in stark contrast to the significant prevalence and toll of brain-based diseases. Though these setbacks pose a challenge to drug development, we maintain optimism that OT-based therapeutics may provide relief, though as discussed below, development of OT-targeted therapeutics has its own challenges.
In this review, we approach OT from the perspective of researcher-clinicians interested in the development of OT-targeted pharmaceuticals for the abridgement of human psychiatric disease. Given intense interest in this molecule and the ever-mushrooming literature, this review has a necessarily limited scope. Herein, we constrain our discussion to arenas of significant, direct relevance to the development of OT-based therapeutics, and direct interested readers to several recent, well-referenced reviews on other vital aspects of OT including details of its neurophysiology and interaction with arginine vasopressin (AVP) (Stoop, 2012), implications of genetic variations in the OT receptor (OTR; Ebstein et al., 2012; Kumsta and Heinrichs, 2013), and OT's role in human development (Gordon et al., 2011; Feldman, 2012).
Brain Disorders for which Oxytocin may have Therapeutic Efficacy
As mentioned above, as the result of its revealed effects on behavior and brain processes observed in both animal studies and translational studies in humans, OT has been proposed as a potential treatment for a wide range of brain disorders: addiction, anxiety disorders, autism-spectrum and other developmental disorders, borderline personality disorder, mood disorders, and schizophrenia. For a more complete background on OT's preclinical profile and the justification for these therapeutic speculations see the following recent, extensive reviews (Slattery and Neumann, 2010; MacDonald and Feifel, 2012a; McGregor and Bowen, 2012; Modi and Young, 2012; Neumann and Landgraf, 2012). As of November 2012, we note ongoing treatment trials of intranasal (IN) OT in autism, schizophrenia and schizoaffective disorder, frontotemporal dementia, major depressive disorder and treatment resistant depression, post-traumatic stress disorder, borderline personality disorder, and drug dependence (i.e., alcohol, marijuana) (www.clinicaltrials.gov). Therewith, we anticipate that the next several years will show a corresponding increase in actual clinical data. To date, however, there have been a relatively limited number of patient-targeted clinical OT trials, with the majority being single-dose (Table 1). Given the limited number of studies that have been conducted evaluating OT's potential as a bona-fide treatment for clinical brain disorders, one could conclude that almost all of the much-anticipated therapeutic potential of this neuropeptide remains to be proven.
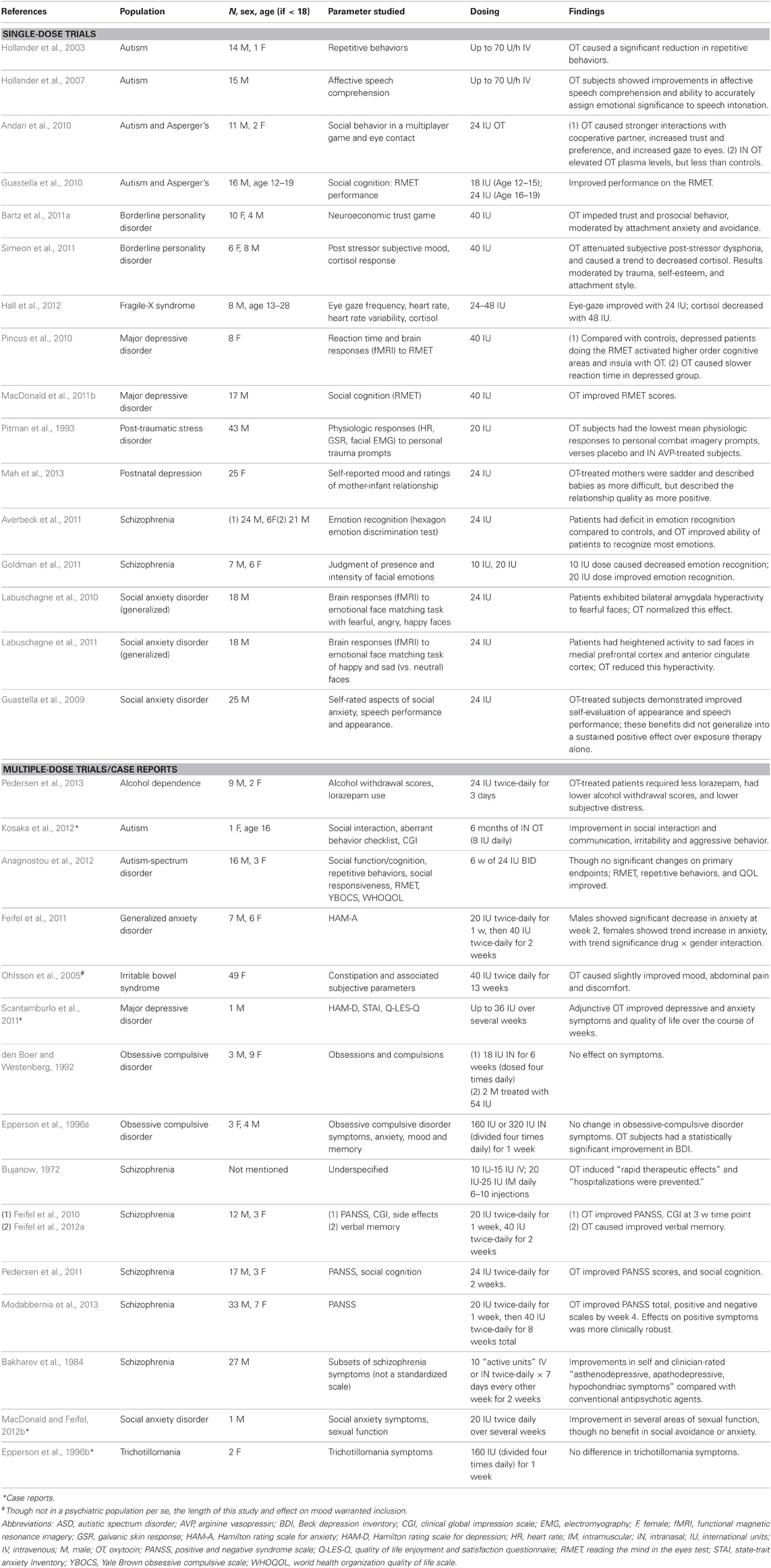
Table 1. Studies using oxytocin in patients with brain-based illness.
Important Questions for Developing Oxytocin-Targeted Therapeutics
Most preclinical and translational studies conducted to date—as well as single-dose studies in normals—attempt to answer the question: “what does OT do?” A pragmatic, treatment-oriented clinician may wish to restate the question, asking: “what does OT do when used as drug?” More specifically, “what effects does OT have when given chronically to patients with psychiatric illness?” Sadly, in spite of a decade of high-profile studies, we would be hard-pressed to answer this question for the majority of putative indications. Delving deeper, three-linked facts make this clinically oriented query even more incisive: (1) as mentioned, the vast majority of published OT studies are single-dose studies in normals; (2) in normals, the single-dose effects of lifesaving psychiatric medications [i.e., antipsychotics, serotonin reuptake inhibitors (SSRIs)] are often either negligible (Harmer et al., 2003; Murphy et al., 2009) or aversive (Belmaker and Wald, 1977; Harmer et al., 2008); (3) in psychiatric patients, the short-term effects of some medications are the opposite of their effect when given chronically [i.e., short-term anxiogenesis with SSRIs (Kent et al., 1998)]. As such, though in some cases single-dose effects in normals can be linked to longer-term benefits in patient populations [i.e., enhancement of emotional processing as a biomarker for antidepressant activity (Harmer et al., 2009a; Tranter et al., 2009)], we should be circumspect when extrapolating too directly from these studies to the effects of chronic dosing in psychiatrically ill samples. For all these reasons, multi-week, daily dose, randomized placebo-controlled trials—the mainstay for evaluating the therapeutic efficacy and safety of investigational psychotropic drugs—are needed to advance the field from the stage of optimistic speculation into the realm where definitive verdicts can be obtained. Only then will we be able to decisively answer the vital question asked by our treatment-seeking clinician.
Beyond these sorely needed proof-of-concept clinical trials, the development of OT-targeted therapeutics for CNS disease faces a significant number of challenges. In the sections below—in the form of 10 questions—we attempt to identify and describe them (Table 2). En toto, these questions span a wide spectrum of issues that need to be addressed in order to complete the bench-to-bedside arc with OT. Of note, the question of the role of several important individual factors (variations in the OT and CD38 receptor, sex, and early experience) in clinical response to OT is covered in an accompanying mini-review (MacDonald, 2012) in this special section.

Table 2. Ten questions for the development of oxytocin-targeted therapeutics for brain disorders.
How does Acute and Chronic Oxytocin Administration Differ?
The vast majority of published studies of IN OT—even those done in patient samples (Table 1)—have used only a single-application dosing paradigm. In stark contrast, almost all treatments for the most debilitating brain disorders are delivered chronically, with most achieving their maximal clinical effects after weeks of daily administration. Furthermore, as intimated above, the acute and chronic effects of medications are often diametrically opposite, as seen in the case of SSRIs, which are a first-line chronic treatment for anxiety disorders, yet can cause anxiety after a single-dose (Spigset, 1999; Birkett et al., 2011). One process that contributes to the difference between acute and chronic drug administration is “tachyphylaxis” or “tolerance” in which the acute effects of a drug dissipate with repeated administration. At a cellular level, persistently stimulated receptors like the OTR may become desensitized or may be expressed in smaller numbers on cell surfaces, via several processes, including one called internalization. Notably, internalization has been demonstrated to occur with the OTR (Gimpl and Fahrenholz, 2001).
Specifically in the case of OT, basic science research supports the fact that that there are often significant differences between the effects of acute and chronic administration of OT (Kramer et al., 2003; Bowen et al., 2011; Keebaugh and Young, 2011; Bales and Perkeybile, 2012). Furthermore, though the neurobiological mechanism of certain of OT's effects (i.e., acute anxiolysis) have been carefully dissected in animal models (Viviani and Stoop, 2008; Yoshida et al., 2009; Viviani et al., 2011; Knobloch et al., 2012; Stoop, 2012, for review), the mechanism of action of therapeutic later-onset effects of chronic OT treatment—the mode of treatment most salient to human brain disorders—remains unknown.
On this latter point, the small body of research in which chronic IN OT has been given to psychiatrically ill patients indicates that like currently used medications from other classes, IN OTs antipsychotic (Feifel et al., 2010, 2012a; Pedersen et al., 2011; Modabbernia et al., 2013) and anxiolytic (Feifel et al., 2011) effects may take weeks to emerge to a clinically meaningful degree. Unfortunately for the development of OT-targeted therapeutics, then, the large number of extant IN OT studies may not speak directly to key issues relevant to the effects of chronic OT administration. As discussed below in section “What is the Role of Vasopressin Receptors in Oxytocin's Effects?” however, the discovery and development of biomarkers which track with clinical outcomes and syndromes would significantly improves the clinical gain from single-dose trials (de Oliveira et al., 2012a). Examples relevant to OT include the abovementioned single-dose antidepressant effects on emotional processing (Harmer, 2008) and the attenuation of the anxiogenic effects of CO2 inhalation (Bailey et al., 2007). We anticipate that future biomarker and pharmacokinetic studies in humans treated with acute and chronic OT will build on the few single-dose, functional-imaging trials in humans with psychiatric illness (Labuschagne et al., 2010, 2011; Pincus et al., 2010) to illuminate these important clinical questions.
How do Oxytocin's Therapeutic-Like Effects in Healthy Subjects Translate to Patient with Brain Disorders?
A second pharmacodynamic issue that requires more study concerns the difference between OT's effects in normal vs. psychiatrically ill samples. Just as a host of individual differences significantly influence response to OT in normal samples (MacDonald, 2012), so it is likely that OT's effects will differ between healthy and clinical populations. For example, in a functional imaging study of the effects of OT on brain activity during the reading the mind in the eyes test (RMET), OT-mediated alteration in brain activity differed significantly between untreated patients with depression and normal subjects (Pincus et al., 2010). A second, unpublished set of data from our group found that patients with depression had an anxiogenic response to single-dose IN OT given in a psychotherapy context, in contrast to acute anxiolytic effects reported in normals (Heinrichs et al., 2003; de Oliveira et al., 2012a,b). Additionally, Bartz et al. has demonstrated that many patients with borderline personality disorder have divergent responses to OT that those seen in normals, with OT-decreasing trust and cooperation (Bartz et al., 2011a). On the other hand, some acute neural responses (attenuation of amygdala activity) are seen in both patient groups (Labuschagne et al., 2010) and normals (Zink and Meyer-Lindenberg, 2012) (Figure 1). Notwithstanding these similarities, the findings from several single-dose studies indicating that the effects of OT may differ between patients with psychiatric disease and those without calls for caution when extrapolating clinical effects of OT in patients from the study of its effects in normals.
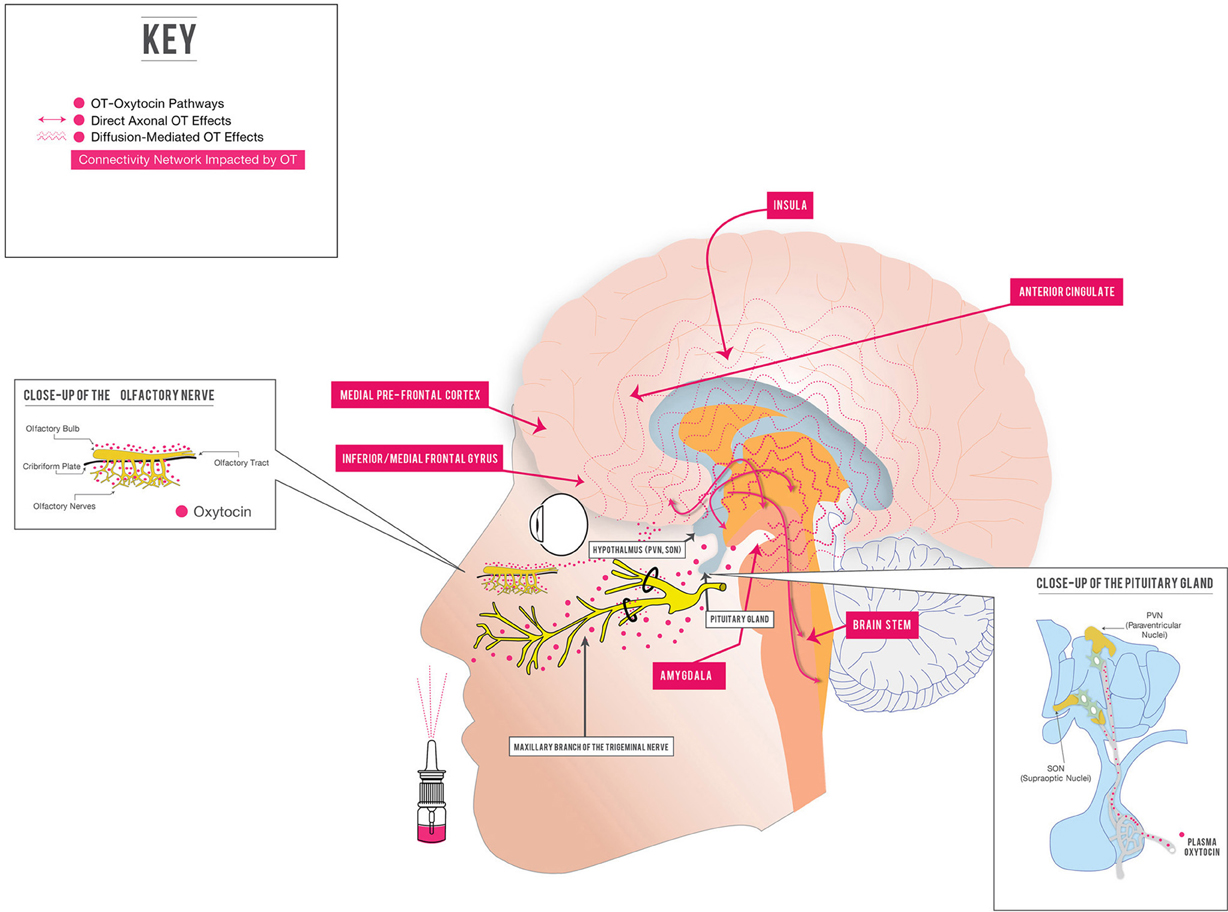
Figure 1. Intranasal oxytocin: potential therapeutic regulation of brain function in psychiatric illness. Several important aspects of intranasally delivered OT (IN OT) treatment of brain-based illness are represented. One potential way that IN OT may cross the blood-brain barrier and cause central effects is represented: directly via extraneuronal/perineuronal routes along trigeminal or olfactory nerve pathways (Thorne and Frey, 2001; Ross et al., 2004; Dhuria et al., 2010; Renner et al., 2012a,b). Other mechanisms of entry (bulk flow, lymphatic channels, intraneuronal transport, active or passive transport from vasculature) are discussed in references and the text. IN OT may cause some of its central effects by stimulating the endogenous OT system, which secretes OT into the peripheral circulation (right pullout), and has both direct, “wired” and diffusion-mediated central effects (Landgraf and Neumann, 2004; Stoop, 2012). Through these mechanisms, IN OT impacts the function of amygdala-anchored connectivity networks in normals (Kirsch et al., 2005; Sripada et al., 2013), as well as important brain regions (amygdala, insula, anterior cingulate, medial prefrontal cortex) in patients with psychiatric illness (Labuschagne et al., 2010, 2011; Pincus et al., 2010). For simplicity, not all brain areas impacted by OT are shown; see Bethlehem et al. (2012); Zink and Meyer-Lindenberg (2012) for recent, detailed reviews.
There may be, moreover, significant associations between the OT system and certain psychiatric disease states or endophenotypes. One component of this distinction is covered in an accompanying mini-review (MacDonald, 2012), which discusses the clinical import of research on variations in the OTR and the CD38 ectoenzyme. Though many of the studies in this growing literature are in normals, certain genetic variations in aspects of the OT system have been associated with disease states (Kumsta and Heinrichs, 2013). In addition to these known variations in the OT system, one could assume that certain individuals (and perhaps certain diagnostic groups) have an as-yet undocumented state of more significant functional OT deficiency akin to that found in central diabetes insipidus (DI). Specifically, in the genetic form of central DI called familial neurohypophyseal diabetes insipidus (FNDI), variations in the AVP prohormone gene (AVP-neurophysin II) on chromosome 20 result in inadequate protein folding and dimerization. These changes cause the aberrant protein to be retained in the neuron, ultimately leading to cell death of hypothalamic magnocellular neurons in the supraoptic nucleus and paraventricular nucleus (Bergeron et al., 1991; Ito and Jameson, 1997). Given endogenous AVPs role in natriuresis, these patients present clinically with symptoms of progressive functional AVP deficiency, including polyuria, and polydipsia (Robertson, 1995). Currently, more than 60 clinically relevant genetic variants of the AVP prohormone have been identified (Christensen et al., 2013). Returning to OT, then, variants of “OT deficient” animals have been created via genetic alterations of the OT gene or its receptor (Young et al., 1996; Bernatova et al., 2004; Lee et al., 2008; Nishimori et al., 2008): these animals display a range of behavioral abnormalities. As such, although no OT-related genetic syndrome akin to FNDI has yet been characterized in humans, the abovementioned findings raise the interesting question of the possibility of an “OT-deficiency syndrome” which manifests with deficits in the production and/or function of either the hormone, the receptor, or other components of the system (i.e., the ectoenzyme CD38).
How do Oxytocin's Therapeutically Relevant Effects Differ in Men and Women?
Given OT's intimate evolutionary involvement with reproductive function, it should not be surprising to find that is has distinct effects on the brains of males and females. Indeed, the histological structure for OT neurons is sexually dimorphic (de Vries, 2008) and sex biases in behavioral responses to OT have been frequently found in animal studies (Williams et al., 1994; Cho et al., 1999; Bales and Carter, 2003; Bales et al., 2007a). Estrogen increases OT and OTR production (Patisaul et al., 2003; Windle et al., 2006; Choleris et al., 2008), whereas testosterone promotes hypothalamic OTR-binding (Johnson et al., 1991) as well as production of AVP (Delville et al., 1996), which has many opponent actions to OT (Neumann and Landgraf, 2012).
Though the question of the meaning and measurement of OT levels is still subject to active study (see section “What is Oxytocin's Optimal Therapeutic Dosing Schedule?” below), men and women show differences in plasma OT levels (Ozsoy et al., 2009; Gordon et al., 2010; Holt-Lunstad et al., 2011; Weisman et al., 2012c), as well as sex-specific behavioral correlations with OT (Gordon et al., 2010; Zhong et al., 2012). In addition, amygdala-prefrontal cortical connectivity—which can be impacted by OT in normal subjects (Sripada et al., 2013) and anxiety patients (Labuschagne et al., 2011)—may be related in a gender-specific way to the development of anxiety and depressive disorders (Burghy et al., 2012). Furthermore, numerous studies in the growing OTR literature note sex-specific associations between genetic variants in the OTR gene and personality characteristics (Stankova et al., 2012), neural responses to emotionally salient cues (Tost et al., 2010), pair-bonding (Walum et al., 2012), hypothalamic gray matter volume (Tost et al., 2010), and empathy (Wu et al., 2012). On the other hand, several studies in this area have failed to find a sex bias (Rodrigues et al., 2009; Saphire-Bernstein et al., 2011; Feldman, 2012).
With regards to clinical studies of the effects of IN OT, a sex difference in its effects has been demonstrated in some single-dose studies (Hurlemann et al., 2010), including studies of OT's effects on the amygdala (Domes et al., 2010; Rupp et al., 2012), and interpersonal behavior (Liu et al., 2012). Again, these effects are variable: many other studies in this area have not found an effect of sex [see Bartz et al. (2011b), for review].
Focusing on the few multi-week clinical trials of OT in psychiatric populations, the three published clinical trials in schizophrenia included a disproportionate number of males (62 males treated vs. 13 females), consistent with most clinical trials of this disorder (Feifel et al., 2010, 2012a; Pedersen et al., 2011; Modabbernia et al., 2013). The number of women included in each trial was not sufficient to analyze for a sex-by-drug effect. Though schizophrenia is the clinical disorder with the largest number of separate randomized trials using IN OT, the first study to intimate a sex moderation effect of OT was a randomized, double-blind, within-subjects crossover study of OT (40 IU BID for 3 weeks) in patients with generalized anxiety disorder (GAD) (Feifel et al., 2011). This trial demonstrated a trend-level dose-by-gender effect such that males treated with OT showed a significant clinical improvement in HAM-A scores with OT, whereas females did not. En toto, the abovementioned sex differences indicate that delineation of the role of sex and sex hormones in the response to chronic OT treatment will be critical.
What is the Optimal Therapeutic Dose Range for Oxytocin?
Despite the groundswell of IN OT research, we know very little about either the optimal dose or dosing parameters of IN OT for any CNS indication, and single-dose studies, though informative, speak somewhat peripherally to this important issue. Noteworthy is that animal research indicates a discrepancy between the effects of both dose (Windle et al., 1997; Kramer et al., 2003; Bales et al., 2007b) and single vs. chronic dosing of OT (Bales and Perkeybile, 2012). Aside from optimizing therapeutic effect, dosing issues are also important in terms of side effects, given that OT has some cross-affinity with AVP receptors which mediate its potential diuretic and natriuretic effects (Gimpl and Fahrenholz, 2001), and have been noted in a single case report of high-dose IN OT (Ansseau et al., 1987). An illuminating primate study in this regard indicated that perhaps due to cross-reactivity with AVP, [which potentiates stress responses (Legros, 2001)], chronic higher-dose IN OT (200 IU) did not attenuate cortisol (ACTH) responses, whereas a lower-dose (50 IU) did (Parker et al., 2005).
Largely due to prior precedent (vs. pharmacological rationale), the majority of published human studies have tested OT in single doses in the 20–40 IU range (MacDonald et al., 2011a), though doses as low at 10 IU (Goldman et al., 2011) and as high as 160 IU daily (Epperson et al., 1996a,b) have been reported. The few chronic-dosing studies cited herein have used a dose range of 24–40 IU BID (Feifel et al., 2010, 2012a; Pedersen et al., 2011; Modabbernia et al., 2013). Importantly, very few studies have directly compared the effects of two or more doses, a standard strategy in dose-finding clinical trials.
In one the first clinical study to examine effects of multiple doses of OT in the same clinical subject, Goldman et al. demonstrated that in patients with schizophrenia, 10 IU caused a decrement in ability to identify facial emotions (due to increased false-response rate), whereas 20 IU improved emotion recognition in polydipsic relative to non-polydipsic patients (Goldman et al., 2011). This dose-related finding is interesting given that more placebo-controlled IN OT trials have been done in schizophrenia than any other indication. In a study of men with fragile X syndrome, 24 IU but not 48 IU IN OT improved eye gaze frequency, whereas 48 IU but not 24 IU decreased social-stress induced salivary cortisol levels (Hall et al., 2012). A study in normals on the effect of OT on exercise-induced increased in salivary cortisol levels demonstrated that 24 IU but not 48 IU attenuated this effect (Cardoso et al., 2012). Another related study in normal men found that IV OT (titrated to a level 10 times higher than physiologically normal baseline levels) demonstrated a linear, dose-response OT effect on ACTH and cortisol (Legros et al., 1984). Finally, a recent study documented that salivary OT levels in normal subjects remained similarly elevated for up to 7 h, regardless of which of 2 doses of IN OT patients received (16 or 24 IU) (van Ijzendoorn et al., 2012). In light of these findings suggesting that OT's effects may be dose-dependent, more studies are needed in which more than 1 dose—preferably a range of doses—are directly compared. Clearly, we need to understand whether a dose-response relationship exists regarding the effects of OT on core disease symptoms, and whether such dose-response relationships are disease specific.
What is Oxytocin's Optimal Therapeutic Dosing Schedule?
In addition to an inadequate understanding regarding the dose-response curve for most therapeutically relevant effects of OT, there is very little known regarding its optimal therapeutic-dosing frequency (e.g., once daily, twice daily, etc.). In addition to knowing the optimal dose range, dose frequency data is critical for successful design of future proof-of-concept clinical trials. Typically, dosing schedule is based upon the plasma half-life of the drug in question. However, this heuristic is likely not applicable to the CNS effects of OT, particularly OT delivered IN, given its putative direct access to the brain via this route of administration (Born et al., 2002). Specifically, whereas the plasma half-life of IV OT is less than 10 min (Mens et al., 1983), it lasts much longer in the CSF, and studies measuring plasma OT have found elevated levels of the peptide lasting more than 1 h after a single IN administration (Burri et al., 2008; Gossen et al., 2012).
Relevant here are single-dose studies measuring both salivary (Huffmeijer et al., 2012; Weisman et al., 2012a) and plasma OT levels (Burri et al., 2008; Andari et al., 2010; Domes et al., 2010; Gossen et al., 2012), which indicate that IN OT quickly elevates peripheral OT levels, and that, these levels remain above baseline for some time. In terms of mechanism, in addition to direct absorption via the nasal vasculature, it is thought that IN OT may also elevate peripheral OT levels by entering the brain and stimulating OT neurons to secrete endogenous OT from central stores into the peripheral circulation (Neumann et al., 1996) (Figure 1). This suggestion is supported by the fact that OT neurons operate in a feed-forward “bursting” mechanism, such that exogenous or endogenous OT stimulates further pulsatile OT release (Renaud et al., 1984; Rossoni et al., 2008), part of a locally regulated positive-feedback mechanism (Neumann et al., 1996). This ongoing release from endogenous OT stores, mediated partially through glutamatergic mechanisms (Jourdain et al., 1998; Israel et al., 2003), could certainly contribute to the sustained peripheral blood levels seen in IN OT studies. After IN delivery, peripheral OT levels are elevated starting between 10 (Andari et al., 2010) and 30 (Gossen et al., 2012) min and stay elevated for between 150 min (Gossen et al., 2012) to several hours (Burri et al., 2008; van Ijzendoorn et al., 2012), in spite of OTs short plasma half-life (Mens et al., 1983). Vitally, it is currently impossible to distinguish between transnasally absorbed exogenous OT and endogenously secreted OT, so the relative contribution of these two sources to subsequently measured OT levels is unknown. Moreover, though animal studies have demonstrated some concordance between intraneuronal levels of OT and peripheral OT levels (Wotjak et al., 1998; Cushing and Carter, 2000; Wigger and Neumann, 2002), and though several human studies show concordance between peripheral OT levels and naturalistic, centrally mediated behaviors (i.e., parenting, breastfeeding) (Feldman, 2012; Weisman et al., 2012b; for review), the concordance between OT measured in different body spaces (saliva, plasma, CSF) and central effects is still a matter of active debate (Carter et al., 2007; Neumann, 2007).
A variety of important controversies and questions surround the measurement of OT levels. Though space does not permit a full elaboration on the topic, its relevance to many of the questions in this review warrants a discussion. First, we note that there have been controversies regarding the validity and reliability of measurement of OT levels in urine (Anderson, 2006; Young and Anderson, 2010), saliva (Horvat-Gordon et al., 2005), and plasma (Szeto et al., 2011) [and see references in Carter et al. (2007); Szeto et al. (2011); Weisman et al. (2012c); Zhong et al. (2012)]. One aspect of this controversy regards the measurement of OT via immunoassay: the most cost-effective measurement technique for large samples. Notably, a recent report questioned both (1) the accuracy of both radioimmunoassays (RIA) and enzyme immunoassays (EIAs) and (2) the necessity of the technical step of sample extraction which can changes by up to 100-fold the measured levels of the peptide (Szeto et al., 2011). Most recent studies that measure either plasma or saliva OT levels use a commercial OT-Elisa kit (Assay-Design, MI, USA), which has been validated for linearity, cross reactivity, matrix effects, accuracy, precision, and recovery (Carter et al., 2007) though the use of extraction techniques in different studies is variable. In support of the validity of this assay, across a broad range of different studies—including several very large samples (Weisman et al., 2012c; Zhong et al., 2012)—these techniques have produced congruent results, with most of them finding reasonable correlations between saliva and plasma OT levels (Grewen et al., 2010; Feldman et al., 2011; Hoffman et al., 2012), and between OT levels and a wide range of OT-dependent biological processes (White-Traut et al., 2009; Grewen et al., 2010; Feldman, 2012).
A second, related issue surrounding the measurement and meaning of peripheral OT levels is that of endogenous fluctuations in OT levels. Though OT has a diurnal rhythm of daytime rise and night-time decline in mice (Zhang and Cai, 2011) and primates (Amico et al., 1990), the bulk of data does not support significant diurnal variations in plasma OT in humans (Amico et al., 1983; Kuwabara et al., 1987; Challinor et al., 1994; Kostoglou-Athanassiou et al., 1998; Turner et al., 2002; Graugaard-Jensen et al., 2008), [but see Forsling et al. (1998), Landgraf et al. (1982) for evidence of a nocturnal nadir]. CSF levels may differ, as there is some evidence for a diurnal variation in this body space (Amico et al., 1983; Kuboyama et al., 1988). These data on circadian fluctuations stand apart from studies of dynamic fluctuations in OT levels in states like pregnancy (Kuwabara et al., 1987; Fuchs et al., 1992; Lindow et al., 1996), breastfeeding (Jonas et al., 2009; Grewen et al., 2010), orgasm (Carmichael et al., 1987), parenting (Feldman, 2012), and certain stressors (Nussey et al., 1988; Sanders et al., 1990). Also related are documented increases in peripheral OT levels due to both natural variations in estrogen levels (Mitchell et al., 1981; Shukovski et al., 1989) and the ingestion of exogenous estrogen which is known to increase the magnocellular release of OT (Wang et al., 1995) and plasma OT levels (Amico et al., 1981; Silber et al., 1987; Uvnas-Moberg et al., 1989; Michopoulos et al., 2011). Adding complexity is that data on fluctuations in plasma OT levels across the menstrual cycle are mixed, with studies in different healthy and clinical populations showing both variation (Shukovski et al., 1989; Salonia et al., 2005; Liedman et al., 2008) and lack of variation (Stock et al., 1991; Kostoglou-Athanassiou et al., 1998; Light et al., 2005) in normally cycling women, with both estrogen and progesterone levels playing a role.
A third, yoked pair of topics related to OT levels are (1) the correlations between peripheral and central OT levels (see discussion above) and (2) the correlation of OT levels and different disease states. Regarding the latter, investigators have studied OT levels and their relationship to aspects of autism (Modahl et al., 1998; Al-Ayadhi, 2005), eating disorders (Hoffman et al., 2012; Lawson et al., 2012), post-traumatic stress disorder (Seng et al., 2013), schizophrenia (Goldman et al., 2008; Keri et al., 2009; Rubin et al., 2011), social anxiety disorder (Hoge et al., 2008, 2012) and depression (Scantamburlo et al., 2007; Parker et al., 2010). An important but uninvestigated clinical question is whether a transient or chronic increase of peripheral OT levels via treatment with IN OT (Andari et al., 2010; Gossen et al., 2012) correlates with OT-responsive clinical symptoms or treatment-related symptomatic improvement (e.g., whether OT levels may function as a biomarker).
Returning, then, to the clinical issue of OT dose and frequency: though the abovementioned studies of OT levels,—including post-dose OT levels—are somewhat informative regarding the task of determining an optimal OT dose and frequency, to date, there have been no published studies examining the time course of the brain-mediated effects of IN OT, nor of their correlation with peripheral OT levels. Such studies would greatly enhance our ability to optimize OT dose frequency for therapeutic ends. In fact, most studies of IN OT in humans examine its effects at a single time point, typically 30–60 min after administration. For these reasons, in addition to studies examining a range of doses of IN OT, studies examining OT's brain effects over a range of time points are needed to inform optimal OT treatment design.
Can Native Oxytocin be Improved Upon?
Though the native nonapeptide OT has significant advantages in terms of therapeutic modulation of the central OT system, there are problems with peptides. Specifically, neuropeptides like OT lack many “drug-like” properties, especially as regards CNS indications. Though they have certain advantages over other chemical medicinal classes (i.e., evolved specificity for unique functions and receptors, limited drug–drug interactions, little accumulation in tissues, few side-effects), neuropeptides also carry unique liabilities as medications related to their molecular nature (Manning et al., 2012; McGonigle, 2012). These shortcomings include a brief plasma half-life and poor oral bioavailability due to their degradation by plasma and gastric proteases, as well as limited penetrance of the blood-brain barrier due to their large size and hydrophilic nature (McGonigle, 2012).
Technological advances in medicinal chemistry, however, are providing specific solutions to these challenges. In a few cases, medicinal chemists have managed to design small non-peptidergic molecules that bind a specific peptide receptor and are either inactive at that receptor (antagonist) or mimic the actions of the endogenous peptide (agonist). In general this strategy has been much more successful in producing antagonists than agonists (Manning et al., 2012). However, the corpus of preclinical and translation research with OT suggests that it is OT agonists, not antagonists, that have promise as treatments for several psychiatric disorders. With regards to OT, several low-molecular weight non-peptidergic OT agonists have been developed that penetrate the brain after peripheral administration (Pitt et al., 2004; Ring et al., 2010). The only non-peptide OT agonist with experimental evidence for an OT-like behavioral profile is WAY-277464, which had 87% of the binding affinity of OT and significant greater selectivity for the OTR (Ring et al., 2010). Though it exhibited an OT-like anxiolytic behavioral and physiological profile in several animal tests (four-plate test, elevated zero maze, stress-induced hyperthermia), and also an OT-like preclinical antipsychotic profile [reversing amphetamine- and MK-801-induced disruption of prepulse inhibition (PPI)], it did not have an OT-like antidepressant profile [no reduced immobility in the tail suspension test (TST)] (Ring et al., 2010). An interesting corollary finding in this study—one that speaks to the mechanism of action of OT's antidepressant-like effects in the TST—was that a selective OTR antagonist failed to block these antidepressant effects, indicating WAY-27744's effect may be mediated through a different receptor system (i.e., AVPR: also infra vida section “Is Intranasal Delivery of Oxytocin the Optimal Route?”) (Ring et al., 2010) and raising similar questions for OT. In any case, as a result of both pharmacological and market factors, development of WAY-277464 was not pursued by Wyeth (Manning et al., 2012).
Because of the difficulty of developing a non-peptide OT agonist, medicinal chemists have utilized another approach to the problem of stimulating the OT system: chemical modification of the native peptide or an active fragment to increase its resistance to enzymatic degradation and increase metabolic stability. This process has produced carbetocin: an uterotonic OT analog with a peripheral half-life of 85–100 min, significantly longer than OT's (Hunter et al., 1992). Carbetocin—produced by Ferring Pharmaceuticals—is approved in 23 countries outside the United States for post-partum hemorrhage, but there are no published studies investigating its CNS effects in humans. Though a potent uterotonic agent, carbetocin has about 10-fold lower affinity for the OTR than OT (Engstrom et al., 1998; Gimpl et al., 2005), and has been shown to lack anxiolytic efficacy (elevated plus maze) when delivered peripherally, vs. OT, which has anxiolytic efficacy when delivered peripherally (McCarthy et al., 1996; Ring et al., 2006). In another experiment, peripheral carbetocin failed to produce antipsychotic-like effects on PPI (Feifel et al., 2012b). Interestingly, carbetocin did demonstrate an antidepressant-like profile in the forced swim test when administered peripherally and centrally (Chaviaras et al., 2010), and does have short-term anxiolytic effects when delivered centrally (Mak et al., 2012). It would be very instructive to determine carbetocin's effects on centrally mediated processes, especially on clinical symptoms of pyschiatric disorders in humans.
Besides biochemical modifications of the molecules themselves, alternative drug delivery systems (i.e., patches, microspheres, liposomes) can also improve the pharmacokinetic profile of peptides and represent another approach to addressing the challenges of therapeutic modulation of endogenous peptide systems (Patil and Sawant, 2008; Manning et al., 2012; McGonigle, 2012). As an example, a mucoadhesive buccal OT patch has been tested in animals and was able to deliver OT continuously over 3 h (Li et al., 1997). Notably, the efficacy of this or other alternative OT delivery systems on centrally mediated processes has not yet been tested in human or animal studies.
Aside from the delivery of OT or non-peptide OT analogs to the CNS, there are several other ways to impact the central OT system [reviewed in Modi and Young (2012)]. These include inhibition of the non-specific enzyme that degrades OT in the CSF [aminopeptidase placental-leucineaminopeptidase (P-LAP) (Chai et al., 2008; Albiston et al., 2011)] and the use of drugs that may stimulate OT release from endogenous stores via the serotonergic (Jorgensen et al., 2003a) and melanocortin receptors (Sabatier, 2006) found on OT neurons. Drugs like the serotonin 1a agonist buspirone (Bagdy and Kalogeras, 1993; Jorgensen et al., 2003b), 3,4 methlyenedioxymethamphetamine (MDMA) or ecstasy (Thompson et al., 2007; Dumont et al., 2009; Broadbear et al., 2011), and the uniquely effective antipsychotic clozaril (MacDonald and Feifel, 2012a), have been proposed to exert some of their pharmacological activity via stimulation of the central OT system.
Is Intranasal Delivery of Oxytocin the Optimal Route?
As mentioned above, a prominent pharmacokinetic issue with synthetic OT involves getting this relatively large, hydrophilic molecule into the brain, given its poor penetration of the blood-brain barrier (McEwen, 2004). IN application of peptidergic drugs to the CNS has been proven since 1989 (Frey, 1991), and is a delivery system increasingly utilized for a variety of drugs for a range of putative central indications, including memory (Benedict et al., 2007) and multiple sclerosis (Ross et al., 2004). Delivering a peptide IN capitalizes first on the heavily vascularized nasal mucosa, which drains through both fenestrated epithelium and via several facial veins (facial and sphenopalatine), into the peripheral circulation, circumventing first pass metabolism (Zhu et al., 2012). In this way, IN delivery simulates IV delivery: drugs delivered IN may reach the brain via active transport or diffusion from the blood compartment across the blood-CSF or blood-brain barrier (Thorne and Frey, 2001; Morimoto et al., 2009). Beyond this, a direct-to-the-brain path of entry after IN delivery of peptides and other drugs has been proposed via two possible mechanisms (Figure 1): (1) intraneuronal active uptake along the olfactory or trigeminal nerve into the brain; and (2) extraneuronal passive diffusion into the CSF through perineural clefts in the nasal epithelium which provide a gap in BBB (Illum, 2004; Thorne et al., 2004; Renner et al., 2012a,b) and (Dhuria et al., 2010; Chapman et al., 2012; Zhu et al., 2012 for references and details).
In point of fact, direct-to-the-brain delivery of IN OT was extrapolated from studies with OT's sister nanopeptide vasopressin, which differs from OT by the substitution of two amino acids (Riekkinen et al., 1987; Born et al., 2002). These studies found that IN delivery increased both CSF levels and plasma levels of AVP (Riekkinen et al., 1987; Born et al., 2002). Indirect support for this hypothesis comes from findings that both vasopressin and OT administered IN have central effects [Fehm et al. (2000) and references therein]. Though contemporary critiques have raised important questions about the details of whether nasal OT gets directly into the brain, and if so, how (Churchland and Winkielman, 2012), support for the direct-to-brain notion comes from recent studies in primates that found that IN OT doubles CSF OT levels within 35 min (Chang et al., 2012), strong evidence of central penetration, given that extant evidence supports that endogenous OT in the CSF derives from central not peripheral sources (McEwen, 2004). Further support comes from recent, unpublished rodent data indicating that IN OT elevates OT levels in the extracellular fluid in the hippocampus and amygdala (Rainer Landgraf, pers. communication). Studies with other IN-delivered peptides showing perineuronal transport are also of interest in this regard (Chen et al., 1998; Renner et al., 2012b; Zhu et al., 2012). On the other hand, increased central levels of OT after IN administration can occur indirectly via elevated levels of OT in the peripheral circulation, thus the evidence described above does not represent definitive evidence of a direct nose-to-brain mechanism, nor whether IN-administered OT has advantages over peripheral or even orally administered OT in terms of brain penetration or reduced peripheral side effects. Indeed, at least one study examining this issue concluded that Devunetide (an 8-amino acid peptide) administered IN to rats entered the brain via the peripheral blood system (Morimoto et al., 2009). As such, given potential disadvantages of IN delivery (i.e., reliance on patients for consistent dose delivery), the issue of whether the IN route or another route is optimal for OT-targeted therapeutics is still an open question.
What is the Role of Vasopressin Receptors in Oxytocin's Effects?
Due to their close evolutionary relationship, the pharmacological story of the OT system is interleaved with that of its “sister” hormone AVP. Pivotally, though they have evolved to serve very different functions, these two neuropeptides differ by only 2 amino acids (Gimpl and Fahrenholz, 2001). In terms of receptors, 4 G-protein-coupled receptors have been identified that bind these peptides in both humans and rodents: AVPR1a, AVPR1b, AVPR2, and OTR (Gimpl and Fahrenholz, 2001; Grimmelikhuijzen and Hauser, 2012). Of these receptors, AVPR1a is the most abundantly expressed in the brain, whereas AVPR1b has more limited brain expression and AVPR2 exists almost exclusively in the periphery (Stoop, 2012). Pertaining to OT-directed therapeutics, it is important to note that OT is relatively selective, binding to AVP receptors with ~1% the affinity it binds to OTRs, whereas AVP is non-selective, binding with similar affinity to both OTR and AVPRs (Lowbridge et al., 1977; Mouillac et al., 1995; Manning et al., 2012). Also important is that population-level genetic studies of OT and AVPR1a receptors in humans indicate that both systems appear to be responsible for important behavioral phenotypes in humans (Prichard et al., 2007; Walum et al., 2008, 2012; Levin et al., 2009; Meyer-Lindenberg et al., 2009; Kumsta and Heinrichs, 2013). In terms of conceptualizing their role in mammalian behavior, it appears that differences in receptor co-expression and region-specific density—parameters which vary meaningfully in rodents (see Veinante and Freund-Mercier, 1995; Huber et al., 2005; Young et al., 2006; Raggenbass, 2008), and humans (Loup et al., 1991)—influence the different roles of these two nonapeptides in the CNS, and many models suggest these related peptides have somewhat opposing roles in terms of anxiety and behavioral measures of coping (Legros, 2001; Neumann and Landgraf, 2012). Notwithstanding this larger framework, evidence also exists suggesting some of OTs activities, including both its putative central therapeutic effects (Schorscher-Petcu et al., 2010; Sala et al., 2011), as well as some of its potential side effects [i.e., hyponatremia (Seifer et al., 1985; Stratton et al., 1995)] may be mediated by binding to AVP receptors (Liggins, 1963; Li et al., 2008).
A number of specific agonists and antagonists for each of the four different nonapeptide receptors have been developed (Manning et al., 2012), and have allowed more precise delineation of the role of the different receptors in the activities of each of these nonapeptides. Specifically, whereas OT (but not AVP) normalizes deficits in OT and CD38 knockout mice (Ferguson et al., 2000; Jin et al., 2007), both OT and AVP normalize defects in OT-receptor knockout mice, which demonstrate an autism-like profile (deficits in social behavior, seizures) (Sala et al., 2013). This latter experiment indicated that ICV delivery of both OT and AVP reduced some of these autism-like difficulties via the AVPV1a receptor (Sala et al., 2013). A second, related experiment found that some of OTs analgesic activity in mice is related to AVPV1a, as demonstrated by OT's lack of analgesic activity in AVPR1a knockout mice and the ability of an AVPR1a receptor antagonist to block the effect (Schorscher-Petcu et al., 2010).
With regard to any putative therapeutic effect of OT, OT-mediated activation of AVP receptors may have four potential consequences on OT's behavioral effects: potentiation, mediation, attenuation, or no impact. The same holds true for any possible side effects of OT. Elucidating the role AVP receptor activation plays in each of OT's putative therapeutic effects is therefore important in order to inform development of drugs to optimally target central OT/AVP systems. Knowledge gained from this effort would determine whether energy should be directed toward developing OT agonists with greater selectivity for OTR than OT itself, or toward compounds with more balanced affinity for OTR and one or more AVP receptor types. Highlighting this issue, a recent animal study revealed that AVP1a activation (via desmopressin, an AVP receptor agonist) and blockade (via atosiban, an OT/AVP1a receptor antagonist) produced anxiogenic and anxiolytic effect, respectively (Mak et al., 2012). Based on this, an OTR agonist with no cross-affinity for AVPR1a would be expected to have superior anxiolytic efficacy and a compound that acted as a dual OTR agonist/AVPR1a antagonist might have even greater efficacy. Also worth mention here is a very recent human trial of the vasopressin V1b receptor antagonist SSR149415, which showed negligible anxiolytic effects, and antidepressant effects that warrant further study (Griebel et al., 2012).
Monotherapy vs. Augmentation: Can Oxytocin Treat on its Own or is it Better Suited to Augment other Established Treatments?
The discovery of the molecular mechanisms wherein experience becomes written in the nervous system (Kandel and Squire, 1999), and the growing understanding that OT's effects vary significantly based on context (Bartz et al., 2011b), opens the possibility for pharmacological augmentation of learning-based treatments, including computer-based cognitive training programs (i.e., Vinogradov et al., 2012) and many forms of psychotherapy (see Choi et al., 2010). Several medications have already been examined in this capacity, including the NMDA partial agonist d-cycloserine (DCS) (Otto et al., 2010), the NMDA receptor antagonist ketamine, (Krupitsky et al., 2002), the beta-blocker propranolol (Kindt et al., 2009), and the serotonergic-enhancer MDMA (“ecstasy”), an amphetamine-related CNS stimulant which may exert some of its effects via the OT system (Parrott, 2007; Dumont et al., 2009). Evidence that OT enhances neurogenesis (Leuner et al., 2012), the beneficial effects of social support (Heinrichs et al., 2003), social salience and social memory (Hurlemann et al., 2010; Guastella and MacLeod, 2012) also suggest that OT is a good candidate for such “augmentation” trials. In the case of schizophrenia, for example, it may be valuable to examine OTs effects when given in conjunction with cognitive enhancement therapies already demonstrated to have benefit (Chou et al., 2012; Twamley et al., 2012). Aside from OT's ability to augment learning-based treatments, we note that OT's benefits in schizophrenia have all been when it is given in conjunction with established antipsychotics (i.e., a pharmacological “augmentation” strategy) (Feifel et al., 2010, 2012a; Pedersen et al., 2011; Modabbernia et al., 2013) and that the role of OT as a primary antipsychotic needs investigation.
More speculative, but related, is “OT therapy by proxy” wherein OT given to an adult in a social context (i.e., parent and child) may cause OT-driven changes in the child without direct drug administration to this sensitive population (Naber et al., 2010; Weisman et al., 2012b). Current studies of OT's “augmentation” effect in patients have been limited to one single-dose study and have demonstrated limited success on primary clinical endpoints (Guastella et al., 2009).
Are there Identifiable Biomarkers for Oxytocin's Therapeutic Effects?
Biomarkers are objectively measured characteristics that relate to the cause, clinical course, and treatment of illness (Frank and Hargreaves, 2003). In drug development, biomarkers optimize the efficiency of clinical drug studies by facilitating the detection of early signals of drug response (Wiedemann, 2011). Properties of an optimal pharmacological biomarker include: high sensitivity and specificity for clinical outcomes; relatively inexpensive, and low-risk; interpretable by clinicians in many different practice locations; a dose-response relationship; and a plausible link with pharmacology and pathogenesis (Dumont et al., 2005; Wiedemann, 2011; Baskaran et al., 2012; Leuchter et al., 2012). For a variety of reasons—including a chasm between our understanding of the short-term neurobiological effects of drugs and later clinical improvements—these characteristics are of special import in neuropsychiatry research (Wiedemann, 2011). In this field, specifically, a wide variety of biomarkers relevant to OT are in different stages of utilization and development, including laboratory markers (serum markers, genetic tests), electrophysiological markers (EEG, MEG, facial EMG, GSR), brain imaging techniques (fMRI, PET), and behavioral measures (challenge tests, cue exposure tasks, PPI, fear-potentiated startle) (Wiedemann, 2011). Most of these putative biomarkers have been utilized in conjunction with OT, and this is one arena where single-dose OT studies have been invaluable in terms of OT's development as a pharmaceutical. Though there has been relatively little clinically oriented biomarker research with OT (i.e., correlation of a biomarker with meaningful clinical syndromes or outcomes), the extant OT literature contains several promising candidates: heart-rate variability (HRV) (Kemp et al., 2012), skin conductance (GSR) (de Oliveira et al., 2012b), stressed cortisol responses (Ditzen et al., 2009; Quirin et al., 2011; Simeon et al., 2011; Cardoso et al., 2012; Linnen et al., 2012), facial affect recognition (Fu et al., 2007, 2008; Harmer et al., 2009a,b), pupil responses (Leknes et al., 2012), EEG measures (Perry et al., 2010), MEG (Hirosawa et al., 2012), and a variety of functional imaging (fMRI) parameters, including alteration of default-mode network (Sripada et al., 2013), responses to naturalistic social stimuli (Riem et al., 2011, 2012), and stress-induced amygdala responsivity and connectivity patterns (Labuschagne et al., 2010, 2011; Zink and Meyer-Lindenberg, 2012). Regarding functional imaging biomarkers, these techniques would be greatly aided by technical advances, especially a radionucleotide for OTRs. The development of such a tracer—invaluable in the study of clinically relevant aspects of central dopaminergic (Seeman and Tallerico, 1998; Volkow et al., 2001) and opiatergic systems (Greenwald et al., 2003; Mitchell et al., 2012)—would aid in characterizing the relationship between central OTR density, clinical phenotypes, and treatment with OT. In vivo visualization of the human OTR would be particularly fascinating given that (1) in animal species, OTR distribution is a significant determinant of behavior (Hammock and Young, 2006; Ross et al., 2009; Ophir et al., 2012); and (2) OTR density appears to vary dynamically during phases of life (Bale et al., 2001; Meddle et al., 2007). As well, functional imaging studies demonstrating the cortical effects of IN OT (Figure 1) and (Bethlehem et al., 2012) are vital additions to translational OT research, given the significant variability of cortical organization among different species, including those most frequently used in OT research (Preuss, 2000). Though a few studies have examined post-mortem OTR binding in the human CNS (using the same radiolabeled peptide as in rodents) (Loup et al., 1989, 1991), synthesis of small-molecule radioligands for the OTR (Smith et al., 2012), would greatly aid our understanding of the functional role of the OT system in human brain disorders and treatment.
To advance the therapeutic potential of OT, the abovementioned biomarkers need to be refined and applied to clinically ill patients. These studies would clarify several basic pharmacodynamics and pharmacokinetic questions surrounding OT (infra supra), and—most importantly—could be used to predict therapeutic response. Vitally, biomarker-guided clinical trials may optimize the efficiency of future clinical trials, facilitating the optimal use of a shrinking pool of funding for OT research (driven in part by OT's lack of patent exclusivity).
From Dearth to Birth, and Preclinical to Clinical Research—Helping Oxytocin Deliver
We believe the above review supports two broad conclusions about OT as potential therapeutic agent for CNS disease. First, the last decade of translational and clinical research has provided a great deal of reason to be cautiously optimistic that OT-based treatments may be developed to help ease the dearth in novel treatments for psychiatric illness. Secondly, and somewhat in contrast, the translation of OT's therapeutic promise has been remarkably slow, considering clinical studies with OT are not hindered by the typical limitations imposed by non-approved investigational drugs (i.e., costly animal and human safety and toxicity testing before testing in proof-of-concept human trials). As discussed above, single-dose studies in normal subjects—and a much-smaller set of single-dose studies in clinical populations (Table 1)—has left the field pregnant with anticipation about OT's potential therapeutic utility. In our opinion, however, direct tests of this utility are now past due. We need to help OT deliver.
The fact that there are only a few published small, multi-week clinical trials of OT is problematic. More single-dose studies—overwhelmingly in normal subjects—continue to be generated. Some of these add to the body of support for therapeutic effects, while others do the opposite, revealing a more complex role for OT in human behavior, emotion, and cognition (De Dreu et al., 2010, 2011). These complexity-revealing findings in particular have spurred some investigators to suggest that it is premature to speculate about OT's therapeutic potential for neuropsychiatric disorders and opine that before we do clinical trials, the field needs more translational studies to elucidate OT's complex role (e.g., Grillon et al., 2012; Miller, 2013).
As active clinicians and translational researchers, we recognize the value of translational research. Faced daily with individuals and families who have profound and often urgent need for better treatments, however, we also recognize its limitations. While we agree that additional preclinical OT research in animals and humans is vital, we do not believe these trials should be done at the expense of randomized controlled trials in clinical populations. Instead, a stepwise, tandem progression is optimal. Translational research works best in a bi-directional mode, with preclinical studies informing clinical trials and the results of clinical trials—in turn—helping identify which preclinical paradigms have the best predictive validity for a specific disorder and drug class. In this way, translational paradigms can be further leveraged to conduct impactful preclinical research. Animal studies have the ability to efficiently deliver clinically relevant information without the expense, time, and risk-considerations inherent in human trials. Similarly, preclinical human studies in which acute effects of drugs, like OT, are examined on symptom proxies such as functional imaging changes in clinically relevant circuits or laboratory analogs of pathological conditions (e.g., CCK-induced panic) are much easier, less expensive, and less risky than classic randomized clinical trials. However, at present, the predictive validity of both these forms of preclinical drug research with regard to psychiatric disorders is far from perfect. Examples abound in which efficacy or deleterious effects noted in clinical trials were not manifested in preclinical studies and vice versa. Though clearly we support translational research and the ongoing search for reliable biomarkers of clinical response in psychiatric illness, the simple facts are: rodents are not humans, and changes in functional imaging or laboratory tasks are not the same as changes in clinical symptoms.
As one can see in the abovementioned review, there is currently reasonably good evidence from animal and human preclinical trials that OT may have therapeutic benefit in at least three brain-based conditions: schizophrenia, anxiety, and autism. For the myriad reasons discussed above, we believe that additional preclinical studies will not—by themselves—answer the critical therapeutic questions that face clinicians in the field. Therefore, proof-of-concept clinical trials are warranted. As a point of comparison, phase II studies—the kind that are now needed to directly test the various hypotheses regarding OT's therapeutic utility—have been carried out by industry on investigational agents with far less data supporting their efficacy and safety. These facts, together with (1) the profound impairments imposed by brain-based disease; (2) the often-inadequate efficacy of extant treatment; and (3) a disheartening lack of promising, novel treatments in the pipeline, we believe, justifies cautious execution of clinical trials with OT in the abovementioned conditions. In this regard it is noteworthy that—after a long period of gestation—the water seems to have broken on research directly testing OT's clinical promise: several potentially seminal clinical studies of OT appear be underway in several top “target” disorders (www.clinicaltrials.gov).
However, OT will not be developed into a drug for any psychiatric indication with one, two, or even three investigator-initiated clinical trials. These initial trials often produce negative or weak therapeutic effects. Thus, concurrent preclinical studies—both animal and human—are needed to advance the therapeutic development of OT. These additional preclinical studies are needed to inform the design of the inevitable second wave of clinical trials in the current “big three” indications, as well as initial proof-of-concept trials in other emerging candidate OT-responsive disorders. For example, preclinical studies demonstrating dichotomous dose-dependent effects on clinically relevant measures may prompt research emphasis on wider dose ranging in phase-II studies of OT. Likewise, animal studies showing tolerance of clinically relevant preclinical effects after a certain duration of treatment may prompt longer-duration trials to evaluate this effect in humans. Similarly, evidence of adverse effect in animals emerging at certain doses or durations may prompt incorporation of specific safety monitoring features into OT clinical trials. This latter point is particularly important given that randomized controlled trials are costly and labor-intensive, and that negative results due to type-II errors (i.e., missing a significant therapeutic effect) for example, by infelicitous selection of dose(s) or dosing frequency can be devastating to future studies.
To this end, there is a definitely a need for more translational research using animal models with validity—particularly predictive validity—for the specific conditions for which OT is a candidate treatment (e.g., autism, schizophrenia, anxiety, etc.). Such studies will help address the vital questions we have delineated in this paper. These animal studies should be complemented by translational human studies using single doses and non-symptom outcomes. Knowledge derived from both of these approaches will increase the likelihood of success of critical clinical trials. As mentioned above, the third element in the bench-to-beside arc are clinical trials using OT, which can reciprocally provide useful information to evaluate the predictive validity of various animal models and proxy-symptom human paradigms.
In light of these facts, and in light of the prodigious amount of animal and human OT research, it is surprising how little effort has been specifically directed to address the translational questions delineated above. For example, despite good evidence from preliminary clinical trials that OT has therapeutic benefit in schizophrenia (MacDonald and Feifel, 2012a), at the time of this writing, only three published studies have explored exogenous OT's effects in animals models with predictive relevance specifically for schizophrenia (Feifel and Reza, 1999; Lee et al., 2005; Feifel et al., 2012b). In order to help OT deliver on its therapeutic promise, there remains much work across the entire translational spectrum.
Conflict of Interest Statement
David Feifel is a named inventor on a method of use patent for oxytocin submitted by UCSD. K. MacDonald declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Thanks to Maribel Santos for help with the illustration and Tina, Kainoa, and Mataio MacDonald for editorial assistance.
References
Al-Ayadhi, L. Y. (2005). Altered oxytocin and vasopressin levels in autistic children in Central Saudi Arabia. Neurosciences 10, 47–50.
Albiston, A. L., Diwakarla, S., Fernando, R. N., Mountford, S. J., Yeatman, H. R., Morgan, B., et al. (2011). Identification and development of specific inhibitors for insulin-regulated aminopeptidase as a new class of cognitive enhancers. Br. J. Pharmacol. 164, 37–47.
Amico, J. A., Challinor, S. M., and Cameron, J. L. (1990). Pattern of oxytocin concentrations in the plasma and cerebrospinal fluid of lactating rhesus monkeys (Macaca mulatta): evidence for functionally independent oxytocinergic pathways in primates. J. Clin. Endocrinol. Metab. 71, 1531–1535.
Amico, J. A., Seif, S. M., and Robinson, A. G. (1981). Oxytocin in human plasma: correlation with neurophysin and stimulation with estrogen. J. Clin. Endocrinol. Metab. 52, 988–993.
Amico, J. A., Tenicela, R., Johnston, J., and Robinson, A. G. (1983). A time-dependent peak of oxytocin exists in cerebrospinal fluid but not in plasma of humans. J. Clin. Endocrinol. Metab. 57, 947–951.
Anagnostou, E., Soorya, L., Chaplin, W., Bartz, J., Halpern, D., Wasserman, S., et al. (2012). Intranasal oxytocin versus placebo in the treatment of adults with autism spectrum disorders: a randomized controlled trial. Mol. Autism 3:16. doi: 10.1186/2040-2392-3-16
Andari, E., Duhamel, J. R., Zalla, T., Herbrecht, E., Leboyer, M., and Sirigu, A. (2010). Promoting social behavior with oxytocin in high-functioning autism spectrum disorders. Proc. Natl. Acad. Sci. U.S.A. 107, 4389–4394.
Anderson, G. M. (2006). Report of altered urinary oxytocin and AVP excretion in neglected orphans should be reconsidered. J. Autism Dev. Disord. 36, 829–830.
Ansseau, M., Legros, J. J., Mormont, C., Cerfontaine, J. L., Papart, P., Geenen, V., et al. (1987). Intranasal oxytocin in obsessive-compulsive disorder. Psychoneuroendocrinology 12, 231–236.
Averbeck, B. B., Bobin, T., Evans, S., and Shergill, S. S. (2011). Emotion recognition and oxytocin in patients with schizophrenia. Psychol. Med. 1–8. doi: 10.1017/S0033291711001413
Bagdy, G., and Kalogeras, K. T. (1993). Stimulation of 5-HT1A and 5-HT2/5-HT1C receptors induce oxytocin release in the male rat. Brain Res. 611, 330–332.
Bailey, J. E., Kendrick, A., Diaper, A., Potokar, J. P., and Nutt, D. J. (2007). A validation of the 7.5% CO2 model of GAD using paroxetine and lorazepam in healthy volunteers. J. Psychopharmacol. 21, 42–49.
Bakharev, V. D., Tikhomirov, S. M., and Lozhkina, T. K. (1984). Psychotropic properties of oxytocin. Probl. Endokrinol. (Mosk) 30, 37–41.
Bale, T. L., Davis, A. M., Auger, A. P., Dorsa, D. M., and McCarthy, M. M. (2001). CNS region-specific oxytocin receptor expression: importance in regulation of anxiety and sex behavior. J. Neurosci. 21, 2546–2552.
Bales, K. L., and Carter, C. S. (2003). Sex differences and developmental effects of oxytocin on aggression and social behavior in prairie voles (Microtus ochrogaster). Horm. Behav. 44, 178–184.
Bales, K. L., and Perkeybile, A. M. (2012). Developmental experiences and the oxytocin receptor system. Horm. Behav. 61, 313–319.
Bales, K. L., Plotsky, P. M., Young, L. J., Lim, M. M., Grotte, N., Ferrer, E., et al. (2007a). Neonatal oxytocin manipulations have long-lasting, sexually dimorphic effects on vasopressin receptors. Neuroscience 144, 38–45.
Bales, K. L., van Westerhuyzen, J. A., Lewis-Reese, A. D., Grotte, N. D., Lanter, J. A., and Carter, C. S. (2007b). Oxytocin has dose-dependent developmental effects on pair-bonding and alloparental care in female prairie voles. Horm. Behav. 52, 274–279.
Bartz, J., Simeon, D., Hamilton, H., Kim, S., Crystal, S., Braun, A., et al. (2011a). Oxytocin can hinder trust and cooperation in borderline personality disorder. Soc. Cogn. Affect. Neurosci. 6, 556–563.
Bartz, J. A., Zaki, J., Bolger, N., and Ochsner, K. N. (2011b). Social effects of oxytocin in humans: context and person matter. Trends Cogn. Sci. 15, 301–309.
Baskaran, A., Milev, R., and McIntyre, R. S. (2012). The neurobiology of the EEG biomarker as a predictor of treatment response in depression. Neuropharmacology 63, 507–513.
Benedict, C., Hallschmid, M., Schmitz, K., Schultes, B., Ratter, F., Fehm, H. L., et al. (2007). Intranasal insulin improves memory in humans: superiority of insulin aspart. Neuropsychopharmacology 32, 239–243.
Bergeron, C., Kovacs, K., Ezrin, C., and Mizzen, C. (1991). Hereditary diabetes insipidus: an immunohistochemical study of the hypothalamus and pituitary gland. Acta Neuropathol. 81, 345–348.
Bernatova, I., Rigatto, K. V., Key, M. P., and Morris, M. (2004). Stress-induced pressor and corticosterone responses in oxytocin-deficient mice. Exp. Physiol. 89, 549–557.
Bethlehem, R. A. I., van Honk, J., Auyeung, B., and Baron-Cohen, S. (2012). Oxytocin, brain physiology, and functional connectivity: a review of intranasal oxytocin fMRI studies. Psychoneuroendocrinology. Available online at: http://dx.doi.org/10.1016/j.psyneuen.2012.10.011
Birkett, M. A., Shinday, N. M., Kessler, E. J., Meyer, J. S., Ritchie, S., and Rowlett, J. K. (2011). Acute anxiogenic-like effects of selective serotonin reuptake inhibitors are attenuated by the benzodiazepine diazepam in BALB/c mice. Pharmacol. Biochem. Behav. 98, 544–551.
Born, J., Lange, T., Kern, W., McGregor, G. P., Bickel, U., and Fehm, H. L. (2002). Sniffing neuropeptides: a transnasal approach to the human brain. Nat. Neurosci. 5, 514–516.
Bowen, M. T., Carson, D. S., Spiro, A., Arnold, J. C., and McGregor, I. S. (2011). Adolescent oxytocin exposure causes persistent reductions in anxiety and alcohol consumption and enhances sociability in rats. PLoS ONE 6:e27237. doi: 10.1371/journal.pone.0027237
Broadbear, J. H., Tunstall, B., and Beringer, K. (2011). Examining the role of oxytocin in the interoceptive effects of 3, 4-methylenedioxymethamphetamine (MDMA, ‘ecstasy’) using a drug discrimination paradigm in the rat. Addict. Biol. 16, 202–214.
Burghy, C. A., Stodola, D. E., Ruttle, P. L., Molloy, E. K., Armstrong, J. M., Oler, J. A., et al. (2012). Developmental pathways to amygdala-prefrontal function and internalizing symptoms in adolescence. Nat. Neurosci. 15, 1736–1741.
Burri, A., Heinrichs, M., Schedlowski, M., and Kruger, T. H. (2008). The acute effects of intranasal oxytocin administration on endocrine and sexual function in males. Psychoneuroendocrinology 33, 591–600.
Cardoso, C., Ellenbogen, M. A., and Linnen, A. M. (2012). Acute intranasal oxytocin improves positive self-perceptions of personality. Psychopharmacology 220, 741–749.
Carmichael, M. S., Humbert, R., Dixen, J., Palmisano, G., Greenleaf, W., and Davidson, J. M. (1987). Plasma oxytocin increases in the human sexual response. J. Clin. Endocrinol. Metab. 64, 27–31.
Carter, C. S., Pournajafi-Nazarloo, H., Kramer, K. M., Ziegler, T. E., White-Traut, R., Bello, D., et al. (2007). Oxytocin: behavioral associations and potential as a salivary biomarker. Ann. N.Y. Acad. Sci. 1098, 312–322.
Chai, S. Y., Yeatman, H. R., Parker, M. W., Ascher, D. B., Thompson, P. E., Mulvey, H. T., et al. (2008). Development of cognitive enhancers based on inhibition of insulin-regulated aminopeptidase. BMC Neurosci. 9(Suppl. 2):S14. doi: 10.1186/1471-2202-9-S2-S14
Challinor, S. M., Winters, S. J., and Amico, J. A. (1994). Pattern of oxytocin concentrations in the peripheral blood of healthy women and men: effect of the menstrual cycle and short-term fasting. Endocr. Res. 20, 117–125.
Chang, S. W., Barter, J. W., Ebitz, R. B., Watson, K. K., and Platt, M. L. (2012). Inhaled oxytocin amplifies both vicarious reinforcement and self reinforcement in rhesus macaques (Macaca mulatta). Proc. Natl. Acad. Sci. U.S.A. 109, 959–964.
Chapman, C. D., Frey, W. H. 2nd., Craft, S., Danielyan, L., Hallschmid, M., Schioth, H. B., et al. (2012). Intranasal treatment of central nervous system dysfunction in humans. Pharm. Res. doi: 10.1007/s11095-012-0915-1. [Epub ahead of print].
Chaviaras, S., Mak, P., Ralph, D., Krishnan, L., and Broadbear, J. H. (2010). Assessing the antidepressant-like effects of carbetocin, an oxytocin agonist, using a modification of the forced swimming test. Psychopharmacology 210, 35–43.
Chen, X. Q., Fawcett, J. R., Rahman, Y. E., Ala, T. A., and Frey, I. W. (1998). Delivery of nerve growth factor to the brain via the olfactory pathway. J. Alzheimers Dis. 1, 35–44.
Cho, M. M., DeVries, A. C., Williams, J. R., and Carter, C. S. (1999). The effects of oxytocin and vasopressin on partner preferences in male and female prairie voles (Microtus ochrogaster). Behav. Neurosci. 113, 1071–1079.
Choi, D. C., Rothbaum, B. O., Gerardi, M., and Ressler, K. J. (2010). Pharmacological enhancement of behavioral therapy: focus on posttraumatic stress disorder. Curr. Top. Behav. Neurosci. 2, 279–299.
Choleris, E., Devidze, N., Kavaliers, M., and Pfaff, D. W. (2008). Steroidal/neuropeptide interactions in hypothalamus and amygdala related to social anxiety. Prog. Brain Res. 170, 291–303.
Chou, H. H., Twamley, E., and Swerdlow, N. R. (2012). Towards medication-enhancement of cognitive interventions in schizophrenia. Handb. Exp. Pharmacol. 213, 81–111.
Christensen, J., Kvistgaard, H., Knudsen, J., Shaikh, G., Tolmie, J., Cooke, S., et al. (2013). A novel deletion partly removing the AVP gene causes autosomal recessive inheritance of early-onset neurohypophyseal diabetes insipidus. Clin. Genet. 83, 44–52.
Churchland, P. S., and Winkielman, P. (2012). Modulating social behavior with oxytocin: how does it work? What does it mean? Horm. Behav. 61, 392–399.
Cushing, B. S., and Carter, C. S. (2000). Peripheral pulses of oxytocin increase partner preferences in female, but not male, prairie voles. Horm. Behav. 37, 49–56.
De Dreu, C. K., Greer, L. L., Handgraaf, M. J., Shalvi, S., Van Kleef, G. A., Baas, M., et al. (2010). The neuropeptide oxytocin regulates parochial altruism in intergroup conflict among humans. Science 328, 1408–1411.
De Dreu, C. K., Greer, L. L., Van Kleef, G. A., Shalvi, S., and Handgraaf, M. J. (2011). Oxytocin promotes human ethnocentrism. Proc. Natl. Acad. Sci. U.S.A. 108, 1262–1266.
Delville, Y., Mansour, K. M., and Ferris, C. F. (1996). Testosterone facilitates aggression by modulating vasopressin receptors in the hypothalamus. Physiol. Behav. 60, 25–29.
den Boer, J. A., and Westenberg, H. G. (1992). Oxytocin in obsessive compulsive disorder. Peptides 13, 1083–1085.
de Oliveira, D. C., Chagas, M. H., Garcia, L. V., Crippa, J. A., and Zuardi, A. W. (2012a). Oxytocin interference in the effects induced by inhalation of 7.5% CO2 in healthy volunteers. Hum. Psychopharmacol. 27, 378–385.
de Oliveira, D. C., Zuardi, A. W., Graeff, F. G., Queiroz, R. H., and Crippa, J. A. (2012b). Anxiolytic-like effect of oxytocin in the simulated public speaking test. J. Psychopharmacol. 26, 497–504.
de Vries, G. J. (2008). Sex differences in vasopressin and oxytocin innervation of the brain. Prog. Brain Res. 170, 17–27.
Dhuria, S. V., Hanson, L. R., and Frey, W. H. 2nd. (2010). Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J. Pharm. Sci. 99, 1654–1673.
Ditzen, B., Schaer, M., Gabriel, B., Bodenmann, G., Ehlert, U., and Heinrichs, M. (2009). Intranasal oxytocin increases positive communication and reduces cortisol levels during couple conflict. Biol. Psychiatry 65, 728–731.
Domes, G., Lischke, A., Berger, C., Grossmann, A., Hauenstein, K., Heinrichs, M., et al. (2010). Effects of intranasal oxytocin on emotional face processing in women. Psychoneuroendocrinology 35, 83–93.
Dumont, G. J., de Visser, S. J., Cohen, A. F., and van Gerven, J. M. (2005). Biomarkers for the effects of selective serotonin reuptake inhibitors (SSRIs) in healthy subjects. Br. J. Clin. Pharmacol. 59, 495–510.
Dumont, G. J., Sweep, F. C., van der Steen, R., Hermsen, R., Donders, A. R., Touw, D. J., et al. (2009). Increased oxytocin concentrations and prosocial feelings in humans after ecstasy (3,4-methylenedioxymethamphetamine) administration. Soc. Neurosci. 4, 359–366.
Ebstein, R. P., Knafo, A., Mankuta, D., Chew, S. H., and Lai, P. S. (2012). The contributions of oxytocin and vasopressin pathway genes to human behavior. Horm. Behav. 61, 359–379.
Engstrom, T., Barth, T., Melin, P., and Vilhardt, H. (1998). Oxytocin receptor binding and uterotonic activity of carbetocin and its metabolites following enzymatic degradation. Eur. J. Pharmacol. 355, 203–210.
Epperson, C. N., McDougle, C. J., and Price, L. H. (1996a). Intranasal oxytocin in obsessive-compulsive disorder. Biol. Psychiatry 40, 547–549.
Epperson, C. N., McDougle, C. J., and Price, L. H. (1996b). Intranasal oxytocin in trichotillomania. Biol. Psychiatry 40, 559–560.
Fehm, H. L., Perras, B., Smolnik, R., Kern, W., and Born, J. (2000). Manipulating neuropeptidergic pathways in humans: a novel approach to neuropharmacology? Eur. J. Pharmacol. 405, 43–54.
Feifel, D., MacDonald, K., Cobb, P., and Minassian, A. (2012a). Adjunctive intranasal oxytocin improves verbal memory in people with schizophrenia. Schizophr. Res. 139, 207–210.
Feifel, D., Shilling, P. D., and Belcher, A. M. (2012b). The effects of oxytocin and its analog, carbetocin, on genetic deficits in sensorimotor gating. Eur. Neuropsychopharmacol. 22, 374–378.
Feifel, D., MacDonald, K., McKinney, R., Heisserer, N., and Serrano, V. (2011). A randomized, placebo-controlled investigation of intranasal oxytocin in patients with anxiety. Neuropsychopharmacology 36, S324–S449.
Feifel, D., MacDonald, K., Nguyen, A., Cobb, P., Warlan, H., Galangue, B., et al. (2010). Adjunctive intranasal oxytocin reduces symptoms in schizophrenia patients. Biol. Psychiatry 68, 678–680.
Feifel, D., and Reza, T. (1999). Oxytocin modulates psychotomimetic-induced deficits in sensorimotor gating. Psychopharmacology 141, 93–98.
Feldman, R., Gordon, I., and Zagoory-Sharon, O. (2011). Maternal and paternal plasma, salivary, and urinary oxytocin and parent-infant synchrony: considering stress and affiliation components of human bonding. Dev. Sci. 14, 752–761.
Ferguson, J. N., Young, L. J., Hearn, E. F., Matzuk, M. M., Insel, T. R., and Winslow, J. T. (2000). Social amnesia in mice lacking the oxytocin gene. Nat. Genet. 25, 284–288.
Fibiger, H. C. (2012). Psychiatry, the pharmaceutical industry, and the road to better therapeutics. Schizophr. Bull. 38, 649–650.
Forsling, M. L., Montgomery, H., Halpin, D., Windle, R. J., and Treacher, D. F. (1998). Daily patterns of secretion of neurohypophysial hormones in man: effect of age. Exp. Physiol. 83, 409–418.
Frank, R., and Hargreaves, R. (2003). Clinical biomarkers in drug discovery and development. Nat. Rev. Drug Discov. 2, 566–580.
Fu, C. H., Mourao-Miranda, J., Costafreda, S. G., Khanna, A., Marquand, A. F., Williams, S. C., et al. (2008). Pattern classification of sad facial processing: toward the development of neurobiological markers in depression. Biol. Psychiatry 63, 656–662.
Fu, C. H., Williams, S. C., Brammer, M. J., Suckling, J., Kim, J., Cleare, A. J., et al. (2007). Neural responses to happy facial expressions in major depression following antidepressant treatment. Am. J. Psychiatry 164, 599–607.
Fuchs, A. R., Behrens, O., and Liu, H. C. (1992). Correlation of nocturnal increase in plasma oxytocin with a decrease in plasma estradiol/progesterone ratio in late pregnancy. Am. J. Obstet. Gynecol. 167, 1559–1563.
Gimpl, G., and Fahrenholz, F. (2001). The oxytocin receptor system: structure, function, and regulation. Physiol. Rev. 81, 629–683.
Gimpl, G., Postina, R., Fahrenholz, F., and Reinheimer, T. (2005). Binding domains of the oxytocin receptor for the selective oxytocin receptor antagonist barusiban in comparison to the agonists oxytocin and carbetocin. Eur. J. Pharmacol. 510, 9–16.
Goldman, M., Marlow-O'Connor, M., Torres, I., and Carter, C. S. (2008). Diminished plasma oxytocin in schizophrenic patients with neuroendocrine dysfunction and emotional deficits. Schizophr. Res. 98, 247–255.
Goldman, M. B., Gomes, A. M., Carter, C. S., and Lee, R. (2011). Divergent effects of two different doses of intranasal oxytocin on facial affect discrimination in schizophrenic patients with and without polydipsia. Psychopharmacology 216, 101–110.
Gordon, I., Martin, C., Feldman, R., and Leckman, J. F. (2011). Oxytocin and social motivation. Dev. Cogn. Neurosci. 1, 471–493.
Gordon, I., Zagoory-Sharon, O., Leckman, J. F., and Feldman, R. (2010). Oxytocin and the development of parenting in humans. Biol. Psychiatry 68, 377–382.
Gossen, A., Hahn, A., Westphal, L., Prinz, S., Schultz, R. T., Grunder, G., et al. (2012). Oxytocin plasma concentrations after single intranasal oxytocin administration—A study in healthy men. Neuropeptides 46, 211–215.
Graugaard-Jensen, C., Hvistendahl, G. M., Frokiaer, J., Bie, P., and Djurhuus, J. C. (2008). The influence of high and low levels of estrogen on diurnal urine regulation in young women. BMC Urol. 8:16. doi: 10.1186/1471-2490-8-16
Greenwald, M. K., Johanson, C. E., Moody, D. E., Woods, J. H., Kilbourn, M. R., Koeppe, R. A., et al. (2003). Effects of buprenorphine maintenance dose on mu-opioid receptor availability, plasma concentrations, and antagonist blockade in heroin-dependent volunteers. Neuropsychopharmacology 28, 2000–2009.
Grewen, K. M., Davenport, R. E., and Light, K. C. (2010). An investigation of plasma and salivary oxytocin responses in breast- and formula-feeding mothers of infants. Psychophysiology 47, 625–632.
Griebel, G., Beeske, S., and Stahl, S. M. (2012). The vasopressin V(1b) receptor antagonist SSR149415 in the treatment of major depressive and generalized anxiety disorders: results from 4 randomized, double-blind, placebo-controlled studies. J. Clin. Psychiatry 73, 1403–1411.
Grillon, C., Krimsky, M., Charney, D. R., Vytal, K., Ernst, M., and Cornwell, B. (2012). Oxytocin increases anxiety to unpredictable threat. Mol. Psychiatry. doi: 10.1038/mp.2012.156. [Epub ahead of print].
Grimmelikhuijzen, C. J., and Hauser, F. (2012). Mini-review: the evolution of neuropeptide signaling. Regul. Pept. 177(Suppl.), S6–S9.
Guastella, A. J., Einfeld, S. L., Gray, K. M., Rinehart, N. J., Tonge, B. J., Lambert, T. J., et al. (2010). Intranasal oxytocin improves emotion recognition for youth with autism spectrum disorders. Biol. Psychiatry 67, 692–694.
Guastella, A. J., Howard, A. L., Dadds, M. R., Mitchell, P., and Carson, D. S. (2009). A randomized controlled trial of intranasal oxytocin as an adjunct to exposure therapy for social anxiety disorder. Psychoneuroendocrinology 34, 917–923.
Guastella, A. J., and MacLeod, C. (2012). A critical review of the influence of oxytocin nasal spray on social cognition in humans: evidence and future directions. Horm. Behav. 61, 410–418.
Hall, S. S., Lightbody, A. A., McCarthy, B. E., Parker, K. J., and Reiss, A. L. (2012). Effects of intranasal oxytocin on social anxiety in males with fragile X syndrome. Psychoneuroendocrinology 37, 509–518.
Hammock, E. A., and Young, L. J. (2006). Oxytocin, vasopressin and pair bonding: implications for autism. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 2187–2198.
Harmer, C. J. (2008). Serotonin and emotional processing: does it help explain antidepressant drug action? Neuropharmacology 55, 1023–1028.
Harmer, C. J., Bhagwagar, Z., Perrett, D. I., Vollm, B. A., Cowen, P. J., and Goodwin, G. M. (2003). Acute SSRI administration affects the processing of social cues in healthy volunteers. Neuropsychopharmacology 28, 148–152.
Harmer, C. J., Goodwin, G. M., and Cowen, P. J. (2009a). Why do antidepressants take so long to work? A cognitive neuropsychological model of antidepressant drug action. Br. J. Psychiatry 195, 102–108.
Harmer, C. J., O'Sullivan, U., Favaron, E., Massey-Chase, R., Ayres, R., Reinecke, A., et al. (2009b). Effect of acute antidepressant administration on negative affective bias in depressed patients. Am. J. Psychiatry 166, 1178–1184.
Harmer, C. J., Heinzen, J., O'Sullivan, U., Ayres, R. A., and Cowen, P. J. (2008). Dissociable effects of acute antidepressant drug administration on subjective and emotional processing measures in healthy volunteers. Psychopharmacology (Berl.) 199, 495–502.
Heinrichs, M., Baumgartner, T., Kirschbaum, C., and Ehlert, U. (2003). Social support and oxytocin interact to suppress cortisol and subjective responses to psychosocial stress. Biol. Psychiatry 54, 1389–1398.
Hirosawa, T., Kikuchi, M., Higashida, H., Okumura, E., Ueno, S., Shitamichi, K., et al. (2012). Oxytocin attenuates feelings of hostility depending on emotional context and individuals' characteristics. Sci. Rep. 2:384. doi: 10.1038/srep00384
Hoffman, E. R., Brownley, K. A., Hamer, R. M., and Bulik, C. M. (2012). Plasma, salivary, and urinary oxytocin in anorexia nervosa: a pilot study. Eat. Behav. 13, 256–259.
Hoge, E. A., Lawson, E. A., Metcalf, C. A., Keshaviah, A., Zak, P. J., Pollack, M. H., et al. (2012). Plasma oxytocin immunoreactive products and response to trust in patients with social anxiety disorder. Depress. Anxiety 29, 924–930.
Hoge, E. A., Pollack, M. H., Kaufman, R. E., Zak, P. J., and Simon, N. M. (2008). Oxytocin levels in social anxiety disorder. CNS Neurosci. Ther. 14, 165–170.
Hollander, E., Bartz, J., Chaplin, W., Phillips, A., Sumner, J., Soorya, L., et al. (2007). Oxytocin increases retention of social cognition in autism. Biol. Psychiatry 61, 498–503.
Hollander, E., Novotny, S., Hanratty, M., Yaffe, R., DeCaria, C. M., Aronowitz, B. R., et al. (2003). Oxytocin infusion reduces repetitive behaviors in adults with autistic and Asperger's disorders. Neuropsychopharmacology 28, 193–198.
Holma, K. M., Holma, I. A., Melartin, T. K., Rytsala, H. J., and Isometsa, E. T. (2008). Long-term outcome of major depressive disorder in psychiatric patients is variable. J. Clin. Psychiatry 69, 196–205.
Holt-Lunstad, J., Birmingham, W., and Light, K. C. (2011). The influence of depressive symptomatology and perceived stress on plasma and salivary oxytocin before, during and after a support enhancement intervention. Psychoneuroendocrinology 36, 1249–1256.
Horvat-Gordon, M., Granger, D. A., Schwartz, E. B., Nelson, V. J., and Kivlighan, K. T. (2005). Oxytocin is not a valid biomarker when measured in saliva by immunoassay. Physiol. Behav. 84, 445–448.
Huber, D., Veinante, P., and Stoop, R. (2005). Vasopressin and oxytocin excite distinct neuronal populations in the central amygdala. Science 308, 245–248.
Huffmeijer, R., Alink, L. R., Tops, M., Grewen, K. M., Light, K. C., Bakermans-Kranenburg, M. J., et al. (2012). Salivary levels of oxytocin remain elevated for more than two hours after intranasal oxytocin administration. Neuro Endocrinol. Lett. 33, 21–25.
Hunter, D. J., Schulz, P., and Wassenaar, W. (1992). Effect of carbetocin, a long-acting oxytocin analog on the postpartum uterus. Clin. Pharmacol. Ther. 52, 60–67.
Hurlemann, R., Patin, A., Onur, O. A., Cohen, M. X., Baumgartner, T., Metzler, S., et al. (2010). Oxytocin enhances amygdala-dependent, socially reinforced learning and emotional empathy in humans. J. Neurosci. 30, 4999–5007.
Illum, L. (2004). Is nose-to-brain transport of drugs in man a reality? J. Pharm. Pharmacol. 56, 3–17.
Israel, J. M., Le Masson, G., Theodosis, D. T., and Poulain, D. A. (2003). Glutamatergic input governs periodicity and synchronization of bursting activity in oxytocin neurons in hypothalamic organotypic cultures. Eur. J. Neurosci. 17, 2619–2629.
Ito, M., and Jameson, J. L. (1997). Molecular basis of autosomal dominant neurohypophyseal diabetes insipidus. Cellular toxicity caused by the accumulation of mutant vasopressin precursors within the endoplasmic reticulum. J. Clin. Invest. 99, 1897–1905.
Jin, D., Liu, H. X., Hirai, H., Torashima, T., Nagai, T., Lopatina, O., et al. (2007). CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 446, 41–45.
Johnson, A. E., Coirini, H., Insel, T. R., and McEwen, B. S. (1991). The regulation of oxytocin receptor binding in the ventromedial hypothalamic nucleus by testosterone and its metabolites. Endocrinology 128, 891–896.
Jonas, K., Johansson, L. M., Nissen, E., Ejdeback, M., Ransjo-Arvidson, A. B., and Uvnas-Moberg, K. (2009). Effects of intrapartum oxytocin administration and epidural analgesia on the concentration of plasma oxytocin and prolactin, in response to suckling during the second day postpartum. Breastfeed. Med. 4, 71–82.
Jorgensen, H., Kjaer, A., Knigge, U., Moller, M., and Warberg, J. (2003a). Serotonin stimulates hypothalamic mRNA expression and local release of neurohypophysial peptides. J. Neuroendocrinol. 15, 564–571.
Jorgensen, H., Riis, M., Knigge, U., Kjaer, A., and Warberg, J. (2003b). Serotonin receptors involved in vasopressin and oxytocin secretion. J. Neuroendocrinol. 15, 242–249.
Jourdain, P., Dupouy, B., Bonhomme, R., Theodosis, D. T., Poulain, D. A., and Israel, J. M. (1998). Electrophysiological studies of oxytocin neurons in organotypic slice cultures. Adv. Exp. Med. Biol. 449, 135–145.
Kandel, E. R., and Squire, L. R. (1999). Memory: From Mind to Molecules. New York, NY: Henry Holt and Company.
Keebaugh, A. C., and Young, L. J. (2011). Increasing oxytocin receptor expression in the nucleus accumbens of pre-pubertal female prairie voles enhances alloparental responsiveness and partner preference formation as adults. Horm. Behav. 60, 498–504.
Kemp, A. H., Quintana, D. S., Kuhnert, R. L., Griffiths, K., Hickie, I. B., and Guastella, A. J. (2012). Oxytocin increases heart rate variability in humans at rest: implications for social approach-related motivation and capacity for social engagement. PLoS ONE 7:e44014. doi: 10.1371/journal.pone.0044014
Kent, J. M., Coplan, J. D., and Gorman, J. M. (1998). Clinical utility of the selective serotonin reuptake inhibitors in the spectrum of anxiety. Biol. Psychiatry 44, 812–824.
Keri, S., Kiss, I., and Kelemen, O. (2009). Sharing secrets: oxytocin and trust in schizophrenia. Soc. Neurosci. 4, 287–293.
Kimura, T., Tanizawa, O., Mori, K., Brownstein, M. J., and Okayama, H. (1992). Structure and expression of a human oxytocin receptor. Nature 356, 526–529.
Kindt, M., Soeter, M., and Vervliet, B. (2009). Beyond extinction: erasing human fear responses and preventing the return of fear. Nat. Neurosci. 12, 256–258.
Kirsch, P., Esslinger, C., Chen, Q., Mier, D., Lis, S., Siddhanti, S., et al. (2005). Oxytocin modulates neural circuitry for social cognition and fear in humans. J. Neurosci. 25, 11489–11493.
Knobloch, H. S., Charlet, A., Hoffmann, L. C., Eliava, M., Khrulev, S., Cetin, A. H., et al. (2012). Evoked axonal oxytocin release in the central amygdala attenuates fear response. Neuron 73, 553–566.
Kosaka, H., Munesue, T., Ishitobi, M., Asano, M., Omori, M., Sato, M., et al. (2012). Long-term oxytocin administration improves social behaviors in a girl with autistic disorder. BMC Psychiatry 12:110. doi: 10.1186/1471-244X-12-110
Kostoglou-Athanassiou, I., Athanassiou, P., Treacher, D. F., Wheeler, M. J., and Forsling, M. L. (1998). Neurohypophysial hormone and melatonin secretion over the natural and suppressed menstrual cycle in premenopausal women. Clin. Endocrinol. 49, 209–216.
Kramer, K. M., Cushing, B. S., and Carter, C. S. (2003). Developmental effects of oxytocin on stress response: single versus repeated exposure. Physiol. Behav. 79, 775–782.
Krupitsky, E., Burakov, A., Romanova, T., Dunaevsky, I., Strassman, R., and Grinenko, A. (2002). Ketamine psychotherapy for heroin addiction: immediate effects and two-year follow-up. J. Subst. Abuse Treat. 23, 273–283.
Kuboyama, T., Hashimoto, H., Ueguchi, T., Yamaki, T., Hirakawa, K., Noto, T., et al. (1988). Diurnal changes in vasopressin and oxytocin levels in cerebrospinal fluid of post-operative patients with intracranial aneurysms. Endocrinol. Jpn. 35, 249–254.
Kumsta, R., and Heinrichs, M. (2013). Oxytocin, stress and social behavior: neurogenetics of the human oxytocin system. Curr. Opin. Neurobiol. 23, 11–16.
Kuwabara, Y., Takeda, S., Mizuno, M., and Sakamoto, S. (1987). Oxytocin levels in maternal and fetal plasma, amniotic fluid, and neonatal plasma and urine. Arch. Gynecol. Obstet. 241, 13–23.
Labuschagne, I., Phan, K. L., Wood, A., Angstadt, M., Chua, P., Heinrichs, M., et al. (2010). Oxytocin attenuates amygdala reactivity to fear in generalized social anxiety disorder. Neuropsychopharmacology 35, 2403–2413.
Labuschagne, I., Phan, K. L., Wood, A., Angstadt, M., Chua, P., Heinrichs, M., et al. (2011). Medial frontal hyperactivity to sad faces in generalized social anxiety disorder and modulation by oxytocin. Int. J. Neuropsychopharmacol. 1–14. doi: 10.1017/S1461145711001489
Landgraf, R., Hacker, R., and Buhl, H. (1982). Plasma vasopressin and oxytocin in response to exercise and during a day-night cycle in man. Endokrinologie 79, 281–291.
Landgraf, R., and Neumann, I. D. (2004). Vasopressin and oxytocin release within the brain: a dynamic concept of multiple and variable modes of neuropeptide communication. Front. Neuroendocrinol. 25, 150–176. doi: 10.1016/j.yfrne.2004.05.001
Lawson, E. A., Holsen, L. M., Santin, M., Meenaghan, E., Eddy, K. T., Becker, A. E., et al. (2012). Oxytocin secretion is associated with severity of disordered eating psychopathology and insular cortex hypoactivation in anorexia nervosa. J. Clin. Endocrinol. Metab. 97, E1898–E1908.
Lee, H. J., Caldwell, H. K., Macbeth, A. H., and Young, W. S., 3rd. (2008). Behavioural studies using temporal and spatial inactivation of the oxytocin receptor. Prog. Brain Res. 170, 73–77.
Lee, P. R., Brady, D. L., Shapiro, R. A., Dorsa, D. M., and Koenig, J. I. (2005). Social interaction deficits caused by chronic phencyclidine administration are reversed by oxytocin. Neuropsychopharmacology 30, 1883–1894.
Legros, J. J. (2001). Inhibitory effect of oxytocin on corticotrope function in humans: are vasopressin and oxytocin ying-yang neurohormones? Psychoneuroendocrinology 26, 649–655.
Legros, J. J., Chiodera, P., Geenen, V., Smitz, S., and von Frenckell, R. (1984). Dose-response relationship between plasma oxytocin and cortisol and adrenocorticotropin concentrations during oxytocin infusion in normal men. J. Clin. Endocrinol. Metab. 58, 105–109.
Leknes, S., Wessberg, J., Ellingsen, D. M., Chelnokova, O., Olausson, H., and Laeng, B. (2012). Oxytocin enhances pupil dilation and sensitivity to ‘hidden’ emotional expressions. Soc. Cogn. Affect. Neurosci. doi: 10.1093/scan/nss062. [Epub ahead of print].
Leuchter, A. F., Cook, I. A., Hunter, A. M., Cai, C., and Horvath, S. (2012). Resting-state quantitative electroencephalography reveals increased neurophysiologic connectivity in depression. PLoS ONE 7:e32508. doi: 10.1371/journal.pone.0032508
Leuner, B., Caponiti, J. M., and Gould, E. (2012). Oxytocin stimulates adult neurogenesis even under conditions of stress and elevated glucocorticoids. Hippocampus 22, 861–868.
Levin, R., Heresco-Levy, U., Bachner-Melman, R., Israel, S., Shalev, I., and Ebstein, R. P. (2009). Association between arginine vasopressin 1a receptor (AVPR1a) promoter region polymorphisms and prepulse inhibition. Psychoneuroendocrinology 34, 901–908.
Li, C., Bhatt, P. P., and Johnston, T. P. (1997). Transmucosal delivery of oxytocin to rabbits using a mucoadhesive buccal patch. Pharm. Dev. Technol. 2, 265–274.
Li, C., Wang, W., Summer, S. N., Westfall, T. D., Brooks, D. P., Falk, S., et al. (2008). Molecular mechanisms of antidiuretic effect of oxytocin. J. Am. Soc. Nephrol. 19, 225–232.
Lieberman, J. A., and Stroup, T. S. (2011). The NIMH-CATIE schizophrenia study: what did we learn? Am. J. Psychiatry 168, 770–775.
Liedman, R., Hansson, S. R., Howe, D., Igidbashian, S., McLeod, A., Russell, R. J., et al. (2008). Reproductive hormones in plasma over the menstrual cycle in primary dysmenorrhea compared with healthy subjects. Gynecol. Endocrinol. 24, 508–513.
Liggins, G. C. (1963). Antidiuretic effects of oxytocin, morphine and pethidine in pregnancy and labour. Aust. N.Z. J. Obstet. Gynaecol. 3, 81–83.
Light, K. C., Grewen, K. M., and Amico, J. A. (2005). More frequent partner hugs and higher oxytocin levels are linked to lower blood pressure and heart rate in premenopausal women. Biol. Psychol. 69, 5–21.
Lindow, S. W., Newham, A., Hendricks, M. S., Thompson, J. W., and van der Spuy, Z. M. (1996). The 24-hour rhythm of oxytocin and beta-endorphin secretion in human pregnancy. Clin. Endocrinol. 45, 443–446.
Linnen, A. M., Ellenbogen, M. A., Cardoso, C., and Joober, R. (2012). Intranasal oxytocin and salivary cortisol concentrations during social rejection in university students. Stress 15, 393–402.
Liu, J. C., Guastella, A. J., and Dadds, M. R. (2012). Effects of oxytocin on human social approach measured using intimacy equilibriums. Horm. Behav. 62, 585–591.
Loup, F., Tribollet, E., Dubois-Dauphin, M., and Dreifuss, J. J. (1991). Localization of high-affinity binding sites for oxytocin and vasopressin in the human brain. an autoradiographic study. Brain Res. 555, 220–232.
Loup, F., Tribollet, E., Dubois-Dauphin, M., Pizzolato, G., and Dreifuss, J. J. (1989). Localization of oxytocin binding sites in the human brainstem and upper spinal cord: an autoradiographic study. Brain Res. 500, 223–230.
Lowbridge, J., Manning, M., Haldar, J., and Sawyer, W. H. (1977). Synthesis and some pharmacological properties of [4-threonine 7-glycine]oxytocin, [1-(L-2-hydroxy-3-mercaptopropanoic acid), 4-threonine 7-glycine]oxytocin (hydroxy[Thr4, Gly7]oxytocin), and [7-Glycine]oxytocin, peptides with high oxytocic-antidiuretic selectivity. J. Med. Chem. 20, 120–123.
MacDonald, K. (2012). Sex, receptors, and attachment: a review of individual factors influencing response to oxytocin. Front. Neurosci. 6:194. doi: 10.3389/fnins.2012.00194
MacDonald, E., Dadds, M. R., Brennan, J. L., Williams, K., Levy, F., and Cauchi, A. J. (2011a). A review of safety, side-effects and subjective reactions to intranasal oxytocin in human research. Psychoneuroendocrinology 36, 1114–1126.
MacDonald, K., MacDonald, T., Wilson, M. P., and Feifel, D. (2011b). Oxytocin improves social cognition in patients with depression. Biol. Psychiatry 69, 279S–280S.
MacDonald, K., and Feifel, D. (2012a). Oxytocin in schizophrenia: a review of evidence for its therapeutic effects. Acta Neuropsychiatr. 24, 130–146.
MacDonald, K., and Feifel, D. (2012b). Dramatic improvement in sexual function induced by intranasal oxytocin. J. Sex. Med. 9, 1407–1410.
MacDonald, K., and MacDonald, T. M. (2010). The peptide that binds: a systematic review of oxytocin and its prosocial effects in humans. Harv. Rev. Psychiatry 18, 1–21.
Mah, B. L., Van Ijzendoorn, M. H., Smith, R., and Bakermans-Kranenburg, M. J. (2013). Oxytocin in postnatally depressed mothers: its influence on mood and expressed emotion. Prog. Neuropsychopharmacol. Biol. Psychiatry 40, 267–272.
Mak, P., Broussard, C., Vacy, K., and Broadbear, J. H. (2012). Modulation of anxiety behavior in the elevated plus maze using peptidic oxytocin and vasopressin receptor ligands in the rat. J. Psychopharmacol. 26, 532–542.
Manning, M., Misicka, A., Olma, A., Bankowski, K., Stoev, S., Chini, B., et al. (2012). Oxytocin and vasopressin agonists and antagonists as research tools and potential therapeutics. J. Neuroendocrinol. 24, 609–628.
McCarthy, M. M., McDonald, C. H., Brooks, P. J., and Goldman, D. (1996). An anxiolytic action of oxytocin is enhanced by estrogen in the mouse. Physiol. Behav. 60, 1209–1215.
McEwen, B. B. (2004). Brain-fluid barriers: relevance for theoretical controversies regarding vasopressin and oxytocin memory research. Adv. Pharmacol. 50, 531–592, 655–708.
McGregor, I. S., and Bowen, M. T. (2012). Breaking the loop: oxytocin as a potential treatment for drug addiction. Horm. Behav. 61, 331–339.
Meddle, S. L., Bishop, V. R., Gkoumassi, E., van Leeuwen, F. W., and Douglas, A. J. (2007). Dynamic changes in oxytocin receptor expression and activation at parturition in the rat brain. Endocrinology 148, 5095–5104.
Mens, W. B., Witter, A., and van Wimersma Greidanus, T. B. (1983). Penetration of neurohypophyseal hormones from plasma into cerebrospinal fluid (CSF): half-times of disappearance of these neuropeptides from CSF. Brain Res. 262, 143–149.
Meyer-Lindenberg, A., Kolachana, B., Gold, B., Olsh, A., Nicodemus, K. K., Mattay, V., et al. (2009). Genetic variants in AVPR1A linked to autism predict amygdala activation and personality traits in healthy humans. Mol. Psychiatry 14, 968–975.
Michopoulos, V., Checchi, M., Sharpe, D., and Wilson, M. E. (2011). Estradiol effects on behavior and serum oxytocin are modified by social status and polymorphisms in the serotonin transporter gene in female rhesus monkeys. Horm. Behav. 59, 528–535.
Mitchell, J. M., O'Neil, J. P., Janabi, M., Marks, S. M., Jagust, W. J., and Fields, H. L. (2012). Alcohol consumption induces endogenous opioid release in the human orbitofrontal cortex and nucleus accumbens. Sci. Transl. Med. 4:116ra6. doi: 10.1126/scitranslmed.3002902
Mitchell, M. D., Haynes, P. J., Anderson, A. B., and Turnbull, A. C. (1981). Plasma oxytocin concentrations during the menstrual cycle. Eur. J. Obstet. Gynecol. Reprod. Biol. 12, 195–200.
Modabbernia, A., Rezaei, F., Salehi, B., Jafarinia, M., Ashrafi, M., Tabrizi, M., et al. (2013). Intranasal oxytocin as an adjunct to risperidone in patients with schizophrenia: an 8-week, randomized, double-blind, placebo-controlled study. CNS Drugs 27, 57–65.
Modahl, C., Green, L., Fein, D., Morris, M., Waterhouse, L., Feinstein, C., et al. (1998). Plasma oxytocin levels in autistic children. Biol. Psychiatry 43, 270–277.
Modi, M. E., and Young, L. J. (2012). The oxytocin system in drug discovery for autism: animal models and novel therapeutic strategies. Horm. Behav. 61, 340–350.
Morimoto, B., De Lannoy, I., Fox, A. W., Gozes, I., and Stewart, A. (2009). Davunetide: pharmacokinetics and distribution to brain after intravenous or intranasal administration to rat. Chem. Today 27, 16–20.
Mouillac, B., Chini, B., Balestre, M. N., Jard, S., Barberis, C., Manning, M., et al. (1995). Identification of agonist binding sites of vasopressin and oxytocin receptors. Adv. Exp. Med. Biol. 395, 301–310.
Murphy, S. E., Norbury, R., O'Sullivan, U., Cowen, P. J., and Harmer, C. J. (2009). Effect of a single dose of citalopram on amygdala response to emotional faces. Br. J. Psychiatry 194, 535–540.
Naber, F., van Ijzendoorn, M. H., Deschamps, P., van Engeland, H., and Bakermans-Kranenburg, M. J. (2010). Intranasal oxytocin increases fathers' observed responsiveness during play with their children: a double-blind within-subject experiment. Psychoneuroendocrinology 35, 1583–1586.
Neumann, I., Douglas, A. J., Pittman, Q. J., Russell, J. A., and Landgraf, R. (1996). Oxytocin released within the supraoptic nucleus of the rat brain by positive feedback action is involved in parturition-related events. J. Neuroendocrinol. 8, 227–233.
Neumann, I. D. (2007). Stimuli and consequences of dendritic release of oxytocin within the brain. Biochem. Soc. Trans. 35(Pt 5), 1252–1257.
Neumann, I. D., and Landgraf, R. (2012). Balance of brain oxytocin and vasopressin: implications for anxiety, depression, and social behaviors. Trends Neurosci. 35, 649–659.
Nishimori, K., Takayanagi, Y., Yoshida, M., Kasahara, Y., Young, L. J., and Kawamata, M. (2008). New aspects of oxytocin receptor function revealed by knockout mice: sociosexual behaviour and control of energy balance. Prog. Brain Res. 170, 79–90.
Nussey, S. S., Page, S. R., Ang, V. T., and Jenkins, J. S. (1988). The response of plasma oxytocin to surgical stress. Clin. Endocrinol. 28, 277–282.
Ohlsson, B., Truedsson, M., Bengtsson, M., Torstenson, R., Sjolund, K., Bjornsson, E. S., et al. (2005). Effects of long-term treatment with oxytocin in chronic constipation; a double blind, placebo-controlled pilot trial. Neurogastroenterol. Motil. 17, 697–704.
Ophir, A. G., Gessel, A., Zheng, D. J., and Phelps, S. M. (2012). Oxytocin receptor density is associated with male mating tactics and social monogamy. Horm. Behav. 61, 445–453.
Otto, M. W., Tolin, D. F., Simon, N. M., Pearlson, G. D., Basden, S., Meunier, S. A., et al. (2010). Efficacy of d-cycloserine for enhancing response to cognitive-behavior therapy for panic disorder. Biol. Psychiatry 67, 365–370.
Ozsoy, S., Esel, E., and Kula, M. (2009). Serum oxytocin levels in patients with depression and the effects of gender and antidepressant treatment. Psychiatry Res. 169, 249–252.
Parker, K. J., Buckmaster, C. L., Schatzberg, A. F., and Lyons, D. M. (2005). Intranasal oxytocin administration attenuates the ACTH stress response in monkeys. Psychoneuroendocrinology 30, 924–929.
Parker, K. J., Kenna, H. A., Zeitzer, J. M., Keller, J., Blasey, C. M., Amico, J. A., et al. (2010). Preliminary evidence that plasma oxytocin levels are elevated in major depression. Psychiatry Res. 178, 359–362.
Parrott, A. C. (2007). The psychotherapeutic potential of MDMA (3, 4-methylenedioxymethamphetamine): an evidence-based review. Psychopharmacology (Berl.) 191, 181–193.
Patil, S. B., and Sawant, K. K. (2008). Mucoadhesive microspheres: a promising tool in drug delivery. Curr. Drug Deliv. 5, 312–318.
Patisaul, H. B., Scordalakes, E. M., Young, L. J., and Rissman, E. F. (2003). Oxytocin, but not oxytocin receptor, is regulated by oestrogen receptor beta in the female mouse hypothalamus. J. Neuroendocrinol. 15, 787–793.
Pedersen, C. A., Gibson, C. M., Rau, S. W., Salimi, K., Smedley, K. L., Casey, R. L., et al. (2011). Intranasal oxytocin reduces psychotic symptoms and improves theory of mind and social perception in schizophrenia. Schizophr. Res. 132, 50–53.
Pedersen, C. A., Smedley, K. L., Leserman, J., Jarskog, L. F., Rau, S. W., Kampov-Polevoi, A., et al. (2013). Intranasal oxytocin blocks alcohol withdrawal in human subjects. Alcohol. Clin. Exp. Res. 37, 484–489.
Perry, A., Bentin, S., Shalev, I., Israel, S., Uzefovsky, F., Bar-On, D., et al. (2010). Intranasal oxytocin modulates EEG mu/alpha and beta rhythms during perception of biological motion. Psychoneuroendocrinology 35, 1446–1453.
Pincus, D., Kose, S., Arana, A., Johnson, K., Morgan, P. S., Borckardt, J., et al. (2010). Inverse effects of oxytocin on attributing mental activity to others in depressed and healthy subjects: a double-blind placebo controlled FMRI study. Front. Psychiatry 1:134. doi: 10.3389/fpsyt.2010.00134
Pitman, R. K., Orr, S. P., and Lasko, N. B. (1993). Effects of intranasal vasopressin and oxytocin on physiologic responding during personal combat imagery in Vietnam veterans with posttraumatic stress disorder. Psychiatry Res 48, 107–117.
Pitt, G. R., Batt, A. R., Haigh, R. M., Penson, A. M., Robson, P. A., Rooker, D. P., et al. (2004). Non-peptide oxytocin agonists. Bioorg. Med. Chem. Lett. 14, 4585–4589.
Preuss, T. M. (2000). Taking the measure of diversity: comparative alternatives to the model-animal paradigm in cortical neuroscience. Brain Behav. Evol. 55, 287–299.
Prichard, Z. M., Mackinnon, A. J., Jorm, A. F., and Easteal, S. (2007). AVPR1A and OXTR polymorphisms are associated with sexual and reproductive behavioral phenotypes in humans. Mutation in brief no. 981. Online. Hum. Mutat. 28, 1150.
Quirin, M., Kuhl, J., and Dusing, R. (2011). Oxytocin buffers cortisol responses to stress in individuals with impaired emotion regulation abilities. Psychoneuroendocrinology 36, 898–904.
Raggenbass, M. (2008). Overview of cellular electrophysiological actions of vasopressin. Eur. J. Pharmacol. 583, 243–254.
Renaud, L. P., Day, T. A., and Ferguson, A. V. (1984). CNS regulation of reproduction: peptidergic mechanisms. Brain Res. Bull. 12, 181–186.
Renner, D. B., Frey, W. H. 2nd., and Hanson, L. R. (2012a). Intranasal delivery of siRNA to the olfactory bulbs of mice via the olfactory nerve pathway. Neurosci. Lett. 513, 193–197.
Renner, D. B., Svitak, A. L., Gallus, N. J., Ericson, M. E., Frey, W. H. 2nd., and Hanson, L. R. (2012b). Intranasal delivery of insulin via the olfactory nerve pathway. J. Pharm. Pharmacol. 64, 1709–1714.
Riekkinen, P., Legros, J. J., Sennef, C., Jolkkonen, J., Smitz, S., and Soininen, H. (1987). Penetration of DGAVP (Org 5667) across the blood-brain barrier in human subjects. Peptides 8, 261–265.
Riem, M. M., Bakermans-Kranenburg, M. J., Pieper, S., Tops, M., Boksem, M. A., Vermeiren, R. R., et al. (2011). Oxytocin modulates amygdala, insula, and inferior frontal gyrus responses to infant crying: a randomized controlled trial. Biol. Psychiatry 70, 291–297.
Riem, M. M., van, I. M. H., Tops, M., Boksem, M. A., Rombouts, S. A., and Bakermans-Kranenburg, M. J. (2012). No laughing matter: intranasal oxytocin administration changes functional brain connectivity during exposure to infant laughter. Neuropsychopharmacology 37, 1257–1266.
Ring, R. H., Malberg, J. E., Potestio, L., Ping, J., Boikess, S., Luo, B., et al. (2006). Anxiolytic-like activity of oxytocin in male mice: behavioral and autonomic evidence, therapeutic implications. Psychopharmacology (Berl.) 185, 218–225.
Ring, R. H., Schechter, L. E., Leonard, S. K., Dwyer, J. M., Platt, B. J., Graf, R., et al. (2010). Receptor and behavioral pharmacology of WAY-267464, a non-peptide oxytocin receptor agonist. Neuropharmacology 58, 69–77.
Rodrigues, S. M., Saslow, L. R., Garcia, N., John, O. P., and Keltner, D. (2009). Oxytocin receptor genetic variation relates to empathy and stress reactivity in humans. Proc. Natl. Acad. Sci. U.S.A. 106, 21437–21441.
Ross, H. E., Freeman, S. M., Spiegel, L. L., Ren, X., Terwilliger, E. F., and Young, L. J. (2009). Variation in oxytocin receptor density in the nucleus accumbens has differential effects on affiliative behaviors in monogamous and polygamous voles. J. Neurosci. 29, 1312–1318.
Ross, T. M., Martinez, P. M., Renner, J. C., Thorne, R. G., Hanson, L. R., and Frey, W. H., 2nd. (2004). Intranasal administration of interferon beta bypasses the blood-brain barrier to target the central nervous system and cervical lymph nodes: a non-invasive treatment strategy for multiple sclerosis. J. Neuroimmunol. 151, 66–77.
Rossoni, E., Feng, J., Tirozzi, B., Brown, D., Leng, G., and Moos, F. (2008). Emergent synchronous bursting of oxytocin neuronal network. PLoS Comput. Biol. 4:e1000123. doi: 10.1371/journal.pcbi.1000123
Rubin, L. H., Carter, C. S., Drogos, L., Jamadar, R., Pournajafi-Nazarloo, H., Sweeney, J. A., et al. (2011). Sex-specific associations between peripheral oxytocin and emotion perception in schizophrenia. Schizophr. Res. 130, 266–270.
Rupp, H. A., James, T. W., Ketterson, E. D., Sengelaub, D. R., Ditzen, B., and Heiman, J. R. (2012). Amygdala response to negative images in postpartum vs nulliparous women and intranasal oxytocin. Soc. Cogn. Affect. Neurosci. doi: 10.1093/scan/nss100. [Epub ahead of print].
Sabatier, N. (2006). alpha-Melanocyte-stimulating hormone and oxytocin: a peptide signalling cascade in the hypothalamus. J. Neuroendocrinol. 18, 703–710.
Sala, M., Braida, D., Donzelli, A., Martucci, R., Busnelli, M., Bulgheroni, E., et al. (2013). Mice heterozygous for the oxytocin receptor gene (Oxtr(+/−)) show impaired social behaviour but not increased aggression or cognitive inflexibility: evidence of a selective haploinsufficiency gene effect. J. Neuroendocrinol. 25, 107–118.
Sala, M., Braida, D., Lentini, D., Busnelli, M., Bulgheroni, E., Capurro, V., et al. (2011). Pharmacologic rescue of impaired cognitive flexibility, social deficits, increased aggression, and seizure susceptibility in oxytocin receptor null mice: a neurobehavioral model of autism. Biol. Psychiatry 69, 875–882.
Salonia, A., Nappi, R. E., Pontillo, M., Daverio, R., Smeraldi, A., Briganti, A., et al. (2005). Menstrual cycle-related changes in plasma oxytocin are relevant to normal sexual function in healthy women. Horm. Behav. 47, 164–169.
Sanders, G., Freilicher, J., and Lightman, S. L. (1990). Psychological stress of exposure to uncontrollable noise increases plasma oxytocin in high emotionality women. Psychoneuroendocrinology 15, 47–58.
Saphire-Bernstein, S., Way, B. M., Kim, H. S., Sherman, D. K., and Taylor, S. E. (2011). Oxytocin receptor gene (OXTR) is related to psychological resources. Proc. Natl. Acad. Sci. U.S.A. 108, 15118–15122.
Scantamburlo, G., Ansseau, M., Geenen, V., and Legros, J. J. (2011). Intranasal oxytocin as an adjunct to escitalopram in major depression. J. Neuropsychiatry Clin. Neurosci. 23:E5. doi: 10.1176/appi.neuropsych.23.2.E5
Scantamburlo, G., Hansenne, M., Fuchs, S., Pitchot, W., Marechal, P., Pequeux, C., et al. (2007). Plasma oxytocin levels and anxiety in patients with major depression. Psychoneuroendocrinology 32, 407–410.
Schorscher-Petcu, A., Sotocinal, S., Ciura, S., Dupre, A., Ritchie, J., Sorge, R. E., et al. (2010). Oxytocin-induced analgesia and scratching are mediated by the vasopressin-1A receptor in the mouse. J. Neurosci. 30, 8274–8284.
Seeman, P., and Tallerico, T. (1998). Antipsychotic drugs which elicit little or no parkinsonism bind more loosely than dopamine to brain D2 receptors, yet occupy high levels of these receptors. Mol. Psychiatry 3, 123–134.
Seifer, D. B., Sandberg, E. C., Ueland, K., and Sladen, R. N. (1985). Water intoxication and hyponatremic encephalopathy from the use of an oxytocin nasal spray. A case report. J. Reprod. Med. 30, 225–228.
Seng, J., Miller, J., Sperlich, M., van de Ven, C. J., Brown, S., Carter, C. S., et al. (2013). Exploring dissociation and oxytocin as pathways between trauma exposure and trauma-related hyperemesis gravidarum: a test-of-concept pilot. J. Trauma Dissociation 14, 40–55.
Shukovski, L., Healy, D. L., and Findlay, J. K. (1989). Circulating immunoreactive oxytocin during the human menstrual cycle comes from the pituitary and is estradiol dependent. J. Clin. Endocrinol. Metab. 68, 455–460.
Silber, M., Almkvist, O., Larsson, B., Stock, S., and Uvnas-Moberg, K. (1987). The effect of oral contraceptive pills on levels of oxytocin in plasma and on cognitive functions. Contraception 36, 641–650.
Simeon, D., Bartz, J., Hamilton, H., Crystal, S., Braun, A., Ketay, S., et al. (2011). Oxytocin administration attenuates stress reactivity in borderline personality disorder: a pilot study. Psychoneuroendocrinology 36, 1418–1421.
Slattery, D. A., and Neumann, I. (2010). Oxytocin and major depressive disorder: experimental and clinical evidence for links to aetiology and possible treatment. Pharmaceuticals 3, 702–724.
Smith, A. L., Freeman, S. M., Stehouwer, J. S., Inoue, K., Voll, R. J., Young, L. J., et al. (2012). Synthesis and evaluation of C-11, F-18 and I-125 small molecule radioligands for detecting oxytocin receptors. Bioorg. Med. Chem. 20, 2721–2738.
Spigset, O. (1999). Adverse reactions of selective serotonin reuptake inhibitors: reports from a spontaneous reporting system. Drug Saf. 20, 277–287.
Sripada, C. S., Phan, K. L., Labuschagne, I., Welsh, R., Nathan, P. J., and Wood, A. G. (2013). Oxytocin enhances resting-state connectivity between amygdala and medial frontal cortex. Int. J. Neuropsychopharmacol. 16, 255–260.
Stankova, T., Eichhammer, P., Langguth, B., and Sand, P. G. (2012). Sexually dimorphic effects of oxytocin receptor gene (OXTR) variants on Harm Avoidance. Biol. Sex Differ. 3:17. doi: 10.1186/2042-6410-3-17
Stock, S., Bremme, K., and Uvnas-Moberg, K. (1991). Plasma levels of oxytocin during the menstrual cycle, pregnancy and following treatment with HMG. Hum. Reprod. 6, 1056–1062.
Stratton, J. F., Stronge, J., and Boylan, P. C. (1995). Hyponatraemia and non-electrolyte solutions in labouring primigravida. Eur. J. Obstet. Gynecol. Reprod. Biol. 59, 149–151.
Swaab, D. F., Pool, C. W., and Nijveldt, F. (1975). Immunofluorescence of vasopressin and oxytocin in the rat hypothalamo-neurohypophypopseal system. J. Neural Trans. 36, 195–215.
Szeto, A., McCabe, P. M., Nation, D. A., Tabak, B. A., Rossetti, M. A., McCullough, M. E., et al. (2011). Evaluation of enzyme immunoassay and radioimmunoassay methods for the measurement of plasma oxytocin. Psychosom. Med. 73, 393–400.
Thompson, M. R., Callaghan, P. D., Hunt, G. E., Cornish, J. L., and McGregor, I. S. (2007). A role for oxytocin and 5-HT(1A) receptors in the prosocial effects of 3, 4 methylenedioxymethamphetamine ("ecstasy"). Neuroscience 146, 509–514.
Thorne, R. G., and Frey, W. H. 2nd. (2001). Delivery of neurotrophic factors to the central nervous system: pharmacokinetic considerations. Clin. Pharmacokinet. 40, 907–946.
Thorne, R. G., Pronk, G. J., Padmanabhan, V., and Frey, W. H. 2nd. (2004). Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience 127, 481–496.
Tost, H., Kolachana, B., Hakimi, S., Lemaitre, H., Verchinski, B. A., Mattay, V. S., et al. (2010). A common allele in the oxytocin receptor gene (OXTR) impacts prosocial temperament and human hypothalamic-limbic structure and function. Proc. Natl. Acad. Sci. U.S.A. 107, 13936–13941.
Tranter, R., Bell, D., Gutting, P., Harmer, C., Healy, D., and Anderson, I. M. (2009). The effect of serotonergic and noradrenergic antidepressants on face emotion processing in depressed patients. J. Affect. Disord. 118, 87–93.
Turner, R. A., Altemus, M., Yip, D. N., Kupferman, E., Fletcher, D., Bostrom, A., et al. (2002). Effects of emotion on oxytocin, prolactin, and ACTH in women. Stress 5, 269–276.
Twamley, E. W., Vella, L., Burton, C. Z., Heaton, R. K., and Jeste, D. V. (2012). Compensatory cognitive training for psychosis: effects in a randomized controlled trial. J. Clin. Psychiatry 73, 1212–1219.
Uvnas-Moberg, K., Sjogren, C., Westlin, L., Andersson, P. O., and Stock, S. (1989). Plasma levels of gastrin, somatostatin, VIP, insulin and oxytocin during the menstrual cycle in women (with and without oral contraceptives). Acta Obstet. Gynecol. Scand. 68, 165–169.
van Ijzendoorn, M., Bhandari, R., van der Veen, R., Grewen, K., and Bakermans-Kranenburg, M. J. (2012). Elevated salivary levels of oxytocin persist more than seven hours after intranasal administration. Front. Neurosci. 6:174. doi: 10.3389/fnins.2012.00174
Veinante, P., and Freund-Mercier, M. J. (1995). Histoautoradiographic detection of oxytocin- and vasopressin-binding sites in the amygdala of the rat. Adv. Exp. Med. Biol. 395, 347–348.
Vinogradov, S., Fisher, M., and de Villers-Sidani, E. (2012). Cognitive training for impaired neural systems in neuropsychiatric illness. Neuropsychopharmacology 37, 43–76.
Viviani, D., Charlet, A., van den Burg, E., Robinet, C., Hurni, N., Abatis, M., et al. (2011). Oxytocin selectively gates fear responses through distinct outputs from the central amygdala. Science 333, 104–107.
Viviani, D., and Stoop, R. (2008). Opposite effects of oxytocin and vasopressin on the emotional expression of the fear response. Prog. Brain Res. 170, 207–218.
Volkow, N. D., and Skolnick, P. (2012). New medications for substance use disorders: challenges and opportunities. Neuropsychopharmacology 37, 290–292.
Volkow, N. D., Wang, G., Fowler, J. S., Logan, J., Gerasimov, M., Maynard, L., et al. (2001). Therapeutic doses of oral methylphenidate significantly increase extracellular dopamine in the human brain. J. Neurosci. 21, RC121.
Walum, H., Lichtenstein, P., Neiderhiser, J. M., Reiss, D., Ganiban, J. M., Spotts, E. L., et al. (2012). Variation in the oxytocin receptor gene is associated with pair-bonding and social behavior. Biol. Psychiatry 71, 419–426.
Walum, H., Westberg, L., Henningsson, S., Neiderhiser, J. M., Reiss, D., Igl, W., et al. (2008). Genetic variation in the vasopressin receptor 1a gene (AVPR1A) associates with pair-bonding behavior in humans. Proc. Natl. Acad. Sci. U.S.A. 105, 14153–14156.
Wang, H., Ward, A. R., and Morris, J. F. (1995). Oestradiol acutely stimulates exocytosis of oxytocin and vasopressin from dendrites and somata of hypothalamic magnocellular neurons. Neuroscience 68, 1179–1188.
Weisman, O., Zagoory-Sharon, O., and Feldman, R. (2012a). Intranasal oxytocin administration is reflected in human saliva. Psychoneuroendocrinology 37, 1582–1586.
Weisman, O., Zagoory-Sharon, O., and Feldman, R. (2012b). Oxytocin administration to parent enhances infant physiological and behavioral readiness for social engagement. Biol. Psychiatry 72, 982–989.
Weisman, O., Zagoory-Sharon, O., Schneiderman, I., Gordon, I., and Feldman, R. (2012c). Plasma oxytocin distributions in a large cohort of women and men and their gender-specific associations with anxiety. Psychoneuroendocrinology. Available online at: http://dx.doi.org/10.1016/j.psyneuen.2012.08.011
White-Traut, R., Watanabe, K., Pournajafi-Nazarloo, H., Schwertz, D., Bell, A., and Carter, C. S. (2009). Detection of salivary oxytocin levels in lactating women. Dev. Psychobiol. 51, 367–373.
Wiedemann, K. (2011). Biomarkers in development of psychotropic drugs. Dialogues Clin. Neurosci. 13, 225–234.
Wigger, A., and Neumann, I. D. (2002). Endogenous opioid regulation of stress-induced oxytocin release within the hypothalamic paraventricular nucleus is reversed in late pregnancy: a microdialysis study. Neuroscience 112, 121–129.
Williams, J. R., Insel, T. R., Harbaugh, C. R., and Carter, C. S. (1994). Oxytocin administered centrally facilitates formation of a partner preference in female prairie voles (Microtus ochrogaster). J. Neuroendocrinol. 6, 247–250.
Windle, R. J., Gamble, L. E., Kershaw, Y. M., Wood, S. A., Lightman, S. L., and Ingram, C. D. (2006). Gonadal steroid modulation of stress-induced hypothalamo-pituitary-adrenal activity and anxiety behavior: role of central oxytocin. Endocrinology 147, 2423–2431.
Windle, R. J., Judah, J. M., and Forsling, M. L. (1997). Effect of oxytocin receptor antagonists on the renal actions of oxytocin and vasopressin in the rat. J. Endocrinol. 152, 257–264.
Wotjak, C. T., Ganster, J., Kohl, G., Holsboer, F., Landgraf, R., and Engelmann, M. (1998). Dissociated central and peripheral release of vasopressin, but not oxytocin, in response to repeated swim stress: new insights into the secretory capacities of peptidergic neurons. Neuroscience 85, 1209–1222.
Wu, N., Li, Z., and Su, Y. (2012). The association between oxytocin receptor gene polymorphism (OXTR) and trait empathy. J. Affect. Disord. 138, 468–472.
Yoshida, M., Takayanagi, Y., Inoue, K., Kimura, T., Young, L. J., Onaka, T., et al. (2009). Evidence that oxytocin exerts anxiolytic effects via oxytocin receptor expressed in serotonergic neurons in mice. J. Neurosci. 29, 2259–2271.
Young, S. N., and Anderson, G. N. (2010). Bioanalytical inaccuracy: a threat to the integrity and efficiency of research. J. Psychiatry Neurosci. 35, 3–6.
Young, W. S. 3rd., Shepard, E., Amico, J., Hennighausen, L., Wagner, K. U., LaMarca, M. E., et al. (1996). Deficiency in mouse oxytocin prevents milk ejection, but not fertility or parturition. J. Neuroendocrinol. 8, 847–853.
Young, W. S., Li, J., Wersinger, S. R., and Palkovits, M. (2006). The vasopressin 1b receptor is prominent in the hippocampal area CA2 where it is unaffected by restraint stress or adrenalectomy. Neuroscience 143, 1031–1039.
Zhang, G., and Cai, D. (2011). Circadian intervention of obesity development via resting-stage feeding manipulation or oxytocin treatment. Am. J. Physiol. Endocrinol. Metab. 301, E1004–E1012.
Zhong, S., Monakhov, M., Mok, H. P., Tong, T., Lai, P. S., Chew, S. H., et al. (2012). U-shaped relation between plasma oxytocin levels and behavior in the trust game. PLoS ONE 7:e51095. doi: 10.1371/journal.pone.0051095
Zhu, J., Jiang, Y., Xu, G., and Liu, X. (2012). Intranasal administration: a potential solution for cross-BBB delivering neurotrophic factors. Histol. Histopathol. 27, 537–548.
Keywords: oxytocin, pharmacology, humans, intranasal administration, psychiatry, drug development
Citation: MacDonald K and Feifel D (2013) Helping oxytocin deliver: considerations in the development of oxytocin-based therapeutics for brain disorders. Front. Neurosci. 7:35. doi: 10.3389/fnins.2013.00035
Received: 06 December 2012; Paper pending published: 08 January 2013;
Accepted: 28 February 2013; Published online: 15 March 2013.
Edited by:
Idan Shalev, Duke University, USACopyright © 2013 MacDonald and Feifel. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: K. MacDonald, Department of Psychiatry, University of California, San Diego, 200 W. Arbor Dr. MC 8216, San Diego, CA 92103, USA. e-mail:a2Fpc21hY2RvbmFsZEBtZS5jb20=