

- Department of Neuroscience, Carleton University, Ottawa, ON, Canada
Stress was once defined as the non-specific result of the body to any demand or challenge to homeostasis. A more current view of stress is the behavioral and physiological responses generated in the face of, or in anticipation of, a perceived threat. The stress response involves activation of the sympathetic nervous system and recruitment of the hypothalamic-pituitary-adrenal (HPA) axis. When an organism encounters a stressor (social, physical, etc.), these endogenous stress systems are stimulated in order to generate a fight-or-flight response, and manage the stressful situation. As such, an organism is forced to liberate energy resources in attempt to meet the energetic demands posed by the stressor. A change in the energy homeostatic balance is thus required to exploit an appropriate resource and deliver useable energy to the target muscles and tissues involved in the stress response. Acutely, this change in energy homeostasis and the liberation of energy is considered advantageous, as it is required for the survival of the organism. However, when an organism is subjected to a prolonged stressor, as is the case during chronic stress, a continuous irregularity in energy homeostasis is considered detrimental and may lead to the development of metabolic disturbances such as cardiovascular disease, type II diabetes mellitus and obesity. This concept has been studied extensively using animal models, and the neurobiological underpinnings of stress induced metabolic disorders are beginning to surface. However, different animal models of stress continue to produce divergent metabolic phenotypes wherein some animals become anorexic and lose body mass while others increase food intake and body mass and become vulnerable to the development of metabolic disturbances. It remains unclear exactly what factors associated with stress models can be used to predict the metabolic outcome of the organism. This review will explore a variety of rodent stress models and discuss the elements that influence the metabolic outcome in order to further extend our understanding of stress-induced obesity.
Introduction
It is now generally accepted that obesity has reached epidemic proportions. To some medical professionals, obesity is considered the leading preventable cause of death worldwide. More worrisome is the fact that the prevalence of obesity has drastically increased in the past 30 years, where ~10% of the population was considered overweight in the 1970's, a number that has now risen to over 35% in the United States (Flegal et al., 2010, 2012). This estimate is accompanied by a wealth of statistics demonstrating that individuals who are overweight or obese have a much higher susceptibility to diseases such as Type II diabetes, cardiovascular disease and metabolic syndrome. In fact, some studies show that life expectancy decreases significantly as a function of weight gain, and that individuals suffering from obesity die up to a decade sooner than those of a normal weight (Peeters et al., 2003). For this reason, obesity is one of the most pressing health issues seen in the twenty-first century.
It is clear that there is a strong genetic predisposition to obesity and metabolic disorders. Nevertheless, environmental and social factors influence amounts of food consumed or energy spent and ultimately impact phenotype. Arguably, stress may be the most important environmental factor affecting metabolism and feeding behavior to promote obesity. Indeed, there is an association between adiposity, particularly abdominal adiposity, and psychological distress (Raikkonen et al., 1996; Chrousos and Gold, 1998; Bjorntorp, 2001). Furthermore, and in parallel with the increasing rates of obesity, subjects from industrialized communities report higher levels of workplace stress and sleep deprivation in recent years relative to the past (Bjorntorp, 2001; Caruso, 2006; Grosch et al., 2006).
The physiological and behavioral responses to stress are necessary for survival. During an acute stress response, activation of the hypothalamic-pituitary-adrenal (HPA) axis entails a cascade of events that begins with the release of corticotropin-releasing factor (CRF) from specialized neurons in the paraventricular nucleus of the hypothalamus (PVN) into the portal blood neighboring the anterior pituitary gland. CRF secretion in turn leads to the synthesis and release of adrenocorticotropic hormone (ACTH) from the anterior pituitary into the general circulation, which ultimately stimulates the release of glucocorticoids from the adrenal cortex. ACTH exerts negative feedback control over CRF at the level of the hypothalamus and, along with CRF, orchestrates the body's response to a homeostatic challenge (Aguilera, 1994). Glucocorticoid release from the adrenal glands is the final output stage of the HPA axis, which represents, amongst other things, an organism's attempt to mobilize energy stores to fuel the brain, skeletal and cardiac muscles.
The primary glucocorticoids secreted in response to stress are cortisol and corticosterone in humans and rodents, respectively. In many tissues, glucocorticoids serve an opposing function to insulin's attempt at storing energy, in part by suppressing insulin secretion (Dinneen et al., 1993). By inducing a transient state of insulin resistance, glucocorticoids restrain hepatic glucose production and regulate glucose delivery to peripheral tissue thereby producing an acute state of hyperglycemia (Rizza et al., 1982; Mangos et al., 2000). The peripheral tissues involved in the stress response use the acute hyperglycemic environment to meet the energetic demands of the stressor.
The aforementioned secretion of CRF and subsequent release of glucocorticoids is under the control of a finely tuned negative feedback loop, wherein elevated circulating glucocorticoids travel back to the hypothalamus (Jones et al., 1977; Di et al., 2003, 2005), hippocampus (Sapolsky et al., 1984, 2000), medial prefrontal cortex (Diorio et al., 1993; Radley et al., 2008) and pituitary gland (Mahmoud et al., 1984; Cole et al., 2000) to inhibit further stimulation of the HPA axis (McEwen, 2007; Ulrich-Lai and Herman, 2009). At the level of the hypothalamus, glucocorticoids have been shown to cause rapid suppression of excitatory synaptic inputs to CRF neurons within the PVN (Di et al., 2003), an effect that is thought to be mediated by the retrograde action of endocannabinoids on upstream glutamate neurons (Di et al., 2005). In contrast, glucocorticoid feedback at the level of the hippocampus increases excitatory post-synaptic potentials via modulation of membrane-bound mineralocorticoid receptors (Olijslagers et al., 2008). Rapid excitation of efferent hippocampal neurons, in turn, leads to the activation of inhibitory GABAergic input onto the neuroendocrine PVN neurons (Boudaba et al., 1996; Herman et al., 2002) and is therefore implicated in the negative feedback regulation of the HPA axis (Furay et al., 2008). Furthermore, the medial prefrontal cortex (mPFC) is also responsive to stress-induced secretion of glucocorticoids and is capable of directly modulating the activity of secretory PVN neurons (Radley et al., 2008). Injections of corticosterone (CORT) into the mPFC results in significant reductions of CRF and ACTH release following restraint stress in rats (Diorio et al., 1993), demonstrating a role for the mPFC in the regulation of HPA axis activity. After answering the energetic demands posed by a stressor, the negative feedback loop of the HPA axis ensures an efficient return to homeostatic balance following cessation of the stressor and it is therefore considered a mediator of allostasis.
The acute response to stress is one that is both necessary and beneficial because it mediates the behavioral and physiological adaptations necessary for homeostatic maintenance under stressful conditions. Protracted stressors, on the other hand, lead to an excessive energetic burden resulting in long lasting physiological changes, a process referred to as allostatic overload (McEwen, 2004). Failure to adjust to the energetic demands posed by this overload, or failure to reduce the stressor itself, may lead to negative health consequences, both metabolic and psychiatric. Increasing rates of chronic daily stress, regardless of their origin, in combination with increased availability of foods high in calories creates an environment conducive to the development of obesity and other metabolic disturbances. In turn, the rising incidence of obesity is detrimental to health care as it exerts a financial burden to taxpayers while saturating medical services that would otherwise be available. The interaction between neural circuits involved in regulating energy homeostasis and body composition with the physiological responses to stressors has therefore become pertinent and clinically relevant. In attempt to shed light on this matter, scientists must turn to one of the most widely used tools for studying the molecular underpinnings of disease, which is the use of animal models. The present review aims to investigate the use of animal models in the study of stress and to highlight some of the factors that contribute to the metabolic outcome of these various stress paradigms.
Stress, Eating and Energy Homeostasis
Stress influences feeding behaviors and energy homeostasis in both humans and rodents. However, the neural mechanisms that are recruited by the stress response are complex and involve a number of circuits that include hypothalamic networks regulating energy balance, brain stem networks regulating visceral function, sympathetic and parasympathetic mechanisms, midbrain nuclei involved in reward and arousal, and corticolimbic structures that mediate emotional and cognitive aspects related to both stress and food intake. Furthermore, stress produces dramatic endocrine and neuroendocrine responses that in themselves affect energy balance and food intake. Among these, the best-characterized responses are the activation of the sympathetic nervous system and that of the HPA axis. The former is elicited to generate rapid physiological responses including increased heart rate and respiration, glucose uptake in muscle and decreased visceral function, all important for generating a fight or flight response. Subsequent activation of the HPA axis, as previously discussed, represents a hormonal cascade that culminates in the secretion of glucocorticoids. These steroid hormones have a multitude of effects, both in the periphery and the brain that among other things alter metabolism, promote appetite and increase the locomotor activities associated with food seeking behaviors in rodents (McEwen, 2004).
Numerous studies have shown that stress modulates metabolism, but these effects are not always consistent and can range from marked decreases in both body weight and food intake, to increases in body weight and calorie consumption (Dallman et al., 2005; Depke et al., 2008; Nieuwenhuizen and Rutters, 2008). For example, acute psychogenic or systemic stressors results in a rapid decline of food intake and the successive breakdown of nutrient stores (Depke et al., 2008). Similarly, exposure to chronic stressors can also lead to decreased body weight and food intake (Torres and Nowson, 2007; Depke et al., 2008). While these animals appear to lose weight, they do by using nutrients derived from tissues other than fat in spite of decreasing their body weight (Akana et al., 1999; Depke et al., 2008). Metabolically, these animals show altered glucose regulation, an early indicator of insulin resistance (Black, 2006).
Interestingly repeated exposure to psychogenic stressors, in particular social stressors, generates increases in body weight, adiposity, and in the intake of high calorie “comfort” meals in a number of species including non-human and human primates (Pecoraro et al., 2004; Dallman et al., 2005; Tamashiro et al., 2007a; Coccurello et al., 2008; O'Connor et al., 2008; Wilson et al., 2008). These data are of particular relevance considering that in human populations stress is highly associated with social interactions (Tamashiro et al., 2007a,b; Michaud et al., 2008; O'Connor et al., 2008; Wilson et al., 2008). The mechanisms underlying metabolic changes in response to social stressors have commonly been associated with the negative consequences of repeated stimulation of the HPA axis (Anisman and Matheson, 2005). In the case of obesity, elevated levels of glucocorticoids appear to be associated with abdominal adiposity (Black, 2006). Indeed, clinical conditions characterized by high cortisol levels, as seen in patients with Cushing's or Prader–Willi syndrome, are also associated with abdominal obesity and type II diabetes (Nieuwenhuizen and Rutters, 2008; Weaver, 2008). In contrast, removal of the adrenal glands in experimental animals results in decreased body weight and food intake that can be restored by corticosterone replacement (Saito and Bray, 1984; Pralong et al., 1993; Makimura et al., 2000).
Finally stress also affects psychological processes that could lead to the preference of foods that have a comfort value. For instance, some stressors have been associated with a loss of control over food intake and an increased drive to eat highly palatable non-nutritious foods (Groesz et al., 2012). This active seeking of highly palatable foods is thought to be a form of self-medication for both humans and rodents, wherein HPA axis activation is down regulated in response to the rewarding properties of such foods ultimately creating a preference, and potentially cravings, for these foods (Dallman et al., 2003, 2005; Pecoraro et al., 2004; Tomiyama et al., 2011). Interestingly, while all stressors are known to simulate the HPA axis, there is a documented, bi-directional effect on food intake. Physical stress, for example, may cause a reduction in food intake (Popper et al., 1989) while psychological stress can increase or decrease food intake (Huhman, 2006).
Regulation of Food Intake and Energy Balance: A Look Inside the Brain
To understand how stress influences metabolism and feeding we first need to consider how feeding and metabolism are regulated and then how stress can influence these mechanisms. Within the brain there exists a very complex organization of nuclei that collectively control food intake and energy homeostasis. For a more comprehensive review on the many brain circuits regulating energy homeostasis and the integration of afferent metabolic signals see Horvath et al, (2004) Abizaid and Horvath (2008).
Traditionally, the hypothalamus is considered the focal point for processing and integrating both afferent and efferent metabolic information (Elmquist, 2000; Schwartz et al., 2000; Dhillo and Bloom, 2001; Cummings and Shannon, 2003). Within the hypothalamus, these nuclei can be roughly divided into two distinct groups based on their tendencies to promote (lateral hypothalamus, LH) or inhibit food intake (ventromedial nucleus and paraventricular nucleus; VMH and PVN, respectively). Furthermore, at the base of the hypothalamus the arcuate nucleus (ARC) lies within an area that is less protected by the blood brain barrier and is thus capable of detecting peripherally circulating metabolic signals (Ciofi, 2011; Morita and Miyata, 2012, 2013). With this real estate, and the expression of both orexigenic and anorectic peptides, the ARC is thought to play an intimate role in the regulation of food intake and energy homeostasis (Schwartz et al., 2000; Cone et al., 2001; Cowley et al., 2001, 2003). The influence of the ARC on feeding behaviors is mediated by a sub-set of first-order neurons that either co-express the orexigenic transcripts neuropeptide Y (NPY) and agouti-related peptide (AgRP), or the anorectic transcripts proopiomelanocortin (POMC) and cocaine-amphetamine related transcript (CART). NPY and AgRP are two potent orexigenic peptides found predominantly in the ARC and whose expression and secretions are mediated through impairments in energy balance such as depletions of body fat stores, reductions in circulating glucose and/or altered metabolic hormone signaling in the brain (White and Kershaw, 1990; Kalra et al., 1991; Wilding et al., 1993; Schwartz et al., 2000). POMC and CART are also major regulators of energy balance, however, in contrast to NPY and AgRP, they produce a potent and long-lasting anorexigenic effect (Kristensen et al., 1998; Hagan et al., 1999; Larsen et al., 2000; Cowley et al., 2001). The expression and secretions of POMC and CART are mediated by signals of an energy surplus such as overfeeding and increases in body weight (Hagan et al., 1999). NPY/AgRP and POMC/CART neurons are profoundly interconnected and differentially regulated by other hypothalamic nuclei involved in energy homeostasis (Broberger et al., 1997; Parker and Herzog, 1999). Recently, it has been shown that selective activation of NPY/AgRP neurons in the ARC causes a potent suppression of POMC/CART neuronal activity, and a subsequent increase in long-term food intake (Atasoy et al., 2012). Thus, in addition to being influenced by peripheral signals indicating energy availability, NPY/AgRP neurons are capable of directly regulating activity of other neuronal subpopulations involved in food intake (Wahlestedt et al., 1986; Broberger et al., 1997; Parker and Herzog, 1999; Cowley et al., 2001; Atasoy et al., 2012). Furthermore, satiety centers such as the VMH have been shown to send excitatory inputs onto POMC/CART neurons, providing a more complex inhibition of food intake (Sternson et al., 2005). Interestingly, the excitatory glutamanergic neurons in the VMH express CRF receptors (Chalmers et al., 1995) and are thus stimulated by stress-induced CRF secretion, as well as the structurally related urocortin peptides (Chen et al., 2010; Kuperman et al., 2010). Once stimulated, these neurons project from the VMH to arcuate POMC/CART neurons and therefore initiate an anorectic signaling cascade downstream of the VMH (Chen et al., 2010). In contrast, NPY/AgRP neurons are regulated by weak inhibitory inputs from within the ARC but do not respond to the VMH (Sternson et al., 2005) and are not controlled by anorectic signals.
Peripheral signals ascending via the third ventricle and/or the vagus nerve are capable of directly and/or indirectly updating the first order arcuate neurons with information pertaining to the organism's nutritional status. Once stimulated, these first order NPY/AgRP and POMC/CART neurons project to second order hypothalamic centers located predominantly in the PVN, LH, and brain stem nuclei (Clark et al., 1984; Ramos et al., 2005; Sternson et al., 2005; Palmiter, 2007; Atasoy et al., 2012; Wu et al., 2012) to regulate meal frequency (Marin Bivens et al., 1998) and meal size, respectively (Kalra et al., 1988). NPY/AgRP neurons, for example, project directly to the PVN where they are capable of producing prolonged inhibitory post-synaptic currents and a robust feeding bout (Atasoy et al., 2012). In addition, NPY/AgRP neurons project to the LH where they serve to promote the secretion of other orexigenic transcripts such as hypocretin/orexin in response to hunger signals (Cone et al., 2001). POMC/CART neurons, on the other hand, are capable of down regulating activity in these same areas. POMC/CART project to the LH and PVN to reduce food intake and increase energy expenditure, respectively (Cone et al., 2001; Cowley et al., 2001).
Stress and Autonomic Function
Integration of circulating metabolic and stress signals also occurs in several brain stem nuclei that are part of the autonomic and enteric nervous system (Berthoud et al., 2006). As such, these nuclei are critical for the responses that follow the activation of the stress axis by psychogenic, metabolic, or immunological challenges (Smith and Vale, 2006). Within the brain stem, the nucleus of the solitary tract (NTS) is the main target for information ascending from the gut via the vagus nerve and the enteric nervous system. The NTS contains noradrenergic neurons that modulate the activity of hypothalamic and limbic structures implicated in feeding, including the PVN and central nucleus of the amygdala (CeA) (Treece et al., 2000; Grill and Kaplan, 2001, 2002). Another brain stem region important for the regulation of food intake and stress is the area postrema (AP). The AP lies outside of the blood brain barrier and is sensitive to blood born nutritional signals including hormones like leptin, insulin, cholecystokinin (CCK) and ghrelin, as well as being sensitive to changes in circulating levels of glucose. Signals are relayed from the AP to the NTS where they are integrated with ascending visceral signals, as well as to the PVN where they may elicit feeding and autonomic responses (Ellenberger and Feldman, 1990; Woulfe et al., 1990; Ferguson, 1991; Castaneyra-Perdomo et al., 1992; Diaz-Regueira and Anadon, 1992; Cai et al., 1996). In addition to the regulation of feeding responses, the brain stem plays a key role in the generation of the autonomic responses, including increases in blood pressure, respiration, hepatic glucose production, vasoconstriction, lipolysis, and thermogenesis (Bamshad et al., 1998; Krukoff, 1998; Dunn et al., 2004).
Given these roles, it is not surprising that noradrenergic cells in the brain stem are affected by chronic stress. Indeed, many of the effects of chronic stress on this system are controlled by medications that block noradrenergic receptors (i.e., β-blockers) (Grassi, 2007). Moreover, NPY released from sympathetic cells into peripheral circulation may be critical for the adipogenic effects of stress, primarily by acting directly on NPY-Y2 receptors located on adipocytes (Kuo et al., 2007).
Stress and Comfort Foods: The Role of Reward
The midbrain contains a number of cell groups implicated in brain functions underlying reward seeking, learning and memory, affective states and the generation of locomotor responses. The ventral tegmentum area (VTA) has re-emerged as an important structure in the modulation of food intake (Simerly, 2006). Dopamine (DA) cells within this region project to the striatum, prefrontal cortex, hippocampus and amygdala (Margolis et al., 2006; Fields et al., 2007; Robinson et al., 2007) where they produce behaviors to get rewards such as food and sex, or behaviors to escape aversive stimuli (Wang et al., 2004; Wise, 2004, 2006; Volkow and Wise, 2005; Baler and Volkow, 2006). These cells also play a key role in the development of addiction (Wise, 2004, 2006; Volkow and Wise, 2005). Interestingly, fasting increases DA tone and re-feeding produces an increase of DA release into the nucleus accumbens (Wang et al., 2002). Anticipation of food and other rewards also produce DA release from the VTA into the nucleus accumbens (Blackburn et al., 1992; Richardson and Gratton, 1996). In addition to DA-related processes, serotonergic components of the raphe nucleus have been implicated in the regulation of food intake (Heisler et al., 2003; Halford et al., 2007). Indeed, there are serotonin (5-HT) projections to hypothalamic and limbic centers involved in the regulation of food intake (Heym and Gladfelter, 1982; Vertes et al., 1999; Brown and Molliver, 2000; Silva et al., 2002; Jankowski and Sesack, 2004). Several lines of evidence demonstrate that 5-HT has an inhibitory effect on food intake, including clinical and experimental data on the anorectic effects of 5-HT reuptake inhibitors (i.e., SSRIs) (Halford et al., 2007). The effects of 5-HT on food intake and metabolic function are mediated, at least in part, through the stimulation of POMC cells in the hypothalamic ARC, as well as through direct effects of 5-HT onto cells of the PVN (Heisler et al., 2003).
Stress influences these midbrain regions, and chronic stress produces long lasting changes in the monoaminergic neurotransmitters produced and released by cells in these structures (Anisman et al., 2008). The way in which these systems are changed by chronic stress are numerous and may include inflammatory responses, direct and indirect glucocorticoid action and a number of associative processes that enhance the salience of external stimuli related to the stressor (Anisman and Matheson, 2005; Anisman et al., 2008). Nevertheless, stimulation of DA and 5-HT seem to be counter regulatory responses to the stress and appear to promote behaviors associated with coping including the consumption of palatable comfort foods to reduce stress (Dallman et al., 2006).
Stress and Metabolic Hormones Interact to Regulate Energy Balance
In response to stress, there are several peptides and hormones released both centrally and peripherally that are capable of acting on many hypothalamic centers, which will inevitably dictate the feeding behaviors generated in response to a stressor. There are countless transmitters, peptides and hormones released directly, or indirectly, in response to stressful stimuli (See Anisman and Matheson, 2005 or Black, 2006 for reviews). Here, we will review a subset of these messengers and discuss briefly how they relate to feeding responses following stress.
Glucocorticoids
Both human and rodent obesity, with an emphasis on visceral obesity, have been associated with increased HPA axis activity and subsequent rises in glucocorticoid concentrations that can affect both the brain and peripheral tissues (Weaver et al., 1993). Excess production of glucocorticoids, as seen in patients with Cushing's disease for example, leads to increases in central adiposity and additional metabolic complications (Bjorntorp and Rosmond, 1999, 2000). Like other steroid hormones, the lipophilic glucocorticoids gain access to the brain via the blood brain barrier (Castonguay, 1991) and influence the expression of hypothalamic peptides involved in the regulation of food intake and energy homeostasis (Cavagnini et al., 2000; Savontaus et al., 2002). Expression of glucocorticoid receptors (GR's) has been shown in many brain regions heavily implicated in energy homeostasis such as the ARC, LH and PVN (Morimoto et al., 1996). Given that glucocorticoids are capable of regulating behaviors that control energy input and energy expenditure, they are regarded as a key factor in the link between stress and obesity. To strengthen this association, it has been shown that bilateral adrenalectomy in rats decreases glucocorticoid concentrations as well as food intake (Germano et al., 2007), and glucocorticoid replacement in adrenalectomized animals restores food intake (Green et al., 1992; Jacobson, 1999). However, the association between glucocorticoid concentration and obesity may not be as straightforward as it was once thought to be. Glucocorticoids have been shown to stimulate expression, inhibit expression and interact with, other metabolically active hormones (e.g., insulin, leptin, CRF, and others), in turn providing a much more complex control over feeding behaviors (Brindley and Rolland, 1989; Cavagnini et al., 2000; La Fleur, 2006; Jahng et al., 2008). As previously mentioned, all stressors possess the ability to elicit glucocorticoid secretion. No two stressors, however, elicit the same response from the HPA axis or the same feeding response. The degree to which the HPA axis becomes activated, and thus the amount of glucocorticoids produced in response to the stressor, depends in large part on the length and severity of the stressor at hand (Marti et al., 1994; Harris et al., 1998; Valles et al., 2000).
Ghrelin
Despite glucocorticoids being considered the primary hormonal culprit behind the development of stress-induced metabolic disturbances, other metabolically activate hormones may also play a role in this process. One of these potential hormones is the gut derived hormone ghrelin. Ghrelin is a 28 amino acid hormone peptide produced in the X/A-like cells of the gastric oxyntic mucosa lining the stomach (Dornonville De La Cour et al., 2001). Acutely, ghrelin has the unique ability to promote food intake (Nakazato et al., 2001; Currie et al., 2005; Date et al., 2006) an effect that is mediated by the stimulation of distinct hypothalamic, brain stem, and midbrain nuclei (Cowley et al., 2003; Seoane et al., 2003; Abizaid et al., 2006). While there are countless neuropeptides that carry out anorexic functions, to date, ghrelin is the only known gut-brain peptide that encourages food intake. In addition, ghrelin contributes to the control of long-term energy homeostasis by regulating body weight and adiposity presumably by reducing lipid oxidation (Tschop et al., 2000; Choi et al., 2003; Theander-Carrillo et al., 2006; Patterson et al., 2013).
The only known ghrelin receptor, growth hormone secretagogue receptor-1a (GHSR-1a) is widely expressed in both rodents and humans, with the highest expression found in the hypothalamus and the pituitary gland, both are areas heavily invested in energy homeostasis and the physiological response to stress. Interestingly, plasma ghrelin levels have been shown to rise in parallel with glucocorticoids in response to both acute and chronic stress, and ghrelin levels remain elevated for an extended period of time after the cessation of the stressor (Asakawa et al., 2001; Kristenssson et al., 2006; Lutter et al., 2008b; Ochi et al., 2008; Patterson et al., 2010, 2013; Chuang et al., 2011). For example, acute water avoidance stress in rats has been shown to elicit an 85% increase in circulating ghrelin 1 h after cessation of the stressor (Kristenssson et al., 2006). Similarly, 24 h after the cessation of a 2-week chronic unpredictable stress paradigm stressed animals show a significant increase in both CORT and plasma acylated ghrelin compared to non-stressed animals (Patterson et al., 2010). Finally, chronic social defeat stress has been shown to increase circulating acylated ghrelin levels (Lutter et al., 2008b; Chuang et al., 2011; Patterson et al., 2013), a change that persists up to 30 days after the stress paradigm has been terminated (Lutter et al., 2008b). Administration of ghrelin, both centrally and peripherally, has been shown to indirectly stimulate hypothalamic CRF neurons, leading to HPA axis activation (Cabral et al., 2012). Ghrelin deficient mice show a reduction in the number of CRF expressing cells in the PVN and demonstrate a hyporesponsive HPA axis in response to acute stress (Spencer et al., 2012). Interruption of ghrelin signaling, both genetically and pharmacologically, during times of chronic stress can improve the metabolic outcome of these animals suggesting that ghrelin does in fact contribute to the development of stress-induced metabolic disturbances (Lutter et al., 2008b; Chuang et al., 2011; Patterson et al., 2013). Although it is generally accepted that ghrelin stimulates food intake in response to a stressor, it remains unclear what type of food is encouraged following a stress-induced secretion of ghrelin. The caloric content of foods ingested in response to a stressor will ultimately shape the metabolic phenotype of that organism. Data from our laboratory suggests that ghrelin promotes the intake of carbohydrates while simultaneously decreasing the intake of a high-fat diet, an effect thought to reflect an acute need for rapid energy resources (Patterson et al., 2013). This idea originates from findings that stressed mice display a shift toward preferentially breaking down carbohydrates as a fuel source, while protecting adipose tissue, in a ghrelin-dependent manner (Patterson et al., 2013). In contrast, others have demonstrated ghrelin-mediated increases in high-fat diet consumption, along with a stronger conditioned place preference toward a high-fat diet, following a chronic stressor (Chuang et al., 2011). Differences between these data may reflect differences in diet availability during the stress paradigm as well as differences in the time in which the food was presented relative to the stressor itself. Thus, mice may engage in reward-based eating of comfort foods and develop a conditioned place preference to such foods after being stressed (Chuang et al., 2011), however, this may not be the case if they are tested during the stress paradigms, as is the case in our laboratory where they show a marked preference toward a carbohydrate rich diet (Patterson et al., 2013). Furthermore, an association between the presentation of the high-fat diet and the onset of the social stressor may contribute the lack of high-fat consumption seen in wild-type animals (Patterson et al., 2013).
Leptin
Leptin is another peripherally derived hormone, expressed in accordance to the size and amounts of adipocytes that is known to exert some control of feeding behaviors and energy homeostasis. Once secreted from peripheral adipocytes, leptin acts on hypothalamic centers (e.g., ARC) to reduce food intake and increase energy expenditure, thus encouraging a negative energy balance (Friedman, 2002). It has been suggested that leptin is also involved in the metabolic consequences of stress. Circulating glucocorticoids, for example, increase leptin mRNA expression and its secretion from adipocytes (Hardie et al., 1996; Russell et al., 1998; Williams et al., 2000). This pattern of direct glucocorticoid-mediated stimulation of leptin secretion has been shown in both humans and rodents (Miell et al., 1996; Mostyn et al., 2001). Once secreted, leptin directly stimulates anorectic POMC/CART neurons in the ARC and mediates the ability of other metabolically active hormones like ghrelin, for example, to depolarize these neurons (Elias et al., 1998; Cowley et al., 2001). Furthermore, it has been shown that leptin shares an overlapping physiological and intracellular signaling cascade with hormones such as insulin (Niswender and Schwartz, 2003) thus exerting control of feeding circuits from multiple standpoints. There is, however, contradicting data regarding interactions with leptin and glucocorticoids thus making it difficult to interpret the effects of leptin in the context of stress-induced feeding and/or changes in body weight. One group has shown that IP administration of glucocorticoids (mimicking levels seen following an acute stressor) increases leptin concentrations and therefore resulted in a decrease in food intake and body weight (Zakrzewska et al., 1999a). However, the same group demonstrated an opposing role for leptin when glucocorticoids were administered centrally, wherein NPY expression was increased and so too were food intake and body weight (Zakrzewska et al., 1999b). It remains unclear how leptin contributes to stress induced metabolic disorders and more studies are required in order to dissociate the interactions between central vs. peripheral leptin and glucocorticoids in response to stressful stimuli.
Others
In addition to corticosterone, ghrelin and leptin, there are a host of hormones that are stimulated by stress and that contribute directly to the regulation of food intake and energy balance. For instance, cytokines, both inflammatory and anti-inflammatory, are secreted following acute stressors (Black, 2006; Anisman et al., 2008). Chronic stressors, however, result in high levels of cytokines like IL-6 and TNF-α, both of which may contribute to obesity by causing peripheral and central leptin resistance (Velloso et al., 2008). Furthermore, while insulin may not be directly related to the stress response, its interactions with glucocorticoids allow it to participate in shaping an organism's phenotype in response to stress. For example, in the presence of insulin glucocorticoids shift their focus from mobilization of lipids to lipid accumulation (Ottosson et al., 2000). In additions, the interaction between insulin and elevated levels of glucocorticoids has been linked to increased obesity following overconsumption of comfort foods (Dallman et al., 2005). For a more detailed review on the neurochemical control of food intake see (Maniam and Morris, 2012).
Stress Models
There are many disorders/diseases that are studied through the use of animal models, and obesity is no different. Over the past century, many different stress models have been designed, some in attempt to study the effects of stress on depression or anxiety, and others to directly study the effects of stress on food intake and obesity. While it is well-documented that stress is associated with changes in body weight in rodents (Barnett, 1958; Barnett et al., 1960; Marti et al., 1994) it is not always the case that animal models mimic human reality and the basis of dissimilar findings remains poorly understood. Here, we will examine several different stress models used in the context of obesity and discuss the benefits and pitfalls of each.
Visible Burrow System
The Visible Burrow System (VBS) was originally developed as a naturalistic social stressor in attempt to investigate agonistic behaviors in rats (Blanchard et al., 1977, 1995). This model reproduces an underground burrow system, which is thought to mimic the natural habitat of a rat. Traditionally, four adult male rats, and two adult female rats, are group housed in the VBS and a social hierarchy is rapidly formed within days yielding one dominant and three submissive male animals (Tamashiro et al., 2006). This provides a very unique scenario for scientists interested in studying aggressive behaviors in rodents in a “naturalistic” setting wherein there is little requirement for intervention by the experimenter. Since its inception, others who are more interested in studying the neuroendocrine systems involved with stress, feeding and energy homeostasis have adopted the VBS. Inside the VBS, submissive animals show increased basal CORT (Blanchard et al., 1993, 1995; McKittrick et al., 1995, 2000), increases in testosterone (Blanchard et al., 1995; Hardy et al., 2002; Tamashiro et al., 2004) and reliably lose ~10–15% of their body weight (Blanchard et al., 1995; Hardy et al., 2002; Tamashiro et al., 2004). This reduction in body weight has been carefully attributed to decreases in food intake and changes in metabolic rate, while other factors such as basal locomotor activity have been eliminated as contributing factors (Tamashiro et al., 2004, 2006). The molecular underpinnings of such a robust and reliable reduction of food intake are not entirely clear. The VBS has been shown to elicit no change in the expression of orexigenic peptides or hormones, but rather elicits a reduction of leptin and insulin in submissive animals. More experiments are required for a detailed description of changes in hypothalamic feedings circuits that underlie the metabolic outcome of this stress paradigm. As a result of decreased food intake and changes in metabolic rate, submissive animals in the VBS show reductions of adiposity and a decline in lean body mass (Tamashiro et al., 2004). Interestingly, if the animals are removed from the VBS and allowed to recover, in attempt to correct their metabolic phenotype, they show a restoration of most physiological and endocrine measures, but will not regain body weight to levels equivalent to dominant or non-stressed control animals (Tamashiro et al., 2006). Animals immediately become hyperphagic and it has been shown that the gradual accumulation of body weight during a 3 week recovery from the VBS is the result of increased visceral adipose tissue (Tamashiro et al., 2006). Interestingly, the animals display persistent anhedonia and will consume less sucrose during the recovery period, compared to controls (Tamashiro et al., 2007a).
In this regard, it would appear that the VBS is not an ideal stress paradigm for the study of obesity. While there are certainly some well-documented increases in adipose tissue observed in submissive animals, this is a relative measurement to where the animals were following the participation in the VBS. In absolute terms, these animals show an overall reduction of food intake, body weight and visceral adipose tissue and therefore do not mimic human cases of stress-induced obesity. Perhaps these findings are of no surprise given the endocrine profile of these animals. Increased testosterone, for example, as seen in submissive animals has been shown to predict reduced adipose tissue (Krotkiewski et al., 1980). Interestingly, in the absence of females inside the VBS, submissive animals do not show any changes in body weight (Tamashiro et al., 2007a). It is possible that competition for resources (food, water, enrichment, females, etc.) results in submissive animals lacking energetic resources required to develop an obese-like phenotype.
Resident Intruder
The resident intruder paradigm, also known as social defeat stress, has long been used as a method for studying the effects of social dominance and subordination in animals (Ginsburg and Allee, 1942; Miczek, 1979; Miczek et al., 1982). Social hierarchies result in the expression of dominant and submissive behavioral and neuroendocrine patterns that can be used to study the effects of chronic stressors. When compared to other types of stress paradigms such as foot shock or restraint stress, the resident intruder paradigm appears to elicit the most robust physiological response, as measured by changes in hormonal and cardiovascular function (Koolhaas et al., 1997; Sgoifo et al., 1999; Keeney et al., 2006). The territorial nature of rodents allows experimenters to manipulate social hierarchies in attempt to study the physiological consequences of chronic social stressors. When an animal is single housed for ~1 week, there is a rapid development of territorial behavior within the home cage and this animal is said to be the resident (Bartolomucci et al., 2001). Traditionally, in the resident intruder paradigm, the experimenter presents a novel, non-littermate rodent to the resident inside the home cage. The novel animal is thus considered an intruder and will have to fight for his (typically performed in males only) position in the social hierarchy within that cage. It has been shown that housing animals with littermates, while permitting the formation of a social hierarchy, has no influence on CORT production or anxiety-like behaviors in submissive animals and is thus considered non-stressful (Bartolomucci et al., 2001). In contrast, when housed with a novel, non-sibling rodent, submissive animals show signs of stress such as decreased immune responses (Bartolomucci et al., 2001), increased food intake, increased body weight and fat mass (Haller et al., 1999; Bhatnagar and Vining, 2003; Foster et al., 2006; Patterson et al., 2013). It is therefore necessary to use non-familiar animals when attempting to elicit metabolic changes as a consequence of social defeat stress.
Normally, this paradigm consists of two male rodents, one resident and one intruder. There are several variants of the resident intruder paradigm, all of which reflect differences in predictability, habituation, intensity and ultimately the consequent phenotype. Typically, the intruder is presented in the home cage of the resident and they are allowed to interact until one animal subdues the other. Following interaction, animals are separated by a central divider hemisecting the home cage of the resident (Bartolomucci et al., 2001; Patterson et al., 2013). This divider permits the exchange of sensory information but forbids any physical contact between the animals. By lifting this divider and allowing the two animals to interact, the experiment presents an opportunity for the animals to establish a social hierarchy, creating four possible groups: resident-dominants, resident-subordinates, intruder-dominant and intruder-subordinate (Bartolomucci et al., 2001). The physiological outcome in animals exposed to the resident intruder paradigm seems highly dependent on the nature of the social hierarchy, in particular the change in position within the social hierarchy (Bartolomucci et al., 2001, 2004, 2005; Solomon et al., 2007). Reversal of social status in mice, for example, seen when a dominant resident becomes submissive to the intruder has been associated with immunosuppression (Devoino et al., 2003) and dramatic increases in body weight (Bartolomucci et al., 2004), while residents who did not lose their dominant status showed no changes in immune function (Devoino et al., 2003) and in some cases show reductions in body weight (Bartolomucci et al., 2004). The emergence of aggressive and dominant behavior in non-defeated intruders is considered an immunostimulant (Devoino et al., 2003) and these animals show resistance to the development of obesity, as well as predictable allocation of adipose depots (Solomon et al., 2007). Therefore, status in the social hierarchy within the resident-intruder paradigm, as seen in the VBS, can be used to predict the metabolic outcome of this stress paradigm and can even predict the location wherein animals will store adipose tissue (Solomon et al., 2007). In the context of chronic social defeat stress, some laboratories choose novel residents each day of defeat (Bhatnagar and Vining, 2003; Devoino et al., 2003; Foster et al., 2006; Keeney et al., 2006; Krishnan et al., 2007; Lutter et al., 2008a,b; Chuang et al., 2011). By employing novel residents, the experimental subjects are faced with a new social hierarchy within each unique episode of defeat. In contrast, other laboratories reuse the resident, thus maintaining an exclusive social hierarchy for the duration of the experiment (Bartolomucci et al., 2001, 2004, 2010; Moles et al., 2006; Solomon et al., 2007; Patterson et al., 2013). The former prevents habituation through predictability and the latter leads to eventual habituation and is thus considered a milder form of chronic social defeat.
Interestingly, the metabolic phenotypes generated in concert with positions in social hierarchies can be substantiated by the hormonal profile of the animals. Submissive animals in the resident intruder paradigm, for example show elevations in CORT and blood glucose, but also increases in circulating orexigenic/adipogenic plasma ghrelin (Lutter et al., 2008b; Patterson et al., 2013) and increased expression of hypothalamic NPY and AgRP (Patterson et al., 2013). Increases in ghrelin signaling leading to subsequent increases in hypothalamic NPY/AgRP results in stimulation of food intake and adiposity, with an emphasis on visceral adiposity leading to obesity. Interruption in ghrelin signaling, via pharmacological blockage or genetic manipulation, can blunt the stress-induced increases of food intake, body weight gain and adiposity seen in submissive animals (Patterson et al., 2013). Furthermore, hormonal changes that influence metabolic phenotype may vary across time throughout a chronic social defeat stress paradigms. Some physiological changes, for example, are seen immediately following an episode of defeat (Bhatnagar and Vining, 2003; Bartolomucci et al., 2004, 2005; Foster et al., 2006; Keeney et al., 2006; Lutter et al., 2008b) whereas others do not emerge until the subject has been removed from the stress paradigm and allowed to recover (Keeney et al., 2006; Moles et al., 2006; Solomon et al., 2007; Lutter et al., 2008b; Chuang et al., 2011; Patterson et al., 2013).
There are several advantages to using the resident intruder paradigm in attempt to study the physiological consequences of chronic stress. One distinct advantage is there has been a clear dissociation between food intake and body weight changes, where stressed animas will show distinct changes in body weight relative to non-stressed controls with no changes in food intake, thus suggesting that stress-induced changes in body weight and body composition are not solely attributed to increases or decreases in food intake (Haller et al., 1999; Bhatnagar and Vining, 2003; Bartolomucci et al., 2004; Moles et al., 2006; Solomon et al., 2007). Some groups have taken this one step further and dissociated the social stress aspect from the physical stress aspect of an episode of defeat and shown that social stress alone can produce changes in caloric intake (Moles et al., 2006). Interestingly, these changes in metabolism appear to extend beyond the last social interaction between resident and intruder. Subordinate animals in the resident intruder paradigm, for example, continue to show increases in body mass even weeks after last social defeat (Solomon et al., 2007; Patterson et al., 2013), when levels of food intake were equivalent to non-stressed controls (Solomon et al., 2007) suggesting that this stress paradigm left an imprint on the metabolism of these animals. This increase in body mass has been attributed to the ghrelin-dependent shift in carbohydrate utilization resulting in an increase in adipose visceral adipose tissue (Bartolomucci et al., 2001, 2004; Patterson et al., 2013) and increase overall lipid content (Moles et al., 2006; Solomon et al., 2007).
Another advantage to using the resident intruder paradigm is its apparent external validity. It has been well-documented in human cases that a higher position on a social hierarchy (as measured by socioeconomic status, for example) is associated with decreased reported stress levels (Cohen et al., 2006; Sujoldzic and De Lucia, 2007; Burdette and Hill, 2008), healthier diets (McEwen, 1998; Cartwright et al., 2003; Burdette and Hill, 2008), decreased body mass index (BMI) (George et al., 2005; Brunner et al., 2007; Burdette and Hill, 2008), etc. On the contrary, being lower on the socioeconomic hierarchy has been associated with increased stress and increased BMI (Hellerstedt and Jeffery, 1997; Laitinen et al., 2002; George et al., 2005; Cohen et al., 2006), thus shaping the trends in obesity such that humans are likely to be healthier and more fit then those beneath them on the socioeconomic ladder, regardless of their starting position. While the VBS also involves a similar type of stressor, one major difference between these two paradigms is food availability. Because a divider in the resident intruder paradigm separates the animals, they are granted unimpeded access to ad libitum calories, as is seen in human cases of chronic social stress.
Finally, while some stressors have anorectic effects, this particular stress paradigm causes submissive animals to overeat (Patterson et al., 2013). Overall, animals placed in the resident intruder paradigm eat more foods that are high in fat relative to non-stressed controls, but subordinate animals show increases in high fat diet intake relative to their dominant counterparts (Moles et al., 2006). However, it should be noted that the consumption of a high fat diet as a consequence of chronic stress varies from one study to another and may depend on other factors such as diet availability (time and location), gender and animal strain.
While acute social defeat is considered to be a potent stressor by means of HPA axis activity (ACTH, CORT, immune function, etc.) within hours of a defeat episode (Koolhaas et al., 1997; Sgoifo et al., 1999; Keeney et al., 2006) chronic social defeat stress results in significant habituation (Keeney et al., 2006; Patterson et al., 2013) wherein defeat stress elicits a bi-phasic response from the HPA axis. Within the first 24 h, a dramatic increase in CORT is evident following an episode of defeat. If the paradigm is continued, the rise in CORT in response to an episode of defeat is blunted for the next 2 weeks, but will show another spike if the paradigm is continued beyond 2 weeks (Keeney et al., 2006). Furthermore, there is a discrepancy in food intake, hypothalamic CRF expression and glucocorticoid secretion between acute vs. chronically defeated animals (Keeney et al., 2006; Moles et al., 2006; Patterson et al., 2013). These data are indicative of an adaptive stress response, whose ability to downplay the potency of the stressor is limited and may become detrimental in response to chronic social defeat (Keeney et al., 2006).
Chronic Mild Stressors
Chronic mild stress (CMS) has been well-documented for its ability to produce depression and the wide range of accompanying physical, behavioral and neurochemical changes that accompany depression (e.g., Anhedonia) (Willner et al., 1992; Willner, 1997; Stohr et al., 2000). Generally speaking, chronic mild stress is composed of daily stressors generated randomly from a select list of possible stressors, however, CMS protocols can vary substantially from one laboratory to the next. Daily stressors could be housing the animal in a small cage or a cage with wet bedding, housing in an overcrowded cage, cage tilt, foot shock, tail pinch, changes to light/dark cycle (24 h of light, for example), housing in a soiled cage, predator odor, loud noises and more (Willner, 1991, 1997; Muscat and Willner, 1992; Willner et al., 1992; Stohr et al., 2000; Duncko et al., 2001; Westenbroek et al., 2003; Patterson et al., 2010).
The most notable physiological consequence of CMS is the induction of anhedonia, however, this model is also capable of producing impairments in conditioned place preference, reductions in brain stimulation reward, decreased sexual behavior, compromised immune function and irregular sleep architecture (Willner, 1997). These consequences of CMS are reminiscent of the physiological and neurochemical/neuroendocrine changes observed in human cases of depression, and many consequences of CMS can be corrected with chronic anti-depressant administration (Willner et al., 1992; Moreau et al., 1994). Because chronic, but not acute, treatment with generic antidepressants can consistently reduce the consequences of CMS, it is considered one of the best animal models of depression.
However, very few studies that use CMS elicit an obese like state in their experimental animals. Instead, it has been shown that CMS elicits decreases in the consumption of highly palatable foods (Muscat and Willner, 1992; Patterson et al., 2010) and subsequently body weight (Muscat and Willner, 1992; Westenbroek et al., 2003; Patterson et al., 2010). It is difficult to map out the hormonal and neurochemical responses as a consequence of CMS due to the inherent variability of stress paradigms. Within any given CMS protocol exists multiple forms of stressors, each capable of eliciting a unique feeding response. Furthermore, it is difficult to draw similarities between animal models of CMS and any type of human chronic stress. As previously mentioned, any given stressor poses a distinct physiological challenge to an organism and therefore elicits a distinct response. One must consider what type of CMS can be applied to a human scenario before attempting to model human obesity.
Physical Stressors
Despite the growing body of evidence suggesting that psychological stress is one of the largest sources of human stress and may in fact contribute to the obesity epidemic, most animals models aimed at investigating the role of stress on metabolic processes focus on physical, rather then psychosocial, stressors (Tamashiro et al., 2006). Physical stressors are often used in tandem with other forms of stressors in chronic stress models (e.g., UCMS) or, more frequently, as acute stressors on their own.
Tail pinch/foot shock
Tail pinch and foot shock, for example, are two physical stressors used commonly and have both been shown to have an impact on feeding behaviors in rodents. It was shown very early on that chronic tail pinch in rats is capable of eliciting the consumption of an energy rich diet (Rowland and Antelman, 1976). In this particular experiment the rats were encouraged to consume calories through the presence of a highly palatable, energy rich diet. It is unclear if tail pinch is sufficient enough to induce physiological changes capable of influencing dietary habits. Rather, it has been suggested that these feeding responses are a product of general arousal as opposed to an induction of motivated feeding behaviors (Greeno and Wing, 1994). Furthermore, it has been shown that an increase in feeding behavior is not observed in all subjects exposed to tail pinch, and is therefore thought to produce unreliable feeding responses in rodents (Levine and Morley, 1981).
Foot shock, on the other hand, has been shown to increase circulating plasma glucose, insulin and leptin in rats (Farias-Silva et al., 2002; Solomon et al., 2007) suggesting an anorectic hormonal profile. The hyperglycemia and hyperleptinemia observed in animals exposed to foot shock has been attributed to insulin subsensitivity in adipose tissue (Farias-Silva et al., 2002). Despite the anorectic hormonal profile in response to repeated foot shock, animals show no change in food intake but still maintain a positive energy balance and show greater cumulative weight gain relative to non-stressed controls (Solomon et al., 2007). The changes in body mass and body composition following repeated foot shocks are said to be reminiscent of the metabolic consequences of subordinate animals in the intruder resident paradigm, albeit occurred at a much faster rate (Solomon et al., 2007).
Restraint stress/immobilization
Physical restraint and immobilization are other commonly used methods of acute physical stressors. Restraint stress is regarded as a form of mild stress, as indicated by physiologic and neuroendocrine responses, however, it has been shown to increase expression of orexigenic peptides in the hypothalamus nonetheless (Chagra et al., 2011). Interestingly, the expression of orexigenic peptides seems to increase when restraint stress is repeated over time (Chagra et al., 2011) and it has been shown that feeding responses to restraint stress are maintained in the absence of the stressor (Tamashiro et al., 2007a). Mild restraint stress has the ability to increase the number of AgRP-expressing neurons, while simultaneously reducing α-MSH receptors, in the ARC (Chagra et al., 2011). The pattern of hypothalamic activation seems to change with repeated exposure to restraint stress. Specifically, cFos activation in the PVN, ARC and LH can be observed following acute restraint, but activation is subsided with repeated exposures suggesting habituation to this stress paradigm (Chagra et al., 2011).
Exposure to an immobilization stress, on the other hand, elicits a dramatic increase in CORT secretion and is considered to be one of the most potent stressors with respect to HPA axis hyperactivity (Haque et al., 2012). Following immobilization stress, rats have been shown to decrease their food-intake and body weight (Haque et al., 2012; Wang et al., 2012), an indication of the stressors potency. Similarly, immobilization stress has been shown to increase anorectic leptin and leptin receptor (ob-R) expression in the rat hypothalamus, particularly the ARC (Wang et al., 2012). Furthermore, no changes in NPY expression are observed following immobilization stress (Wang et al., 2012), providing hormonal data to support the feeding responses to this type of stress paradigm. It is unclear as to whether chronic exposure to immobilization stress exaggerates or blunts the feeding responses seen following acute exposure. There exists conflicting data pertaining to the potential for immobilization stress habituation (Marquez et al., 2002; Dal-Zotto et al., 2004; Wang et al., 2012).
Cold stress
Exposure to cold (cold stress) has been used in both rodent and human models of stress. In humans, a cold stress produces a significant rise in cortisol levels at about 15 min following the onset of the stressor (Geliebter et al., 2012). Normally, in stress protocols that are capable of eliciting an obese like phenotype, cortisol secretion can be correlated to subsequent caloric intake. However, the cortisol secretion observed in human patients following a cold stress do not correlate with subsequent caloric intake and these subjects show an overall decrease in the amount of calories consumed following the stressor relative to non-stressed controls (Geliebter et al., 2012). In contrast, cold stress in rodents has been shown to increase CORT secretion and CORT secretion was directly correlated to the upregulation of hypothalamic orexigenic peptides (e.g., NPY/AgRP) (Kuo et al., 2007). When cold stress was combined with the presence of a high-fat diet, there appears to be a significant increase in NPY and its receptor (Y2R) in white adipose tissue (Kuo et al., 2007). Furthermore, these changes were thought to elicit proliferation and differentiation of adipocytes, ultimately producing obesity and a metabolic syndrome-like condition (Kuo et al., 2007). Similar increases in Y2R in white adipose tissue have been shown following subordination stress (Patterson et al., 2013), however, it remains unclear if changes in NPY circuitry in response to cold stress are an attempt to insulate/thermoregulate or if they are due to marked changes in metabolism leading to an obese like state.
Given that cold stress can be consistently applied to both human and animal experimental subjects, but produce opposing metabolic outcomes, it is not considered to be a good model for studying stress-induced obesity. Decreasing body temperature, as can be expected during a cold stress paradigm, is metabolically expensive to an organism, as autonomic processes will immediately attempt to thermoregulate. Thermoregulation is metabolically expensive and might therefore elicit a feeding response to replenish the organisms energy stores. This in itself can be considered a major confound when attempting to study stress-induced obesity. Further studies are required to better define the role of orexigenic transcripts in response to cold stress.
Factors Influencing Metabolic Outcome
One hurdle faced by those using animal models to study stress-induced obesity is choosing the right model, but also the right parameters. One of the biggest difficulties when comparing results between laboratories is that their stress paradigms are often associated with different parameters (predictability, presence/absence of a calorically dense diet, presence/absence of a recovery period, time of day, etc.) Undoubtedly, there are many variables that can be manipulated by an experimenter when using any of the aforementioned stress paradigms, all of which appear to influence the metabolic outcome of the stress paradigm.
Predictability
Predictability of a stressor will occur when experimental routines are repeated multiple times, at the same time of day in accordance with the light/dark schedule. It is well-known that the stress response adapts quickly to the presence of a stressor (McEwen, 1998, 2004; McEwen and Wingfield, 2010), and it has therefore been suggested that unpredictability of stressors will exaggerate the normal physiological response to that stressor. Intermittent bouts of social defeat, for example, have been shown to elicit very large CORT responses (Koolhaas et al., 1997; Sgoifo et al., 1999; Moles et al., 2006) and greater increases in adipose tissue deposition in rodents when compared to repeated bouts in chronic social defeat (Foster et al., 2006). Repeated bouts, on the other hand, become habituated to and elicit a dampened HPA axis response and metabolic adaptations (Keeney et al., 2006; Patterson et al., 2013). By varying the time lapse between aggressive episodes in the resident-intruder paradigm, habituation of the physiological response to the stressor cannot occur. If sustained for an extended period of time, this response may overwhelm the homeostatic mechanisms and increase the potency of that stress paradigm.
Similarly, intermittent, or acute, foot shock has been shown to cause a significant reduction in food intake and body weight, as do most physical stressors. Interestingly, when chronic foot shocks are delivered, animals are able to predict their presence, and they show no changes in caloric consumption or body weight (Griffiths et al., 1992), again lending the experimenter control over the metabolic outcome through predictability.
One must consider the evolutionary purpose of the physiological response to stress when discussing how predictability influences the outcome of a particular stressor. The physiological responses to stress are generated in attempt to meet the energetic demands of the particular stressor, liberate the appropriate energy resources while temporarily down regulating any non-pertinent bodily functions. As previously discussed, the acute stress response is an adaptive response and is one that is necessary for the survival and well-being of an organism. However, this may not be the case when dealing with protracted stressors and even more so to unpredictable protracted stressors. Habituation of the CORT response and other secreted hormones may be indicative that the demand for energy in response to the stressor may also have familiarized, thus avoiding any unnecessary changes to metabolism. However, if the animal cannot predict the presence of the stressor, then metabolic habituation is not possible which exerts the largest expense on the organism and ultimately produces the greatest metabolic consequence.
Intensity
The intensity of a stressor can be gauged through several objective measures such as glucocorticoid secretion (i.e., CORT), circulating CRF/ACTH levels, cFos activation in stress related brain regions, etc. There are limited number of studies investigating the relationship between stress severity and metabolic outcome, however, as a rule of thumb, the magnitude of food intake reduction as a consequence of stress can be used as a general marker of stress severity. For example, in an experiment wherein 3 types of physical stressors were used to represent low, medium and high intensity (handling, restraint, and immobilization) there was a graduated anorexic response as the intensity of the stressors increased (Marti et al., 1994). Interestingly, in this particular experiment they also employed a restraint stress (medium intensity) for different durations of time and showed no differences in reactivity, suggesting that it is in fact dependent on the stressor itself and not the length of time it is administered (Marti et al., 1994). The same pattern has been shown when comparing feeding responses between a mild food shock (1.5 mA) and a potent immobilization stressor, where immobilization leads to a significantly greater reduction in food intake, body weight and increased CORT secretion (Rabasa et al., 2011). Similar findings have been reported from human cases, wherein eating patterns of US Marine's are negatively correlated with severity of stress exposure (Popper et al., 1989). Stress severity not only influences feeding response during time of stress, but it may also predict the persistent feeding behaviors during the recovery period (Harris et al., 1998; Valles et al., 2000). This suggests that stress severity, even in acute stressors, may in fact shape metabolic outcome of stress paradigm for the long-term.
Physiologically speaking, what makes one stressor more potent then the next? Of course one would have to consider varying levels of glucocorticoid secretion, or other metabolically active hormones, given the extent to which each stressor activates CRF neurons in the PVN. However, this may not be the case. It has been shown that many different stress paradigms, both physical and psychological stressors, with seemingly different potencies elicit indiscriminate activation of the HPA axis, with measurable differences in amygdala activation (Dayas et al., 2001). Furthermore, it is well-documented that the melanocortin system is an integral component to the anorexic responses to stress. Interestingly MC4 receptors are densely expressed in the amygdala, and when activated, these particular receptors potently inhibit food intake (Liu et al., 2013). Therefore, perhaps analysis of regional activation, outside of the HPA axis, in areas like the amygdala can better predict the intensity of a stressor and therefore the metabolic outcome. More experiments need to be conducted to better understand this phenomena.
Gender Differences
Like many other disorders (e.g., schizophrenia, major depression, anxiety, etc.), gender seems to play a key role in determining the overarching consequences of stress on body weight management. While there exists evidence suggesting that, in humans, there is no sex difference in stress exposure (Young and Korszun, 2010), there also exists evidence suggesting that women are more likely to acknowledge and/or recognize an event as being stressful (Goldman et al., 2005). In humans, women often report more stress-related eating (Greeno and Wing, 1994) and more consumption of foods high in fats and sugars in response to stressful situations (Wansink et al., 2003) compared to men. In support of these data, it has been suggested that perhaps the feeding responses to stressors is different between men and women as a result of their coping strategies (Grunberg and Straub, 1992). Men, for example, tend to display the typical fight or flight response to a stressor while women are more likely to seek out social support following stress, a strategy termed “tend-befriend” (Taylor et al., 2000).
Once again, we look to the use of animal models to further clarify the role of gender in stress induced metabolic dysfunctions. Under basal conditions, female rats show higher levels of CORT then do males (Stohr et al., 2000) which sets the stage for divergent phenotypes in response to stress. There is an abundance of evidence demonstrating gender-specific effects of social defeat stress (Haller et al., 1999), social instability stress (Haller et al., 1999), chronic mild stress (Stohr et al., 2000; Dalla et al., 2005, 2008; Shors et al., 2007), foot shock (Iwasaki-Sekino et al., 2009) acute forced swim test and the development of learned helplessness (Shors et al., 2007; Dalla et al., 2008, 2010). There is very limited data pertaining to the effect of social stressors on females. In the context of social defeat, males seem to be more vulnerable to changes in body weight and food intake then do females (Haller et al., 1999). It is possible that the aggressive nature of males renders them more susceptible to detrimental effects of social stressors with an aggressive component. In comparison, social stressors that lack aggression (e.g., overcrowded housing or social isolation, unpredictable social environments) elicit a greater response in female rats compared to males (Haller et al., 1999; Schmidt et al., 2010).
Given that, in humans, women are twice as likely to develop major depression then men (Kendler et al., 2002; Marcus et al., 2005), the majority of animal work done investigating gender differences in response to stress have employed the use of CMS and acute physical stressors due to their superiority in modeling human depression. It has been shown that both CMS and acute foot shock elicits an exaggerated CORT response in females compared to males (Dalla et al., 2005; Iwasaki-Sekino et al., 2009), suggesting that gender affects stress-related regions of the brain. Additionally, female rats show disruption of estrous cycles, increased floating time in a forced swim test, no reduction of body weight and dramatic changes in hippocampal serotonergic and dopaminergic neurotransmission compared to male rats, in response to CMS (Dalla et al., 2005). Interestingly, these data reflect, to some extent, human conditions of depression wherein hypo-serotonergic function in the hippocampus may lead to depressive like symptoms and correction with antidepressants, whom increase serotonergic neurotransmission, can alleviate atrophy seen in hippocampal neurons. Here we see that female rats showed reduced hippocampal serotonin turnover in response to CMS, whereas males showed no changes (Dalla et al., 2005). As would be expected, these same female rats show reduced locomotion in an open field test and increased immobility in a forced swim test (Dalla et al., 2005), behaviors reminiscent of human depression.
Genetics and Genetic Models
It is not surprising that genetics also contribute to the metabolic outcome of some stress paradigms. When discussing rodent models of stress, different rodent strains will reliably acquire distinct metabolic phenotypes in response to equivalent stressors. It is well-documented that different strains of rodents show different effects of social stress (Berton et al., 1998) and unpredictable chronic mild stressors including both physical and psychological components (Stohr et al., 2000; Mineur et al., 2003; Pothion et al., 2004; Yalcin et al., 2008). Under basal conditions, for example, Fischer rats show a hyperactive HPA axis, as evidenced by elevated CORT levels, compared to the hypoactive HPA axis of the Lewis rat (Stohr et al., 2000). This provides a unique opportunity for experimenters to investigate the relationship between HPA axis activity and metabolic outcome of stress paradigms. For example, it has been shown that Fischer rats are more susceptible to the anxiogenic effects of CMS compared to Lewis rats, as would be expected based on their genetic differences (Stohr et al., 2000). Fischer rats also demonstrate differences in their peak CORT response and negative feedback loops associated with the HPA axis as measured by a two fold increase in latency to return to basal CORT levels following a stressor (Stohr et al., 2000). Interestingly, in this study Lewis rats showed greater startle responses and more risk assessment behaviors as compared to Fischer rats (Stohr et al., 2000), an effect thought to be mediated by CRF.
The same phenomena has been demonstrated in different mouse strains, wherein BALB/c and C3H/He strains appear more sensitive to an unpredictable CMS compared to C57BL/6J mice, for example (Mineur et al., 2003). Similarly, differences in body weight changes, sucrose consumption, water maze performance, grooming behaviors, coat characteristics and responses to forced swim following unpredictable CMS vary significantly over different strains of mice (Mineur et al., 2003; Pothion et al., 2004; Yalcin et al., 2008). In a study by Pothion et al. (2004), it was demonstrated that sucrose preference over water, and baseline body weights, were consistent across 11 different strains of mice. When exposed to an unpredictable CMS, however, some obvious strain differences were observed wherein stressed mice consumed significantly less sucrose (CBA/H and C57 BL/6J) and gained significantly less weight (CBA/H only) relative to control mice, a phenomena that did not occur in any other strain (Pothion et al., 2004). Similarly, others have shown a reduction in sucrose consumption in CD-1, DBA/2J, C57 BL/6J and BALB/C mice in response to acute footshock while this effect was entirely absent in A/J mice (Griffiths et al., 1992). Furthermore, the extent of sucrose consumption decrease varied across different strains of mice where CD-1, DBA/2J and BALB/C mice showed a greater reduction compared to that of C57BL/6J and A/J mice (Griffiths et al., 1992).
In addition to comparing how different rodent strains respond to a variety of stress paradigms, many have used knock-out models, wherein the gene encoding a specific peptide or receptor has been removed, to explore the roles and contributions of these peptide signaling systems in response to stress. The use of ghrelin and ghrelin receptor knockout mice has been instrumental in determining the role of central ghrelin in mediating the orexigenic, adipogenic and anxiolytic effects of ghrelin following chronic social defeat stress (Lutter et al., 2008b; Patterson et al., 2013). Others have used the well-defined leptin deficient ob/ob mouse model to determine leptin's role in stress-induced feeding and anxiety. The obese-like phenotype in ob/ob mice is characterized by hypercorticosteronemia and leptin injections given to ob/ob mice reverse this hormonal profile (Ahima et al., 1996) suggesting that leptin plays a key role in the regulation of the HPA axis. It should be of no surprise that knock-out models that interrupt normal ghrelin and/or leptin signaling will influence the metabolic response to a stressor given the dense expression of ghrelin and leptin receptors on neurons containing NPY/AgRP, POMC/CART, CRF, ACTH and more. Similarly, urocortin knock-out animals have been used to help characterize the role or urocortins in mediating the physiological response to stress. Urocortin-2 deficient mice, for example, show elevations in basal stress hormones (i.e., ACTH) and do not show a rebound feeding in response to a 2 h caloric restriction test (Chen et al., 2006). Furthermore, urocortin-2 deficient mice display an antidepressant-like phenotype when exposed to both a forced swim test and a tail-suspension test (Chen et al., 2006). CRF2 receptor deficient mice also show premature inactivation of the HPA axis (Coste et al., 2000), both illustrating urocortin's influence on HPA axis activation as well as stress induced food intake and anxiety (Coste et al., 2000; Chen et al., 2006). The same patterns of diverse physiological responses can be expected from other rodent models wherein the normal expression of hypothalamic peptides is interrupted genetically.
Interestingly, the responses to antidepressants can also vary across different rodent strains and genetically modified rodent models (Yalcin et al., 2008) suggesting not only a genetic component to stress susceptibility but to the treatment of stress-related disorders as well. For example, it has been shown that hypothalamic orexigenic peptides such as ghrelin and orexin mediate the depressive-like symptoms and antidepressant-like effects of calorie restriction following chronic stress (Lutter et al., 2008a,b). These findings are relevant given the association between stress, depression and feeding. Calorically restricted orexin-null mice, for example, show anxiety like behaviors following chronic social defeat where wild-type littermates do not (Lutter et al., 2008a). Similarly ghrelin is capable of dampening the depressive like symptoms that normally arise following chronic social defeat (Lutter et al., 2008b). These data highlight the ability of hypothalamic feeding circuitry to influence the physiological response to stress and alter the behavioral and metabolic responses generated.
Conclusion
It has been well-established that stressful environments are conducive to the development of obesity and related metabolic disorders. Indeed, the stress response represents a metabolic challenge that is followed by physiological and behavioral responses in attempt to meet this challenge. Evolutionary pressures have shaped these homeostatic and allostatic responses, yet continued exposure to stressors and the constant struggle to meet these energetic demands may ultimately serve as detrimental. Chronic stress may therefore alter the response to stressors and tilt the energy homeostatic scale toward an increase in energy consumption and adiposity. A detailed understanding of the interaction between the stress response, the nature of the stressors and their impact on food intake and energy balance regulation may allow for more personalized treatments of obesity, be it pharmacologically or psychologically based. The use of animal models will certainly increase our understanding of these types of interactions and aid in defining the neurobiological underpinnings of consequent metabolic disorders. However, a comprehensive understanding of the wide array of stress paradigms poses its own challenges. Using variants, or derivatives, of established stress paradigms makes any type of direct comparison of data between laboratories difficult. As a result, many different stressors are broadly categorized (e.g., Table 1) and may not reflect the physiological differences between them. Perhaps an attempt can be made to better distinguish the details surrounding these stress paradigms or implement some type of standardization. One thing is for certain, however, in that many different stressors pose distinctive energetic challenges on an organism, and while metabolic processes are adjusted to compensate for these challenges, many result in the conservation of adipose tissue.
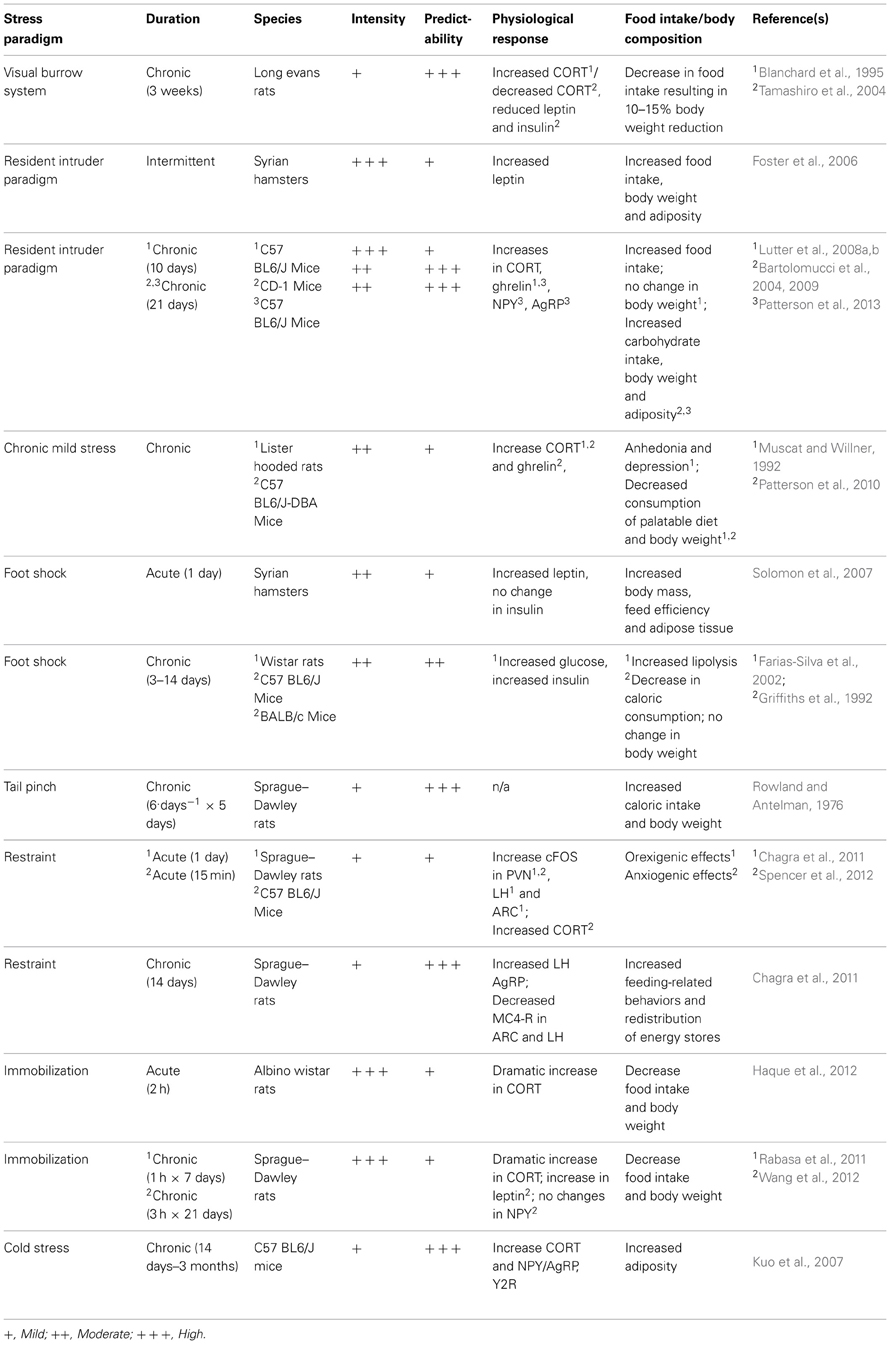
Table 1. Summary of stress paradigms, hormonal responses and physiological outcome.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abizaid, A., and Horvath, T. L. (2008). Brain circuits regulating energy homeostasis. Regul. Pept. 149, 3–10. doi: 10.1016/j.regpep.2007.10.006
Abizaid, A., Liu, Z. W., Andrews, Z. B., Shanabrough, M., Borok, E., Elsworth, J. D., et al. (2006). Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J. Clin. Invest. 116, 3229–3239. doi: 10.1172/JCI29867
Aguilera, G. (1994). Regulation of pituitary ACTH secretion during chronic stress. Front. Neuroendocrinol. 15, 321–350. doi: 10.1006/frne.1994.1013
Ahima, R. S., Prabakaran, D., Mantzoros, C., Qu, D., Lowell, B., Maratos-Flier, E., et al. (1996). Role of leptin in the neuroendocrine response to fasting. Nature 382, 250–252. doi: 10.1038/382250a0
Akana, S. F., Strack, A. M., Hanson, E. S., Horsley, C. J., Milligan, E. D., Bhatnagar, S., et al. (1999). Interactions among chronic cold, corticosterone and puberty on energy intake and deposition. Stress 3, 131–146. doi: 10.3109/10253899909001118
Anisman, H., and Matheson, K. (2005). Stress, depression, and anhedonia: caveats concerning animal models. Neurosci. Biobehav. Rev. 29, 525–546. doi: 10.1016/j.neubiorev.2005.03.007
Anisman, H., Merali, Z., and Hayley, S. (2008). Neurotransmitter, peptide and cytokine processes in relation to depressive disorder: comorbidity between depression and neurodegenerative disorders. Prog. Neurobiol. 85, 1–74. doi: 10.1016/j.pneurobio.2008.01.004
Asakawa, A., Inui, A., Kaga, T., Yuzuriha, H., Nagata, T., Fujimiya, M., et al. (2001). A role of ghrelin in neuroendocrine and behavioral responses to stress in mice. Neuroendocrinology 74, 143–147. doi: 10.1159/000054680
Atasoy, D., Betley, J. N., Su, H. H., and Sternson, S. M. (2012). Deconstruction of a neural circuit for hunger. Nature 488, 172–177. doi: 10.1038/nature11270
Baler, R. D., and Volkow, N. D. (2006). Drug addiction: the neurobiology of disrupted self-control. Trends Mol. Med. 12, 559–566. doi: 10.1016/j.molmed.2006.10.005
Bamshad, M., Aoki, V. T., Adkison, M. G., Warren, W. S., and Bartness, T. J. (1998). Central nervous system origins of the sympathetic nervous system outflow to white adipose tissue. Am. J. Physiol. 275, R291–R299.
Barnett, S. A. (1958). Physiological effects of social stress in wild rats. I. The adrenal cortex. J. Psychosom. Res. 3, 1–11. doi: 10.1016/0022-399990012-6
Barnett, S. A., Eaton, J. C., and McCallum, C. H. (1960). Physiological effects of “social stress” in wild rats–II. Liver glycogen and blood glucose. J. Psychosom. Res. 4, 251–260. doi: 10.1016/0022-399990001-5
Bartolomucci, A., Cabassi, A., Govoni, P., Ceresini, G., Cero, C., Berra, D., et al. (2009). Metabolic consequences and vulnerability to diet-induced obesity in male mice under chronic social stress. PLoS ONE 4:e4331. doi: 10.1371/journal.pone.0004331
Bartolomucci, A., Carola, V., Pascucci, T., Puglisi-Allegra, S., Cabib, S., Lesch, K. P., et al. (2010). Increased vulnerability to psychosocial stress in heterozygous serotonin transporter knockout mice. Dis. Model. Mech. 3, 459–470. doi: 10.1242/dmm.004614
Bartolomucci, A., Palanza, P., Gaspani, L., Limiroli, E., Panerai, A. E., Ceresini, G., et al. (2001). Social status in mice: behavioral, endocrine and immune changes are context dependent. Physiol. Behav. 73, 401–410. doi: 10.1016/S0031-938400453-X
Bartolomucci, A., Palanza, P., Sacerdote, P., Panerai, A. E., Sgoifo, A., Dantzer, R., et al. (2005). Social factors and individual vulnerability to chronic stress exposure. Neurosci. Biobehav. Rev. 29, 67–81. doi: 10.1016/j.neubiorev.2004.06.009
Bartolomucci, A., Pederzani, T., Sacerdote, P., Panerai, A. E., Parmigiani, S., and Palanza, P. (2004). Behavioral and physiological characterization of male mice under chronic psychosocial stress. Psychoneuroendocrinology 29, 899–910. doi: 10.1016/j.psyneuen.2003.08.003
Berthoud, H. R., Sutton, G. M., Townsend, R. L., Patterson, L. M., and Zheng, H. (2006). Brainstem mechanisms integrating gut-derived satiety signals and descending forebrain information in the control of meal size. Physiol. Behav. 89, 517–524. doi: 10.1016/j.physbeh.2006.08.018
Berton, O., Aguerre, S., Sarrieau, A., Mormede, P., and Chaouloff, F. (1998). Differential effects of social stress on central serotonergic activity and emotional reactivity in Lewis and spontaneously hypertensive rats. Neuroscience 82, 147–159. doi: 10.1016/S0306-452200282-0
Bhatnagar, S., and Vining, C. (2003). Facilitation of hypothalamic-pituitary-adrenal responses to novel stress following repeated social stress using the resident/intruder paradigm. Horm. Behav. 43, 158–165. doi: 10.1016/S0018-506X(02)00011-9
Bjorntorp, P. (2001). Do stress reactions cause abdominal obesity and comorbidities. Obes. Rev. 2, 73–86. doi: 10.1046/j.1467-789x.2001.00027.x
Bjorntorp, P., and Rosmond, R. (1999). Hypothalamic origin of the metabolic syndrome X. Ann. N.Y. Acad. Sci. 892, 297–307. doi: 10.1111/j.1749-6632.1999.tb07803.x
Bjorntorp, P., and Rosmond, R. (2000). The metabolic syndrome–a neuroendocrine disorder. Br. J. Nutr. 83(Suppl. 1), S49–S57. doi: 10.1017/S0007114500000957
Black, P. H. (2006). The inflammatory consequences of psychologic stress: relationship to insulin resistance, obesity, atherosclerosis and diabetes mellitus, type II. Med. Hypotheses 67, 879–891. doi: 10.1016/j.mehy.2006.04.008
Blackburn, J. R., Pfaus, J. G., and Phillips, A. G. (1992). Dopamine functions in appetitive and defensive behaviours. Prog. Neurobiol. 39, 247–279. doi: 10.1016/0301-008290018-A
Blanchard, D. C., Sakai, R. R., McEwen, B., Weiss, S. M., and Blanchard, R. J. (1993). Subordination stress: behavioral, brain, and neuroendocrine correlates. Behav. Brain Res. 58, 113–121. doi: 10.1016/0166-432890096-9
Blanchard, D. C., Spencer, R. L., Weiss, S. M., Blanchard, R. J., McEwen, B., and Sakai, R. R. (1995). Visible burrow system as a model of chronic social stress: behavioral and neuroendocrine correlates. Psychoneuroendocrinology 20, 117–134. doi: 10.1016/0306-4530E0045-B
Blanchard, R. J., Blanchard, D. C., Takahashi, T., and Kelley, M. J. (1977). Attack and defensive behaviour in the albino rat. Anim. Behav. 25, 622–634. doi: 10.1016/0003-347290113-0
Boudaba, C., Szabo, K., and Tasker, J. G. (1996). Physiological mapping of local inhibitory inputs to the hypothalamic paraventricular nucleus. J. Neurosci. 16, 7151–7160.
Brindley, D. N., and Rolland, Y. (1989). Possible connections between stress, diabetes, obesity, hypertension and altered lipoprotein metabolism that may result in atherosclerosis. Clin. Sci. (Lond.) 77, 453–461.
Broberger, C., Landry, M., Wong, H., Walsh, J. N., and Hokfelt, T. (1997). Subtypes Y1 and Y2 of the neuropeptide Y receptor are respectively expressed in pro-opiomelanocortin- and neuropeptide-Y-containing neurons of the rat hypothalamic arcuate nucleus. Neuroendocrinology 66, 393–408. doi: 10.1159/000127265
Brown, P., and Molliver, M. E. (2000). Dual serotonin (5-HT) projections to the nucleus accumbens core and shell: relation of the 5-HT transporter to amphetamine-induced neurotoxicity. J. Neurosci. 20, 1952–1963.
Brunner, E. J., Chandola, T., and Marmot, M. G. (2007). Prospective effect of job strain on general and central obesity in the Whitehall II Study. Am. J. Epidemiol. 165, 828–837. doi: 10.1093/aje/kwk058
Burdette, A. M., and Hill, T. D. (2008). An examination of processes linking perceived neighborhood disorder and obesity. Soc. Sci. Med. 67, 38–46. doi: 10.1016/j.socscimed.2008.03.029
Cabral, A., Suescun, O., Zigman, J. M., and Perello, M. (2012). Ghrelin indirectly activates hypophysiotropic CRF neurons in rodents. PLoS ONE 7:e31462. doi: 10.1371/journal.pone.0031462
Cai, Y., Hay, M., and Bishop, V. S. (1996). Synaptic connections and interactions between area postrema and nucleus tractus solitarius. Brain Res. 724, 121–124. doi: 10.1016/0006-899300282-X
Cartwright, M., Wardle, J., Steggles, N., Simon, A. E., Croker, H., and Jarvis, M. J. (2003). Stress and dietary practices in adolescents. Health Psychol. 22, 362–369. doi: 10.1037/0278-6133.22.4.362
Caruso, C. C. (2006). Possible broad impacts of long work hours. Ind. Health 44, 531–536. doi: 10.2486/indhealth.44.531
Castaneyra-Perdomo, A., Perez-Delgado, M. M., Montagnese, C., and Coen, C. W. (1992). Brainstem projections to the medial preoptic region containing the luteinizing hormone-releasing hormone perikarya in the rat. An immunohistochemical and retrograde transport study. Neurosci. Lett. 139, 135–139. doi: 10.1016/0304-394090875-8
Castonguay, T. W. (1991). Glucocorticoids as modulators in the control of feeding. Brain Res. Bull. 27, 423–428. doi: 10.1016/0361-923090136-8
Cavagnini, F., Croci, M., Putignano, P., Petroni, M. L., and Invitti, C. (2000). Glucocorticoids and neuroendocrine function. Int. J. Obes. Relat. Metab. Disord. 24(Suppl. 2), S77–S79. doi: 10.1038/sj.ijo.0801284
Chagra, S. L., Zavala, J. K., Hall, M. V., and Gosselink, K. L. (2011). Acute and repeated restraint differentially activate orexigenic pathways in the rat hypothalamus. Regul. Pept. 167, 70–78. doi: 10.1016/j.regpep.2010.11.006
Chalmers, D. T., Lovenberg, T. W., and De Souza, E. B. (1995). Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: comparison with CRF1 receptor mRNA expression. J. Neurosci. 15, 6340–6350.
Chen, A., Zorrilla, E., Smith, S., Rousso, D., Levy, C., Vaughan, J., et al. (2006). Urocortin 2-deficient mice exhibit gender-specific alterations in circadian hypothalamus-pituitary-adrenal axis and depressive-like behavior. J. Neurosci. 26, 5500–5510. doi: 10.1523/JNEUROSCI.3955-05.2006
Chen, P., Vaughan, J., Donaldson, C., Vale, W., and Li, C. (2010). Injection of Urocortin 3 into the ventromedial hypothalamus modulates feeding, blood glucose levels, and hypothalamic POMC gene expression but not the HPA axis. Am. J. Physiol. Endocrinol. Metab. 298, E337–E345. doi: 10.1152/ajpendo.00402.2009
Choi, K., Roh, S. G., Hong, Y. H., Shrestha, Y. B., Hishikawa, D., Chen, C., et al. (2003). The role of ghrelin and growth hormone secretagogues receptor on rat adipogenesis. Endocrinology 144, 754–759. doi: 10.1210/en.2002-220783
Chrousos, G. P., and Gold, P. W. (1998). A healthy body in a healthy mind–and vice versa–the damaging power of “uncontrollable” stress. J. Clin. Endocrinol. Metab. 83, 1842–1845. doi: 10.1210/jc.83.6.1842
Chuang, J. C., Perello, M., Sakata, I., Osborne-Lawrence, S., Savitt, J. M., Lutter, M., et al. (2011). Ghrelin mediates stress-induced food-reward behavior in mice. J. Clin. Invest. 121, 2684–2692. doi: 10.1172/JCI57660
Ciofi, P. (2011). The arcuate nucleus as a circumventricular organ in the mouse. Neurosci. Lett. 487, 187–190. doi: 10.1016/j.neulet.2010.10.019
Clark, J. T., Kalra, P. S., Crowley, W. R., and Kalra, S. P. (1984). Neuropeptide Y and human pancreatic polypeptide stimulate feeding behavior in rats. Endocrinology 115, 427–429. doi: 10.1210/endo-115-1-427
Coccurello, R., D'amato, F. R., and Moles, A. (2008). Chronic social stress, hedonism and vulnerability to obesity: lessons from Rodents. Neurosci. Biobehav. Rev. 31, 623–633.
Cohen, S., Doyle, W. J., and Baum, A. (2006). Socioeconomic status is associated with stress hormones. Psychosom. Med. 68, 414–420. doi: 10.1097/01.psy.0000221236.37158.b9
Cole, M. A., Kim, P. J., Kalman, B. A., and Spencer, R. L. (2000). Dexamethasone suppression of corticosteroid secretion: evaluation of the site of action by receptor measures and functional studies. Psychoneuroendocrinology 25, 151–167. doi: 10.1016/S0306-453000045-1
Cone, R. D., Cowley, M. A., Butler, A. A., Fan, W., Marks, D. L., and Low, M. J. (2001). The arcuate nucleus as a conduit for diverse signals relevant to energy homeostasis. Int. J. Obes. Relat. Metab. Disord. 25(Suppl. 5), S63–S67. doi: 10.1038/sj.ijo.0801913
Coste, S. C., Kesterson, R. A., Heldwein, K. A., Stevens, S. L., Heard, A. D., Hollis, J. H., et al. (2000). Abnormal adaptations to stress and impaired cardiovascular function in mice lacking corticotropin-releasing hormone receptor-2. Nat. Genet. 24, 403–409. doi: 10.1038/74255
Cowley, M. A., Smart, J. L., Rubinstein, M., Cerdan, M. G., Diano, S., Horvath, T. L., et al. (2001). Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411, 480–484. doi: 10.1038/35078085
Cowley, M. A., Smith, R. G., Diano, S., Tschop, M., Pronchuk, N., Grove, K. L., et al. (2003). The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37, 649–661. doi: 10.1016/S0896-627300063-1
Cummings, D. E., and Shannon, M. H. (2003). Roles for ghrelin in the regulation of appetite and body weight. Arch. Surg. 138, 389–396. doi: 10.1001/archsurg.138.4.389
Currie, P. J., Mirza, A., Fuld, R., Park, D., and Vasselli, J. R. (2005). Ghrelin is an orexigenic and metabolic signaling peptide in the arcuate and paraventricular nuclei. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, R353–R358. doi: 10.1152/ajpregu.00756.2004
Dalla, C., Antoniou, K., Drossopoulou, G., Xagoraris, M., Kokras, N., Sfikakis, A., et al. (2005). Chronic mild stress impact: are females more vulnerable. Neuroscience 135, 703–714. doi: 10.1016/j.neuroscience.2005.06.068
Dalla, C., Antoniou, K., Kokras, N., Drossopoulou, G., Papathanasiou, G., Bekris, S., et al. (2008). Sex differences in the effects of two stress paradigms on dopaminergic neurotransmission. Physiol. Behav. 93, 595–605. doi: 10.1016/j.physbeh.2007.10.020
Dalla, C., Pitychoutis, P. M., Kokras, N., and Papadopoulou-Daifoti, Z. (2010). Sex differences in animal models of depression and antidepressant response. Basic Clin. Pharmacol. Toxicol. 106, 226–233. doi: 10.1111/j.1742-7843.2009.00516.x
Dallman, M. F., Pecoraro, N., Akana, S. F., La Fleur, S. E., Gomez, F., Houshyar, H., et al. (2003). Chronic stress and obesity: a new view of “comfort food”. Proc. Natl. Acad. Sci. U.S.A. 100, 11696–11701. doi: 10.1073/pnas.1934666100
Dallman, M. F., Pecoraro, N. C., and La Fleur, S. E. (2005). Chronic stress and comfort foods: self-medication and abdominal obesity. Brain Behav. Immun. 19, 275–280. doi: 10.1016/j.bbi.2004.11.004
Dallman, M. F., Pecoraro, N. C., La Fleur, S. E., Warne, J. P., Ginsberg, A. B., Akana, S. F., et al. (2006). Glucocorticoids, chronic stress, and obesity. Prog. Brain Res. 153, 75–105. doi: 10.1016/S0079-612353004-3
Dal-Zotto, S., Marti, O., Delgado, R., and Armario, A. (2004). Potentiation of glucocorticoid release does not modify the long-term effects of a single exposure to immobilization stress. Psychopharmacology (Berl.) 177, 230–237. doi: 10.1007/s00213-004-1939-y
Date, Y., Shimbara, T., Koda, S., Toshinai, K., Ida, T., Murakami, N., et al. (2006). Peripheral ghrelin transmits orexigenic signals through the noradrenergic pathway from the hindbrain to the hypothalamus. Cell Metab. 4, 323–331. doi: 10.1016/j.cmet.2006.09.004
Dayas, C. V., Buller, K. M., Crane, J. W., Xu, Y., and Day, T. A. (2001). Stressor categorization: acute physical and psychological stressors elicit distinctive recruitment patterns in the amygdala and in medullary noradrenergic cell groups. Eur. J. Neurosci. 14, 1143–1152. doi: 10.1046/j.0953-816x.2001.01733.x
Depke, M., Fusch, G., Domanska, G., Geffers, R., Volker, U., Schuett, C., et al. (2008). Hypermetabolic syndrome as a consequence of repeated psychological stress in mice. Endocrinology 149, 2714–2723. doi: 10.1210/en.2008-0038
Devoino, L., Alperina, E., and Pavina, T. (2003). Immunological consequences of the reversal of social status in C57BL/6J mice. Brain Behav. Immun. 17, 28–34. doi: 10.1016/S0889-159100037-5
Dhillo, W. S., and Bloom, S. R. (2001). Hypothalamic peptides as drug targets for obesity. Curr. Opin. Pharmacol. 1, 651–655. doi: 10.1016/S1471-489200110-2
Di, S., Malcher-Lopes, R., Halmos, K. C., and Tasker, J. G. (2003). Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J. Neurosci. 23, 4850–4857.
Di, S., Malcher-Lopes, R., Marcheselli, V. L., Bazan, N. G., and Tasker, J. G. (2005). Rapid glucocorticoid-mediated endocannabinoid release and opposing regulation of glutamate and gamma-aminobutyric acid inputs to hypothalamic magnocellular neurons. Endocrinology 146, 4292–4301. doi: 10.1210/en.2005-0610
Diaz-Regueira, S., and Anadon, R. (1992). Central projections of the vagus nerve in Chelon labrosus Risso (Teleostei, O. Perciformes). Brain. Behav. Evol. 40, 297–310. doi: 10.1159/000113920
Dinneen, S., Alzaid, A., Miles, J., and Rizza, R. (1993). Metabolic effects of the nocturnal rise in cortisol on carbohydrate metabolism in normal humans. J. Clin. Invest. 92, 2283–2290. doi: 10.1172/JCI116832
Diorio, D., Viau, V., and Meaney, M. J. (1993). The role of the medial prefrontal cortex (cingulate gyrus) in the regulation of hypothalamic-pituitary-adrenal responses to stress. J. Neurosci. 13, 3839–3847.
Dornonville De La Cour, C., Bjorkqvist, M., Sandvik, A. K., Bakke, I., Zhao, C. M., Chen, D., et al. (2001). A-like cells in the rat stomach contain ghrelin and do not operate under gastrin control. Regul. Pept. 99, 141–150. doi: 10.1016/S0167-011500243-9
Duncko, R., Kiss, A., Skultetyova, I., Rusnak, M., and Jezova, D. (2001). Corticotropin-releasing hormone mRNA levels in response to chronic mild stress rise in male but not in female rats while tyrosine hydroxylase mRNA levels decrease in both sexes. Psychoneuroendocrinology 26, 77–89. doi: 10.1016/S0306-453000040-8
Dunn, A. J., Swiergiel, A. H., and Palamarchouk, V. (2004). Brain circuits involved in corticotropin-releasing factor-norepinephrine interactions during stress. Ann. N.Y. Acad. Sci. 1018, 25–34. doi: 10.1196/annals.1296.003
Elias, C. F., Lee, C., Kelly, J., Aschkenasi, C., Ahima, R. S., Couceyro, P. R., et al. (1998). Leptin activates hypothalamic CART neurons projecting to the spinal cord. Neuron 21, 1375–1385. doi: 10.1016/S0896-627380656-X
Ellenberger, H. H., and Feldman, J. L. (1990). Brainstem connections of the rostral ventral respiratory group of the rat. Brain Res. 513, 35–42. doi: 10.1016/0006-899391086-V
Elmquist, J. K. (2000). Anatomic basis of leptin action in the hypothalamus. Front. Horm. Res. 26, 21–41. doi: 10.1159/000061020
Farias-Silva, E., Sampaio-Barros, M. M., Amaral, M. E., Carneiro, E. M., Boschero, A. C., Grassi-Kassisse, D. M., et al. (2002). Subsensitivity to insulin in adipocytes from rats submitted to foot-shock stress. Can. J. Physiol. Pharmacol. 80, 783–789. doi: 10.1139/y02-104
Ferguson, A. V. (1991). The area postrema: a cardiovascular control centre at the blood-brain interface. Can. J. Physiol. Pharmacol. 69, 1026–1034. doi: 10.1139/y91-153
Fields, H. L., Hjelmstad, G. O., Margolis, E. B., and Nicola, S. M. (2007). Ventral tegmental area neurons in learned appetitive behavior and positive reinforcement. Annu. Rev. Neurosci. 30, 289–316. doi: 10.1146/annurev.neuro.30.051606.094341
Flegal, K. M., Carroll, M. D., Kit, B. K., and Ogden, C. L. (2012). Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999-2010. JAMA 307, 491–497. doi: 10.1001/jama.012.39
Flegal, K. M., Carroll, M. D., Ogden, C. L., and Curtin, L. R. (2010). Prevalence and trends in obesity among US adults, 1999-2008. JAMA 303, 235–241. doi: 10.1001/jama.2009.2014
Foster, M. T., Solomon, M. B., Huhman, K. L., and Bartness, T. J. (2006). Social defeat increases food intake, body mass, and adiposity in Syrian hamsters. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R1284–R1293. doi: 10.1152/ajpregu.00437.2005
Friedman, J. M. (2002). The function of leptin in nutrition, weight, and physiology. Nutr. Rev. 60, S1–S14. discussion S68–S84, 85–17.
Furay, A. R., Bruestle, A. E., and Herman, J. P. (2008). The role of the forebrain glucocorticoid receptor in acute and chronic stress. Endocrinology 149, 5482–5490. doi: 10.1210/en.2008-0642
Geliebter, A., Gibson, C. D., Hernandez, D. B., Atalayer, D., Kwon, A., Lee, M. I., et al. (2012). Plasma cortisol levels in response to a cold pressor test did not predict appetite or ad libitum test meal intake in obese women. Appetite 59, 956–959. doi: 10.1016/j.appet.2012.08.025
George, G. C., Milani, T. J., Hanss-Nuss, H., and Freeland-Graves, J. H. (2005). Compliance with dietary guidelines and relationship to psychosocial factors in low-income women in late postpartum. J. Am. Diet. Assoc. 105, 916–926. doi: 10.1016/j.jada.2005.03.009
Germano, C. M., De Castro, M., Rorato, R., Laguna, M. T., Antunes-Rodrigues, J., Elias, C. F., et al. (2007). Time course effects of adrenalectomy and food intake on cocaine- and amphetamine-regulated transcript expression in the hypothalamus. Brain Res. 1166, 55–64. doi: 10.1016/j.brainres.2007.05.077
Ginsburg, B., and Allee, W. C. (1942). Some effects of conditioning on social dominance and subordination in inbred strains of mice. Physiol. Zool. 15, 485–506.
Goldman, N., Glei, D. A., Seplaki, C., Liu, I. W., and Weinstein, M. (2005). Perceived stress and physiological dysregulation in older adults. Stress 8, 95–105. doi: 10.1080/10253890500141905
Grassi, G. (2007). Adrenergic overdrive as the link among hypertension, obesity, and impaired thermogenesis: lights and shadows. Hypertension 49, 5–6. doi: 10.1161/01.HYP.0000251688.60327.37
Green, P. K., Wilkinson, C. W., and Woods, S. C. (1992). Intraventricular corticosterone increases the rate of body weight gain in underweight adrenalectomized rats. Endocrinology 130, 269–275. doi: 10.1210/en.130.1.269
Greeno, C. G., and Wing, R. R. (1994). Stress-induced eating. Psychol. Bull. 115, 444–464. doi: 10.1037/0033-2909.115.3.444
Griffiths, J., Shanks, N., and Anisman, H. (1992). Strain-specific alterations in consumption of a palatable diet following repeated stressor exposure. Pharmacol. Biochem. Behav. 42, 219–227. doi: 10.1016/0091-305790519-L
Grill, H. J., and Kaplan, J. M. (2001). Interoceptive and integrative contributions of forebrain and brainstem to energy balance control. Int. J. Obes. Relat. Metab. Disord. 25(Suppl 5), S73–S77. doi: 10.1038/sj.ijo.0801917
Grill, H. J., and Kaplan, J. M. (2002). The neuroanatomical axis for control of energy balance. Front. Neuroendocrinol. 23, 2–40. doi: 10.1006/frne.2001.0224
Groesz, L. M., McCoy, S., Carl, J., Saslow, L., Stewart, J., Adler, N., et al. (2012). What is eating you. Stress and the drive to eat. Appetite 58, 717–721. doi: 10.1016/j.appet.2011.11.028
Grosch, J. W., Caruso, C. C., Rosa, R. R., and Sauter, S. L. (2006). Long hours of work in the U.S.: associations with demographic and organizational characteristics, psychosocial working conditions, and health. Am. J. Ind. Med. 49, 943–952. doi: 10.1002/ajim.20388
Grunberg, N. E., and Straub, R. O. (1992). The role of gender and taste class in the effects of stress on eating. Health Psychol. 11, 97–100. doi: 10.1037/0278-6133.11.2.97
Hagan, M. M., Rushing, P. A., Schwartz, M. W., Yagaloff, K. A., Burn, P., Woods, S. C., et al. (1999). Role of the CNS melanocortin system in the response to overfeeding. J. Neurosci. 19, 2362–2367.
Halford, J. C., Harrold, J. A., Boyland, E. J., Lawton, C. L., and Blundell, J. E. (2007). Serotonergic drugs: effects on appetite expression and use for the treatment of obesity. Drugs 67, 27–55. doi: 10.2165/00003495-200767010-00004
Haller, J., Fuchs, E., Halasz, J., and Makara, G. B. (1999). Defeat is a major stressor in males while social instability is stressful mainly in females: towards the development of a social stress model in female rats. Brain Res. Bull. 50, 33–39. doi: 10.1016/S0361-923000087-8
Haque, Z., Akbar, N., Yasmin, F., Haleem, M. A., and Haleem, D. J. (2012). Inhibition of immobilization stress-induced anorexia, behavioral deficits, and plasma corticosterone secretion by injected leptin in rats. Stress 6, 353–362. doi: 10.3109/10253890.2012.736047
Hardie, L. J., Guilhot, N., and Trayhurn, P. (1996). Regulation of leptin production in cultured mature white adipocytes. Horm. Metab. Res. 28, 685–689. doi: 10.1055/s-2007-979878
Hardy, M. P., Sottas, C. M., Ge, R., McKittrick, C. R., Tamashiro, K. L., McEwen, B. S., et al. (2002). Trends of reproductive hormones in male rats during psychosocial stress: role of glucocorticoid metabolism in behavioral dominance. Biol. Reprod. 67, 1750–1755. doi: 10.1095/biolreprod.102.006312
Harris, R. B., Zhou, J., Youngblood, B. D., Rybkin, Ii, Smagin, G. N., et al. (1998). Effect of repeated stress on body weight and body composition of rats fed low- and high-fat diets. Am. J. Physiol. 275, R1928–R1938.
Heisler, L. K., Cowley, M. A., Kishi, T., Tecott, L. H., Fan, W., Low, M. J., et al. (2003). Central serotonin and melanocortin pathways regulating energy homeostasis. Ann. N.Y. Acad. Sci. 994, 169–174. doi: 10.1111/j.1749-6632.2003.tb03177.x
Hellerstedt, W. L., and Jeffery, R. W. (1997). The association of job strain and health behaviours in men and women. Int. J. Epidemiol. 26, 575–583. doi: 10.1093/ije/26.3.575
Herman, J. P., Tasker, J. G., Ziegler, D. R., and Cullinan, W. E. (2002). Local circuit regulation of paraventricular nucleus stress integration: glutamate-GABA connections. Pharmacol. Biochem. Behav. 71, 457–468. doi: 10.1016/S0091-305700681-5
Heym, J., and Gladfelter, W. E. (1982). Locomotor activity and ingestive behavior after damage to ascending serotonergic systems. Physiol. Behav. 29, 459–467. doi: 10.1016/0031-938490267-0
Horvath, T. L., Diano, S., and Tschop, M. (2004). Brain circuits regulating energy homeostasis. Neuroscientist 10, 235–246. doi: 10.1177/1073858403262151
Huhman, K. L. (2006). Social conflict models: can they inform us about human psychopathology. Horm. Behav. 50, 640–646. doi: 10.1016/j.yhbeh.2006.06.022
Iwasaki-Sekino, A., Mano-Otagiri, A., Ohata, H., Yamauchi, N., and Shibasaki, T. (2009). Gender differences in corticotropin and corticosterone secretion and corticotropin-releasing factor mRNA expression in the paraventricular nucleus of the hypothalamus and the central nucleus of the amygdala in response to footshock stress or psychological stress in rats. Psychoneuroendocrinology 34, 226–237. doi: 10.1016/j.psyneuen.2008.09.003
Jacobson, L. (1999). Glucocorticoid replacement, but not corticotropin-releasing hormone deficiency, prevents adrenalectomy-induced anorexia in mice. Endocrinology 140, 310–317. doi: 10.1210/en.140.1.310
Jahng, J. W., Kim, N. Y., Ryu, V., Yoo, S. B., Kim, B. T., Kang, D. W., et al. (2008). Dexamethasone reduces food intake, weight gain and the hypothalamic 5-HT concentration and increases plasma leptin in rats. Eur. J. Pharmacol. 581, 64–70. doi: 10.1016/j.ejphar.2007.11.029
Jankowski, M. P., and Sesack, S. R. (2004). Prefrontal cortical projections to the rat dorsal raphe nucleus: ultrastructural features and associations with serotonin and gamma-aminobutyric acid neurons. J. Comp. Neurol. 468, 518–529. doi: 10.1002/cne.10976
Jones, M. T., Hillhouse, E. W., and Burden, J. L. (1977). Structure-activity relationships of corticosteroid feedback at the hypothalamic level. J. Endocrinol. 74, 415–424. doi: 10.1677/joe.0.0740415
Kalra, S. P., Dube, M. G., and Kalra, P. S. (1988). Continuous intraventricular infusion of neuropeptide Y evokes episodic food intake in satiated female rats: effects of adrenalectomy and cholecystokinin. Peptides 9, 723–728. doi: 10.1016/0196-978190112-X
Kalra, S. P., Dube, M. G., Sahu, A., Phelps, C. P., and Kalra, P. S. (1991). Neuropeptide Y secretion increases in the paraventricular nucleus in association with increased appetite for food. Proc. Natl. Acad. Sci. U.S.A. 88, 10931–10935. doi: 10.1073/pnas.88.23.10931
Keeney, A., Jessop, D. S., Harbuz, M. S., Marsden, C. A., Hogg, S., and Blackburn-Munro, R. E. (2006). Differential effects of acute and chronic social defeat stress on hypothalamic-pituitary-adrenal axis function and hippocampal serotonin release in mice. J. Neuroendocrinol. 18, 330–338. doi: 10.1111/j.1365-2826.2006.01422.x
Kendler, K. S., Gardner, C. O., and Prescott, C. A. (2002). Toward a comprehensive developmental model for major depression in women. Am. J. Psychiatry 159, 1133–1145. doi: 10.1176/appi.ajp.159.7.1133
Koolhaas, J. M., Meerlo, P., De Boer, S. F., Strubbe, J. H., and Bohus, B. (1997). The temporal dynamics of the stress response. Neurosci. Biobehav. Rev. 21, 775–782. doi: 10.1016/S0149-763400057-7
Krishnan, V., Han, M. H., Graham, D. L., Berton, O., Renthal, W., Russo, S. J., et al. (2007). Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131, 391–404. doi: 10.1016/j.cell.2007.09.018
Kristensen, P., Judge, M. E., Thim, L., Ribel, U., Christjansen, K. N., Wulff, B. S., et al. (1998). Hypothalamic CART is a new anorectic peptide regulated by leptin. Nature 393, 72–76. doi: 10.1038/29993
Kristenssson, E., Sundqvist, M., Astin, M., Kjerling, M., Mattsson, H., Dornonville De La Cour, C., et al. (2006). Acute psychological stress raises plasma ghrelin in the rat. Regul. Pept. 134, 114–117. doi: 10.1016/j.regpep.2006.02.003
Krotkiewski, M., Kral, J. G., and Karlsson, J. (1980). Effects of castration and testosterone substitution on body composition and muscle metabolism in rats. Acta Physiol. Scand. 109, 233–237. doi: 10.1111/j.1748-1716.1980.tb06592.x
Krukoff, T. L. (1998). Central regulation of autonomic function: no brakes. Clin. Exp. Pharmacol. Physiol. 25, 474–478. doi: 10.1111/j.1440-1681.1998.tb02238.x
Kuo, L. E., Kitlinska, J. B., Tilan, J. U., Li, L., Baker, S. B., Johnson, M. D., et al. (2007). Neuropeptide Y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat. Med. 13, 803–811. doi: 10.1038/nm1611
Kuperman, Y., Issler, O., Regev, L., Musseri, I., Navon, I., Neufeld-Cohen, A., et al. (2010). Perifornical Urocortin-3 mediates the link between stress-induced anxiety and energy homeostasis. Proc. Natl. Acad. Sci. U.S.A. 107, 8393–8398. doi: 10.1073/pnas.1003969107
La Fleur, S. E. (2006). The effects of glucocorticoids on feeding behavior in rats. Physiol. Behav. 89, 110–114. doi: 10.1016/j.physbeh.2006.01.028
Laitinen, J., Ek, E., and Sovio, U. (2002). Stress-related eating and drinking behavior and body mass index and predictors of this behavior. Prev. Med. 34, 29–39. doi: 10.1006/pmed.2001.0948
Larsen, P. J., Vrang, N., Petersen, P. C., and Kristensen, P. (2000). Chronic intracerebroventricular administration of recombinant CART(42-89) peptide inhibits and causes weight loss in lean and obese Zucker (fa/fa) rats. Obes. Res. 8, 590–596. doi: 10.1038/oby.2000.76
Levine, A. S., and Morley, J. E. (1981). Stress-induced eating in rats. Am. J. Physiol. 241, R72–R76.
Liu, J., Garza, J. C., Li, W., and Lu, X. Y. (2013). Melanocortin-4 receptor in the medial amygdala regulates emotional stress-induced anxiety-like behaviour, anorexia and corticosterone secretion. Int. J. Neuropsychopharmacol. 16, 105–120. doi: 10.1017/S146114571100174X
Lutter, M., Krishnan, V., Russo, S. J., Jung, S., McClung, C. A., and Nestler, E. J. (2008a). Orexin signaling mediates the antidepressant-like effect of calorie restriction. J. Neurosci. 28, 3071–3075. doi: 10.1523/JNEUROSCI.5584-07.2008
Lutter, M., Sakata, I., Osborne-Lawrence, S., Rovinsky, S. A., Anderson, J. G., Jung, S., et al. (2008b). The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat. Neurosci. 11, 752–753. doi: 10.1038/nn.2139
Mahmoud, S. N., Scaccianoce, S., Scraggs, P. R., Nicholson, S. A., Gillham, B., and Jones, M. T. (1984). Characteristics of corticosteroid inhibition of adrenocorticotrophin release from the anterior pituitary gland of the rat. J. Endocrinol. 102, 33–42. doi: 10.1677/joe.0.1020033
Makimura, H., Mizuno, T. M., Roberts, J., Silverstein, J., Beasley, J., and Mobbs, C. V. (2000). Adrenalectomy reverses obese phenotype and restores hypothalamic melanocortin tone in leptin-deficient ob/ob mice. Diabetes 49, 1917–1923. doi: 10.2337/diabetes.49.11.1917
Mangos, G. J., Walker, B. R., Kelly, J. J., Lawson, J. A., Webb, D. J., and Whitworth, J. A. (2000). Cortisol inhibits cholinergic vasodilation in the human forearm. Am. J. Hypertens. 13, 1155–1160. doi: 10.1016/S0895-706101201-2
Maniam, J., and Morris, M. J. (2012). The link between stress and feeding behaviour. Neuropharmacology 63, 97–110. doi: 10.1016/j.neuropharm.2012.04.017
Marcus, S. M., Young, E. A., Kerber, K. B., Kornstein, S., Farabaugh, A. H., Mitchell, J., et al. (2005). Gender differences in depression: findings from the STAR*D study. J. Affect. Disord. 87, 141–150. doi: 10.1016/j.jad.2004.09.008
Margolis, E. B., Lock, H., Hjelmstad, G. O., and Fields, H. L. (2006). The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons. J. Physiol. 577, 907–924. doi: 10.1113/jphysiol.2006.117069
Marin Bivens, C. L., Thomas, W. J., and Stanley, B. G. (1998). Similar feeding patterns are induced by perifornical neuropeptide Y injection and by food deprivation. Brain Res. 782, 271–280. doi: 10.1016/S0006-899301289-4
Marquez, C., Belda, X., and Armario, A. (2002). Post-stress recovery of pituitary-adrenal hormones and glucose, but not the response during exposure to the stressor, is a marker of stress intensity in highly stressful situations. Brain Res. 926, 181–185. doi: 10.1016/S0006-899303112-2
Marti, O., Marti, J., and Armario, A. (1994). Effects of chronic stress on food intake in rats: influence of stressor intensity and duration of daily exposure. Physiol. Behav. 55, 747–753. doi: 10.1016/0031-938490055-8
McEwen, B. S. (1998). Stress, adaptation, and disease. Allostasis and allostatic load. Ann. N.Y. Acad. Sci. 840, 33–44. doi: 10.1111/j.1749-6632.1998.tb09546.x
McEwen, B. S. (2004). Protection and damage from acute and chronic stress: allostasis and allostatic overload and relevance to the pathophysiology of psychiatric disorders. Ann. N.Y. Acad. Sci. 1032, 1–7. doi: 10.1196/annals.1314.001
McEwen, B. S. (2007). Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol. Rev. 87, 873–904. doi: 10.1152/physrev.00041.2006
McEwen, B. S., and Wingfield, J. C. (2010). What is in a name. Integrating homeostasis, allostasis and stress. Horm. Behav. 57, 105–111. doi: 10.1016/j.yhbeh.2009.09.011
McKittrick, C. R., Blanchard, D. C., Blanchard, R. J., McEwen, B. S., and Sakai, R. R. (1995). Serotonin receptor binding in a colony model of chronic social stress. Biol. Psychiatry 37, 383–393. doi: 10.1016/0006-322300152-S
McKittrick, C. R., Magarinos, A. M., Blanchard, D. C., Blanchard, R. J., McEwen, B. S., and Sakai, R. R. (2000). Chronic social stress reduces dendritic arbors in CA3 of hippocampus and decreases binding to serotonin transporter sites. Synapse 36, 85–94.
Michaud, K., Matheson, K., Kelly, O., and Anisman, H. (2008). Impact of stressors in a natural context on release of cortisol in healthy adult humans: a meta-analysis. Stress 11, 177–197. doi: 10.1080/10253890701727874
Miczek, K. A. (1979). A new test for aggression in rats without aversive stimulation: differential effects of d-amphetamine and cocaine. Psychopharmacology (Berl.) 60, 253–259. doi: 10.1007/BF00426664
Miczek, K. A., Thompson, M. L., and Shuster, L. (1982). Opioid-like analgesia in defeated mice. Science 215, 1520–1522. doi: 10.1126/science.7199758
Miell, J. P., Englaro, P., and Blum, W. F. (1996). Dexamethasone induces an acute and sustained rise in circulating leptin levels in normal human subjects. Horm. Metab. Res. 28, 704–707. doi: 10.1055/s-2007-979882
Mineur, Y. S., Prasol, D. J., Belzung, C., and Crusio, W. E. (2003). Agonistic behavior and unpredictable chronic mild stress in mice. Behav. Genet. 33, 513–519. doi: 10.1023/A:1025770616068
Moles, A., Bartolomucci, A., Garbugino, L., Conti, R., Caprioli, A., Coccurello, R., et al. (2006). Psychosocial stress affects energy balance in mice: modulation by social status. Psychoneuroendocrinology 31, 623–633. doi: 10.1016/j.psyneuen.2006.01.004
Moreau, J. L., Bourson, A., Jenck, F., Martin, J. R., and Mortas, P. (1994). Curative effects of the atypical antidepressant mianserin in the chronic mild stress-induced anhedonia model of depression. J. Psychiatry Neurosci. 19, 51–56.
Morimoto, M., Morita, N., Ozawa, H., Yokoyama, K., and Kawata, M. (1996). Distribution of glucocorticoid receptor immunoreactivity and mRNA in the rat brain: an immunohistochemical and in situ hybridization study. Neurosci. Res. 26, 235–269. doi: 10.1016/S0168-010201105-4
Morita, S., and Miyata, S. (2012). Different vascular permeability between the sensory and secretory circumventricular organs of adult mouse brain. Cell Tissue Res. 349, 589–603. doi: 10.1007/s00441-012-1421-9
Morita, S., and Miyata, S. (2013). Accessibility of low-molecular-mass molecules to the median eminence and arcuate hypothalamic nucleus of adult mouse. Cell Biochem. Funct. doi: 10.1002/cbf.2953. [Epub ahead of print].
Mostyn, A., Keisler, D. H., Webb, R., Stephenson, T., and Symonds, M. E. (2001). The role of leptin in the transition from fetus to neonate. Proc. Nutr. Soc. 60, 187–194. doi: 10.1079/PNS200086
Muscat, R., and Willner, P. (1992). Suppression of sucrose drinking by chronic mild unpredictable stress: a methodological analysis. Neurosci. Biobehav. Rev. 16, 507–517. doi: 10.1016/S0149-763480192-7
Nakazato, M., Murakami, N., Date, Y., Kojima, M., Matsuo, H., Kangawa, K., et al. (2001). A role for ghrelin in the central regulation of feeding. Nature 409, 194–198. doi: 10.1038/35051587
Nieuwenhuizen, A. G., and Rutters, F. (2008). The hypothalamic-pituitary-adrenal-axis in the regulation of energy balance. Physiol. Behav. 94, 169–177. doi: 10.1016/j.physbeh.2007.12.011
Niswender, K. D., and Schwartz, M. W. (2003). Insulin and leptin revisited: adiposity signals with overlapping physiological and intracellular signaling capabilities. Front. Neuroendocrinol. 24, 1–10. doi: 10.1016/S0091-302200105-X
O'Connor, D. B., Jones, F., Conner, M., McMillan, B., and Ferguson, E. (2008). Effects of daily hassles and eating style on eating behavior. Health Psychol. 27, S20–S31. doi: 10.1037/0278-6133.27.1.S20
Ochi, M., Tominaga, K., Tanaka, F., Tanigawa, T., Shiba, M., Watanabe, T., et al. (2008). Effect of chronic stress on gastric emptying and plasma ghrelin levels in rats. Life Sci. 82, 862–868. doi: 10.1016/j.lfs.2008.01.020
Olijslagers, J. E., De Kloet, E. R., Elgersma, Y., Van Woerden, G. M., Joels, M., and Karst, H. (2008). Rapid changes in hippocampal CA1 pyramidal cell function via pre- as well as postsynaptic membrane mineralocorticoid receptors. Eur. J. Neurosci. 27, 2542–2550. doi: 10.1111/j.1460-9568.2008.06220.x
Ottosson, M., Lonnroth, P., Bjorntorp, P., and Eden, S. (2000). Effects of cortisol and growth hormone on lipolysis in human adipose tissue. J. Clin. Endocrinol. Metab. 85, 799–803. doi: 10.1210/jc.85.2.799
Palmiter, R. D. (2007). Is dopamine a physiologically relevant mediator of feeding behavior. Trends Neurosci. 30, 375–381. doi: 10.1016/j.tins.2007.06.004
Parker, R. M., and Herzog, H. (1999). Regional distribution of Y-receptor subtype mRNAs in rat brain. Eur. J. Neurosci. 11, 1431–1448. doi: 10.1046/j.1460-9568.1999.00553.x
Patterson, Z. R., Ducharme, R., Anisman, H., and Abizaid, A. (2010). Altered metabolic and neurochemical responses to chronic unpredictable stressors in ghrelin receptor-deficient mice. Eur. J. Neurosci. 32, 632–639. doi: 10.1111/j.1460-9568.2010.07310.x
Patterson, Z. R., Khazall, R., Mackay, H., Anisman, H., and Abizaid, A. (2013). Central ghrelin signaling mediates the metabolic response of C57BL/6 male mice to chronic social defeat stress. Endocrinology 154, 1080–1091. doi: 10.1210/en.2012-1834
Pecoraro, N., Reyes, F., Gomez, F., Bhargava, A., and Dallman, M. F. (2004). Chronic stress promotes palatable feeding, which reduces signs of stress: feedforward and feedback effects of chronic stress. Endocrinology 145, 3754–3762. doi: 10.1210/en.2004-0305
Peeters, A., Barendregt, J. J., Willekens, F., Mackenbach, J. P., Al Mamun, A., Bonneux, L., et al. (2003). Obesity in adulthood and its consequences for life expectancy: a life-table analysis. Ann. Intern. Med. 138, 24–32. doi: 10.7326/0003-4819-138-1-200301070-00008
Popper, R., Smits, G., Meiselman, H. L., and Hirsch, E. (1989). Eating in combat: a survey of U.S. Marines. Mil. Med. 154, 619–623.
Pothion, S., Bizot, J. C., Trovero, F., and Belzung, C. (2004). Strain differences in sucrose preference and in the consequences of unpredictable chronic mild stress. Behav. Brain Res. 155, 135–146. doi: 10.1016/j.bbr.2004.04.008
Pralong, F. P., Corder, R., and Gaillard, R. C. (1993). The effects of chronic glucocorticoid excess, adrenalectomy and stress on neuropeptide Y in individual rat hypothalamic nuclei. Neuropeptides 25, 223–231. doi: 10.1016/0143-417990107-L
Rabasa, C., Munoz-Abellan, C., Daviu, N., Nadal, R., and Armario, A. (2011). Repeated exposure to immobilization or two different footshock intensities reveals differential adaptation of the hypothalamic-pituitary-adrenal axis. Physiol. Behav. 103, 125–133. doi: 10.1016/j.physbeh.2011.02.022
Radley, J. J., Williams, B., and Sawchenko, P. E. (2008). Noradrenergic innervation of the dorsal medial prefrontal cortex modulates hypothalamo-pituitary-adrenal responses to acute emotional stress. J. Neurosci. 28, 5806–5816. doi: 10.1523/JNEUROSCI.0552-08.2008
Raikkonen, K., Keltikangas-Jarvinen, L., Adlercreutz, H., and Hautanen, A. (1996). Psychosocial stress and the insulin resistance syndrome. Metab. Clin. Exp. 45, 1533–1538. doi: 10.1016/S0026-049590184-5
Ramos, E. J., Meguid, M. M., Campos, A. C., and Coelho, J. C. (2005). Neuropeptide, Y, alpha-melanocyte-stimulating hormone, and monoamines in food intake regulation. Nutrition 21, 269–279. doi: 10.1016/j.nut.2004.06.021
Richardson, N. R., and Gratton, A. (1996). Behavior-relevant changes in nucleus accumbens dopamine transmission elicited by food reinforcement: an electrochemical study in rat. J. Neurosci. 16, 8160–8169.
Rizza, R. A., Mandarino, L. J., and Gerich, J. E. (1982). Cortisol-induced insulin resistance in man: impaired suppression of glucose production and stimulation of glucose utilization due to a postreceptor detect of insulin action. J. Clin. Endocrinol. Metab. 54, 131–138. doi: 10.1210/jcem-54-1-131
Robinson, S., Rainwater, A. J., Hnasko, T. S., and Palmiter, R. D. (2007). Viral restoration of dopamine signaling to the dorsal striatum restores instrumental conditioning to dopamine-deficient mice. Psychopharmacology (Berl.) 191, 567–578. doi: 10.1007/s00213-006-0579-9
Rowland, N. E., and Antelman, S. M. (1976). Stress-induced hyperphagia and obesity in rats: a possible model for understanding human obesity. Science 191, 310–312. doi: 10.1126/science.1246617
Russell, C. D., Petersen, R. N., Rao, S. P., Ricci, M. R., Prasad, A., Zhang, Y., et al. (1998). Leptin expression in adipose tissue from obese humans: depot-specific regulation by insulin and dexamethasone. Am. J. Physiol. 275, E507–E515.
Saito, M., and Bray, G. A. (1984). Adrenalectomy and food restriction in the genetically obese (ob/ob) mouse. Am. J. Physiol. 246, R20–R25.
Sapolsky, R. M., Krey, L. C., and McEwen, B. S. (1984). Glucocorticoid-sensitive hippocampal neurons are involved in terminating the adrenocortical stress response. Proc. Natl. Acad. Sci. U.S.A. 81, 6174–6177. doi: 10.1073/pnas.81.19.6174
Sapolsky, R. M., Romero, L. M., and Munck, A. U. (2000). How do glucocorticoids influence stress responses. Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr. Rev. 21, 55–89. doi: 10.1210/er.21.1.55
Savontaus, E., Conwell, I. M., and Wardlaw, S. L. (2002). Effects of adrenalectomy on AGRP, POMC, NPY and CART gene expression in the basal hypothalamus of fed and fasted rats. Brain Res. 958, 130–138. doi: 10.1016/S0006-899303674-0
Schmidt, M. V., Scharf, S. H., Liebl, C., Harbich, D., Mayer, B., Holsboer, F., et al. (2010). A novel chronic social stress paradigm in female mice. Horm. Behav. 57, 415–420. doi: 10.1016/j.yhbeh.2010.01.010
Schwartz, M. W., Woods, S. C., Porte, D. Jr., Seeley, R. J., and Baskin, D. G. (2000). Central nervous system control of food intake. Nature 404, 661–671.
Seoane, L. M., Lopez, M., Tovar, S., Casanueva, F. F., Senaris, R., and Dieguez, C. (2003). Agouti-related peptide, neuropeptide Y, and somatostatin-producing neurons are targets for ghrelin actions in the rat hypothalamus. Endocrinology 144, 544–551. doi: 10.1210/en.2002-220795
Sgoifo, A., Koolhaas, J., De Boer, S., Musso, E., Stilli, D., Buwalda, B., et al. (1999). Social stress, autonomic neural activation, and cardiac activity in rats. Neurosci. Biobehav. Rev. 23, 915–923. doi: 10.1016/S0149-763400025-1
Shors, T. J., Mathew, J., Sisti, H. M., Edgecomb, C., Beckoff, S., and Dalla, C. (2007). Neurogenesis and helplessness are mediated by controllability in males but not in females. Biol. Psychiatry 62, 487–495. doi: 10.1016/j.biopsych.2006.10.033
Silva, R. C., Cruz, A. P., Avanzi, V., Landeira-Fernandez, J., and Brandao, M. L. (2002). Distinct contributions of median raphe nucleus to contextual fear conditioning and fear-potentiated startle. Neural Plast. 9, 233–247. doi: 10.1155/NP.2002.233
Simerly, R. (2006). Feeding signals and drugs meet in the midbrain. Nat. Med. 12, 1244–1246. doi: 10.1038/nm1106-1244
Smith, S. M., and Vale, W. W. (2006). The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin. Neurosci. 8, 383–395.
Solomon, M. B., Foster, M. T., Bartness, T. J., and Huhman, K. L. (2007). Social defeat and footshock increase body mass and adiposity in male Syrian hamsters. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R283–R290. doi: 10.1152/ajpregu.00330.2006
Spencer, S. J., Xu, L., Clarke, M. A., Lemus, M., Reichenbach, A., Geenen, B., et al. (2012). Ghrelin regulates the hypothalamic-pituitary-adrenal axis and restricts anxiety after acute stress. Biol. Psychiatry 72, 457–465. doi: 10.1016/j.biopsych.2012.03.010
Sternson, S. M., Shepherd, G. M., and Friedman, J. M. (2005). Topographic mapping of VMH –> arcuate nucleus microcircuits and their reorganization by fasting. Nat. Neurosci. 8, 1356–1363. doi: 10.1038/nn1550
Stohr, T., Szuran, T., Welzl, H., Pliska, V., Feldon, J., and Pryce, C. R. (2000). Lewis/Fischer rat strain differences in endocrine and behavioural responses to environmental challenge. Pharmacol. Biochem. Behav. 67, 809–819. doi: 10.1016/S0091-305700426-3
Sujoldzic, A., and De Lucia, A. (2007). A cross-cultural study of adolescents–BMI, body image and psychological well-being. Coll. Antropol. 31, 123–130.
Tamashiro, K. L., Hegeman, M. A., and Sakai, R. R. (2006). Chronic social stress in a changing dietary environment. Physiol. Behav. 89, 536–542. doi: 10.1016/j.physbeh.2006.05.026
Tamashiro, K. L., Hegeman, M. A., Nguyen, M. M., Melhorn, S. J., Ma, L. Y., Woods, S. C., et al. (2007a). Dynamic body weight and body composition changes in response to subordination stress. Physiol. Behav. 91, 440–448. doi: 10.1016/j.physbeh.2007.04.004
Tamashiro, K. L., Nguyen, M. M., Ostrander, M. M., Gardner, S. R., Ma, L. Y., Woods, S. C., et al. (2007b). Social stress and recovery: implications for body weight and body composition. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R1864–R1874. doi: 10.1152/ajpregu.00371.2007
Tamashiro, K. L., Nguyen, M. M., Fujikawa, T., Xu, T., Yun Ma, L., Woods, S. C., and Sakai, R. R. (2004). Metabolic and endocrine consequences of social stress in a visible burrow system. Physiol. Behav. 80, 683–693. doi: 10.1016/j.physbeh.2003.12.002
Taylor, S. E., Klein, L. C., Lewis, B. P., Gruenewald, T. L., Gurung, R. A., and Updegraff, J. A. (2000). Biobehavioral responses to stress in females: tend-and-befriend, not fight-or-flight. Psychol. Rev. 107, 411–429. doi: 10.1037/0033-295X.107.3.411
Theander-Carrillo, C., Wiedmer, P., Cettour-Rose, P., Nogueiras, R., Perez-Tilve, D., Pfluger, P., et al. (2006). Ghrelin action in the brain controls adipocyte metabolism. J. Clin. Invest. 116, 1983–1993. doi: 10.1172/JCI25811
Tomiyama, A. J., Dallman, M. F., and Epel, E. S. (2011). Comfort food is comforting to those most stressed: evidence of the chronic stress response network in high stress women. Psychoneuroendocrinology 36, 1513–1519. doi: 10.1016/j.psyneuen.2011.04.005
Torres, S. J., and Nowson, C. A. (2007). Relationship between stress, eating behavior, and obesity. Nutrition 23, 887–894. doi: 10.1016/j.nut.2007.08.008
Treece, B. R., Ritter, R. C., and Burns, G. A. (2000). Lesions of the dorsal vagal complex abolish increases in meal size induced by NMDA receptor blockade. Brain Res. 872, 37–43. doi: 10.1016/S0006-899302432-X
Tschop, M., Smiley, D. L., and Heiman, M. L. (2000). Ghrelin induces adiposity in rodents. Nature 407, 908–913. doi: 10.1038/35038090
Ulrich-Lai, Y. M., and Herman, J. P. (2009). Neural regulation of endocrine and autonomic stress responses. Nat. Rev. Neurosci. 10, 397–409. doi: 10.1038/nrn2647
Valles, A., Marti, O., Garcia, A., and Armario, A. (2000). Single exposure to stressors causes long-lasting, stress-dependent reduction of food intake in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R1138–R1144.
Velloso, L. A., Araujo, E. P., and De Souza, C. T. (2008). Diet-induced inflammation of the hypothalamus in obesity. Neuroimmunomodulation 15, 189–193. doi: 10.1159/000153423
Vertes, R. P., Fortin, W. J., and Crane, A. M. (1999). Projections of the median raphe nucleus in the rat. J. Comp. Neurol. 407, 555–582.
Volkow, N. D., and Wise, R. A. (2005). How can drug addiction help us understand obesity. Nat. Neurosci. 8, 555–560. doi: 10.1038/nn1452
Wahlestedt, C., Yanaihara, N., and Hakanson, R. (1986). Evidence for different pre-and post-junctional receptors for neuropeptide Y and related peptides. Regul. Pept. 13, 307–318. doi: 10.1016/0167-011590048-0
Wang, G. J., Volkow, N. D., and Fowler, J. S. (2002). The role of dopamine in motivation for food in humans: implications for obesity. Expert Opin. Ther. Targets 6, 601–609. doi: 10.1517/14728222.6.5.601
Wang, G. J., Volkow, N. D., Thanos, P. K., and Fowler, J. S. (2004). Similarity between obesity and drug addiction as assessed by neurofunctional imaging: a concept review. J. Addict. Dis. 23, 39–53. doi: 10.1300/J069v23n03_04
Wang, S. X., Chen, J. X., Yue, G. X., Bai, M. H., Kou, M. J., and Jin, Z. Y. (2012). Xiaoyaosan decoction regulates changes in neuropeptide y and leptin receptor in the rat arcuate nucleus after chronic immobilization stress. Evid. Based. Complement. Alternat. Med. 2012, 381278.
Wansink, B., Cheney, M. M., and Chan, N. (2003). Exploring comfort food preferences across age and gender. Physiol. Behav. 79, 739–747. doi: 10.1016/S0031-938400203-8
Weaver, J. U. (2008). Classical endocrine diseases causing obesity. Front. Horm. Res. 36, 212–228. doi: 10.1159/000115367
Weaver, J. U., Kopelman, P. G., McLoughlin, L., Forsling, M. L., and Grossman, A. (1993). Hyperactivity of the hypothalamo-pituitary-adrenal axis in obesity: a study of ACTH, AVP, beta-lipotrophin and cortisol responses to insulin-induced hypoglycaemia. Clin. Endocrinol. (Oxf.) 39, 345–350. doi: 10.1111/j.1365-2265.1993.tb02375.x
Westenbroek, C., Ter Horst, G. J., Roos, M. H., Kuipers, S. D., Trentani, A., and Den Boer, J. A. (2003). Gender-specific effects of social housing in rats after chronic mild stress exposure. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 21–30. doi: 10.1016/S0278-584600310-X
White, J. D., and Kershaw, M. (1990). Increased hypothalamic neuropeptide Y expression following food deprivation. Mol. Cell. Neurosci. 1, 41–48. doi: 10.1016/1044-743190040-B
Wilding, J. P., Gilbey, S. G., Bailey, C. J., Batt, R. A., Williams, G., Ghatei, M. A., et al. (1993). Increased neuropeptide-Y messenger ribonucleic acid (mRNA) and decreased neurotensin mRNA in the hypothalamus of the obese (ob/ob) mouse. Endocrinology 132, 1939–1944. doi: 10.1210/en.132.5.1939
Williams, L. B., Fawcett, R. L., Waechter, A. S., Zhang, P., Kogon, B. E., Jones, R., et al. (2000). Leptin production in adipocytes from morbidly obese subjects: stimulation by dexamethasone, inhibition with troglitazone, and influence of gender. J. Clin. Endocrinol. Metab. 85, 2678–2684. doi: 10.1210/jc.85.8.2678
Willner, P. (1991). Animal models as simulations of depression. Trends Pharmacol. Sci. 12, 131–136. doi: 10.1016/0165-614790529-2
Willner, P. (1997). Validity, reliability and utility of the chronic mild stress model of depression: a 10-year review and evaluation. Psychopharmacology (Berl.) 134, 319–329. doi: 10.1007/s002130050456
Willner, P., Muscat, R., and Papp, M. (1992). Chronic mild stress-induced anhedonia: a realistic animal model of depression. Neurosci. Biobehav. Rev. 16, 525–534. doi: 10.1016/S0149-763480194-0
Wilson, M. E., Fisher, J., Fischer, A., Lee, V., Harris, R. B., and Bartness, T. J. (2008). Quantifying food intake in socially housed monkeys: social status effects on caloric consumption. Physiol. Behav. 94, 586–594. doi: 10.1016/j.physbeh.2008.03.019
Wise, R. A. (2004). Dopamine, learning and motivation. Nat. Rev. Neurosci. 5, 483–494. doi: 10.1038/nrn1406
Wise, R. A. (2006). The parsing of food reward. Am. J. Physiol. Regul. Integr. Comp. Physiol. 291, R1234–R1235. doi: 10.1152/ajpregu.00443.2006
Woulfe, J. M., Flumerfelt, B. A., and Hrycyshyn, A. W. (1990). Efferent connections of the A1 noradrenergic cell group: a DBH immunohistochemical and PHA-L anterograde tracing study. Exp. Neurol. 109, 308–322. doi: 10.1016/S0014-488680022-6
Wu, Q., Clark, M. S., and Palmiter, R. D. (2012). Deciphering a neuronal circuit that mediates appetite. Nature 483, 594–597. doi: 10.1038/nature10899
Yalcin, I., Belzung, C., and Surget, A. (2008). Mouse strain differences in the unpredictable chronic mild stress: a four-antidepressant survey. Behav. Brain Res. 193, 140–143. doi: 10.1016/j.bbr.2008.04.021
Young, E., and Korszun, A. (2010). Sex, trauma, stress hormones and depression. Mol. Psychiatry 15, 23–28. doi: 10.1038/mp.2009.94
Zakrzewska, K. E., Cusin, I., Stricker-Krongrad, A., Boss, O., Ricquier, D., Jeanrenaud, B., et al. (1999a). Induction of obesity and hyperleptinemia by central glucocorticoid infusion in the rat. Diabetes 48, 365–370. doi: 10.2337/diabetes.48.2.365
Keywords: stress, obesity, animal models, hormones, feeding behavior, hypothalamus
Citation: Patterson ZR and Abizaid A (2013) Stress induced obesity: lessons from rodent models of stress. Front. Neurosci. 7:130. doi: 10.3389/fnins.2013.00130
Received: 29 May 2013; Paper pending published: 13 June 2013;
Accepted: 08 July 2013; Published online: 24 July 2013.
Edited by:
Zane B. Andrews, Monash University, AustraliaReviewed by:
Alessandro Bartolomucci, University of Minnesota, USAYael Kuperman, Weizmann Instituf of Science, Israel
Copyright © 2013 Patterson and Abizaid. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Zachary R. Patterson, Department of Neuroscience, Carleton University, 1125 Colonel By Drive, Life Sciences Research Building, Ottawa, ON K1S 5B6, Canada e-mail:emFjay5wYXR0ZXJzb25AZ21haWwuY29t